

Abstract
The Saccharomyces cerevisiae FAB1 gene encodes a 257-kD protein that contains a cysteine-rich RING-FYVE domain at its NH2-terminus and a kinase domain at its COOH terminus. Based on its sequence, Fab1p was initially proposed to function as a phosphatidylinositol 4-phosphate (PtdIns(4)P) 5-kinase (Yamamoto et al., 1995). Additional sequence analysis of the Fab1p kinase domain, reveals that Fab1p defines a subfamily of putative PtdInsP kinases that is distinct from the kinases that synthesize PtdIns(4,5)P2. Consistent with this, we find that unlike wild-type cells, fab1Δ, fab1tsf, and fab1 kinase domain point mutants lack detectable levels of PtdIns(3,5)P2, a phosphoinositide recently identified both in yeast and mammalian cells. PtdIns(4,5)P2 synthesis, on the other hand, is only moderately affected even in fab1Δ mutants. The presence of PtdIns(3)P in fab1 mutants, combined with previous data, indicate that PtdIns(3,5)P2 synthesis is a two step process, requiring the production of PtdIns(3)P by the Vps34p PtdIns 3-kinase and the subsequent Fab1p- dependent phosphorylation of PtdIns(3)P yielding PtdIns(3,5)P2. Although Vps34p-mediated synthesis of PtdIns(3)P is required for the proper sorting of hydrolases from the Golgi to the vacuole, the production of PtdIns(3,5)P2 by Fab1p does not directly affect Golgi to vacuole trafficking, suggesting that PtdIns(3,5)P2 has a distinct function. The major phenotypes resulting from Fab1p kinase inactivation include temperature-sensitive growth, vacuolar acidification defects, and dramatic increases in vacuolar size. Based on our studies, we hypothesize that whereas Vps34p is essential for anterograde trafficking of membrane and protein cargoes to the vacuole, Fab1p may play an important compensatory role in the recycling/turnover of membranes deposited at the vacuole. Interestingly, deletion of VAC7 also results in an enlarged vacuole morphology and has no detectable PtdIns(3,5)P2, suggesting that Vac7p functions as an upstream regulator, perhaps in a complex with Fab1p. We propose that Fab1p and Vac7p are components of a signal transduction pathway which functions to regulate the efflux or turnover of vacuolar membranes through the regulated production of PtdIns(3,5)P2.
Keywords: vacuole; lipid kinase; PtdIns(3,5)P2 ; VAC7; VPS34
Acharacteristic feature of eucaryotic cells is the presence of multiple intracellular organelles that are specialized to carry out specific functions. Each organelle is defined by its unique protein and lipid composition that is maintained despite the continuous flow of cargo molecules between compartments. This necessitates having an accurate and efficient sorting system that guarantees the directional and spatial specificity of vesicle-mediated trafficking in the secretory and endocytic pathways (Palade, 1975; Rothman, 1994). Secretory proteins synthesized at the endoplasmic reticulum are transported to the Golgi complex where proteins destined for the cell surface must be sorted from those trafficking to the lysosome (or yeast vacuole; Kornfeld and Mellman, 1989; Stack et al., 1995). Significant progress has been made in understanding the machinery responsible for the sorting, transport, and docking/fusion of carrier vesicles with the appropriate target organelle (Schekman and Orci, 1996). The current data supports a model where both proteins and lipids function as regulators of these processes. The proteins directly involved in these pathways range from enzymes such as AAA-type ATPases and rab GTPases to polypeptide complexes that serve as membrane vesicle coats or vesicle docking components (Babst et al., 1997; Novick and Zerial, 1997; Schmid, 1997). Phospholipids, in particular phosphorylated derivatives of phosphatidylinositol (PtdIns),1 also appear to play a critical role in regulating these transport events (De Camilli et al., 1996; Martin, 1997). Given the numerous modification sites present on the inositol headgroup and the combinatorial manner in which these sites can be modified, phosphoinositides are well suited for the specific regulation of this complex trafficking process. Phosphoinositides also function in a variety of signal transduction pathways ranging from cell growth and differentiation, to apoptosis and cytoskeletal rearrangement (De Camilli et al., 1996; Toker and Cantley, 1997).
A requirement for phosphoinositides in Golgi to vacuole (lysosome) trafficking in yeast was established by the characterization of the VPS34 gene product as a PtdIns(3)-kinase, responsible for phosphatidylinositol 3-phosphate (PtdIns(3)P) synthesis (Schu et al., 1993). VPS34 was identified in a screen for vacuolar protein sorting mutants which missort and secrete proteins that are normally targeted to the yeast vacuole (Banta et al., 1988; Herman and Emr, 1990). Vps34p is recruited from the cytosol to a membrane-bound complex by the protein kinase Vps15p (Stack et al., 1993). Together, Vps15p and Vps34p facilitate protein sorting in both the Golgi to endosome and endosome to vacuole transport pathways through the regulated synthesis of PtdIns(3)P (Munn and Riezman, 1994; Stack et al., 1995). This lipid product appears to be required for the proper localization and/or activation of other proteins essential for Golgi to vacuole transport. Recently, candidate effectors of PtdIns(3)P signaling were identified in yeast as a set of proteins that contain a cysteine-rich RING-FYVE finger domain that binds PtdIns(3)P and not other phosphoinositides (Burd and Emr, 1998). Two of these proteins Vac1p/Vps19p and Vps27p are known to function in the Golgi to endosome and endosome to vacuole transport reactions, respectively (Weisman and Wickner, 1992; Piper et al., 1995; Burd et al., 1997). Similarly, in mammalian cells, wortamannin-induced inhibition of phosphoinositide 3-kinase activities results in lysosomal hydrolase missorting, cathepsin D is secreted into the media (Brown et al., 1995; Davidson, 1995).
Additional polyphosphoinositides have also been implicated in membrane trafficking. PEP1 and PEP3 proteins are required in the Ca++-dependent exocytosis of secretory granules from semi-intact PC12 cells (Hay and Martin, 1993; Hay et al., 1995). PEP1 is a PtdIns(4)P 5-kinase, responsible for PtdIns(4,5)P2 synthesis. PEP3, a PtdIns transfer protein (PITP), exchanges PtdIns and phosphatidylcholine between distinct intracellular membranes, thereby increasing local lipid concentrations. PITPs may also directly present lipid substrates to lipid-modifying enzymes (e.g., PEP1; Liscovitch and Cantley, 1995; Kearns et al., 1998). The activities of these two proteins indicate that the ultimate synthesis of PtdIns(4,5)P2 plays a direct role in the release of secretory granules, perhaps by recruiting essential regulatory proteins to the proper membrane. Mutation of the yeast PITP, SEC14, also results in a block of secretory traffic from the late Golgi (Bankaitis et al., 1990; Kearns et al., 1998).
PtdIns(3)P was the only known D-3 phosphoinositide in yeast, until recently, when PtdIns(3,5)P2 was identified in yeast, mouse fibroblasts, and plants (Dove et al., 1997; Whiteford et al., 1997). Neither the kinase responsible for PtdIns(3,5)P2 synthesis nor the biological role of this lipid have been defined. In this report, we provide evidence that yeast Fab1p, a protein that bears homology to a general family of PtdInsP kinases, functions as a PtdIns(3)P 5-kinase. FAB1 deletion mutants completely lack PtdIns(3,5)P2 and are viable, but exhibit severe growth defects and have an extremely enlarged vacuole that occupies the majority of the cell (Yamamoto et al., 1995). Proper localization of the vacuolar ATPase (V-ATPase) and alkaline phosphatase (ALP) in fab1 mutants indicates that Fab1p, and therefore PtdIns(3,5)P2, is not required for biosynthetic protein and membrane trafficking to the vacuole. A similar enlarged vacuole morphology and severe depletion of PtdIns(3,5)P2 in vac7 mutants, indicates that Vac7p functions as a possible regulator or cofactor of the Fab1p kinase. Consistent with the vacuolar phenotypes of fab1 mutants as well as the vacuolar localization of Vac7p, a large pool of Fab1p cofractionates with the V-ATPase. We propose that Fab1p, together with Vac7p, function downstream of Vps34p to regulate vacuolar membrane recycling/turnover through the production of the polyphosphoinositide PtdIns(3,5)P2.
Materials and Methods
Strains and Media
The Escherichia coli strain XL1-Blue (supE44 thi-1 lac endA1 gyrA96 hsdR17 relA1 F′ proAB LaqIq ZΔM15) was grown with standard LB media. Unless otherwise specified, the Saccharomyces cerevisiae strains used in this study were grown in standard YPD and SD minimal media with the necessary auxotrophic supplements. S. cerevisiae strains used in this study were as follows: (a) SEY6210 (MATα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9; Robinson et al., 1988). (b) fab1Δ1 (SEY6210 fab1Δ::LEU2, this study). (c) fab1Δ2 (SEY6210 fab1Δ::HIS3, this study). (d) LWY1481 (MATα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 pep4-Δ1137 vac7Δ::HIS3; Bonangelino et al., 1997). (e) LWY2365 (MATα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 pep4-Δ1137 vac14-1; Bonangelino et al., 1997). (f) EMY119 (MATa leu2-3,112 ura3-52 fab1-2; lab strain).
Genetic and DNA Manipulations
All restriction enzymes were purchased from Boehringer Mannheim Biochemicals (Indianapolis, IN). T4 DNA ligase was received from GIBCO BRL (Gaithersburg, MD). PCR was performed using synthetic oligonucleotide primers and dNTPs purchased from GIBCO BRL. All PCR reactions used a 2:1 mixture of KlenTaq polymerase (Ab Peptides, Inc., St. Louis, MO) and Pfu polymerase (Stratagene, La Jolla, CA) as described in (Barnes, 1994). Otherwise standard molecular biology techniques were used (Maniatis et al., 1982). Yeast transformations were performed using the lithium acetate method (Ito et al., 1983) and genomic DNA was isolated using a modified version of the procedure described by Hoffman and Winston (1987).
Disruption of FAB1.
The entire open reading frame of FAB1 was deleted in SEY6210 through transformation with a 1.1-kbp PCR fragment containing the HIS3 marker flanked by 52 nucleotides upstream and downstream of the FAB1 sequence. Histidine prototrophs were selected on histidine-free minimal media plates and confirmation of the disruption was done by PCR using two different sets of primers.
pfab1-2 Construction.
A 6,350 bp AatII-NruI fragment of the wild-type FAB1 gene was excised from pAY60 (pRS416-FAB1; Yamamoto et al., 1995). The resulting linearized 9,100-bp pRS416 vector containing ∼2,000 bp of flanking genomic sequence 5′ of the AatII site and 3′ of the NruI site was then transformed into EMY119, a strain carrying the fab1-2 allele (Yamamoto et al., 1995) and selected on uracil-free plates. Through homologous recombination and the subsequent purification of the gap-repaired plasmid, the fab1-2 allele was rescued from the genome.
Generation of Fab1p Kinase Domain Point Mutations.
The G2042/2045V point mutant construct was made using the gene SOE (splicing by overlap extension) and plasmid gap repair techniques (Muhlrad et al., 1992; Horton et al., 1993). For the gene SOE, two successive rounds of PCR were used to generate the final 1.9-kbp fragment containing the desired mutations. In the first round, the 5′ and 3′-PCR fragments were synthesized in two separate PCR reactions. For the second round of PCR, the products from the 5′ and 3′ reactions were diluted 100-fold and amplified in the presence of both outside primers to create the full-length fragment containing the desired mutations. pAY60 (Yamamoto et al., 1995) was then digested with NheI and NruI and the gapped vector was purified and cotransformed into the fab1Δ2 strain with the full-length gene SOE fragment. Plasmid DNA was rescued from yeast cells that had become uracil prototrophs, indicating successful gap repair of the pRS416-based plasmid. The gene SOE fragment containing the D2134R point mutation was made in a similar fashion. However, after synthesis of the full-length gene SOE fragment, it was digested with NheI and NruI and introduced into the gapped vector by direct ligation. Once transformed into E. coli the plasmid constructs for both point mutations were sequenced to confirm the presence of only the desired mutation. DNA sequencing was done at the UCSD CFAR core facility using the ABI Prism BigDye chemistry with Amplitaq DNA Polymerase FS. Gels were analyzed with a 373XL DNA Sequencer (PE Applied Biosystems, Foster City, CA). Correct constructs were then retransformed into fab1Δ2 for phenotypic analysis.
Construction of HA-tagged Fab1p.
Primers specific to FAB1 were used to separately amplify a 350-bp and a 1,300-bp region upstream and downstream of codon 8, while also engineering a BglII site at codon 8. The 350-bp fragment was cut with SnaBI/BglII while the 1,300-bp FAB1 fragment was cut with BglII/Bsu36I. These fragments were then simultaneously ligated into pAY60 (Yamamoto et al., 1995) and pEMY105 (pRS426-FAB1) digested with SnaBI/Bsu36I. The resulting pRS416-FAB1-BglII and pRS426-FAB1-BglII constructs contain the full-length FAB1 gene with a unique BglII site engineered at codon 8. A triple-HA epitope repeat was also amplified using PCR primers that introduced BglII sites into both ends of the PCR product to make the triple-HA repeat in frame with the BglII site engineered into FAB1. This HA insert was ligated into pRS416-FAB1-BglII and pRS426-FAB1-BglII via the BglII sites.
In Vivo Labeling and Immunoprecipitation of Fab1p
Approximately 6 OD600 units of yeast were harvested by centrifugation and resuspended in 2 ml of minimal media and allowed to recover at 24°C for 10 min. Express [35S]-protein labeling mix (DuPont NEN, Boston, MA) was then added to a final concentration of 5 μCi/OD600 unit. After a 20-min incubation at 24°C, cells were chased for 45 min at either 24 or 38°C with 5 mM methionine, 1 mM cysteine, 0.4% yeast extract and 2% glucose. The chase was ended with the addition trichloroacetic acid was added to a final concentration of 10%. Trichloroacetic acid precipitates were subjected to glass bead lysis and Fab1p was then immunoprecipitated as previously described (Gaynor et al., 1994) with antisera raised against a TrpE fusion of the Fab1 protein.
Subcellular Fractionation
Spheroplasted wild-type cells were subjected to gentle osmotic lysis as previously described (Gaynor et al., 1994). The resulting cellular extract was then sequentially centrifuged at 325 g for 5 min to remove unbroken cells, 13,000 g for 10 min to generate pelletable, P13 and soluble S13 fractions and 100,000 g for 30 min to yield P100 and S100 fractions. Protein present in all fractions was then precipitated using TCA to a final concentration of 5%. In the case of Fab1p and glucose 6-phosphate dehydrogenase, the extracts were derived from wild-type cells pulse-labeled with Express [35S]-protein labeling mix for 20 min and chased with 5 mM methionine, 1 mM cysteine, 0.4% yeast extract for 40 min. Fractions were then immunoprecipitated for Fab1p and glucose 6-phosphate dehydrogenase using antiserum raised against these proteins as described (Gaynor et al., 1994). The presence of Kex2p and the 100-kD subunit of the vacuolar ATPase in specific fractions were determined using antisera specific to these proteins for immunoblotting and ECL as described in (Babst et al., 1997).
In Vivo Analysis of Phosphoinositides
To label strains with myo-[2-3H]inositol (Nycomed Amersham Inc., Princeton, NJ), cells were grown for 24 h in YPD or SD minimal media (if selecting for a plasmid). 0.2–0.5 OD600 units of cells were harvested, washed with 1 ml of synthetic media lacking inositol and used to inoculate 5 ml of inositol-free media containing 75 μCi myo-[2-3H]inositol. Cells were labeled 12 h at 22°C, harvested by centrifugation, washed with fresh synthetic media and then resuspended in 100 to 200 μL of synthetic media. An equal volume of 1.2 M NaCl was then added to the cells which were then incubated for a 10-min period to osmotically shock cells. This was carried out at 22°C unless otherwise indicated. Following this treatment, labeled cells were lysed in 1.5 ml of 1 M HCl/chloroform/methanol (1:1:1) by vortexing 10 × 30 s periods in the presence of 0.5 g of 0.25-mm glass beads. As previously described, cellular lipids were extracted, deacylated and analyzed by HPLC (Schu et al., 1993; Stack et al., 1995). Fractions eluting from an HPLC column (catalog no. 4611-1505; Whatman Inc., Clifton, NJ) were collected every 0.66 min. [3H]-PtdIns(3,5)P2 standards were used to determine the elution point of PtdIns(3,5)P2 from our column were a generous gift of Robert H. Michell (University of Birmingham, AL). The data presented are representative of multiple experiments. A variation of 10% was observed between experiments.
FM4-64 Labeling of Yeast Vacuoles
Approximately 1 OD600 of yeast were harvested at an OD600 of 0.6–0.8, and then labeled with the vital vacuolar dye FM4-64 as previously described (Vida and Emr, 1995). FM4-64 [N-(3-triethylammoniumpropyl)- 4-(p-diethylaminophenylhexatrienyl) pyridinium dibromide] was obtained from Molecular Probes Inc. (Eugene, OR). For temperature-shift experiments, cells were labeled at the permissive temperature and then shifted to the elevated temperature for the remainder of the incubation in the absence of the fluorophore.
Vacuolar Protein Sorting Assays
For analysis of CPS, CPY, ALP, and AP1 processing, whole cells were labeled as previously described (Gaynor et al., 1994). Express [35S]-protein labeling mix was added to a final concentration of 10 μCi/μl and cells were labeled during a 10-min pulse at 24°C. For temperature-shift experiments, cells were preshifted to 38°C for 30 min before labeling. The cultures were kept at the identical temperatures and chased as described above. After the chase, cells were harvested and lysates generated as previously described (Gaynor et al., 1994). Extracts were immunoprecipitated with antisera against CPY, ALP, CPS, and AP1, that have been previously characterized (Klionsky and Emr, 1989; Cowles et al., 1997b ; antibody to API was a generous gift from Dan Klionsky, University of California, Davis, CA; Klionsky et al., 1992). Radioactive immunoprecipitates of CPS were treated with endoglycosidase H (DuPont NEN) as previously described (Gaynor et al., 1994). SDS-PAGE was done as described above however, gels were run at 25 mA constant current.
Microscopy
For observing the indirect immunofluorescence of the 60-kD subunit of the vacuolar ATPase, the method of (Redding et al., 1997) was used. In brief, exponentially growing cells were fixed in 4% formaldehyde and 0.1 M potassium phosphate, pH 6.5 for 16 h. The cell wall was removed by incubating cells with 45 μg/ml Zymolyase 100T (Seikagaku Kogyo, Tokyo, Japan) and cells were then treated with 1% SDS for 10 min. The fixed spheroplasts were first incubated in a 1:40 dilution of monoclonal antibody specific for the 60-kD subunit of the vacuolar ATPase (Molecular Probes Inc.), followed by successive incubations in goat anti–mouse IgG (1:3,000), and donkey anti–goat (1:200; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). GFP-ALP was transformed into the fab1Δ2 strain and observed as previously described (Cowles et al., 1997a ). 50 OD600 units of cells grown to mid-log phase were harvested for each sample analyzed by electron microscopy. All cells were grown in YNB supplemented with the required amino acids and 2% dextrose. Cells were then fixed and processed for electron microscopy as described (Rieder et al., 1996).
Immunofluorescence localization of an HA-tagged version of Fab1p was performed as previously described (Bonangelino et. al., 1997). In brief, cells overexpressing Fab1p-HA were resuspended in 0.1 M sodium phosphate buffer (pH 6.5) and fixed in 4.4% formaldehyde for 40 min at 30°C. Fixed cells were converted to spheroplasts by incubation in 1.2 M sorbitol in 0.1 M phosphate buffer (pH 6.5) with 1% mercaptoethanol and 150 g of oxalyticase (Enzogenetics, Eugene, OR) per ml for 15 min at 30°C. After adhering the fixed spheroplasts to 1% polyethyleneimine-coated wells, cells were incubated with monoclonal anti-HA (MMSR101; Berkeley Antibody Co., Inc., Richmond, CA) at a 1:200 dilution overnight (16 h), followed by affinity-purified rabbit anti-Vac8p (Wang et al., 1998) at a 1:50 dilution for 1 h. The primary mouse antibodies were detected by using a 1:200 dilution of Oregon-Green-488 conjugated goat anti–mouse (Molecular Probes) and the rabbit antibodies were detected by 1:200 dilution of Rhodamine red–conjugated goat anti–rabbit (Jackson ImmunoResearch Laboratories Inc.). Incubations with the fluorophore-conjugated secondary antibodies were performed in a dark humidity chamber for 1 h. Images were collected using an MRC 1024 Scanning Confocal head mounted on a Nikon Optiphot equipped with a 100× oil immersion lens.
Results
Fab1p Is a Member of a Distinct Family of PtdInsP Kinases
Many of the phosphoinositides and phosphoinositide kinases (Fig. 1 A) present in mammalian cells are also found in the yeast S. cerevisiae. Homologs of the PtdIns 4-kinase (Stt4p and Pik1p; Flanagan et al., 1993; Yoshida et al., 1994), PtdIns 3-kinase (Vps34p; Schu et al., 1993), and PtdIns(4)P 5-kinase (Mss4p; Desrivieres et al., 1998; Homma et al., 1998) have been identified in yeast. Two groups recently have identified a novel lipid, PtdIns(3,5)P2, in yeast and mammalian cells, that is produced by the sequential phosphorylation of PtdIns at the 3′ and 5′-positions (Dove et al., 1997; Whiteford et al., 1997). The PtdIns(3)P 5-kinase required for the synthesis of this previously undocumented lipid has not yet been identified.
Figure 1.

Phosphatidylinositol derivatives and their kinases in yeast. (A) A diagram of the D-3 and D-4 phosphorylated phosphoinositides present in the yeast S. cerevisiae. Above the arrows, the specific kinases determined to be involved in their biosynthesis are listed. (B) A phylogenetic comparison of the Fab1p lipid kinase domain to the analogous domain of other PtdInsP kinases. Using the DNAStar program MegAlign, the S. cerevisiae Fab1p kinase domain (a.a. 2022–2263) and its homologues SPBC6B1.11c (AL021838; a.a. 358–595) and C05E7.5 (Z67879; a.a. 924–1170) from S. pombe and C. elegans, respectively, were aligned and a phylogenetic tree was constructed with the corresponding kinase domains of human PtdIns(4)P 5-kinases Type Iα (U78575; a.a. 125–449), Type Iβ (U52376; a.a. 1–288), Type IIβ (U85245; a.a. 100–416), Type IIc (P53807; a.a. 110–406), and S. cerevisiae Mss4p (D13716; a.a. 439–765). The multiple sequence alignment was performed using the Jotun-Hein method with default program parameters, using the PAM250 residue weight table.
Based on initial sequence analyses, Fab1p from S. cerevisiae, as well as its homologs from S. pombe and C. elegans, were proposed to be PtdIns(4)P 5-kinases (Yamamoto et al., 1995; Boronenkov and Anderson, 1995). A BLASTP search (NCBI; Altschul et al., 1997) with a COOH-terminal region of Fab1p (a.a. 2,000–2,278), containing the lipid kinase domain, reveals that the most closely related sequences with known enzymatic activities belong to a subset of PtdInsP kinases which include the Type I and Type II mammalian PtdIns(4)P 5-kinases. The Fab1p kinase domain therefore, is more closely related to the kinase domains of PtdInsP kinases (Fig. 1 B) than PtdIns kinases such as the 3-kinase Vps34p, the 4-kinases Stt4p and Pik1p, and the PtdIns kinase homologs Tor1p and Tor2p (Kunz et al., 1993).
Recent evidence supports the reclassification of the Type II PtdIns(4)P 5-kinases as PtdIns(5)P 4-kinases (Rameh et al., 1997) and this biochemical distinction of enzymatic activity is also borne out in sequence comparisons. Construction of a phylogenentic tree based on the pairwise alignment of the kinase domains of these groups, separates the Type I and Type II kinase domains into two classes (Fig. 1 B; Loijens and Anderson, 1996). Interestingly, Fab1p belongs to a third, distinct class of PtdInsP kinases when included in this analysis (Fig. 1 B; Loijens and Anderson, 1996). Consistent with this phylogenetic analysis, the yeast Mss4 protein has recently been identified as a PtdIns(4)P 5-kinase (Desrivieres et al., 1998; Homma et al., 1998; Fig. 1 B), indicating that this additional class does not appear to have been established simply on the basis of comparing lipid kinases from different species. These results suggest that the lipid kinase domain of Fab1p is distinct from analogous domains of known PtdIns(4)P 5-kinases and PtdIns(5)P 4-kinases, raising the possibility that Fab1p phosphorylates a PtdInsP substrate distinct from either PtsIns(4)P or PtdIns(5)P, perhaps PtdIns(3)P.
Inactivation of Fab1p Results in Undetectable Levels of PtdIns(3,5)P2
To determine if Fab1p is required for the production of specific phosphatidylinositol derivatives in vivo, we analyzed phosphoinositide levels in wild-type yeast cells and in a strain deleted for FAB1. SEY6210 (wild-type) and fab1Δ1 cells were labeled with myo-[2-3H]inositol for 12 h. At the end of this labeling period, cells were osmotically shocked with 0.9 M NaCl for 10 min. Hyperosmotic shock facilitates the analysis of PtdIns(3,5)P2, which is difficult to detect in the absence of this treatment, without dramatically affecting the levels of other observable lipids (Dove et al., 1997). The labeled cells were then lysed in acidified chloroform/methanol and the cellular lipids were deacylated and identified by HPLC analysis (see Materials and Methods). This method enables the resolution and quantification of cellular PtdIns(3)P, PtdIns(4)P, PtdIns(3,5)P2 and PtdIns(4,5)P2 (Auger et al., 1989; Schu et al., 1993; Dove et al., 1997).
When this analysis is carried out using wild-type yeast cells, four distinct glycero-phosphoinositides corresponding to PtdIns(3)P (13,700 cpm, fraction 32-33), PtdIns(4)P (13,700 cpm, fraction 35-36), PtdIns(3,5)P2 (2,600 cpm, fraction 66-67) and PtdIns(4,5)P2 (4,100 cpm, fraction 73-74) are clearly separated (Fig. 2 A). The identities of the deacylated lipid products were confirmed using tritiated standards (data not shown). When an equal quantity of labeled cellular lipids isolated from fab1Δ1 cells are assayed in the same manner, the most dramatic effects are observed on PtdIns(3,5)P2 levels, which are undetectable (<50 cpm; Fig. 2 A). Relative to wild-type cells, other phosphoinositide levels are also influenced in the fab1Δ1 mutant cells: PtdIns(3)P increases ∼100% (25,000 cpm), PtdIns(4)P decreases ∼50% (7,300 cpm) whereas PtdIns(4,5)P2 decreases 60% (1,600 cpm).
Figure 2.

PtdIns(3,5)P2 is undetectable in fab1 mutant cells. (A) Wild-type (SEY6210) and fab1Δ1 cells were labeled with myo- [2-3H]inositol for 12 h at 22°C and hyperosmotically shocked with 0.9 M NaCl for 10 min. Cells were then glass bead lysed and the cellular lipids extracted, deacylated and separated by HPLC (see Materials and Methods). Fractions (0.67 min) were collected and counted for [3H] radioactivity. Deacylated (glycero-) PtdIns(3)P, PtdIns(4)P, PtdIns(3,5)P2, and PtdIns(4,5)P2 are indicated. Arrows indicate the expected position of gPtdIns(3,5)P2. (B) Wild-type and fab1tsf cells were labeled for 12 h with myo-[2-3H]inositol at 22°C, after which both cultures were separated into two equal aliquots. One aliquot of each strain was incubated with 0.9 M NaCl for 10 min at 22°C. The remaining wild-type and fab1tsf cells were incubated with 0.9 M NaCl for 5 min at 22°C and, for the final 5 min of hyperosmotic shock, shifted to the nonpermissive temperature of 38°C. The peaks representing PtdIns(3,5)P2 and PtdIns(4,5)P2 are indicated. The arrow highlights the elution point of the gPtdIns(3,5)P2 standard. The data presented are representative of multiple experiments.
To determine if the dramatic changes in PtdIns(3,5)P2 levels seen in the fab1Δ1 strain resulted directly from a loss of Fab1p activity, we assayed PtdIns(3,5)P2 in a fab1Δ2 + pfab1-2 (fab1tsf) strain briefly shifted to nonpermissive temperature (Yamamoto et al., 1995; see Materials and Methods). At the permissive temperature, fab1tsf cells have normal growth characteristics and wild-type vacuole morphology. However, shifting these cells to 37°C results in growth defects and aberrant vacuole morphologies (Bonangelino et al., 1997; Yamamoto et al., 1995). Wild-type and fab1tsf cells were labeled for 12 h with myo-[2-3H]inositol at 22°C, as before. At the end of this labeling period, half of the wild-type and fab1tsf cells were incubated with 0.9 M NaCl for 10 min at permissive temperature. The remaining cells were exposed to 0.9 M NaCl for 5 min at 22°C and shifted to the nonpermissive temperature of 38°C for the final 5 min of osmotic shock. Even at the permissive temperature (22°C) PtdIns(3,5)P2 levels are reduced sevenfold (800 cpm) in the fab1tsf cells compared to the wild-type control (Fig. 2), whereas the levels of other phosphoinositides remain only moderately affected (data not shown). In the fab1tsf strain at the nonpermissive temperature, PtdIns(3,5)P2 synthesis is dramatically affected, reaching undetectable levels (<50 cpm) after a 5-min shift. Compared to the wild-type control, the high temperature incubation results in no significant changes in the other lipids. Given the homology of Fab1p to the family of PtdInsP kinases and the fact that in yeast Vps34p is the only source of PtdIns(3)P, the rapid depletion of PtdIns(3,5)P2 observed in the fab1 mutant cells suggests that Fab1p encodes a PtdIns(3)P 5-kinase.
Mutations within the Kinase Domain of Fab1p Cause a Dramatic Decrease in Cellular PtdIns(3,5)P2 Levels
The results demonstrate that Fab1p is required for normal PtdIns(3,5)P2 levels. To address the role of the putative kinase domain of Fab1p in phosphatidylinositol metabolism, we used site-directed mutagenesis to make substitutions in highly conserved amino acid positions of the kinase domain. Specifically, Gly2042 and Gly2045 were simultaneously changed to valine residues (G2042/2045V) and Asp2134 was replaced with an arginine residue (D2134R; Fig. 3 A; see Materials and Methods). The GGxxG glycine triad motif containing Gly2042 and Gly2045 of Fab1p is reminiscent of the highly conserved GxGxxG motif present in the nucleotide binding site of numerous protein kinases (Bossemeyer et al., 1993; Hemmer et al., 1997; Sicheri et al., 1997; Xu et al., 1997). In the protein kinases, these glycine residues are included in the conserved glycine-rich loop and mutation of these residues to alanine or serine residues results in decreases in enzymatic activity (Hemmer et al., 1997; Grant et al., 1998). This comparison predicts that the substitution of these glycine residues to valine residues in Fab1p would result in commensurate impairment of lipid kinase activity. By similar sequence comparisons, Asp2134 of Fab1p appears to be analogous to the catalytically important Asp386 of the c-Src protein kinase (Xu et al., 1997) and mutation of this aspartate residue would be expected to severely affect phosphotransfer (Bossemeyer et al., 1993). An analogous mutation of this aspartate residue to an alanine residue in the Vps34p lipid kinase also lacked detectable amounts of PtdIns(3)P synthesis (Schu et al., 1993). Because of the apparent importance of this residue in catalysis, we decided to make the corresponding mutation in Fab1p.
Figure 3.


Point mutations within the Fab1p kinase domain. (A) A conserved region of the Fab1p kinase domain is aligned with several lipid kinase homologues identified from a BLAST search. Residues identical to those in Fab1p are outlined in black. Above the alignment, the amino acid substitutions engineered to make the Fab1p point mutant constructs are shown. (B) Fab1p was immunoprecipitated from a wild-type (SEY6210), fab1Δ2, fab1tsf, as well as the two kinase domain point mutant strains (fab1G2042/2045V and fab1D2134R). Cells were metabolically labeled with Express [35S]-protein labeling mix and then chased in an excess of methionine and cysteine for 45 min at 24°C (see Materials and Methods). The immunoprecipitated protein was visualized after SDS-PAGE by fluorography (7-d exposure at −80°C).
Both the CEN-based G2042/2045V and the D2134R point mutant constructs were transformed into fab1Δ2 (strains fab1G2042/2045V and fab1D2134R, respectively). Immunoprecipitation of metabolically labeled Fab1p indicates that the proteins derived from the fab1G2042/2045V and fab1D2134R strains are stably expressed, as the levels of the Fab1p do not vary over the course of a 10- and 45-min chase at either 24°C (Fig. 3 B) or 38°C (data not shown). Interestingly, Fab1p from the fab1tsf strain is also stable after a 45-min chase at the nonpermissive temperature (data not shown), indicating that despite its loss of kinase activity, the protein is not susceptible to rapid proteolytic degradation.
To determine the effect Fab1p kinase domain point mutations have on PtdIns(3,5)P2 synthesis, the Fab1p G2042/ 2045V and Fab1p D2134R expressing strains were labeled for 12 h with myo-[2-3H]-inositol and hyperosmotically shocked with 0.9 M NaCl, as described above. Relative to the wild-type control (8,000 cpm of the PtdIns(3,5)P2), the Fab1p G2042/2045V point mutant exhibits a 9-fold decrease in PtdIns(3,5)P2 (900 cpm; Fig. 4). This reduced level of PtdIns(3,5)P2 production in the fab1G2042/2045V strain is consistent with the reduced levels of kinase activity observed when similar mutations are made in the analogous glycine residues of cAPK (Hemmer et al., 1997; Grant et al., 1998). PtdIns(3,5)P2 is virtually undetectable (<50 cpm) in the fab1D2134R strain containing the D2134R change in the putative catalytic loop of Fab1p (Fig. 4). In the same assay, levels of cellular PtdIns(4)P (data not shown) and PtdIns(4,5)P2 (Fig. 4) did not change dramatically (<30%) as a result of these Fab1p kinase domain point mutations. However, increases of four- and sixfold were observed for the Fab1p substrate PtdIns(3)P in the fab1G2042/2045V and fab1D2134R strains, respectively.
Figure 4.

fab1 point mutant strains are compromised for PtdIns(3,5)P2 synthesis. Wild-type (SEY6210), fab1G2042/2045V, and fab1D2134R cells were labeled with myo- [2-3H]inositol for 12 h at 24°C and hyperosmotically shocked with 0.9 M NaCl for 10 min (see Fig. 2). The deacylated PtdIns derivatives were separated by HPLC, only the region of the elution profile separating gPtdIns(3,5)P2 and gPtdIns(4,5)P2 are shown. The arrow highlights the elution point of the glycero-PtdIns(3,5)P2 standard.
Fab1p Cofractionates with PtdIns(3)P-enriched Membrane Compartments
PtdIns(3)P is synthesized from PtdIns in Golgi/endosomal membranes, and we have recently discovered that it is transported in a Vps-dependent manner from these compartments to the vacuole (Wurmser and Emr, 1998). Using GFP-RING-FYVE domain fusion constructs which, when expressed in vivo, specifically interact with PtdIns(3)P-containing membranes, the localization of PtdIns(3)P to vacuolar and endosomal membranes was confirmed (Burd and Emr, 1998). Thus, all organelles which contain PtdIns(3)P (Golgi, endosome, and vacuole) are potential sites of Fab1p activity. To localize Fab1p we examined wild-type cells by differential centrifugation.
Cells were converted to spheroplasts, osmotically lysed and cleared of unbroken cells by a low speed spin. The cell extract was then sequentially centrifuged at 13,000 g and 100,000 g yielding soluble fractions (S13 and S100) and pelletable fractions (P13 and P100; see Materials and Methods). All fractions were assayed for the presence of Fab1p, the 100-kD vacuolar ATPase subunit, the Golgi/endosome localized protease, Kex2p and the cytosolic marker glucose 6-phosphate dehydrogenase (G6PDH) using antibodies specific to these proteins. The majority of Fab1p is detected in the pellet fractions (30% P13, 40% P100) and the remainder (30%) is found in the S100 fraction (Table I). In this experiment, vacuolar membranes, identified by the presence of the vacuolar ATPase 100-kD subunit, are found predominantly in the P13 fraction (75%) and also in the P100 (25%). Golgi and endosomal compartments containing Kex2p are enriched in the P100 fraction whereas the cytosolic fraction (G6PDH) is predominantly S100. Fab1p, therefore, appears to be enriched in fractions that contain endosomal and vacuolar membranes, suggesting that Fab1p functions at one (or more) of these compartments.
Table I.
Cellular Distribution of Fab1 Protein
| P13 | P100 | S100 | ||||
|---|---|---|---|---|---|---|
| Fab1p | 30% | 40% | 30% | |||
| V-ATPase | 70% | 25% | <5% | |||
| Kex2p | 10% | 90% | <5% | |||
| G6PDH | 5% | 5% | 90% |
To refine the localization of native Fab1p, CEN and 2μ versions of HA-tagged Fab1p (Fab1p-HA) have been constructed (see Materials and Methods). These constructs complement the growth and vacuolar morphology defects of the fab1 deletion strain (data not shown). Indirect immunofluorescence using the CEN construct did not produce a discernible signal above background (data not shown). However, at multicopy levels (8–10-fold overexpression; data not shown), we find that a pool of Fab1p-HA colocalizes with Vac8p (Fig. 5), a protein associated with the vacuolar membrane (Wang et al., 1998). Cytoplasmic accumulations of the Fab1p-HA protein are also evident. In some cells, no signal could be detected above background, presumably due to variable levels of Fab1p-HA overexpression within the cell population. Consistent with the subcellular fractionation results, which suggest that Fab1p is distributed among multiple membranes (Table I), this data indicates that Fab1p is associated with vacuolar and prevacuolar compartments.
Figure 5.

Localization of HA-tagged Fab1p by indirect immunofluorescence. Cells overexpressing HA-tagged Fab1 or wild-type Fab1 protein from multi-copy plasmids were fixed, converted to spheroplasts and then incubated with monoclonal anti-HA or anti-Vac8p antibodies as described in the Materials and Methods. The localization of Fab1p-HA and Vac8p were determined by indirect immunofluorescence. HA-tagged Fab1p and Vac8p signals are indicated.
Point Mutations within the Fab1p Kinase Domain Result in Abnormal Vacuolar Morphology
Transformation of the fab1 null background with either of the Fab1p point mutant constructs results in a differential suppression of the fab1 phenotypes. As shown previously (Yamamoto et al., 1995), these defects include: (a) temperature-sensitive growth, (b) vacuole acidification defects and, (c) a 2.5-fold increase in vacuolar surface area, which leads to inappropriate nuclear segregation during mitosis. The temperature-sensitive growth of the fab1Δ2 at 30°C is complemented in cells expressing either of the Fab1p point mutant constructs. However, although the fab1G2042/ 2045V strain grows even at 38°C, the fab1D2134R strain does not. Similarly, rescue of the vacuole acidification defect as measured by quinacrine staining of the vacuoles in the fab1 mutants also varied. Vacuolar quinacrine fluorescence in both the fab1Δ2 and the fab1D2134R strains is extremely weak, indicating a severe acidification defect. However, the fab1G2042/2045V vacuole appears to have the same fluorescent intensity as the wild-type control (data not shown). By FM4-64 staining of the vacuolar membrane and Nomarski optics, the yeast vacuole appears as a multilobed structure. However, the vacuolar phenotype of the fab1G2042/2045V and fab1D2134R strains are similar to that of the fab1 null strain (Yamamoto et al., 1995) or the fab1tsf after a 30-min shift to the nonpermissive temperature (Fig. 6). The vacuole in these mutants appears as a single grossly enlarged organelle that comprises the majority of the cell volume. Smaller compartments were also revealed immediately adjacent to the vacuole in the fab1D2134R strain (Fig. 6) At the permissive temperature however, the vacuole morphology of the fab1tsf strain is indistinguishable from the wild-type control (Fig. 6).
Figure 6.

fab1 mutant cells have an abnormal vacuole morphology. In the right panels, vacuoles were observed by Nomarski optics. On the left, the same fields are shown under fluorescent illumination (rhodamine channel). Cells were grown to mid-log phase at 24°C and the vacuoles labeled with the vital stain FM4-64 for 15 min (see Materials and Methods). After labeling, all of the strains were chased in the absence of dye for 1 h at 24°C except a duplicate of the fab1tsf strain that was chased at the nonpermissive temperature of 38°C.
Electron microscopic analysis not only highlights the enlarged vacuolar phenotype resulting from decreases in PtdIns(3,5)P2 synthesis, but also shows the differential effects of Fab1p kinase domain point mutations on vacuole morphology. The electron-dense vacuole of the wild-type and fab1G2042/2045V strains contrasts greatly with the electron-transparent nature of the vacuole in the fab1Δ1 and fab1D2134R strains (Fig. 7). The opacity of the vacuole is likely to be a measure of vacuole acidification, as inactivation of the vacuolar ATPase results in electron-transparent vacuoles (Wurmser and Emr, 1998). Thus, the relatively severe acidification defect in the fab1D2134R mutant compared to the fab1G2042/2045V mutant strain correlates with the levels of PtdIns(3,5)P2 synthesis. Fab1p kinase activity is therefore not only essential for maintenance of vacuolar size and acidification, but may also influence temperature-sensitive growth.
Figure 7.
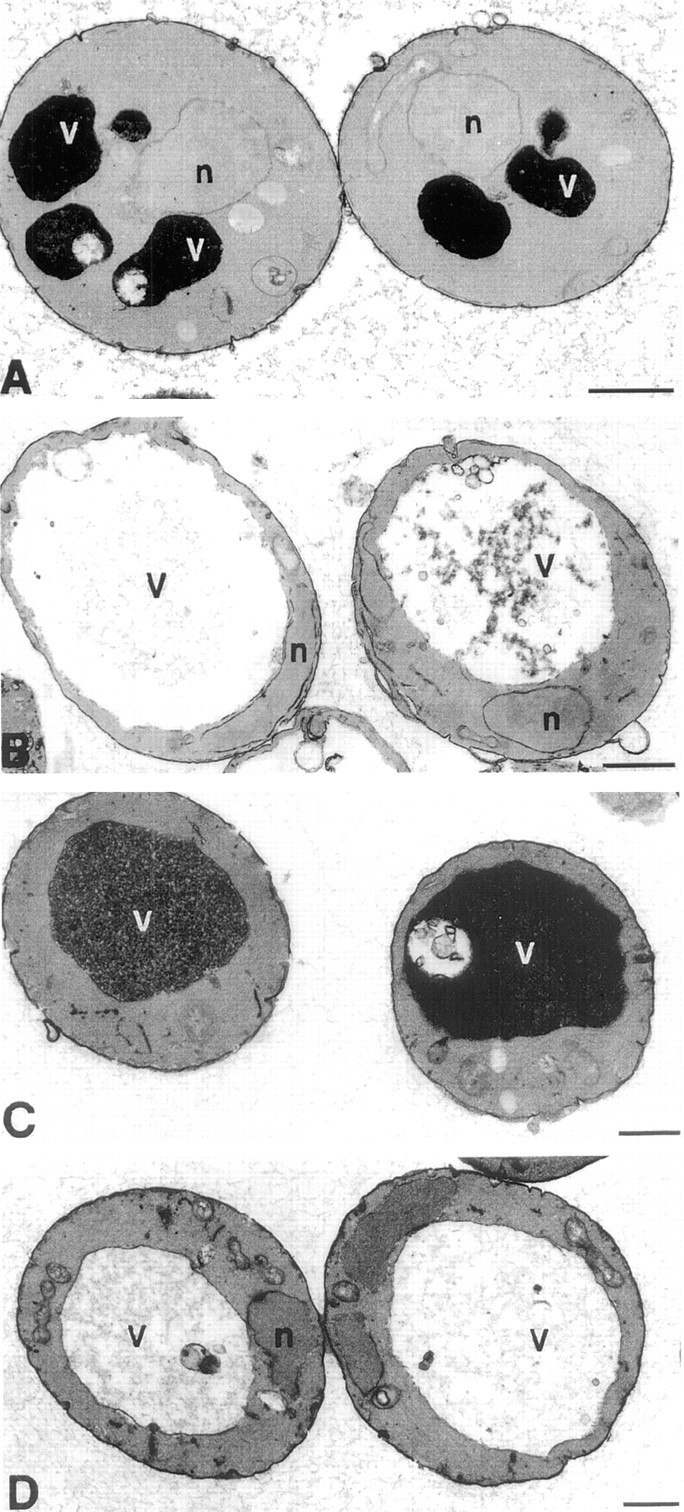
Electron microscopy of fab1 mutants. Cells were grown to mid-log phase and fixed with 3% glutaraldehyde. Cells were then converted to spheroplasts and visualized by electron microscopy (see Materials and Methods). (A) Wild-type (SEY6210) cells. (B) fab1Δ1 cells. (C) fab1G2042/2045V cells. (D) fab1D2134R cells. Bar, 0.5 μm.
Fab1p Kinase Activity Is Not Required for Proper Vacuolar Hydrolase Sorting
The Vps34p PtdIns 3-kinase is crucial in Golgi to vacuole transport of hydrolases, and recent evidence suggests that one function of PtdIns(3)P is to regulate RING-FYVE-containing proteins (i.e., Vac1p and Vps27p) that are essential for hydrolase transport from the Golgi to the vacuole (Weisman and Wickner, 1992; Piper et al., 1995; Burd et al., 1997). The finding that PtdIns(3)P can serve as a substrate for a PtdIns(3)P 5-kinase raises the possibility that the requirement for Vps34p in vacuolar protein transport arises through its role in the production of PtdIns(3,5)P2 (Dove et al., 1997). Our findings that PtdIns(3,5)P2 levels are specifically and dramatically decreased in fab1 mutant strains provides a direct means to assess this hypothesis.
Several trafficking routes from the Golgi to the yeast vacuole have been characterized. Soluble CPY and the type II integral membrane protein carboxypeptidase S, CPS, transit to the vacuole via an endosomal-dependent trafficking route, the CPY pathway (Banta et al., 1988). Alkaline phosphatase (ALP) however, arrives at the vacuole by a distinct pathway that bypasses the endosome, the ALP pathway (Cowles et al., 1997a ,b). A third route for protein delivery to the vacuole delivers the protease aminopeptidase I (API) from the cytoplasm to the vacuole through autophagy (Scott et al., 1997). To address the effects that the loss of PtdIns(3,5)P2 synthesis may have on each pathway, we assayed the proteolytic maturation of representative hydrolases from the three known pathways in the fab1 mutant strains. The conversion of the precursor hydrolase to the mature form occurs upon delivery of the hydrolase to a proteolytically competent vacuole.
Data from the original fab1-2 temperature sensitive allele supports the idea that PtdIns(3,5)P2 is not required for anterograde flow of traffic to the vacuole via the CPY pathway. After a 60-min preshift at the nonpermissive temperature, CPY was properly sorted and processed (Yamamoto et al., 1995). Our data with the fab1tsf strain also indicate that the other transport pathways to the vacuole do not depend on Fab1p function. As shown for CPY, ALP is also properly matured (Fig. 8 A) after a 30-min preshift to the nonpermissive temperature. The processing of two additional vacuolar hydrolases, CPS and API, was also found to proceed as in wild-type cells (data not shown). This sorting data from the fab1tsf strain (Fig. 8 A) at the nonpermissive temperature combined with the PtdIns(3,5)P2 quantitation (Fig. 2 B) from the same strain under similar temperature shift conditions indicates that Fab1p and PtdIns(3,5)P2 are not directly involved in the CPY, ALP, or API sorting pathways. In contrast, after only a 5-min preshift to the nonpermissive temperature, the vps34tsf missorts and secretes >95% of the pulse-labeled CPY (Stack et al., 1995).
Figure 8.

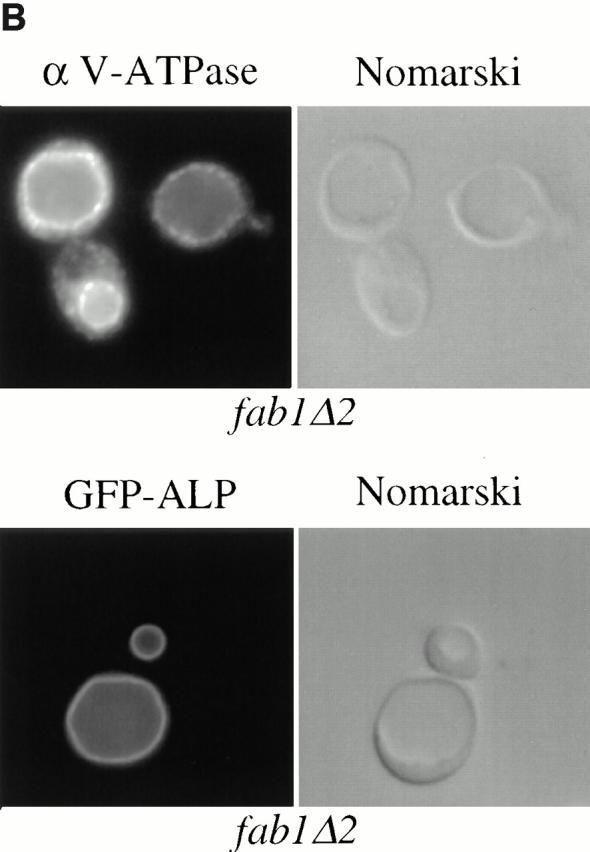
Intracellular sorting of vacuolar hydrolases in fab1 mutant cells. Yeast cells were metabolically labeled with Express [35S]-protein labeling mix during a 10-min pulse and then chased in the presence of an excess of nonradiolabeled methionine and cysteine for 0, 15, or 30 min (see Materials and Methods). Each of the vacuolar enzymes was immunoprecipitated, followed by SDS-PAGE and autoradiography. The migration positions of the precursor and mature forms of the hydrolases are indicated on the left side of the figure panels. (A) The maturation of CPY and ALP was examined in the fab1tsf strain compared with wild-type, as described in Materials and Methods. However, for the nonpermissive temperature, cells were preshifted to 38°C for 30 min before labeling and maintained at the elevated temperature throughout the chase period. (B) The upper panel, on the left, shows indirect immunofluorescence localization of the 60-kD subunit of the vacuolar ATPase using a monoclonal antibody in the fab1Δ2 strain. For the immunofluorescence, cells were fixed overnight, spheroplasted, and then incubated with antibody as described in Materials and Methods. The lower panel, on the left, shows GFP-ALP localization in a fab1Δ2 strain, observed by fluorescence microscopy (see Materials and Methods). On the right side for both panels are the identical fields observed under Nomarski optics. (C) The maturation of the vacuolar hydrolases CPS and ALP in the two point mutant strains (fab1G2042/2045V and fab1D2134R) and the wild-type strain SEY6210. The labeling and chase were done as in A however, the cells were maintained at 24°C.
As an additional test for protein trafficking to the vacuole, we used indirect immunofluorescence in fab1Δ2 cells with monoclonal antibody against the 60-kD vacuolar ATPase subunit (V1 complex). This subunit is found distributed on the vacuolar membrane in wild-type cells and arrives at the vacuole via the CPY pathway (Rieder et al., 1996; Piper et al., 1997). In fab1Δ2 cells, the fluorescence for the 60-kD vacuolar ATPase subunit is coincident with the vacuolar membrane observed by Nomarski optics (Fig. 8 B). The ATPase subunit is similarly localized in the original fab1-2 temperature-sensitive strain after incubation at the nonpermissive temperature for 2 h (Bonangelino et al., 1997). Similarly, we used a GFP-ALP construct to localize ALP in the fab1Δ2 mutants. GFP-ALP also was properly localized to the vacuolar membrane in the fab1Δ2 cells (Fig. 8 B). Therefore, Fab1p function, and PtdIns(3,5)P2 synthesis, is not essential for delivery of biosynthetic cargo to the vacuole via either the CPY or ALP transport pathways.
In the fab1G2042/2045V and fab1D2134R strains, with reduced and undetectable steady-state levels of PtdIns(3,5)P2, respectively, we assessed the proteolytic processing of both CPS and ALP (Fig. 8 C). In both mutant strains, ALP maturation occurs at a rate indistinguishable from wild-type, even at the 15-min chase point (Fig. 8 C). Compared to the control, CPS processing also occurs with wild-type kinetics in fab1G2042/2045V mutant cells (Fig. 8 C). However, there is a significant kinetic delay in CPS maturation in fab1D2134R cells (Fig. 8 C). Based on chase times extending to 120 min (data not shown), the half-time for processing CPS to the mature form in wild-type and fab1G2042/2045V cells is approximately 20 min at 24°C, whereas in the fab1D2134R strain it is >120 min. Mature API is also detectable in both of these mutants, however with the fab1G2042/2045V mutant, there is a slight kinetic delay in maturation that is more severe in the fab1D2134R mutant (data not shown). These data indicate that all three vacuolar transport pathways are generally intact, as a significant pool of hydrolases delivered to the vacuole by these routes are matured independent of Fab1p kinase activity.
The kinetic delay in hydrolase maturation observed in the fab1Δ (Yamamoto et al., 1995) or point mutant strains (Fig. 8 C) may be due to vacuolar acidification defects that result from the loss of Fab1p kinase activity. Loss of vacuole acidification is known to reduce the activity of proteinase A (Sorensen et al., 1994), a vacuolar protease that plays a key role in the maturation of many vacuolar hydrolases (Morano and Klionsky, 1994; Van Den Hazel et al., 1996). As a result, cells lacking vacuolar ATPase function exhibit kinetic delays in the conversion of many hydrolases to their mature forms (Morano and Klionsky, 1994). As assessed by quinacrine staining, the lipid kinase activity of Fab1p is also required for proper vacuole acidification. The normal quinacrine fluorescence in the fab1G2042/2045Vstrain (data not shown), indicates that even low levels of PtdIns(3,5)P2 are sufficient to maintain vacuolar pH. However, the fab1D2134R mutant strain shows a severe vacuole acidification defect (data not shown). The processing delays observed in the fab1 point mutants are likely to be directly due to the slow maturation of certain hydrolases in a compromised vacuole. Consistent with this hypothesis, the degree of vacuole acidification in the two point mutants correlates well with the hydrolase maturation delays observed in these strains.
Vac7p Is Required for PtdIns(3,5)P2 Synthesis
A number of observations suggest that an upstream regulatory factor(s) may be required for Fab1p activity. First, overexpression of FAB1 from a multicopy vector fails to cause elevations in endogenous PtdIns(3,5)P2 (data not shown), suggesting that the concentration of Fab1-mediated synthesis of PtdIns(3,5)P2 requires a limiting cofactor. Furthermore, immunoprecipitations of Fab1p from yeast protein extracts did not contain detectable PtdIns(3)P 5-kinase activity (data not shown), possibly due to the disassociation of Fab1p from an activator protein(s) during immunopurification. Finally, several of the recently described vac mutants exhibit phenotypes which are strikingly similar to the fab1 mutants, VAC7 is the best characterized. Like fab1, vac7 mutants exhibit acidification defects and an enlarged vacuolar phenotype (Bonangelino et al., 1997). This raises the possibility that Fab1p and Vac7p function together to regulate a common cell function (i.e., vacuole membrane homeostasis). To further assess whether Vac7p functions in the Fab1p pathway, we examined the morphology of vac7Δ cells by electron microscopy and assayed a vac7 mutant for changes in PtdIns(3,5)P2 levels.
Relative to wild-type cells, which exhibited two to three electron-dense staining vacuoles when examined by electron microscopy, the vac7Δ (LWY1481) strain has a single enlarged vacuole that appears less electron dense (Fig. 7 and 9 A). Heterogeneous intravacuolar structures appear more prominently in vac7Δ cells than in fab1Δ1 cells (Fig. 7 B and 9 A). It is also important to note that aberrant membranes (vesicles and endosomal intermediates) do not appear to accumulate in the cytoplasm of the fab1Δ1 or vac7Δ cells, a hallmark of many mutants which have Golgi to vacuole transport defects (Cowles et al., 1994; Babst et al., 1997).
As the similar morphological defects of fab1Δ1 and vac7Δ cells suggest, if Fab1p and Vac7p function in the same pathway, then the mutation of VAC7 might also have effects on PtdIns(3,5)P2 levels. Therefore, we analyzed PtdIns(3,5)P2 in a vac7 mutant strain. Wild-type and vac7Δ cells were labeled 12 h with myo-[2-3H]inositol, osmotically shocked for 10 min with 0.9 M NaCl, lysed and analyzed by HPLC as described earlier. PtdIns(3,5)P2 (5,300 cpm total) was observed in wild-type cells, however, PtdIns(3,5)P2 was undetectable in vac7Δ cells (<50 cpm; Fig. 9 B). As seen in fab1Δ1 cells (Fig. 2 A), the vac7Δ mutant also exhibits a ∼50% decrease PtdIns(4,5)P2 (Fig. 9 B), 100% increase in PtdIns(3)P and a 50% decrease in PtdIns(4)P (data not shown). Vac7p is not homologous to any other known protein and does not contain kinase motifs (Bonangelino et al., 1997). These data suggest that Vac7p, possibly in association with Fab1p, is required for PtdIns(3,5)P2 synthesis.
Figure 9.


The vac7Δ strain displays phenotypes similar to the fab1Δ1 strain. (A) Electron microscopy of vac7Δ cells prepared as in Fig. 6. (B) Wild-type and vac7Δ cells were labeled 12 h with myo-[2-3H]inositol and osmotically shocked with 0.9 M NaCl for 10 min at 22°C. Each strain was then lysed and the extracted cellular lipids deacylated and separated by HPLC as in Fig. 2. Deacylated products representing PtdIns(3,5)P2 and PtdIns(4,5)P2 are indicated. The arrow indicates the expected position of PtdIns(3,5)P2. The data are representative of several experiments. Bar, 0.5 μm.
Discussion
Fab1p Kinase Activity Is Required for PtdIns(3,5)P2 Synthesis in S. cerevisiae
Sequence comparisons of the putative lipid kinase domain of Fab1p (a.a. 2,022–2,263) with other kinases of known enzymatic activities categorized Fab1p into a unique subclass of general PtdInsP kinases (Fig. 1 B). Two distinct doubly phosphorylated derivatives of PtdIns have been identified in yeast, PtdIns(4,5)P2 and PtdIns(3,5)P2, and the enzyme activities involved in their stepwise synthesis already have been suggested (Fig. 1 A). Although its identity was not known, the existence of a specific PtdIns(3)P 5-kinase is supported by the sequential synthesis of PtdIns(3,5)P2 in murine Ph cells from PtdIns(3)P, as well as the requirement for Vps34p activity in yeast for PtdIns(3,5)P2 production (Dove et al., 1997; Whiteford et al., 1997). Given that the sequence of Fab1p kinase domain distinguishes it from the Type I and Type II PtdIns(4)P 5-kinases, Fab1p is the likely PtdIns(3)P 5-kinase in yeast.
Consistent with this hypothesis, our data show that fab1Δ1 cells have undetectable levels of PtdIns(3,5)P2, whereas the amounts of other phosphoinositides are only moderately affected (Fig. 2 A). To determine if this decrease in PtdIns(3,5)P2 is the direct result of Fab1p inactivation, we assessed the phosphoinositide levels in a strain harboring a temperature-sensitive allele of FAB1. Despite the fact that this mutant protein is stable during the shift to the nonpermissive temperature, cellular PtdIns(3,5)P2 levels show a rapid decrease, reaching undetectable levels within 5 min at 38°C, whereas the amounts of other phosphoinositides do not change relative to wild-type cells treated in the same manner (Fig. 2 B). The differential loss of PtdIns(3,5)P2 in cells expressing either of the two kinase domain point mutant constructs also provides evidence that Fab1p is the PtdIns(3)P 5-kinase (Fig. 4). By comparison with known structures of protein kinases, the glycine residues replaced in the Fab1p G2042/2045V mutant are located in a conserved glycine-rich loop region between two β-strands that form the ATP-positioning motif (Sicheri et al., 1997; Xu et al., 1997). Mutagenesis of these residues in cAPK and the insulin receptor results in reduced kinase activity (Hemmer et al., 1997; Odawara et al., 1989). Therefore, the residual PtdIns(3,5)P2 synthesis observed in the fab1G2042/2045V strain is consistent with Fab1p directly catalyzing the synthesis of PtdIns(3,5)P2 from PtdIns(3)P. Additionally, mutation of the aspartate residue at position 2,134 eliminates the production of PtdIns(3,5)P2. This aspartate is analogous to the catalytically critical Asp166 of cAPK (Bossemeyer et al., 1993). Mutation of the comparable residue in the v-fps and c-kit protein kinases as well as the yeast lipid kinase Vps34p results in the loss of kinase activity (Moran et al., 1988; Tan et al., 1990; Schu et al., 1993).
Although the in vivo lipid quantitation data from the fab1 mutants provide strong evidence that Fab1p is the functionally relevant PtdIns(3)P 5-kinase in vivo, it has not yet been possible to demonstrate the activity in vitro. Immunopurification of Fab1p from native yeast extracts did not contain detectable PtdIns(3)P 5-kinase activity (data not shown), nor were we able to demonstrate the production of PtdIns(3,5)P2 in crude yeast lysates (data not shown). We suggest that these negative in vitro results may be due to the instability or lack of necessary cofactors required for Fab1p 5-kinase activity (see below) or the relative inefficiency of the enzyme under our in vitro conditions.
Vac7p as an Upstream Regulator of Fab1p
The catalytic activity and localization of most if not all phosphoinositide kinases are controlled by interactions with regulatory proteins in vivo. The p85/p110 phosphoinositide 3-kinase translocates to the plasma membrane through its interaction with the cytosolic tail of ligand-bound tyrosine kinase receptors (Hu et al., 1992), whereas the p110 phosphoinositide 3-kinase γ-isoform is activated in response to heterotrimeric G-protein–coupled receptor stimulation (Stoyanov et al., 1995; Stephens et al., 1997). Similarly, the Vps34p PtdIns 3-kinase binds a membrane associated serine/threonine protein kinase, Vps15p (Stack et al., 1993, 1995). This interaction localizes Vps34p to an intracellular membrane compartment and activates its PtdIns 3-kinase activity (Schu et al., 1993; Stack et al., 1995). We have made several observations which point to the existence of a protein cofactor(s) that is required for Fab1p activity. First, cells overexpressing Fab1p failed to exhibit increases in PtdIns(3,5)P2 levels, suggesting that a protein critical for Fab1p activation may be limiting. Similarly, overexpression of Vps34p does not elevate cellular PtdIns(3)P levels (data not shown), presumably due to the limiting pool of activated Vps15p available to properly recruit and regulate Vps34p activity. Second, the lack of detectable PtdIns(3)P 5-kinase activity in Fab1p in vitro experiments also suggests that stimulatory signals from an upstream regulator may be absent from immunoprecipitated Fab1p or unstable in crude lysates.
The identification of a possible upstream activator for Fab1p came from a genetic screen in yeast for mutants exhibiting aberrant vacuole segregation structures during cytokinesis (Wang et al., 1996). This screen revealed several complementation groups displaying the enlarged vacuolar phenotypes of fab1 cells. Indeed FAB1 was isolated numerous times as well as a previously uncharacterized gene, VAC7 (Bonangelino et al., 1997). To test the possibility that Vac7p and Fab1p function in the same pathway, we assayed phosphoinositide levels in vac7Δ cells and found that PtdIns(3,5)P2 was undetectable in this mutant strain (Fig. 9 B). Thus, Vac7p activity, like Fab1p, is required for PtdIns(3,5)P2 production, suggesting that Vac7p functions upstream of Fab1p. Vac7p encodes a 1,165 a.a. protein that is enriched in vacuole membranes and bears no homology to lipid kinases (Bonangelino et al., 1997). It contains a potential transmembrane domain (a.a. 919–943) and is not stripped from the membrane fraction with high NaCl, urea, or Na2CO3 treatment indicating that Vac7p is an intergral membrane protein. Vac7p however, is unlikely to be critical for localizing Fab1p to the membrane as its deletion failed to increase the soluble pool of Fab1p (unpublished observation). This suggests that Vac7p may function to stimulate Fab1p PtdIns(3)P 5-kinase activity rather than recruit Fab1p to the membrane.
Other members of the FAB1/VAC7 gene family are likely to exist. In fact, a complementation group distinct from fab1 and vac7, designated vac14-1, has been identified that shares the vacuole acidification and morphological defects observed in fab1 and vac7 mutants (Bonangelino et al., 1997). Furthermore, in the vac14-1 mutant, both these defects are suppressed by the overexpression of FAB1 (Bonangelino et al., 1997). Therefore, Vac14p represents another protein that is likely to function upstream of Fab1p. Consistent with this model, vac14-1 mutant cells are also severely compromised in their ability to synthesize PtdIns(3,5)P2 (unpublished observations). Efforts to clone VAC14 are currently underway. Interestingly, overexpression of FAB1 does not suppress the mutant phenotypes of vac7Δ cells (Bonangelino et al., 1997). Collectively, these data suggest that Fab1p, Vac7p and Vac14p function in a common signal transduction pathway which, through the activation of Fab1p, results in the localized production of PtdIns(3,5)P2.
It has been previously demonstrated that a hyperosmotic shock is required for maximal PtdIns(3,5)P2 accumulation in yeast. This was suggested to be due to the activation of a PtdIns(3)P 5-kinase (Dove et al., 1997). Our data does not exclude the possibility that Fab1p, or its putative upstream activators Vac7p and Vac14p, are sensitive to hyperosmotic stress. However, enzymes dedicated to the turnover of PtdIns(3,5)P2, possibly corresponding to one or more of the four PtdIns(5)P 5-phosphatase homologs in yeast (Srinivasan et al., 1997; Stolz et al., 1998), also represent candidate activities that may be sensitive to hyperosmotic stress. The further characterization of upstream activators, such as Vac7p and Vac14p, and the biologically relevant PtdIns(3,5)P2 turnover activity should help in distinguishing between these possibilities.
Potential Intracellular Roles for PtdIns(3,5)P2
With the identification of PtdIns(3,5)P2 in yeast, it was suggested that this lipid, rather than PtdIns(3)P, plays a role in Golgi to vacuole trafficking (Dove et al., 1997). Although previous data indicates the essential nature of PtdIns(3)P in anterograde traffic to the vacuole (Schu et al., 1993), it was argued that the apparent requirement for Vps34p in this pathway reflects the need for PtdIns(3)P solely as a substrate for the 5-kinase activity. However, our data show that Fab1p, and therefore PtdIns(3,5)P2 synthesis, appears not to play a role in anterograde protein traffic, as the delivery of proteins along all known vacuolar transport pathways are unaffected in fab1 mutants. fab1 mutant cells do give rise to several phenotypes including, temperature-sensitive growth, a vacuolar acidification defect, as well as an inability to properly segregate genomic and vacuolar material upon cytokinesis (Yamamoto et al., 1995). The later phenotypes appear to be secondary consequences resulting from the enlargement of the vacuole, which is the most immediate defect caused by the inactivation of the fab1tsf. Indeed, the normal vacuole morphology observed in the fab1tsf at the permissive temperature contrasts dramatically with the enlarged vacuole phenotype of the fab1G2042/2045V strain, suggesting that a threshold level of PtdIns(3,5)P2 production is required for proper vacuolar morphology.
We envision three possible models for how Fab1p activity is responsible for maintenance of vacuolar size. (a) PtdIns(3,5)P2 may be required for vacuole ion homeostasis. Aside from its function as a site for macromolecular degradation, the vacuole is an important ion storage site which plays a role in regulating intracellular ion concentrations (Klionsky et al., 1990). Therefore, PtdIns(3,5)P2 could be an activator of a vacuolar ion transporter and loss of this lipid could affect vacuolar morphology through changes in its osmolarity with respect to the cytoplasm. In mammalian cells, PtdIns(4,5)P2 has been shown to be a positive regulator of inward-rectifying potassium channels (Huang et al., 1998; Sui et al., 1998). (b) Alternatively, PtdIns(3,5)P2 could regulate the turnover of vacuolar membranes by mediating its invagination at the vacuole (or endosome), allowing for degradation within the vacuole lumen. Invaginations at the vacuole or endosome have been suggested by the presence of lumenal vesicles within the vacuole (Wurmser and Emr, 1998) and endosome (multivesicular body; Gruenberg and Maxfield, 1995). Consistent with this idea, fab1Δ1 and fab1D2134R kinase domain mutant strains have significantly fewer intravacuolar structures. (c) PtdIns(3,5)P2 may direct the efflux of vacuolar membranes to earlier secretory compartments. According to this model vacuolar membrane homeostasis requires a balance between anterograde traffic (mediated by PtdIns(3)P) and retrograde traffic (perhaps through PtdIns(3,5)P2; Fig. 10). Disruption of PtdIns(3,5)P2 synthesis might prevent membrane recycling and result in the accumulation of membranes at the vacuole. This predicts that PtdIns(3,5)P2 may be required for the formation or docking/fusion of retrograde carrier vesicles emanating from the vacuole. Indeed, the existence of a retrograde pathway from the vacuolar membrane to earlier secretory compartments has also been suggested based on the recycling of a recombinant protein construct from the vacuole to the Golgi (Bryant et al., 1998).
Figure 10.

(A) The pathway for the biosynthesis of PtdIns(3,5)P2 in yeast. The Vps15/34p-dependent phosphorylation of PtdIns leads to the production of PtdIns(3)P. A pool of this lipid then either traffics to the lumen of the vacuole for degradation or remains cytoplasmically exposed, serving as a substrate for a second, Fab1p kinase domain-dependent phosphotransfer reaction resulting in the synthesis of PtdIns(3,5)P2. Below the phosphorylated lipid products, roles for these lipids are indicated. PtdIns(3)P is required in anterograde traffic from the Golgi to the vacuole, whereas PtdIns(3,5)P2 may regulate the efflux of vacuolar membrane material. (B) A model for Fab1p-mediated regulation of vacuolar membrane recycling/turnover. Activation of Vps34p by the membrane associated lipid kinase Vps15p, leads to the site-specific synthesis of PtdIns(3)P at the Golgi/endosomal membrane. The production of PtdIns(3)P is not only required for this biosynthetic trafficking route (highlighted by the solid arrows), but also is used by the Fab1p lipid kinase for the synthesis of PtdIns(3,5)P2. A large pool of PtdIns(3)P is turned over in a hydrolase-dependent manner, possibly by being sorted into the inner leaflet of endosomal invaginations which are transported to the lumen of the vacuole and, together with PtdIns(3)P, degraded. A separate pool of PtdIns(3)P must remain in the cytoplasmic membrane leaflet in order to be available to Fab1p. The presence of PtdIns(3,5)P2 may then serve to recruit factors involved in the efflux of membrane to earlier compartments (top, dashed lines) or cause the internalization of vacuolar membrane into the lumen for degradation.
Therefore, this model suggests that PtdIns(3)P may itself act as an internal membrane marker which is synthesized early in the Golgi to vacuole transport pathway and imprints the carrier vesicles with a tag that is subsequently decoded/recognized at a distal site in the pathway. Fab1p may recognize and convert this anterograde-specific lipid tag into a second signaling lipid that then directs vacuolar membrane efflux or turnover. Recently, however, it has been shown that a significant fraction of PtdIns(3)P is delivered to the vacuole as cargo where it is degraded by lumenal hydrolases (Wurmser and Emr, 1998). This necessitates that while a pool of PtdIns(3)P is transferred to the lumen of the vacuole for degradation, a separate or earlier pool of PtdIns(3)P remains available as a substrate for Fab1p in the cytoplasmic leaflet (Wurmser and Emr, 1998; Burd and Emr, 1998). This sequential signaling system at distinct organelles in the Golgi to vacuolar signaling pathway would allow the cell to maintain vacuole size/function (homeostasis).
The involvement of phosphoinositides in the spatial and temporal regulation of membrane trafficking argues that the various phosphorylated derivatives must be synthesized and degraded at specific sites along the pathway. For instance, the lipid PtdIns(4,5)P2 may not only direct vesicle fusion (Hay and Martin, 1993; Hay et al., 1995), but its conversion by a PtdIns(5)P 5-phosphatase may also play a role in synaptic vesicle endocytosis (McPherson et al., 1994, 1996). Similarly, mutations within the PDGF tyrosine kinase receptor that prevent the recruitment of the mammalian phosphoinositide 3-kinase cause defects in the sorting and downregulation of this receptor (Kapeller et al., 1994). Interestingly, the internalization of the receptor occurs normally, indicating that the subsequent endosome to lysosome trafficking step is affected. Thus, the regulated synthesis and turnover of specific lipids at distinct organelle sites is required for the regulation of membrane trafficking. The isolation of Vps34p as a vacuolar protein sorting mutant lead to the hypothesis that the regulated production PtdIns(3)P on a specific membrane directly effects or recruits necessary factors for proper trafficking (Stack et al., 1993). Candidate downstream targets of PtdIns(3)P in yeast have already been identified, in part, based on the observation that the mammalian EEA1 gene product specifically interacts with PtdIns(3)P-containing liposomes (Patki et al., 1997). This interaction has now been determined to be mediated by the RING-FYVE finger domain (Burd and Emr, 1998) that is also present in Vps27p and Vac1p, proteins previously shown to be required for Golgi to vacuole protein trafficking (Piper et al., 1995; Burd et al., 1997). The binding of PtdIns(3)P by this domain may serve to recruit these proteins to a specific membrane site and/or modulate the activity of these and other transport factors. Interestingly, this domain is also present at the NH2 terminus of Fab1p and may function to localize (and/or activate) Fab1p to membranes containing its substrate, PtdIns(3)P.
Specific downstream effects of phosphoinositides are often mediated by proteins which bind directly to specific lipid ligands (Martin, 1997). Regulation of membrane homeostasis by Fab1p is likely to be mediated by a set of proteins that specifically interact with PtdIns(3,5)P2. These PtdIns(3,5)P2-specific binding proteins could correspond with proteins that commonly play roles in membrane trafficking events, such as coat proteins, SNAREs, or rab GTPases. As the ongoing VAC mutant screen has been successful in defining candidate upstream regulators of Fab1p activity, it also promises to be instrumental in identifying downstream effectors of Fab1p. Our results with Fab1p confirm the use of yeast in defining signal transduction pathways on the basis of an observable phenotype and ordering these factors within the pathway based on in vivo biochemical analyses.
Acknowledgments
We thank Michael McCaffrey and Chris Hofeditz for carrying out electron microscope analysis and figure preparation (Immunoelectron microscopy Core B of Program Project grant CA58689 headed by M. Farquar). We also appreciate the helpful scientific discussions with Chris Burd, Markus Babst, and Tamara Darsow.
This work was supported by NIH grants CA58689 (S.D. Emr and A.E. Wurmser) and GM50403 (L.S. Weisman). S.D. Emr is supported as an investigator of the Howard Hughes Medical Institute.
Abbreviations used in this paper
- a.a.
amino acid
- ALP
alkaline phosphatase
- API
aminopeptidase I
- CPS
carboxypeptidase S
- CPY
carboxypeptidase Y
- G6PHD
glucose 6-phosphate dehydrogenase
- PITP
PtdIns transfer protein
- PtdIns(4)P
phosphatidylinositol 4-phosphate
Footnotes
The first two authors contributed equally to this work.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger KR, Carpenter CL, Cantley LC, Varticovski L. Phosphatidylinositol 3-kinase and its novel product, phosphatidylinositol 3-phosphate, are present in Saccharomyces cerevisiae. . J Biol Chem. 1989;264:20181–20184. [PubMed] [Google Scholar]
- Babst M, Sato TK, Banta LM, Emr SD. Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. EMBO (Eur Mol Biol Organ) J. 1997;16:1820–1831. doi: 10.1093/emboj/16.8.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis VA, Aitken JR, Cleves AE, Dowhan W. An essential role for a phospholipid transfer protein in yeast Golgi function. Nature. 1990;347:561–562. doi: 10.1038/347561a0. [DOI] [PubMed] [Google Scholar]
- Banta LM, Robinson JS, Klionsky DJ, Emr SD. Organelle assembly in yeast: characterization of yeast mutants defective in vacuolar biogenesis and protein sorting. J Cell Biol. 1988;107:1369–1383. doi: 10.1083/jcb.107.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes WM. PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates. Proc Natl Acad Sci USA. 1994;91:2216–2220. doi: 10.1073/pnas.91.6.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonangelino CJ, Catlett NL, Weisman LS. Vac7p, a novel vacuolar protein, is required for normal vacuole inheritance and morphology. Mol Cell Biol. 1997;17:6847–6858. doi: 10.1128/mcb.17.12.6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boronenkov IV, Anderson RA. The sequence of phosphatidylinositol-4-phosphate 5-kinase defines a novel family of lipid kinases. J Biol Chem. 1995;270:2881–2884. doi: 10.1074/jbc.270.7.2881. [DOI] [PubMed] [Google Scholar]
- Bossemeyer D, Engh RA, Kinzel V, Ponstingl H, Huber R. Phosphotransferase and substrate binding mechanism of the cAMP-dependent protein kinase catalytic subunit from porcine heart as deduced from the 2.0 A structure of the complex with Mn2+adenylyl imidodiphosphate and inhibitor peptide PKI(5-24) EMBO (Eur Mol Biol Organ) J. 1993;12:849–859. doi: 10.1002/j.1460-2075.1993.tb05725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WJ, DeWald DB, Emr SD, Plutner H, Balch WE. Role for phosphatidylinositol 3-kinase in the sorting and transport of newly synthesized lysosomal enzymes in mammalian cells. J Cell Biol. 1995;130:781–796. doi: 10.1083/jcb.130.4.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant NJ, Piper RC, Weisman LS, Stevens TH. Retrograde traffic out of the yeast vacuole to the TGN occurs via the prevacuolar/endosomal compartment. J Cell Biol. 1998;142:651–663. doi: 10.1083/jcb.142.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CG, Emr SD. Phosphatidylinositol(3)-phosphate signaling mediated by specific binding to RING FYVE domains. Mol Cell. 1998;2:157–162. doi: 10.1016/s1097-2765(00)80125-2. [DOI] [PubMed] [Google Scholar]
- Burd CG, Peterson M, Cowles CR, Emr SD. A novel Sec18p/ NSF-dependent complex required for Golgi-to-endosome transport in yeast. Mol Biol Cell. 1997;8:1089–1104. doi: 10.1091/mbc.8.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles C, Emr S, Horazdovsky B. Mutations in the VPS45 gene, a SEC1 homologue, result in vacuolar protein sorting defects and accumulation of membrane vesicles. J Cell Sci. 1994;107:3449–3459. doi: 10.1242/jcs.107.12.3449. [DOI] [PubMed] [Google Scholar]
- Cowles CR, Odorizzi G, Payne GS, Emr SD. The AP-3 adaptor complex is essential for cargo-selective transport to the yeast vacuole. Cell. 1997a;91:109–118. doi: 10.1016/s0092-8674(01)80013-1. [DOI] [PubMed] [Google Scholar]
- Cowles CR, Snyder WB, Burd CG, Emr SD, Rieder SE, Emr SD. Novel Golgi to vacuole delivery pathway in yeast: identification of a sorting determinant and required transport component. EMBO (Eur Mol Biol Organ) J. 1997b;16:2769–2782. doi: 10.1093/emboj/16.10.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson HW. Wortmannin causes mistargeting of procathepsin D. evidence for the involvement of a phosphatidylinositol 3-kinase in vesicular transport to lysosomes. J Cell Biol. 1995;130:797–805. doi: 10.1083/jcb.130.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli P, Emr SD, McPherson PS, Novick P. Phosphoinositides as regulators in membrane traffic. Science. 1996;271:1533–1539. doi: 10.1126/science.271.5255.1533. [DOI] [PubMed] [Google Scholar]
- Desrivieres S, Cooke FT, Parker PJ, Hall MN. MSS4, a phosphatidylinositol-4-phosphate 5-kinase required for organization of the actin cytoskeleton in Saccharomyces cerevisiae. . J Biol Chem. 1998;273:15787–15793. doi: 10.1074/jbc.273.25.15787. [DOI] [PubMed] [Google Scholar]
- Dove SK, Cooke FT, Douglas MR, Sayers LG, Parker PJ, Michell RH. Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature. 1997;390:187–192. doi: 10.1038/36613. [DOI] [PubMed] [Google Scholar]
- Flanagan CA, Schnieders EA, Emerick AW, Kunisawa R, Admon A, Thorner J. Phosphatidylinositol 4-kinase: gene structure and requirement for yeast cell viability. Science. 1993;262:1444–1448. doi: 10.1126/science.8248783. [DOI] [PubMed] [Google Scholar]
- Gaynor EC, te Heesen S, Graham TR, Aebi M, Emr SD. Signal-mediated retrieval of a membrane protein from the Golgi to the ER in yeast. J Cell Biol. 1994;127:653–665. doi: 10.1083/jcb.127.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BD, Hemmer W, Tsigelny I, Adams JA, Taylor SS. Kinetic analyses of mutations in the glycine-rich loop of cAMP-dependent protein kinase. Biochemistry. 1998;37:7708–7715. doi: 10.1021/bi972987w. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Maxfield FR. Membrane transport in the endocytic pathway. Curr Opin Cell Biol. 1995;7:552–563. doi: 10.1016/0955-0674(95)80013-1. [DOI] [PubMed] [Google Scholar]
- Hay JC, Fisette PL, Jenkins GH, Fukami K, Takenawa T, Anderson RA, Martin TF. ATP-dependent inositide phosphorylation required for Ca(2+)-activated secretion. Nature. 1995;374:173–177. doi: 10.1038/374173a0. [DOI] [PubMed] [Google Scholar]
- Hay JC, Martin TF. Phosphatidylinositol transfer protein required for ATP-dependent priming of Ca(2+)-activated secretion. Nature. 1993;366:572–575. doi: 10.1038/366572a0. [DOI] [PubMed] [Google Scholar]
- Hemmer W, McGlone M, Tsigelny I, Taylor SS. Role of the glycine triad in the ATP-binding site of cAMP-dependent protein kinase. J Biol Chem. 1997;272:16946–16954. doi: 10.1074/jbc.272.27.16946. [DOI] [PubMed] [Google Scholar]
- Herman PK, Emr SD. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Saccharomyces cerevisiae. . Mol Cell Biol. 1990;10:6742–6754. doi: 10.1128/mcb.10.12.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman CS, Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. . Gene. 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- Homma K, Terui S, Minemura M, Qadota H, Anraku Y, Kanaho Y, Ohya Y. Phosphatidylinositol-4-phosphate 5-kinase localized on the plasma membrane is essential for yeast cell morphogenesis. J Biol Chem. 1998;273:15779–15786. doi: 10.1074/jbc.273.25.15779. [DOI] [PubMed] [Google Scholar]
- Horton RM, Ho SN, Pullen JK, Hunt HD, Cai Z, Pease LR. Gene splicing by overlap extension. Methods Enzymol. 1993;217:270–279. doi: 10.1016/0076-6879(93)17067-f. [DOI] [PubMed] [Google Scholar]
- Hu P, Margolis B, Skolnik EY, Lammers R, Ullrich A, Schlessinger J. Interaction of phosphatidylinositol 3-kinase-associated p85 with epidermal growth factor and platelet-derived growth factor receptors. Mol Cell Biol. 1992;12:981–990. doi: 10.1128/mcb.12.3.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapeller R, Chakrabarti R, Cantley L, Fay F, Corvera S. Internalization of activated platelet-derived growth factor receptor-phosphatidylinositol-3′ kinase complexes: potential interactions with the microtubule cytoskeleton. J Biol Chem. 1994;269:6052–6063. doi: 10.1128/mcb.13.10.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns BG, Alb JG, Jr, Bankaitis VA. Phosphatidylinositol transfer proteins: the long and winding road to physiological function. Trends Cell Biol. 1998;8:276–281. doi: 10.1016/s0962-8924(98)01281-1. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Cueva R, Yaver DS. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J Cell Biol. 1992;119:287–299. doi: 10.1083/jcb.119.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD. Membrane protein sorting: biosynthesis, transport and processing of yeast vacuolar alkaline phosphatase. EMBO (Eur Mol Biol Organ) J. 1989;8:2241–2250. doi: 10.1002/j.1460-2075.1989.tb08348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Herman PK, Emr SD. The fungal vacuole: composition, function, and biogenesis. Microbiol Rev. 1990;54:266–292. doi: 10.1128/mr.54.3.266-292.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld S, Mellman I. The biogenesis of lysosomes. Annu Rev Cell Biol. 1989;5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73:585–596. doi: 10.1016/0092-8674(93)90144-f. [DOI] [PubMed] [Google Scholar]
- Liscovitch M, Cantley LC. Signal transduction and membrane traffic: the PITP/phosphoinositide connection. Cell. 1995;81:659–662. doi: 10.1016/0092-8674(95)90525-1. [DOI] [PubMed] [Google Scholar]
- Loijens JC, Anderson RA. Type I phosphatidylinositol-4-phosphate 5-kinases are distinct members of this novel lipid kinase family. J Biol Chem. 1996;271:32937–32943. doi: 10.1074/jbc.271.51.32937. [DOI] [PubMed] [Google Scholar]
- Maniatis, T., E.F. Fritsch, and J. Sambrook. 1982. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Martin TF. Phosphoinositides as spatial regulators of membrane traffic. Curr Opin Neurobiol. 1997;7:331–338. doi: 10.1016/s0959-4388(97)80060-8. [DOI] [PubMed] [Google Scholar]
- McPherson PS, Garcia EP, Slepnev VI, David C, Zhang X, Grabs D, Sossin WS, Bauerfeind R, Nemoto Y, De Camilli P, et al. A presynaptic inositol-5-phosphatase p145, a major Grb2-binding protein in brain, is co-localized with dynamin in nerve terminals where it undergoes activity-dependent dephosphorylation. Nature. 1996;379:353–357. doi: 10.1038/379353a0. [DOI] [PubMed] [Google Scholar]
- McPherson PS, Takei K, Schmid SL, De Camilli P. p145, a major Grb2-binding protein in brain, is co-localized with dynamin in nerve terminals where it undergoes activity-dependent dephosphorylation. J Biol Chem. 1994;269:30132–30139. [PubMed] [Google Scholar]
- Moran MF, Koch CA, Sadowski I, Pawson T. Mutational analysis of a phosphotransfer motif essential for v-fps tyrosine kinase activity. Oncogene. 1988;3:665–672. [PubMed] [Google Scholar]
- Morano KA, Klionsky DJ. Differential effects of compartment deacidification on the targeting of membrane and soluble proteins to the vacuole in yeast. J Cell Sci. 1994;107:2813–2824. doi: 10.1242/jcs.107.10.2813. [DOI] [PubMed] [Google Scholar]
- Muhlrad D, Hunter R, Parker R. A rapid method for localized mutagenesis of yeast genes. Yeast. 1992;8:79–82. doi: 10.1002/yea.320080202. [DOI] [PubMed] [Google Scholar]
- Munn AL, Riezman H. Endocytosis is required for the growth of vacuolar H(+)-ATPase-defective yeast: identification of six new END genes. J Cell Biol. 1994;127:373–386. doi: 10.1083/jcb.127.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P, Zerial M. The diversity of Rab proteins in vesicle transport. Curr Opin Cell Biol. 1997;9:496–504. doi: 10.1016/s0955-0674(97)80025-7. [DOI] [PubMed] [Google Scholar]
- Odawara M, Kadowaki T, Yamamoto R, Shibasaki Y, Tobe K, Accili D, Bevins C, Mikami Y, Matsuura N, Akanuma Y, et al. Human diabetes associated with a mutation in the tyrosine kinase domain of the insulin receptor. Science. 1989;245:66–68. doi: 10.1126/science.2544998. [DOI] [PubMed] [Google Scholar]
- Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:347–358. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- Patki V, Virbasius J, Lane WS, Toh BH, Shpetner HS, Corvera S. Identification of an early endosomal protein regulated by phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1997;94:7326–7330. doi: 10.1073/pnas.94.14.7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper RC, Cooper AA, Yang H, Stevens TH. VPS27 controls vacuolar and endocytic traffic through a prevacuolar compartment in Saccharomyces cerevisiae. J Cell Biol. 1995;131:603–617. doi: 10.1083/jcb.131.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper RC, Bryant NJ, Stevens TH. The membrane protein alkaline phosphatase is delivered to the vacuole by a route that is distinct from the VPS-dependent pathway. J Cell Biol. 1997;138:531–545. doi: 10.1083/jcb.138.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameh LE, Tolias KF, Duckworth BC, Cantley LC. A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature. 1997;390:192–196. doi: 10.1038/36621. [DOI] [PubMed] [Google Scholar]
- Redding K, Holcomb C, Fuller RS. Immunolocalization of Kex2 protease identifies a putative late Golgi compartment in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 1997;8:527–538. doi: 10.1083/jcb.113.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder SE, Banta LM, Kohrer K, McCaffery JM, Emr SD. Multilamellar endosome-like compartment accumulates in the yeast vps28 vacuolar protein sorting mutant. Mol Biol Cell. 1996;7:985–999. doi: 10.1091/mbc.7.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JS, Klionsky DJ, Banta LM, Emr SD. Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol Cell Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Schmid SL. Clathrin-coated vesicle formation and protein sorting: an integrated process. Annu Rev Biochem. 1997;66:511–548. doi: 10.1146/annurev.biochem.66.1.511. [DOI] [PubMed] [Google Scholar]
- Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 1993;260:88–91. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- Scott SV, Baba M, Ohsumi Y, Klionsky DJ. Aminopeptidase I is targeted to the vacuole by a nonclassical vesicular mechanism. J Cell Biol. 1997;138:37–44. doi: 10.1083/jcb.138.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- Sorensen SO, Van Den Hazel HB, Kielland-Brandt MC, Winther JR. pH-dependent processing of yeast procarboxypeptidase Y by proteinase A in vivo and in vitro. Eur J Biochem. 1994;220:19–27. doi: 10.1111/j.1432-1033.1994.tb18594.x. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Seaman M, Nemoto Y, Daniell L, Suchy SF, Emr S, De Camilli P, Nussbaum R. Disruption of three phosphatidylinositol-polyphosphate 5-phosphatase genes from Saccharomyces cerevisiaeresults in pleiotropic abnormalities of vacuole morphology, cell shape, and osmohomeostasis. Eur J Cell Biol. 1997;74:350–360. [PubMed] [Google Scholar]
- Stack JH, DeWald DB, Takegawa K, Emr SD. Vesicle-mediated protein transport: regulatory interactions between the Vps15 protein kinase and the Vps34 PtdIns 3-kinase essential for protein sorting to the vacuole in yeast. J Cell Biol. 1995;129:321–334. doi: 10.1083/jcb.129.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack JH, Herman PK, Schu PV, Emr SD. A membrane-associated complex containing the Vps15 protein kinase and the Vps34 PI 3-kinase is essential for protein sorting to the yeast lysosome-like vacuole. EMBO (Eur Mol Biol Organ) J. 1993;12:2195–2204. doi: 10.1002/j.1460-2075.1993.tb05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack JH, Horazdovsky B, Emr SD. Receptor-mediated protein sorting to the vacuole in yeast: roles for a protein kinase, a lipid kinase and GTP-binding proteins. Annu Rev Cell Dev Biol. 1995;11:1–33. doi: 10.1146/annurev.cb.11.110195.000245. [DOI] [PubMed] [Google Scholar]
- Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka AS, Thelen M, Cadwallader K, Tempst P, Hawkins PT. The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell. 1997;89:105–114. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Stolz LE, Huynh CV, Thorner J, York JD. Identification and characterization of an essential family of inositol polyphosphate 5-phosphatases (INP51, INP52 and INP53 gene products) in the yeast Saccharomyces cerevisiae. . Genetics. 1998;148:1715–1729. doi: 10.1093/genetics/148.4.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoyanov B, Volinia S, Hanck T, Rubio I, Loubtchenkov M, Malek D, Stoyanova S, Vanhaesebroeck B, Dhand R, Nurnberg B, et al. Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science. 1995;269:690–693. doi: 10.1126/science.7624799. [DOI] [PubMed] [Google Scholar]
- Sui JL, Petit-Jacques J, Logothetis DE. Activation of the atrial KACh channel by the βγ subunits of G proteins or intracellular Na+ions depends on the presence of phosphatidylinositol phosphates. Proc Natl Acad Sci USA. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JC, Nocka K, Ray P, Traktman P, Besmer P. The dominant W42 spotting phenotype results from a missense mutation in the c-kit receptor kinase. Science. 1990;247:209–212. doi: 10.1126/science.1688471. [DOI] [PubMed] [Google Scholar]
- Toker A, Cantley LC. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- Van Den Hazel HB, Kielland-Brandt MC, Winther JR. Review: biosynthesis and function of yeast vacuolar proteases. Yeast. 1996;12:1–16. doi: 10.1002/(sici)1097-0061(199601)12:1<1::aid-yea902>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-U, Catlett NL, Weisman LS. Vac8p, a vacuolar protein with armadillo repeats, functions in both vacuole inheritance and protein targeting from the cytoplasm to the vacuole. J Cell Biol. 1998;140:1063–1074. doi: 10.1083/jcb.140.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Zhao H, Harding TM, Gomes de Mesquita DS, Woldringh CL, Klionsky DJ, Munn AL, Weisman LS. Multiple classes of yeast mutants are defective in vacuole partitioning yet target vacuole proteins correctly. Mol Biol Cell. 1996;7:1375–1389. doi: 10.1091/mbc.7.9.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman LS, Wickner W. Molecular characterization of VAC1, a gene required for vacuole inheritance and vacuole protein sorting. J Biol Chem. 1992;267:618–623. [PubMed] [Google Scholar]
- Whiteford CC, Brearley CA, Ulug ET. Phosphatidylinositol 3,5-bisphosphate defines a novel PI 3-kinase pathway in resting mouse fibroblasts. Biochem J. 1997;323:597–601. doi: 10.1042/bj3230597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurmser A, Emr SD. Phosphoinositide signaling and turnover: PtdIns(3)P, a regulator of membrane traffic, is transported to the vacuole and degraded by a process that requires lumenal vacuolar hydrolase activities. EMBO (Eur Mol Biol Organ) J. 1998;17:4930–4942. doi: 10.1093/emboj/17.17.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, DeWald DB, Boronenkov IV, Anderson RA, Emr SD, Koshland D. Novel PI(4)P 5-kinase homologue, Fab1p, essential for normal vacuole function and morphology in yeast. Mol Biol Cell. 1995;6:525–539. doi: 10.1091/mbc.6.5.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Ohya Y, Goebl M, Nakano A, Anraku Y. A novel gene, STT4, encodes a phosphatidylinositol 4-kinase in the PKC1 protein kinase pathway of Saccharomyces cerevisiae. J Biol Chem. 1994;269:1166–1172. [PubMed] [Google Scholar]