

Abstract
Activation energies for the individual steps of secretory and viral fusion are reported to be large [Oberhauser, A. F., Monck, J. R. & Fernandez, J. M. (1992) Biophys. J. 61, 800–809; Clague, M. J., Schoch, C., Zech, L. & Blumenthal, R. (1990) Biochemistry 29, 1303–1308]. Understanding the cause for these large activation energies is crucial to defining the mechanisms of these two types of biological membrane fusion. We showed recently that the fusion of protein-free model lipid bilayers mimics the sequence of steps observed during secretory and viral fusion, suggesting that these processes may involve common lipid, rather than protein, rearrangements. To test for this possibility, we determined the activation energies for the three steps that we were able to distinguish as contributing to the fusion of protein-free model lipid bilayers. Activation energies for lipid rearrangements associated with formation of the reversible first intermediate, with conversion of this to a semi-stable second intermediate, and with irreversible fusion pore formation were 37 kcal/mol, 27 kcal/mol, and 22 kcal/mol, respectively. The first and last of these were comparable to the activation energies observed for membrane lipid exchange (42 kcal/mol) during viral fusion and for the rate of fusion pore opening during secretory granule release (23 kcal/mol). This striking similarity suggests strongly that the basic molecular processes involved in secretory and viral fusion involve a set of lipid molecule rearrangements that also are involved in model membrane fusion.
Membrane fusion is an essential event during viral infection and during such key eukaryotic cell functions as compartmentalization, endocytosis, secretion, and synaptic transmission (1). Despite the importance of this process, little is known about the molecular mechanism(s) by which cell membrane fusion is accomplished. There are three primary views of the mechanism of pore formation during secretory and viral fusion. In one widely held view (2), the initial, transient pore is formed by a ring of protein, and this expands slowly by recruiting lipid so as to break the protein ring and form an irreversible fusion pore (FP) comprised primarily of lipid. In another view (3), the initial FP consists of a protein–lipid complex that transforms slowly until the fusion proteins dissociate from the complex to form an irreversible lipidic pore. In these two views, the initial pore that is formed is primarily a proteinaceous structure. In a third view (4, 5), the initial pore is a transient or fluctuating lipid junction between two stressed and closely opposed lipid bilayers held in place by a “protein scaffold.” This transient pore fluctuates around a certain size before either expanding irreversibly or closing. In this view, the transient pore is lipidic in nature. There has as yet been no means of distinguishing experimentally between these three hypotheses. As an approach to this problem, at least two laboratories have developed protein-free, model membrane systems with which to define the mechanism of lipid bilayer fusion (6, 7). In one of these systems, we have reported that the time course of fusion of highly curved phospholipid vesicles in the presence of poly(ethylene glycol) (PEG) (6) parallels that of secretory (8–10) and viral (11) fusion. This suggested the hypothesis that lipid bilayer structural changes occurring during PEG-mediated vesicle fusion mimic the membrane structural changes associated with secretory and viral fusion (6).
When phospholipid vesicles are treated with PEG, they aggregate and their bilayer membranes come into near molecular contact (12). Reduced lipid packing density in the contacting bilayer leaflets of the aggregated vesicles is then necessary and sufficient to induce vesicle fusion (13). Reduced lipid packing density can be achieved by high bilayer curvature (14–16), acyl chain unsaturation within fusion-prone small unilamellar vesicles (SUVs) (17), very small surface mole fractions (0.5 mol%) of certain amphipaths (16), and imperfect outer leaflet lipid packing (13, 18).
We have proposed a three-step mechanism for PEG-mediated phospholipid vesicle fusion (6), as shown in Fig. 1. The first step was shown (6) to involve formation of a reversible intermediate (I1), depicted in Fig. 1 as consisting of a dynamic mixture of two unstable forms, a hemi-fused “stalk” (19) and a transient, small pore (6, 7). This unstable intermediate reverts to unfused vesicles when PEG is diluted (6). If inter-bilayer contact is maintained, intermediate I1 matures in a second step to the semi-stable intermediate (I2) envisioned as a hemi-fused “septum” (third down from top left) (6, 19). The existence of a second intermediate was established clearly by demonstrating that dilution of PEG at increasing times led to reduced recovery of original SUV and increased observation of fusion product (6). The third step involves irreversible formation of a FP, presumed here to result from spontaneous “popping” of the septum once it grows to a point where it is destabilized by the large, unfavorable annulus surrounding it. Rapid, irreversible pore formation well after observation of transient pores is also a feature of secretory vesicle (8) and viral protein-mediated cell membrane (20) fusion. To follow the time courses of these three steps, we made measurements of membrane lipid and trapped aqueous proton redistribution between fusing vesicles (6). Here, we report the rates and activation energies of each of the three steps of PEG-mediated phospholipid vesicle fusion and show that the first and last of these steps have activation energies that correspond to those reported for the analogous kinetic events recorded for viral and secretory vesicle fusion.
Figure 1.

Schematic diagram of proposed sequential steps leading to phospholipid vesicle fusion and biomembrane fusion. Outer monolayers brought to close contact by PEG are destabilized by high bilayer curvature induced by sonication. For protein-mediated biomembrane fusion, protein scaffolds may allow for close bilayer contact and protein conformational changes may provide local curvature stress (see Discussion). In the first step, a dynamic mixture of stalk (second down from left) and transient pore (top right) structures forms (I1) and accounts for outer-leaflet lipid transfer and transient proton transfer. This transient structure is presumed to account for the flickering pores seen by electrophysiological methods during the very early stages of both planar membrane and biomembrane fusion (see Discussion). The unstable intermediate I1 matures to the semi-stable intermediate I2 (third down from top left) that we show as a single bilayer septum separating the two aqueous compartments. This conversion accounts for the observed delay in both lipid transfer and proton transfer. Disintegration of the mature septum produces the FP (FP; bottom left) and allows complete lipid and proton transfer. Dilation of the FP (bottom right) has been discussed (but not unequivocally demonstrated) for biomembranes (37) and has not been demonstrated for model membranes. Phospholipid symbols with filled heads represent probe lipids that report lipid transfer during fusion. Dilution of probe lipids during inter-vesicle lipid transfer resulted in an increase in probe fluorescence lifetime (see Fig. 2). HPTS was used to measure proton transfer between two compartments at different pHs. Proton transfer was observed during both transient pore and FP formation, whereas mixing of larger molecules, Tb3+ and dipicolinic acid, was observed only during FP formation (6). Rate constants for the formation of intermediate I1, conversion to I2, and FP formation were defined as k1, k2, and 1/σ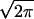 respectively (see Materials and Methods).
respectively (see Materials and Methods).
MATERIALS AND METHODS
Vesicle Preparation and Fluorescence Fusion Assays.
SUVs were prepared as described (6) by using a mixture (85/15 mol%) of 1,2-dioleoyl-sn-phosphatidylcholine and 1,2-dilinolenoyl-sn-phosphatidylcholine purchased from Avanti Polar Lipids. SUVs were 450 Å in diameter, as determined by quasi-elastic light scattering (6). Poly(ethylene glycol) (PEG 8000) was purchased from Fisher Scientific and further purified to remove impurities including ions, peroxides, aldehyde, and anti-oxidant (21). Fluorescence probes, 1-palmitoyl-2-[[[2-[4-(phenyl-trans-1, 3, 5-hexatrienyl)phenyl]ethyl]oxyl]carbonyl]-3-sn-phosphatidylcholine (DPHpPC), and 8-hydroxypyrene-1,3,6-trisulfonic acid (HPTS) were purchased from Molecular Probes and used for monitoring membrane lipid mixing and proton redistribution, respectively. Rapid mixing of vesicles and PEG, to a final lipid concentration of 0.5–0.6 mM and a PEG concentration of 17.5 wt%, was accomplished by using a pneumatically driven stopped-flow accessory (Model RX1000 from Applied Photophysics, Surrey, U.K.). The temperature was controlled by water circulation through both the fluorometer chamber and the stopped-flow device.
Movement of lipids.
The movement of lipids between PEG-aggregated SUVs was detected by a change in the fluorescence lifetime of DPHpPC (22). This fluorescent lipid displays a much smaller fluorescent lifetime when incorporated into vesicles at high surface concentration than it reports at dilute surface concentrations (23). Thus, this assay reports movement of DPHpPC from probe-rich to probe-free vesicles as an increase in average lifetime (22). It has the advantage of allowing a direct measure of the probe concentrations in different membrane compartments in a sample and thus can distinguish between true membrane fusion and mixing of lipids in contacting membrane outer leaflets (24). The fluorescence lifetimes of DPHpPC were measured by using the UV multiline (351.1–363.8 nm) from a Coherent Inova 90 argon-ion laser (Coherent Auburn Group, Auburn, CA). Phase shifts and modulation ratios used to calculate the average fluorescence lifetimes were collected at 30 frequencies (with a base frequency of 4 MHz) on an SLM 48000 MHF spectrofluorometer (Spectronic, Westbury, NY) by using a 25- to 50-Hz correlation frequency, 0.1364- to 0.5-s analysis window, and 30–50 acquisition average.
Movement of protons.
Movement of protons between fusing SUVs was followed by trapping HPTS in one SUV population prepared at pH 8 and fusing these with vesicles prepared at pH 5.5. A decrease in HPTS fluorescence intensity indicated movement of protons between these two vesicular compartments (6). HPTS fluorescence was excited with 460 nm of light from a 450-W Xenon short arc lamp (Ushio, Japan). A full description of these methods is reported elsewhere (6).
Kinetic Model.
Each of the steps of PEG-mediated fusion is an average of fusion events involving an ensemble of individual vesicle pairs. Thus, we treated each step as a kinetic event with a start time described by a Gaussian distribution. A single-exponential with a unique start time at t = 0 provided a good description of the first step, formation of I1. The start time for the second step, formation of I2, distributed as a narrow Gaussian whose mean value could not be precisely fixed, although it was always large enough that the first step was essentially complete before the second step began. Thus, we simply fixed the delay between the start times for steps 1 and 2 as 3/k1 so that step 2 began after 95% of step 1 was complete. The third step, FP formation, was treated as very rapid septum “popping” that was distributed randomly about a mean “pop time” (t3) with a Gaussian distribution width of σ. The integral of the Gaussian distribution [approximated by the Gaussian cumulative distribution or “error” function (25)] was used to obtain the probability of FP opening (to which fluorescence signals would be proportional) as a function of time after initiation by addition of PEG. The rate constant for the third step was determined from the peak height (
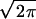 ) of the distribution, which is the frequency of FP opening. The detailed functional forms used to determine the rate constant for each step by fitting to membrane lipid mixing (Eq. 1) and proton transfer (Eq. 2) data were:
) of the distribution, which is the frequency of FP opening. The detailed functional forms used to determine the rate constant for each step by fitting to membrane lipid mixing (Eq. 1) and proton transfer (Eq. 2) data were:
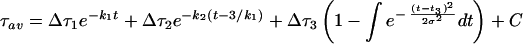 |
1 |
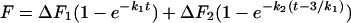 |
2 |
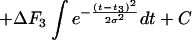 |
where Δτ1 (ΔF1), Δτ2 (ΔF2), and Δτ3 (ΔF3) are the fluorescence lifetime (or intensity) changes associated with formation of I1, I2, and FP, respectively. The parameter C is the fluorescence lifetime at time zero just before PEG addition for lipid mixing data and is the fluorescence intensity after complete fusion (t = ∞) for proton transfer data. These expressions were fit by nonlinear least-squares analysis to the data obtained at four temperatures to obtain estimates of k1, k2, σ, and t3. All three terms of Eqs. 1 and 2 were required to obtain an adequate description of the data sets. Values of the Δτs, ΔFs, and Cs also were treated as parameters but, except for Δτ2 and ΔF2, were assigned largely by inspection. There were, therefore, really only six parameters, including Δτ2 and ΔF2, that could be adjusted to fit both sets of data. Either set of data could be described well by adjusting five of these. The fact that only one set of kinetic and Gaussian parameters fit both curves so well lends credence to the parameter values obtained. The fluorescence intensity data sets were limited to every fifth point to reduce the computing time needed to obtain a fit.
RESULTS AND DISCUSSION
Kinetics of Fusion Between Pure Lipid Bilayers.
Previously, we showed that the individual steps of the fusion process could be continuously monitored by lipid mixing and proton transfer between PEG-aggregated vesicles (6). The time courses of fluorescence changes reporting lipid (Fig. 2A) and proton (Fig. 2B) movements during vesicle fusion are shown in Fig. 2 for four temperatures. The initial increase/decrease corresponds to outer leaflet lipid mixing/proton transfer due to the formation of intermediate I1 (6). The delay after the initial increase/decrease is presumed to be due to a slow lipid rearrangement associated with formation of intermediate I2. A broad sigmoidal increase/decrease in signal occurred after the slow process had proceeded for some time. This represented inner leaflet mixing and complete contents mixing via a FP and corresponded to irreversible formation of fused product (FP) (6). The time courses of lipid mixing and proton transfer are parallel and are well described by a common, three-step, sequential process. Increasing temperature in the range of 15–35°C accelerated each step of the process but did not alter this parallelism between proton and lipid movement.
Figure 2.

Time courses of lipid and proton transfer during PEG-mediated vesicle fusion at different temperatures. PEG and vesicles were mixed at time zero. Lipid mixing (A) and proton transfer (B) were monitored by an increase in DPHpPC fluorescence lifetime and a decrease in HPTS fluorescence intensity, respectively (6). The initial lipid mixing/proton transfer indicates outer leaflet mixing and initial pore formation caused by the formation of intermediate I1. The delay suggests closing of initial pores and a transition to more stable intermediate I2. A slow lipid mixing and proton transfer during the delay was discussed in terms of lipid mass movement from outer to inner leaflet and occasional transient pore formation through the septum (see Discussion). Formation of FP causes the final step of lipid mixing/proton transfer: inner leaflet mixing, and complete proton transfer. The origins of each curve have been displaced to display data obtained at all four temperatures on the same plot. Each data set was fit to the kinetic model described in Materials and Methods, and the fitted curves are shown as solid lines. The activation energies (see Fig. 3) and rate constants at 35°C are listed for each step in Table 1.
The rates of each step were obtained by fitting the data to the 3-step model described in Materials and Methods (curves, Fig. 2). Physically, the model corresponds to rapid, first-order formation of a transient intermediate I1, which slowly converts in a first-order fashion to an irreversible intermediate I2, which, once it is mature, spontaneously degrades or “pops” to form a FP. The time at which “pop” occurs distributes as a Gaussian. The rate constants resulting from fitting our data obtained at 35°C to this model were 2.16–2.43 s−1 for the formation of I1, 0.031–0.044 s−1 for the conversion of I1 to I2, and 0.017–0.020 s−1 for the formation of FP. The rate constants determined at 35°C by using lipid mixing data were very close to those determined by using proton transfer data for all three steps (see Table 1). For the second process, however, the rates of lipid movement and proton movement diverged somewhat at low temperatures. This could indicate that lipid mixing and proton transfer were mechanistically dependent on some common set of molecular events in the cases of the first and third steps, although this might not be the case for the second step.
Table 1.
Rates and activation energies for individual steps of PEG-mediated vesicle and biomembrane fusion
| Process | Rate constant, s−1 | Activation energy, kcal/mol | ||
|---|---|---|---|---|
| Formation of I1 in vesicle fusion | 2.43 (LM) | 2.16 (PT) | 37 (LM) | 35 (PT) |
| Conversion of I1 to I2 in vesicle fusion | 0.044 (LM) | 0.031 (PT) | 27 (LM) | 33 (PT) |
| Formation of FP in vesicle fusion | 0.020* (LM) | 0.017* (PT) | 21 (LM) | 22 (PT) |
| Lipid mixing in viral fusion† | 1.77‡ (pH 6.05) 4.21 (pH 5.87) 20.6‡ (pH 5.59) | 42 | ||
| Lipid flip-flop§ | ≈3.53 × 10−5 | 24 | ||
| Pore opening in secretory fusion¶ | ≈0.010* | 23 | ||
The rate constants listed for PEG-mediated SUV fusion were determined at 35°C. Rates for biomembrane fusion and flip-flop were interpolated from published data to estimate rates at 35°C.
Fraction of total pores formed per second.
Fusion between vesicular stomatitis virus and erythrocyte ghost (32).
Rates calculated from values reported for 37°C (2.75 and 32.1 s−1) using the activation energy measured at pH 5.85.
For dimyristoylphosphatidylcholine SUV’s in the fluid phase (26).
Fusion between secretory granules and mouse mast cell plasma membrane (10).
The Arrhenius plots shown in Fig. 3 yielded the activation energies for each step, which are listed in Table 1. Both lipid mixing and proton transfer data provided remarkably large activation energies for the first step. The fact that these were nearly identical supports our earlier hypothesis that intermediate I1 is a dynamic equilibrium between a “stalk” and a transient pore (ref. 6; Fig. 1). The activation energy for lipid rearrangements during the second step (27 kcal/mol) is comparable to the activation energy reported for lipid “flip-flop” (24 kcal/mol) in liquid-crystalline SUVs (26). However, the activation energy associated with proton movement during this second phase of the fusion process was considerably larger (33 kcal/mol) and comparable to that observed for proton movement (35 kcal/mol) during the first step. The nearly identical values for the activation energy of FP formation from lipid mixing and proton transfer data indicate that both inner leaflet lipid mixing and proton transfer were caused by the same process, which we view as irreversible septum popping leading to FP formation.
Figure 3.

Arrhenius plots for each of the three steps that we have identified as occurring during PEG-mediated vesicle fusion: (A) formation of I1, (B) conversion of I1 to I2, and (C) septum popping to form FP. Circles correspond to the rates obtained from lipid mixing data (LM) and squares correspond to the rates from proton transfer data (PT). The slopes determined the activation energies summarized in Table 1.
Interpretation of Activation Energies.
The formation of I1 is expected to be a complex process involving several elemental lipid motions. Possibly reflecting this, the activation energy for lipid rearrangements during the first step of SUV fusion (37 kcal/mol) is much larger than that associated even with lipid desorption from a membrane (22 kcal/mol; ref. 27) or with lipid flip-flop across a membrane (24 kcal/mol; ref. 26). These activation energies are the largest heretofore reported for elementary lipid rearrangements in a lipid bilayer. It has been pointed out, however, that the activation energy of a complex and reversible kinetic process such as fusion should reflect a complex sum or difference of the activation energies of the elementary processes constituting the complex process (28). This suggests that formation of intermediate I1 may require some combination of many elementary lipid rearrangements, such as lipid desorption, lipid flip-flop, lateral lipid diffusion (8 kcal/mol; ref. 29), whole molecule “wobble” (7–9 kcal/mol; ref. 30), along with others that have not yet been recognized and characterized.
Lipid movement from the outer to the inner leaflet is associated with PEG-mediated fusion of highly curved, sonicated vesicles (31). This is expected because the conversion from SUV to large unilamellar vesicles requires this directed trans-bilayer lipid movement. I1 to I2 conversion should also require transmembrane lipid movement from the outer to the inner leaflet if this process involves conversion of a stalk to a septum, as depicted in Fig. 1. Consistent with this, the activation energy for DPHpPC movement during I1 → I2 conversion (27 kcal/mol; Fig. 3B; Table 1) was similar to that observed for lipid flip-flop in highly curved vesicles (24 kcal/mol) (26). The rate of spontaneous lipid flip-flop (≈3.5 × 10−5 s−1 at 35°C) (26) is much slower than that for I1 → I2 conversion (3–4 × 10−2 s−1; Table 1), suggesting that lipid movement during conversion to intermediate I2 is driven by the highly stressed nature of the initial lipidic intermediate. In this regard, it is particularly interesting that the activation energy for proton movement during intermediate maturation was not equal to that of lipid flip-flop. Because proton transfer presumably is caused by occasional transient pores forming through the septum, the activation energy would not be expected to correspond to that for lipid flip-flop but should be close to that observed for proton transfer data during the first step, as is observed.
Relationship of Pure Lipid Bilayer Fusion to Biomembrane Fusion.
The pH-induced fusion between vesicular stomatitis virus and erythrocyte ghosts has been followed at several temperatures by lipid mixing measurements (32). Data were fit to a multi-exponential model, and it was suggested that a multi-step protonation of fusion protein might be necessary for fusion. The activation energy of the first process was determined by using the initial slope of outer leaflet lipid mixing and was found to be very large (42 kcal/mol at pH 5.85). The fact that this activation energy was so much larger than that of recognized lipid rearrangements was interpreted as reflective of protein rearrangements being rate limiting for the lipid transfer associated with the first step of viral fusion (32). However, we report here a similarly large activation energy (37 kcal/mol) associated with the lipid rearrangements needed for formation of intermediate I1 during PEG-mediated fusion of pure lipid bilayer membranes. We are warned appropriately of the dangers associated with making a simple interpretation of the activation energy for a reversible, complex process such as I1 formation (28). Nonetheless, the striking similarity in their activation energies suggests that the rate-determining event for lipid rearrangements associated with the initial step of viral fusion may be the same as that observed during PEG-mediated fusion of pure lipid bilayers.
The initial steps of the fusion events associated with both viral entry and secretory granule release are reported to involve formation of transient “flickering” pores (9, 11, 33–35). Our measurements of proton movement associated with pure lipid bilayer fusion establish the initial formation of a transient pore but cannot establish the flickering demonstrated by electrophysiological measurements on whole cells. Chanturiya et al. (7) recently reported, by electrophysiological measurements, transient flickering pore formation before irreversible FP formation during the fusion of protein-free phospholipid vesicles with planar phospholipid films. As in our earlier report on vesicle fusion (6), transient pores were associated with initial lipid movement between vesicles and planar lipid films. Although one can argue that planar lipid films contain solvents and are thus not “pure” lipid bilayers, the agreement between the observations of Chanturiya et al. (7) and our studies implies that the transient pores we observe during the fusion of pure lipid bilayers are also flickering pores. These observations, coupled with the correspondence of activation energies reported here, strongly argue that the initial flickering pores reported for both secretory and viral fusion correspond to the initial transient pores we report for pure lipid bilayer fusion (6).
In our model membrane system, even the large activation energy we have observed for the initial step in the fusion process was achieved only after raising the free energy of the initial, unfused state by curving the bilayer (6, 17). Membrane curvature has been hypothesized as a possible initiation mechanism for viral fusion (36). Because of the similarity in activation energies observed for the initial lipid mixing associated with viral and PEG-mediated vesicle fusion, our results support this hypothesis. Highly curved model membrane vesicles are created by sonic energy; in viral fusion, high membrane curvature could be caused locally by conformational changes in fusion proteins. The rate observed for initial intermediate formation during PEG-mediated fusion (2.2–2.4 s−1 at 35°C) was an order of magnitude smaller than that observed for viral fusion (≈20.6 s−1 at pH 5.59, 35°C; see Table 1). This suggests that the probability of contact between viral and target membranes may be enhanced by a fusion protein bridge. A strong pH dependency of lipid mixing rate in viral fusion (32) indicates that protonation of viral fusion proteins may either increase the membrane contact probability or decrease the activation energy for this process.
The kinetics of the final step of the fusion process, FP formation, have been characterized carefully in the case of fusion between secretory granules and mouse mast cell plasma membrane (10). Oberhauser et al. (10) made detailed analyses of capacitance tracings for this release process. Each trace was viewed as the sum of thousands of irreversible “FP” formations. The interval between irreversible FP steps distributed as a Gaussian, with the inverse of the mean interval being taken as the “rate of FP formation.” By analyzing the temperature dependence of the frequency of single pore openings, these authors determined the activation energy of FP formation to be 23 kcal/mol (10). This large activation energy also was suggested to be due to protein conformational changes.
In our analysis of PEG-mediated vesicle fusion, the frequency of pore opening (or septum “popping”) is given by 1/σ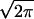 , where σ is the width of the Gaussian function describing the distribution of pop times. We note a fundamental difference between this frequency and the first order rate constants reported for the lipid rearrangements associated with the first two steps in bilayer fusion. The use of distinct start-times for these two processes (t1 = 0; t2 = 3/k1) implies that all vesicles in the observed population were undergoing similar and fairly slow lipid rearrangements roughly simultaneously. Septum popping, on the other hand, is viewed as being essentially instantaneous. The distributions of pop times can reflect two features of our experiments. First, the frequency of popping should reflect both the rate and extent of large-scale fluctuations in stressed septum structure. When one such fluctuation becomes sufficiently energetic, a new and lower energy lipid structure (the FP) can be viewed as replacing it. This “pop” frequency reflects the basic lipid structure within the I2 intermediate. However, our estimate of pop frequency derives from the width of the pop time distribution. This can reflect as well diversity of vesicle structure within our SUV preparations. Although we have used fractionated SUVs, it is well established that these contain vesicles of varying size and curvature. This diversity is expected to broaden our distribution of pop times, making the pop frequency we report a lower limit to the intrinsic pop frequency of the I2 structures that form under the conditions of our experiment. However, because SUV diversity should not vary with temperature of the fusion experiment, the activation energies derived from our pop frequencies should reflect the basic lipid structural fluctuations that lead to septum popping in our pure lipid bilayer system.
, where σ is the width of the Gaussian function describing the distribution of pop times. We note a fundamental difference between this frequency and the first order rate constants reported for the lipid rearrangements associated with the first two steps in bilayer fusion. The use of distinct start-times for these two processes (t1 = 0; t2 = 3/k1) implies that all vesicles in the observed population were undergoing similar and fairly slow lipid rearrangements roughly simultaneously. Septum popping, on the other hand, is viewed as being essentially instantaneous. The distributions of pop times can reflect two features of our experiments. First, the frequency of popping should reflect both the rate and extent of large-scale fluctuations in stressed septum structure. When one such fluctuation becomes sufficiently energetic, a new and lower energy lipid structure (the FP) can be viewed as replacing it. This “pop” frequency reflects the basic lipid structure within the I2 intermediate. However, our estimate of pop frequency derives from the width of the pop time distribution. This can reflect as well diversity of vesicle structure within our SUV preparations. Although we have used fractionated SUVs, it is well established that these contain vesicles of varying size and curvature. This diversity is expected to broaden our distribution of pop times, making the pop frequency we report a lower limit to the intrinsic pop frequency of the I2 structures that form under the conditions of our experiment. However, because SUV diversity should not vary with temperature of the fusion experiment, the activation energies derived from our pop frequencies should reflect the basic lipid structural fluctuations that lead to septum popping in our pure lipid bilayer system.
The activation energies (Table 1) of this process obtained from Arrhenius plots of lipid mixing and proton transfer (Fig. 3C) were in excellent agreement with each other and with that obtained by Oberhauser et al. (10) for pore formation during secretory granule release. Again, the similarity of the activation energies of model membrane (22 kcal/mol) and biomembrane (23 kcal/mol) FP formation argues that pore formation proceeds by the same lipidic mechanism in both cases. The frequency of pore formation between lipid vesicles (0.017–0.020 s−1 at 35°C) is slightly greater than that seen in secretory vesicle fusion (≈0.010 s−1 at 35°C) (10). Because our frequencies represent lower limits, it may be that the septum structures formed during PEG-mediated SUV fusion are more stressed than those formed within the protein scaffolds that direct secretory vesicle release.
Although it is impossible to completely exclude the protein pore model on the basis of our observations, the parallelism between kinetic events and activation energies presented in this report strongly supports the hypothesis that the mechanism of membrane fusion during secretory and viral fusion involves lipid bilayer structural rearrangements that correspond to those we have proposed for PEG-mediated lipid vesicle fusion (6).
Acknowledgments
This work was supported by U.S. Public Health Service Grant GM32707 to B.R.L.
ABBREVIATIONS
- PEG
poly(ethylene glycol)
- SUV
small unilamellar vesicles
- FP
fusion pore
- DPHpPC
1-palmitoyl-2-[[[2-[4-(phenyl-trans-1, 3, 5-hexatrienyl)phenyl]ethyl]oxyl]carbonyl]-3-sn-phosphatidylcholine
- HPTS
8-hydroxypyrene-1,3,6-trisulfonic acid
References
- 1.White J M. Science. 1992;258:917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- 2.Lindau M, Almers W. Curr Opin Cell Biol. 1995;7:509–517. doi: 10.1016/0955-0674(95)80007-7. [DOI] [PubMed] [Google Scholar]
- 3.Zimmerberg J, Curran M, Cohen F C. Anal N Y Acad Sci. 1991;635:307–317. doi: 10.1111/j.1749-6632.1991.tb36501.x. [DOI] [PubMed] [Google Scholar]
- 4.Monck J R, Fernandez J M. J Cell Biol. 1992;119:1395–1404. doi: 10.1083/jcb.119.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monck J R, Oberhauser A F, Fernandez J M. Mol Membr Biol. 1995;12:800–809. doi: 10.3109/09687689509038511. [DOI] [PubMed] [Google Scholar]
- 6.Lee J, Lentz B R. Biochemistry. 1997;36:6251–6259. doi: 10.1021/bi970404c. [DOI] [PubMed] [Google Scholar]
- 7.Chanturiya A N, Chernomordik L V, Zimmerberg J. Proc Natl Acad Sci USA. 1997;94:14423–14428. doi: 10.1073/pnas.94.26.14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez de Toledo G, Fernandez-Chacon R, Fernandez J M. Nature (London) 1993;363:554–558. doi: 10.1038/363554a0. [DOI] [PubMed] [Google Scholar]
- 9.Spruce A E, Breckenridge L J, Lee A K, Almers W. Neuron. 1990;4:643–654. doi: 10.1016/0896-6273(90)90192-i. [DOI] [PubMed] [Google Scholar]
- 10.Oberhauser A F, Monck J R, Fernandez J M. Biophys J. 1992;61:800–809. doi: 10.1016/S0006-3495(92)81884-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimmerberg J, Blumenthal R, Sarkar D P, Curran M, Morris S J. J Cell Biol. 1994;127:1885–1894. doi: 10.1083/jcb.127.6.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burgess S W, McIntosh T J, Lentz B R. Biochemistry. 1992;31:2653–2661. doi: 10.1021/bi00125a004. [DOI] [PubMed] [Google Scholar]
- 13.Lee J, Lentz B R. Biochemistry. 1997;36:421–431. doi: 10.1021/bi9622332. [DOI] [PubMed] [Google Scholar]
- 14.Suurkuusk J, Lentz B R, Barenholtz R L, Biltonen R L, Thompson T E. Biochemistry. 1976;15:1393–1401. doi: 10.1021/bi00652a007. [DOI] [PubMed] [Google Scholar]
- 15.Lentz B R, Carpenter T J, Alford D R. Biochemistry. 1987;26:5389–5397. doi: 10.1021/bi00391a026. [DOI] [PubMed] [Google Scholar]
- 16.Lentz B R, McIntyre G F, Parks D J, Yates J C, Massenburg D. Biochemistry. 1992;31:2643–2653. doi: 10.1021/bi00125a003. [DOI] [PubMed] [Google Scholar]
- 17.Talbot W A, Zheng L-X, Lentz B R. Biochemistry. 1997;36:5827–5836. doi: 10.1021/bi962437i. [DOI] [PubMed] [Google Scholar]
- 18.Wu H, Zheng L-X, Lentz B R. Biochemistry. 1996;35:12602–12611. doi: 10.1021/bi960168q. [DOI] [PubMed] [Google Scholar]
- 19.Kozlov M M, Leikin S L, Chernomordik L V, Markin V S, Chizmedzhev Y A. Eur Biophys J. 1989;17:121–129. doi: 10.1007/BF00254765. [DOI] [PubMed] [Google Scholar]
- 20.Melikyan G B, Niles W D, Cohen F S. J Gen Physiol. 1993;102:1151–1170. doi: 10.1085/jgp.102.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ray W J, Jr, Puvathingal J M. Anal Biochem. 1985;146:307–312. doi: 10.1016/0003-2697(85)90544-5. [DOI] [PubMed] [Google Scholar]
- 22.Burgess S W, Lentz B R. Methods Enzymol. 1993;220:42–50. doi: 10.1016/0076-6879(93)20072-b. [DOI] [PubMed] [Google Scholar]
- 23.Lentz B R, Burgess S W. Biophys J. 1989;56:723–733. doi: 10.1016/S0006-3495(89)82720-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J R, Lentz B R. J Fluoresc. 1994;4:153–163. doi: 10.1007/BF01881883. [DOI] [PubMed] [Google Scholar]
- 25.Feller W. An Introduction to Probability Theory and Its Applications. 3rd Ed. New York: Wiley; 1968. [Google Scholar]
- 26.De Kruijff B, Van Zoelen E J J. Biochim Biophys Acta. 1978;511:105–115. doi: 10.1016/0005-2736(78)90068-8. [DOI] [PubMed] [Google Scholar]
- 27.Jones J D, Thompson T E. Biochemistry. 1990;29:1593–1600. doi: 10.1021/bi00458a034. [DOI] [PubMed] [Google Scholar]
- 28.Bentz J. Biophys J. 1992;63:448–459. doi: 10.1016/S0006-3495(92)81622-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sassaroli M, Vauhkonen M, Perry D, Eisinger J. Biophys J. 1990;57:281–290. doi: 10.1016/S0006-3495(90)82530-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore B M, Lentz B R, Meissner G. Biochemistry. 1978;17:5248–5255. doi: 10.1021/bi00617a026. [DOI] [PubMed] [Google Scholar]
- 31.Lentz B R, Talbot W A, Lee J, Zheng L-X. Biochemistry. 1997;36:2076–2083. doi: 10.1021/bi9623340. [DOI] [PubMed] [Google Scholar]
- 32.Clague M J, Schoch C, Zech L, Blumenthal R. Biochemistry. 1990;29:1303–1308. doi: 10.1021/bi00457a028. [DOI] [PubMed] [Google Scholar]
- 33.Fernandez J M, Neher E, Gomperts B D. Nature (London) 1984;312:453–455. doi: 10.1038/312453a0. [DOI] [PubMed] [Google Scholar]
- 34.Breckenridge L J, Almers W. Proc Natl Acad Sci USA. 1987;84:1945–1949. doi: 10.1073/pnas.84.7.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spruce A E, Iwata A, Almers W. Proc Natl Acad Sci USA. 1991;88:3623–3627. doi: 10.1073/pnas.88.9.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rafalski M, Ortiz A, Rockwell A, van Ginkel L C, Lear J D, DeGrado W F, Wilschut J. Biochemistry. 1991;30:10211–10220. doi: 10.1021/bi00106a020. [DOI] [PubMed] [Google Scholar]
- 37.Blumenthal R, Sarkar D P, Durell S, Howard D E, Morris S J. J Cell Biol. 1996;135:63–71. doi: 10.1083/jcb.135.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]