

Abstract
Vasoactive effects of soluble matrix proteins and integrin-binding peptides on arterioles are mediated by αvβ3 and α5β1 integrins. To examine the underlying mechanisms, we measured L-type Ca2+ channel current in arteriolar smooth muscle cells in response to integrin ligands. Whole-cell, inward Ba2+ currents were inhibited after application of soluble cyclic RGD peptide, vitronectin (VN), fibronectin (FN), either of two anti–β3 integrin antibodies, or monovalent β3 antibody. With VN or β3 antibody coated onto microbeads and presented as an insoluble ligand, current was also inhibited. In contrast, beads coated with FN or α5 antibody produced significant enhancement of current after bead attachment. Soluble α5 antibody had no effect on current but blocked the increase in current evoked by FN-coated beads and enhanced current when applied in combination with an appropriate IgG. The data suggest that αvβ3 and α5β1 integrins are differentially linked through intracellular signaling pathways to the L-type Ca2+ channel and thereby alter control of Ca2+ influx in vascular smooth muscle. This would account for the vasoactive effects of integrin ligands on arterioles and provide a potential mechanism for wound recognition during tissue injury.
Keywords: voltage-gated Ca2+ channel, vascular smooth muscle, wound repair, extracellular matrix, integrin-mediated signaling
Integrins are heterodimeric receptors (α, β) that mediate cell–extracellular matrix (ECM)1 and cell–cell adhesion events. The cytoskeleton is mechanically linked to the ECM by integrins so that cytoskeletal stiffening increases in direct proportion to applied stress (Wang et al., 1993). Integrins can therefore serve as mechanochemical transducers (Ingber, 1991). Integrins can also function as signaling receptors that transduce biochemical signals both into and out of cells (Clark and Brugge, 1995; Sjaastad and Nelson, 1997). Intracellular signals known to be linked to integrins include pH, Ca2+, protein kinase C activation, and protein tyrosine phosphorylation (Schwartz et al., 1991a ; Schwartz, 1993).
Integrin signaling pathways are generally believed to be initiated by integrin clustering through interactions with insoluble ECM ligands (Clark and Brugge, 1995). These signals are initiated by cell interactions with ECM-coated substrates or with beads coated with ECM proteins or antiintegrin antibodies (Miyamoto et al., 1995b ; Plopper et al., 1995). When soluble ECM protein or antibody is added, minimal or no signaling is thought to occur, but if soluble antibody is followed by a cross-linking antibody, signaling pathways are activated (Yamada and Geiger, 1997). A widely studied recognition site on ECM proteins, including vitronectin (VN) and fibronectin (FN) (Schwarzbauer, 1991), is the tripeptide Arg-Gly-Asp (RGD), which is recognized by a common subset of integrins, including αvβ3, α5β1, αvβ5, and αIIbβ3. RGD peptides are known to disrupt integrin-dependent cell adhesive events (Akiyama, 1996) as well as produce inhibitory effects on major cellular processes such as platelet aggregation and angiogenesis (Weiss et al., 1997). For this reason, RGD peptides are potential therapeutic agents for thrombotic diseases and cancer. One important, unresolved issue is whether RGD peptides act solely by disrupting cell–ECM contacts or whether they provide direct signals to cells by binding to unoccupied integrins. Recent data from our laboratories suggest that soluble RGD peptides may provide vasoactive signals to cells in the vascular wall (Mogford et al., 1996, 1997; D'Angelo et al., 1997). Thus, RGD peptides may be capable of directly stimulating integrin-dependent intracellular signaling pathways.
In rat cremaster muscle arterioles, integrin-binding RGD peptides and fragments of denatured collagen type I cause dilation through an interaction with the αvβ3 integrin on vascular smooth muscle (Mogford et al., 1996). Dilation is associated with a decrease in intracellular Ca2+ concentration ([Ca2+]i) (D'Angelo et al., 1997) and can be prevented by a function-blocking antibody specific for the β3 integrin (Mogford et al., 1996). In addition to these prolonged effects, RGD peptides also cause a transient, endothelium-independent constriction of arterioles, mediated by the α5β1 integrin (Mogford et al., 1997). In rat afferent arterioles, RGD peptide causes a sustained constriction that is associated with an increase in smooth muscle cell [Ca2+]i (Yip and Marsh, 1997). The signaling mechanisms downstream from integrin-ligand binding are poorly understood, particularly in vascular smooth muscle cells (SMCs). We hypothesized that the L-type, voltage-gated calcium channel was involved in the vasoactive responses of arterioles since this channel is known to be a major pathway for calcium entry into vascular SMCs. To test this hypothesis, we isolated single SMCs from rat cremaster arterioles and selectively measured whole-cell calcium current before and after application of integrin ligands in both soluble and insoluble form.
Materials and Methods
Cell Isolation Techniques
Male Sprague-Dawley rats (120–200 g) were anesthetized with intraperitoneal injection of pentobarbital sodium (120 mg/kg). All animal handling procedures followed institutional guidelines. The two cremaster muscles were excised and pinned flat for vessel dissection in a 4°C silastic-coated Plexiglas chamber containing Ca2+-free saline solution. The composition was (in mM) 147 NaCl, 8.6 KCl, 1.17 MgSO4, 1.2 NaH2PO4, 5.0 d-glucose, 2.0 pyruvate, 0.02 EDTA, and 3 MOPS (pH adjusted to 7.4 with NaOH), with BSA (0.1 mg/ml; Amersham Life Science, Arlington Heights, IL) added to maintain cell integrity. Dissected segments of first- and second-order arterioles were transferred to a tube of low-Ca2+ saline solution containing (in mM) 144 NaCl, 5.6 KCl, 0.1 CaCl2, 1.0 MgCl2, 0.42 Na2HPO4, 0.44 NaH2PO4, 10 Hepes, 4.17 NaHCO3, and 1 mg/ml BSA (pH adjusted to 7.4 with NaOH) at room temperature for 10 min. After allowing the vessels to settle to the bottom of the tube, the solution was decanted and replaced with low-Ca2+ saline containing 26 U/ml papain (Sigma Chemical Co., St. Louis, MO) and 1 mg/ml dithioerythritol (Sigma Chemical Co.). The vessels were incubated for 30 min at 37°C with occasional agitation, after which vessel fragments were transferred to low-Ca2+ saline solution containing 1.95 collagenase (FALGPA U/ml; Sigma Chemical Co.), 1 mg/ml soybean trypsin inhibitor (Sigma Chemical Co.), and 75 U/ml elastase (Calbiochem, La Jolla, CA) for 15 min at 37°C. After further digestion, the remaining fragments were rinsed two times with low-Ca2+ saline solution and gently triturated using a fire-polished Pasteur pipette to release single cells.
Patch Clamp Techniques
Perforated, whole-cell recordings were made as described previously (Rae and Fernandez, 1991). Micropipettes were pulled from 1.5-mm glass tubing (Corning No. 8161; Warner Instruments, Hamden, CT) on a programmable puller and fire polished. Pipette resistances ranged from 1 to 3 MΩ. The pipettes were dipped for 2–3 s in Cs+ pipette solution (high Cs+) containing (in mM) 110 CsCl, 20 TEA chloride, 10 EGTA, 2 MgCl2, 10 Hepes, and 1 CaCl2 (pH adjusted to 7.2 with CsOH) and then backfilled with the same solution containing 240 μg/ml amphotericin B. An EPC-7 amplifier (HEKA, Darmstadt-Eberstadt, Germany) was used to record current, and hydraulic manipulators (model M0-102; Narishige, Tokyo, Japan) were used for fine control of the micropipettes. Analog to digital conversions were made using a TL-1 DMA interface (Axon Instruments, Foster City, CA) and stored on a Pentium computer for subsequent analysis. Data were sampled at 5–10 kHz and filtered at 1–2 kHz using an eight-pole Bessel filter. Series resistance varied from 2 to 6 MΩ. Current records were analyzed using pClamp (version 6.0.3; Axon Instruments). Currents through the L-type calcium channel were elicited by voltage ramps (from −100 mV to +80 mV, duration = 200 ms) or by voltage steps (from −80 to +60 mV in 10 mV increments, duration = 300 ms). All experiments were performed at 22°C.
A suspension of freshly dispersed cells was plated onto a thin glass coverslip in a recording chamber on the stage of an inverted microscope. The coverslip was not usually treated, but in some experiments it was coated with FN (120 kD, 20 μg/ml) before addition of cells (Schwartz, 1993). Current recordings were made from individual cells between 30 min and 3 h after plating. Cells harvested using the digestion procedure were elongated with tapered ends in physiological saline solution (PSS), refractile under interference contrast optics, and contractile in solutions containing 140 mM K+ or 20 mM Ba2+. At the beginning of each experiment, the recording chamber was suffused with PSS from a gravity-fed reservoir at a rate of 1.5 ml/min. PSS had the following composition (in mM): 136 NaCl, 5.9 KCl, 10 Hepes, 1.16 NaH2PO4, 1.2 MgCl2, 1.8 CaCl2, 18 d-glucose, 0.02 EGTA, and 2 pyruvate (pH adjusted to 7.4 with NaOH). To record whole-cell current through the calcium channel, Ba2+ (20 mM) was used as the charge carrier in place of K+ and Na+ in the bath solution. This procedure is known to increase the size of the inward currents elicited by depolarization, and to minimize calcium-dependent inactivation of these currents (Griffith et al., 1994). The Ba2+ bath solution (20 Ba2+) contained (in mM) 20 BaCl2, 124 choline chloride, 10 Hepes, and 15 d-glucose (pH adjusted to 7.4 with TEA-OH).
Both ramp and step voltage protocols elicited inward, whole-cell Ba2+ currents (IBa) that peaked at +30 mV (range = 3.0–10.4 pA/pF); typically, these currents were stable for more than 30 min. Since current–voltage (I-V) relations for the ramp and step protocols were nearly identical, the average of five voltage ramps was used to measure IBa in most experiments. The activation portion of the I-V curve (from −60 to +30 mV) increased smoothly to a single maximum with no secondary “hump” in its voltage dependence, which is the pattern consistent with activation of only a single type Ca2+ channel (L-type) in this tissue (Nelson et al., 1990; Cox et al., 1992). As noted previously (Hill et al., 1996), the entire I-V relation was shifted about 30 mV to the right in 20 mM Ba2+ solution. This behavior is typical for voltage-gated calcium channels because of the fact that the equilibrium potential for the permeable ion shifts to the right with increasing extracellular ion concentration. When physiological Ca2+ is used as the charge carrier, the peak of the I-V curve occurs between −10 mV and 0 mV, and the activation threshold occurs at approximately −50 mV, as demonstrated in other SMC preparations (Aaronson et al., 1988).
Ligand Application
VN, FN (120 kD), lyophilized cyclic GPenGRGDSPCA (cRGD, with Pen indicating penicillamine), and the control GRGESP peptide (RGE) were obtained from GIBCO-BRL (Gaithersburg, MD). The anti–β3 integrin function-blocking antibodies (F11; anti–rat monoclonal), 2C9.G2 (monoclonal), and the anti–α5 integrin function-blocking antibody (HMα5-1; anti–rat monoclonal raised in Armenian hamster) were obtained from PharMingen (San Diego, CA). Anti–rat MHC class I monoclonal antibody (MHC; clone R4-8B1) was obtained from Seikagaku Inc. (Tokyo, Japan). Anti–Armenian hamster monoclonal IgG was obtained from Sigma Chemical Co. Monovalent antibodies were made by digesting F11 (in stock solution) with papain, followed by subsequent extraction of Fc fragments using a column of anti–mouse Fc coupled to Sephadex. The resulting Fab digest displayed a prominent band at 50 kD with no evidence of intact F11 at 150 kD.
For application to single cells, each agent was added to 20 Ba2+ solution and ejected from a picospritzer pipette (General Valve Corp., Fairfield, NJ) positioned ∼50 μm away from a cell (Fig. 1 A).
Figure 1.
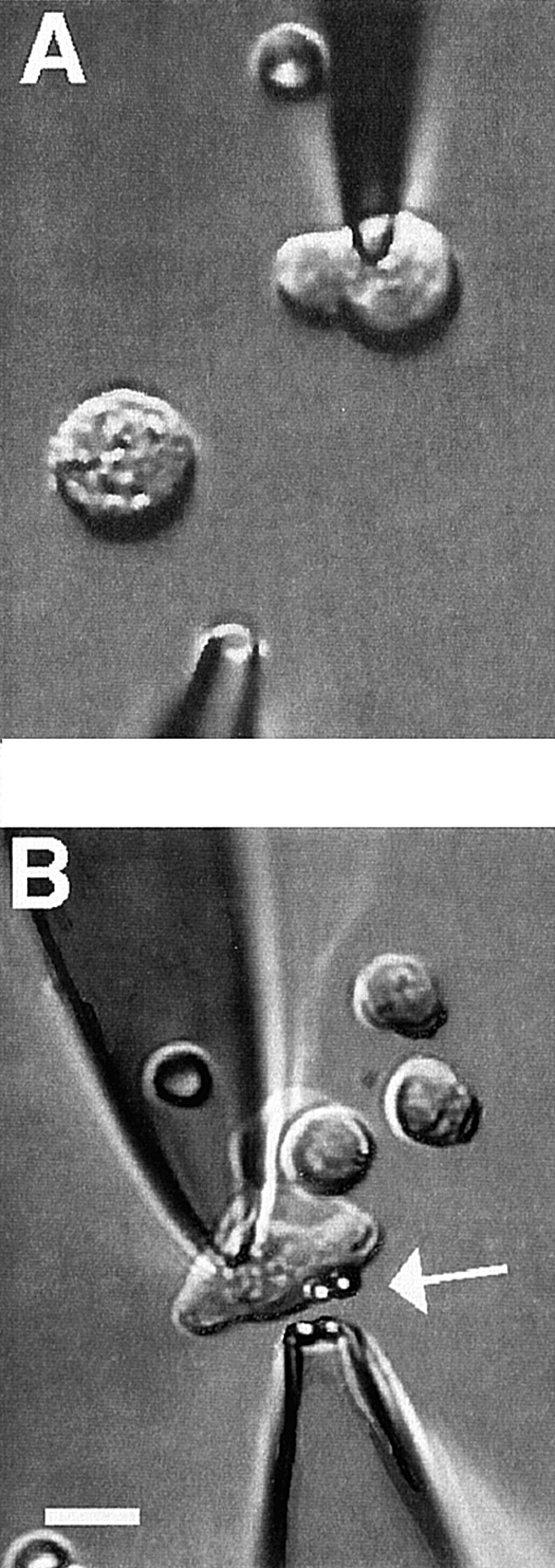
Methods for application of integrin ligands to smooth muscle cells. The images show voltage-clamped SMCs during application of (A) soluble integrin ligand from a picospritzer pipette or (B) protein-coated beads using gentle aspiration from a pipette. In both panels, the pipette at the top is a recording pipette used to hold the cell at −80 mV. Cells are contracted because of the solutions used. In A, the lower pipette is connected to a picospritzer for rapid application of solutions to the area surrounding the cell. In B, a red blood cell can be seen on the chamber bottom (underneath, not inside, the patch pipette). The pipette at the bottom is a large-tip pipette containing FN-coated beads. Two beads become attached to the cell (white arrow), and two remain in the pipette. Both panels: bath solution is 20 Ba2+; patch pipette solution is high Cs+; second pipette solution: 20 Ba2+. Bar, 10 μm.
Application of Protein-coated Beads
Streptavidin-coated microspheres (3.2 μm in diameter) were obtained from Bangs Laboratories (Fishers, IN). Before each experiment, the beads were coated with protein using a biotinylation procedure. Biotinylated FN, F11, VN, HMα5-1, and MHC were prepared using a method similar to that described previously (Hnatowich et al., 1987; Larson et al., 1992). The molar ratio of NHS-LC-Biotin (Pierce Chemical Co., Rockford, IL) to protein (10 μg/ml) was 20:1. To remove unreacted biotin, ultrafree-MC filters were used (Millipore Corp., Bedford, MA). Nonspecific sites on the beads were blocked by incubation with 0.1% heat-denatured BSA in PSS. A dilute suspension of beads in Ba2+ bath solution was then used to backfill micropipettes for application to single cells. These pipettes were positioned 5–10 μm away from the cells and fashioned so that their tip diameters were approximately twice the diameter of the beads; gentle pressure from a glass syringe (<2 cm H2O) was used to eject the beads (Fig. 1 B).
Data Analysis
Whole-cell recordings were made from cells with capacitances varying from 4 to 16 pF. We used data only from cells in which stable gigaseals were maintained. In most analyses, the raw current value was normalized to cell capacitance (an index of cell size) and expressed as current density (pA/pF). Statistical comparisons were performed with repeated-measures analysis of variance followed by post hoc tests, or with an independent two-tail t test, as appropriate. Averaged values are expressed as mean ± SEM. Values of P < 0.05 were considered to be statistically significant.
Results
Effect of cRGD on IBa
The effect of soluble cRGD peptide (100 μM for 1 min) on inward Ba2+ current is shown in Fig. 2. This dose of peptide was reported to produce near-maximal dilation of isolated cremaster arterioles (Mogford et al., 1996). Currents from single arteriolar myocytes were elicited every 15 s by a depolarizing pulse to +30 mV (300-ms duration) from a holding potential of −80 mV. The time course of the response from a representative cell is shown on the left side of Fig. 2 A, and individual current traces at the indicated time points are shown on the right side. Before peptide application, peak current ranged from −76 pA to −77 pA. Within 15 s after application of soluble cRGD peptide (100 μM) from a picospritzer pipette, current was inhibited (to −60 pA) and maximal inhibition (to −51 pA) was achieved 45 s after cRGD application. Nearly complete recovery from inhibition (to −75 pA) was observed within 30 s after peptide washout.
Figure 2.
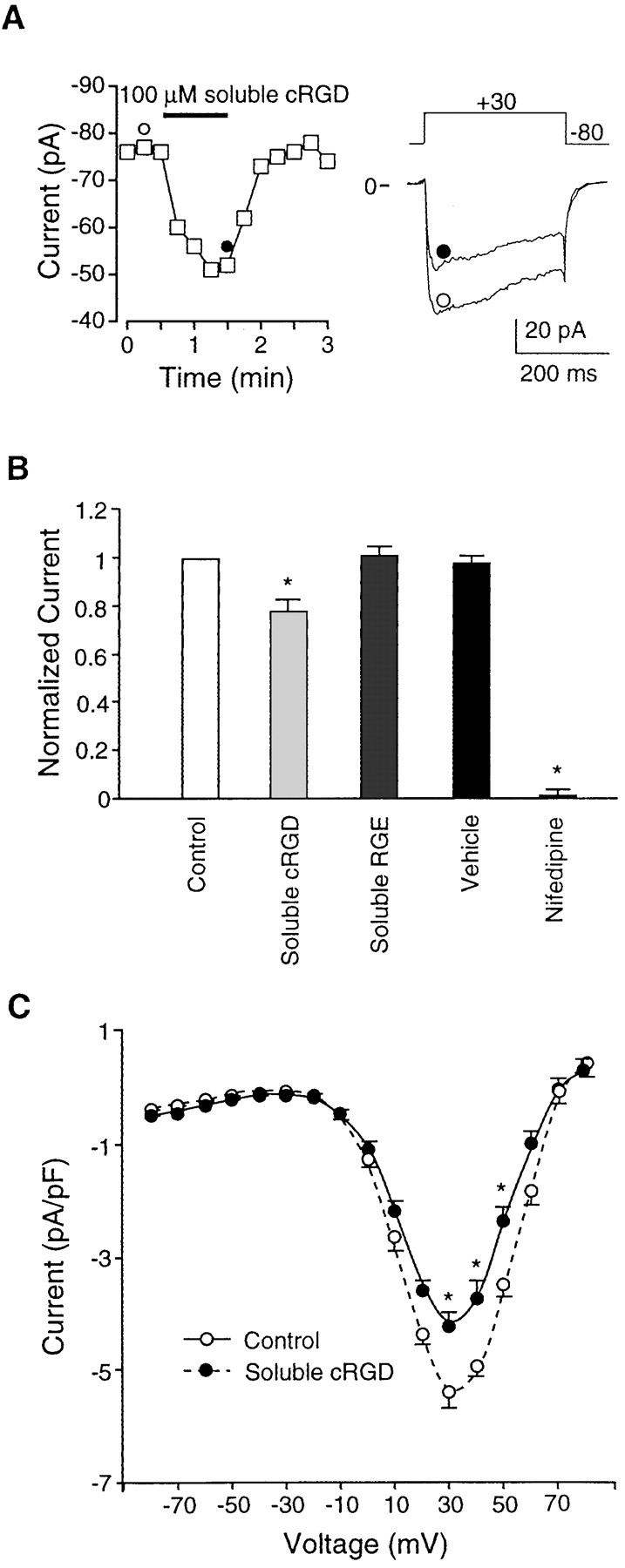
Effects of soluble cRGD on IBa. (A) Time course of changes in IBa (measurements made at 15-s intervals) for a single arteriolar SMC in response to application of soluble cRGD peptide (100 μM). Test potential was +30 mV in each case. Individual traces at right are leak-subtracted current traces at the time points indicated by the symbols. Cell capacitance was 12 pF. (B) Bar graph summarizing data for effects of soluble cRGD (n = 9), soluble RGE (n = 4), vehicle (n = 4), and nifedipine (1 μM, n = 7). For each cell, the data represent peak currents 1 min after application, as normalized to the current at the peak of the control I-V relationship (usually +20 or +30 mV). (C) Summary I-V curves for Ba2+ current before or 60 s after application of soluble cRGD (100 μM). Data from nine cells. All panels: bath solution is 20 Ba2+; pipette solution is high Cs+; HP = −80 mV. *P < 0.05 vs. control.
The average response of nine cells to soluble RGD peptide is summarized in Fig. 2 B, where the data for each cell have been normalized to the peak Ba2+ current recorded just before peptide application. On average, 100 μM cRGD produced 22% inhibition of IBa at +30 mV (measurements taken immediately before peptide washout). Also illustrated in the bar graph are the effects of vehicle, RGE peptide (which does not interact with integrin receptors), and nifedipine, a dihydropyridine calcium channel blocker. Neither vehicle nor RGE peptide (80 μM; n = 4) had a significant effect on IBa. Nifedipine (1 mM; n = 7) produced nearly 100% inhibition of current at this dose, which is consistent with the behavior of an L-type Ca2+ channel. A comparison of current–voltage relationships recorded before and during cRGD application (Fig. 2 C) indicates that inhibition of IBa occurred across the entire range of voltages associated with activation of the L-type Ca2+ channel. Thus, there appeared to be no significant effect of RGD peptide on the threshold or reversal potential of the current.
Effect of Vitronectin on IBa
VN is known to interact with several integrins, including αvβ3 (the VN receptor). Fig. 3 A illustrates the effect of soluble VN on IBa. Before application, peak current in this representative cell was stable between −86 and −87 pA. Within 15 s after ejection of soluble VN (0.04 μM) from the picospritzer pipette, IBa decreased to −69 pA, with a further inhibition to −49 pA at 60 s after application. Recovery of current was complete within 60 s after VN washout. The bar graph in Fig. 3 A summarizes results from seven cells. On average, this concentration of soluble VN inhibited current by 39 ± 5%. Although not illustrated in this figure, inhibition of IBa by VN was sustained during longer periods of application (48 ± 7% inhibition at 4 min).
Figure 3.
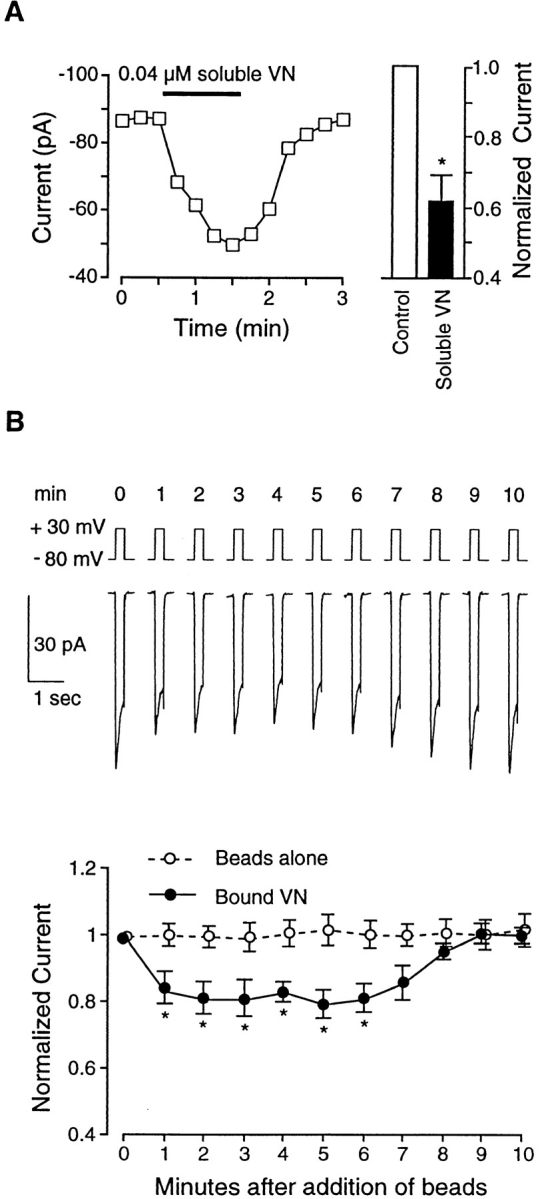
Effects of VN on IBa. (A) Graph at left shows time course of changes in IBa for a single cell before and during application of soluble VN (0.04 μM). Test potential was +30 mV in each case, and measurements were made at 15-s intervals. Cell capacitance was 15 pF. Bar graph at right shows summary data (n = 7) for IBa 60 s after application of soluble VN compared with control current (just before VN application). Currents were normalized to the current at the peak of the control I-V relationship. (B) Top traces show time course of changes in IBa (note compressed time scale for each trace compared with traces in Fig. 2 A) for a single cell before and after application of four VN-coated beads (at t = 0 min). Lower graph shows summary time course of IBa changes in response to VN-coated beads (filled circles; 2–5 beads/cell; n = 6) or uncoated beads (open circles; n = 5). All values were normalized to the peak value of IBa at t = 0 min. Both panels: bath solution is 20 Ba2+; pipette solution is high Cs+; HP = −80 mV. *P < 0.05 vs. control.
Fig. 3 B shows the effect of VN-coated beads on IBa. The top trace shows the time course of changes in current before (time = 0 min) and after attachment of four beads to a representative cell. Note that both peak and steady-state currents were inhibited within 1 min of bead attachment, remained inhibited for ∼5 min, and then gradually returned toward control levels even though the beads appeared to remain attached. Data from six cells are summarized in the lower portion of Fig. 3 B. On average, a 20% inhibition of IBa was observed in response to bead attachment. As a control for nonspecific mechanical effects associated with bead application, the response to uncoated beads was also tested (open circles); no significant changes in IBa were noted with uncoated beads (n = 5) or with BSA-coated beads (n = 4).
Inhibition of IBa after attachment of VN-coated beads was proportional to the number of beads that attached to a given cell, a process over which we had only partial control. Regression analysis of the percent inhibition of IBa as a function of the number of attached beads gave a correlation coefficient of 0.86 (ΔIBa = −0.8 pA − 5.2 × No. of beads). For the purpose of determining the average responses of cells to coated beads in this and subsequent protocols, data were therefore pooled from cells to which between two and five beads attached.
Effect of β3 Antibody on IBa
To test the hypothesis that the effects of cRGD and VN were mediated through the αvβ3 receptor, a function-blocking, monoclonal antibody to the rat β3 integrin (F11) was applied to the cells. F11 is known to block the dilatory effects of cRGD peptide on isolated arterioles (Mogford et al., 1996). β3 integrins are known to associate with two different α subunits (Hemler, 1990), but only one of those, αv, has been identified in vascular smooth muscle (Yip and Marsh, 1997). Fig. 4 A shows the time course of changes in IBa after application of soluble F11 (0.03 μM) to a representative cell. In this cell, soluble F11 inhibited current from −70 pA to −45 pA by 1 min after application. Data from nine cells are summarized in the bar graph of Fig. 4 A and show that this dose of soluble F11 inhibited IBa by an average of 33 ± 5%. We also tested the effect of a second β3 integrin antibody, 2C9.G2, which is reported to block adhesion (Schultz and Armant, 1995). After 60 s of application, soluble 2C9.G2 (0.03 μM) inhibited IBa by 22 ± 4.5% (n = 8). In addition, we made Fab fragments of F11 to test the effect of a monovalent integrin ligand on Ca2+ current. After dilution to 0.03 μM in PSS, Fab fragments caused a 29 ± 5% inhibition of IBa 1 min after application (n = 7). As a control for nonspecific effects of antibody, a nonintegrin binding antibody (anti–rat MHC, 0.2 μM) was also tested; MHC had no significant effect on current (n = 4), as shown in the right portion of Fig. 4 A.
Figure 4.
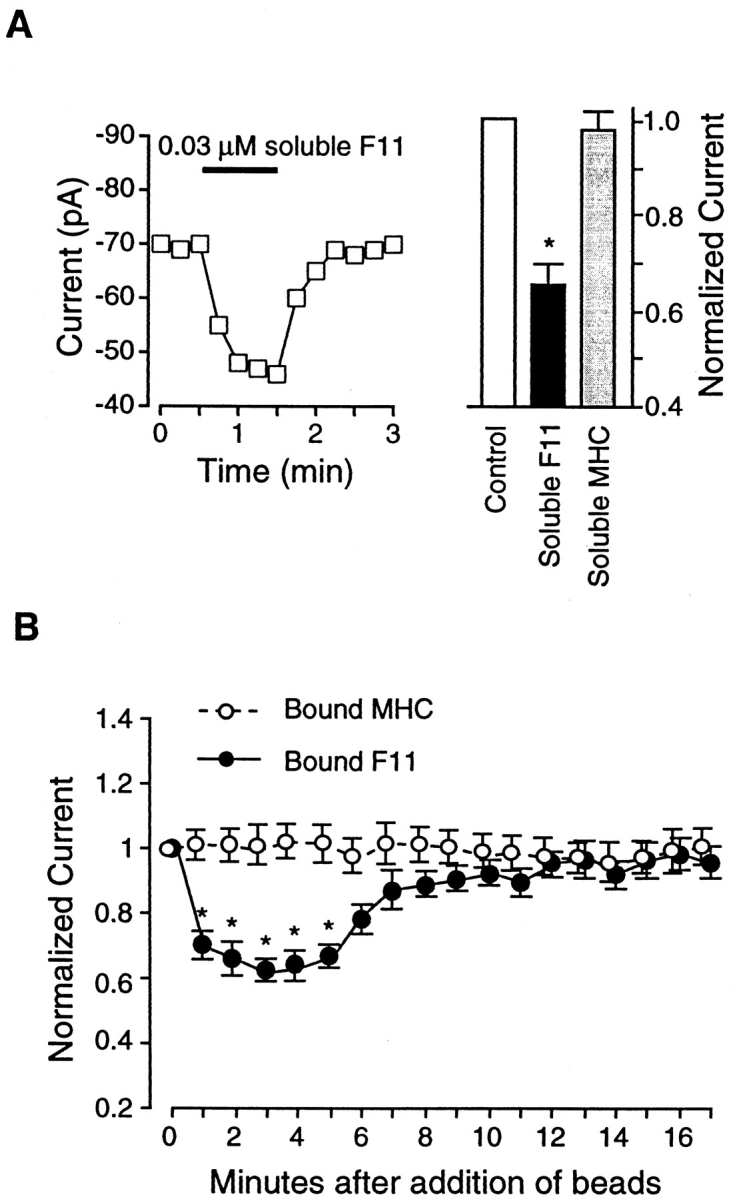
Effects of the β3 antibody, F11, on IBa. (A) Graph at left shows time course of changes in IBa for a single cell before and during application of soluble F11 (0.03 μM). Test potential was +30 mV in each case, and measurements were made at 15-s intervals. Cell capacitance was 15 pF. Bar graph at right shows summary data for IBa 60 s after application of soluble F11 (n = 9) or soluble MHC (0.2 μM; n = 4), compared with control current. Currents were normalized to the current at the peak of the control I-V relationship. (B) Summary time course of IBa changes in response to F11-coated beads (filled circles; n = 6) or MHC-coated beads (open circles; n = 9). All values were normalized to the peak value of IBa at t = 0 min. Both panels: bath solution is 20 Ba2+; pipette solution is high Cs+; HP = −80 mV. *P < 0.05 vs. control.
When F11-coated beads were applied to cells, IBa was inhibited (Fig. 4 B, closed circles). IBa was reduced to 61% of control at 1.5 min after F11 bead attachment. The inhibition lasted ∼5 min, after which current gradually and spontaneously returned toward control values, even though the beads remained attached. As a control for nonspecific effects of antibody-coated beads, we tested the responses of cells to MHC-coated beads, which had no significant effect on IBa (Fig. 4 B, open circles).
Effect of Fibronectin on IBa
Next, we examined the effect of FN on current. FN is known to interact with both αvβ3 and α5β1 receptors present in this cell type, as well as with a number of other integrins (Hynes, 1992). Fig. 5 A shows the time course of changes in IBa in response to soluble FN (0.1 μM). In this cell, soluble FN inhibited IBa from −80 pA to −56 pA within 60 s after application. The response of seven cells to soluble FN is summarized by the bar graph in Fig. 5 A. On average, this concentration of soluble FN reduced IBa to 75% of control at 1 min. The inhibition was maintained for at least 10 min, when current was still reduced to 80 ± 5% (n = 5; data not shown).
Figure 5.
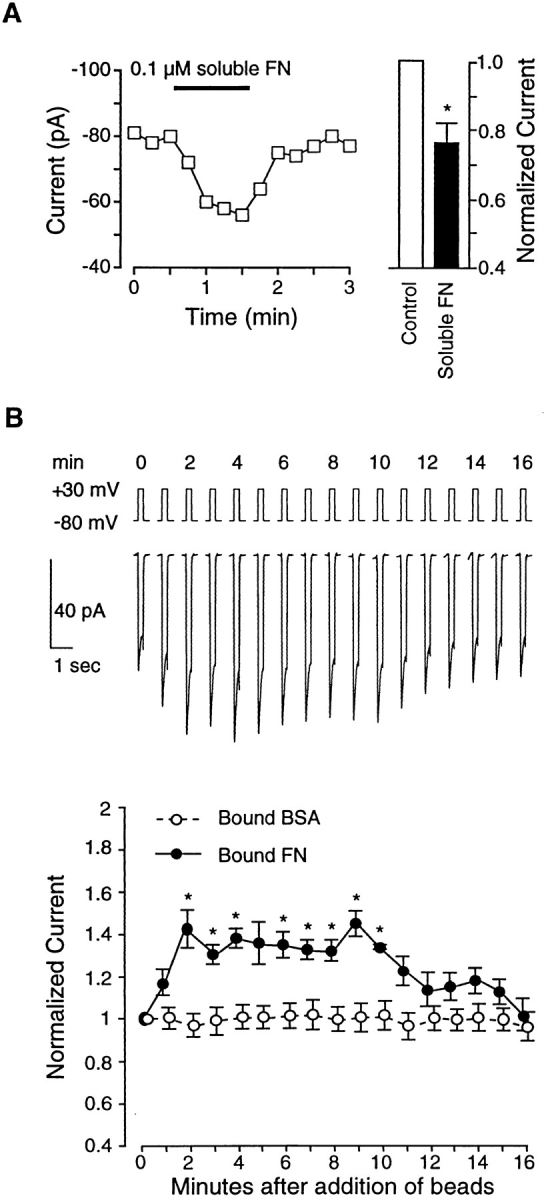
Effects of FN on IBa. (A) Graph at left shows time course of changes in IBa for a single cell before and during application of soluble FN (0.1 μM). Test potential was +30 mV in each case, and measurements were made at 15-s intervals. Cell capacitance was 12 pF. Bar graph at right shows summary data (n = 7 cells) for IBa 60 s after application of soluble FN, compared with control current (just before FN application). Currents were normalized to the current at the peak of the control I-V relationship. (B) Top traces show time course of changes in IBa for a single cell before and after application of three FN-coated beads (at t = 0 min). Lower graph shows summary time course of IBa changes in response to FN-coated beads (filled circles; n = 9) or BSA-coated beads (open circles; n = 4). All values were normalized to the peak value of IBa at t = 0 min. Both panels: bath solution is 20 Ba2+; pipette solution is high Cs+; HP = −80 mV. *P < 0.05 vs. control.
To test the effect of insoluble FN on IBa, FN-coated beads were applied to single cells. The top trace in Fig. 5 B shows the response of a representative cell to attachment of three FN-coated beads. Interestingly, FN-coated beads had the opposite effect on current compared with VN-coated beads or F11-coated beads. Attachment of FN-coated beads led to an enhancement of IBa as early as 1 min after bead attachment. This enhancement peaked at 2 min (∼135% of control), remained stable for 10 min, and then declined gradually by 16 min, even though the beads remained attached. For reference, the time course of changes in IBa in response to BSA-coated beads (which had no significant effect on current) is shown.
Effect of α5 Antibody on IBa
The fact that insoluble VN and insoluble FN had opposite effects on current suggests that an integrin other than αvβ3 might mediate the enhancement of IBa in response to FN-coated beads. Experiments by Mogford et al. (1997) also suggest a role for the α5β1 receptor in vasoactive responses of arterioles because RGD peptide-mediated dilation was converted to constriction after blockade of β3 integrins: the steady-state portion of that constriction was mediated by endothelin and blocked by α5 antibody, but the initial transient constriction was an endothelium-independent response.
To test for the involvement of the α5β1 integrin in our preparation, we used the anti–rat α5 antibody, HMα5-1. The α5 subunit is known to associate only with β1, making this antibody specific for the α5β1 heterodimer (Hynes, 1992). Fig. 6 A shows the effect of applying soluble HMα5-1 to a representative cell: no significant change in IBa was observed. The bar graph in Fig. 6 A summarizes the response of nine cells to application of soluble HMα5-1, which on average produced less than a 2% change in IBa.
Figure 6.
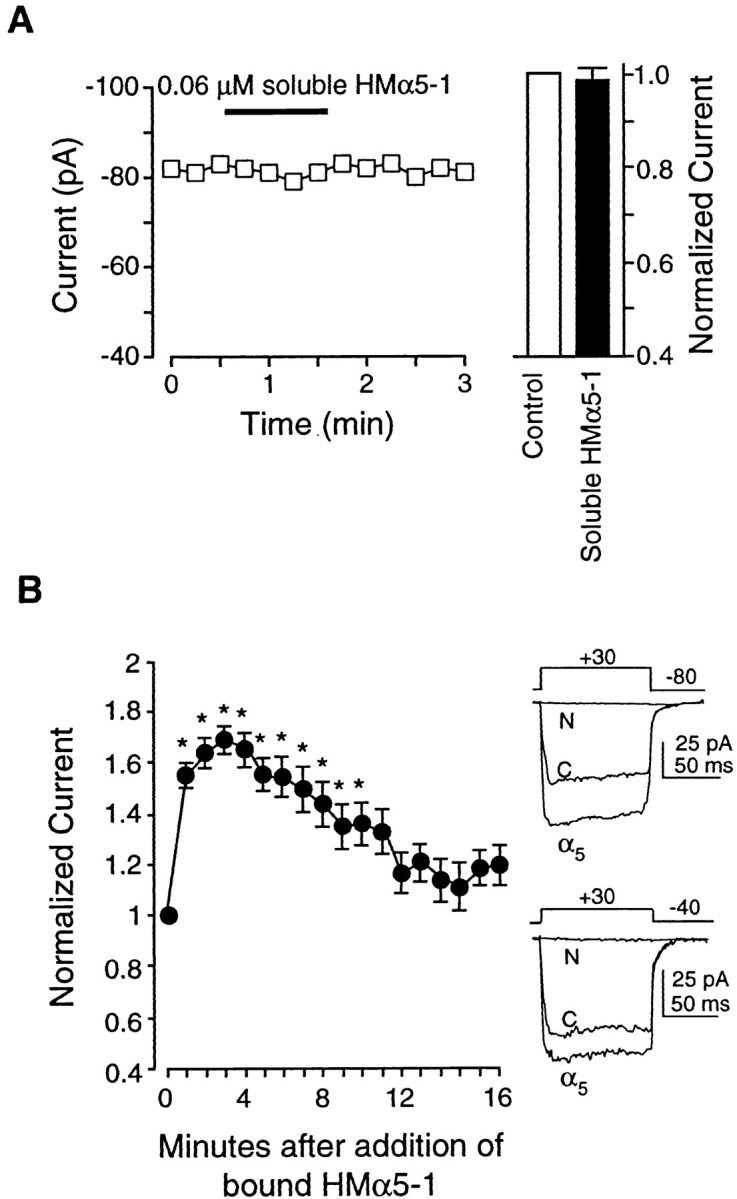

Effects of the α5 antibody, HMα5-1, on IBa. (A) Left trace shows time course of changes in IBa for a single cell before and during application of soluble HMα5-1 (0.06 μM). Test potential was +30 mV in each case, and measurements were made at 15-s intervals. Cell capacitance was 8 pF. Bar graph at right shows summary data for IBa 60 s after application of soluble HMα5-1 (n = 9), compared with control current. Currents were normalized to the current at the peak of the control I-V relationship. (B) Left graph shows time course of average IBa changes in response to HMα5-1–coated beads (n = 5). HMα5-1–coated beads caused a biphasic change in IBa with a large, significant increase lasting about 4 min, followed by a slow return toward control while the beads remained attached. All values were normalized to the peak value of IBa at t = 0 min. Right traces show current traces evoked by a depolarizing pulse to +30 mV from a holding potential of −80 mV (top) or −40 mV (bottom). C, control; α5, HMα5-1-coated beads attached; N, HMα5-1–coated beads attached in the presence of nifedipine (1 μM). (C) I-V curve for currents before (control) or 4 min after attachment of HMα5-1–coated beads. All panels: bath solution is 20 Ba2+; pipette solution is high Cs+; *P < 0.05 vs. control.
However, when beads coated with HMα5-1 were applied to cells, a large and significant increase in IBa was consistently observed, as summarized in Fig. 6 B (left). Within the first minute after attachment of α5-coated beads, IBa had increased to 158% of control. IBa peaked at 170% of control ∼3 min after bead application and then progressively declined toward control; however, IBa did not completely recover even by 17 min after α5-coated bead attachment. Individual current recordings before and after HMα5-1 application are shown in Fig. 6 B (right). The two sets of tracings represent currents evoked from a holding potential of −80 mV (top) or −40 mV (bottom) before and after attachment of HMα5-1–coated beads. As is evident from these recordings, the current stimulated by HMα5-1 was completely inhibited by nifedipine (1 μM), which is consistent with the conclusion that it flowed through L-type calcium channels. Although there is no selective blocker of T-type calcium channels, the possibility that some current might be contributed by T-type channels is ruled out by the fact that the time course of the current recordings evoked from the two different holding potentials are virtually identical.
Fig. 6 C compares the current–voltage relationships for control current and current stimulated by bound HMα5-1. There appeared to be no significant effect on either the threshold or reversal potential of the current.
Effect of Antibody Pretreatment on IBa Response to Coated Beads
To test the idea that clustering of receptors was required to initiate signaling through the α5β1 integrin, soluble α5 antibody was first applied to cells, and then anti–hamster IgG was subsequently added. As shown in Fig. 7 A, there was no response to either agent alone, but when both agents were applied in combination, a significant enhancement in current was noted. The time course of this enhancement was approximately the same as that seen in response to insoluble α5 antibody (compare to Fig. 6 B).
Figure 7.

Effects of antibody pretreatment on response to coated beads. (A) Enhancement in current (n = 5) is observed during application of soluble HMα5-1 in combination with anti–Armenian hamster IgG. Neither IgG or soluble HMα5-1 alone altered current, but when both were added in combination, current was enhanced over approximately the same time course as with bound HMα5-1. (B) Time course of change in IBa when FN-coated beads were applied to cells in the presence of soluble F11 (0.03 μM; n = 7). Currents are normalized to the value of IBa at t = 0 min. Soluble F11 failed to inhibit the enhancement in IBa after bead attachment, although it inhibited current alone, as in Fig. 4 A. In fact, the magnitude of the change in current after FN bead attachment was greater than in the absence of F11 (Fig. 5 B) and similar to that observed in response to HMα5-1–coated beads (Fig. 6 B). (C) Time course of change in IBa when FN-coated beads were applied to cells after application of soluble HMα5-1 (0.06 μM; n = 12). Soluble HMα5-1 alone had no effect on current but blocked the rise in current after FN bead attachment; in fact, a slight but significant decrease in current was observed. (D) Time course of change in IBa when FN-coated beads were applied to cells after pretreatment with soluble HMα5-1 (0.06 μM) and soluble F11 (0.03 μM) in combination. An initial decrease in current was observed (presumably due to the effect of F11), and very little change in current was seen after application of insoluble FN. All panels: bath solution is 20 Ba2+; pipette solution is high Cs+; HP = −80 mV. *P < 0.05 vs. control.
If the response of arteriolar smooth muscle cells to FN-coated beads involves interaction of insoluble FN with both αvβ3 and α5β1 integrins, we predicted that pretreatment with antibody specific to one integrin would result in changes in current characteristic of selective activation of the other integrin. To test this hypothesis, cells were treated with either F11 to block β3 or HMα5-1 to block α5 before application of FN-coated beads. Fig. 7, B–D, shows the results of these experiments.
In Fig. 7 B, application of soluble F11 caused a 30% inhibition of IBa, an effect which is comparable to that observed previously (compare to Fig. 4 A). From this new baseline, application of FN-coated beads increased current from 70% of control to 116% of control. Although interpretation of this response is complicated by the shift in baseline, the absolute change in IBa appeared to be larger than that observed in response to FN-coated beads alone (average increase = 35%; Fig. 5 B) and nearly as large as that produced by insoluble HMα5-1 (Fig. 6 B).
In Fig. 7 C, application of soluble HMα5-1 again produced no change in IBa, but subsequent application of FN-coated beads caused a significant inhibition of current, which is the opposite response observed to FN-coated beads alone (Fig. 5). This observation is consistent with the hypothesis that insoluble FN activates both β3 and α5 integrins in these cells. Importantly, it also indicates that soluble HMα5-1 was indeed interacting with the α5β1 receptor even though no change in IBa was noted in response to soluble HMα5-1 alone. Interestingly, simultaneous application of F11 and HMα5-1 resulted in a 34 ± 8% decrease in current (n = 5), and little change in current was noted after subsequent application of FN-coated beads (Fig. 7 D).
Discussion
To investigate the mechanisms underlying the vasoactive effects of ECM proteins and integrin-specific peptides on rat skeletal muscle arterioles (Mogford et al., 1996, 1997), we measured the response of L-type Ca2+ channel current in arteriolar myocytes to integrin ligands. Soluble αvβ3 ligands (cRGD, VN, FN, bivalent or monovalent β3 antibodies) caused significant inhibition of calcium current, as did beads coated with VN or β3 antibody. In contrast, beads coated with α5β1 ligands (FN or α5 antibody) caused significant enhancement of current. Soluble α5 antibody alone had no effect on current but blocked the increase in current evoked by FN-coated beads and enhanced current when applied in combination with an appropriate IgG. This is the first electrophysiological evidence for regulation of a Ca2+ channel by integrin–ligand interactions and demonstrates that αvβ3 and α5β1 integrins in smooth muscle are differentially linked through intracellular signaling pathways to the L-type calcium channel.
The implications of our findings are threefold: (a) part of the resting current through L-type Ca2+ channels in vascular smooth muscle, and therefore blood vessel tone, is dependent on integrin-matrix interactions; (b) bidirectional regulation of Ca2+ influx in this cell type can be achieved through preferential ligation of αvβ3 or α5β1 integrins; (c) soluble integrin ligands can initiate signaling through the αvβ3 receptor. Since ECM protein denaturation and fragmentation can provide soluble integrin-specific signals to cells (Davis, 1992), this mechanism is likely to be important in the microvascular response to injury (Mogford et al., 1996). Inhibition of smooth muscle cell Ca2+ current could account for integrin-mediated vasodilation of arterioles (Mogford et al., 1996).
Integrin Signaling in Response to Tissue Injury
Local vasodilation is one of the initial responses to tissue injury, resulting in an increase in blood flow to the affected area. This response is mediated primarily by arterioles, which are the strategic control point for local regulation of pressure and flow in every tissue. Increased flow contributes to injury repair by enhancing delivery of inflammatory cells to the injured site. Classic mediators of injury-induced arteriolar dilation include reactive oxygen species (Wei et al., 1981), tachykinins, and histamine (Treede et al., 1990). Recently, Mogford et al. (1996) described an additional mechanism by which RGD-containing peptides induce vasodilation by interacting with the αvβ3 integrin on smooth muscle cells of rat skeletal muscle arterioles. Involvement of the αvβ3 integrin was implicated by the findings that (a) cRGD and GRGDSP peptide were more potent vasodilators than GRGDNP peptide (enhancement of RGD potency by cyclization implicates the involvement of αv integrins [Pierschbacher and Ruoslahti, 1987]) and (b) dilations were attenuated in the presence of a function-blocking β3 monoclonal antibody (Mogford et al., 1996). In addition to synthetic peptides, fragments of denatured collagen type I were potent vasodilators of arterioles (Mogford et al., 1996). While RGD sequences are not exposed in native collagen, cryptic RGD sites become exposed after collagen denaturation and proteolysis, allowing for their interaction with RGD-binding integrins. Exposure of cryptic RGD sites has been proposed to be a potential wound recognition signal during tissue injury (Davis, 1992). Thus, a certain proportion of the αvβ3 receptors may normally be unoccupied on vascular smooth muscle, and after tissue injury, generation of RGD peptide signals that bind the receptor result in decreased Ca2+ current, arteriolar dilation, and increased blood flow to the injured tissue.
Arteriolar dilations to RGD-containing peptides and proteins are mediated by direct effects on vascular smooth muscle integrins rather than on endothelial cell integrins (Mogford et al., 1996). However, none of the downstream signaling mechanisms in smooth muscle have been identified, except that dilation to soluble cyclo-RGD peptide is preceded by a significant decrease in smooth muscle [Ca2+]i (D'Angelo et al., 1997). Our finding that cRGD caused an inhibition of current through the L-type Ca2+ channel in the same cell type (Fig. 2) is consistent with data from intact vessels. In isolated arterioles (Mogford et al., 1996; D'Angelo et al., 1997), dilation was the result of inhibition of myogenic tone, which in resistance vessels (Nelson et al., 1990; Hill and Meininger, 1994) is dependent on basal influx of Ca2+ through L-type Ca2+ channels and can be antagonized by dihydropyridines (Hill and Meininger, 1994). We confirmed that the dihydropyridine nifedipine completely blocked current in our cells (Figs. 2 B and 6 B). Mogford et al. (1996) found that cRGD peptide also inhibited phenylephrine- and KCl-induced vascular tone, but the primary actions of both agents are known to be mediated by Ca2+ influx through voltage-gated Ca2+ channels as well (Nelson et al., 1988).
Integrin-mediated [Ca2+]i Signaling
Integrin-mediated [Ca2+]i signaling has been demonstrated in a number of cell types, including endothelium (Schwartz and Denninghoff, 1994) and vascular smooth muscle (McNamee et al., 1993). Integrins including αIIbβ3, αvβ6, αvβ5, α5β1, and αvβ3 (Hynes, 1992) are known to be involved in [Ca2+]i signaling responses; these integrins also recognize the RGD sequence common to many ECM (FN, osteopontin, and collagens) and plasma proteins (FN, VN, and fibrinogen). Thus, our finding that Ca2+ channel current (and by direct extension Ca2+ influx [Ganitkevich and Isenberg, 1991]) is modulated after αvβ3 and α5β1 receptor ligation is consistent with previous reports in the literature.
Changes in [Ca2+]i initiated by integrin ligation involve a number of mechanisms that result in Ca2+ release from intracellular stores and/or Ca2+ influx (McNamee et al., 1993; Somogyi et al., 1994; Sjaastad et al., 1996). In endothelial cells, αv integrins mediate a rise in [Ca2+]i after adhesion to FN (Schwartz and Denninghoff, 1994). The mechanism underlying this response was not determined, but [Ca2+]i increases did not occur in the absence of extracellular Ca2+. Likewise, both Ca2+ release and Ca2+ influx contributed to the [Ca2+]i rise after adhesion of MDCK cells to RGD-coated beads (Sjaastad et al., 1996), but the influx component was more important for feedback regulation of integrin-mediated adhesion. No mechanism for integrin-mediated Ca2+ influx in nonexcitable cells has been identified, although a role for a 50-kD integrin-associated protein, not yet characterized electrophysiologically, has been postulated (Brown, 1993; Schwartz et al., 1993).
Our data represent the first electrophysiological evidence that integrin ligation can modulate a plasma membrane Ca2+ channel. To make these measurements, Ba2+ was used instead of Ca2+ to carry current through the L-type Ca2+ channel because (a) Ba2+ is more permeable than Ca2+ through this channel, resulting in larger current; (b) Ba2+ blocks the large, outward K+ current that normally masks Ca2+ current in these cells; and (c) Ba2+ currents do not exhibit the rapid inactivation observed when Ca2+ is used (Griffith et al., 1994). Nifedipine, a dihydropyridine that is a selective antagonist of L-type calcium channels (as opposed to other types of voltage-gated calcium channels [Birnbaumer et al., 1994]) at concentrations less than 10−5 M, produced essentially a complete block of basal Ca2+ current (Fig. 2 B) as well as inhibited the enhanced current in response to insoluble α5-antibody (Fig. 6 B).
Although we have not directly measured [Ca2+]i in our preparation, it is highly likely that any treatment causing a significant change in IBa would lead to a similar directional change in [Ca2+]i; this relationship has been clearly demonstrated for visceral (Ganitkevich and Isenberg, 1991) and vascular (Fleischmann et al., 1994) smooth muscle. The previously reported decrease in arteriolar smooth muscle [Ca2+]i in response to soluble RGD peptide (D'Angelo et al., 1997) is consistent with inhibition of IBa by soluble RGD peptide (Fig. 2). In another vascular bed, RGD peptide caused a constriction that was associated with an increase in SMC [Ca2+]i (Yip and Marsh, 1997).
The direct effect of cRGD, VN, FN, and F11 on Ca2+ channel current in isolated SMCs provides strong support for the concept that interaction of αvβ3 with soluble ligands transduces an intracellular signal in this cell type. It remains to be determined if αvβ3 expressed in other cell types, such as endothelium, delivers a similar or different signal. However, endothelial cells (with one exception [Bossu et al., 1989]) lack voltage-gated calcium channels and, in some ways, use opposite mechanisms of controlling calcium entry than smooth muscle. Therefore, it is not surprising that ligation of β3 integrins might lead to increases in endothelial cell [Ca2+]i (Schwartz and Denninghoff, 1994) but opposite changes in SMC [Ca2+]i.
Effects of Soluble and Insoluble Integrin Ligands on Ba2+ Current
A number of possible explanations may account for the differences between the effects of soluble and insoluble integrin ligands on Ca2+ channel current. An obvious possibility is that inhibition of current by soluble FN may be mediated by competitive antagonism of existing integrin– matrix interactions, as suggested for other systems (Poole and Watson, 1995). This would require constitutive phosphorylation of the channel through an integrin-dependent pathway. Indeed, the L-type calcium channel in vascular smooth muscle has been shown to require tyrosine phosphorylation for normal function (Wijetunge et al., 1992; Wijetunge and Hughes, 1996), but whether integrins regulate this pathway is not known. If they do, then disruption of existing integrin–matrix interactions by soluble ligands would produce inhibition of current while clustering of receptors by insoluble ligands (Altieri et al., 1990; Schwartz, 1993), including antibodies (Miyamoto et al., 1995a ), would produce enhancement of current. In our system, soluble ligands of the αvβ3 receptor did produce inhibition of current; however, insoluble β3 ligands (VN and F11) also produced inhibition of current (Figs. 3 and 4). Thus, it seems likely that these effects resulted from activation of a signaling pathway rather than competition for existing β3– matrix interactions. Likewise, since insoluble α5 caused enhancement of current, the competition hypothesis would predict that soluble α5 should reduce current, which it did not.
A more tenable explanation for our results is the possibility that αvβ3 and α5β1 integrins provide distinct and opposing signals to regulate calcium current. As illustrated in Fig. 8 A, we propose that selective ligands of the αvβ3 receptor (F11, 2C9.G2, VN) cause inhibition of current, selective ligands of the α5β1 receptor (HMα5-1) cause enhancement of current, and ligands for both receptors (FN) cause an intermediate response. Our hypothesis requires that several conditions be met: (a) β1 and β3 integrins must signal through different mechanisms in smooth muscle cells. This is supported by the different responses of current to selective ligands of the two respective integrins (Fig. 4 B vs. Fig. 6 B). In endothelial cells as well (Leavesley et al., 1993), β1 and β3 integrins play different roles in regulating Ca2+ entry (Leavesley et al., 1993). Our hypothesis also requires that (b) the α5β1 integrin can only be activated by insoluble ligands. This is consistent with the observation that soluble α5 antibody had no effect on current (Fig. 6 A), yet was an effective blocker of the response to insoluble FN (Fig. 7 C). Experiments by other groups have also shown that soluble α5 antibody failed to increase pHi unless it was cross-linked with a secondary antibody to induce integrin clustering (Schwartz et al., 1991b ). Our hypothesis requires that (c) the αvβ3 integrin must be capable of signaling when ligands are supplied in either a soluble or insoluble form. In support of this is the observation that six different soluble β3 ligands (cRGD, VN, FN, bivalent F11, monovalent F11, and 2C9.G2 antibody) all caused inhibition of IBa, as did two different insoluble β3 ligands (VN and F11). According to our hypothesis, (d) soluble signals must be transmitted only through the αvβ3 integrin and not the α5β1 integrin. This is supported by the observation that soluble FN (a proposed ligand only for αvβ3) inhibited current, while insoluble FN (a known ligand for both αvβ3 and α5β1) enhanced current. Finally, our hypothesis predicts that (e) selective ligands of the α5β1 receptor should produce a larger enhancement in current than a common ligand for both integrins (Fig. 8 B). Accordingly, the magnitude of the increased current was nearly twofold greater when cells were presented with α5 antibody–coated beads compared with FN-coated beads (compare Figs. 5 and 6). Collectively, our data are consistent with the hypothesis that αvβ3 ligation leads to inhibition of the Ca2+ channel, whereas α5β1 ligation leads to stimulation of the Ca2+ channel. Further work will be needed to thoroughly test this hypothesis and to determine if other integrins in vascular smooth muscle are also linked to this channel.
Figure 8.

Diagram of hypothesized interactions between αvβ3 and α5β1 integrins and the voltage-gated Ca2+ channel in vascular smooth muscle. See Discussion for details.
Mechanisms of Calcium Current Modulation
The mechanisms by which αvβ3 and α5β1 ligands modulate this calcium channel are not yet clear, but the possibility that the ligands exert a direct effect on the channel seems unlikely for several reasons: (a) selective antibodies for β3 and α5 integrins modulate Ca2+ current, suggesting that regulation occurs in a signaling pathway upstream from the channel rather than at the channel itself; (b) there is no reported RGD binding sequence in the structure of α1c, the L-type subunit found in vascular smooth muscle (Koch et al., 1990); and (c) antagonists such as dihydropyridines inhibit Ca2+ channels within seconds, and divalent cations block within a fraction of a second (Dolphin, 1995; Hughes, 1995), while inhibition of current by soluble αvβ3 ligands (Figs. 2–5) required ∼60 s to achieve >90% of its maximal effect.
In terms of mechanisms, a more likely possibility is that modulation of current after integrin ligation involves clustering of integrin receptors, recruitment of cytoskeletal proteins, and tyrosine phosphorylation of cytoplasmic signaling molecules, such as FAK, Src, or paxillin, as they are brought into close proximity (Clark and Brugge, 1995). One difference between the effects of soluble and insoluble ligands in our experiments is that soluble ligands had sustained effects on current (soluble FN inhibited current for at least 10 min), while insoluble ligands elicited changes in current that lasted between 6 and 14 min followed by spontaneous recovery. The latter observation would be consistent with a phosphorylation-dependent signaling step that is subject to negative feedback control. This could occur at the level of the receptor, at the channel, or at an intermediate step. In this regard, the affinity of the α5β1 integrin for ligand has been shown to be controlled by the Ca2+-dependent phosphatase CaMKII (Bouvard et al., 1998), such that inhibition of CaMKII preserves the high affinity state of α5β1. A link between integrin signaling and CaMKII has also been demonstrated in vascular smooth muscle (Bilato et al., 1997). Activation of CaMKII after Ca2+ influx through L-type channels could reduce α5β1 affinity and reverse the enhancement of current stimulated by α5β1 ligation. However, other possibilities for initiating signals downstream from integrin ligation may also exist, including pathways involving phospholipase C and protein kinase C (Somogyi et al., 1994).
It is likely that αvβ3 and α5β1 integrins associate directly with one of the L-type Ca2+ channel subunits (e.g., α1c) or with another protein that controls gating or modulates channel activity. A number of cytoplasmic signaling molecules are potential candidates to interact with the calcium channel. Data from recent experiments on the L-type Ca2+ channel in visceral smooth muscle (Hu et al., 1998) have shown that PDGF, which activates a receptor tyrosine kinase, enhances L-type Ca2+ current and this effect is blocked after dialysis of the cells with anti-FAK or anti-Src antibodies. Furthermore, α1c coprecipitates with c-Src in that tissue and has a potential tyrosine phosphorylation site (Koch et al., 1990). Dialysis of SMCs with c-Src (Wijetunge and Hughes, 1995) or with a peptide that activates c-Src (Wijetunge and Hughes, 1996) results in enhancement of Ca2+ current. Taken together, these results and our own preliminary data showing that tyrosine kinase inhibitors reverse the enhancement of current in response to insoluble FN (Wu, X., G.A. Meininger, G.E. Davis, J.E. Mogford, S.H. Platts, and M.J. Davis. 1997. Microcirculation. 4:136a) suggest that the pore-containing subunit of the L-type Ca2+ channel may be tyrosine phosphorylated by c-Src, which in turn is regulated by integrin ligation. Additional experiments will be needed to directly test this idea.
Acknowledgments
The authors thank Judy A. Davidson for technical assistance and Drs. Cindy Meininger and Emily Wilson for advice on various aspects of the experimental design.
This study was supported by National Institutes of Health grants HL-46502 to M.J. Davis and HL-33324 and HL-55050 to G.A. Meininger. Address for reprint requests: M.J. Davis, Dept. of Medical Physiology, 346 Reynolds Medical Building, Texas A & M University Health Science Center, College Station, TX 77843-1114.
Abbreviations used in this paper
- ECM
extracellular matrix
- F11
β3 integrin monoclonal antibody
- FAK
focal adhesion kinase
- FN
fibronectin
- HMα5-1
α5 integrin monoclonal antibody
- IBa
whole-cell Ba2+ current
- MHC
anti–rat IgG monoclonal antibody
- PSS
physiological saline solution
- SMC
smooth muscle cell(s)
- VN
vitronectin
Footnotes
Address all correspondence to Michael J. Davis, Department of Medical Physiology, 336 Reynolds Medical Building, Texas A & M University Health Science Center, College Station, TX 77843-1114. Tel.: (409) 845-7816. Fax: (409) 847-8635. E-mail: mjd@tamu.edu
References
- Aaronson PI, Bolton TB, Lang RJ, MacKenzie I. Calcium currents in single isolated smooth muscle cells from the rabbit ear artery in normal-calcium and high-barium solutions. J Physiol. 1988;405:57–75. doi: 10.1113/jphysiol.1988.sp017321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama SK. Integrins in cell adhesion and signaling. Hum Cell. 1996;9:181–186. [PubMed] [Google Scholar]
- Altieri DC, Wiltse WL, Edgington TS. Signal transduction initiated by extracellular nucleotides regulates the high affinity ligand recognition of the adhesive receptor CD11b/CD18. J Immunol. 1990;145:662–670. [PubMed] [Google Scholar]
- Bilato C, Curto KA, Monticone RE, Pauly RR, White AJ, Crow MT. The inhibition of vascular smooth muscle cell migration by peptide and antibody antagonists of the αvβ3integrin complex is reversed by activated calcium/calmodulin-dependent protein kinase II. J Clin Invest. 1997;100:693–704. doi: 10.1172/JCI119582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaumer L, Campbell KP, Catterall WA, Harpold MM, Hofmann F, Horne WA, Mori Y, Schwartz A, Snutch TP, Tanabe T, Tsien RW. The naming of voltage-gated calcium channels. Neuron. 1994;13:505–506. doi: 10.1016/0896-6273(94)90021-3. [DOI] [PubMed] [Google Scholar]
- Bossu JL, Feltz A, Rodeau JL, Tanzi F. Voltage-dependent transient calcium currents in freshly dissociated capillary endothelial cells. FEBS Lett. 1989;255:377–380. doi: 10.1016/0014-5793(89)81126-3. [DOI] [PubMed] [Google Scholar]
- Bouvard D, Molla A, Block MR. Calcium/calmodulin-dependent protein kinase II controls α5β1integrin–mediated inside-out signaling. J Cell Sci. 1998;111:657–665. doi: 10.1242/jcs.111.5.657. [DOI] [PubMed] [Google Scholar]
- Brown, E.J. 1993. Signal transduction from leukocyte integrins. In Cell Adhesion Molecules. M.E. Hemler and E. Mihich, editors. Plenum Press, New York. 105–125.
- Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Cox RH, Katzka D, Morad M. Characteristics of calcium currents in rabbit portal vein myocytes. Am J Physiol. 1992;263:H453–H463. doi: 10.1152/ajpheart.1992.263.2.H453. [DOI] [PubMed] [Google Scholar]
- D'Angelo G, Mogford JE, Davis GE, Davis MJ, Meininger GA. Integrin-mediated reduction in vascular smooth muscle [Ca2+]iinduced by RGD-containing peptide. Am J Physiol. 1997;272:H2065–H2070. doi: 10.1152/ajpheart.1997.272.4.H2065. [DOI] [PubMed] [Google Scholar]
- Davis GE. Affinity of integrins for damaged extracellular matrix: αvβ3binds to denatured collagen type I through RGD sites. Biochem Biophys Res Commun. 1992;182:1025–1031. doi: 10.1016/0006-291x(92)91834-d. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. Voltage-dependent calcium channels and their modulation by neurotransmitters and G proteins. Exp Physiol. 1995;80:1–36. doi: 10.1113/expphysiol.1995.sp003825. [DOI] [PubMed] [Google Scholar]
- Fleischmann BK, Murray RK, Kotlikoff MI. Voltage window for sustained elevation of cytosolic calcium in smooth muscle cells. Proc Natl Acad Sci USA. 1994;91:11914–11918. doi: 10.1073/pnas.91.25.11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganitkevich VY, Isenberg G. Depolarization-mediated intracellular calcium transients in isolated smooth muscle cells of guinea-pig urinary bladder. J Physiol. 1991;435:187–205. doi: 10.1113/jphysiol.1991.sp018505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith WH, Taylor L, Davis MJ. Whole-cell and single-channel calcium currents in guinea pig basal forebrain neurons. J Neurophysiol. 1994;71:2359–2376. doi: 10.1152/jn.1994.71.6.2359. [DOI] [PubMed] [Google Scholar]
- Hemler ME. VLA proteins in the integrin family: structures, functions, and their role on leukocytes. Annu Rev Immunol. 1990;8:365–400. doi: 10.1146/annurev.iy.08.040190.002053. [DOI] [PubMed] [Google Scholar]
- Hill MA, Meininger GA. Calcium entry and myogenic phenomena in skeletal muscle arterioles. Am J Physiol. 1994;267:H1085–H1092. doi: 10.1152/ajpheart.1994.267.3.H1085. [DOI] [PubMed] [Google Scholar]
- Hill MA, Davis MJ, Song JB, Zou H. Calcium dependence of indolactam-mediated contractions in resistance vessels. J Pharmacol Exp Ther. 1996;276:867–874. [PubMed] [Google Scholar]
- Hnatowich DJ, Virzi F, Ruschowski M. Investigations of avidin and biotin for imaging applications. J Nucl Med. 1987;28:1294–1302. [PubMed] [Google Scholar]
- Hu X-Q, Singh N, Mukhopadhyay D, Akbarali HI. Modulation of voltage-dependent Ca2+channels in rabbit colonic smooth muscle cells by c-Src and focal adhesion kinase. J Biol Chem. 1998;273:5337–5342. doi: 10.1074/jbc.273.9.5337. [DOI] [PubMed] [Google Scholar]
- Hughes AD. Calcium channels in vascular smooth muscle cells. J Vasc Res. 1995;32:353–370. doi: 10.1159/000159111. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Ingber D. Integrins as mechanochemical transducers. Curr Opin Cell Biol. 1991;3:841–848. doi: 10.1016/0955-0674(91)90058-7. [DOI] [PubMed] [Google Scholar]
- Koch WJ, Ellinor PT, Schwartz A. cDNA cloning of a dihydropyridine-sensitive calcium channel from rat aorta. Evidence for the existence of alternatively spliced forms. J Biol Chem. 1990;265:17786–17791. [PubMed] [Google Scholar]
- Larson RC, Ignotz GG, Currie WB. Effect of fibronectin on early embryo development in cows. J Reprod Fert. 1992;96:289–297. doi: 10.1530/jrf.0.0960289. [DOI] [PubMed] [Google Scholar]
- Leavesley DI, Schwartz MA, Rosenfeld M, Cheresh DA. Integrin β1- and β3-mediated endothelial cell migration is triggered through distinct signaling mechanisms. J Cell Biol. 1993;121:163–170. doi: 10.1083/jcb.121.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamee HP, Ingber DE, Schwartz MA. Adhesion to fibronectin stimulates inositol lipid synthesis and enhances PDGF-induced inositol lipid breakdown. J Cell Biol. 1993;121:673–678. doi: 10.1083/jcb.121.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Akiyama SK, Yamada KM. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science. 1995a;267:883–885. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Teramoto H, Coso OA, Gutkind JS, Burbelo PD, Akiyama SK, Yamada KM. Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol. 1995b;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogford JE, Davis GE, Platts SH, Meininger GA. Vascular smooth muscle αvβ3integrin mediates arteriolar vasodilation in response to RGD peptides. Circ Res. 1996;79:821–826. doi: 10.1161/01.res.79.4.821. [DOI] [PubMed] [Google Scholar]
- Mogford JE, Davis GE, Meininger GA. RGDN peptide interaction with endothelial α5β1integrin causes sustained endothelin-dependent vasoconstriction of rat skeletal muscle arterioles. J Clin Invest. 1997;100:1647–1653. doi: 10.1172/JCI119689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Standen NB, Brayden JE, Worley JF., III Noradrenaline contracts arteries by activating voltage-dependent calcium channels. Nature. 1988;336:382–385. doi: 10.1038/336382a0. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol. 1990;259:C3–C18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- Pierschbacher MD, Ruoslahti E. Influence of stereochemistry of the sequence Arg-Gly-Asp-Xaa on binding specificity in cell adhesion. J Biol Chem. 1987;262:17294–17298. [PubMed] [Google Scholar]
- Plopper GE, McNamee HP, Dike LE, Bojanowski K, Ingber DE. Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol Biol Cell. 1995;6:1349–1365. doi: 10.1091/mbc.6.10.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AW, Watson SP. Regulation of cytosolic calcium by collagen in single human platelets. Br J Pharmacol. 1995;115:101–106. doi: 10.1111/j.1476-5381.1995.tb16326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae JL, Fernandez J. Perforated patch recordings in physiology. NIPS (News Physiol Sci) 1991;6:273–277. [Google Scholar]
- Schultz JF, Armant DR. β1- and β3-class integrins mediate fibronectin binding activity at the surface of developing mouse peri-implantation blastocysts. J Biol Chem. 1995;270:11522–11531. doi: 10.1074/jbc.270.19.11522. [DOI] [PubMed] [Google Scholar]
- Schwartz MA. Spreading of human endothelial cells on fibronectin or vitronectin triggers elevation of intracellular-free calcium. J Cell Biol. 1993;120:1003–1010. doi: 10.1083/jcb.120.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, Denninghoff K. αvintegrins mediate the rise in intracellular calcium in endothelial cells on fibronectin even though they play a minor role in adhesion. J Biol Chem. 1994;269:11133–11137. [PubMed] [Google Scholar]
- Schwartz MA, Ingber DE, Lawrence M, Springer TA, Lechene C. Multiple integrins share the ability to induce elevation of intracellular pH. Exp Cell Res. 1991a;195:533–535. doi: 10.1016/0014-4827(91)90407-l. [DOI] [PubMed] [Google Scholar]
- Schwartz MA, Lechene C, Ingber DE. Insoluble fibronectin activates the Na/H antiporter by clustering and immobilizing integrin α5β1, independent of cell shape. Proc Natl Acad Sci USA. 1991b;88:7849–7853. doi: 10.1073/pnas.88.17.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, Brown EJ, Fazeli B. A 50 kDa integrin-associated protein is required for integrin-regulated calcium entry in endothelial cells. J Biol Chem. 1993;268:19931–19934. [PubMed] [Google Scholar]
- Schwarzbauer JE. Fibronectin: from gene to protein. Curr Opin Cell Biol. 1991;3:786–791. doi: 10.1016/0955-0674(91)90051-y. [DOI] [PubMed] [Google Scholar]
- Sjaastad MD, Nelson WJ. Integrin-mediated calcium signaling and regulation of cell adhesion by intracellular calcium. BioEssays. 1997;19:47–55. doi: 10.1002/bies.950190109. [DOI] [PubMed] [Google Scholar]
- Sjaastad MD, Lewis RS, Nelson WJ. Mechanisms of integrin-mediated calcium signaling in MDCK cells: regulation of adhesion by IP3- and store-independent calcium influx. Mol Biol Cell. 1996;7:1025–1041. doi: 10.1091/mbc.7.7.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi L, Lasic Z, Vukicevic S, Banfic H. Collagen type IV stimulates an increase in intracellular Ca2+in pancreatic acinar cells via activation of phospholipase C. Biochem J. 1994;299:603–611. doi: 10.1042/bj2990603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treede RD, Meyer RA, Davis KD, Campbell JN. Intradermal injections of bradykinin or histamine cause a flare-like vasodilation in monkey: evidence from laser Doppler studies. Neurosci Lett. 1990;115:201–206. doi: 10.1016/0304-3940(90)90455-i. [DOI] [PubMed] [Google Scholar]
- Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- Wei EP, Kontos HA, Dietrich WD, Povlishock JT, Ellis EF. Inhibition by free radical scavengers and by cyclooxygenase inhibitors of pial arteriolar abnormalities from concussive brain injury in cats. Circ Res. 1981;48:95–103. doi: 10.1161/01.res.48.1.95. [DOI] [PubMed] [Google Scholar]
- Weiss DJ, Evanson OA, Wells RE. Evaluation of arginine-glycine-aspartate-containing peptides as inhibitors of equine platelet function. Am J Vet Res. 1997;58:457–460. [PubMed] [Google Scholar]
- Wijetunge S, Hughes AD. pp60c-srcincreases voltage-operated calcium channel currents in vascular smooth muscle cells. Biochem Biophys Res Commun. 1995;217:1039–1044. doi: 10.1006/bbrc.1995.2874. [DOI] [PubMed] [Google Scholar]
- Wijetunge S, Hughes AD. Activation of endogenous c-Src or a related tyrosine kinase by intracellular (PY)EEI peptide increases voltage-operated calcium channel currents in rabbit ear artery cells. FEBS Lett. 1996;399:63–66. doi: 10.1016/s0014-5793(96)01177-5. [DOI] [PubMed] [Google Scholar]
- Wijetunge S, Aalkjaer C, Schachter M, Hughes AD. Tyrosine kinase inhibitors block calcium channel currents in vascular smooth muscle cells. Biochem Biophys Res Commun. 1992;189:1620–1623. doi: 10.1016/0006-291x(92)90262-j. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Geiger B. Molecular interactions in cell adhesion complexes. Curr Opin Cell Biol. 1997;9:76–85. doi: 10.1016/s0955-0674(97)80155-x. [DOI] [PubMed] [Google Scholar]
- Yip KP, Marsh DJ. An Arg-Gly-Asp peptide stimulates constriction in rat afferent arteriole. Am J Physiol. 1997;273:F768–F776. doi: 10.1152/ajprenal.1997.273.5.F768. [DOI] [PubMed] [Google Scholar]