

Abstract
The spindle checkpoint prevents the metaphase to anaphase transition in cells containing defects in the mitotic spindle or in chromosome attachment to the spindle. When the checkpoint protein Xmad2 is depleted from Xenopus egg extracts, adding Xmad2 to its endogenous concentration fails to restore the checkpoint, suggesting that other checkpoint component(s) were depleted from the extract through their association with Xmad2. Mass spectrometry provided peptide sequences from an 85-kD protein that coimmunoprecipitates with Xmad2 from egg extracts. This information was used to clone XMAD1, which encodes a homologue of the budding yeast (Saccharomyces cerevisiae) checkpoint protein Mad1. Xmad1 is essential for establishing and maintaining the spindle checkpoint in egg extracts. Like Xmad2, Xmad1 localizes to the nuclear envelope and the nucleus during interphase, and to those kinetochores that are not bound to spindle microtubules during mitosis. Adding an anti-Xmad1 antibody to egg extracts inactivates the checkpoint and prevents Xmad2 from localizing to unbound kinetochores. In the presence of excess Xmad2, neither chromosomes nor Xmad1 are required to activate the spindle checkpoint, suggesting that the physiological role of Xmad1 is to recruit Xmad2 to kinetochores that have not bound microtubules.
Keywords: mitosis, checkpoint, kinetochore, Xenopus, Xmad1
Accurate sister chromatid segregation during anaphase is pivotal for the faithful transmission of genetic information during each cell division. Mistakes in this process result in missegregation of chromosomes, leading to aneuploidy that can cause birth defects and contribute to tumor formation and progression. The spindle checkpoint keeps cells from entering anaphase until all of the chromosomes are attached to the spindle through their kinetochores (Rieder et al., 1995; for review see Hardwick, 1998). Lesions in this checkpoint have recently been found in colorectal tumors, suggesting that they play an important role in tumor progression (Cahill et al., 1998).
Components of the spindle checkpoint were first identified in the budding yeast Saccharomyces cerevisiae. When treated with microtubule-depolymerizing drugs, budding yeast cells arrest in mitosis with unseparated sister chromatids. Mutations in the MAD (mitotic arrest-deficient)1 (Li and Murray, 1991) and BUB (budding uninhibited by benzimidazole) (Hoyt et al., 1991) genes abolish this cell cycle arrest and allow cells to enter anaphase in the absence of a functional spindle. This premature entry into anaphase leads to rapid cell death (Hoyt et al., 1991; Li and Murray, 1991). Although MAD and BUB genes are not essential for cell viability, mutations in these genes increase the chromosome loss rate, suggesting that they regulate the metaphase to anaphase transition during normal cell cycles (Hoyt et al., 1991; Li and Murray, 1991). All of the corresponding MAD and BUB genes have been isolated. Mad1 is a nuclear protein whose phosphorylation increases greatly upon spindle depolymerization and rises transiently during normal mitosis (Hardwick and Murray, 1995). Phosphorylation of Mad1 appears to be important for its checkpoint function, because all three checkpoint-defective mad1 alleles fail to be phosphorylated during mitosis and phosphorylation of Mad1 requires the function of Mad2, Bub1, and Bub3 proteins (Hardwick and Murray, 1995). However, hyperphosphorylation of Mad1 is not absolutely required for mitotic arrest, since overexpression of a dominant BUB1-5 mutant leads to mitotic arrest without Mad1 hyperphosphorylation (Farr and Hoyt, 1998). Genetic and biochemical evidence suggests that Mad1 is phosphorylated by Mps1, whose function is also required for the checkpoint (Hardwick et al., 1996; Weiss and Winey, 1996). MAD2 is a novel gene and its gene product forms a tight complex with Mad1 (Chen, R.-H., K.G. Hardwick, and A.W. Murray, unpublished results). Both MAD3 (Hardwick, K.G., and A.W. Murray, unpublished results) and BUB2 (Hoyt et al., 1991) encode novel proteins of unknown function. BUB1 encodes a serine/ threonine protein kinase that associates with Bub3 (Hoyt et al., 1991; Roberts et al., 1994). Homologues of Bub2 (Murone and Simanis, 1996) and Mad2 (He et al., 1997; Kim et al., 1998) have been identified in the fission yeast Schizosaccharomyces pombe and shown to be important for checkpoint function.
Homologues of spindle checkpoint components have also been identified in vertebrates. Mad2 homologues in the frog Xenopus laevis (Xmad2) (Chen et al., 1996) and humans (Hmad2) (Li and Benezra, 1996) are essential for checkpoint function in frog egg extracts (Chen et al., 1996) and in cultured human cells (Li and Benezra, 1996). The protein also controls the metaphase to anaphase transition during normal cell division (Gorbsky et al., 1998). At mitosis, a fraction of vertebrate Mad2 associates with kinetochores that are not bound to the mitotic spindle, but not with kinetochores that are associated with spindle microtubules (Chen et al., 1996; Li and Benezra, 1996; Gorbsky et al., 1998; Waters et al., 1998). In meiotic cells, lack of tension on kinetochores that bind to only one pole of the mitotic spindle plays a role in inhibiting anaphase onset (Li and Nicklas, 1995). Kinetochore tension does not appear to regulate Mad2 binding, since the protein can be found on only very few kinetochores in taxol-treated cells, all of whose kinetochores lack tension (Waters et al., 1998). The localization of the vertebrate Mad2 homologues suggests that they play a role in activating the checkpoint in response to kinetochores that have not associated with microtubules. A mouse homologue of Bub1 also associates with unattached kinetochores (Taylor and McKeon, 1997) in a Bub3-dependent manner (Taylor et al., 1998). Human homologues of Bub1 have been identified and mutations in these genes are found in colorectal tumors that show high levels of chromosome missegregation (Cahill et al., 1998), suggesting that lesions in the spindle checkpoint contribute to tumor progression. The conservation of the spindle checkpoint proteins in eukaryotes indicates that this checkpoint plays an important role in cell division and that its mechanism has been conserved throughout evolution.
Anaphase is triggered by ubiquitin-mediated proteolysis of mitotic cyclins (cyclin B) and proteins that regulate sister chromatid separation, such as Pds1 in budding yeast (Cohen-Fix et al., 1996) and Cut2 in fission yeast (Funabiki et al., 1996). The degradation of mitotic cyclins leads to the inactivation of the mitotic form of Cdc2, while the destruction of Pds1/Cut2 leads to sister chromatid separation. The destruction of these substrates requires the anaphase-promoting complex (APC), a large ubiquitin ligase complex (Irniger et al., 1995; King et al., 1995; Sudakin et al., 1995; Cohen-Fix et al., 1996). The target that the spindle checkpoint inhibits appears to be Cdc20, a protein required for exit from mitosis (Dawson et al., 1995; Schwab et al., 1997; Visintin et al., 1997), which associates with and activates the APC (Fang et al., 1998; Kallio et al., 1998). In fission and budding yeast, Cdc20 binds to Mad2, and Cdc20 mutants that disrupt this binding act as dominant checkpoint-defective mutations (Hwang et al., 1998; Kim et al., 1998).
We have studied the spindle checkpoint in frog egg extracts, which provide a synchronous cell cycle and an excellent source for the biochemical dissection and reconstitution of various cellular processes (Murray, 1991). Using this system, we have previously demonstrated that Xmad2 is important for spindle checkpoint function (Chen et al., 1996). We report the isolation of Xmad1, the frog homologue of Mad1, as a protein that binds tightly and specifically to Xmad2. Like Xmad2, Xmad1 is essential for establishing and maintaining the checkpoint, and is localized to unattached kinetochores during mitosis. The major role of Xmad1 appears to be to target Xmad2 to kinetochores that lack attached microtubules.
Materials and Methods
Preparation of Egg Extracts and Activation of the Spindle Checkpoint in Egg Extracts
Demembranated frog sperm nuclei, as well as cytostatic factor (CSF)- arrested and cycling egg extracts were prepared as described (Murray, 1991), except that dejellied eggs were activated by incubation with the calcium ionophore A23187 at 0.5 μg/ml for 4–5 min at room temperature. After activation, eggs were incubated at room temperature for another 15–20 min to ensure complete inactivation of CSF activity. Activation of the spindle checkpoint in CSF-arrested extracts was performed as described previously (Minshull et al., 1994; Chen et al., 1996). To examine the effect of recombinant 6H-Xmad2 (Chen et al., 1996), extracts were incubated on ice for 1 h with the recombinant protein at concentrations indicated in the figures before the addition of any nuclei or nocodazole.
Cell Cultures and Cell Lysates
The maintenance of the Xenopus fibroblast cell line XTC and human HeLa cells as well as the preparation the cell lysates were performed as described (Chen et al., 1996).
Immunoblot Analysis
Proteins were transferred to nitrocellulose membrane after SDS-PAGE. The membrane was first preblocked with PBS (2.7 mM potassium chloride, 137 mM sodium chloride, 1.5 mM potassium phosphate, 4.3 mM sodium phosphate, pH 7.2) containing 2% BSA and 0.2% Tween-20, and then probed with affinity-purified anti-Xmad1 (1 μg/ml) or with anti-Xmad2 (0.5 μg/ml) antibodies in the same buffer for 1 h at room temperature. The blot was washed three times with PBST (PBS + 0.2% Tween-20), incubated with peroxidase-conjugated anti–rabbit antibody (Amersham Corp., Arlington Heights, IL) for 30 min in PBST, washed three times with PBST, and then developed by enhanced chemiluminescene detection (ECL; Amersham Corp.).
Purification of p85-Xmad1
Xmad1 was purified from Xmad2 immunoprecipitates. To immunoprecipitate Xmad2, Affi-Prep protein A support beads (Bio-Rad Laboratories, Hercules, CA) were washed twice with 1 ml of extract buffer (XB; 10 mM Hepes, pH 7.8, 50 mM sucrose, 100 mM potassium chloride, 10 mM magnesium chloride, 1 mM calcium chloride) containing 10 μg/ml each of leupeptin, pepstatin, and chymostatin (LPC). The beads were then incubated with 5 μg of affinity-purified anti-Xmad2 antibodies (Chen et al., 1996) or with control IgG (Harlow and Lane, 1988) at 4°C for 30 min. After washing the beads three times with 1 ml of XB plus LPC, 500 μl of CSF- arrested egg extracts were added to the antibody-coated beads and gently mixed on a mixer at 4°C for 1 h. The Xmad2 protein complex was isolated by centrifugation at 16,000 g for 1 min, and the immunoprecipitates were washed five times with 1 ml of XB plus LPC and 1% Triton X-100 or 0.1% SDS. Proteins bound to the antibodies were solubilized in SDS sample buffer, resolved by SDS-PAGE, and then visualized by Coomassie blue staining. The 85-kD protein that was specifically isolated in the Xmad2 immunoprecipitates was then subject to mass spectrometry analysis to determine its sequence.
Mass Spectrometry
The protein band was excised from the gel and in-gel digested with trypsin (unmodified, sequencing grade; Boehringer Mannheim Corp., Indianapolis, IN) as described (Shevchenko et al., 1996). Digestion buffer contained 50% (vol/vol) H2 18O. H2 18O was purchased from Cambridge Isotope Laboratories (Andover, MA) and was additionally purified by microdistillation using a sealed glass microdistillator constructed in house. The unseparated pool of tryptic peptides recovered from the gel matrix was sequenced on a experimental prototype of a hybrid quadrupole Time-Of-Flight tandem mass spectrometer developed by PE Sciex (Ontario, Canada) and University of Manitoba (Winnipeg, Canada) (Shevchenko et al., 1997). The mass spectrometer was equipped with a nanoelectrospray ion source developed in house (Wilm and Mann, 1996).
Isolation of XMAD1 cDNA
Eight different peptide sequences were obtained from mass spectrometry (see Fig. 5). Degenerate oligonucleotides were made corresponding to the sequences EDQAAQ, TPTEH, AQEQDAA, EDNTTV, NDSEDNT, and DAAI/LVK (mass spectrometry can not differentiate I from L). All oligonucleotides were made in both orientations, and used in PCR reactions in all possible pairwise combinations (10 pmol/μl each) using a Xenopus egg cDNA library (20 ng/μl, a gift of J. Minshull, formerly at University of California, San Francisco) as templates. The 550-bp PCR product generated from a reaction containing oligonucleotides for the peptides EDQAAQ and AQEQDAA was successfully cloned into the plasmid pBLUESCRIPT KS(−) (Stratagene, La Jolla, CA). The PCR product was sequenced to verify its authenticity, and was used to generate a probe by PCR using digoxigenin-labeled nucleotides (Boehringer Mannheim Corp.). The digoxigenin-labeled probe was then used to screen a Xenopus ovary library (a gift of J. Minshull). The phage plaques were lifted with nylon filters (Hybond-N; Amersham Corp.) as described (Sambrook et al., 1989). Phage DNA was denatured by autoclaving the filters for 1 min. Filters were washed briefly in 0.15 M sodium phosphate, pH 7.0, with 2% SDS, and then preblocked for 30 min at 65°C in hybridization buffer (0.5 M sodium phosphate, pH 7.0, 1% BSA, 7% SDS, 1 mM EDTA). Filters were then probed with the digoxigenin-labeled probe at 50 ng/ml in hybridization buffer at 65°C for 16 h. After hybridization, the filters were washed three times with 0.15 M sodium phosphate, 2% SDS at 65°C for 10 min each, three times with TTBS (25 mM Tris, pH 7.5, 0.5 M NaCl, 0.1% Tween 20) at room temperature for 5 min each, and once with TTBS containing 4% nonfat dry milk. The filters were then probed for 1 h with alkaline phosphatase-labeled anti-digoxigenin antibodies (Boehringer Mannheim Corp.) at 1:5,000 dilution in TTBS plus 4% milk. After washing the filters three times with TTBS for 10 min each and once with alkaline phosphatase color development buffer (100 mM sodium chloride, 5 mM magnesium chloride, 100 mM Tris-Cl, pH 9.5), the filters were then developed with 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) as described (Sambrook et al., 1989). The positive clones were picked and used for secondary screening with the same procedures. Three clones of 1, 1.4, and 1.6 kb were isolated and the largest one was sequenced using Sequenase Version 2.0 (United States Biochemical Corp., Cleveland, OH).
Figure 5.


p85 is Xmad1. (A) Alignment of amino acid sequences of Xenopus p85 (X. laevis), human (H. sapiens), budding yeast (S. cerevisiae), and fission yeast (S. pombe) Mad1. The peptide sequences obtained by mass spectrometry analysis of p85 are indicated as dots on top of the Xenopus sequence. The common residues shared by two or more sequences are shaded. The alignment was performed with DNASTAR “MegAlign” program. (B) Predicted coiled-coil regions in p85 (X.l.), human (H.s.), budding yeast (S.c.), and fission yeast (S.p.) Mad1. The scale of the amino acid sequence is indicated on top. The prediction was performed with DNASTAR “PROTEAN” program.
Purification of 6H-Xmad1 and Anti-Xmad1 Antibody
The full-length Xmad1 coding sequence was tagged with six histidines at the NH2-terminus, and expressed in Escherichia coli using the Qiaexpress vector pQE9 (QIAGEN Inc., Valencia, CA). Protein expression was induced with 0.1 mM isopropyl β-d-thiogalactopyranoside (IPTG) for 4 h at 37°C. Bacteria were collected by centrifugation, resuspended, and then sonicated in sonication buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 0.5% Triton X-100, 1 mg/ml lysozyme, 1 mM PMSF, 10 μg/ml LPC). The inclusion bodies containing Xmad1 were pelleted by centrifugation at 8.5 krpm for 30 min at 4°C. After extensive washing with high salt buffer (10 mM Tris, pH 7.2, 100 mM sodium phosphate, pH 7.2, 0.5 M NaCl), RIPA buffer (10 mM Tris, pH 7.2, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1 mM sodium orthovanadate), and water, the inclusion bodies were then solubilized in SDS sample buffer and resolved by SDS-PAGE. The gel slice containing Xmad1 band was diced, and the protein was eluted into a buffer of 10 mM Hepes, pH 7.2, and 1 mM DTT. The purified protein was used to raise antisera in rabbits and mice (Berkeley Antibody Co., Richmond, CA), and to affinity purify antibodies. Affinity purification of anti-Xmad1 antibodies was performed as described (Chen et al., 1992). Control IgG was purified from preimmune serum over a protein A column made from Affi-Prep Protein A (156-0006; Bio-Rad Laboratories) as described (Chen et al., 1992). Purified antibodies were dialyzed and concentrated in Antibody Dilution Buffer (10 mM sodium phosphate, pH 7.4, 100 mM potassium chloride, 1 mM magnesium chloride, 50% glycerol).
Immunofluorescent Staining of XTC Cells and of Chromosomes Assembled in Egg Extracts
Immunofluorescent staining of Xmad1 and Xmad2 in XTC cells was performed as described (Chen et al., 1996). Assembly of metaphase chromosomes in egg extracts was performed with an in vitro metaphase to anaphase transition assay as described (Chen and Murray, 1997; Shamu and Murray, 1992). In brief, 20 μl of CSF-arrested extracts were induced to enter interphase by adding calcium chloride, and sperm nuclei (1,500 per μl of extract) were allowed to replicate in these interphase extracts for 80–90 min at 23°C. One half volume (10 μl) of CSF-arrested extracts was then added to drive the extracts into mitosis, and the reactions were incubated for another 80-90 min to establish metaphase arrest. For experiments shown in Fig. 3, 6H-Xmad2 protein was supplemented at 0.1 μg/μl after the addition of CSF-arrested extracts. After metaphase arrest was achieved, calcium chloride was added to inactivate the CSF activity and to induce anaphase. To examine the nuclear and spindle morphology, 1 μl of each sample was removed and fixed in Fix solution (1.1% formaldehyde, 48% glycerol, 1 mg/ml Hoechst 33258 made in MMR [100 mM sodium chloride, 2 mM potassium chloride, 1 mM magnesium sulfate, 2 mM calcium chloride, 5 mM Hepes, 0.1 mM EDTA]). Specimens were viewed with a Nikon Microphot-FXA microscope equipped with a 60×, 1.4 NA oil immersion objective, and photographed with Kodak Tri-X pan 400 film.
Figure 3.

Excess Xmad2 inhibits sister chromatid segregation. Metaphase spindle and chromosomes were assembled in egg extracts treated with buffer (top, Control) or Xmad2 protein (100 ng/μl extract; bottom, Xmad2) as described in Materials and Methods. To initiate anaphase, calcium chloride was added and samples were taken immediately before (0′) or at the indicated times after calcium addition to visualize the nuclear (DNA) and spindle morphology (Spindle). The reduced microtubule intensity in the control experiment at 40 and 60 min reflects the reduced microtubule density typical of interphase extracts.
To study the effect of antibodies on Xmad1 and Xmad2 localization as shown in Fig. 9, anti-Xmad1 (30 ng/μl of extracts) or anti-Xmad2 (8 ng/μl of extracts) antibodies were added immediately after the addition of CSF-arrested extracts in the in vitro metaphase to anaphase assay. After metaphase arrest was established, the mitotic spindle was disrupted by adding nocodazole to 10 μg/ml. (To avoid inactivation of experimental extracts by excess DMSO, a 10 mg/ml stock of nocodazole made in DMSO was first diluted 50-fold in a separate aliquot of extract before being added to the samples.) After 10 min of incubation, reactions were diluted with 9 vol of XB plus 0.5% Triton X-100, and incubated at room temperature for another 10 min. Samples were then layered over 5 ml of 30% glycerol cushion made in XB plus 0.5% Triton X-100 with a coverslip placed at the bottom, and the chromosomes were collected onto the coverslip by centrifugation at 10 krpm in a HB-4 rotor (Sorvall, Newtown, CT) for 10 min at 4°C. Chromosomes were fixed in PBS containing 1% formaldehyde for 10 min. Immunofluorescent staining of Xmad1 and Xmad2 was performed as described for cultured cells (Chen et al., 1996). For Figs. 8 and 9, images were collected using a charge-coupled device (CCD) camera (KAF1400 chip, 5 MHz controller; Princeton Laboratories, Inc., Princeton, NJ) attached to a fluorescence microscope (model BX50; Olympus America, Lake Success, NY). Images were collected and processed with the Metamorph Imaging System (version 3.0; Universal Imaging Corporation, West Chester, PA) and converted to Photoshop format (Adobe Systems Inc., Mountain View, CA).
Figure 9.

Anti-Xmad1 antibodies interfere with Xmad1 and Xmad2 binding to kinetochores. Metaphase chromosomes were assembled in egg extracts and treated with (B–D) or without (A) nocodazole. C and D, anti-Xmad1 and anti-Xmad2 antibodies, respectively, were added to the extracts 1 h before the addition of nocodazole. The chromosomes were isolated through a sucrose cushion and stained for Xmad1 and Xmad2. For D, Xmad2 staining is essentially the same if the anti-Xmad2 antibody was omitted during the immunofluorescent staining procedures (data not shown). Aggregates of chromosomes from several sperm nuclei are shown.
Figure 8.
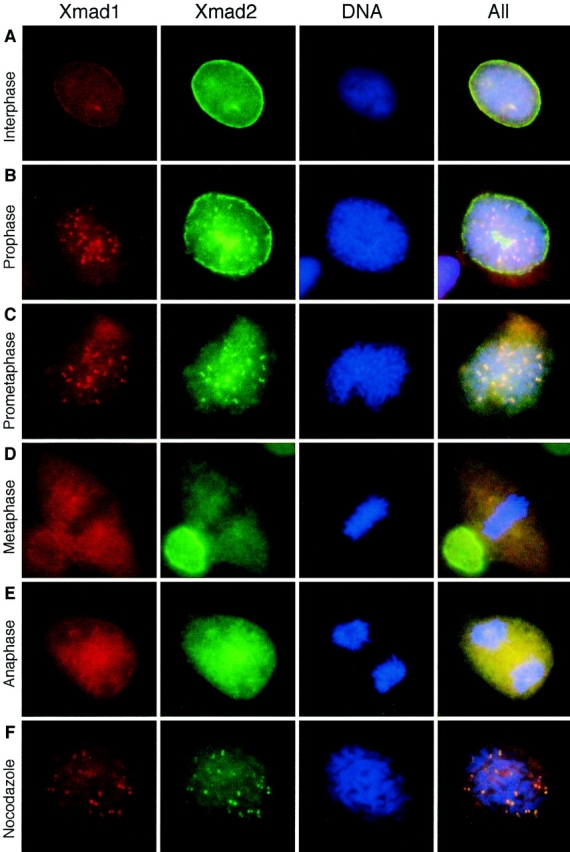
Xmad1 localized to unattached kinetochores during mitosis. Asynchronously growing XTC cells (A–E) or cells treated with nocodazole (F) were fixed in 3% paraformaldehyde and stained with affinity-purified mouse anti-Xmad1 and rabbit anti-Xmad2 antibodies as indicated on top. Texas red– conjugated anti–mouse and fluorescein-conjugated anti– rabbit IgG antibodies were used as secondary antibodies. The nuclei were viewed by staining with the DNA-binding dye Hoechst 33258 (DNA). The merges of all three fluorochromes are also shown (All). The cell cycle stages were determined by the morphology of the cell and its chromosomes. A, interphase; B, prophase; C, prometaphase; D, metaphase; E, anaphase; F, a nocodazole-arrested cell. Representative pictures of each cell type are shown.
Immunodepletion
For immunodepletion of Xmad1 or Xmad2, 15 μl of Affi-Prep protein A beads were washed twice in 1 ml of XB plus LPC. The beads were then mixed with affinity-purified anti-Xmad1 (12 μg) or anti-Xmad2 (3 μg) antibodies on a rocker for 30 min at 4°C. After washing the beads twice with 1 ml of XB plus LPC, as much residual buffer was removed as possible to prevent dilution of the extracts. The beads were further rinsed quickly with 60 μl of egg extracts, spun at 16,000 g in a microcentrifuge for 1 min at 4°C, and supernatants were removed. 60 μl of extracts were then incubated with the antibody-coated beads for 1 h on a rocker at 4°C. At the end of the incubation, the beads were spun at 16,000 g in a microcentrifuge for 1 min at 4°C. The supernatants were removed and subjected to another centrifugation to avoid any contamination with the beads. This procedure removed >95% of Xmad1 or Xmad2 protein from the extracts. For mock depletion, the beads were prepared in the same manner, except that control IgG was used.
Results
Excess Xmad2 Induces Metaphase Arrest
Mature eggs from the frog Xenopus laevis are naturally arrested at metaphase of meiosis II by CSF (Sagata et al., 1989), and are released from this arrest by a transient increase in the cytoplasmic calcium concentration that is triggered by fertilization. Extracts made from unfertilized eggs maintain the CSF arrest and can be induced to enter anaphase by adding calcium to inactivate CSF activity, leading to degradation of cyclin B and inactivation of Cdc2. When the spindle checkpoint is activated in these extracts, by adding a high density of sperm nuclei and nocodazole, the mitotic arrest cannot be overcome by calcium addition (Minshull et al., 1994). We have previously shown that adding a neutralizing anti-Xmad2 antibody overcomes the spindle checkpoint (Chen et al., 1996), and we expected that immunodepletion of Xmad2 would also inactivate the checkpoint. Fig. 1 B shows that egg extracts depleted of Xmad2 were not able to activate the checkpoint; despite the presence of a high concentration of sperm nuclei and nocodazole, calcium addition inactivated the histone H1 kinase activity of Cdc2 and led to the formation of interphase nuclei (data not shown), indicating that Xmad2 and/or its associated proteins are required for the checkpoint. If Xmad2 was the only checkpoint component removed from the egg extracts during immunodepletion, adding recombinant Xmad2 should restore the checkpoint. When recombinant Xmad2 was added back to depleted extracts to restore the level of Xmad2 to normal levels, egg extracts failed to maintain a mitotic arrest (Fig. 1 B), suggesting that some additional checkpoint components were also removed during Xmad2 immunodepletion, or that recombinant Xmad2 protein was not fully functional. When recombinant Xmad2 protein was added to higher levels, it prevented exit from mitosis in the presence of nocodazole (Fig. 1 B). In addition, it also blocked the inactivation of H1 kinase activity in the absence of nocodazole, a condition where the checkpoint is not normally active (Fig. 1 C). This effect of recombinant Xmad2 is dose-dependent. At low concentrations, Xmad2 partially maintained H1 kinase activity. At 100 μg/ml Xmad2, ∼17-fold the endogenous level of Xmad2, inactivation of H1 kinase was completely inhibited (Fig. 1 C) and the added sperm nuclei remained condensed (data not shown), indicating that the extracts remained arrested in mitosis. These results suggest that recombinant Xmad2 protein is functional and at high levels induces mitotic arrest. The finding that overexpression of Mad2 in fission yeast arrests cells in mitosis (He et al., 1997; Kim et al., 1998) is consistent with our results and argues that the effects of high levels of Xmad2 are due to an increased concentration of the protein rather than an aberrant activity of the recombinant protein.
Figure 1.

Excess Xmad2 protein inhibits exit from mitosis in CSF-arrested extracts. (A) Immunoblot of Xmad2 in egg extracts (lane 1), in mock-depleted extracts (lane 2), and in Xmad2-depleted extracts (lane 3). Extracts shown in lanes 2 and 3 were used in the experiment shown in B. The migration of molecular size standards and Xmad2 is indicated on the left and right, respectively. (B) Autoradiogram of histone H1 kinase assay. Mock- or Xmad2-depleted CSF-arrested egg extracts were treated as indicated on the left: low 6H-Xmad2, recombinant 6H-Xmad2 protein supplemented to the level of endogenous protein; high 6H-Xmad2, 6H-Xmad2 added to 17-fold the concentration of endogenous protein. After 1 h of incubation on ice, sperm nuclei (10,000/μl) were added to all samples and incubated for 10 min at 23°C, followed by incubation with nocodazole (lower three panels) for another 10 min. The metaphase arrest was then released by the addition of calcium chloride to inactivate CSF activity. Aliquots were taken immediately before calcium was added (time 0) and every 15 min thereafter to determine Cdc2-associated histone H1 kinase activity. (C) Dose-dependent inhibition of Cdc2 inactivation by Xmad2. Xmad2 protein at the indicated concentrations (□, 0; ⋄, 25; ○, 50; ▵, 100 ng/μl of extract, respectively) was preincubated with CSF-arrested extracts, followed by incubation with sperm nuclei (500/μl) in the absence of nocodazole. H1 kinase assays were performed as described in B and incorporation of 32P into H1 was quantified using a PhosphorImager.
The inability of calcium addition to trigger mitotic exit in CSF-arrested extracts in the presence of excess Xmad2 is similar to the phenomenon seen after activation of the spindle checkpoint. We examined the effect of excess Xmad2 in various conditions that normally do not activate the checkpoint. When extracts were incubated with a low density of nuclei or with nocodazole alone or in the absence of both nuclei and nocodazole, excess Xmad2 prevented calcium from inactivating H1 kinase activity (Fig. 2 A). The persistently high level of H1 kinase activity correlated with a high level of cyclin B (Fig. 2 A), suggesting that Xmad2 inhibits Cdc2 inactivation by preventing cyclin B degradation. These results show that excess Xmad2 prevents mitotic exit under conditions where the spindle checkpoint is normally not active. The effect of excess Xmad2 was also observed in cycling extracts prepared from activated eggs, which lack CSF activity (Fig. 2 B), demonstrating that excess Xmad2 has the general effect of preventing mitotic exit rather than simply inhibiting the ability of calcium to overcome the metaphase arrest induced by CSF.
Figure 2.

(A) Excess Xmad2 sustains Cdc2 activity by maintaining cyclin B levels in the absence of any nuclei and nocodazole. The addition of Xmad2 (100 ng/μl of extract), sperm nuclei (500/μl of extract), and/or nocodazole to CSF-arrested extracts is indicated on the left. Samples were taken for H1 kinase measurement and for cyclin B Western blots at the indicated times after calcium addition. (B) Excess Xmad2 arrests cycling extracts at the first mitosis. Eggs were activated in vitro to start the embryonic cell cycle and extracts were prepared. This cycling extract entered mitosis at 30 and 90 min upon incubation at 23°C (top) and became arrested at first mitosis in the presence of excess 6H-Xmad2 (bottom). Autoradiograms of H1 kinase assays are shown.
At the onset of anaphase, ubiquitin-mediated protein degradation triggers the destruction of cyclin B (Glotzer et al., 1991; Hershko et al., 1991; King et al., 1995; Sudakin et al., 1995) and proteins involved in sister chromatid cohesion (Holloway et al., 1993; Surana et al., 1993; Cohen-Fix et al., 1996; Funabiki et al., 1996). Because sister chromatid separation is independent of cyclin B degradation and inactivation of Cdc2 (Holloway et al., 1993), we used egg extracts to test whether excess Xmad2 inhibits degradation of the proteins involved in sister chromatid cohesion as well as of cyclin B. Extracts can be induced to replicate the DNA of sperm nuclei and then to enter a metaphase arrest that can be released by adding calcium (Shamu and Murray, 1992). In control extracts, the sister chromatids started to separate 10–15 min after calcium addition, and chromosomes decondensed at 40–60 min as extracts entered interphase (Fig. 3). The presence of excess Xmad2 prevented calcium from inducing anaphase (Fig. 3). The intact mitotic spindle and alignment of chromosomes at the metaphase plate indicated that excess Xmad2 did not disrupt spindle structure or interfere with kinetochore attachment to the spindle. Thus excess Xmad2 prevents three hallmarks of the metaphase to anaphase transition: Cdc2 inactivation, cyclin B degradation, and sister chromatid separation. This mitotic arrest is independent of any chromosome component or microtubule perturbation (Fig. 2 A), suggesting that excess Xmad2 constitutively activates the spindle checkpoint.
Isolation and Characterization of Xmad1
The failure of exogenous Xmad2 at normal concentration to restore the checkpoint activity in Xmad2-depleted extracts (Fig. 1 B) suggested that other checkpoint components bound to Xmad2 and were removed during the immunodepletion. To test this possibility, we examined the anti-Xmad2 immunoprecipitates for Xmad2-associated proteins. Immunoprecipitates of Xmad2 from CSF-arrested extracts contained an 85-kD protein (p85) that was absent in immunoprecipitates made with a control antibody (Fig. 4 A). This protein was still associated with Xmad2 when the immunoprecipitates were washed with a buffer containing 0.1% SDS (Fig. 4 A), indicating that it formed a tight complex with Xmad2.
Figure 4.


(A) Xmad2 associates with an 85-kD protein in CSF-arrested extracts. Immunoprecipitation was performed by using a control antibody (lanes 1 and 3) or an anti-Xmad2 antibody (lanes 2 and 4) as indicated. The immunoprecipitates were washed with a buffer containing 1% Triton X-100 (lanes 1 and 2) or 0.1% SDS (lanes 3 and 4). The proteins were resolved by SDS-PAGE and stained with Coomassie blue. The migration of p85, IgG, and Xmad2 is indicated on the left, and that of molecular weight standards on the right. (B) Sequencing of Xmad1. The unseparated pool of tryptic peptides recovered from the gel matrix was analyzed using a novel quadrupole TOF mass spectrometer. (a) Mass spectrum of the unseparated tryptic digest of the Xmad1 band. The peptide ions designated with T were in turn isolated by a quadrupole mass analyzer, fragmented in the collision cell, and then their tandem mass spectra were acquired with a reflector time-of-flight module. Peaks designated with asterisks are trypsin autolysis products. The peptide ion designated with A belongs to the antibody used in the purification of Xmad1. (b) Tandem mass spectrum of the doubly charged peptide ion T8 (marked with an arrow in a). Upon collisional fragmentation, tryptic peptides tend to produce a continuous series of fragment ions containing the COOH terminus (Y″ ions; Roepstorff and Fohlman, 1984). The peptide sequence (shown above) is deduced by considering precise mass differences between adjacent Y″ ions. M*, N-acetylated methionine sulfoxide amino acid residue. The spectrum also contains minor series of the NH2-terminal fragment ions (B ions) as well as internal fragment ions. Therefore to deduce the sequence with high confidence it is necessary to distinguish Y″ ions from the other ions in the spectrum. This has been achieved by selective isotopic labeling of the COOH-terminal carboxyl group of the peptides. (c) A zoom of the region in the tandem mass spectrum of T8 shows the principal of sequence readout of isotopically labeled peptides. Upon tryptic cleavage of the protein in a buffer containing H2 16O/H2 18O 1:1 (vol/vol) COOH-terminal carboxyl groups of peptide molecules incorporate 16O and 18O atoms in a 1:1 ratio. Thus the fragment ions containing the COOH terminus of the peptide (mostly Y″ ions) appear in the fragment spectrum as a characteristic isotopic pattern—a doublet split by 2 D. B ions or other fragment ions not containing COOH-terminal carboxyl group are observed as ions having normal isotopic pattern.
We sequenced p85 by mass spectrometry. To simplify the readout of the peptide sequences the protein was in-gel digested in a buffer containing H2 16O and H2 18O in equal proportion. The digestion resulted in characteristic isotopic labeling of COOH-terminal carboxyl groups of tryptic peptides (Fig. 4 B, a). The unseparated protein digest was then analyzed by nanoelectrospray tandem mass spectrometry using a novel hybrid Quadrupole Time-Of-Flight instrument. Eight peptide ions originating from p85 were fragmented in the mass spectrometer (Fig. 4 B, b). Seven tryptic peptides were sequenced completely and one peptide was sequenced partially, covering altogether 90 amino acid residues (Fig. 5 A). We used this sequence information to isolate Xenopus cDNA clones that encoded p85. The largest cDNA clone we isolated had an open reading frame of 718 amino acids and contained exact matches to six of the eight peptide sequences (Fig. 5 A). Two of the peptide sequences contained mismatches at residues Gln323Ser324 and Gly656Glu657. Because these residues are conserved in a recently isolated human Mad1 homologue (Jin et al., 1998) that shares 57.9% identity with p85, these mismatches likely reflect errors in mass spectrometry resulting from low intensity of the particular fragment ions. The sequence of Xmad1 predicts that much of the protein forms a coiled coil. In budding yeast we found that Mad2 tightly associated with Mad1 (Chen, R.-H., K. Hardwick, and A. Murray, unpublished result), which is also predicted to be a coiled-coil protein. Although sequence alignment showed that Mad1 and p85 shared only 19% identity, the sequences of p85 and Mad1 gave similar coiled-coil plots (Fig. 5 B), indicating that they are alike in structure. The human Mad1 homologue also associated with the human Mad2 (Jin et al., 1998) and strikingly resembled p85 in coiled-coil plots (Fig. 5 B). Based on these characteristics, we predict that p85 is the Xenopus homologue of Mad1, and refer to it as Xmad1. A search in the data base also revealed that a Schizosaccharomyces pombe Mad1 homologue (these sequence data are available from GenBank/EMBL/DDBJ under accession No. Z95620) shared 23.9% identity with Xmad1 (Fig. 5 A).
To investigate the function of Xmad1, we generated antiserum against full-length recombinant Xmad1. The affinity-purified antibody specifically recognized an 85-kD protein in frog egg extracts (Fig. 6 A), which was also present in Xmad2 immunoprecipitates (Fig. 6 A), consistent with the idea that this protein is the Xmad2-binding protein that we originally identified. The antibody also recognized an 84-kD protein in the frog cultured cell line XTC (Fig. 6 A), indicating that the somatic Xmad1 may be a different isoform or modified differently from the protein in oocytes. In HeLa cells, the antibody recognized an 86-kD Mad2-binding protein (Fig. 6 A), indicating the presence of a Xmad1 homologue in human cells. The anti-Xmad1 antibodies were used to examine the abundance of Xmad1 throughout the cell cycle in egg extracts. Like Xmad2, Xmad1 level remained unchanged when the CSF-arrested extract was induced to exit mitosis by calcium addition (Fig. 6 B). The protein level also stayed constant in an extract that went through two cell cycles (Fig. 6 C). These results demonstrate that Xmad1 and Xmad2 levels are not regulated during the cell cycle, including at the metaphase to anaphase transition, and that the proteins are not subject to proteolysis at specific cell cycle stages. These findings are similar to those seen in budding yeast, in which Mad1 (Hardwick and Murray, 1995) and Mad2 (Chen, R.-H., unpublished results) levels are constant throughout the cell cycle.
Figure 6.

Xmad1 abundance is constant throughout the cell cycle. (A) Specificity of anti-Xmad1 antibodies in immunoblots of frog egg extracts (lanes E), frog cultured cells XTC (lanes X), and HeLa cells (lanes H). An affinity-purified anti-Xmad1 antibody (lanes 4–9) or the same antibody preblocked with recombinant Xmad1 protein (lanes 1–3) were used. Lanes 1–6, egg extracts or cell lysates. Lanes 7–12, anti-Xmad2 immunoprecipitates prepared from egg extracts or cell lysates were immunoblotted with anti-Xmad1 (lanes 7–9) or anti-Xmad2 (lanes 10–12) antibodies. (B) CSF-arrested extract (time 0) was incubated with calcium for the time indicated on top, and immunoblotted for Xmad1 (middle) or Xmad2 (bottom). Top, histone H1 kinase assay. (C) Immunoblots of cycling extracts using antibodies specific to Xmad1 (middle) or Xmad2 (bottom). Mitosis occurs at ∼50 and 120 min as determined by histone H1 kinase assay (top) and by the nuclear morphology (data not shown).
We used the anti-Xmad1 antibodies to determine whether Xmad1 is involved in the spindle checkpoint in frog egg extracts. When an extract was preincubated with anti-Xmad1 antibodies, it failed to establish mitotic arrest in the presence of a high concentration of sperm nuclei and nocodazole, indicating that the antibody interfered with the spindle checkpoint (Fig. 7 A). This effect was specific to Xmad1, because antibody preblocked with recombinant Xmad1 had no effect on the checkpoint (Fig. 7 A). When added to egg extracts after the checkpoint was first activated, the antibody still abolished the checkpoint function (Fig. 7 B). These results show that, like Xmad2 (Fig. 7 B; Chen et al., 1996), Xmad1 is critical for both establishing and maintaining the spindle checkpoint.
Figure 7.
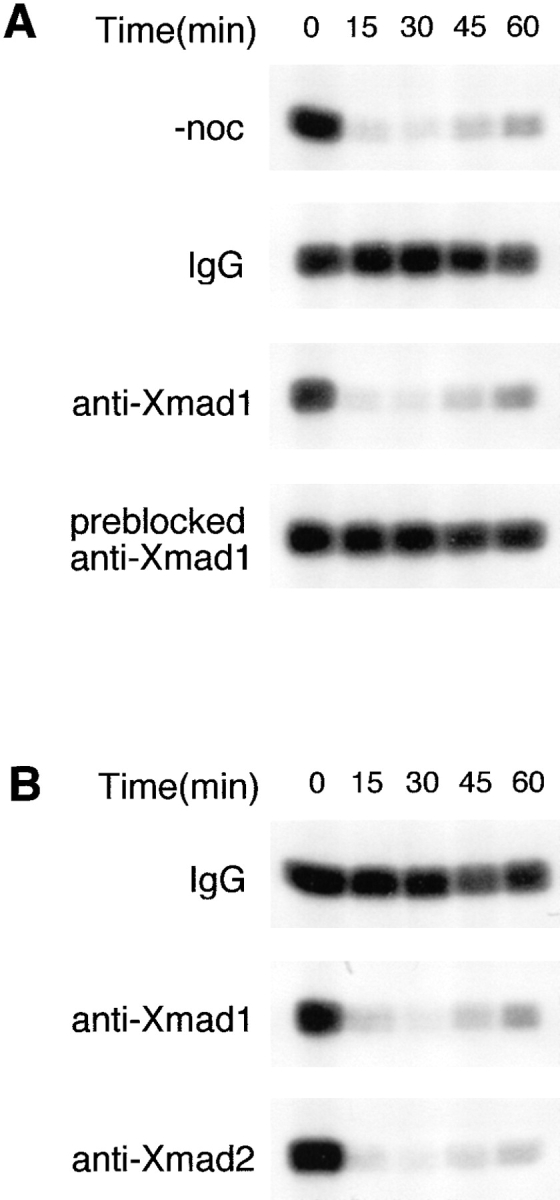
Xmad1 is important for establishing and maintaining the spindle checkpoint in frog egg extracts. (A) Xmad1 is important for spindle checkpoint function. CSF-arrested extracts were preincubated with a control antibody (IgG), an anti-Xmad1 antibody, or an anti-Xmad1 antibody preblocked with recombinant Xmad1 protein as indicated. Nocodazole and sperm nuclei were added to activate the spindle checkpoint for the lower three panels, whereas nocodazole was omitted for the top panel. H1 kinase activities were determined immediately before and at the indicated times after calcium addition as described in Fig. 1 B. (B) Xmad1 is required for maintaining the spindle checkpoint. The spindle checkpoint was first activated in CSF-arrested extracts with nocodazole and sperm nuclei followed by incubation with control (top), anti-Xmad1 (middle), or anti-Xmad2 (bottom) antibodies. H1 kinase activities were determined as in A.
Immunofluorescent staining of Xmad1 in XTC cells revealed that Xmad1 and Xmad2 localized to the nuclear envelope and the nucleus during interphase (Fig. 8 A). At prophase, Xmad1 dissociated from the nuclear envelope before all of the Xmad2 did, and a fraction of both Xmad1 and Xmad2 localized to kinetochores (Fig. 8 B). The kinetochore staining of both anti-Xmad1 and anti-Xmad2 antibodies persisted at prometaphase (Fig. 8 C), and the staining was absent from the kinetochores at metaphase and anaphase (Fig. 8, D and E). In cells arrested at mitosis by nocodazole, both anti-Xmad1 and anti-Xmad2 antibodies stained the kinetochores (Fig. 8 F). The colocalization of Xmad1 and Xmad2 likely reflects the binding of a Xmad1/ Xmad2 complex to the kinetochores. These results suggest that Xmad1 is part of the complex that senses unattached kinetochores and activates the spindle checkpoint.
Xmad1 Targets Xmad2 to Kinetochores
We also investigated the association of Xmad1 and Xmad2 with the kinetochores of chromosomes assembled in egg extracts. We first allowed frog egg extracts to assemble metaphase spindles and then added nocodazole to the extracts to induce microtubule depolymerization. When chromosomes were isolated during the metaphase arrest, neither Xmad1 nor Xmad2 was found on the kinetochores (Fig. 9 A), whereas anti-Xmad1 and anti-Xmad2 antibodies both stained paired dots on chromosomes that had been isolated after nocodazole addition (Fig. 9 B). The punctate staining colocalized with staining from anti– CENP-E antibodies (data not shown), indicating that it represented kinetochore staining. These results are consistent with the in vivo data that both Xmad1 and Xmad2 bind kinetochores that are not associated with microtubules, and dissociate after microtubules attach to kinetochores. We next asked whether the anti-Xmad1 and anti-Xmad2 antibodies that block the checkpoint in egg extracts have an effect on the localization of these proteins. When antibodies made against full-length Xmad2 (Chen et al., 1996) were added before nocodazole, both Xmad1 and Xmad2 still associated with kinetochores (Fig. 9 D), showing that our anti-Xmad2 antibodies did not prevent Xmad1 or Xmad2 from binding to kinetochores. This result demonstrates that binding of Xmad1 and Xmad2 to kinetochores is not sufficient to activate the checkpoint, and suggests that Xmad2 interacts with proteins other than Xmad1 at kinetochores and that our anti-Xmad2 antibodies blocked this interaction. On the other hand, adding anti-Xmad1 antibodies kept both Xmad1 and Xmad2 from localizing to kinetochores (Fig. 9 C), suggesting that anti-Xmad1 antibodies abolish the checkpoint by preventing Xmad1 from binding to unattached kinetochores. Even when Xmad1 and Xmad2 were first allowed to bind to unattached kinetochores, the addition of anti-Xmad1 antibodies displaced both proteins from these sites (data not shown). These results indicate that Xmad1 is essential for kinetochore localization of Xmad2 and that Xmad1 must reside on kinetochores to maintain the checkpoint.
We next investigated the dependence of Xmad2 function on Xmad1. If the major role of Xmad1 were to recruit Xmad2 to unattached kinetochores, Xmad1 would be dispensable when the checkpoint is constitutively activated by a high level of Xmad2. We tested this hypothesis by immunodepleting Xmad1 from egg extracts. When >95% of Xmad1 was removed (Fig. 10 A), the extracts were unable to activate the checkpoint in response to microtubule depolymerization at high concentrations of sperm nuclei (Fig. 10 B), even though 60% of Xmad2 remained in these extracts (Fig. 10 A). However, addition of excess recombinant Xmad2 could still trigger mitotic arrest in Xmad1-depleted extracts (Fig. 10 B). Similarly, when >95% of Xmad1 was removed by Xmad2-immunodepletion (Fig. 10 A), recombinant Xmad2 also induced a mitotic arrest (Fig. 10 B). In addition, the dose response curve to Xmad2 in extracts depleted for Xmad1 was the same as that in mock-depleted extracts (data not shown), consistent with the notion that the effect of excess Xmad2 is independent of Xmad1. Taken together, these observations suggest that Xmad1 recruits Xmad2 to the kinetochore but is not involved in signaling downstream of Xmad2.
Figure 10.

Mitotic arrest induced by excess Xmad2 is independent of Xmad1. (A) Immunoblots of Xmad1 (top) and Xmad2 (bottom) in CSF-arrested extracts (lane 1), mock- (lane 2), Xmad1- (lane 3), or Xmad2-depleted (lane 4) extracts. (B) Excess Xmad2 can still induce a mitotic arrest in the absence of Xmad1. The extracts shown in lanes 2–4 of A were used. Top panels, extracts were incubated with sperm nuclei alone. Middle panels, nocodazole and sperm nuclei were added to activate the spindle checkpoint. Bottom panels, excess 6H-Xmad2 was added to the extracts in the absence of sperm nuclei and nocodazole. H1 kinase activities were determined before and at various times after calcium addition as described for Fig. 1 B.
Discussion
We have identified a Xenopus homologue of the budding yeast Mad1 protein and shown that it is a component of the spindle checkpoint. Xmad1 was isolated as a protein that coimmunoprecipitated with Xmad2. When Xmad2 is depleted from extracts, adding back physiological doses does not restore the checkpoint. This observation suggests that other components of the checkpoint bind to Xmad2 and are removed with it from the extract upon immunodepletion. The observation that Xmad1 coimmunoprecipitates with Xmad2 strongly suggests that the additional depleted component is Xmad1, although we cannot exclude the possibility that there are other checkpoint components bound to Xmad2. One protein that has been reported to bind to Mad2 is Cdc20 (Fang et al., 1998; Hwang et al., 1998; Kallio et al., 1998; Kim et al., 1998), a protein that is necessary for the metaphase to anaphase transition (Dawson et al., 1995; Visintin et al., 1997; Kallio et al., 1998). Since Xmad2-depleted extracts can still exit from mitosis (data not shown), they must still contain functional Cdc20.
As expected, Xmad1 shares several characteristics with Xmad2. First, Xmad1 is important for establishing and maintaining the spindle checkpoint in egg extracts. Second, Xmad1 localizes to the nuclear envelope and the nucleus during interphase, and a fraction of the protein binds to the kinetochores of chromosomes that are not attached to microtubules during prophase and prometaphase. Despite these similarities and the tight interaction between these two proteins, Xmad1 and Xmad2 appear to play distinct roles in the checkpoint. Our studies indicate that Xmad1 likely recruits Xmad2 to unattached kinetochores, where Xmad2 is converted into a form that can prevent the onset of anaphase.
Our results extend previous experiments on the function of Mad1 and Mad2 in the spindle checkpoint. Experiments in budding yeast show that Mad1 and Mad2 bind tightly to each other (Chen, R.-H., K. Hardwick, and A. Murray, unpublished results), and that the presence of Mad2 is required for the hyperphosphorylation of Mad1 that correlates with the activation of the checkpoint (Hardwick and Murray, 1995). Mad2 has not been localized in yeast cells, and Mad1 shows a punctate localization within the nucleus (Hardwick and Murray, 1995). It remains a possibility that some of the punctate staining is at kinetochores. We now demonstrate that Xmad1 binds to unattached kinetochores in both tissue culture cells and in frog egg extracts. This is the same pattern of localization for vertebrate homologues of Mad2 (Figs. 8 and 9; Chen et al., 1996; Li and Benezra, 1996), Bub1 (Taylor and McKeon, 1997), Bub3 (Taylor et al., 1998), and Mad3 (Taylor et al., 1998). The result that anti-Xmad1 antibodies block the association of both Xmad1 and Xmad2 with the kinetochores in egg extracts suggests that the binding of Xmad2 to unattached kinetochores is dependent on Xmad1. Even when Xmad1 and Xmad2 are allowed to bind to unattached kinetochores first, the subsequent addition of anti-Xmad1 antibodies removes both proteins from kinetochores (data not shown), consistent with the idea that both proteins must reside on kinetochores to maintain the checkpoint. Immunodepletion experiments showed that almost all of the Xmad1 is bound with Xmad2, whereas only 40% of Xmad2 is bound with Xmad1 (Fig. 10 A), suggesting that Xmad1 is the limiting factor in the Xmad1–Xmad2 complex.
Although the spindle checkpoint is normally triggered by unattached kinetochores, we have demonstrated that a high level of Xmad2 in frog egg extracts also inhibits the metaphase to anaphase transition under conditions where the checkpoint is not normally activated. Excess Xmad2 blocks three hallmarks of anaphase: degradation of cyclin B, inactivation of Cdc2, and sister chromatid segregation. Consistent with our result, overexpression of Mad2 homologue in fission yeast also blocks anaphase onset (He et al., 1997; Kim et al., 1998), and addition of human Mad2 protein to frog egg extracts also prevents cyclin B degradation (Li et al., 1997; Fang et al., 1998). Furthermore, deletion of as few as 10 amino acids from COOH terminus of Xmad2 abolishes the ability of excess protein to induce metaphase arrest (data not shown). Introduction of a high level of the recombinant human Mad2 of similar truncation also fails to induce a mitotic arrest in Xenopus embryos and to inhibit ubiquitination of cyclin B in vitro (Fang et al., 1998). Taken together, these results indicate that the phenotypes we observed were not due to some aberrant activity of the recombinant protein. We can rule out several possibilities for how excess Xmad2 induces metaphase arrest. The arrest cannot be dependent on binding of Xmad2 to kinetochores, since it occurs in extracts without any nuclei. In addition, it is unlikely that Xmad2 acts as a competitive inhibitor of proteolysis mediated by the anaphase-promoting complex (APC), because Xmad2 lacks any trace of the destruction boxes found in other APC substrates (Glotzer et al., 1991; review in Hershko, 1997 and Cohen-Fix and Koshland, 1997) and Xmad2 levels remain constant during the cell cycle (Fig. 6).
Increases in the level or activity of a number of proteins other than Mad2 can also activate the checkpoint in cells that have normal spindles. Adding an active form of Ste11, a budding yeast MAPK kinase family member, arrests frog egg extracts at mitosis (Takenaka et al., 1997). In budding yeast, overexpression of Mps1, the kinase that is thought to phosphorylate Mad1, arrests cells at mitosis without spindle defects (Hardwick et al., 1996), and overexpression of Mph1, a S. pombe homologue of Mps1, also induces metaphase arrest (He et al., 1998). All these observations suggest that the activity or the level of the spindle checkpoint components must be tightly regulated in order for the metaphase to anaphase transition to progress properly.
How is Xmad2 activated by unattached kinetochores and how does it inhibit the onset of anaphase? We speculate that once Xmad1 binds to kinetochores and activates its associated Xmad2, Xmad2 is converted into a form that no longer binds Xmad1 and thus diffuses throughout the cell preventing the onset of anaphase. The free, inactive Xmad2 can then interact with and become activated by kinetochore-bound Xmad1, making the kinetochore a catalytic center for activating the checkpoint (Fig. 11). The active molecules of Xmad2 would be slowly inactivated, thus ensuring that the checkpoint would be turned off once all the kinetochores had bound to microtubules. The molecular basis of Xmad2 activation is obscure since we have been unable to detect posttranslational modifications on this protein (unpublished data). One possible form of activation is changes in the state of Xmad2, as indicated by the finding that monomers and tetramers of the recombinant human Mad2 protein differ in their ability to inhibit cyclin degradation in egg extracts (Fang et al., 1998).
Figure 11.

A model for how Xmad1 and Xmad2 work to activate the spindle checkpoint. Binding of Xmad1 to unattached kinetochores enables its associated Xmad2 to interact with a downstream checkpoint component X. This interaction converts X into a form that is capable of directly or indirectly inhibiting the anaphase-promoting complex (APC). The interaction between Xmad2 and X is unstable when Xmad2 is not associated with kinetochores, so that the checkpoint is not activated without unattached kinetochores. Increasing the global Xmad2 concentration drives the complex formation by mass action even in the absence of kinetochores. At a high enough concentration of Xmad2, the level of Xmad2–X complex becomes comparable to that induced through kinetochore-associated Xmad2 and results in constitutive activation of the spindle checkpoint. A likely candidate for X is Cdc20.
The most likely target of Xmad2 is Cdc20. In budding yeast, Cdc20 binds to Mad2 and Mad3 proteins, and mutations in Cdc20 that block this binding act as dominant checkpoint-defective mutants (Hwang et al., 1998), suggesting that Cdc20 is the target of the checkpoint. Similar results have also been obtained in fission yeast (Kim et al., 1998). Cdc20 homologues also mediate the association of Mad2 with APC in vertebrates (Gorbsky et al., 1998; Fang et al., 1998). A simple model is that interacting with unattached kinetochores converts Xmad2 into a form that can interact and inhibit Cdc20. In the absence of kinetochores, Xmad2 may assume the active conformation at a very slow rate, but this small amount of active Xmad2 cannot efficiently inhibit Cdc20 function. As the level of Xmad2 is increased, the concentration of the active form rises by mass action until it is sufficient to inhibit Cdc20, thus arresting extracts in metaphase (Fig. 11). In such a scheme, kinetochore-bound molecules that convert Xmad2 into its active form would be dispensable for excess Xmad2 to arrest the cell cycle, whereas the arrest would require molecules that functioned with Xmad2 to inhibit Cdc20. Our data show that excess Xmad2 can still activate the checkpoint when >95% of Xmad1 is immunodepleted, suggesting that Xmad1 is not involved in signaling events downstream of Xmad2, and that the major role of Xmad1 in the checkpoint is to recruit Xmad2 to unattached kinetochores. Future experiments will be required to determine how kinetochores monitor their interactions with microtubules and control the binding of Xmad1, how Xmad1 binding activates Xmad2, and how the activated Xmad2 inhibits Cdc20 and any other targets of the spindle checkpoint.
Acknowledgments
We thank T. Huffaker and M. Goldberg for critically reading the manuscript; T. Matsumoto for sharing the fission yeast Mad1 sequence before its publication; and A. Hyman for bringing together the labs of A.W. Murray and M. Mann.
This work was supported by grants from National Institutes of Health (NIH), the Packard Foundation, the Markey Foundation, and the March of Dimes (to A.W. Murray), and a Start-Up Fund from Cornell University, as well as an additional NIH grant (to R.-H. Chen).
Abbreviations used in this paper
- APC
anaphase-promoting complex
- BUB
budding unihibited by benzimidazole
- CSF
cytostatic factor
- MAD
mitotic arrest-deficient
Footnotes
Address all correspondence to Rey-Huei Chen, Section of Biochemistry, Molecular and Cell Biology, Biotechnology Building, Cornell University, Ithaca, NY 14853. Tel.: (607) 255-6542. Fax: (607) 255-2428. E-mail: rc70@cornell.edu
M. Mann's present address is Center for Experimental BioInformatics (CEBI), Odense University, Odense, Denmark.
References
- Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- Chen R-H, Murray AW. Characterization of spindle assembly checkpoint in Xenopus egg extracts. Methods Enzymol. 1997;283:572–584. doi: 10.1016/s0076-6879(97)83045-5. [DOI] [PubMed] [Google Scholar]
- Chen R-H, Sarnecki C, Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R-H, Waters JC, Salmon ED, Murray AW. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science. 1996;274:242–246. doi: 10.1126/science.274.5285.242. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Koshland D. The metaphase-to-anaphase transition: avoiding a mid-life crisis. Curr Opin Cell Biol. 1997;9:800–806. doi: 10.1016/s0955-0674(97)80080-4. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Peters JM, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996;10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- Dawson IA, Roth S, Artavanis-Tsakonas S. The Drosophila cell cycle gene fizzy is required for normal degradation of cyclins A and B during mitosis and has homology to the CDC20 gene of Saccharomyces cerevisiae. . J Cell Biol. 1995;129:725–737. doi: 10.1083/jcb.129.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 1998;12:1871–1883. doi: 10.1101/gad.12.12.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr KA, Hoyt MA. Bub1p kinase activates the Saccharomyces cerevisiae spindle assembly checkpoint. Mol Cell Biol. 1998;18:2738–2747. doi: 10.1128/mcb.18.5.2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funabiki H, Yamano H, Kumada K, Nagao K, Hunt T, Yangida M. Cut2 proteolysis required for sister-chromatid separation in fission yeast. Nature. 1996;381:438–441. doi: 10.1038/381438a0. [DOI] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Gorbsky GJ, Chen RH, Murray AW. Microinjection of antibody to mad2 protein into mammalian cells in mitosis induces premature anaphase. J Cell Biol. 1998;141:1193–1205. doi: 10.1083/jcb.141.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick K, Murray AW. Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J Cell Biol. 1995;131:709–720. doi: 10.1083/jcb.131.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG. The spindle checkpoint. Trends Genet. 1998;14:1–4. doi: 10.1016/S0168-9525(97)01340-1. [DOI] [PubMed] [Google Scholar]
- Hardwick KG, Weiss E, Luca FC, Winey M, Murray AW. Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science. 1996;273:953–956. doi: 10.1126/science.273.5277.953. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Antibodies. A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York. 309–310.
- He X, Patterson TE, Sazer S. The Schizosaccharomyces pombe spindle checkpoint protein mad2p blocks anaphase and genetically interacts with the anaphase-promoting complex. Proc Natl Acad Sci USA. 1997;94:7965–7970. doi: 10.1073/pnas.94.15.7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Jones MH, Winey M, Sazer S. mph1, a member of the Mps1-like family of dual specificity protein kinases, is required for the spindle checkpoint in S. pombe. J Cell Sci. 1998;111:1635–1647. doi: 10.1242/jcs.111.12.1635. [DOI] [PubMed] [Google Scholar]
- Hershko A. Roles of ubiquitin-mediated proteolysis in cell cycle control. Curr Opin Cell Biol. 1997;9:788–799. doi: 10.1016/s0955-0674(97)80079-8. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ganoth D, Pehrson J, Palazzo RE, Cohen LH. Methylated ubiquitin inhibits cyclin degradation in clam embryo extracts. J Biol Chem. 1991;266:16376–16379. [PubMed] [Google Scholar]
- Holloway SL, Glotzer M, King RW, Murray AW. Anaphase is initiated by proteolysis rather than by the inactivation of MPF. Cell. 1993;73:1393–1402. doi: 10.1016/0092-8674(93)90364-v. [DOI] [PubMed] [Google Scholar]
- Hoyt MA, Trotis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–517. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- Hwang LH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW. Budding yeast Cdc20: a target of the spindle checkpoint. Science. 1998;279:1041–1044. doi: 10.1126/science.279.5353.1041. [DOI] [PubMed] [Google Scholar]
- Irniger S, Piatti S, Michaelis C, Nasmyth K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 1995;77:1037–1050. doi: 10.1016/0092-8674(95)90337-2. [DOI] [PubMed] [Google Scholar]
- Jin DY, Spencer F, Jeang KT. Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell. 1998;93:81–91. doi: 10.1016/s0092-8674(00)81148-4. [DOI] [PubMed] [Google Scholar]
- Kallio M, Weinstein J, Daum JR, Burke DJ, Gorbsky GJ. Mammalian p55CDC mediates association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase-promoting complex, and is involved in regulating anaphase onset and late mitotic events. J Cell Biol. 1998;141:1393–1406. doi: 10.1083/jcb.141.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Lin DP, Matsumoto S, Kitazono A, Matsumoto T. Fission yeast Slp1: an effector of the Mad2-dependent spindle checkpoint. Science. 1998;279:1045–1047. doi: 10.1126/science.279.5353.1045. [DOI] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–531. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- Li X, Nicklas RB. Mitotic forces control a cell cycle checkpoint. Nature. 1995;373:630–632. doi: 10.1038/373630a0. [DOI] [PubMed] [Google Scholar]
- Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 1996;274:246–248. doi: 10.1126/science.274.5285.246. [DOI] [PubMed] [Google Scholar]
- Li Y, Gorbea C, Mahaffey D, Rechsteiner M, Benezra R. MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. Proc Natl Acad Sci USA. 1997;94:12431–12436. doi: 10.1073/pnas.94.23.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshull J, Sun H, Tonks NK, Murray AW. MAP-kinase dependent mitotic feedback arrest in Xenopus egg extracts. Cell. 1994;79:475–486. doi: 10.1016/0092-8674(94)90256-9. [DOI] [PubMed] [Google Scholar]
- Murone M, Simanis V. The fission yeast dma1 gene is a component of the spindle assembly checkpoint, required to prevent septum formation and premature exit from mitosis if spindle function is compromised. EMBO (Eur Mol Biol Organ) J. 1996;15:6605–6616. [PMC free article] [PubMed] [Google Scholar]
- Murray AW. Cell cycle extracts. Methods Cell Biol. 1991;36:573–597. [PubMed] [Google Scholar]
- Rieder CL, Cole RW, Khodjakov A, Sluder G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 1995;130:941–948. doi: 10.1083/jcb.130.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RT, Farr KA, Hoyt MA. The Saccharomyces cerevisiae checkpoint gene BUB1encodes a novel protein kinase. Mol Cell Biol. 1994;14:8282–8291. doi: 10.1128/mcb.14.12.8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagata N, Watanabe N, Vande GF, Woude, Ikawa Y. The c-mos proto-oncogene product is a cytostatic factor responsible for meiotic arrest in vertebrate eggs. Nature. 1989;342:512–518. doi: 10.1038/342512a0. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning, A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Schwab M, Lutum AS, Seufert W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 1997;90:683–693. doi: 10.1016/s0092-8674(00)80529-2. [DOI] [PubMed] [Google Scholar]
- Shamu CE, Murray AW. Sister chromatid separation in frog egg extracts requires DNA topoisomerase II activity during anaphase. J Cell Biol. 1992;117:921–934. doi: 10.1083/jcb.117.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins from silver stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Chernushevich I, Ens W, Standing KG, Thomson B, Wilm M, Mann M. Rapid ‘de novo' peptide sequencing by a combination of nanoelectrospray, isotopic labeling and a quadrupole/time-of-flight mass spectrometer. Rapid Commun Mass Spectrom. 1997;11:1015–1024. doi: 10.1002/(SICI)1097-0231(19970615)11:9<1015::AID-RCM958>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hersko J, Luca F, Ruderman JV, Hershko A. The cyclosome, a large complex containing cyclin-selective ubiquitination ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–198. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surana U, Amon A, Dowzer C, McGrew J, Byers B, Nasmyth K. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO (Eur Mol Biol Organ) J. 1993;12:1969–1978. doi: 10.1002/j.1460-2075.1993.tb05846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka K, Gotoh Y, Nishida E. MAP kinase is required for the spindle assembly checkpoint but is dispensable for the normal M phase entry and exit in Xenopusegg cell cycle extracts. J Cell Biol. 1997;136:1091–1097. doi: 10.1083/jcb.136.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89:727–735. doi: 10.1016/s0092-8674(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Ha E, McKeon F. The human homologue of Bub3 is required for kinetochore localization of bub1 and a Mad3/Bub1-related protein kinase. J Cell Biol. 1998;142:1–11. doi: 10.1083/jcb.142.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A. CDC20 and CDH1: a family of substrate-specific activators of APC- dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- Waters JC, Chen RH, Murray AW, Salmon ED. Localization of mad2 to kinetochores depends on microtubule attachment, not tension. J Cell Biol. 1998;141:1181–1191. doi: 10.1083/jcb.141.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss E, Winey M. The S. cerevisiae SPB duplication gene MPS1is part of a mitotic checkpoint. J Cell Biol. 1996;132:111–123. doi: 10.1083/jcb.132.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm M, Mann M. Analytical properties of the nano electrospray ion source. Anal Chem. 1996;66:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]