

Abstract
We have compared cytoplasmic extracts from chicken DU249 cells at various stages along the apoptotic pathway. Extracts from morphologically normal “committed stage” cells induce apoptotic morphology and DNA cleavage in substrate nuclei but require ongoing caspase activity to do so. In contrast, extracts from frankly apoptotic cells induce apoptotic events in added nuclei in a caspase-independent manner. Biochemical fractionation of these extracts reveals that a column fraction enriched in endogenous active caspases is unable to induce DNA fragmentation or chromatin condensation in substrate nuclei, whereas a caspase-depleted fraction induces both changes. Further characterization of the “execution phase” extracts revealed the presence of an ICAD/DFF45 (inhibitor of caspase-activated DNase/DNA fragmentation factor)- inhibitable nuclease resembling CAD, plus another activity that was required for the apoptotic chromatin condensation. Despite the presence of active caspases, committed stage extracts lacked these downstream activities, suggesting that the caspases and downstream factors are segregated from one another in vivo during the latent phase. These observations not only indicate that caspases act in an executive fashion, serving to activate downstream factors that disassemble the nucleus rather than disassembling it themselves, but they also suggest that activation of the downstream factors (rather than the caspases) is the critical event that occurs at the transition from the latent to active phase of apoptosis.
Keywords: apoptosis, caspases, CAD, ICAD, DFF
Apoptosis is a cellular disassembly pathway that is activated by members of a distinct family of cysteine proteases called caspases (Cohen, 1997; Nicholson and Thornberry, 1997; Villa et al., 1997; Cryns and Yuan, 1998). Genetic analyses show clearly that caspases such as CED-3 and caspase-3 are essential for apoptotic death (Yuan and Horvitz, 1990; Kuida et al., 1996; Woo et al., 1998). Nonetheless, the role of these enzymes in the death pathway remains unclear. One recent study has suggested that cells can activate caspases without undergoing apoptosis (Boise and Thompson, 1997). Conversely, even though caspase inhibitors usually rescue cells from apoptosis (for review see Villa et al., 1997), cell death can occur in response to proapoptotic stimuli in the presence of these inhibitors (Xiang et al., 1996; McCarthy et al., 1997; Lavoie et al., 1998).
Careful examination has revealed that cells dying in the presence of caspase inhibitors display membrane blebbing and cell surface alterations but no changes in nuclear morphology (McCarthy et al., 1997). This observation suggests that certain cytoplasmic hallmarks of apoptosis may be triggered by enzymes other than caspases, but that nuclear events require caspase activity. Consistent with this view, cells from caspase-3–null mice (Woo et al., 1998) have been reported to display plasma membrane changes and cleavage of the nuclear protein poly(ADP-ribose) polymerase (PARP)1 when undergoing apoptosis, but not the chromatin condensation and DNA cleavage that are characteristic of apoptosis (Wyllie et al., 1980).
Although the preceding observations suggest that caspases play a role in certain apoptotic events, particularly those occurring in the nucleus, it is not known whether caspases function in an executive role to initiate the apoptotic pathway and leave the work of actually disassembling the cell to other downstream factors, or whether they are workhorses whose cleavage of key substrates drives cellular disassembly. Support for an executive role comes from the observation that expression of the caspase cleavage product of the actin-binding protein GAS2 triggers cytoskeletal changes similar to those seen in apoptosis (Brancolini et al., 1995). Likewise, caspase cleavage of ICAD/DFF (inhibitor of caspase-activated DNase/DNA fragmentation factor) releases the nuclease CAD, presumably allowing it to enter the nucleus and degrade genomic DNA (Liu et al., 1997; Enari et al., 1998; Sakahira et al., 1998). Thus, certain products of caspase cleavage play important downstream roles in apoptotic events. On the other hand, numerous important structural and nonstructural proteins are also directly cleaved by caspases (Cohen, 1997; Nicholson and Thornberry, 1997; Porter et al., 1997; Villa et al., 1997), supporting the alternative hypothesis that caspases do the bulk of the work of cutting the cell apart themselves (Martin and Green, 1995).
Current understanding of caspase function has been facilitated by the development of cell-free systems for the study of apoptosis (Lazebnik et al., 1993; Solary et al., 1993; Newmeyer et al., 1994; Enari et al., 1995a ; Martin et al., 1995; Schlegel et al., 1995; Liu et al., 1996). One such system uses extracts from DU249 chicken hepatoma cells that become committed to apoptosis after perturbation of the cell cycle (Lazebnik et al., 1993). Highly concentrated cytosolic extracts prepared from these morphologically normal cells (S/M extracts) reproduce all of the biochemical features of apoptosis in substrate nuclei (Lazebnik et al., 1993), including genome digestion (Wyllie et al., 1980; Lazebnik et al., 1993); cleavage of a subset of nuclear proteins, including PARP and lamins (Kaufmann, 1989; Ucker et al., 1992a ; Lazebnik et al., 1994, 1995); chromatin condensation; and nuclear fragmentation into apoptotic bodies (Lazebnik et al., 1993). This entire program of apoptotic events is inhibited in vitro by caspase inhibitors (Lazebnik et al., 1994) or millimolar concentrations of Zn2+ (Lazebnik et al., 1993) just as in intact cells. Although nuclei are used as the substrate in these studies, the extracts themselves are derived from the cytoplasm of the DU249 cells (Lazebnik et al., 1993), thus supporting the view that cytoplasmic factors and events have an essential role in the apoptotic pathway (Jacobson et al., 1994; Schulze-Osthoff et al., 1994; Nakajima et al., 1995; Martin et al., 1996; Kroemer, 1997).
The aim of the present study was to extend this approach by preparing extracts from cells at various stages of the apoptotic pathway to further evaluate caspase involvement in nuclear disassembly. Interestingly, extracts prepared from morphologically normal cells in the latent phase (S/M extracts) and those prepared from frankly apoptotic cells (execution phase extracts) induced similar apoptotic events in exogenous nuclei but exhibited fundamental biochemical differences. In particular, apoptotic events in the S/M extracts were abolished by caspase inhibitors as previously reported (Lazebnik et al., 1994), whereas the same events in execution phase extracts were not. Further examination revealed that execution phase extracts contain at least two caspase-activated factors required for nuclear disassembly, one of which appears to be the nuclease CAD. These experiments not only support the view that caspases act in an executive role in nuclear apoptosis by activating downstream factors that disassemble nuclei but also suggest that activation of the downstream factors (rather than the caspases) accompanies the transition between the latent and execution phases of apoptosis.
Materials and Methods
Cell Treatment and Preparation of Extracts
Chicken DU249 cells were presynchronized in S phase with aphidicolin for 12 h, released from the block for 6 h, and synchronized in mitosis with nocodazole for 3 h as described previously (Wood and Earnshaw, 1990; Lazebnik et al., 1993). DU249 cells start to undergo apoptosis asynchronously during and after the aphidicolin treatment, presumably as a result of the cell cycle disruption. Execution phase (E/X) extracts were prepared from the floating cells (mostly apoptotic) obtained from the flasks just before the addition of nocodazole. S/M extracts were prepared from floating cells (>60% mitotic) obtained from the same flasks by selective detachment after the nocodazole treatment. After harvesting of cells for S/M extract production, cells for the preparation of condemned phase (C/D) extracts were obtained from the attached (interphase) cells by rinsing the same flasks with PBS-EDTA and trypsinization. Large scale “roller S/M” extract was prepared as described above for S/M extract except that cells were grown in roller bottles.
In each case, the cells were then washed with KPM buffer (50 mM Pipes-KOH, pH 7.0, 50 mM KCl, 10 mM EGTA, 2 mM MgCl2, 20 μM cytochalasin B [Sigma Chemical Co., St. Louis, MO], 1 mM DTT, 0.1 mM PMSF, 1 μg/ml each chymostatin, leupeptin, antipain, pepstatin A) (Wood and Earnshaw, 1990) and centrifuged in a small glass Dounce homogenizer. The cells were subjected to several cycles of freezing and thawing and further disrupted by grinding during each thawing cycle. The cell lysate was then centrifuged at 139,000 g for 2 h, yielding clear cytosolic extracts. Protein concentration of each extract was measured by the Bradford assay (Bradford, 1976). Extract concentrations ranged between 12 and 18 mg/ml.
Time Course of Caspase Activation
DU249 cells were subjected to the synchrony procedure used in preparation of S/M extracts. At the indicated times (0, 5, 10, 15, and 20 h) after the addition of aphidicolin, both floating and attached cells were harvested from two T150 flasks (the former by shake-off, the latter by trypsinization). Cells were washed with MDB buffer, and the number of cells in each sample was counted using a hemacytometer. The ratio of interphase, mitotic, and apoptotic cells in each sample was determined by examination of the nuclear morphology after cells (n > 400 for each time point) were fixed in methanol/acetic acid (3:1) and stained with 0.5 μg/ml 4,6-diamidino-2-phenylindole (DAPI; Calbiochem, La Jolla, CA). Cells were lysed by the freeze/thaw/grinding protocol described above. Lysates (the supernatants after centrifugation at 13,000 g for 15 min at 4°C) were affinity labeled with z-EK(biotin)D-aomk as described below.
Fractionation of S/M Extracts with Heparin–Agarose Resin
Roller S/M extract (typically 7 mg of protein) prepared in KPM buffer containing 60 mM KCl was mixed with 360 μl of heparin–agarose resin (HiTrapHeparin; Pharmacia Biotech, Piscataway, NJ), which was preequilibrated with KPM buffer. This mixture was rotated at 4°C for 1 h and centrifuged in a microcentrifuge at 5,000 rpm for 5 min, and the supernatant was recovered (fraction 1; data not shown). Three cycles of absorption with heparin–agarose were needed to completely absorb all DNase activities from S/M extracts. After extensive washing of the resin with KPM buffer containing 60 mM KCl, bound proteins were eluted first with 450 μl of KPM containing 0.2 M KCl (fraction 2), and subsequently with 450 μl of KPM containing 0.6 M KCl (fraction 3). The eluted fractions were desalted and concentrated to 4–8 mg/ml protein by centrifugation using Ultrafree-0.5 filters (5K cut; Millipore, Bedford, MA).
In Vitro Apoptosis Reaction
Apoptotic and control extracts were preincubated at 37°C for 15 min with 100 μM caspase inhibitors (YVAD-cmk or DEVD-fmk) or diluent. HeLa nuclei prepared as previously described (Wood and Earnshaw, 1990; Lazebnik et al., 1993) were then added (up to 1.0 × 106 nuclei/10 μl of extract) and incubated at 37°C for up to 2 h in the presence of an ATP regeneration system (Wood and Earnshaw, 1990; Lazebnik et al., 1993). Nuclei were either stained with DAPI to observe chromatin condensation, solubilized in SDS–sample buffer for analysis of protein cleavage, or lysed for analysis of DNA ladder formation.
Caspase Labeling and Fluorogenic Assays
Labeling.
Stock solutions (10 mM in DMSO) of caspase inhibitors (YVAD-cmk or DEVD-fmk from Calbiochem) were diluted immediately before use with MDB buffer (Wood and Earnshaw, 1990). Extracts were preincubated at 37°C for 15 min with 100 μM YVAD-cmk or DEVD-fmk or diluent. After Z-EK(bio)D-aomk (Martins et al., 1997a ) was added to a final concentration of 1 μM from a 100× stock solution in DMSO, extracts were incubated at 37°C for 15 min. Labeled proteins were subjected to conventional 16% SDS-PAGE (Laemmli, 1970), transferred to nitrocellulose membrane, probed with peroxidase-coupled streptavidin, and visualized by ECL (Amersham Corp., Arlington Heights, IL).
Fluorogenic Assays.
DEVD-AFC cleavage activity was determined by a slight modification of previously described methods (Martins et al., 1997a ). Extracts were preincubated with caspase inhibitors or diluent for 15 min at 37°C. Samples containing 20–30 μg of various fractions or 25 μg of cytosolic protein (estimated by the Bradford assay) from etoposide-treated K562 leukemia cells (a positive control) were diluted to 50 μl with buffer A (25 mM Hepes, pH 7.5, 5 mM MgCl2, 5 mM EDTA, 1 mM EGTA supplemented immediately before use with 1 mM PMSF, 1 mM DTT, 10 μg/ml pepstatin A, and 10 μg/ml leupeptin), mixed with 225 μl freshly prepared buffer B (25 mM Hepes, pH 7.5, 0.1% [wt/vol] CHAPS, 10 mM DTT, 100 U/ml aprotinin, 1 mM PMSF) containing 100 μM DEVD-AFC (Enzyme System Products, Dublin, CA), and incubated for 4 h at 37°C. Reactions were terminated by addition of 1.225 ml ice-cold buffer B. Fluorescence was measured in a Sequoia-Turner spectrofluorometer using an excitation wavelength of 360 nm and emission wavelength of 475 nm. After subtraction of fluorescence in blank samples (lacking protein), amounts of the liberated fluorophore were determined by comparison to a standard curve containing 0–1,500 pmol of 7-amino-4-trifluoromethylcoumarin. Control experiments indicated that product release was linear with respect to incubation time and extract protein under the conditions used.
Protein and DNA Gel Electrophoresis
PARP and Lamin Cleavage.
After HeLa nuclei (5 × 105 per loading) were incubated in extract for 2 h at 37°C, the reaction was stopped by the addition of sample buffer. Samples were boiled at 95°C for 5 min, sonicated briefly, subjected to 10% conventional SDS-PAGE (Laemmli, 1970; Wood and Earnshaw, 1990; Lazebnik et al., 1993), and transferred to nitrocellulose. PARP and its 89-kD cleavage product were detected with the C-2-10 monoclonal antibody (Lamarre et al., 1988). Lamins A/C and their cleavage product were detected with a rabbit polyclonal antibody recognizing the NH2 terminus of the protein (gift of Larry Gerace). Bound antibody was detected by ECL.
DNA Ladder Formation.
HeLa nuclei (5 × 105 per loading) were incubated for 1–2 h in extract, centrifuged, and lysed in DNA lysis buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 0.5% Sarcosyl, 0.5 mg/ml proteinase K) at 50°C for 1 h. DNA was treated with RNase at 50°C for 1 h, phenol/chloroform extracted, ethanol-precipitated overnight at −70°C, resuspended in TE, and loaded onto 1% agarose gels containing 0.5 μg/ml ethidium bromide.
Plasmid DNA Digestion Assay.
Extracts (18–36 μg protein in 10 μl of KPM buffer) supplemented with an ATP regeneration system (Wood and Earnshaw, 1990) and 100 μM DEVD-fmk (or diluent) were incubated at 37°C for 15 min. After addition of 160 ng purified glutathione-S-transferase–ICAD (GST-ICAD) (Sakahira et al., 1998), incubation was continued for an additional 10 min at 37°C. Upon addition of substrate (pBluescript, 1.2 μg), incubation was continued for 30 min at 37°C. DNA extraction and electrophoresis were then performed as above. The results of the preincubation assay (Fig. 5) were best seen by Southern blotting. DNA agarose gels were denatured for 30 min with denaturing buffer (1.5 M NaCl, 0.5 M NaOH), neutralized for 30 min with neutralizing buffer (0.5 M Tris-HCl, pH 7.5, 1.5 M NaCl, 1 mM EDTA), and transferred to nylon membrane (Hybond-N; Amersham Corp.) with 20× SSC buffer. The nylon membrane was UV cross-linked and hybridized (Church and Gilbert, 1984) with a pBluescript probe that was labeled with 32P using the megaprimer system (Amersham Corp.). The film was exposed for 1 h at −80°C with an intensifying screen.
Figure 5.
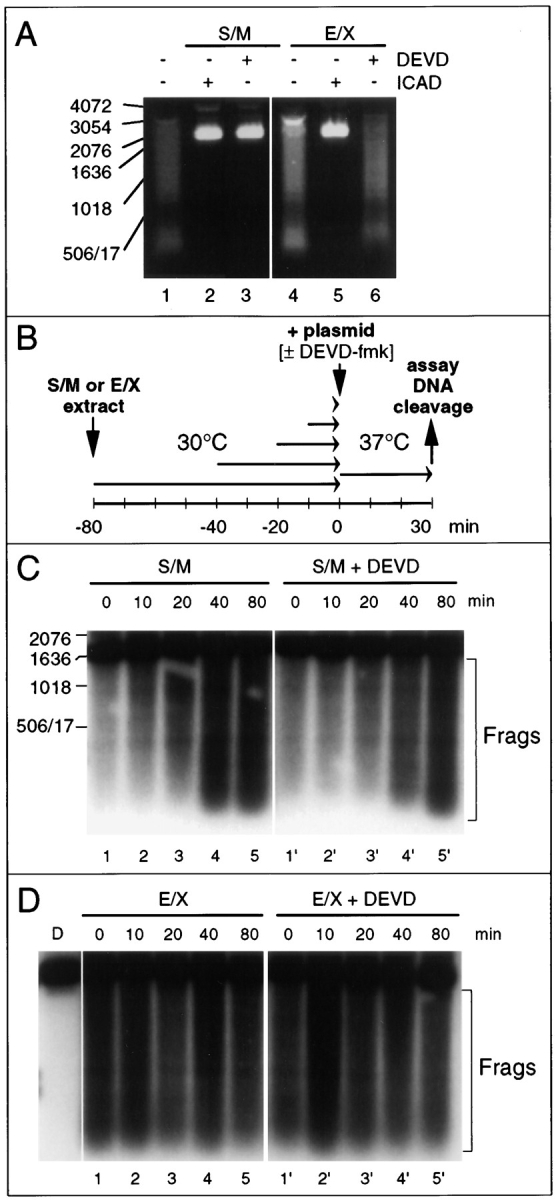
Differences in the activity of the CAD-like nuclease in S/M and E/X extracts. (A) CAD-like nuclease in S/M extracts is sensitive to DEVD-fmk, whereas that in E/X extracts is not. Apoptotic extracts cleave a plasmid substrate at 37°C (lanes 1 and 4). The nuclease responsible is inhibited by purified murine GST-ICAD (lanes 2 and 5) and therefore is functionally related to murine CAD (Enari et al., 1998). In S/M extracts, but not E/X extracts, the nuclease is abolished by addition of DEVD-fmk together with the plasmid DNA (lanes 3 and 6). (B) Diagram of experimental protocol designed to test whether the CAD-like enzyme is active in both S/M and E/X extracts before the incubation with DNA. (C) In S/M extracts, the CAD-like activity increases during preincubation of the extract at 30°C before addition of the plasmid DNA (lanes 1–5). This activity is now insensitive to the addition of DEVD-fmk at the time of plasmid addition (lanes 1′– 5′). Bracket labeled “Frags” indicates the products of caspase cleavage. The DNA cleavage activity in lane 1′ may arise from some limited activation of CAD before all caspases were inhibited since DNA and DEVD-fmk were added at the same time. (D) In contrast, preincubation has no effect on the cleavage of plasmid substrate by E/X extracts (lanes 1–5), which is likewise resistant to inhibition with DEVD-fmk (lanes 1′–5′). Lane D, added plasmid DNA alone. Bracket labeled “Frags” indicates the products of caspase cleavage.
Pulsed-Field Gel Electrophoresis.
HeLa nuclei (5 × 105) were incubated with each fraction (10 μl) for the specified time and embedded in 1.5% low melting agarose (Sea Plaque GTG agarose, FMK). Gel blocks were soaked in nuclei lysis buffer (10 mM Tris-HCl, pH 9.5, 500 mM EDTA, 1 mg/ml proteinase K, 1% sarkosyl) for 20 h at 50°C and then stored in TE buffer at 4°C. Blocks were transferred into the wells of an agarose gel and sealed in place by the addition of a small volume of agarose. Contour-clamped homogeneous (CHEF) electrophoresis was performed using a CHEF system purchased from Bio-Rad Laboratories Ltd. (Watford, England) with a model 200/2.0 power supply and a Pulsewave 760 switcher. Horizontal gels (1% agarose; Sigma Chemical Co.) were run at 14°C at 200 V for 20 h with a pulse ramp of 4–40 s and stained with ethidium bromide.
Electron Microscopy.
Isolated HeLa cell nuclei incubated in extract or MDB buffer were centrifuged, washed with MDB buffer, placed on adhesion slides (Marienfeld) for 5 min, and fixed for 30 min with 2% glutaraldehyde in Dulbecco's PBS, pH 7.4. After fixation the nuclei were washed in 0.1 M cacodylate buffer, and postfixed with 4% OsO4 in 0.1 M sodium cacodylate, pH 7.4, for 30 min. Prestaining was done with 3% uranyl acetate in H2O for 1 h. The nuclei were dehydrated in ethanol (30–100%) and embedded in Araldite resin (Agar). Gold sections were cut with a Reichert (Vienna, Austria) microtome and placed on copper grids. Images were photographed on an electron microscope (model CM100 Biotwin; Philips Electron Optics, Mahwah, NJ).
Expression and Purification of Double Mutant His6-ICAD
The method used for mutation was the PCR-based megaprimer strategy (Seraphin and Kandels-Lewis, 1996). Primer 1 (T7 primer), primer 2 (5′-GCCCTGCTCTCAGGCTCATC-3′), and primer 3 (5′-CAGCTCTGCACATGGGATGTC-3′) were used to generate the DEPD117E mutation in the ICAD cDNA in pBluescript. Primer 4 (5′-CTGCTGTCAGAAGAGGACCTC-3′), primer 5 (5′-GCCGACGCCTGTCTCAACTGC-3′), and primer 6 (T3 primer) were used to generate the DAVD224E mutation in ICAD. Each single mutant was digested with Eco47III (New England Biolabs, Hitchin, UK) and XbaI (New England Biolabs) and then ligated to generate double mutant ICAD in pBluescript. This double mutant ICAD was digested with SpeI, blunt-ended with T4 DNA polymerase (New England Biolabs) plus 100 μM dNTPs, and digested with KpnI (New England Biolabs). The resulting fragment was ligated into pRSET B (Invitrogen, Carlsbad, CA) that had been digested with HindIII, blunt-ended with T4 DNA polymerase, and digested with KpnI. Double mutant ICAD in pRSETB was transformed into Escherichia coli BL21 (DE3)Lys S cells. Transformed cells were grown to OD600 = 0.5–0.7, and protein expression was induced with IPTG (1 mM) for 3–4 h. Cells were collected by centrifugation at 5,000 g for 10 min and frozen at −80°C. The cell pellet was thawed on ice for 15 min and resuspended in lysis buffer (50 mM NaH2PO4 pH 7.5, 300 mM NaCl, 10 mM imidazole). Lysozyme was added to 1 mg/ml, and the suspension was incubated on ice for 30 min, sonicated on ice until 80% of the cells were disrupted, and then centrifuged at 4,000 g for 20 min at 4°C. The supernatant was incubated on a rotating mixer for 1 h at 4°C with 0.5 ml of Ni-agarose (Qiagen, Chatsworth, CA) that had been preequilibrated with lysis buffer. The resin was then loaded onto a polyprep chromatography column (Bio-Rad Laboratories) and washed twice with 4 ml of wash buffer (50 mM NaH2PO4 pH 7.5, 300 mM NaCl, 20 mM imidazole). Protein was eluted with 2 ml of elution buffer (50 mM NaH2PO4 pH 7.5, 300 mM NaCl, 250 mM imidazole). All samples were subjected to SDS-PAGE and examined by Coomassie blue staining. The eluted protein was dialyzed for at least 3 h against two changes of CAD buffer (10 mM Hepes, pH 7.4, 50 mM NaCl, 5 mM EGTA, 2 mM MgCl2, 1 mM DTT), aliquoted, and frozen in N2(l).
Expression and Purification of His6-ICAD/CAD
To express active CAD in E. coli, we constructed a bicistronic expression vector in which His6-ICAD was expressed upstream of CAD. In addition to the NH2-terminal histidine tag, the ICAD open reading frame was engineered to change the stop codon to TAA and to insert a Shine-Delgarno sequence (GGAAT) downstream of the stop codon. The wild-type ICAD cDNA in pBluescript was digested with SpeI, blunt-ended with T4 DNA polymerase, and digested with KpnI. The resulting fragment was ligated into pRSET B that had been digested with HindIII, blunt-ended with T4 DNA polymerase, and digested with KpnI. The coding region of the CAD cDNA was extracted from the full-length CAD cDNA in pBluescript by PCR using Vent polymerase (New England Biolabs) with primer 7 (5′-GGAATTCATGTGCGCGGTGCTCCG-3′) and primer 8 (5′-GCGAAGCTTTCACTAGCGCTTCCGAG-3′). The PCR product was digested with EcoRI and HindIII, and the resulting fragment was ligated into pBluescript.
To create the COOH terminus of ICAD (from the BsmI site at nucleotide 909) engineering in a TAA stop codon and Shine-Delgarno sequence, primer 9 (5′-GGAAGATCTGCATTCACTCAGGAATC-3′) and primer 10 (5′-GGAATTCCTCCTTACGAGGAGTCTCGTTTG-3′) were used with Vent polymerase. The PCR product was digested with BglII (New England Biolabs) and EcoRI and ligated into pRSETB that had been digested with BglII and EcoRI. The CAD coding sequence in pBluescript was digested with EcoRI and HindIII and ligated into pRSET B containing the newly modified COOH terminus of ICAD with the Shine-Delgarno sequence. This intermediate was then digested with NheI and BsmI, and into it was inserted the His-tagged NH2-terminal portion of ICAD, obtained from ICAD in pRSETB that had likewise been digested with NheI and BsmI. This bicistronic vector His6-ICAD/CAD in pRSET B was transformed into E. coli BL21 (DE3)Lys S cells. Protein was expressed and purified by nickel chelate chromatography as described above for double mutant ICAD. The dialyzed ICAD/CAD protein was frozen in N2(l) either directly or following addition of glycerol to 40%.
In Vitro Apoptosis with Purified CAD
50-μl reactions contained various combinations of the following reagents added sequentially (see legend to Fig. 6): 10 μl ICAD/CAD protein (stored in CAD buffer plus 40% glycerol), 5 μl caspase-3, an ATP regeneration system (final concentration 0.8 mM ATP, 4.5 mM creatine phosphate, 22.5 μg/ml creatine kinase), DEVD-fmk (final concentration 300 μM), double mutant ICAD protein (2 μg), and CAD buffer as needed to make up the final volume. ICAD/CAD complexes were preincubated with caspase-3 at room temperature (∼25°C) for 30 min to cleave wild-type ICAD and release active CAD. At the end of this preincubation, diluent, DEVD-fmk, or double mutant ICAD were added (defined as t = 0), and the mixture was divided into three aliquots to assay various apoptotic events. To examine ICAD cleavage during the preincubation, a 10-μl aliquot was mixed with sample buffer, boiled, resolved by SDS-PAGE, transferred to nitrocellulose membranes, and probed with ICAD antibody (Samejima and Earnshaw, 1998), which was detected by ECL (Amersham Corp.). To assay CAD activity against a plasmid DNA substrate, a 6-μl aliquot was supplemented with 0.35 μg pBluescript, BSA (final concentration 1 mg/ml), and 4 μl of CAD buffer. This mixture was further incubated at 37°C for 30 min, extracted with phenol-chloroform, and analyzed on a 1% agarose gel containing ethidium bromide. To examine the ability of the active CAD to induce apoptotic events, HeLa nuclei (1.3 × 106 per sample) in 10 μl CAD buffer were combined with the remaining 34 μl and incubated at 37°C for 2 h. At the end of this incubation, 1 μl of the reaction mixture was stained with DAPI so that nuclear morphology could be examined by fluorescence microscopy (>100 nuclei counted per slide). The remaining HeLa nuclei in 43 μl of reaction mixture were centrifuged and prepared for agarose gel electrophoresis as described above.
Figure 6.
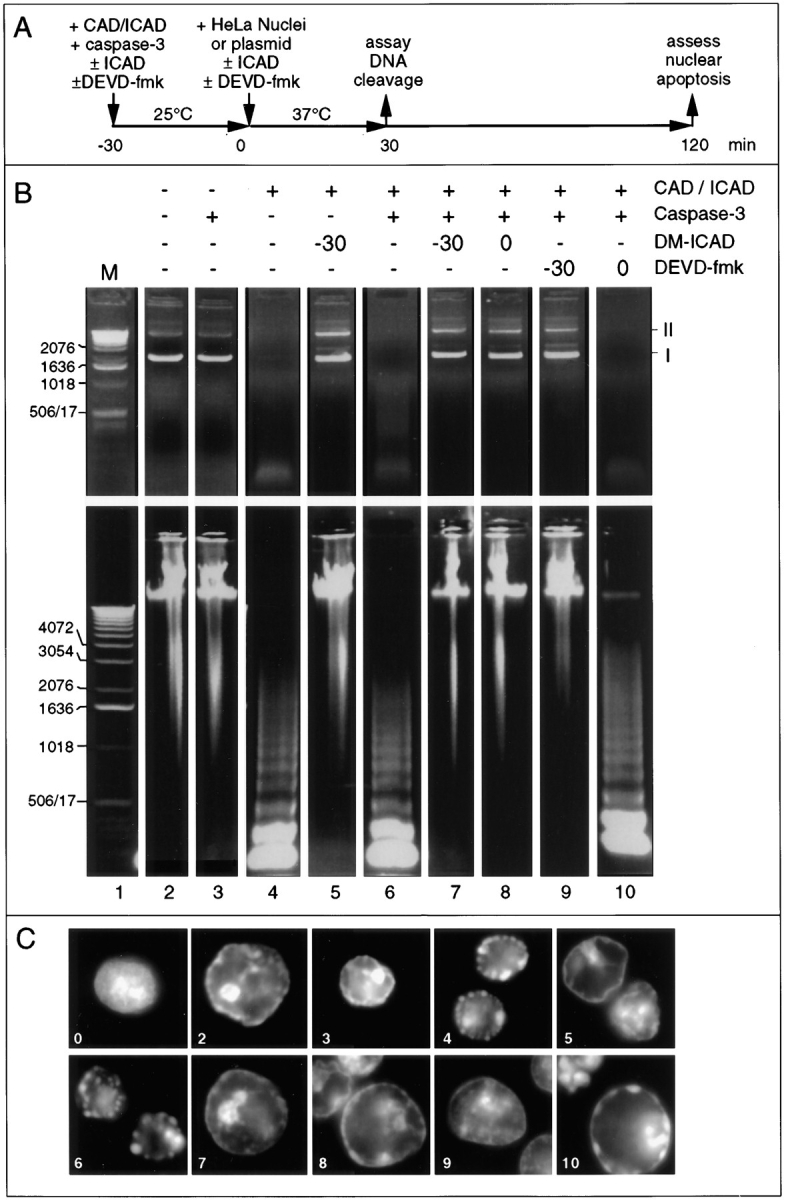
Bacterially expressed CAD is capable of inducing apoptotic morphology in added nuclei in the absence of apoptotic extract. (A) Diagram of the experimental protocol. After CAD was activated during a preincubation with purified caspase-3 at 25°C, substrates were added and incubated for various times with the active enzyme at 37°C. (B) Effects of various incubations on CAD nuclease activity using either a purified plasmid substrate (pBluescript, upper panels) or added nuclei (lower panels). The morphology of the nuclei in these same incubations is shown in C, where the panel numbers refer to lanes in B. Buffer and caspase-3 do not possess nuclease activity or induce apoptotic morphology in added nuclei (lanes and panels 2 and 3). Preincubation of CAD alone induces nuclease activity and apoptotic morphology (lane and panel 4), presumably because of the presence of a contaminating protease. ICAD is cleaved during this preincubation (data not shown). This spontaneous activation of CAD is not observed if noncleavable ICAD is present from the start of the preincubation (lane and panel 5). Preincubation of CAD with caspase-3 induces nuclease activity and apoptotic morphology (lane and panel 6). This activity is not observed if noncleavable ICAD is present either from the start of the preincubation, or if it is added at the end of the preincubation together with the substrate nuclei (lanes and panels 7 and 8). Addition of DEVD-fmk to the preincubation blocks CAD activation but has no effect on nuclease activity or induction of apoptotic morphology if added once the preincubation is complete (lanes and panels 9 and 10). The induction of apoptotic morphology by CAD in the presence of DEVD-fmk was slightly, but reproducibly, reduced (panel 10). These results demonstrate that CAD can induce nuclear apoptosis in the absence of caspase-3 activity. I and II at the right of B indicate the migration of form I (supercoiled) and form II (nicked circular) plasmid DNA.
Results
Extracts from Three Different Stages of Apoptosis
The apoptotic pathway can be conceptually divided into at least three stages (Fig. 1 A). Upon receipt of a proapoptotic signal, cells enter a “condemned” stage: the death program is initiated, but cells can be rescued by various survival factors. Once cells pass a point of no return, they are in the “committed” stage and can no longer be rescued. During both of these stages, preapoptotic cells appear morphologically normal. Eventually, committed cells undergo an abrupt transition into apoptotic execution, a period lasting from 5 min to 1 h, during which cellular disassembly and death occur. It is not known where along this pathway caspases are activated and at what point the caspases activate other downstream factors that act during disassembly of the cell.
Figure 1.

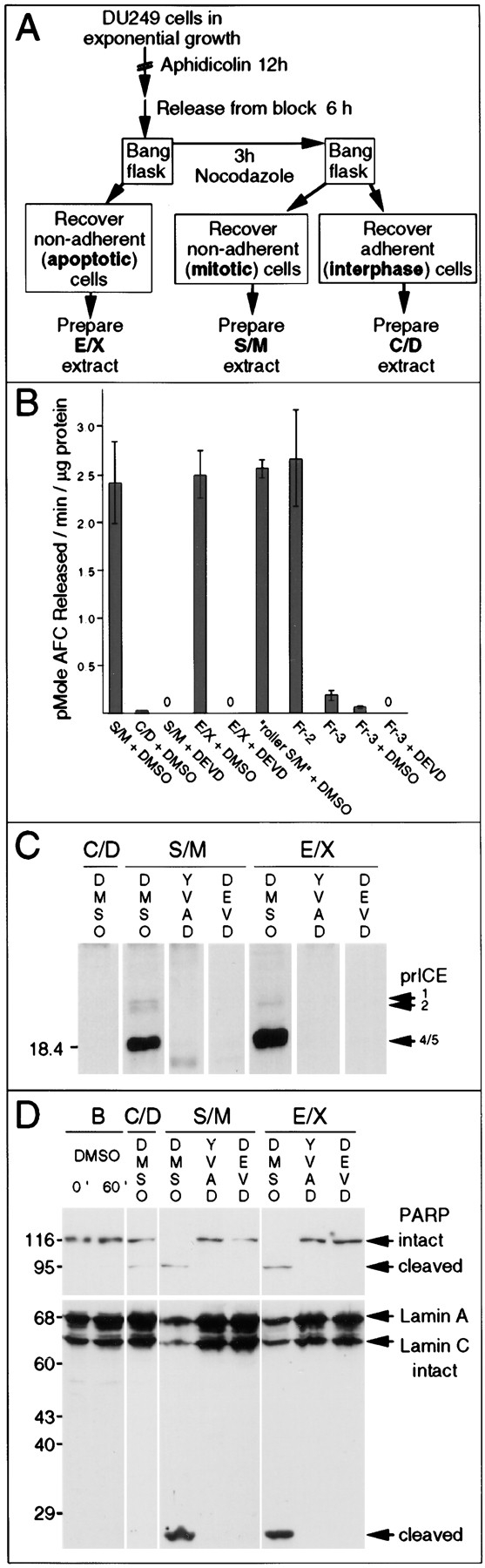
(A) Diagram of apoptosis as a three stage process, with the latent phase being subdivided into condemned and committed stages. (B) The protocol used for harvesting of samples for examination of caspase activation. (C) The cells harvested at each time point were scored for their nuclear morphology, based on DAPI staining. (D) Caspase activity in whole cell lysates prepared from cells harvested at various times points after the addition of aphidicolin to the culture (time in hours shown at bottom). Left, attached cells; right, floating cells. The left-most lane shows the profile of active caspases in S/M extract.
We previously noted that active caspases could be detected in extracts from etoposide-treated HL-60 cells several hours before the bulk of the cells in the culture exhibited an overtly apoptotic morphology (Martins et al., 1997a ). To study this phenomenon in greater detail, we harvested chicken DU249 cells at various times after subjecting cultures to a synchronization protocol (Fig. 1 B) shown previously to induce an apoptotic response in this cell line (Lazebnik et al., 1993). Floating cells obtained 10– 15 h after the addition of aphidicolin were largely (∼ 50%) apoptotic and contained high levels of active caspases, as detected by reactivity with the affinity-labeling reagent z-EK(biotin)D-aomk (Martins et al., 1997a ) (Fig. 1, C and D). Floating cells harvested after a change of medium and a 2-h treatment with nocodazole to induce a mitotic block were predominantly (>60%) mitotic, with only 10–20% of apoptotic cells. Despite the normal appearance of the vast majority of these cells, extracts prepared from them also contained high levels of active caspases. Cells that remained attached throughout the protocol were almost entirely in interphase, and extracts prepared from them lacked caspases detectable with z-EK(biotin)D-aomk (Fig. 1 D, left), although low levels of caspase activity were detectable when more sensitive assays were used (see below).
The results of this experiment suggested that it might be possible to use a similar protocol to prepare extracts sequentially from the same flasks of cells at different stages of apoptosis (Fig. 2 A). C/D (condemned phase) extracts were prepared from the morphologically normal attached cells that did not enter mitosis or apoptosis during the synchrony procedure. Previous studies have indicated that these cells ultimately undergo apoptosis if left in culture (Lazebnik et al., 1993). S/M (committed phase) extracts (Lazebnik et al., 1993) were prepared from morphologically normal mitotic cells at the conclusion of the synchrony procedure. Because these cells are destined to rapidly undergo apoptosis if left in culture (Lazebnik et al., 1993), we postulate that S/M extracts reproduce events from the committed stage of apoptosis. E/X (execution phase) extracts were prepared from cells that were frankly apoptotic after a 12-h exposure to aphidicolin followed by a 6-h recovery period in medium. (Note that nonadherent cells were discarded at the end of the aphidicolin treatment, so these cells must have entered apoptosis during the 6-h recovery period).
Figure 2.
Apoptotic extracts contain active caspases that are functionally inactivated by specific inhibitors. (A) Protocol used to prepare C/D, S/M, and E/X extracts. (B) Quantitative analysis of DEVD-AFC cleavage activity in the various extracts. DEVD-fmk treatment of S/M and E/X extracts reduces caspase activity by at least 250- and 150-fold, respectively. Note the low but nonzero level of caspase activity in the C/D extracts. Fr-3 (see Fig. 4) has ∼8% the caspase activity seen in S/M extract. Treatment of Fr-3 with DEVD-fmk further reduces this activity by ≥130-fold relative to the S/M extract. (C) Active caspases were labeled with zEK(biotin)D-aomk in S/M and E/X extracts. This labeling was completely blocked by prior incubation of extracts with YVAD-cmk and DEVD-fmk (both at 100 μM). (D) PARP and lamin A/C cleavage by caspases in the cell-free extracts is abolished after caspase inactivation with YVAD-cmk and DEVD-fmk. Note that low levels of caspases in the C/D extract cause some PARP cleavage but fail to cleave lamins.
To examine the spectrum of active caspases during the three stages of apoptosis, extracts were assayed for their ability to cleave DEVD-AFC, for affinity labeling with z-EK(bio)D-aomk (Martins et al., 1997a ), and for the ability to cleave known caspase substrates in added nuclei. Collectively, these assays detect all known caspases. DEVD-AFC contains the preferred cleavage site of caspases-3 and -7 (Talanian et al., 1997; Duan et al., 1996b ) but is also cleaved by caspases-1, -2, -4, -6, -8, and -10 (Fernandes-Alnemri et al., 1995a , 1996; Boldin et al., 1996; Srinivasula et al., 1996; Talanian et al., 1997). z-EK(bio)D-aomk covalently modifies all caspases tested to date (Martins et al., 1997a ) and can detect over 30 active caspase species in apoptotic human leukemia cells (Martins, L.M., and W.C. Earnshaw, unpublished observations). PARP is a documented substrate of caspases-3, -7, -8, and -9 (Nicholson et al., 1995; Tewari et al., 1995; Fernandes-Alnemri et al., 1995a ,b; Duan et al., 1996b ; Muzio et al., 1996), while lamin A is a substrate of caspase-6 (Takahashi et al., 1996). Application of these assays to C/D extracts revealed low but detectable levels of active caspases (125-fold less than S/M or E/X extracts in the DEVD-AFC assay; Fig. 2 B) and some ability to cleave PARP (Fig. 2 D, lane C/D), but no evidence of z-EK(biotin)D-aomk labeling or lamin cleavage, suggesting that the latter assays are less sensitive. In contrast, S/M and E/X extracts contained high levels of active caspases that cleaved DEVD-AFC as well as PARP and lamin A (Fig. 2, B and D), and both had a similar pattern of active caspases after affinity-labeling with z-EK(bio)D-aomk (Fig. 2 C). These correspond primarily to active forms of caspases-3 and -6 (Martins et al., 1997a ; Faleiro et al., 1997). All caspase activity detectable in these assays was quantitatively inactivated by treatment with YVAD-cmk (Fig. 2, C and D) or DEVD-fmk (Fig. 2, B–D).
Differences in Dependence of S/M and E/X Extracts on Ongoing Caspase Activity
C/D extracts were unable to induce internucleosomal DNA fragmentation and apoptotic morphological changes in exogenous nuclei (Fig. 3, A and B). In contrast, S/M and E/X extracts induced hallmark biochemical and morphological changes of apoptosis in added nuclei. Further studies focused on these latter two extracts.
Figure 3.
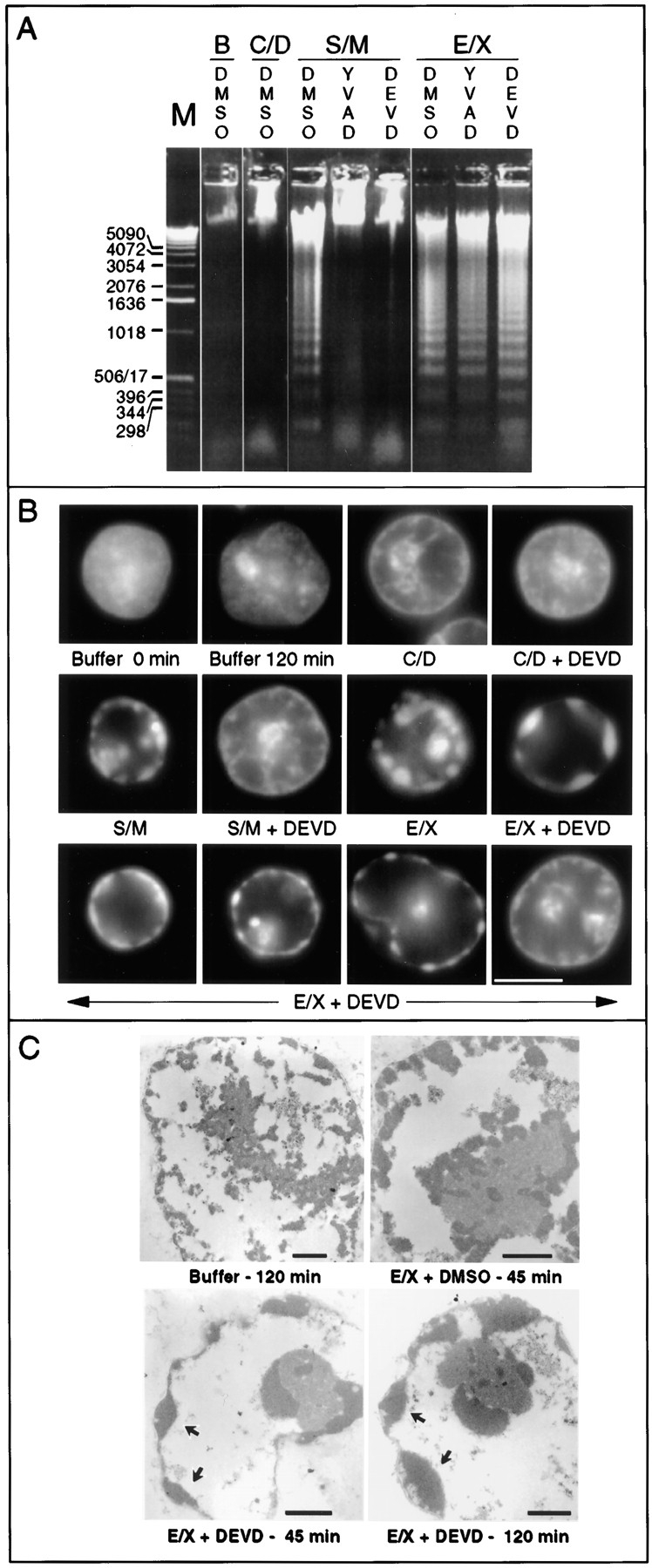
Caspase inhibitors block apoptosis in S/M but not E/X extracts. (A) Inhibition of caspases blocks nuclease activity in S/M extracts but not E/X extracts. As expected, C/D extracts lack detectable nuclease activity. Experiments shown in Fig. 8 confirm that the nuclease activity is inhibitable by the specific CAD inhibitor ICAD/DFF45 (Enari et al., 1995b ; Liu et al., 1997). Lane B, buffer control. (B) Inhibition of caspases abolishes morphological apoptosis in vitro in S/M extracts but not execution phase extracts. Nuclei shown in B were selected at random. (C) Morphological changes characteristic of apoptosis occur in E/X extracts independently of caspase activity: confirmation of apoptotic morphology by electron microscopy. Arrows indicate regions of condensed chromatin. Bars: (B) 5 μm; (C) 1 μm.
Despite the similarities of the S/M and E/X extracts in terms of caspase activity (Fig. 2) and effects on exogenous nuclei (Fig. 3, A and B), the two extracts displayed strikingly different properties after inhibition of the endogenous caspase activity. Pretreatment of S/M extracts with DEVD-fmk or YVAD-cmk before addition of nuclei not only inhibited caspase activity (Fig. 2, B–D) but also completely abolished their ability to produce internucleosomal DNA fragmentation (Fig. 3 A) and induce morphological apoptotic changes (Fig. 3 B). In striking contrast, E/X extracts that had been pretreated with caspase inhibitors continued to strongly induce internucleosomal DNA degradation (Fig. 3 A), chromatin condensation, and fragmentation of added nuclei (shown both by light and electron microscopy; Fig. 3, B and C) despite a lack of detectable caspase activity (Fig. 2, B–D).
Fractionation of Active Extracts
Further evidence for the ability of extracts to induce nuclear apoptotic events in the absence of caspase activity was obtained through biochemical fractionation of large-scale roller S/M extracts prepared using cells growing in roller bottles. These extracts appeared to functionally resemble a mixture of S/M and E/X extracts (i.e., were only partly inhibited by DEVD-fmk or YVAD-cmk). We believe that this is because a much higher percentage of apoptotic cells was present among the committed phase cells at the time of harvest. When roller S/M extracts were fractionated on heparin agarose (Fig. 4 A), proteins eluted from the column at 0.2 M KCl (fraction 2 [Fr-2]) included many of the active endogenous caspases, as shown by their ability to cleave the peptide substrate DEVD-AFC (Fig. 2 B), by activity labeling with z-EK(biotin)D-aomk (Fig. 4 B), and by cleavage of PARP and lamin A (Fig. 4 C). Factors eluted from the column at 0.6 M KCl (fraction 3 [Fr-3]) were substantially depleted of caspases, and this fraction resembled C/D extracts in several regards. DEVD- AFC cleavage activity in Fr-3 was reduced 13-fold relative to roller S/M extract (Fig. 2 B), and only trace levels of PARP cleavage activity were detected (compare Fig. 4 C, lanes Fr-3, with Fig. 2 D, lane C/D). Both zEK(biotin)D-aomk binding activity and lamin A cleaving activity were undetectable in Fr-3 (Fig. 4, B and C).
Figure 4.

A fraction rich in endogenous caspases fails to induce apoptotic events in added nuclei. (A) Protocol used to prepare fractions 1–3 by heparin–agarose chromatography of roller S/M extract. (B) Active caspases were labeled with zEK(biotin)D-aomk in roller S/M extract and fraction 2, but could not be detected in Fr-3. The bands in the upper portion of the gel are cellular proteins that bind to the streptavidin probe. (C) PARP and lamin A are efficiently cleaved by roller S/M extract and by Fr-2. Fr-3 has low levels of PARP cleavage activity that are abolished by pretreatment with DEVD-fmk. (D) Pulsed field gel electrophoresis. High molecular weight DNA fragments were produced in HeLa nuclei incubated in roller S/M extracts and Fr-3 (caspase-deficient), but not in nuclei incubated in buffer or Fr-2, which contains high levels of active caspases. Lane M, DNA markers (sizes shown in kilobase pairs). (E) Conventional agarose gel electrophoresis. An oligonucleosomal DNA ladder is formed in nuclei incubated in roller S/M extracts and Fr-3, but not in nuclei incubated in buffer or Fr-2. Lane M, DNA markers (sizes shown in base pairs). (F) Fr-3 induces very strong morphological apoptosis even when all detectable caspase activity is abolished by pretreatment with DEVD- fmk. In contrast, Fr-2, which has a distribution and concentration of caspases essentially identical to S/M extract, does not induce morphological apoptosis in HeLa nuclei.
Although fraction 2 contained high levels of endogenous caspase activity, it was unable to trigger either internucleosomal DNA fragmentation or morphological apoptosis in added nuclei. In contrast, fraction 3 was a more potent inducer of apoptosis than the starting roller S/M extract, inducing both high molecular weight and oligonucleosomal DNA fragmentation (Fig. 4, D and E) as well as morphological apoptosis in substrate nuclei (Fig. 4 F). This activity was not due to the low levels of residual caspases present in fraction 3. Pretreatment with DEVD-fmk, which abolished all detectable caspase activity (Fig. 2 B), had no effect on the ability of fraction 3 to induce DNA fragmentation (not shown) or apoptotic morphology in added nuclei (Fig. 4 F). Thus, a column fraction rich in endogenous apoptotic caspases (Fr-2) was unable to induce morphological apoptosis in isolated nuclei, while a second fraction prepared on the same column from the same extract (Fr-3) strongly induced apoptotic events despite being severely depleted or devoid (after inhibitor treatment) of detectable caspase activity.
Comparison of CAD Activation in S/M and E/X Extracts
Because the results presented in Figs. 2–4 suggest that the program of nuclear disassembly—one of the distinguishing features of apoptotic cell death under physiological conditions (Wyllie et al., 1980)—is initiated by caspase activity but does not require the ongoing participation of caspases for its successful execution, we next turned our attention to caspase-activated downstream activities that might participate in nuclear disassembly. One of these activities is CAD/CPAN, the recently identified caspase-activated deoxyribonuclease identified in murine and human cells undergoing apoptosis (Enari et al., 1998; Halenbeck et al., 1998). Both S/M and E/X extracts contain an endogenous DNase activity that degrades added plasmid DNA (Fig. 5 A, lanes 1 and 4). Addition of 160 ng of murine ICAD to the extracts abolished this DNase activity (lanes 2 and 5), indicating that this activity is functionally homologous to CAD.
Even though CAD-like activity could be inhibited by ICAD in both S/M and E/X extracts, this activity appeared to be regulated differently in the two extracts. DEVD-fmk pretreatment abolished CAD-like activity in S/M extracts (Fig. 5 A, lanes 1 and 3) but had no effect on CAD activity in E/X extracts (Fig. 5 A, lanes 4 and 6). There are at least two potential explanations for this difference. First, active CAD might be unstable in S/M extracts and might therefore require a constant source of caspase activity to continue to generate active enzyme from CAD/ICAD complexes. Second, despite the presence of high levels of endogenous caspases in S/M extracts, the CAD in these extracts might initially be largely in the form of inactive CAD/ICAD complexes. Active CAD might be released from these complexes by caspase activity during the incubation with DNA substrates.
To distinguish between these possibilities, we preincubated S/M and E/X extracts at 30°C for various times to allow cleavage of endogenous ICAD by endogenous caspases before adding DNA in the presence or absence of DEVD-fmk (see the experimental protocol in Fig. 5 B). This experiment indicated that CAD activity was initially low in S/M extracts but increased gradually with incubation at 30°C (Fig. 5 C, lanes 1–5). Once activated, CAD was stable in the presence of DEVD-fmk (lanes 1′–5′). In contrast, CAD was fully active at all times in E/X extracts in the presence and absence of DEVD-fmk (Fig. 5 D). Collectively, these results indicate that CAD is present but inactive in extracts prepared from committed stage cells and is activated in equivalent extracts prepared from execution phase cells. In addition, this result confirms that the apoptotic activities present in S/M extracts are not due solely to the presence of low levels of contaminating apoptotic cells since these cells would be expected to have high levels of active CAD.
CAD Alone Is Also Capable of Inducing Apoptotic Morphology in Added Nuclei
Collectively, the results presented in Figs. 3 and 5 suggest that CAD might be a major factor that acts downstream of caspases to induce changes in nuclear structure during apoptotic execution. To examine this possibility in greater detail, we exposed isolated nuclei to partly purified cloned murine CAD. Active CAD was expressed in E. coli using a bicistronic vector, which ensured that the CAD was translated in the presence of an excess of ICAD (Enari et al., 1998). The resulting CAD/ICAD complex was purified by nickel chelate chromatography and then tested for activity against plasmid and nuclear substrates according to the experimental protocol shown in Fig. 6 A. Results obtained with the plasmid substrate are shown in Fig. 6 B. The CAD/ICAD complex purified from E. coli became activated after a preincubation of 30 min at 25°C either in the absence (Fig. 6 B, lane 4) or presence (lane 6) of purified recombinant caspase-3. We assume that activation in the absence of added caspase is due to the presence of contaminating protease activity from the E. coli lysate. Addition of double mutant ICAD (Sakahira et al., 1998) at the start of the preincubation blocked CAD activation in both the absence (lane 5) and presence (lane 7) of caspase-3. Double mutant ICAD also blocked CAD activity if added at the end of the preincubation (lane 8). In contrast, DEVD-fmk blocked CAD activation if added at the start of the preincubation (lane 9) but had no effect if added after cleavage of ICAD (lane 10).
The results obtained with the plasmid substrate were exactly duplicated when we examined the ability of bacterially expressed CAD to induce DNA ladders and morphological changes in HeLa nuclei (Fig. 6, B and C). These morphological changes were confirmed by electron microscopy, where condensed chromatin domains could be seen to be closely apposed to the nuclear envelope (Fig. 7). When CAD was fully active, as detected using either plasmid or nuclear substrates, the enzyme strongly induced condensation of chromatin at the periphery of added nuclei (Fig. 6, B and C, lanes and panels 4 and 6). Higher levels of CAD induced the complete disassembly of nuclei into apoptotic bodies (data not shown); however, under these conditions nucleosomal ladders were no longer seen (the DNA was fully degraded). Interestingly, the addition of DEVD-fmk at time t = 0 slightly inhibited the morphological apoptosis (Fig. 6 C, panel 10), although examples of fully apoptotic nuclei could still be seen. It is important to note that caspase-3 alone was unable to induce either DNA cleavage or apoptotic morphology in added nuclei (lane and panel 3), consistent with the results obtained with fraction 2 described above (Fig. 4 F).
Figure 7.
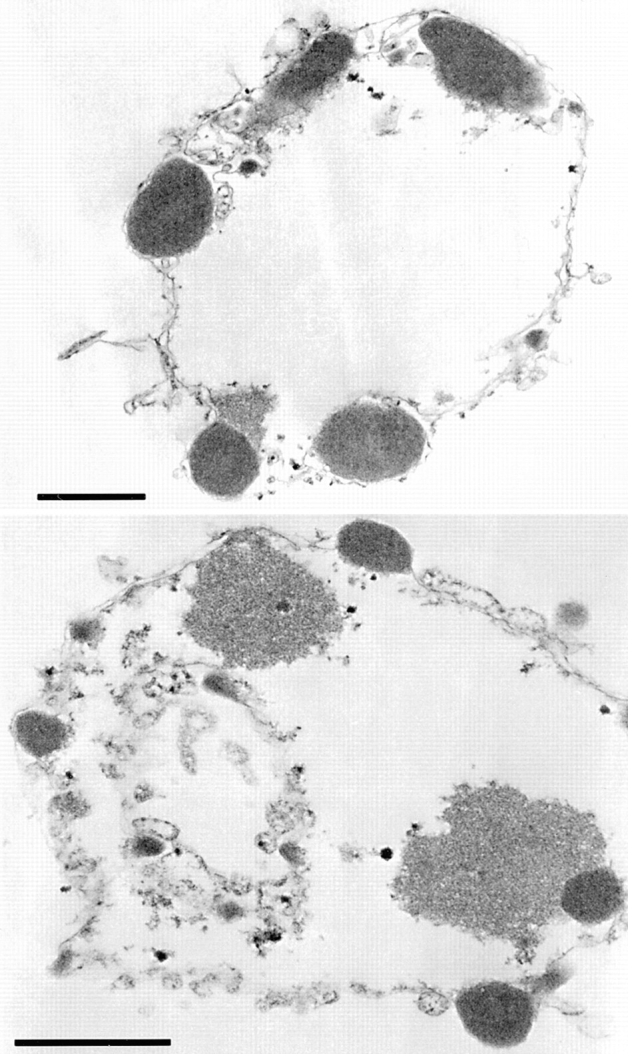
Induction of apoptotic morphology in HeLa nuclei by bacterially expressed CAD: analysis by electron microscopy. Nuclei treated with CAD—as shown in Fig. 6 B, lane and panel 6— were embedded in plastic, thin sectioned, and examined in the electron microscope. Regions of condensed chromatin are seen to abut the nuclear envelope, and often protrude as though beginning to bud outwards through the envelope. These images are indistinguishable from previously published images of nuclei treated with complete apoptotic extract (Lazebnik et al., 1993). Bar, 1 μm.
Evidence for a Nuclear Disassembly Factor Distinct from CAD
To determine whether the CAD-like enzyme was the sole activity downstream of caspases that drives nuclear disassembly in these extracts, S/M and E/X extracts were treated with ICAD before addition of exogenous nuclei. Although addition of excess ICAD abolished production of a nucleosomal ladder in nuclei added to S/M and E/X extracts (Fig. 8 A, lanes 3–5 and 9–11), ICAD did not block the ability of the extracts to induce chromatin condensation and nuclear fragmentation (Fig. 8 B, panels 3–5 and 9–11). The ability of the extracts to induce morphological changes in the presence of ICAD was even more striking when the number of nuclei in the incubation was reduced approximately fourfold (Fig. 8 C). These observations indicate that CAD-like activity is not essential for apoptotic morphological changes in nuclei added to either S/M or E/X extracts.
Figure 8.
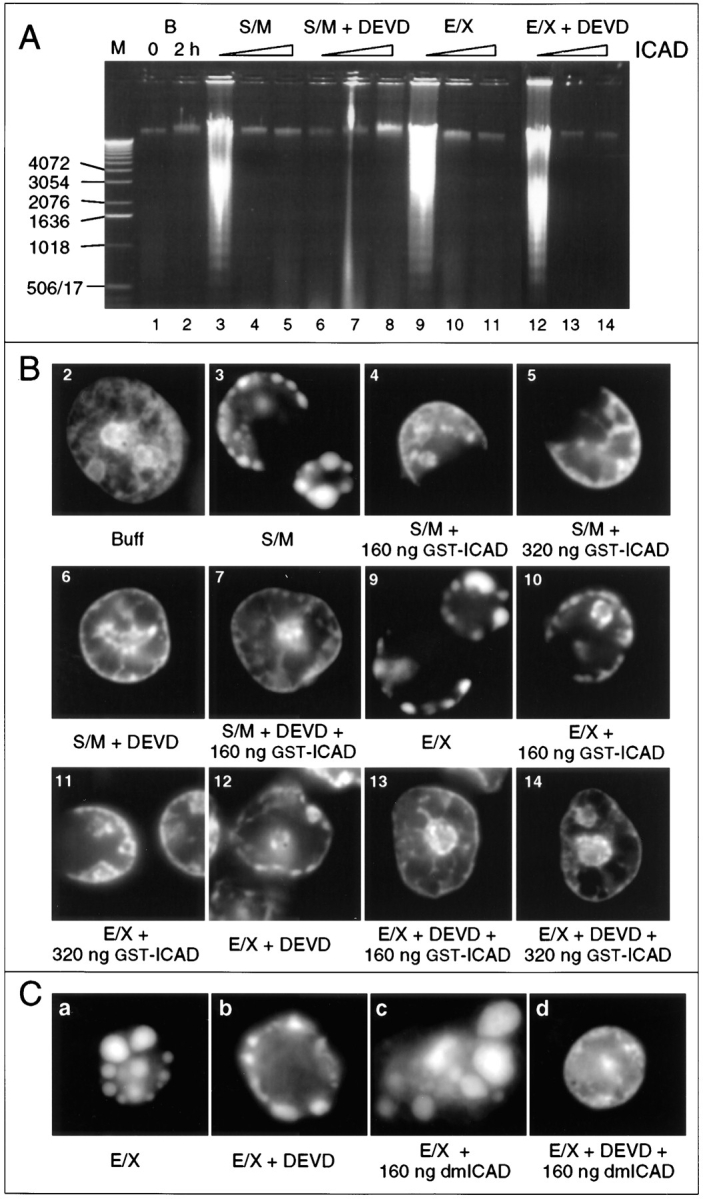
Induction of apoptotic morphology in cell extracts is not blocked by inhibition of the CAD-like nuclease. (A) Induction of nucleosomal ladders in added nuclei by S/M extract is sensitive to ICAD and DEVD-fmk (lanes 3–8). Induction of nucleosomal ladders in added nuclei by E/X extract is sensitive only to ICAD (lanes 9–14). (DEVD-fmk added: none, lanes 1–5 and 9–11; 100 μM, lanes 6–8 and 12–14. ICAD added: none, lanes 3, 6, 9, and 12; 160 ng, lanes 4, 7, 10, and 13; 320 ng, lanes 5, 8, 11, and 14.) (B) Both S/M and E/X extracts can induce apoptotic events under conditions where the CAD-like nuclease is inhibited, provided that they retain active caspases (panels 3–5 and 9–11). E/X extracts, which normally induce apoptosis in the absence of caspase activity (panel 12), are unable to do so if ICAD is added along with DEVD-fmk (panels 13 and 14). Panel numbers refer to the gel lanes in A. These images come from the same incubations shown in A. Time-lapse microscopy analysis reveals that the C-shaped nuclei in panels 3–5 and 9–11 are apoptotic nuclei in which the nuclear envelope has ruptured after collapse of the chromatin against the nuclear periphery. (C) E/X extracts can fully induce apoptotic morphology in the absence of CAD-like activity, but this requires ongoing caspase activity. In a different experiment from that shown in A and B, the number of nuclei per microliter of extract was reduced. This gives a stronger induction of apoptotic morphology (compare a with B, panel 9), even when caspases (b) or CAD are inhibited (c). Extracts produce a range of morphologies in added nuclei. As in the experiment of A and B, simultaneous inhibition of both CAD and caspases abolishes morphological apoptosis in the extracts (d). These images were selected from images taken at random.
Interestingly, simultaneous inhibition of both CAD and caspases did block the induction of apoptotic morphology by E/X extracts (Fig. 8 B, panels 13 and 14; Fig. 8 C, d). As indicated above, the caspase-rich column fraction Fr-2 lacked the ability to induce apoptotic morphological changes in exogenous nuclei (Fig. 4 F). Moreover, addition of purified caspases-3 and -6, the major active caspases in cytosol (Faleiro et al., 1997; Martins et al., 1997a ) and nuclei (Martins et al., 1997a ,b), failed to induce apoptotic morphological changes in purified nuclei (data not shown; identical to Fig. 6 C, panel 5). These observations appear to rule out the possibility that caspases themselves are capable of inducing chromatin condensation by acting alone on endogenous nuclear substrates. When coupled with these results, the observation in Fig. 8 C, d, not only suggests that there is a second chromatin condensation factor present in the extracts but also raises the possibility that ongoing caspase activity is required for activity of this factor.
Discussion
The experiments described above have led to a number of novel observations: (a) It is possible to prepare cell-free extracts specific for the latent and execution phases of apoptosis. (b) These extracts, both of which contain active caspases and induce apoptosis in substrate nuclei, exhibit significant biochemical and functional differences that are best explained if the transition from the latent to the execution phase of apoptosis is accompanied by a transition from caspase-dependent to caspase-independent mechanisms. (c) One aspect of this transition appears to involve activation of a CAD-like nuclease, which is initially latent in S/M extracts but is fully active in E/X extracts. (d) Murine CAD can induce an apoptotic morphology in isolated nuclei. (e) In addition to CAD, apoptotic extracts contain a second activity that can also induce apoptotic chromatin condensation in added nuclei in the absence of DNA fragmentation. Each of these points is discussed in greater detail below.
At present, there is no accepted biochemical marker to rigorously distinguish between the condemned and committed stages of the latent phase. Here, we define latent phase cells as those that have received a proapoptotic stimulus but look morphologically normal. We then operationally define condemned cells as those that yield extracts that lack significant caspase activity and do not induce apoptosis in substrate nuclei. We operationally define committed stage cells as those that yield extracts that contain high levels of active caspases together with low levels of active CAD and that strongly induce apoptosis in substrate nuclei. As expected, these cells contain only low levels of fragmented DNA themselves. Both of these latent phase populations are readily distinguishable from overtly apoptotic cells, which have detached from the substratum and have sustained internucleosomal DNA degradation in response to a proapoptotic stimulus. The various extracts prepared from these three operationally defined cell populations turned out to have remarkably distinct biochemical and functional characteristics.
The most significant difference was the finding that induction of apoptotic events by committed stage extracts is dependent on ongoing caspase activity, whereas induction of apoptotic events by execution phase extracts is independent of ongoing caspase activity. This observation is best explained by a model in which caspases activate downstream factors that are responsible for condensation of the chromatin during apoptosis. Such a model predicts that if we were able to purify the active caspases from apoptotic cells away from the downstream factors, then the caspases on their own should be insufficient to induce apoptotic events in substrate nuclei. To test this hypothesis, so-called roller S/M extracts (large scale extracts functionally intermediate between S/M and E/X extracts) were fractionated on heparin–agarose. The fraction eluting with 0.2 M KCl (Fr-2) appeared to not only encompass the entire range of endogenous caspases activated during apoptosis in these cells but was also highly active in the cleavage of caspase target proteins. Nonetheless, this fraction was completely unable to induce either DNA fragmentation or apoptotic morphology in substrate nuclei. We have independently shown that mixtures of purified caspases-3 and -6 are also unable to induce apoptotic morphology in purified nuclei (unpublished results). Thus, although recent experiments indicate that caspase-3 is essential for nuclear apoptosis (Jänicke et al., 1998; Woo et al., 1998), our experiments indicate that this enzyme is likely to function by activating downstream factors rather than directly disassembling the nucleus itself.
A subsequent 0.6 M KCl wash of the same heparin–agarose column yielded a fraction (Fr-3) that is rich in downstream apoptosis-promoting activities. Fr-3 strongly induces DNA fragmentation and apoptotic morphology despite the fact that it contains only very low levels of caspases, similar to those seen in C/D extracts (which do not induce apoptotic events in substrate nuclei). Furthermore, treatment of Fr-3 with caspase inhibitors (as well as other protease inhibitors; see Materials and Methods) had no effect on its apoptosis-promoting activity. Thus, this fraction is a potential source of downstream factors activated during the apoptotic pathway.
One such downstream factor has recently been identified in murine cell extracts. CAD, the caspase-activated DNase (Enari et al., 1998), interacts with an inhibitory subunit termed ICAD/DFF45 (Sakahira et al., 1998; Liu et al., 1997) to form an inactive complex that is thought to be sequestered in the cytoplasm. Caspase cleavage of ICAD is proposed to cause the release and subsequent nuclear translocation of CAD, leading to digestion of the chromatin (Sakahira et al., 1998). Here, we have shown that both S/M and E/X extracts from chicken DU249 cells contain a nuclease similar or identical to CAD, as defined by its ability to be quantitatively inhibited by exogenous purified mouse ICAD. The fact that ICAD of mouse origin inhibits the apoptotic nuclease in avian (chicken) cell extracts suggests that the CAD nuclease and its interactions with ICAD are widely conserved in phylogeny.
Because the same cohort of caspases is present and active in the S/M and E/X extracts, the transition from the latent to the execution phase of apoptosis may not hinge upon caspase action alone. Instead, the transition might involve activation of downstream factors. The CAD-like nuclease appears to be largely inactive in freshly prepared S/M extracts, becoming active only as the extracts are incubated at 30 or 37°C. This activation presumably reflects the cleavage of ICAD/DFF45 by caspases since CAD activation is blocked by caspase inhibitors. In contrast, the CAD-like nuclease activity appears to be fully active in freshly prepared E/X extracts. Collectively, these observations suggest that caspase activation occurs during the latent phase of apoptosis, whereas CAD activation accompanies the transition from the latent to execution phase of apoptosis. One implication of this conclusion is that cells might simultaneously contain active caspases and inactive complexes of ICAD/DFF bound to CAD and possibly other downstream factors. This would be possible if caspases were segregated from their downstream targets such as ICAD/DFF, possibly by being sequestered in a different cellular compartment. The onset of apoptotic execution might then be triggered not by activation of the caspases but by a change in the localization of either caspases or targets. An alternative explanation of our data, that caspases are inactive in the cells from which S/M extracts are made and become activated during preparation of the extract (Zapata et al., 1998), is unlikely for two reasons. First, we previously demonstrated that caspase activity in similar extracts prepared from latent phase HL-60 cells was paralleled by cleavage of PARP and procaspase-3 in vivo (Martins et al., 1997a ). Second, the caspase-3 activation observed during extract preparation in the previous study was explained by the detergent-induced release of granzyme B from cytoplasmic vesicles (Zapata et al., 1998). In our study, detergents were avoided during extract preparation, and cells lacking granzyme B were used.
In support of the notion that caspases and their targets might be segregated during the latent phase, we note that procaspases have largely been observed in the cytoplasm (Duan et al., 1996a ; Krajewska et al., 1997; Mancini et al., 1998) (Mesner, P.W., and S.H. Kaufmann, unpublished observations) or mitochondria (Mancini et al., 1998), whereas our subcellular fractionation studies have revealed significant levels of active caspases within the nucleus (Martins et al., 1997a ,b). An additional study has indicated that interference with nuclear transport renders cells resistant to Fas-mediated apoptosis (Yasuhara et al., 1997).
We have developed a novel vector to coordinately express CAD and ICAD in E. coli and have used this partially purified CAD to demonstrate that CAD can induce an apoptotic morphology in isolated nuclei, as confirmed by both light and electron microscopy. This reaction does not require ongoing caspase activity. This result is consistent with earlier observations, indicating that addition of micrococcal nuclease to isolated nuclei can cause clumping of chromatin (Arends et al., 1990), although the pattern of condensation seen with CAD more closely resembles that seen in apoptotic cells. However, CAD does not appear to be the only factor downstream of caspases that is capable of inducing apoptotic nuclear changes. Addition of purified ICAD to either S/M or E/X extracts abolished all detectable DNase activity against both chromosomal and plasmid DNA substrates but did not abolish the ability of those extracts to induce an apoptotic morphology in substrate nuclei. This observation is consistent with previous reports that morphologically normal apoptosis can occur in the absence of internucleosomal DNA fragmentation in vivo (Ucker et al., 1992b ; Oberhammer et al., 1993; Sakahira et al., 1998). At present, the nature of the second downstream factor(s) that promotes apoptotic chromatin condensation is unknown. Although two recent studies suggest that serine (Shimizu and Pommier, 1997) or cysteine proteases (Vaux et al., 1997) may be involved, the fact that we can detect apoptotic morphological changes in the presence of DEVD-fmk as well as a battery of serine, cysteine, and acid protease inhibitors (see Materials and Methods) suggest that the chromatin-condensing activity might not be a protease. On the other hand, simultaneous addition of both DEVD-fmk and ICAD to E/X extracts abolished the induction of apoptotic morphology in substrate nuclei. This result suggests that the morphogenic factor either binds to ICAD or is regulated by other molecules that interact with ICAD.
In summary, the experiments reported here demonstrate that it is possible to use cell-free extracts for a biochemical and functional dissection of the sequential phases of the apoptotic pathway. Our studies reveal that the transition from the latent to the execution phase of apoptosis appears to be accompanied by a change from caspase-dependent to caspase-independent mechanisms. The discovery that caspases are capable of cleaving abundant nuclear proteins like PARP (Lazebnik et al., 1994) and the lamins (Lazebnik et al., 1995; Takahashi et al., 1996; Orth et al., 1996; Rao et al., 1996), along with the observation that apoptotic nuclear fragmentation is absent in caspase-3–null cells (Woo et al., 1998; Janicke et al., 1998), led us and others to previously assume that caspases are directly responsible for nuclear disassembly. However, the inability of purified caspases to produce apoptotic morphological changes in isolated nuclei, coupled with the inability of caspase inhibitors to abolish the induction of these morphological changes by E/X extracts, argues that caspases are unlikely to be workhorses that are solely responsible for nuclear disassembly. Instead, the present results provide support for a view in which caspases play an executive role in nuclear disassembly by activating factors that in turn disassemble the nucleus. At least one of the downstream activities is a CAD-like nuclease; but another downstream activity capable of inducing nuclear morphological changes appears to be distinct from CAD.
Acknowledgments
We would like to thank David Dryden and Peter Warburton for advice on using heparin agarose; Dr. Richard Hayward for advice on the design of the bicistronic vector; our colleagues Drs. David Boyle, Joe Lewis, David Leach, Chris French, and Brian Kilby for helpful discussions; Larry Gerace (The Scripps Research Institute, La Jolla, CA) for anti–lamin A antibody; and Drs. A. Ainsztein, J. Craig, and S. Wheatley for their comments on the manuscript.
This work was supported by grants from the Wellcome Trust (W.C. Earnshaw) and The National Institutes of Health (S.H. Kaufmann and W.C. Earnshaw). S.H. Kaufmann was a Scholar of the Leukemia Society of America. W.C. Earnshaw is a Principal Research Fellow of the Wellcome Trust.
Abbreviations used in this paper
- CAD
caspase-activated DNase
- C/D extract
nonapoptotic extract from cells in the condemned phase
- DAPI
4,6-diamidino-2-phenylindole
- DFF
DNA fragmentation factor
- E/X extract
apoptotic extract from execution phase cells
- Fr-2 and Fr-3
fractions 2 and 3 from heparin–agarose chromatography of S/M extract
- ICAD
inhibitor of CAD
- S/M extract
proapoptotic extract from committed phase cells
- PARP
poly(ADP-ribose) polymerase
Footnotes
K. Samejima and S. Toné contributed equally to this work.
Correspondence should be addressed to W.C. Earnshaw, Institute of Cell and Molecular Biology, University of Edinburgh, Kings' Buildings, Mayfield Road, Edinburgh, EH9 3JR, Scotland, UK. Tel.: 44-(0)131-650-7101. Fax: 44-(0)131-650-7100. E-mail: bill.earnshaw@ed.ac.uk
References
- Arends MJ, Morris RG, Wyllie AH. Apoptosis: the role of the endonuclease. Am J Pathol. 1990;136:593–608. [PMC free article] [PubMed] [Google Scholar]
- Boise LH, Thompson CB. Bcl-x(L) can inhibit apoptosis in cells that have undergone Fas-induced protease activation. Proc Natl Acad Sci USA. 1997;94:3759–3764. doi: 10.1073/pnas.94.8.3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brancolini C, Benedetti M, Schneider C. Microfilament reorganization during apoptosis: the role of Gas2, a possible substrate for ICE-like proteases. EMBO (Eur Mol Biol Organ) J. 1995;14:5179–5190. doi: 10.1002/j.1460-2075.1995.tb00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church GM, Gilbert W. Genomic sequencing. Proc Natl Acad Sci USA. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns V, Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- Duan H, Chinnaiyan AM, Hudson PL, Wing JP, He W-W, Dixit VM. ICE-LAP3, a novel mammalian homologue of the Caenorhabditis eleganscell death protein Ced-3 is activated during Fas- and tumor necrosis factor-induced apoptosis. J Biol Chem. 1996a;271:1621–1625. doi: 10.1074/jbc.271.3.1621. [DOI] [PubMed] [Google Scholar]
- Duan H, Orth K, Chinnaiyan AM, Poirier GG, Froelich CJ, He W-W, Dixit VM. ICE-LAP6, a novel member of the ICE/Ced-3 gene family, is activated by the cytotoxic T cell protease granzyme B. J Biol Chem. 1996b;271:16720–16724. doi: 10.1074/jbc.271.28.16720. [DOI] [PubMed] [Google Scholar]
- Enari M, Hase A, Nagata S. Apoptosis by a cytosolic extract from Fas-activated cells. EMBO (Eur Mol Biol Organ) J. 1995a;14:5201–5208. doi: 10.1002/j.1460-2075.1995.tb00204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enari M, Hug H, Nagata S. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature. 1995b;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Faleiro L, Kobayashi R, Fearnhead H, Lazebnik Y. Multiple species of CPP32 and Mch2 are the major active caspases present in apoptotic cells. EMBO (Eur Mol Biol Organ) J. 1997;16:2271–2281. doi: 10.1093/emboj/16.9.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Litwack G, Alnemri E. Mch2, a new member of the apoptotic Ced-3/ICE cysteine protease gene family. Cancer Res. 1995a;55:2737–2742. [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Takahashi A, Armstrong R, Krebs J, Fritz L, Tomaselli K, Wang L, Yu Z, Croce CM, Salvesen G, et al. b. Mch3, a novel human apoptotic cysteine protease highly related to CPP32. Cancer Res. 1995;55:6045–6052. [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Armstrong RC, Krebs J, Srinivasula SM, Wang L, Bullrich F, Fritz LC, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc Natl Acad Sci USA. 1996;93:14486–14491. doi: 10.1073/pnas.93.15.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halenbeck R, MacDonald H, Roulston A, Chen TT, Conroy L, Williams LT. CPAN, a human nuclease regulated by the caspase-sensitive inhibitor DFF-45. Curr Biol. 1998;8:537–540. doi: 10.1016/s0960-9822(98)79298-x. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Burne JF, Raff MC. Programmed cell death and Bcl-2 protection in the absence of a nucleus. EMBO (Eur Mol Biol Organ) J. 1994;13:1899–1910. doi: 10.1002/j.1460-2075.1994.tb06459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jänicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH. Induction of endonucleolytic DNA cleavage in human acute myelogenous leukemia cells by etoposide, camptothecin, and other cytotoxic anticancer drugs: a cautionary note. Cancer Res. 1989;49:5870–5878. [PubMed] [Google Scholar]
- Krajewska M, Wang HG, Krajewski S, Zapata JM, Shabaik A, Gascoyne R, Reed JC. Immunohistochemical analysis of in vivo patterns of expression of CPP32 (Caspase-3), a cell death protease. Cancer Res. 1997;57:1605–1613. [PubMed] [Google Scholar]
- Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lamarre D, Talbot B, de Murcia G, Laplante C, Leduc Y, Mazen A, Poirier GG. Structural and functional analysis of poly(ADP-ribose) polymerase: an immunological study. Biochim Biophys Acta. 1988;950:147–160. doi: 10.1016/0167-4781(88)90007-3. [DOI] [PubMed] [Google Scholar]
- Lavoie JN, Nguyen M, Marcellus RC, Branton PE, Shore GC. E4orf4, a novel adenovirus death factor that induces p53-independent apoptosis by a pathway that is not inhibited by zVAD-fmk. J Cell Biol. 1998;140:637–645. doi: 10.1083/jcb.140.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazebnik YA, Cole S, Cooke CA, Nelson WG, Earnshaw WC. Nuclear events of apoptosis in vitro in cell-free mitotic extracts: a model system for analysis of the active phase of apoptosis. J Cell Biol. 1993;123:7–22. doi: 10.1083/jcb.123.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a protease with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Lazebnik YA, Takahashi A, Moir R, Goldman R, Poirier GG, Kaufmann SH, Earnshaw WC. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Nat Acad Sci USA. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome C. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Mancini M, Nicholson DW, Roy S, Thornberry NA, Peterson EP, Casciola-Rosen LA, Rosen A. The caspase-3 precursor has a cytosolic and mitochondrial distribution: implications for apoptotic signaling. J Cell Biol. 1998;140:1485–1495. doi: 10.1083/jcb.140.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? . Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Newmeyer DD, Mathias S, Farschon DM, Wang H-G, Reed JC, Kolesnick RN, Green DR. Cell-free reconstitution of Fas-, UV radiation-, and ceramide-induced apoptosis. EMBO (Eur Mol Biol Organ) J. 1995;14:5191–5200. doi: 10.1002/j.1460-2075.1995.tb00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Finucane DM, Amarante-Mendes GP, O'Brien GA, Green DR. Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. J Biol Chem. 1996;271:28753–28756. doi: 10.1074/jbc.271.46.28753. [DOI] [PubMed] [Google Scholar]
- Martins LM, Kottke T, Mesner PW, Basi GS, Sinha S, Frigon NJ, Tatar E, Tung JS, Bryant K, Takahashi A, et al. Activation of multiple Interleukin-1β converting enzyme homologues in cytosol and nuclei of HL-60 human leukemia cells during etoposide-induced apoptosis. J Biol Chem. 1997a;272:7421–7430. doi: 10.1074/jbc.272.11.7421. [DOI] [PubMed] [Google Scholar]
- Martins LM, Mesner PW, Kottke TJ, Basi GS, Sinha S, Tung JS, Svingen PA, Madden BJ, Takahashi A, McCormick DJ, et al. Comparison of caspase activation and subcellular localization in HL-60 and K562 cells undergoing etoposide-induced apoptosis. Blood. 1997b;90:4283–4296. [PubMed] [Google Scholar]
- McCarthy NJ, Whyte MK, Gilbert CS, Evan GI. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Golstein P, Henkart PA. The target cell nucleus is not required for cell-mediated granzyme- or Fas-based cytotoxicity. J Exp Med. 1995;181:1905–1909. doi: 10.1084/jem.181.5.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmeyer DD, Farschon DM, Reed JC. Cell-free apoptosis in Xenopusegg extracts—inhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell. 1994;79:353–364. doi: 10.1016/0092-8674(94)90203-8. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Oberhammer F, Wilson JW, Dive C, Morris ID, Hickman JA, Wakeling AE, Walker PR, Sikorska M. Apoptotic death in epithelial cells: cleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO (Eur Mol Biol Organ) J. 1993;12:3679–3684. doi: 10.1002/j.1460-2075.1993.tb06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth K, Chinnaiyan AM, Garg M, Froelich CJ, Dixit VM. The CED-3/ICE-like protease Mch2 is activated during apoptosis and cleaves the death substrate lamin A. J Biol Chem. 1996;271:16443–16446. [PubMed] [Google Scholar]
- Porter AG, Ng P, Jänicke RU. Death substrates come alive. Bioessays. 1997;19:501–507. doi: 10.1002/bies.950190609. [DOI] [PubMed] [Google Scholar]
- Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during apoptosis. J Cell Biol. 1996;135:1441–1455. doi: 10.1083/jcb.135.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Samejima K, Earnshaw WC. ICAD/DFF regulator of apoptotic nuclease is nuclear. Exp Cell Res. 1998;243:453–459. doi: 10.1006/excr.1998.4212. [DOI] [PubMed] [Google Scholar]
- Schlegel J, Peters I, Orrenius S. Isolation and partial characterization of a protease involved in Fas-induced apoptosis. FEBS Lett. 1995;364:139–142. doi: 10.1016/0014-5793(95)00374-i. [DOI] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Walczak H, Dröge W, Krammer PH. Cell nucleus and DNA fragmentation are not required for apoptosis. J Cell Biol. 1994;127:15–20. doi: 10.1083/jcb.127.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seraphin B, Kandels-Lewis S. An efficient PCR mutagenesis strategy without gel purification step that is amenable to automation. Nucleic Acids Res. 1996;24:3276–3277. doi: 10.1093/nar/24.16.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Pommier Y. Camptothecin-induced apoptosis in p53-null human leukemia HL60 cells and their isolated nuclei: effects of the protease inhibitors Z-VAD-fmk and dichloroisocoumarin suggest an involvement of both caspases and serine proteases. Leukemia. 1997;11:1238–1244. doi: 10.1038/sj.leu.2400734. [DOI] [PubMed] [Google Scholar]
- Solary E, Bertrand R, Kohn KW, Pommier Y. Differential induction of apoptosis in undifferentiated and differentiated HL-60 cells by DNA topoisomerase I and II inhibitors. Blood. 1993;81:1359–1368. [PubMed] [Google Scholar]
- Srinivasula SM, Fernandes-Alnemri T, Zangrill J, Robertson N, Armstrong RC, Wang L, Trapani JA, Tomaselli K, Litwack G, Alnemri ES. The Ced3/interleukin 1β converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2α are substrates for the apoptotic mediator CPP32. J Biol Chem. 1996;271:27099–27106. doi: 10.1074/jbc.271.43.27099. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Alnemri E, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Cleavage of lamin A by Mch2a but not CPP32: multiple ICE-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci USA. 1996;93:8395–8400. doi: 10.1073/pnas.93.16.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Ucker DS, Meyers J, Obermiller PS. Activation driven T cell death. II. Quantitative differences alone distinguish stimuli triggering nontransformed T cell proliferation or death. J Immunol. 1992a;149:1583–1592. [PubMed] [Google Scholar]
- Ucker DS, Obermiller PS, Eckhart W, Apgar JR, Berger NA, Meyers J. Genome digestion is a dispensable consequence of physiological cell death mediated by cytotoxic T lymphocytes. Mol Cell Biol. 1992b;12:3060–3069. doi: 10.1128/mcb.12.7.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Wilhelm S, Hacker G. Requirements for proteolysis during apoptosis. Mol Cell Biol. 1997;17:6502–6507. doi: 10.1128/mcb.17.11.6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa P, Kaufmann SH, Earnshaw WC. Caspases and caspase inhibitors. Trends Biochem Sci. 1997;22:388–393. doi: 10.1016/s0968-0004(97)01107-9. [DOI] [PubMed] [Google Scholar]
- Woo M, Haken R, Soengas MS, Duncan GS, Shahinian A, Kägi D, Hakem A, McCurrach M, Khoo W, Kaufman SA, et al. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1998;12:806–819. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood ER, Earnshaw WC. Mitotic chromatin condensation in vitro using somatic cell extracts and nuclei with variable levels of endogenous topoisomerase II. J Cell Biol. 1990;111:2839–2850. doi: 10.1083/jcb.111.6.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–305. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Xiang J, Chao DT, Korsmeyer SJ. BAX-induced cell death may not require interleukin 1 β-converting enzyme-like proteases. Proc Natl Acad Sci USA. 1996;93:14559–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara N, Eguchi Y, Tachibana T, Imamoto N, Yoneda Y, Tsujimoto Y. Essential role of active nuclear transport in apoptosis. Genes Cells. 1997;2:55–64. doi: 10.1046/j.1365-2443.1997.1010302.x. [DOI] [PubMed] [Google Scholar]
- Yuan JY, Horvitz HR. The Caenorhabditis elegansgenes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol. 1990;138:33–41. doi: 10.1016/0012-1606(90)90174-h. [DOI] [PubMed] [Google Scholar]
- Zapata JM, Takahashi R, Salvesen GS, Reed JC. Granzyme release and caspase activation in activated human T-lymphocytes. J Biol Chem. 1998;273:6916–6920. doi: 10.1074/jbc.273.12.6916. [DOI] [PubMed] [Google Scholar]