

Abstract
Neurofilaments are essential for establishment and maintenance of axonal diameter of large myelinated axons, a property that determines the velocity of electrical signal conduction. One prominent model for how neurofilaments specify axonal growth is that the 660–amino acid, heavily phosphorylated tail domain of neurofilament heavy subunit (NF-H) is responsible for neurofilament-dependent structuring of axoplasm through intra-axonal crossbridging between adjacent neurofilaments or to other axonal structures. To test such a role, homologous recombination was used to generate NF-H–null mice. In peripheral motor and sensory axons, absence of NF-H does not significantly affect the number of neurofilaments or axonal elongation or targeting, but it does affect the efficiency of survival of motor and sensory axons. Loss of NF-H caused only a slight reduction in nearest neighbor spacing of neurofilaments and did not affect neurofilament distribution in either large- or small-diameter motor axons. Since postnatal growth of motor axon caliber continues largely unabated in the absence of NF-H, neither interactions mediated by NF-H nor the extensive phosphorylation of it within myelinated axonal segments are essential features of this growth.
Keywords: neurofilaments, radial growth, axoplasm, motor neurons, sensory neurons
Aseries of preceding efforts (Friede and Samorajski, 1970; Hoffman et al., 1987; Cleveland et al., 1991; Lee and Cleveland, 1994) have proven that neurofilaments are essential elements for establishing the correct diameters (and hence volume) of large myelinated motor and sensory axons. These axons elongate during development with diameters of ∼1–2 μm, but after stable synapse formation in early postnatal life and concomitant with myelination, they grow markedly in diameter. This growth continues at a slow rate throughout adulthood, ultimately yielding axons that in humans reach diameters of up to 14 μm (Kawamura et al., 1981), corresponding to a >100-fold increase from their initial volumes. Establishment of axon caliber is of importance for normal functioning of the nervous system since caliber is a principal determinant of the conduction velocity at which nerve impulses are propagated along the axon (Gasser and Grundfest, 1939; Arbuthnott et al., 1980; Sakaguchi et al., 1993).
Overwhelming evidence has demonstrated that neurofilaments—assembled as obligate heteropolymers (Ching and Liem, 1993; Lee et al., 1993) of three polypeptide subunits, light neurofilament (NF-L; 68 kD),1 mid-sized neurofilament (NF-M; 95 kD), and heavy neurofilament (NF-H; 115 kD)—are essential for establishing the caliber of large myelinated axons. The initial suggestion of this arose from the linear relationship between neurofilament number and axonal cross-sectional area during the phase of rapid growth in diameter (Friede and Samorajski, 1970) and during regrowth after axonal injury (Hoffman et al., 1987). The importance of neurofilaments in specifying normal axonal caliber was proven unequivocally by analysis of a recessive mutation (quv) in a Japanese quail that lacks neurofilaments as the result of a premature translation terminator in the NF-L gene. Radial growth of axons fails completely in these animals (Yamasaki et al., 1992; Ohara et al., 1993), with a consequent reduction in axonal conduction velocity and generalized quivering (Sakaguchi et al., 1993). This requirement for neurofilaments has been confirmed in mice both by expression of a NF-H–β-galactosidase fusion protein that completely inhibits neurofilament transport into axons (Eyer and Peterson, 1994) and by targeted deletion of the NF-L gene (Zhu et al., 1997). In both cases, loss of axonal neurofilaments results in failure of normal radial growth. Moreover, since neurofilaments are obligate heteropolymers of NF-L and substoichiometric levels of NF-M or NF-H (Ching and Liem, 1993; Lee et al., 1993), absence of NF-M caused by gene disruption leads to markedly fewer axonal neurofilaments and a suppression of radial growth (Elder et al., 1998).
Although it is clear that neurofilaments are essential for specifying axonal diameter, the mechanism through which neurofilaments mediate increases in axonal size remains unsettled. The linear correlation between neurofilament number and axonal cross-sectional area initially suggested that the axon expanded or contracted to maintain a constant density of neurofilaments (Hoffman et al., 1987). That radial growth was not simply a function of the number of neurofilaments was proven by elevation of wild-type NF-L levels in transgenic mice. This revealed that a two- to threefold increase in the number of neurofilaments (with an elevated proportion of the NF-L subunit) actually decreases axonal diameter slightly (Monteiro et al., 1990; Xu et al., 1993). This led to one attractive and plausible model for how neurofilaments mediate initial growth and then maintain it: the long, COOH-terminal tail domains of NF-M and NF-H, which extend from the core of the 10-nm-diameter filament (Hirokawa et al., 1984; Hisanaga and Hirokawa, 1988; Troncoso et al., 1990), support axonal growth by crossbridging between adjacent neurofilaments or to other axonal constituents, thereby forming a three-dimensional lattice that determines volume. Consistent with this, transgenic methods that increased filament number (by producing higher levels of NF-L) combined with higher levels of NF-M or NF-H yielded increased axonal volumes by 30–45% (Xu et al., 1996).
Superimposed on contributions to radial growth provided by the number and subunit composition of neurofilaments, it is also now clear that the general relationship of neurofilament content and caliber is apparently regulated by the relative degree of phosphorylation of NF-M and NF-H. The tail domain of NF-H is largely composed of a motif repeated between 43 and 51 times in mammals (Julien et al., 1988; Lees et al., 1988) containing a central KSP tripeptide, the serine of which is nearly stoichiometrically phosphorylated in myelinated axonal segments (Julien and Mushynski, 1982; Carden et al., 1985; Lee et al., 1988). It has been proposed that phosphorylation of NF-H, and to a lesser extent NF-M, increases the total negative charge and lateral extension of their side arms (Glicksman et al., 1987; Myers et al., 1987), thereby mediating the increased neurofilament spacing found in myelinated segments and/or increased crossbridging to other axonal components such as microtubules (Hirokawa, 1982). Phosphorylation of both NF-H and NF-M tail domains is strongly correlated with radial growth, but the more extensive repeat domain in NF-H (which in mice contains 51 KSP repeats versus only 4 for murine NF-M) has focused most attention on NF-H. In the normal setting, unmyelinated initial axonal segments contain dephosphorylated NF-H and display higher filament density and much smaller diameters than the adjacent myelinated segments (Hsieh et al., 1994; Nixon et al., 1994). Consistent with this is the finding that a primary defect in myelination (in the Trembler mouse) decreases phosphorylation of NF-H, increases neurofilament density, and inhibits normal radial growth of axons (de Waegh et al., 1992). Similarly, deletion of the peripheral myelin-associated glycoprotein, which has been proposed to signal from the myelinating Schwann cell to the axon, results in reduced neurofilament phosphorylation, decreased neurofilament spacing, and reduced axonal calibers (Yin et al., 1998). These examples show the direct relationship between neurofilament phosphorylation and axonal diameter within myelinated axonal segments.
That NF-H is a primary component of radial growth has been supported by strong correlative evidence: increases in the level of NF-H mRNA is most pronounced during the earliest phase of radial growth (0–4 wk postnatal) (Schlaepfer and Bruce, 1990), suggesting its importance in the process. Doubling NF-M content in transgenic mice yields a 50% reduction in axonal NF-H and strongly inhibits radial growth, despite a constant level of NF-L (Wong et al., 1995). Furthermore, modest increases in NF-H mildly enhance radial growth in transgenic mice (although higher levels severely retard growth by slowing transport and trapping neurofilaments in neuronal cell bodies) (Marszalek et al., 1996). These three examples lend support to the idea that NF-H levels modulate radial growth of axons.
To examine directly the role of NF-H and its phosphorylation in structuring axoplasm and on neurofilament-dependent radial growth of large myelinated axons, we now report the use of homologous recombination to produce mice devoid of NF-H and to document the consequences of chronic absence of NF-H (and its phosphorylation) on elongation and survival of both large and small motor and sensory neurons, organization of neurofilaments and other organelles in those axons, and establishment and maintenance of axonal caliber.
Materials and Methods
Production and Screening of Mice with an NF-H Gene Disruption
A targeting vector for disrupting the mouse NF-H gene using homologous recombination was constructed (from a mouse NF-H gene cloned from a 129 SVJ library) by inserting a 1.6-kb KpnI-SacI fragment of NF-H (which lies 1.5-kb 5′ to NF-H transcriptional initiation site; see Fig. 1 A) adjacent to the phosphoglycerol kinase–promoted neomycin gene of plasmid pSK (Tybulewicz et al., 1991). Next, the 6.7-kb SmaI-EcoRV fragment of the NF-H gene (containing all three introns and coding sequences from amino acid 34 through most of the KSP repeats in the tail) was ligated just 3′ to the neomycin gene. Finally, a phosphoglycerol kinase–promoted thymidine kinase gene cassette was ligated 3′ to this latter NF-H gene segment to yield a final targeting construct. After linearization with KpnI, the targeting DNA was electroporated into embryonic stem (ES) cells (RI cells kindly provided by Andreas Nagy, University of Toronto, Toronto, Canada), and cells were selected for resistance to 200 μg/ml G418 and 2 μM gancyclovir. All cell manipulations were performed as described (Joyner, 1994). To identify homologous recombinants, drug-resistant colonies were amplified, and DNA was prepared and digested with HindIII or with EcoRV, separated on 0.8% agarose gels, and transferred to Hybond N+ filters (Amersham Corp., Arlington Heights, IL). NF-H fragments were identified by hybridization either with the 1.4-kb EcoRV/AatII fragment of NF-H gene, which encodes some of the KSP multiphosphorylation repeats and part of the NF-H 3′-flanking region, or with the 5′ EcoRI/NdeI fragment (see Fig. 1). DNA labeling was performed by random primer extension using [α-32P]dATP.
Figure 1.
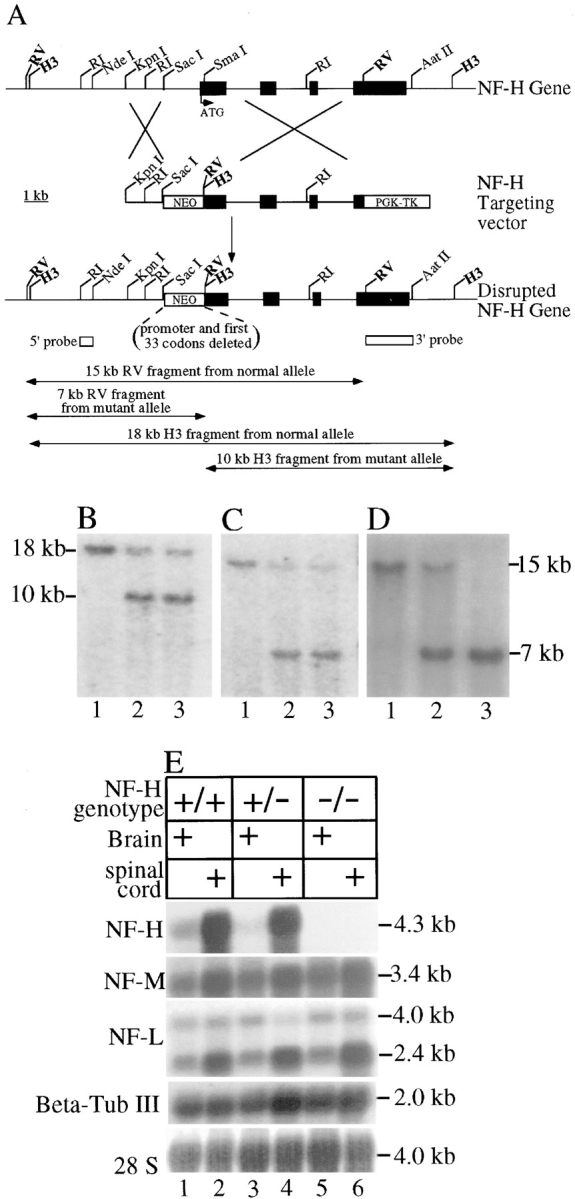
Disruption of the mouse NF-H gene by homologous recombination. (A) Strategy for disruption of the mouse NF-H gene. A targeting construct for disruption of the NF-H gene was constructed by inserting a 1.7-kb gene encoding resistance to neomycin in place of 1.6 kb NF-H putative promoter and the first 34 codons of the gene. The four NF-H exons are indicated by filled boxes interrupted by three introns. ATG denotes the NF-H translation initiation codon. Unique HindIII (H3) and EcoRV (RV) sites were introduced into the disrupted gene allele after homologous recombination. RI, EcoRI; WT, wild type; MT, mutant; PGK, phosphoglycerate kinase promoter; NEO, neomycin phosphotransferase gene; TK, thymidine kinase gene. (B–D) Screening of (B and C) ES and (D) mouse tail DNAs for targeted inactivation of the NF-H gene. (B) Genomic DNA blot of ES cell DNA after digestion with HindIII was probed with a segment 3′ to the targeted domain (the highlighted EcoRV-AatII fragment in A). The normal NF-H allele produces an 18-kb fragment; the targeted allele produces a 10-kb fragment. (C) Genomic DNA blot of ES cell DNA after digestion with EcoRV was probed with a 5′ probe (the EcoRI-NdeI fragment denoted in A). The normal allele produces a 15-kb fragment; the targeted allele produces a 7-kb fragment. (B and C) Lane 1, wild-type ES cell DNA; lanes 2 and 3, DNA from two targeted ES cells. (D) EcoRV-digested mouse tail DNA probed with the 5′ probe. DNAs are from a mouse with (lane 1) two wild-type alleles or (lane 2) heterozygous or (lane 3) homozygous for disruption of the NF-H gene. (E) NF-L, NF-M, NF-H, and βIII-tubulin mRNA levels in mice with zero, one, or two copies of a disrupted NF-H gene. 20 μg of total RNA isolated from 5-wk-old brains and spinal cords of control mice and mice heterozygous or homozygous for disruption of the NF-H gene were fractionated on 1% formaldehyde agarose gels, blotted on to nylon membranes, and probed with radiolabeled cDNA sequences for each subunit (see Materials and Methods). Lanes 1, 3, and 5, brain RNAs from wild-type, heterozygous, and homozygous mice. Lanes 2, 4, and 6, spinal cord RNAs from wild-type, heterozygous, and homozygous mice.
Targeted ES cell clones were injected into C57BL/6J blastocysts to produce chimeric animals. Chimeric animals from two independent ES cell clones were bred to C57BL/6J mice, and mice heterozygous for the disruption were identified by analysis of mouse tail DNAs (prepared as before [Monteiro et al., 1990]).
Analysis of Neurofilament RNA Levels in Nervous Tissues
Brains and spinal cords of 5-wk-old animals were dissected from NF-H deletion, heterozygous, and wild-type mice and immediately frozen at −80°C. Total cellular RNA was purified as described (Chomcznski and Sacchi, 1987). In brief, tissues were homogenized in 4 M guanidinium thiocyanate, 25 mM sodium citrate, pH 7, 0.5% sarcosyl, and 0.1 M 2-mercaptoethanol. Protein was removed by addition of an equal volume of phenol (in 0.2 M sodium citrate, pH 4) and one-fifth volume of chloroform/ isoamyl alcohol (49:1). After mixing well, samples were centrifuged at 10,000 g for 20 min at 4°C. The RNA in the aqueous phase was precipitated (1 h at −20°C) after addition of an equal volume of isopropanol. The precipitate was collected by centrifugation, dissolved in the homogenization solution, and reprecipitated with isopropanol. Final RNA pellets were washed with 70% ethanol, dried, and dissolved in 10 mM Tris, pH 7.4, 1 mM EDTA, reextracted with phenol/chloroform, and reprecipitated with ethanol containing 2.5 M ammonium acetate. Finally, the RNA pellets were dissolved in 0.5% SDS, and RNA concentration was determined by absorbance at 260 nm.
20 μg of total RNA was fractionated on a 1% agarose gel containing 1.85% formaldehyde, transferred to Hybond N+, baked for 2 h at 80°C, and prehybridized for 1 h in 0.5 M sodium phosphate, pH 7.2, 1 mM EDTA, 7% SDS, and 1% bovine albumin. RNAs encoding neurofilament subunits were detected by hybridization with random-primed [α-32P]- dATP-labeled nucleotide probe. Probe to detect NF-H RNA was the EcoRV-AatII fragment of the NF-H gene (containing sequences for codons 798–1087). Probe for NF-M was an AccI-EcoRI fragment of the NF-M gene containing the complete third exon. Probe for NF-L RNA was a complete NF-L cDNA. For identifying the mRNA encoding type III β-tubulin, sequences corresponding to the final 11 codons and the 3′ untranslated region were used as a probe (from mouse EST No. 352879). Filters were then washed in 30 mM Tris, pH 7.4, 300 mM NaCl, for 20 min at room temperature, followed by two washes (30 min each at 60°C) in 3 mM Tris, pH 7.4, 30 mM NaCl, 0.5% SDS. RNA bands corresponding to different neurofilament subunits were visualized by autoradiography using x-ray film (Biomax MS; Eastman Kodak Corp., Rochester, NY) and quantified by phosphorimaging (Molecular Dynamics, Sunnyvale, CA).
SDS-PAGE and Detection of Neurofilament Proteins by Immunoblotting
Sciatic nerves, spinal cords, and brains were homogenized on ice in a buffer containing 50 mM Tris, pH 7.5, 0.5 mM EDTA, pH 8, and 1 mM each of PMSF, leupeptin, aprotinin, and chymostatin. An equal volume of a solution containing 50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 2% SDS was added, and the homogenates were sonicated for 20 s, boiled for 10 min, and clarified by centrifugation at 16,000 g for 5 min. Protein concentration was determined using the bicinchoninic acid (BCA) assay kit (Pierce Chemical Co., Rockford, IL). Protein extracts, as well as known amounts of neurofilament standards, were separated by SDS-PAGE using gels containing 6 or 7.5% polyacrylamide. Proteins separated by SDS-PAGE were transferred to nitrocellulose (Lopata and Cleveland, 1987). The NF-H subunit was identified using an affinity-purified rabbit polyclonal antibody (pAb-NF-HCOOH) prepared to the COOH-terminal 12 amino acids of mouse NF-H (Xu et al., 1993), followed by 125I-conjugated protein A. The NF-L subunit was identified using an affinity-purified rabbit polyclonal antibody (pAb-NF-LCOOH) prepared to the COOH-terminal 12 amino acids of mouse NF-L (Xu et al., 1993), followed by 125I-conjugated protein A. mAbs to phosphorylated epitopes in NF-H and NF-M (SMI-31; Sternberger and Sternberger, 1983), nonphosphorylated NF-H (SMI-32; Sternberger and Sternberger, 1983), NF-M (RM044; Tu et al., 1995), α-tubulin (DM1A; Sigma Chemical Co., St. Louis, MO), or the neuron-specific, class III β-tubulin (TuJ1; Lee et al., 1990) were used to identify each subunit, followed by rabbit anti– mouse IgG (Sigma Chemical Co.) and 125I-conjugated protein A. For plectin, polyclonal antisera (P21; Wiche and Baker, 1982) and mAb 10F6 (Foisner et al., 1991) were used as primary antibodies, and binding was detected with goat anti–rabbit and goat anti–mouse secondary antibodies linked to alkaline phosphatase. Immunoreactive bands were visualized by autoradiography using Kodak Biomax MS film. Quantification was performed by phosphorimaging (Molecular Dynamics) using known amounts of purified mouse spinal cord neurofilaments as standards.
Tissue Preparation and Morphological Analysis
Mice were perfused transcardially with 4% paraformaldehyde, 2.5% glutaraldehyde in 0.1 M sodium phosphate, pH 7.6, and postfixed overnight in the same buffer. Samples were treated with 2% osmium tetroxide, washed, dehydrated, and embedded in Epon-Araldite resin. Thick sections (0.75 μm) for light microscopy were stained with toluidine blue, and thin sections (70 nm) for electron microscopy were stained with uranyl acetate and lead citrate.
Axons were counted in L5 root cross sections from three to four mice of each genotype and of each age group. Axon diameters from two animals of each genotype and age were measured using the Integrated Morphometry Analysis function from the Image 1/Metamorph Imaging System (Universal Imaging Corp., West Chester, PA). Entire roots were imaged, imaging thresholds were selected individually, and the cross sectional area of each axon was calculated and reported as a diameter of a circle of equivalent area. Axon diameters were grouped into 0.5-μm bins.
Analysis of Filament Spacing: Nearest Neighbor Analysis
To measure nearest neighbor distances between neurofilaments, cross sections of axons larger than 3.0 μm in diameter were photographed at a magnification of 20,000 and enlarged an additional 4.25-fold by printing. Neurofilaments were identified in these end-on views as dots ∼10 nm in diameter. Positions of neurofilaments were marked by puncturing the print with a push-pin. By laying the final prints on a light box, neurofilament positions could easily be imaged, and nearest neighbor distances were calculated for each filament using a digital imaging program (Bioquant, Nashville, TN).
Results
Mice Producing no NF-H mRNA or Protein Are Viable, with Elevated Levels of NF-M and Tubulin
To generate mice homozygously deleted for the NF-H gene, a targeting vector was constructed from the mouse NF-H gene (cloned from a mouse 129 SVJ library) by replacing the 1.6-kb segment containing the proximal promoter, methionine initiation codon, and 33 additional codons of the amino-terminal region of NF-H with a neomycin resistance gene (Fig. 1 A; see also Materials and Methods). After electroporation into ES cells and selection for integration by resistance to the neomycin homologue G418, cells in which one NF-H allele was targeted were identified by genomic DNA blotting. With probes corresponding to the gene segments just 5′ and 3′ to that contained in the gene targeting vector, this revealed that 2 of 70 lines examined had been correctly targeted at both 5′ (Fig. 1 C) and 3′ (Fig. 1 B) sites of integration. Both clones were injected into C57BL/6J blastocysts, and both lines produced chimeric animals that when mated to C57BL/6J mice transmitted the disrupted allele to their progeny, as revealed by blotting of genomic tail DNAs from such animals (Fig. 1 D).
Mating of heterozygotes to each other produced animals with both NF-H genes disrupted, as well as littermates that retained one or both normal NF-H alleles in the 1:2:1 ratios expected for Mendelian inheritance. Mice with both disrupted genes were viable and fertile, displaying no overt phenotype, at least up to 6 mo of age. To establish that the disruption eliminated NF-H expression, RNA was prepared from brain and spinal cords of animals with NF-H alleles that were normal or heterozygously or homozygously disrupted. Blotting with a probe corresponding to codons 798 to 1087 of mouse NF-H revealed the complete absence of stable mRNAs carrying NF-H coding sequences in the NF-H–deleted mice (Fig. 1 E, lanes 5 and 6). Relative to ribosomal RNA, phosphorimaging demonstrated that mRNAs coding for NF-L and NF-M were not markedly affected by presence or absence of NF-H mRNA (Fig. 1 E, lanes 1–6).
Immunoblotting with an antibody (Xu et al., 1993) raised to the carboxy terminus of NF-H (Fig. 2 B) or antibodies that recognize either the unphosphorylated NF-H tail domain (SMI-32; Fig. 2 D) or the same domain when phosphorylated (SMI-31; Fig. 2 C) confirmed the complete absence of NF-H in extracts from brain, spinal cords, and sciatic nerves of mice with two disrupted alleles (Fig. 2, B–D, lanes 3, 6, and 9). Animals heterozygous for the disruption had intermediate levels of NF-H, which phosphorimaging revealed to be between ∼60–70% of the level of control samples in brain and sciatic nerves. Parallel immunoblots demonstrated that NF-L levels were unaffected by the presence or absence of NF-H in spinal cord and sciatic nerve extracts from all three genotypes (Fig. 2 F). In contrast, use of an antibody insensitive to the phosphorylation state of NF-M (RMO44; Tu et al., 1995) demonstrated that NF-M levels were elevated after diminution or elimination of NF-H, with phosphorimaging revealing a twofold increase, relative to normal mice, in spinal cords (compare Fig. 2 E, lanes 4 and 6) and sciatic nerves (compare Fig. 2 E, lanes 7 and 9) but not brain of NF-H–disrupted animals. These findings provide additional evidence that in lower motor and sensory neurons, NF-M and NF-H compete in vivo for coassembly with a limiting amount of NF-L (Wong et al., 1995; Marszalek et al., 1996; Elder et al., 1998). Using antibody SMI-31 (Sternberger and Sternberger, 1983) to detect a phosphorylated determinant on NF-M (Fig. 2 C) further revealed that, in the absence of NF-H, NF-M phosphorylation levels increase in spinal cord and sciatic nerve. Phosphorimaging demonstrated that the increased phosphorylation quantitatively matched the increased NF-M subunit abundance; therefore, the proportion of NF-M that is phosphorylated remains unchanged by the loss of NF-H.
Figure 2.
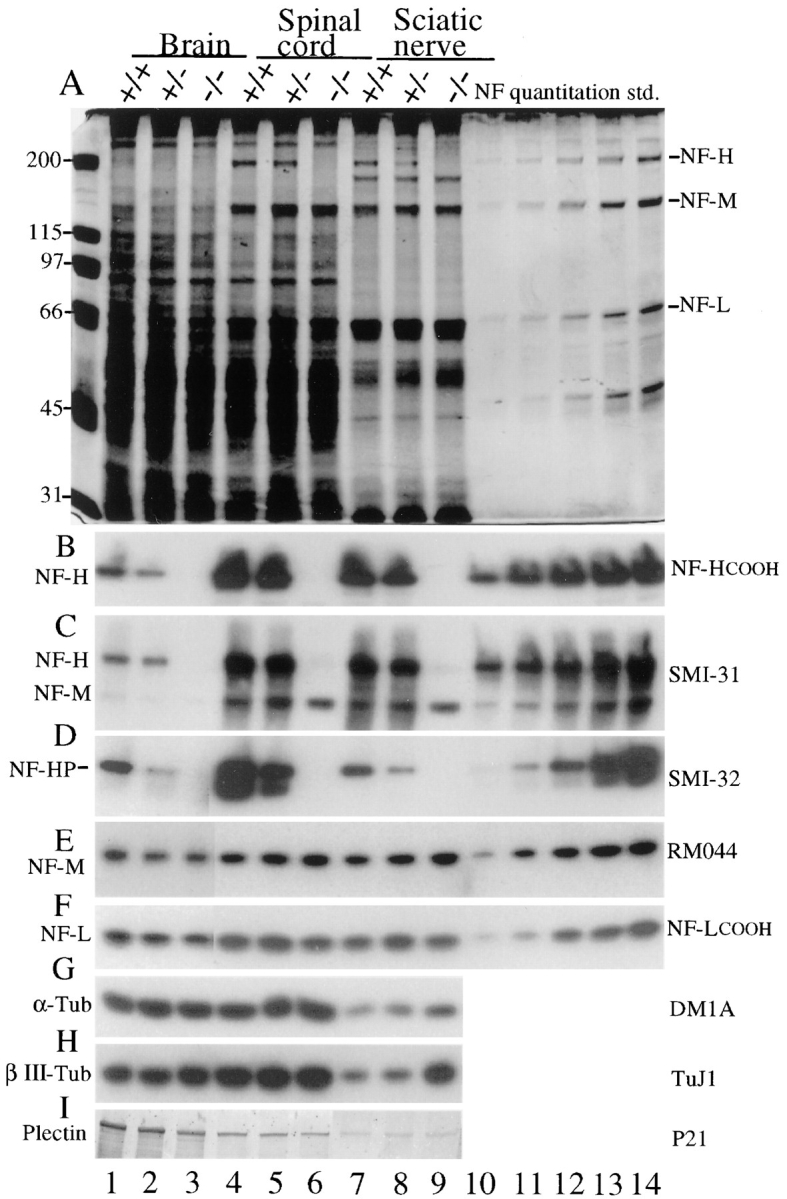
Levels of neurofilament subunits NF-L, NF-M, and NF-H in mice with zero, one, or two copies of a disrupted NF-H gene. (A) Total tissue extracts from 5-wk-old brain, spinal cord, and sciatic nerves were fractionated on 7% SDS–polyacrylamide gels and stained with (A) Coomassie blue or (B–I) electroblotted to nitrocellulose. (B) NF-H detected with a peptide antibody recognizing the extreme COOH terminus of NF-H (Xu et al., 1993); (C) phosphorylated NF-H and NF-M detected with monoclonal antibody SMI-31; (D) nonphosphorylated NF-H detected with monoclonal antibody SMI-32; (E) NF-M detected with monoclonal antibody RM 044 (Tu et al., 1995); (F) NF-L detected with a polyclonal peptide antibody recognizing the COOH terminus of NF-L (Xu et al., 1993); (G) α-tubulin detected with monoclonal antibody DM1A; (H) the neuron-specific class III, β-tubulin isotype with mAb TuJ1 (Lee et al., 1990); and (I) plectin detected with polyclonal antiserum P21 (Wiche and Baker, 1982). (Plectin migrates with a mobility of ∼500 kD in brain and spinal cord but at ∼160 kD in nerve samples using both this antibody and monoclonal antibody 10F6 [Foisner et al., 1991]; not shown.) Lanes 10–14, quantitation standards for the neurofilament subunits provided by a twofold dilution series of a neurofilament preparation. Molecular masses (kD) are indicated at left. (Lanes 1–3 of D–F represent four times longer exposures than lanes 4–14.)
Diminution or loss of NF-H was also accompanied in sciatic nerve (and to a lesser extent spinal cord) by a significant increase in total tubulin levels, as measured by antibodies that react with all α-tubulin (Fig. 2 G) or β-tubulin (not shown) isotypes. Measuring the content of the class III, neuron-specific β-tubulin isotype that comprises ∼25% of β-tubulin in normal nervous tissue (Lopata and Cleveland, 1987) further revealed that the 60% elevation in total tubulin levels seen in nerve samples (Fig. 2 G) was at least in part the result of a selective (fourfold) increase in this neuron-specific isoform (Fig. 2 H). Mechanistically, this increase in tubulin accumulation was due to an approximately twofold elevation in the βIII-tubulin mRNA (85% increase measured by phosphorimaging) in the NF-H–free animals (Fig. 1 E).
No significant change in the level of the large cytoskeletal cross-linker protein plectin was observed in spinal cord and nerve samples from NF-H–free mice compared with normal littermates (Fig. 2 I, compare lanes 4 and 6, 7 and 9). Regardless of the genotype, full-length plectin (∼500 kD) was seen in brain and spinal cord samples, while two plectin antibodies revealed a stable accumulation product that migrated with an apparent size of less than 200 kD in all peripheral nerve samples. (Note that lanes 7–9 of Fig. 2 I represent a lower molecular weight portion of the immunoblot than lanes 1–6.) Since it is retained in neurofilament-enriched nerve fractions, this plectin fragment (or isoform or plectin-related molecule) most probably contains a neurofilament-binding site (such as that found near the carboxy terminus of typical plectin [Elliott et al., 1997]).
Absence of NF-H Has Surprisingly Little Effect on Radial Growth of Motor Axons
Since neurofilaments are known to be a principal determinant of axonal caliber (Hoffman et al., 1987; Yamasaki et al., 1992; Ohara et al., 1993; Zhu et al., 1997) and NF-H phosphorylation has been tightly linked to neurofilament-dependent control of axon diameter (de Waegh et al., 1992; Hsieh et al., 1994; Nixon et al., 1994), we examined how the changes in NF-H content were reflected in changes in axonal caliber of large myelinated motor axons. Light microscopic inspection of the L5 ventral roots (Fig. 3 A) revealed the surprising finding that myelinated axons from wild-type mice, mice heterozygous for NF-H, or mice with both gene copies disrupted were qualitatively similar in caliber to those of wild-type mice (compare Fig. 3 A, left, middle, and right panels).
Figure 3.

Absence of NF-H does not markedly affect growth in motor axon caliber. (A) Cross sections of L5 motor (ventral root) and sensory (dorsal root) axons from wild type (+/+), NF-H heterozygous (+/−), and NF-H homozygous (−/−) mutant mice. Sections are from 4- and 9-wk-old mice as indicated. (B and C) Absence of NF-H yields a partial loss of motor and sensory axons in early postnatal life. Counts of small (<4 μm diameter; black bars) and large (≥4 μm diameter; crosshatched bars) axons in L5 (B) motor and (C) sensory root axons from 9-wk-old mice with zero, one, or two disrupted NF-H alleles. Counts are averages from three to four animals for each genotype and age. Bar, 10 μm.
To quantify potential caliber changes associated with loss of NF-H expression, cross-sectional areas of every axon within each ventral root (Fig. 4) were measured from 4- and 9-wk-old animals (two animals of each genotype at each time point). Each area was then converted to a diameter corresponding to a circle of the equivalent area. As expected, motor axons of wild-type animals showed a typical bimodal distribution of diameters representing the small and large myelinated axons, with initial diameters centered on 1.5 and 5 μm. Both small and large axons grow considerably in size between 4 and 9 wk of age, achieving diameters of 2.5–3 and 8.5 μm, respectively. Summing the areas of all axons after this 5-wk growth period revealed a threefold increase in axonal volume of the larger axons and a doubling of size for the smaller size class. This bimodal axon size distribution and growth phase was also observed in mice heterozygous or homozygous for the NF-H deletion, although at both time points reducing or deleting NF-H clearly decreased or eliminated the very largest diameter axons (e.g., those larger than 9 μm in Fig. 4, E and F) and shifted the distributions toward smaller sizes (Fig. 4, E and F, arrows). However, by 9 wk of age, loss of NF-H resulted in axonal populations of both large and small motor axons that were only slightly (25 and 20%, respectively) reduced in total axon area relative to wild-type animals. Similar findings were also seen in 15-wk-old animals. Additional radial growth of ∼20% continued to equivalent extents in both normal and NF-H–null animals, with a similar loss of the largest population of motor axons found in 15-wk-old NF-H–deleted animals (not shown). Thus, for motor axons, NF-H content (and any interactions provided by it or its long tail domain) are dispensable for the majority of radial growth, despite the fact that it is necessary for achieving the largest diameters.
Figure 4.

Morphometry reveals that the absence of NF-H results in only a small reduction in radial growth and the distribution of calibers of motor axons. Axonal area was determined for all axons within L5 ventral roots using a computerized imaging package and was plotted as the frequency of appearance of diameters corresponding to circles of equivalent areas. Distributions from 4- (A–C) and 9-wk-old (D–F) mice. Arrows indicate the diameter(s) corresponding to the peak(s) of maximum frequency in wild-type animals. Points represent the averaged distributions of axon diameters from the entire roots of two mice for each genotype and age.
Radial Growth of Sensory Axons Requires NF-H
To examine how NF-H contributes to radial growth of sensory axons, calibers were measured in L5 dorsal roots of wild type and animals heterozygous or homozygous for NF-H disruption. While there are many small sensory axons that do not undergo radial growth, light microscopic examination revealed an apparent inhibition of growth of the largest caliber axons (Fig. 3 A). Comparing size distributions of 4- and 9-wk-old wild-type animals demonstrated that radial growth of both larger and smaller axons occurs (Fig. 5 A). For animals with reduced (Fig. 5 B) or no (Fig. 5 C) NF-H content, growth of the smallest axons was eliminated, while growth of the largest class (initially >4 μm) continued, but at a reduced level compared with normal mice, especially for the largest axons (>8 μm in normal mice). Similar findings emerged from analysis of 15-wk-old animals (not shown).
Figure 5.

Morphometry reveals that absence of NF-H results in attenuation of radial growth of both small and large sensory axons. Axonal area was determined for all axons within the L5 dorsal roots using a computerized imaging package and plotted as the frequency of appearance of diameters corresponding to circles of equivalent areas. Distributions from (dashed curves) 4- and (solid curves) 9-wk-old mice from (A) wild-type mice or mice (B) heterozygous or (C) homozygous for the NF-H gene disruption. Arrows indicate the diameter corresponding to the peak of maximum frequency in both 4- and 9-wk wild-type animals. Points represent the averaged distributions of axon diameters from the entire roots of two mice for each genotype and age.
NF-H Is Required for Survival of the Normal Number of Motor and Sensory Axons
The complete absence of neurofilaments (arising from disruption of the NF-L gene) during initial axon elongation, targeting, and myelination has been demonstrated to yield a 13% reduction in the survival of initial motor axons (Zhu et al., 1997). To determine whether NF-H content affects this developmental survival of motor axons, axons were counted in the L5 ventral and sensory roots of wild-type mice and mice with one or both copies of the NF-H gene disrupted. After the initial burst of radial growth, this quantitation revealed that by 9 wk of age, the total number of surviving motor axons was ∼13% lower (n = 6, P = 0.03) in the absence of NF-H than in its presence (Fig. 3 B). This reflects a preferential loss of large (>4 μm in diameter) axons with no significant change in the number of small axons. A similar trend was seen when the sensory axons were counted (Fig. 3 C). Again, the number of large- but not small-caliber axons was reduced by ∼19% (n = 6, P = 0.03).
Nearest Neighbor Spacing between Neurofilaments Is Unaffected by NF-H Content
To examine whether reduction in NF-H content, and the corresponding increase in axonal NF-M, affects neurofilament organization in axons, the nearest neighbor spacing between neurofilaments in cross sections of ventral roots was compared in 9-wk-old wild-type animals and littermates that were heterozygously and homozygously deleted for the NF-H gene. Qualitative inspection of electron micrographs from wild-type versus NF-H–deleted animals revealed no consistent differences in neurofilament spacing (compare Fig. 6, A–C), a point reinforced by marking neurofilament positions in ventral root axons (n = 9, including axons ranging in diameters from 4 to 8 μm) and calculating nearest neighbor spacings. As seen previously (Hsieh et al., 1994; Wong et al., 1995; Marszalek et al., 1996; Xu et al., 1996), measurement of nearest neighbor distances revealed a broad distribution centered on a 45-nm spacing in wild-type mice. Examination of all distances from 10 axons revealed that neither reduction of NF-H nor its complete absence markedly affected the distributions of nearest neighbor distances (Fig. 6 D). A different measure, comparing the mean nearest neighbor distance calculated axon by axon, again revealed very similar average filament spacing, with only a possible shift (P = 0.05 using a two-tail t test) toward very slightly smaller distances in the NF-H–null axons compared with normal axons (Fig. 6 E). This pattern of filament–filament distances was confirmed by examination of overall axonal organization in longitudinal sections of motor axons. Although less organized axoplasm in the NF-H deficient might have been anticipated, especially in light of the demonstration that dephosphorylated NF-H directly binds to microtubules in vitro (Hisanaga and Hirokawa, 1990; Hisanaga et al., 1991; Miyasaka et al., 1993), this was not the case. The absence of NF-H–dependent interactions between adjacent neurofilaments or between neurofilaments and microtubules did not diminish the “straightness” of neurofilaments or their overall orientation along the long axis of the axon.
Figure 6.



Absence of NF-H does not significantly affect nearest neighbor spacing of neurofilaments but does yield increased microtubule density. (A–C) Electron micrographs of axonal cross sections from 9-wk-old (A) wild-type, (B) NF-H heterozygous, and (C) NF-H homozygous mutant mice. (D) Distributions of nearest neighbor distances between neurofilaments in motor axons from 9-wk-old (squares) wild-type, (diamonds) NF-H heterozygous, and (circles) NF-H homozygous mice. Measurements (n > 3,600) were taken from 9 to 13 axons from each genotype. (E) Distributions of nearest neighbor distances between filaments in mice of various genotypes. Each dot represents the average nearest neighbor distance from a single axon. (F) Quantitation of axonal microtubule density in motor axons from (+/+) wild-type mice, (+/−) NF-H heterozygous and (−/−) NF-H homozygous mutant mice. Density was measured by counting all microtubules in a given cross-sectional area and dividing by that area. Counts are averages from four axons of each genotype. Bar, 0.1 μm.
Despite the unchanged spacing and organization of neurofilaments, one difference in axonal organization was apparent by inspection: microtubule density was higher in the animals with diminished or no NF-H. Counting microtubules in axons of each genotype revealed a 60% increased density of microtubules in axons from NF-H–deleted animals (Fig. 6 F). A two-tailed paired t test revealed this difference to be highly significant (P < 0.001). The increase in microtubule number quantitatively corresponds to the similar increase in tubulin content seen in both the spinal cord and sciatic nerve homogenates of NF-H–free animals (Fig. 2, G and H, lanes 6 and 9). In light of the known induction of tubulin expression by nearly an order of magnitude during axonal regeneration (Hoffman et al., 1987) and the reduction in initial survival of motor and sensory axons in the NF-H–null mice, this increase in tubulin levels may reflect partial activation of a regenerative program.
Discussion
Several lines of preceding evidence had implicated a primary role for NF-H in crossbridging between adjacent neurofilaments or between neurofilaments and other axonal structures (Hirokawa et al., 1984; Marszalek et al., 1996; Xu et al., 1996) to promote and maintain axonal caliber. The present analysis of mice with complete absence of NF-H provides unambiguous evidence that neither NF-H nor the nearly stoichiometric phosphorylation of its long, repetitive tail domain in the myelinated, internodal axonal segments are essential features for most of the radial growth of large motor axons. Moreover, neither neurofilament number nor the organization of those neurofilaments (e.g., nearest neighbor distances or longitudinal orientations within domains of axoplasm) are altered by the absence of NF-H. As in previous examples (Wong et al., 1995; Marszalek et al., 1996; Xu et al., 1996), NF-M levels do undergo a compensatory increase in abundance without NF-H, adding further evidence that NF-M and NF-H compete for assembly with a limiting amount of NF-L. Perhaps the most surprising feature of these findings is that they force a reinterpretation of what seemed to be compelling correlative evidence linking a signaling cascade initiated in the myelinating cell as a trigger for nearly stoichiometric phosphorylation of the NF-H and NF-M KSP repeats in myelinated axonal segments to a corresponding increase in neurofilament spacing (de Waegh et al., 1992; Yin et al., 1998). It is now clear that absence of the nearly 25–50 phosphates added to NF-H by this cascade does not markedly affect neurofilament spacing in internodes nor does it affect the preponderance of radial growth, at least of motor axons.
While NF-H and its phosphorylation are not as essential as had been anticipated, it should be emphasized that the collective evidence still supports a role for NF-H in radial growth, certainly an important one in sensory axons and probably a redundant one in motor axons. Indeed, prior work with transgenic mice expressing elevated levels of wild-type NF-L (Monteiro et al., 1990), NF-H (Marszalek et al., 1996; Xu et al., 1996), or epitope-tagged NF-M (Wong et al., 1995) has demonstrated that an excess of any subunit inhibits radial growth to a greater degree than does absence of NF-H, while increasing both NF-L (thereby assembling more filaments) together with either NF-M or NF-H stimulates radial growth (Xu et al., 1996). This lead to the view that radial growth requires contributions from all three subunits: NF-L to support filament assembly and a scaffolding of NF-M and NF-H tails to crossbridge between neurofilaments and/or other axonal components (Hisanaga and Hirokawa, 1990; Xu et al., 1996). What is clear now, however, is that changing the content of NF-H (with a corresponding elevation in NF-M) does not affect nearest neighbor spacing of neurofilaments, a finding that goes hand in hand with the identical situation after doubling axonal NF-M and a corresponding loss in NF-H (Wong et al., 1995).
The current evidence, along with two earlier efforts (Wong et al., 1995; Xu et al., 1996), implicates NF-M (see schematic in Fig. 7) as the subunit that specifies nearest neighbor spacing mediated by its 439–amino acid tail that can extend 30–40 nm from the surface of the filament (Hisanaga and Hirokawa, 1988). Indeed, such crossbridges have been observed after simultaneous expression of NF-L and NF-M (using baculovirus) in insect cells (Nakagawa et al., 1995). Schwann cell–dependent phosphorylation of the KSP repeats in the NF-M tail may lengthen the effective cross-link, so as to force wider filament spacing, a feature potentially necessary, but not sufficient, for radial growth. It must be noted, however, that since nearest neighbor neurofilament spacing is also almost unchanged by the complete absence of NF-M (Elder et al., 1998), the simplest interpretation is that multiple, redundant elements (including NF-M) establish this spacing.
Figure 7.

Model of neurofilament-dependent structuring of axoplasm. Axoplasm is organized into a volume-determining three-dimensional array by a series of linkages that span between adjacent neurofilaments (blue-gray) and between neurofilaments and microtubules (red) or cortical actin (blue) filaments. The NF-M tail (turquoise) sets nearest neighbor spacing of neurofilaments, possibly dependent on phosphorylation of its KSP repeats. (The interaction is drawn here between NF-M tails bound to adjacent neurofilaments, although the alternative of the tail directly contacting the adjacent filament core is equally plausible; see also Nakagawa et al. [1995].) NF-H (purple) normally also crossbridges neurofilaments to each other (Hirokawa et al., 1984), but this is not necessary for establishing nearest neighbor spacing. NF-H may also contribute to a three-dimensional array by crossbridging between neurofilaments and microtubules. Other interfilament linkages are provided by plectin (orange), which is capable of bridging between neurofilaments and cortical actin filaments (blue) and neurofilaments and microtubules (Wiche, 1989; Errante et al., 1994; Svitkina et al., 1996). Final structural stability is contributed by the BPAG1n/dystonin family of cross-linkers that in motor and sensory axons (Dowling et al., 1997; Dalpe et al., 1998) provide additional linkage between neurofilaments and actin filaments (Yang et al., 1996) that are tethered under the cortical membrane. In the absence of NF-H, cross-linking from NF-M, plectin, and BPAG1n/dystonin homologues continue to support a three-dimensional array necessary for acquisition and maintenance of normal axonal volume.
Whatever the interaction between neurofilaments, it must be an attractive one, not simply a repulsion of the negatively charged tails, since even in mice with few axonal neurofilaments (as a consequence of disruption of the NF-M gene [Elder et al., 1998] or trapping most neurofilaments in the cell bodies by expressing high levels of NF-H [Marszalek et al., 1996] or NF-M [Wong et al., 1995]), the few axonal filaments are found in clusters with spacings identical to those in normal mice (Xu et al., 1996). Moreover, since radial growth can be markedly inhibited despite no change in interfilament distance, this gives further weight to the argument that interactions between nearest neighbor filaments do not specify radial growth.
Although it appears unlikely that nearest neighbor filament spacing is a key determinant of radial growth, a great deal of previous evidence still argues for the involvement of neurofilaments in some other manner. The key properties needed to stimulate this growth must include longer-range interactions between neurofilaments that are not nearest neighbors or between neurofilaments and other axonal components. Attractive candidates for these additional interactions include the family of proteins such as BPAG1n/dystonin, proven recently to be a crossbridger between neurofilaments and actin filaments (Yang et al., 1996) that is essential for survival of sensory neurons (Brown et al., 1995; Guo et al., 1995). BPAG1n/dystonin, while expressed at highest levels in sensory neurons, is also found in embryonic and postnatal motor axons (Dowling et al., 1997), and absence of it leads both to rampant sensory axon degeneration with more modest numbers of motor axon abnormalities, including degeneration (Dowling et al., 1997). Another known cross-linker with binding sites for intermediate filaments, actin filaments, and microtubules is plectin (Wiche, 1989), a very large (466-kD), essential (Andra et al., 1997) protein expressed in many neurons, including motor neurons (Errante et al., 1994). While we have not been able to measure BPAG1n/dystonin levels as a function of NF-H content, plectin levels do not change in the absence of NF-H, a finding consistent with plectin as one probable linking component in motor axons that acts in place of NF-H in mediating the bulk of neurofilament-dependent radial growth. However, we found only a plectin fragment (or much smaller isoform or plectin-related polypeptide) to accumulate in peripheral nerves. Definition of precisely what aspects of cross-linking are contributed by this shortened plectin will have to await determination of whether it oligomerizes and whether it retains the actin-binding domain normally situated near plectin's amino terminus and/or a microtubule-binding domain (whose position is not yet mapped within plectin).
Seen from this perspective, a combination of cross-linkers, including NF-H and NF-M, along with members of the plectin and BPAG1n families, interlink between neurofilaments, microtubules, and cortical actin arrays to determine a three-dimensional, space-filling array of connected structural elements that expand axonal volume. Contacts mediated by NF-H are largely, but not completely, dispensable in motor neurons and may, in fact, be compensated by additional interactions mediated by the increased number of microtubules and additional NF-M. This leads to a model (Fig. 7) for structuring axoplasm using elements with overlapping function, with each neuronal type relying upon a different balance of individual components. These findings, along with the discovery of mutations in plectin as the primary cause of one form of muscular dystrophy accompanied by defects in skin and cardiac cell organization (Gache et al., 1996; Andra et al., 1997; Smith et al., 1997), reinforce the essential nature of correct structuring of cytoplasm through establishment and maintenance of a flexible, deformable scaffold of interlinked cytoskeletal elements.
Acknowledgments
We thank Dr.Virginia Lee (University of Pennsylvania, Philadelphia, PA) for providing antibodies to NF-M and Dr. Gerhard Wiche (University of Vienna, Vienna, Austria) for antibodies to plectin. We also thank Ms. Karen Anderson for her expert assistance with some aspects of the microscopy and Mr. Scott Anderson for his assistance with mouse husbandry and genotyping.
This work has been supported by grant NS27036 from the National Institutes of Health to D.W. Cleveland. Salary support for D.W. Cleveland was provided by the Ludwig Institute for Cancer Research. T.L. Williamson was supported, in part, by a postdoctoral fellowship the Muscular Dystrophy Association.
Abbreviations used in this paper
- ES
embryonic stem
- NF-H
NF-M, and NF-L, heavy, mid-sized, and light neurofilament subunits
References
- Andra K, Lassmann H, Bittner R, Shorny S, Fassler R, Propst F, Wiche G. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 1997;11:3143–3156. doi: 10.1101/gad.11.23.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuthnott ER, Boyd IA, Kalu KU. Ultrastructural dimensions of myelinated peripheral nerve fibres in the cat and their relation to conduction velocity. J Physiol. 1980;308:125–137. doi: 10.1113/jphysiol.1980.sp013465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Bernier G, Mathieu M, Rossant J, Kothary R. The mouse dystonia musculorum gene is a neural isoform of bullous pemphigoid antigen 1. Nat Genet. 1995;10:301–306. doi: 10.1038/ng0795-301. [DOI] [PubMed] [Google Scholar]
- Carden MJ, Schlaepfer WW, Lee VM. The structure, biochemical properties, and immunogenicity of neurofilament peripheral regions are determined by phosphorylation state. J Biol Chem. 1985;260:9805–9817. [PubMed] [Google Scholar]
- Ching GY, Liem RK. Assembly of type IV neuronal intermediate filaments in nonneuronal cells in the absence of preexisting cytoplasmic intermediate filaments. J Cell Biol. 1993;122:1323–1335. doi: 10.1083/jcb.122.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomcznski P, Sacchi N. Single step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Monteiro MJ, Wong PC, Gill SR, Gearhart JD, Hoffman PN. Involvement of neurofilaments in the radial growth of axons. J Cell Sci. 1991;15(Suppl.):85–95. doi: 10.1242/jcs.1991.supplement_15.12. [DOI] [PubMed] [Google Scholar]
- Dalpe G, Leclerc N, Vallee A, Messer A, Mattrieu M, Repentigny Y-D, Kothary R. Dystonin is essential for maintaining neuronal cytoskeletal organization. Mol Cell Neurosci. 1998;10:243–257. doi: 10.1006/mcne.1997.0660. [DOI] [PubMed] [Google Scholar]
- de Waegh SM, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-d. [DOI] [PubMed] [Google Scholar]
- Dowling J, Yang Y, Wollmann R, Reichardt J, Fuchs E. Developmental expression of BPAG1-n: insights into the spastic ataxia and gross neurologic degeneration in Dystonia musculorummice. Dev Biol. 1997;187:131–142. doi: 10.1006/dbio.1997.8567. [DOI] [PubMed] [Google Scholar]
- Elder GA, Friedrich VL, Bosco P, Kang C, Goourov A, Tu P-H, Lee VMY, Lazzarini RA. Absence of the mid-sized neurofilament subunit decreases axonal calibers, levels of light neurofilament (NF-L) and neurofilament content. J Cell Biol. 1998;141:727–739. doi: 10.1083/jcb.141.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott CE, Becker B, Oehler S, Castanon MJ, Hauptmann R, Wiche G. Plectin transcript diversity: identification and tissue distribution of variants with distinct first coding exons and rodless isoforms. Genomics. 1997;42:115–125. doi: 10.1006/geno.1997.4724. [DOI] [PubMed] [Google Scholar]
- Errante LD, Wiche G, Shaw G. Distribution of plectin, an intermediate filament-associated protein, in the adult rat central nervous system. J Neurosci Res. 1994;37:515–528. doi: 10.1002/jnr.490370411. [DOI] [PubMed] [Google Scholar]
- Eyer J, Peterson A. Neurofilament-deficient axons and perikaryal aggregates in viable transgenic mice expressing a neurofilament-β-galactosidase fusion protein. Neuron. 1994;12:389–405. doi: 10.1016/0896-6273(94)90280-1. [DOI] [PubMed] [Google Scholar]
- Foisner R, Feldman B, Sander L, Wiche G. Monoclonal antibody mapping of structural and functional plectin epitopes. J Cell Biol. 1991;112:397–405. doi: 10.1083/jcb.112.3.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friede RL, Samorajski T. Axon caliber related to neurofilaments and microtubules in sciatic nerve fibers of rats and mice. Anat Rec. 1970;167:379–387. doi: 10.1002/ar.1091670402. [DOI] [PubMed] [Google Scholar]
- Gache Y, Chavanas S, Lacour JP, Wiche G, Owaribe K, Meneguzzi G, Ortonne JP. Defective expression of plectin/HD1 in epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest. 1996;97:2289–2298. doi: 10.1172/JCI118671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser HS, Grundfest H. Axon diameters in relation to the spike dimensions and the conduction velocity in mammalian A fibers. Am J Physiol. 1939;127:393–414. [Google Scholar]
- Glicksman MA, Soppet D, Willard MB. Posttranslational modification of neurofilament polypeptides in rabbit retina. J Neurobiol. 1987;18:167–196. doi: 10.1002/neu.480180205. [DOI] [PubMed] [Google Scholar]
- Guo L, Degenstein L, Dowling J, Yu QC, Wollmann R, Perman B, Fuchs E. Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell. 1995;81:233–243. doi: 10.1016/0092-8674(95)90333-x. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Cross-linker system between neurofilaments, microtubules, and membranous organelles in frog axons revealed by the quick-freeze, deep-etching method. J Cell Biol. 1982;94:129–142. doi: 10.1083/jcb.94.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Glicksman MA, Willard MB. Organization of mammalian neurofilament polypeptides within the neuronal cytoskeleton. J Cell Biol. 1984;98:1523–1536. doi: 10.1083/jcb.98.4.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisanaga S, Hirokawa N. Structure of the peripheral domains of neurofilaments revealed by low angle rotary shadowing. J Mol Biol. 1988;202:297–305. doi: 10.1016/0022-2836(88)90459-7. [DOI] [PubMed] [Google Scholar]
- Hisanaga S, Hirokawa N. Dephosphorylation-induced interactions of neurofilaments with microtubules. J Biol Chem. 1990;265:21852–21858. [PubMed] [Google Scholar]
- Hisanaga S, Kusubata M, Okumura E, Kishimoto T. Phosphorylation of neurofilament H subunit at the tail domain by CDC2 kinase dissociates the association to microtubules. J Biol Chem. 1991;266:21798–21803. [PubMed] [Google Scholar]
- Hoffman PN, Cleveland DW, Griffin JW, Landes PW, Cowan NJ, Price DL. Neurofilament gene expression: a major determinant of axonal caliber. Proc Natl Acad Sci USA. 1987;84:3472–3476. doi: 10.1073/pnas.84.10.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh S-T, Kidd GJ, Crawford TO, Xu Z-S, Trapp BD, Cleveland DW, Griffin JW. Regional modulation of neurofilament organization by myelination in normal axons. J Neurosci. 1994;14:6392–6401. doi: 10.1523/JNEUROSCI.14-11-06392.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner, A.L. 1994. Gene Targeting. A Practical Approach. IRL Press, New York. 234 pp.
- Julien JP, Mushynski WE. Multiple phosphorylation sites in mammalian neurofilament polypeptides. J Biol Chem. 1982;257:10467–10470. [PubMed] [Google Scholar]
- Julien JP, Cote F, Beaudet L, Sidky M, Flavell D, Grosveld F, Mushynski W. Sequence and structure of the mouse gene coding for the largest neurofilament subunit. Gene. 1988;68:307–314. doi: 10.1016/0378-1119(88)90033-9. [DOI] [PubMed] [Google Scholar]
- Kawamura Y, Dyck PJ, Shimono M, Okazaki H, Tateishi J, Doi H. Morphometric comparison of the vulnerability of peripheral motor and sensory neurons in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1981;40:667–675. doi: 10.1097/00005072-198111000-00008. [DOI] [PubMed] [Google Scholar]
- Lee MK, Cleveland DW. Neurofilament function and dysfunction: involvement in axonal growth and neuronal disease. Curr Opin Cell Biol. 1994;6:34–40. doi: 10.1016/0955-0674(94)90113-9. [DOI] [PubMed] [Google Scholar]
- Lee MK, Tuttle JB, Rebhun LI, Cleveland DW, Frankfurter A. The expression and posttranslational modification of a neuron-specific β-tubulin isotype during chick embryogenesis. Cell Motil Cytoskel. 1990;17:118–132. doi: 10.1002/cm.970170207. [DOI] [PubMed] [Google Scholar]
- Lee MK, Xu Z, Wong PC, Cleveland DW. Neurofilaments are obligate heteropolymers in vivo. J Cell Biol. 1993;122:1337–1350. doi: 10.1083/jcb.122.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Otvos L, Jr, Carden MJ, Hollosi M, Dietzschold B, Lazzarini RA. Identification of the major multiphosphorylation site in mammalian neurofilaments. Proc Natl Acad Sci USA. 1988;85:1998–2002. doi: 10.1073/pnas.85.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JF, Shneidman PS, Skuntz SF, Carden MJ, Lazzarini RA. The structure and organization of the human heavy neurofilament subunit (NF-H) and the gene encoding it. EMBO (Eur Mol Biol Organ) J. 1988;7:1947–1955. doi: 10.1002/j.1460-2075.1988.tb03032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopata MA, Cleveland DW. In vivo microtubules are copolymers of available β-tubulin isotypes: localization of each of six vertebrate β-tubulin isotypes using polyclonal antibodies elicited by synthetic peptide antigens. J Cell Biol. 1987;105:1707–1720. doi: 10.1083/jcb.105.4.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek JR, Williamson TL, Lee MK, Xu Z-S, Crawford TO, Hoffman PN, Cleveland DW. Neurofilament subunit NF-H modulates axonal diameter by affecting the rate or neurofilament transport. J Cell Biol. 1996;135:711–724. doi: 10.1083/jcb.135.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka H, Okabe S, Ishiguro K, Uchida T, Hirokawa N. Interaction of the tail domain of high molecular weight subunits of neurofilaments with the COOH-terminal region of tubulin and its regulation by tau protein kinase II. J Biol Chem. 1993;268:22695–22702. [PubMed] [Google Scholar]
- Monteiro MJ, Hoffman PN, Gearhart JD, Cleveland DW. Expression of NF-L in both neuronal and nonneuronal cells of transgenic mice: increased neurofilament density in axons without affecting caliber. J Cell Biol. 1990;111:1543–1557. doi: 10.1083/jcb.111.4.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MW, Lazzarini RA, Lee VM, Schlaepfer WW, Nelson DL. The human mid-size neurofilament subunit: a repeated protein sequence and the relationship of its gene to the intermediate filament gene family. EMBO (Eur Mol Biol Organ) J. 1987;6:1617–1626. doi: 10.1002/j.1460-2075.1987.tb02409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Chen J, Zhang Z, Kanai Y, Hirokawa N. Two distinct functions of the carboxyl-terminal tail domain of NF-M upon neurofilament assembly; crossbridge formation and longitudinal elongation of filaments. J Cell Biol. 1995;129:411–429. doi: 10.1083/jcb.129.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Paskevich PA, Sihag RK, Thayer CY. Phosphorylation on carboxyl terminus domains of neurofilament proteins in retinal ganglion cell neurons in vivo: influences on regional neurofilament accumulation, interneurofilament spacing, and axon caliber. J Cell Biol. 1994;126:1031–1046. doi: 10.1083/jcb.126.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara O, Gahara Y, Miyake T, Teraoka H, Kitamura T. Neurofilament deficiency in quail caused by nonsense mutation in neurofilament-L gene. J Cell Biol. 1993;121:387–395. doi: 10.1083/jcb.121.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi T, Okada M, Kitamura T, Kawasaki K. Reduced diameter and conduction velocity of myelinated fibers in the sciatic nerve of a neurofilament-deficient mutant quail. Neurosci Lett. 1993;153:65–68. doi: 10.1016/0304-3940(93)90078-y. [DOI] [PubMed] [Google Scholar]
- Schlaepfer WW, Bruce J. Simultaneous up-regulation of neurofilament proteins during the postnatal development of the rat nervous system. J Neurosci Res. 1990;25:39–49. doi: 10.1002/jnr.490250106. [DOI] [PubMed] [Google Scholar]
- Smith FJ, Corden LD, Rugg EL, Ratnavel R, Leigh IM, Moss C, Tidman MJ, Hohl D, Huber M, Kunkeler L, et al. Missense mutations in keratin 17 cause either pachyonychia congenita type 2 or a phenotype resembling steatocystoma multiplex. J Invest Dermatol. 1997;108:220–223. doi: 10.1111/1523-1747.ep12335315. [DOI] [PubMed] [Google Scholar]
- Sternberger LA, Sternberger NH. Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc Natl Acad Sci USA. 1983;80:6126–6130. doi: 10.1073/pnas.80.19.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, Borisy GG. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol. 1996;135:991–1007. doi: 10.1083/jcb.135.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troncoso JC, March JL, Haner M, Aebi U. Effect of aluminum and other multivalent cations on neurofilaments in vitro: an electron microscopic study. J Struct Biol. 1990;103:2–12. doi: 10.1016/1047-8477(90)90080-v. [DOI] [PubMed] [Google Scholar]
- Tu PH, Elder G, Lazzarini RA, Nelson D, Trojanowski JQ, Lee VM. Overexpression of the human NFM subunit in transgenic mice modifies the level of endogenous NFL and the phosphorylation state of NFH subunits. J Cell Biol. 1995;129:1629–1640. doi: 10.1083/jcb.129.6.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tybulewicz VL, Crawford JCE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Wiche G. Plectin: general overview and appraisal of its potential role as a subunit protein of the cytomatrix. Crit Rev Biochem Mol Biol. 1989;24:41–67. doi: 10.3109/10409238909082551. [DOI] [PubMed] [Google Scholar]
- Wiche G, Baker MA. Cytoplasmic network arrays demonstrated by immunolocalization using antibodies to a high molecular weight protein present in cytoskeletal preparations from cultured cells. Exp Cell Res. 1982;138:15–29. doi: 10.1016/0014-4827(82)90086-6. [DOI] [PubMed] [Google Scholar]
- Wong PC, Marszalek J, Crawford TO, Xu Z, Hsieh ST, Griffin JW, Cleveland DW. Increasing neurofilament subunit NF-M expression reduces axonal NF-H, inhibits radial growth, and results in neurofilamentous accumulation in motor neurons. J Cell Biol. 1995;130:1413–1422. doi: 10.1083/jcb.130.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Cork LC, Griffin JW, Cleveland DW. Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell. 1993;73:23–33. doi: 10.1016/0092-8674(93)90157-l. [DOI] [PubMed] [Google Scholar]
- Xu Z-S, Marszalek JR, Lee MK, Wong PC, Folmer J, Crawford TO, Hsieh S-T, Griffin JW, Cleveland DW. Subunit composition of neurofilaments specifies axonal diameter. J Cell Biol. 1996;133:1061–1069. doi: 10.1083/jcb.133.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki H, Bennett GS, Itakura C, Mizutani M. Defective expression of neurofilament protein subunits in hereditary hypotrophic axonopathy of quail. Lab Invest. 1992;66:734–743. [PubMed] [Google Scholar]
- Yang Y, Dowling J, Yu QC, Kouklis P, Cleveland DW, Fuchs E. An essential cytoskeletal linker protein connecting actin microfilaments to intermediate filaments. Cell. 1996;86:655–665. doi: 10.1016/s0092-8674(00)80138-5. [DOI] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu PH, Lee VM-Y, Li C, Roder J, Trapp BD. Myelin-associated glocoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Couillard-Despres S, Julien J-P. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol. 1997;148:299–316. doi: 10.1006/exnr.1997.6654. [DOI] [PubMed] [Google Scholar]