

Abstract
Adherent cells assemble fibronectin into a fibrillar matrix on their apical surface. The fibril formation is initiated by fibronectin binding to the integrins α5β1 and αvβ3, and is completed by a process that includes fibronectin self-assembly. We found that a 76– amino acid fragment of fibronectin (III1-C) that forms one of the self-assembly sites caused disassembly of preformed fibronectin matrix without affecting cell adhesion. Treating attached fibroblasts or endothelial cells with III1-C inhibited cell migration and proliferation. Rho-dependent stress fiber formation and Rho-dependent focal contact protein phosphorylation were also inhibited, whereas Cdc42 was activated, leading to actin polymerization into filopodia. ACK (activated Cdc42-binding kinase) and p38 MAPK (mitogen-activated protein kinase), two downstream effectors of Cdc42, were activated, whereas PAK (p21-activated kinase) and JNK/SAPK (c-Jun NH2-terminal kinase/ stress-activated protein kinase) were inhibited. III1-C treatment also modulated activation of JNK and ERK (extracellular signal-regulated kinases) in response to growth factors, and reduced the activity of the cyclin E–cdk2 complex. These results indicate that the absence of fibronectin matrix causes activation of Cdc42, and that fibronectin matrix is required for Rho activation and cell cycle progression.
Keywords: extracellular matrix, signal transduction, F-actin, mitogen-activated protein kinase (MAPK), cyclin E–cdk2
Fibronectin is a multifunctional component of the extracellular matrix. Intracellular signaling induced by cell adhesion on fibronectin plays a critical role in cytoskeletal organization, cell cycle progression, and cell survival (Hynes, 1990; Frisch and Ruoslahti, 1997). Cells assemble fibronectin into a fibrillar form that accumulates at their apical surface. Fibronectin matrix formation is initiated by fibronectin binding to cell surface receptors, followed by assembly and reorganization of the cell surface–associated fibronectin into fibrils (Mosher et al., 1992; Mosher, 1993; McDonald, 1988). The α5β1 integrin is the major receptor responsible for fibronectin matrix assembly (Ruoslahti, 1991; Wu et al., 1993). The αvβ3 integrin can also direct matrix assembly (Wennerberg et al., 1996; Wu et al., 1996), which may account for fibronectin matrix formation in α5 integrin null mice (Yang et al., 1993). A number of other integrins bind to fibronectin, but do not initiate fibril formation (Busk et al., 1992; Zhang et al., 1993; Wu et al., 1995a ). The first type III repeat of the fibronectin molecule is important in promoting matrix assembly (Morla and Ruoslahti, 1992; Aguirre et al., 1994; Hocking et al., 1994). Small fragments derived from this III1 module induce fibronectin polymerization at moderate concentrations, but inhibit it at high concentrations (Morla and Ruoslahti, 1992; Morla et al., 1994).
Interaction of integrins with the actin cytoskeleton is also essential for matrix assembly (Hynes, 1990). Truncation of the β1 cytoplasmic domain blocks fibronectin matrix assembly by preventing the interaction of the ligand-occupied integrin with the cytoskeleton (Wu et al., 1995b ).
Formation of polymerized actin structures and assembly of associated integrin complexes are controlled by the Rho family of small GTPases: Rho, Rac, and Cdc42 (Hall, 1998). In Swiss 3T3 cells, activation of Rho leads to formation of actin stress fibers and focal adhesion complexes (Kumagai et al., 1993; Rankin et al., 1994; Seufferlein and Rozengurt, 1994), whereas Rac activation induces assembly of an actin meshwork at the cell periphery producing lamellipodia and membrane ruffles (Hotchin and Hall, 1995; Nobes and Hall, 1995). Cdc42 activation causes filopodial protrusions at the cell periphery. A close correlation exists between actin stress fiber formation and fibronectin matrix assembly. Thus, disrupting the cytoskeleton with cytochalasin D inhibits fibronectin matrix assembly, whereas lysophosphatidic acid (LPA)1, which induces stress fiber formation, enhances it (Zhang et al., 1994). Inhibiting Rho with C3 transferase inhibits the LPA-induced matrix assembly (Zhang et al., 1997), suggesting that LPA acts through Rho to increase fibronectin binding at the cell surface. Moreover, fibronectin fibrils coalign with bundles of actin filaments and with focal adhesion-like sites that lack vinculin or talin. These structures are different from the punctate focal adhesion involved in the cell–substratum interaction (Chen and Singer, 1982; Chen et al., 1985). This specialization suggests that fibronectin matrix may activate integrin-linked intracellular signaling pathways and cytoskeletal reorganization in ways that are different from those at canonical focal adhesions.
Here we study the role of the fibronectin matrix in cytoskeletal regulation by disassembling the fibronectin matrix with a 76–amino acid fragment derived from the first type III repeat of fibronectin (Morla and Ruoslahti, 1992; Morla et al., 1994), or by preventing matrix assembly with integrin antibodies. Cell adhesion is not discernibly affected by the III1-C fragment, allowing us to separate the effects of surface matrix disruption from adhesion to a substrate. Removing the fibronectin matrix from the cell surface disrupts the stress fibers and focal contacts, and reorganizes the cytoskeleton into filopodia at the cell periphery, suggesting inactivation of Rho and activation of Cdc42. We also show that III1-C treatment affects growth factor pathways and reduces the activity of the cyclin E–cdk2 complex, thereby impeding the progression of cell cycle.
Materials and Methods
Cell Culture
Primary cultures of human umbilical vein endothelial cells (HUVECs) were purchased from Clonetics (San Diego, CA) and grown in M199 medium supplemented with 20% FCS, 10 ng/ml bFGF, 10 ng/ml EGF, and 1 μg/ml heparin. HUVECs were used between passages 2 and 8. Normal human KD fibroblasts were grown in minimum essential medium (αMEM) supplemented with 10% FCS.
Antibodies
Anti-paxillin, FAK, and phosphotyrosine (PY20) antibodies were purchased from Transduction Laboratories (Lexington, KY); phosphospecific antibodies for p38 MAPK and p44/42 MAPK were purchased from New England Biolabs (Beverly, MA); anti-JNK1 was purchased from PharMingen (San Diego, CA); antibodies against Cdc42, ACK, PAKα, PAKγ, cdk2, cyclin E, ERK1, ERK2, and p38 MAPK were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); anti-α5 blocking antibody (P1D6) was purchased from Calbiochem-Novabiochem Corp. (La Jolla, CA); anti-αvβ3 blocking antibody (LM609) was purchased from Chemicon International, Inc. (Temecula, CA); anti-β1 blocking antibody (P4C10) was purchased from Telios Pharmaceuticals (San Diego, CA); and the anti-fibronectin antibody was generated in our laboratory (Zhang et al., 1993).
Expression and Purification of Recombinant Proteins
III1-C and III-11C (III-CF) fibronectin fragments were expressed in Escherichia coli as histidine-tail fusion proteins, and were purified by metal-chelate affinity chromatography on nickel-agarose beads (Ni-NTA superflow; QIAGEN Inc., Valencia, CA) as previously described (Morla et al., 1994). Purified proteins were dialyzed against TBS and concentrated by centrifugation using Centricon-10 (Amicon Corp., Easton, TX). Purity of protein preparations was verified on Coomassie blue–stained SDS-PAGE gels.
Immunofluorescence Microscopy
Cells were grown to confluence on Lab-Tek 4-well chamber slides (Nunc, Inc., Naperville, IL) in complete medium, washed once in serum-free medium, and incubated in the absence or presence of III1-C for the indicated time. When indicated, the cells were treated for the final 15 min with 5 μM lysophosphatidic acid (LPA; Sigma Chemical Co., St Louis, MO). The cells were fixed in 4% paraformaldehyde in PBS for 30 min, permeabilized in 0.2% Triton X-100 in PBS, treated with PBS containing 0.5% BSA, and incubated for 1 h with the primary antibodies diluted in the PBS/BSA solution. FITC or rhodamine-coupled secondary antibodies (Sigma Chemical Co.) were used for detection. Actin cytoskeleton was stained with rhodamin–phalloidin (Molecular Probes, Inc., Eugene, Oregon).
Fluorescence Assays for Fibronectin Fibril Formation
KD fibroblasts were starved for 24 h, trypsinized to remove their existing fibronectin matrix, and resuspended in serum-free medium containing 100 μg/ml soybean trypsin inhibitor (Sigma Chemical Co.). Cells were washed once in serum-free medium and seeded onto Lab-Tek 4-well chamber slides that had been precoated with a mixture of 10 μg/ml collagen type IV (Sigma Chemical Co.) and 10 μg/ml vitronectin purified from human plasma as described (Yatohgo et al., 1988). Cells were allowed to attach and spread for 1–6 h before fixation for immunostaining. In some experiments, the cells were allowed to attach and spread for 1 h on the matrices before adding inhibitory anti-integrin antibodies. The cells were incubated for an additional 6 h before fixing and staining for surface fibronectin or actin cytoskeleton. The antibodies did not prevent adhesion to or cause detachment from the mixed matrices. In similar experiments, HUVECs, which do not express collagen integrins, were plated on substrates coated with a mixture of 10 μg/ml fibronectin (Boehringer Mannheim Corp., Indianapolis, IN) and 10 μg/ml laminin (Sigma Chemical Co.) and grown in complete medium for 2 d before adding integrin-blocking antibodies for 4 h in serum-free medium. Because fibronectin was one of the substrates, we were only able to stain for actin in these cells.
DNA Replication Assay
Cells were plated in 96-well plates and incubated with the indicated amounts of III1-C in complete medium. After 48 h, HUVEC proliferation was assessed by an MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) incorporation assay. The cells were incubated with 300 μg/ml MTT for 6 h. After overnight extraction with 10% SDS in 0.01 N HCl, the OD was read at 590 nm.
Immunoprecipitation and Immunoblotting
The cells were lysed in an NP-40 based buffer (0.1% NP-40, 50 mM Tris-HCl, pH 7.4, 250 mM NaCl, 5 mM EDTA) containing protease inhibitors (1 mM PMSF, 10 μg/ml leupeptin, 0.1 U/ml aprotinin, 0.4 μg/ml pepstatin) and the phosphatase inhibitor Na3VO4 (1 mM). The insoluble fraction was removed by centrifugation, and the cleared lysates were used for immunoprecipitation with antibodies to FAK and paxillin. ACK immunoprecipitation was performed as previously described (Satoh et al., 1996) in 50 mM Hepes, pH 7.3, 150 mM NaCl, 10% glycerol, 1% NP40, 2 mM MgCl2, 1 mM EDTA, 20 mM glycero-phosphate, 10 mM pyrophosphate, and protease and phosphatase inhibitors. Proteins were separated by SDS-PAGE, transferred to Immobilon-P membranes (Millipore Corp., Bedford, MA), blotted with an anti-phosphotyrosine antibody, and visualized with chemiluminescence substrate (NEN™ Life Science Products, Boston, MA).
Kinase Assays
Kinase assays for ERK2 and JNK1 were performed as described previously (Cavigelli et al., 1995). In brief, cells were lysed on ice in an NP-40 based lysis buffer (50 mM Hepes, pH 7.6, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 0.5% NP-40) containing protease and phosphatase inhibitors. ERK2 or JNK1 were immunoprecipitated using appropriate antibodies and protein A-Sepharose beads (Pharmacia Biotech Sverige, Uppsala, Sweden). For the ERK2 kinase assays, PHAS-1 (Stratagene, La Jolla, CA) was used as the substrate at 5 μg/reaction. GST-c-Jun (1–79) was used as the substrate for the JNK1 kinase assays at 5 μg/reaction. Kinase reactions containing the immunoprecipitated kinase, substrate, 10 μM ATP, and 5 μCi [γ-32P] ATP in the kinase buffer (50 mM Hepes, pH 7.6, 10 mM MgCl2) were incubated at room temperature for 30 min before separation by SDS-PAGE.
For p38 MAP kinase, cells were lysed on ice in a Triton X-100 based lysis buffer (20 mM Tris-HCl, pH 8, 137 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% glycerol, 25 mM glycerophosphate, 2 mM pyrophosphate) containing protease and phosphatase inhibitors as described (Raingeaud et al., 1995). p38 MAPK was immunoprecipitated with the anti-p38 antibody that was preadsorbed to protein-A Sepharose for 30 min. The kinase reaction was performed at 30°C for 30 min in presence of ATF-2 (Santa Cruz Biotechnology, Santa Cruz, CA) as the substrate at 5 μg/reaction, 10 μM ATP, and 5 μCi [γ-32P]ATP in the kinase buffer (25 mM Hepes, pH 7.6, 25 mM MgCl2, 25 mM glycerophosphate, 2 mM DTT, 0.1 mM Na3VO4).
PAKα and PAKγ kinase assays were performed as described (Lamarche et al., 1996). Cells were lysed in 50 mM Tris-HCl, pH 8, 150 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 1 mM PMSF, 10 μg/ml leupeptin, 0.1 U/ml aprotinin, 0.4 μg/ml pepstatin, and 1 mM Na3VO4. The kinase activity of the PAK immunoprecipitates was analyzed by incubating at 30°C for 30 min with MBP (Sigma Chemical Co.) as substrate at 5 μg/reaction, 40 μM ATP, and 5 μCi [γ-32P]ATP in the kinase buffer (20 mM Tris-HCl, pH 7.5, 20 mM MgCl2, 10 mM MnCl2, 1 mM EDTA, 1 mM EGTA).
Cyclin E–cdk2 complex kinase assays were performed as described (Fang et al., 1996). In brief, HUVECs were synchronized by serum starvation in αMEM supplemented with bovine insulin (5 μg/ml), transferrin (5 μg/ml), and sodium selenite (5 ng/ml) for 24 h (Fang et al., 1996). Serum-starved cells (∼70% confluent) were incubated with the indicated amount of fibronectin and III-1C in complete medium. After 18 h, cells were lysed in 0.1% NP-40 buffer (50 mM Tris-HCl, pH 7.4, 5 mM NaF, 250 mM NaCl, 5 mM EDTA, 0.1% NP-40, 15 μg/ml benzamidine, 10 μg/ml trypsin inhibitor, 10 μg/ml leupeptin, 4 μg/ml pepstatin, 0.1 U/ml aprotinin). 50 μg of protein were immunoprecipitated with 2–3 μg of antibody preadsorbed to protein A-Sepharose or protein G-Sepharose (Pharmacia Biotech Sverige) in 0.1% NP-40 buffer. The kinase activity of cyclin E–cdk2 was determined by in vitro phosphorylation of the histone H1 substrate at 100 μg/reaction in kinase buffer (100 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM DTT, 100 μM) for 30 min at room temperature.
All reactions were stopped by adding electrophoresis sample buffer, and proteins were separated on 4–20% SDS-PAGE (Novex, San Diego, CA). Kinase activity was detected by autoradiography (BioMAX MR; Eastman Kodak Co., Rochester, NY) and quantified by scanning the autoradiographs with a flatbed scanner (Hewlett-Packard Co., Palo Alto, CA) and analyzing them with NIH Image 1.57.
Plasmids and Transfections
Dominant negative forms of Cdc42 (Cdc42N17), Rac (RacN17), and the constitutively active forms of Rho (RhoV14) and Rac (RacV12) were gifts from Dr. Alan Hall. The pE-GFP plasmid was from QIAGEN Inc. (Valencia, CA).
One day before transfection, HUVECs were seeded onto Lab-Tek 4-well chamber slides (Nunc, Inc.) at 5 × 104 cells per well in complete medium. Cells were then transfected with 0.2 μg pE-GFP alone, or were cotransfected with 1 μg of the indicated plasmid, by using the Superfect reagent (QIAGEN Inc.) according to the manufacturer's protocols. After 24 h, the cells were treated with III1-C for 4 h in serum-free medium, fixed, and stained with rhodamine-phalloidin as described above.
Results
Cells Treated with Fibronectin Fragment III1-C Lose Fibronectin Matrix
Fragments from the first type III repeat of fibronectin have been shown to promote fibronectin matrix assembly at low concentrations, and to inhibit it at high concentrations (Morla and Ruoslahti, 1992; Morla et al., 1994). We found that high concentrations of III1-C can also disrupt an established fibronectin matrix. Adding the fibronectin III1-C fragment for 4 h to HUVEC or for 16 h to KD fibroblast cultures in serum-free medium caused complete loss of the fibronectin fibrils on the cell apical surface that were detectable by immunostaining of fibronectin (Fig. 1 A). Despite this, the cells remained attached to the culture dish. A fragment derived from the eleventh type III fibronectin repeat (III-CF) that is analogous to III1-C and that has been used previously as a negative control for III1-C (Morla et al., 1994) had no effect on the fibronectin matrix.
Figure 1.


Treatment with III1-C removes surface fibronectin matrix. Immunofluorescence staining of fibronectin: (A, top) on the surface of HUVECs with or without treatment with 20 μM III1-C or with 20 μM of the control fragment III-CF for 4 h in serum-free medium, or (A, bottom) on the surface KD fibroblasts after treatment with or without 20 μM III1-C for 16 h in serum-free medium; (B) on the surface of KD fibroblasts treated with the indicated concentrations of III1-C for 16 h; (C) in Triton X-100– permeabilized KD fibroblasts after treatment for 16 h with 20 μM III1-C in serum free-medium or with the medium alone.
The effect of III1-C on fibronectin matrix was dose-dependent: low concentrations of III1-C (between 0.1 and 1 μM) increased accumulation of fibronectin matrix at the cell surface, higher III1-C concentrations (2.5–10 μM) decreased fibronectin matrix, and at 20 μM the matrix was undetectable by immunofluorescence staining (Fig. 1 B). Removing fibronectin matrix by 20 μM III1-C required several hours. The time course varied depending on the cell type (2–3 h for HUVEC; 4–6 h for KD fibroblasts; not shown), but seemed to depend on the amount of fibronectin matrix that had previously accumulated on the cell surface. The effect of III1-C was reversible; after removing surface fibronectin with 20 μM III1-C, the KD fibroblasts maintained in serum-free medium that did not contain III1-C regenerated their fibronectin matrix within 24 h. Increased intracellular fibronectin staining was seen in the III1-C–treated KD fibroblasts after permeabilization with Triton X-100 (Fig. 1 C). These results show that III1-C disassembles fibronectin fibrils and causes intracellular accumulation of fibronectin, but does not affect cell substrate adhesion.
Fibronectin Matrix Removal Inhibits Cell Migration and Proliferation
As cell substrate adhesion was not discernibly affected by III1-C treatment, we were able to assess the cellular effects of disrupting surface fibronectin matrix without pertubing cell attachment or spreading. Treating CHO cells with high concentrations of III1-C (10–50 μM) has been shown to inhibit their migration in an in vitro wound assay (Morla et al., 1994). HUVECs and KD fibroblasts responded similarly: cell migration assessed by wound assay was inhibited by III1-C in proportion with the reduction of cell surface fibronectin matrix. High concentrations of III1-C also inhibited cell proliferation as measured by an MTT assay, whereas low concentrations enhanced fibronectin deposition and cell proliferation (results not shown). These initial findings encouraged us to use the III1-C treatment to study the effects of fibronectin matrix on various signaling pathways.
Fibronectin Matrix is Required for Rho-mediated Formation of Stress Fibers and Phosphorylation of Focal Adhesion Proteins
Because a strong correlation exists between organization of actin stress fibers and assembly of fibronectin matrix (Zhang et al., 1994; Zhang et al., 1997), we tested whether fibronectin matrix removal by III1-C would affect cytoskeletal organization. HUVECs and KD fibroblasts treated with 20 μM III1-C in serum-free medium redistributed their filamentous actin into filopodia (Fig. 2). The control fragment III-CF had no effect on the cytoskeleton (not shown). Moreover, the Rho-dependent induction of stress fibers by LPA was prevented by the III1-C treatment (Fig. 3), and the III1-C–induced disruption of stress fibers could be obliterated by transfecting cells with a constitutively active form of Rho (result not shown). The cytoskeletal changes correlated with the ability of III1-C to remove cell surface fibronectin matrix; treating the cells with 20 μM III1-C for 15 min, which was not long enough to remove the matrix, did not prevent the cytoskeletal responses to LPA.
Figure 2.
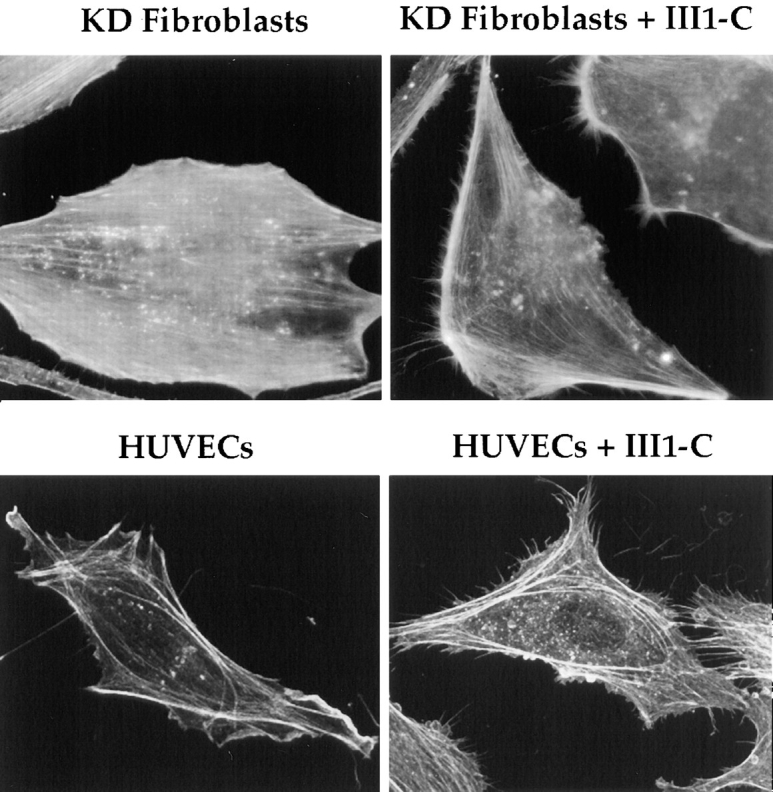
III1-C treatment stimulates formation of filopodial protrusions of the cytoskeleton at the cell periphery. Rhodamine-phalloidin staining of the actin cytoskeleton (top) in KD fibroblasts after treatment with or without 20 μM III1-C for 16 h in serum free medium; (bottom) in HUVECs with or without treatment with 20 μM III1-C for 4 h in serum-free medium. The images were produced with a confocal microscope.
Figure 3.

Fibronectin matrix removal blocks Rho-mediated stress fiber formation. Rhodamine-phalloidin staining of the actin cytoskeleton. HUVECs were treated in serum-free medium in the absence (medium, top) or presence of 20 μM III1-C for 4 h (middle) or 20 μM III1-C for the last 15 min. The cells were then stimulated (right) or left unstimulated (left) with 5 μM LPA for 15 min. The images were produced with a confocal microscope.
III1-C also only slightly reduced the tyrosine phosphorylation of FAK and paxillin in attached cells, prevented the increase in FAK and paxillin phosphorylation elicited by LPA and essentially eliminated the association of FAK and paxillin with one another (Fig. 4 A). These effects were seen only when cells were treated long enough to remove cell surface fibronectin matrix; a 15-min treatment had no effect. Moreover, cells treated with III1-C in suspension before attachment onto various substrates attached equally well as untreated cells and phosphorylated FAK and paxillin to the same extent (Fig. 4 B). These results suggest that Rho-pathways are inhibited in the III1-C treated cells, and that assembling a fibronectin matrix rather than adhering to a substrate, is necessary for cells to initiate Rho-dependent actin stress fiber formation. Substrate adherence seems to elicit most of the focal adhesion protein phosphorylation, but FAK-paxillin association is controlled by the matrix.
Figure 4.

Fibronectin matrix removal blocks Rho-mediated tyrosine phosphorylation of focal contact proteins FAK and paxillin. (A) HUVECs were treated for 4 h with 20 μM III1-C in serum-free medium to remove fibronectin matrix (+) or with the medium alone (−). Where indicated, LPA cells were stimulated with 5 μM LPA for 15 min before lysis. As a control for the effects of III1-C without removing fibronectin matrix, 20 μM III1-C was added for 15 min in absence (III1-C) or presence of 5 μM LPA (III1-C + LPA). FAK or paxillin were immunoprecipitated and immunoblotted with an anti-phosphotyrosine antibody (PY20). The blots were reprobed with anti-FAK or anti-paxillin antibodies to show that similar protein levels were immunoprecipitated and to detect FAK that had coimmunoprecipitated with paxillin. (B) Suspended HUVECs were treated for 1 h with (+) or without (−) 20 μM III1-C in serum-free medium before plating on dishes coated with 10 μg/ml fibronectin, vitronectin, or laminin. The cells were lysed after 30 min, and FAK or paxillin was immunoprecipitated and immunoblotted with an anti-phosphotyrosine antibody (PY20). The experiments were carried out three to five times, and a representative result is shown.
To explore further the dependence of actin organization on the presence of cell surface fibronectin, we removed the fibronectin matrix from KD fibroblasts by trypsinization, plated them onto integrin ligands other than fibronectin—a mixture of collagen IV and vitronectin—and observed accumulation of the fibronectin matrix and the assembly of the actin cytoskeleton at different time points. While integrin-mediated cell adhesion and spreading occurred rapidly (in 15–30 min), fibronectin matrix assembly required several hours (Fig. 5 A). This time course was similar to that required for the cells to form stress fibers (Fig. 5 B).
Figure 5.
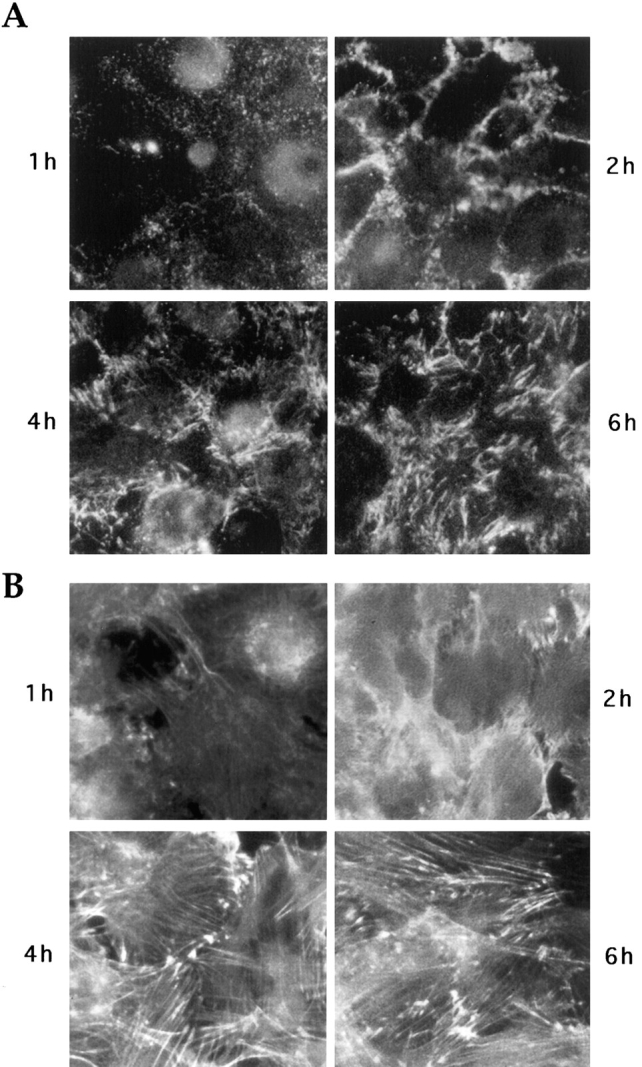
Time course of fibronectin matrix deposition and stress fiber organization after attachment of cells on substrate. Trypsinized KD fibroblasts were plated on a mixture of collagen IV and vitronectin in serum-free medium and stained after 1, 2, 4, or 6 h for cell surface fibronectin (A) or actin cytoskeleton (B).
We also used antibodies to prevent accumulation of surface fibronectin matrix. Integrins, particularly α5β1, and to a some extent, αvβ3 direct fibronectin matrix assembly. We tested whether blocking these integrins would have a similar effect on the actin cytoskeletal organization as III1-C. We allowed KD fibroblasts or endothelial cells to attach and spread on a mixture of collagen and vitronectin or laminin and fibronectin, and then added blocking antibodies directed against the α5β1 and αvβ3 integrins individually or in combination. Because fibroblasts and endothelial cells can use integrins other than α5β1 and αvβ3 to adhere to collagen IV, vitronectin, and laminin, these antibodies did not prevent adhesion and spreading on the substrate. When added individually, both the β1-blocking antibody (P4C10) and the αvβ3-blocking antibody (LM609) reduced fibronectin matrix assembly in the fibroblasts; P4C10 was more efficient than LM609 (Fig. 6 A). Adding both these antibodies together prevented fibronectin matrix accumulation (Fig. 6 A) and reduced stress fiber organization (Fig. 6 B). HUVECs plated on a mixture of laminin and fibronectin and treated with both of the anti-integrin antibodies underwent the same changes as the fibroblasts. They also displayed filopodial protrusions at the cell periphery (Fig. 6 C), similar to those observed in the III1-C-treated cells; however, III1-C was more efficient in both removing fibronectin matrix and inducing filopodia. Thus, it seems that fibronectin matrix assembly, and not cell adhesion per se, is essential for proper stress fiber organization.
Figure 6.


Inhibition of fibronectin matrix assembly with function-blocking integrin antibodies prevents stress fiber formation. Trypsinized KD fibroblasts were plated on a mixture of collagen IV and vitronectin in serum-free medium. The cells were allowed to attach and spread for 1 h before adding function-blocking antibodies directed against the β1 (P4C10, 10 μg/ml) or αvβ3 (LM609, 10 μg/ml) integrins individually or in combination. After an additional 6 h, the cells were stained for cell surface fibronectin (A) or actin cytoskeleton (B). (C) HUVECs were grown on a mixture of fibronectin and laminin in complete medium for 48 h, and were then treated with the function-blocking antibodies directed against the α5 (P1D6, 10 μg/ml) or αvβ3 (LM609, 10 μg/ml) integrins individually or in combination in serum-free medium. The cells were stained after 4 h for actin cytoskeleton.
Fibronectin Matrix Removal Activates Cdc42 and ACK1 but not PAK1
Formation of filopodial protrusions at the cell periphery suggests that Cdc42 may be activated in the III1-C–treated cells. To analyze the involvement of Cdc42, HUVECs were transfected with a dominant-negative form of Cdc42 (Cdc42N17) before treatment with III1-C. Cdc42N17 expression did not affect cytoskeletal organization of stress fibers in control cells, but completely blocked formation of the peripheral filopodia in III1-C-treated cells (Fig. 7), thus indicating that Cdc42 mediates the cytoskeletal reorganization in cells depleted of fibronectin matrix. In contrast, expression of a dominant negative form of Rac (RacN17) had no effect on the III1-C–induced filopodia (not shown).
Figure 7.

Expression of a dominant negative form of Cdc42 prevents the formation of III1-C–induced filopodia. HUVECs cells were transfected GFP alone, or with GFP and Cdc42N17 before treatment with 20 μM III1-C for 4 h in serum-free medium and stained for actin cytoskeleton (top). In the lower panel, the same cells were analyzed for GFP expression to identify the transfected cells.
To develop additional evidence for Cdc42 activation in the III1-C treated cells, we analyzed whether ACK was activated by fibronectin removal. ACK1 and 2 are nonreceptor tyrosine kinases that bind to and are activated by Cdc42 (Manser et al., 1993; Yang and Cerione, 1997). A marked enhancement of ACK1 tyrosine phosphorylation was seen in KD fibroblasts treated with III1-C long enough (16 h) to remove fibronectin matrix (Fig. 8 A). A weaker but consistent induction of ACK tyrosine phosphorylation was observed in HUVECs treated with III1-C for 4 h. In contrast, a 15-min treatment with III1-C did not alter ACK1 tyrosine phosphorylation. Hyperosmotic shock and a decrease in temperature, both of which have been shown to activate ACK1 (Satoh et al., 1996), also increased ACK1 phosphorylation. Activation of ACK1 further suggests that Cdc42 is activated in cells depleted of fibronectin matrix.
Figure 8.

Tyrosine phosphorylation of ACK and inhibition of PAK1 kinase activity in response to fibronectin matrix removal by III1-C. (A) Tyrosine phosphorylation of ACK: KD fibroblasts were treated with 20 μM III1-C for 16 h. HUVECs were treated with 20 μM III1-C for 4 h or 15 min, or were stimulated by hyperosmotic shock (0.8 M NaCl for 10 min) or by decreasing the temperature (25°C for 30 min). Serum-free medium was used as a control (medium). ACK was immunoprecipitated and immunoblotted with an anti-phosphotyrosine antibody (PY20). (B) PAK1 in vitro kinase assay using MBP as the substrate. HUVECs were pretreated with 20 μM III1-C for 4 h in serum-free medium to remove fibronectin matrix (+), or were treated with the medium alone (−), and where indicated, were stimulated by anisomycin (10 μg/ml), bFGF (10 ng/ml), or EGF (10 ng/ml) for 5 min before lysis. This experiment was carried out four times with similar results, and representative results are shown. (C) Controls confirmed that an equivalent amount of PAK protein was immunoprecipitated from the III1-C treated and control cells.
We next tested whether PAK was activated in III1-C treated cells. PAK is a serine/threonine kinase that can be activated in vitro by GTP-bound Cdc42 or Rac (Manser et al., 1995; Manser et al., 1994). Moreover, PAK is a known regulator of JNK/SAPK and p38 MAPK (Retta et al., 1998), and may mediate activation of JNK by Rac and Cdc42 (Joneson et al., 1996; Lamarche et al., 1996; Tang et al., 1997). Surprisingly, we found that basal kinase activity of PAK1 (Fig. 8 B) and PAK2 (not shown) was decreased in III1-C–treated HUVECs. Moreover, III1-C pretreatment attenuated activation of PAK1 by anisomycin or growth factors (bFGF and EGF), suggesting that fibronectin matrix removal inhibits a signaling cascade leading to PAK.
Fibronectin Matrix Removal Causes Activation of p38 MAPK but not JNK or ERK1/2
Cdc42 is known to stimulate a kinase cascade that leads to subsequent activation of the p38 MAPK and JNK/SAPK but not the MAPK ERK1/2 (Coso et al., 1995; Minden et al., 1995). To test whether the activation of Cdc42 in III1-C–treated cells correlates with the activation of these kinases, we examined the activity of p38 MAPK and JNK.
p38 MAPK is activated by dual phosphorylation on tyrosine and threonine residues in cells subjected to various types of stress (Raingeaud et al., 1995). p38 MAPK became phosphorylated in HUVECs treated with III1-C to remove fibronectin matrix (Fig. 9 A). Stimulation with LPA or EGF, but not bFGF, also caused p38 MAPK phosphorylation. Stimulation with III1-C together with LPA or EGF had little or no additive effect, indicating that III1-C alone maximally activated p38 MAPK (Fig. 9 A). A 15-min treatment of HUVECs with III1-C did not induce p38 MAPK phosphorylation, again suggesting that the p38 MAPK activation relates to the loss of fibronectin matrix rather than stimulation by III1-C itself. p38 MAPK activation in the III1-C–treated cells was further confirmed by using an in vitro kinase assay. The stimulation by III1-C was nearly as strong as that of anisomycin, a known activator of p38 MAPK (Fig. 9 B).
Figure 9.

Fibronectin matrix removal activates p38 MAPK. (A) Immunoblotting with a phosphospecific (Tyr182) p38 antibody or conventional p38 antibody. HUVECs were pretreated (+) with 20 μM III1-C for 4 h in serum-free medium to remove fibronectin matrix, or were left untreated (−). Where indicated, the cells were then stimulated with 5 μM LPA for 15 min before lysis (LPA). As a control, HUVECs were treated with 20 μM III1-C for the final 15 min in the absence (III1-C) or the presence of 5 μM LPA (III1-C + LPA). Alternatively, cells were stimulated with 10 ng/ml EGF or bFGF for 15 min before lysis. Treatment with serum-free medium was used as a control (Medium) in both experiments. (B) P38 MAPK in vitro kinase assay using ATF-2 as substrate. HUVECs were treated (+) with 20 μM III1-C for 4 h and, where indicated, were stimulated with bFGF (10 ng/ml) or anisomycin (10 μg/ml) for 15 min or left untreated in serum-free medium (−) and then lysed.
Even though JNK can be activated by Cdc42 and Rac (Coso et al., 1995; Minden et al., 1995), basal kinase activity and growth factor–induced activity of JNK were inhibited in III1-C–treated cells (not shown). Movever, integrin- mediated adhesion is an activator of ERK1 and ERK2 (Chen et al., 1994; Schlaepfer et al., 1994), III1-C had only minor effects on the activation of these kinases and did not affect their expression at the protein level (results not shown).
Fibronectin Matrix Removal Inhibits Cell Proliferation by Inactivating Cyclin E–cdk2 Kinase
Preventing adhesion to an extracellular matrix blocks cell cycle progression by suppressing the activities of cyclin/cyclin-dependent kinase (cdk) complexes such as the cyclin E–cdk2 complex (Fang et al., 1996). We found that cyclin E–cdk2 activity was also completely blocked in HUVECs and in KD fibroblasts treated with III1-C (Fig. 10 and results not shown). The activity of the cyclin D-cdk4 complex, which acts before the cyclin E–cdk2 complex in the cell cycle, was not reduced in cells treated with III1-C (not shown). These results suggest that assembling fibronectin matrix is necessary for proliferation of attached cells and that matrix depletion blocks the cell cycle at the level of cyclin E–cdk2.
Figure 10.

Fibronectin matrix removal inhibits the activity of the cyclin E–cdk2 complex. Synchronized HUVECs and KD fibroblasts were treated with 10 μM III1-C or 10 μg/ml fibronectin (FN) as a control for 18 h in complete medium. Cdk2 was immunoprecipitated and in vitro kinase assays for the cyclin E–cdk2 complex were performed using histone-1 as the substrate. In vitro kinase assays were also performed on lysates that were immunoprecipitated with an unrelated antibody as a control (Control).
Discussion
We show here that disrupting fibronectin matrix causes cytoskeletal reorganization by activating the small GTPase Cdc42, alters growth factor signaling, and blocks cell cycle progression.
We were able to remove the fibronectin matrix by using a small protein fragment derived from the III1-C module of fibronectin. At low concentrations, this fragment promotes fibronectin matrix assembly, but prevents it at high concentrations (Morla and Ruoslahti, 1992; Morla et al., 1994). Furthermore, as we show here, high concentrations of III1-C can disassemble an already formed fibronectin matrix.
Several lines of evidence indicate that the alterations in signaling caused by III1-C treatment are caused by removing fibronectin from the apical surface of attached cells rather than by alterations in cell-substrate adhesion, cell shape, or by some other action of the peptide. First, fibronectin does not accumulate at the basal cell surface (Chen and Singer, 1980; Chen and Singer, 1982), making it unlikely that III1-C would exerts its effect at the site of substrate adhesion. In agreement with this, III1-C did not block adhesion of cells to surfaces coated with extracellular matrix proteins, nor did it change cell shape. Second, as discussed in detail later, we found the cellular changes induced by III1-C to be quite different from those induced by detachment from substrate, Third, treatment of cells with III1-C for short periods of time did not cause the same effect as matrix removal obtained with prolonged exposure of cells to III1-C; the alterations in signaling and cytoskeletal organization always correlated with the amount of surface fibronectin present. Finally, we show that blocking fibronectin matrix assembly with antiintegrin antibodies, while not affecting cell attachment, caused cytoskeletal reorganization similar to that produced by III1-C treatment.
We found that cell growth parallels accumulation of surface fibronectin matrix, and that removal of the matrix prevents growth. Similarly, others have shown that inhibiting of fibronectin matrix assembly in various ways inhibits cell growth (Mercurius and Morla, 1998; Saulnier et al., 1996). Thus, cell proliferation appears to require a fibronectin matrix, at least in normal cells.
Our results show that actin stress fiber organization in spread cells also depends on fibronectin matrix. We found that fibronectin matrix assembly preceded stress fiber formation in attached and spread cells. Moreover, fibronectin matrix removal induced by III1-C or inhibitory integrin antibodies suppressed Rho-dependent stress fiber organization and Rho-dependent tyrosine phosphorylation of focal adhesion proteins. These results indicate that activating Rho GTPase requires the presence of a fibronectin matrix. Rho activity, in turn, promotes fibronectin matrix assembly, and this effect requires an intact cytoskeleton (Zhang et al., 1994; Chrzanowska-Wodnicka and Burridge, 1996; Zhang et al., 1997).
Fibronectin matrix depletion by III1-C treatment, while inhibiting Rho functions, seems to activate the related GTPase Cdc42, as III1-C–treated cells showed the hallmark of Cdc42 activity, formation of filopodia. Perturbing the fibronectin matrix of HUVECs with anti-α5β1 and αvβ3 antibodies showed the same signs of Cdc42 activation, albeit at a lower level. The appearance of filopodia in the III1-C–treated cells was caused by Cdc42 because it could be inhibited by transfecting a dominant negative Cdc42 into the cells.
In most situations, Cdc42, Rac, and Rho are activated sequentially in cells. Thus, formation of filopodia caused by Cdc42 activation is accompanied by the appearance of lamellipodia in cells that contain functional Rac. This is followed by Rho-dependent organization of actin stress fibers (Nobes and Hall, 1995). Our III1-C–treated cells have activated Cdc42, but Rho is inactive. The studies that revealed the existence of the sequential Cdc42/Rac/Rho pathway were done under conditions that would allow fibronectin matrix assembly. Thus, the lack of the Rho activation that would be expected from the earlier results is likely to be due to the absence of fibronectin matrix in the III1-C–treated cells. The increased Cdc42 activity in the cells lacking fibronectin matrix is consistent with their unimpaired attachment properties, as cell-substrate attachment has been shown to increase Cdc42 activity (Price et al., 1998).
Cdc42 and Rac activate JNK and p38 MAPK and, through these kinases, regulate gene transcription (Coso et al., 1995; Minden et al., 1995). However, most studies exploring the effects of the Rho GTPases on the MAP kinases have been performed by expressing mutant forms of these proteins. Because III1-C treatment makes it possible to activate Cdc42, and because it can be achieved without activating Rac or Rho, we were able to study the downstream signaling pathways specifically regulated by endogenous Cdc42. We found that one of the previously identified Cdc42-dependent pathways was activated in the III1-C–treated cells; there was increased tyrosine phosphorylation of ACK1, a protein that interacts with the active form of Cdc42 (Manser et al., 1993). In contrast, PAK1, a potent regulator of both JNK and p38 MAPK that is known to interact not only with active Cdc42, but also with active Rac (Manser et al., 1993), was inhibited in III1-C–treated cells. Downstream activation of JNK was also prevented by III1-C treatment, whereas p38 MAPK was activated. These data suggest that Cdc42 may not directly activate PAK or JNK. Because Rac seems to be inhibited in III1-C–treated cells, Cdc42 may require Rac to activate the JNK pathway, and activation of PAK and JNK in cells expressing a constitutively active form of Cdc42 may be caused by subsequent activation of Rac. Consistent with this idea, JNK activation by Cdc42 can be blocked by dominant negative Rac (Minden et al., 1995). However, both JNK and ERK activation by anisomycin were also inhibited in III1-C–treated cells (not shown). Anisomycin is an inhibitor of protein synthesis commonly used in MAPK activation studies, and it is thought to activate JNK independently of Cdc42 or Rac (Coso et al., 1995). Therefore, it seems likely that a pathway that does not involve the Rho-related proteins can activate JNK, and that this pathway also requires an intact fibronectin matrix.
Detachment of epithelial and endothelial cells from the extracellular matrix leads to programmed cell death; this process is called anoikis (Frisch and Ruoslahti, 1997). JNK is rapidly activated in detached cells; however, its role in induction of apoptosis in these cells is still not clear (Frisch et al., 1996; Khwaja and Downward, 1997). p38 MAPK is also stimulated by detachment from the matrix (Khwaja and Downward, 1997). In contrast, detachment suppresses ERK and the growth factor–mediated activation of ERK requires cell attachment (Renshaw et al., 1997). Interestingly, we found that removal of fibronectin matrix without altering the cell-substrate adhesion of the cells decreased the basal activity of ERK while slightly potentiating ERK response to growth factors. This effect and the activation of P38 MAPK and suppression of JNK in III1-C–treated cells show that the effects of matrix removal on MAPKs are quite different from those of loss of substrate adhesion. In agreement with this, depleting fibronectin matrix had little effect on cell survival (not shown), while inhibiting cell proliferation. Thus, signals from fibronectin matrix seem to control cell proliferation, whereas cell substrate adhesion provides a survival signal. These signals can be modulated separately by removing the matrix or by allowing cells to attach to a substrate in the absence of matrix.
A profound block of cyclin E–cdk2 activity occurred in III1-C–treated cells, apparently accounting for the reduced cell proliferation. These results indicate that fibronectin matrix is required for activation of cyclin E–cdk2 in anchorage-dependent cells. The growth-promoting effects of substrate adhesion requires cytoskeletal organization (Assoian and Zhu, 1997), which is dependent on Rho activity. Moreover, Rac as well as Rho is necessary for the G1/S transition of the cell cycle (Olson et al., 1995; Hall, 1998). In contrast, Cdc42 can inhibit cell cycle progression in G1 through a mechanism that requires p38 MAPK activation (Molnar et al., 1997). Thus, fibronectin matrix removal may be blocking cell cycle progression by activating Cdc42, and through it p38 MAPK, while inhibiting Rac and Rho activation by Cdc42.
In summary, our results reveal a specialized role for cell surface fibronectin matrix in cytoskeletal organization, growth factor responses, and cell cycle control that cannot be substituted for by cell adhesion to a substrate.
Acknowledgments
We thank Drs. T. Hunter, K. Vuori, and E. Pasquale for comments on the manuscript, and Dr. A. Hall for providing plasmids.
This work was supported by grants from the National Institutes of Health (CA67224, CA62042) and Cancer Center Support Grant CA30199. S. Bourdoulous is supported by the Institut National de la Sante et de la Recherche Medicale, and the Centre National de la Recherche Scientifique. D.A. MacKenna was supported by NRSA Fellowship CA67424 from the National Institutes of Health and a fellowship from the Susan G. Komen Breast Cancer Foundation.
Abbreviations used in this paper
- HUVEC
human umbilical vein endothelial cells
- LPA
lysophosphatidic acid
Footnotes
Address all correspondence to Erkki Ruoslahti, M.D., Ph.D., The Burnham Institute, 10901 N. Torrey Pines Road, La Jolla, CA 92037. Tel.: (619) 646-3125. Fax: (619) 646-3198. E-mail: ruoslahti@burnham-inst.org
The present address of Sandrine Bourdoulous is Institut Cochin de Génétique Moléculaire, CNRS UPR415, 22 rue Méchain, 75014 Paris, France.
The present address of Gertraud Orend is Friedrich Miescher Institut, Maulbeerstrasse 66, 058 Basel, Switzerland.
The present address of Deidre A. MacKenna is Tanabe Research Laboratories, 4540 Towne Centre Ct., La Jolla, CA 92121.
References
- Aguirre KM, McCormick RJ, Schwarzbauer JE. Fibronectin self-association is mediated by complementary sites within the amino-terminal one-third of the molecules. J Biol Chem. 1994;269:27863–27868. [PubMed] [Google Scholar]
- Assoian RK, Zhu X. Cell anchorage and the cytoskeleton as partners in growth factor dependent cell cycle progression. Curr Opin Cell Biol. 1997;9:93–98. doi: 10.1016/s0955-0674(97)80157-3. [DOI] [PubMed] [Google Scholar]
- Busk M, Pytela R, Sheppard D. Characterization of the integrin alphavbeta6 as a fibronectin-binding protein. J Biol Chem. 1992;267:5790–5796. [PubMed] [Google Scholar]
- Cavigelli M, Dolfi F, Claret FX, Karin M. Induction of c-fos expression through JNK mediated TCF/Elk-1 phosphorylation. EMBO (Eur Mol Biol Organ) J. 1995;14:5957–5964. doi: 10.1002/j.1460-2075.1995.tb00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Kinch MS, Lin TH, Burridge K, Juliano RL. Integrin-mediated cell adhesion activates mitogen-activated protein kinases. J Biol Chem. 1994;269:26602–26605. [PubMed] [Google Scholar]
- Chen W, Singer S. Fibronectin is not present in the focal adhesions formed between normal cultured fibroblasts and their substrata. Proc Natl Acad Sci USA. 1980;77:7318. doi: 10.1073/pnas.77.12.7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Singer S. Immunoelectron microscopic studies of the sites of cell-substratum and cell-cell contacts in cultured fibroblasts. J Cell Biol. 1982;95:205–222. doi: 10.1083/jcb.95.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WT, Hasegawa E, Hasegawa T, Weinstock C, Yamada K. Development of cell surface linkage complexes in cultured fibroblasts. J Cell Biol. 1985;100:1103–1114. doi: 10.1083/jcb.100.4.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- Fang F, Orend G, Watanabe N, Hunter T, Ruoslahti E. Dependence of cyclin E-CDK2 kinase activity on cell anchorage. Science. 1996;271:499–502. doi: 10.1126/science.271.5248.499. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Kelaita D, Sicks S. A role for Jun-N-terminal kinase in anoikis; suppression by bcl-2 and crmA. J Cell Biol. 1996;135:1377–1382. doi: 10.1083/jcb.135.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hocking DC, Sottile J, McKeown-Longo J. Fibronectin's III-1 module contain a conformation-dependent binding site for the amino-terminal region of fibronectin. J Biol Chem. 1994;269:19183–19191. [PubMed] [Google Scholar]
- Hotchin NA, Hall A. The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J Cell Biol. 1995;131:1857–1865. doi: 10.1083/jcb.131.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes, R.O. 1990. Fibronectins. Springer-Verlag, New York. 546 pp.
- Joneson T, McDonough M, Bar-Sagi D, Van Aelst L. RAC regulation of actin polymerization and proliferation by a pathway distinct from Jun kinase. Science. 1996;274:1374–1376. doi: 10.1126/science.274.5291.1374. [DOI] [PubMed] [Google Scholar]
- Khwaja A, Downward J. Lack of correlation between activation of Jun-NH2-terminal kinase and induction of apoptosis after detachment of epithelial cells. J Cell Biol. 1997;139:1017–1023. doi: 10.1083/jcb.139.4.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai N, Morii N, Fujisawa K, Nemoto Y, Narumiya S. ADP-rybosylation of rho p21 inhibits lysophosphatidic-acid induced protein tyrosine phosphorylation and phosphatidylinositol 3-kinase activation in cultured Swiss 3T3 cells. J Biol Chem. 1993;268:24535–24538. [PubMed] [Google Scholar]
- Lamarche N, Tapon N, Stowers L, Burbelo PD, Aspenstrom P, Bridges T, Chant J, Hall A. Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell. 1996;87:519–529. doi: 10.1016/s0092-8674(00)81371-9. [DOI] [PubMed] [Google Scholar]
- Manser E, Chong C, Zhao ZS, Leung T, Michael G, Hall C, Lim L. Molecular cloning of a new member of the p21-Cdc42/Rac-activated kinase (PAK) family. J Biol Chem. 1995;270:25070–25078. doi: 10.1074/jbc.270.42.25070. [DOI] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Tan L, Lim L. A non-receptor tyrosine kinase that inhibits the GTPase activity of p21cdc42. Nature. 1993;363:364–367. doi: 10.1038/363364a0. [DOI] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- McDonald J. Extracellular matrix assembly. Annu Rev Cell Biol. 1988;4:183–207. doi: 10.1146/annurev.cb.04.110188.001151. [DOI] [PubMed] [Google Scholar]
- Mercurius KO, Morla AO. Inhibition of vascular smooth muscle cell growth by inhibition of fibronectin matrix assembly. Circ Res. 1998;82:548–556. doi: 10.1161/01.res.82.5.548. [DOI] [PubMed] [Google Scholar]
- Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- Molnar A, Theodoras AM, Zon LI, Kyriakis JM. Cdc42Hs, but not Rac1, inhibits serum-stimulated cell cycle progression at G1/S through a mechanism requiring p38/RK. J Biol Chem. 1997;272:13229–13235. doi: 10.1074/jbc.272.20.13229. [DOI] [PubMed] [Google Scholar]
- Morla A, Ruoslahti E. A fibronectin self-assembly site involved in fibronectin matrix assembly: reconstruction in a synthetic peptide. J Cell Biol. 1992;118:421–429. doi: 10.1083/jcb.118.2.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morla A, Zhang Z, Ruoslahti E. Superfibronectin is a functionally distinct form of fibronectin. Nature. 1994;367:193–196. doi: 10.1038/367193a0. [DOI] [PubMed] [Google Scholar]
- Mosher DF. Assembly of fibronectin into extracellular matrix. Curr Opin Struct Biol. 1993;3:214–222. [Google Scholar]
- Mosher DF, Sottile J, Wu C, McDonald JA. Assembly of extracellular matrix. Curr Opin Cell Biol. 1992;4:810–818. doi: 10.1016/0955-0674(92)90104-k. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia and filopodia. Cell. 1995;7:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- Price, L.S., J. Leng, M.A. Schwartz, and G.M. Bockoch. 1998. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Cell. Biol. In press. [DOI] [PMC free article] [PubMed]
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- Rankin S, Morii N, Narumiya S, Rozengurt E. Botulinum C3 exoenzyme blocks the tyrosine phosphorylation of p125FAK and paxillin induced by bombesin and endothelin. FEBS Lett. 1994;354:315–319. doi: 10.1016/0014-5793(94)01148-6. [DOI] [PubMed] [Google Scholar]
- Renshaw MW, Ren XD, Schwartz MA. Growth factor activation of MAP kinase requires cell adhesion. EMBO (Eur Mol Biol Organ) J. 1997;16:5592–5599. doi: 10.1093/emboj/16.18.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retta SF, Balzac F, Ferraris P, Belkin AM, Fassler R, Humphries MJ, De Leo G, Silengo L, Tarone G. Beta1-integrin cytoplasmic subdomains involved in dominant negative function. Mol Cell Biol. 1998;9:715–731. doi: 10.1091/mbc.9.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E. Integrins. J Clin Invest. 1991;87:1–5. doi: 10.1172/JCI114957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Kato J, Nishida K, Kaziro Y. Tyrosine phosphorylation of ACK in response to temperature shift-down, hyperosmotic shock, and epidermal growth factor stimulation. FEBS Lett. 1996;386:230–234. doi: 10.1016/0014-5793(96)00449-8. [DOI] [PubMed] [Google Scholar]
- Saulnier R, Bhardwaj B, Klassen J, Leopold D, Rahimi N, Tremblay E, Mosher D, Elliott B. Fibronectin fibrils and growth factors stimulate anchorage independent growth of a murine mammary carcinoma. Exp Cell Res. 1996;222:360–369. doi: 10.1006/excr.1996.0045. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Seufferlein T, Rozengurt E. Lysophosphatidic acid stimulates tyrosine phosphorylation of focal adhesion kinase, paxillin, and p130. Signaling pathways and cross-talk with platelet-derived growth factor. J Biol Chem. 1994;269:9345–9351. [PubMed] [Google Scholar]
- Tang Y, Chen Z, Ambrose D, Liu J, Gibbs JB, Chernoff J, Field J. Kinase-deficient Pak1 mutants inhibit Ras transformation of Rat-1 fibroblasts. Mol Cell Biol. 1997;17:4454–4464. doi: 10.1128/mcb.17.8.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennerberg K, Lohikangas L, Gullberg D, Pfaff M, Johansson S, Fässler R. beta1 integrin-dependent and -independent polymerization of fibronectin. J Cell Biol. 1996;132:227–238. doi: 10.1083/jcb.132.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Bauer JS, Juliano RL, McDonald JA. The α5β1 integrin fibronectin receptor, but not the α5 cytoplasmic domain, functions in an early and essential step in fibronectin matrix assembly. J Biol Chem. 1993;268:21883–21888. [PubMed] [Google Scholar]
- Wu C, Fields AJ, Kapteijn BAE, McDonald JA. The role of alpha4beta1 integrin in cell motility and fibronectin matrix assembly. J Cell Sci. 1995a;108:821–829. doi: 10.1242/jcs.108.2.821. [DOI] [PubMed] [Google Scholar]
- Wu C, Keivens VM, O'Toole TE, McDonald JA, Ginsberg MH. Integrin activation and cytoskeletal interaction are essential for the assembly of a fibronectin matrix. Cell. 1995b;83:715–724. doi: 10.1016/0092-8674(95)90184-1. [DOI] [PubMed] [Google Scholar]
- Wu C, Hughes PE, Ginsberg MH, McDonald JA. Identification of a new biological function for the integrin alpha v beta 3: initiation of fibronectin matrix assembly. Cell Adh Com. 1996;4:149–158. doi: 10.3109/15419069609014219. [DOI] [PubMed] [Google Scholar]
- Yang JT, Rayburn H, Hynes RO. Embryonic mesoderaml defects in alpha5 integrin-deficient mice. Development. 1993;119:1093–1105. doi: 10.1242/dev.119.4.1093. [DOI] [PubMed] [Google Scholar]
- Yang W, Cerione RA. Cloning and characterization of a novel Cdc42-associated tyrosine kinase, ACK-2, from bovine brain. J Biol Chem. 1997;272:24819–24824. doi: 10.1074/jbc.272.40.24819. [DOI] [PubMed] [Google Scholar]
- Yatohgo T, Izumi M, Kashiwagi H, Hayashi M. Novel purification of vitronectin from human plasma by heparin affinity chromatography. Cell Struct Funct. 1988;13:281–292. doi: 10.1247/csf.13.281. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Checovich WJ, Peters DM, Albrecht RM, Mosher DF. Modulation of cell surface fibronectin assembly sites by lysophosphatidic acid. J Cell Biol. 1994;127:1447–1459. doi: 10.1083/jcb.127.5.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Magnusson MK, Mosher DF. Lysophosphatidic acid and microtubule-destabilizing agents stimulate fibronectin matrix assembly through Rho-dependent actin stress fiber formation and cell contraction. Mol Biol Cell. 1997;8:1415–1425. doi: 10.1091/mbc.8.8.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Morla AO, Vuori K, Bauer JS, Juliano RL, Ruoslahti E. The alphavbeta1 integrin functions as a fibronectin receptor but does not support fibronectin matrix assembly and cell migration on fibronectin. J Cell Biol. 1993;122:235–242. doi: 10.1083/jcb.122.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]