

Abstract
Dividing populations of stratified and simple epithelial tissues express keratins 5 and 14, and keratins 8 and 18, respectively. It has been suggested that these keratins form a mechanical framework important to cellular integrity, since their absence gives rise to a blistering skin disorder in neonatal epidermis, and hemorrhaging within the embryonic liver. An unresolved fundamental issue is whether different keratins perform unique functions in epithelia. We now address this question using transgenic technology to express a K16-14 hybrid epidermal keratin transgene and a K18 simple epithelial keratin transgene in the epidermis of mice null for K14. Under conditions where the hybrid epidermal keratin restored a wild-type phenotype to newborn epidermis, K18 partially but not fully rescued. The explanation does not appear to reside in an inability of K18 to form 10-nm filaments with K5, which it does in vitro and in vivo. Rather, it appears that the keratin network formed between K5 and K18 is deficient in withstanding mechanical stress, leading to perturbations in the keratin network in regions of the skin that are subjected either to natural or to mechanically induced trauma. Taken together, these findings suggest that the loss of a type I epidermal keratin cannot be fully compensated by its counterpart of simple epithelial cells, and that in vivo, all keratins are not equivalent.
Keywords: keratins, intermediate filaments, functional redundancy, epidermis, epithelia
Keratins belong to the superfamily of intermediate filament (IF)1 proteins, which have the remarkable capacity to assemble into 10-nm filaments in vitro, in the absence of auxiliary proteins or factors (for review see Fuchs and Weber, 1994). They are composed of two sequence types, both of which share a common, largely α-helical secondary structure, capable of forming coiled-coil heterodimers, which then further assemble to form obligatory heteropolymers. Type I keratins include K9-K20 and type II keratins encompass K1-K8 (Moll et al., 1982). While most combinations of type I and type II keratins can copolymerize in vitro (Franke et al., 1983), keratins are often coexpressed as specific pairs in vivo, where they are largely restricted to epithelial tissues (Sun et al., 1984).
The innermost, mitotically active (basal) layer of many stratified epithelial tissues, including the epidermis, cornea, and tongue, express the type II keratin K5 and the type I keratin K14 (Fuchs and Green, 1980; Moll et al., 1982; Nelson and Sun, 1983; Byrne et al., 1994). A second natural partner for K5 is K15, which is more abundantly expressed in internal stratified squamous epithelia, such as esophagus (Moll et al., 1982; Leube et al., 1988; Lloyd et al., 1995). As keratinocytes of stratified tissues withdraw from the cell cycle and commit to differentiate, they often downregulate transcription of K5/K14 (K5/K15); concomitantly, as the differentiating cells move into the upper layers, they induce one of five to six different pairs of keratin genes depending upon the particular stratified tissue (Fuchs and Green, 1980; Moll et al., 1982). In contrast to these elaborate patterns in stratified tissues, simple epithelia express K8 and K18, and only under certain circumstances do they express additional keratins K7 and K19 (Moll et al., 1982; Wu et al., 1982).
One role that has been ascribed to the various keratin filament networks of stratified squamous epithelia is to impart mechanical integrity to cells, without which, the cells become fragile and prone to rupturing. This function was initially proposed on the basis of transgenic mice engineered to express a dominant-negative K14 mutant (Vassar et al., 1991), and it has been documented by K14 null mutations identified in both mice (Lloyd et al., 1995) and humans (Chan et al., 1994; Rugg et al., 1994). In all of these cases, the skin displays features of epidermolysis bullosa simplex (EBS), a blistering disorder typified by basal epidermal cell rupturing upon mild mechanical stress (Fitzpatrick et al., 1993; Fuchs and Cleveland, 1998). Similarly, mice expressing a mutant version of the K10 gene, normally expressed in differentiating epidermal cells, exhibit features of epidermolytic hyperkeratosis (EH), a disorder analogous to EBS but involving suprabasal rather than basal cell degeneration (Fuchs et al., 1992; Porter et al., 1996). In humans, EBS and EH are generally autosomal dominant skin disorders, and correlate with the appearance of clumps or aggregates of keratin filaments in the cell cytoplasm (Anton-Lamprecht, 1994).
It is now well established that these mechanically induced blistering disorders of the epidermis involve mutations in keratins (Bonifas et al., 1991; Coulombe et al., 1991; Cheng et al., 1992; Chipev et al., 1992; Lane et al., 1992; Rothnagel et al., 1992). Recently, a number of other keratin disorders have been identified genetically, and they involve additional stratified epithelial tissues, including cornea, esophagus, and hair; they are characterized by cell degeneration and mechanical fragility (for review see Fuchs and Cleveland, 1998). Where tested, the severity of these diseases seems to correlate with the degree to which the keratin mutants perturb IF assembly in vitro (Letai et al., 1993).
Human disorders involving simple epithelial keratins have not yet been established, although a K18 mutation was reported recently in a patient with cryptogenic cirrhosis, a liver disorder (Ku and Omary, 1994; Ku et al., 1997), and transgenic mice expressing a mutant K18 develop chronic hepatitis (Ku et al., 1995, 1996). Simple epithelial keratins may also function to maintain hepatocyte integrity as evidenced by (a) the death of some K8 null mouse embryos resulting from hemorrhaging of the liver at the time of vascularization (Baribault et al., 1993); (b) the liver necrosis in adult K8 mice when subjected to hepatectomy (Loranger et al., 1997); and (c) the signs of hepatitis seen in adult K18 null mice, which also display K8 aggregates in their hepatocytes (Magin et al., 1998). Taken together, these findings suggest a special importance of simple epithelial keratins in the liver. A role for K8/K18 in intestinal epithelia has also surfaced in that one strain of K8 null mice displays marked colorectal hyperplasia and inflammation (Baribault et al., 1994). Whether simple epithelial keratins function to impart mechanical integrity, however, is considerably less clear than it is for stratified epithelial keratins.
If keratins have comparable functions in different tissues, then what is the functional significance of the multiplicity of keratin sequences? Is it that keratin genes have simply duplicated and diverged over evolution, but functionally the gene products are redundant? Or, did different keratin genes evolve to suit the particular functional needs of the tissues in which they are expressed? To begin to explore the answer to this fundamental issue of keratin biology, we have used transgenic technology to replace K14 with K18 gene expression in K14 null mice. We show that while K18 can coassemble with K5 into a keratin filament network in vivo and in vitro, the keratin network is unable to withstand mechanical trauma, giving rise to natural skin blistering in the paws and to mechanically induced blistering in the back. Our findings suggest that keratins within a single family type have specially tailored properties and are not functionally redundant. They further support the notion that keratins have evolved to suit the particular structural and functional needs of different epithelial tissues.
Materials and Methods
Generation of Transgenes
A K14 transgene was generated using 2,100 bp of the human K14 promoter to drive expression of a hybrid human K16-14 cDNA. The hybrid K16-14 cDNA was assembled from human K14 cDNA and human K16 cDNA. A conserved SphI site was used to engineer the hybrid cDNA, resulting in an encoded 49-kD protein containing 368 amino acid (a.a.) residues of human K16 and 105 a.a. of human K14 (Wawersik et al., 1997). A human K18 transgene was generated using 2,100 bp of human K14 promoter to drive expression of a full-length human K18 cDNA, provided by Dr. R. Oshima (Burnham Institute, La Jolla, CA) (Oshima et al., 1986). The 1,425-bp K18 cDNA was placed in a mammalian epidermal expression vector (Vassar et al., 1989), modified to contain a globin intron sequence in the 5′ untranslated region.
Engineering the K14 Null Mice Replaced by Either the K14 or K18 Transgene
Genetically engineered transgenes were microinjected into fertilized, single cell embryos (CD-1 for K18; B6C3 F2 for K16-14), and after culturing the embryos to the two cell stage, they were transferred to the oviducts of pseudopregnant female mice (Vassar et al., 1991; Paladini and Coulombe, 1998). 3 wk after birth, ear or tail samples were taken for DNA analysis. Presence of the transgene was determined by PCR or Southern blotting of these DNAs. Mice expressing the transgenes were then bred to mice that were null for the K14 gene (Lloyd et al., 1995). Genotyping was determined by Southern blot analysis and/or PCR using the conditions and probes described (Lloyd et al., 1995; Paladini and Coulombe, 1998).
Immunoblot Analyses
Intermediate filaments were isolated from skin samples as described (Wu et al., 1982), and keratins were solubilized in a solution of 8 M urea and 10% β-mercaptoethanol. Water-soluble proteins were concentrated at 4°C with a Microcon 10 concentrator (Amicon W.R. Grace & Co., Beverly, MA). After electrophoretic transfer of SDS-PAGE–resolved IF proteins to nitrocellulose, blots were pre-incubated overnight in 1% gelatin (Sigma Chemical Co., St. Louis, MO), 20 mM Tris HCl, pH 7.6, 150 mM NaCl, and 0.1% Tween-20, followed by incubation with primary antibody for 1 h and washing three times for 10 min each in PBS, 0.5% Tween-20. After this procedure, blots were then developed using either a HRP-conjugated secondary antibody and chemiluminescence ECL (Amersham Corp., Arlington Heights, IL) or an alkaline phosphatase–conjugated secondary antibody and color development (Bio-Rad Laboratories, Hercules, CA). Primary antibodies included: (a) mouse CY-90 anti-K18 mAb (Sigma Chemical Co.), (b) rabbit polyclonal anti-K14 COOH-terminal peptide antibody (Stoler et al., 1988), or (c) guinea pig polyclonal anti-K5 COOH-terminal peptide antibody (Lersch et al., 1989). For successive incubations after ECL development, blots were stripped at 70°C in 62.5 mM Tris HCl, pH 7.6, 2% SDS, 0.7% β-mercaptoethanol. To quantitate signals, blots were scanned into a densitometer (Molecular Dynamics, Sunnyvale, CA), and protein levels were determined with ImageQuant software (Molecular Dynamics).
Immunohistochemistry
Animals were killed at 1, 2, or 5 d of age, or as adults, and skin was frozen in isopentane, sectioned (8 μm), and then processed for immunofluorescence immunohistochemistry. Antisera were used at the following dilutions: anti-K14, 1:200 (Stoler et al., 1988); anti-K18, 1:200 (Sigma Chemical Co.); anti-K6, 1:200 (gift of D. Roop, Baylor College of Medicine, Houston, TX); anti-K5, 1:200 (Lersch et al., 1989). All secondary antibodies, conjugated to either FITC or Texas red were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA).
Ultrastructural Analyses
Tissues were obtained from the skin of duplicate K14 null, K14 wild-type, K14 null/K16-14 transgenic, K14 null/K18 transgenic, and K14 wild-type/ K18 transgenic animals, killed at 2–3 d of age. For conventional transmission electron microscopy, tissues were immediately fixed in 2.5% glutaraldehyde and 4% formaldehyde, pH 7.3, for 24 h, followed by postfixation with 1% osmium tetroxide for 1 h. After en bloc staining and dehydration with ethanol, the samples were embedded with LX-112 medium (Ladd Research Industries, Burlington, Vermont). All tissues had been properly oriented to ensure that the entire thickness of epidermis or mucous membrane would be sectioned perpendicular to the normal tissue plane. Semithin sections were stained with toluidine blue. Ultrathin sections of 80 nm were cut with a diamond knife, mounted on uncoated grids, stained with uranyl acetate and lead citrate, and then observed with a JEOL-CXII electron microscope (JEOL USA, Inc., Peabody, MA) operated at 60 kv.
For immunoelectron microscopy, the methods of Warhol et al. (1985) and Coulombe et al. (1989) were used with some modifications. The tissues were (a) fixed with 2% PFA, 0.1% glutaraldehyde in PBS for 6 h, (b) dehydrated with ethanol and embedded with Lowicryl K4M medium (Ladd Research Industries) at −20°C, and then (c) polymerized with UV light for 7 d at −20°C. Ultrathin sections were mounted on Formvar/carbon-coated nickel grids. All sections were first incubated overnight with polyclonal rabbit antisera monospecific for either K14 (1:200), K5 (1:100), or K18 (1:100), followed by a 45-min incubation with a secondary antibody, 20-nm gold–conjugated goat anti–rabbit IgG. All grids were then briefly stained with uranyl acetate and lead citrate, and then examined with a JEOL-CXII electron microscope at 60 kV.
Filament Assembly Studies
The assembly properties of K5-K18 and K5-K14 were compared using assays previously described (Coulombe and Fuchs, 1990; Wawersik et al., 1997). Plasmids pET-K5 and pET-K14 (Coulombe and Fuchs, 1990) and pET-K18 (Ku et al., 1997) were transformed into Escherichia coli strain BL21 (DE3) to generate mg amounts of the corresponding recombinant human keratins. Purified type I and type II keratins were mixed in a ∼45: 55 molar ratio at a final concentration of 500 μg/ml in 6 M urea buffer, incubated for 1 h at room temperature, and then fractionated by anion exchange chromatography on a Pharmacia Mono Q column (Pharmacia Biotech, Inc., Piscataway, NJ). Collected fractions were analyzed by SDS-PAGE and those containing type I–type II heterotypic complexes were pooled.
To assay for tetramer formation, the K5-K18 and K5-K14 samples were dialyzed against 25 mM sodium phosphate and 10 mM β-ME, containing 6 M or 8 M urea at pH 7.4, to remove Tris ions. Protein concentration was adjusted to 200 μg/ml, and chemical cross-linking was performed by adding BS3 (Bis-[sulfosuccinimidyl] suberate; Pierce Chemical Co., Rockford, IL) to a final concentration of 5 mM for 1 h at room temperature. Cross-linked products (6 μg proteins) were resolved on a 4–16% gradient SDS-PAGE, and stained with Coomassie blue.
To assay for polymerization into filaments, the K5-K18 and K5-K14 samples (200 μg/ml) were serially dialyzed against the following buffers at room temperature: (a) 9 M urea, 25 mM Tris-HCl, 10 mM β-ME, pH 8.1; (b) 4 M urea, 25 mM Tris-HCl, 5 mM β-ME, pH 7.4; and (c) 5 mM Tris-HCl, 5 mM β-ME, pH 7.4. Assemblies were examined by negative staining (1% aqueous uranyl acetate) using a Zeiss electron microscope (Carl Zeiss Inc., Thornwood, NY). Polymerization efficiency was determined by subjecting final assemblies (40 μl aliquots, ∼8 μg proteins) to centrifugation at 100,000 g for 60 min at 4°C (Airfuge; Beckman Coulter, Inc., Fullerton, CA). Supernatant and pellet fractions were analyzed by SDS-PAGE and Coomassie blue staining.
Results
A K16-14 Hybrid Transgene Rescues Blistering in K14 Null Mice
The hallmark of K14 null mice is that they display marked blistering over their paws shortly after birth (Lloyd et al., 1995). Our first aim was to determine if we could rescue this phenotype by expressing a K14-like transgene using 2,100 bp of the human K14 promoter (Vassar et al., 1989). Fig. 1 shows the rescue vector, which contains a significant portion of human K16 coding sequence to facilitate its distinction by size from endogenous K14, and to facilitate quantitation of transgene levels relative to endogenous K14 levels. The hybrid retains the COOH-terminal epitope of K14, to which a peptide-specific antibody exists (Stoler et al., 1988). Transgenic mice were generated by standard procedures, and confirmed by Southern blot analysis (Paladini and Coulombe, 1998). Mice from two K16-14–expressing lines were generated. Expression of the hybrid K16-14 at ≤75% relative to endogenous K14 levels does not cause any detectable alteration in the skin of transgenic mice (Paladini and Coulombe, 1998).
Figure 1.

Expression of K16-K14 in transgenic mice. (A) A schematic of the replacement vector, showing 2,100 bp of the human K14 promoter (hK14), followed by the 5′ intron and 5′ untranslated sequence from the rabbit β-globin gene (β int), followed by a K16-14 hybrid transgene followed by the K14 3′ untranslated sequence (see Allen et al., 1996 for expression vector). The dotted vertical lines demarcate the relative portions of K16 versus K14 coding sequences used (see Paladini and Coulombe, 1998). (B) IF proteins were isolated from the skins of K16-K14 transgenic (lane 1) and control (lane 2) neonatal mice. Proteins were then subjected to SDS-PAGE. Gels were subjected to immunoblot analysis using an antibody against the COOH terminus of K14, also present in the 49-kD transgene product. Relative amounts of transgenic versus wild-type K14 were determined by densitometry scanning. Sample shown is from the low expressing K16-14 transgenic mouse.
To quantitate the levels of transgene expression for the present study, we isolated the IF proteins from tail skins and conducted SDS-PAGE, followed by immunoblot analysis to detect the transgene product and the endogenous K14, both of which share identical COOH-terminal sequences (Fig. 1). The antibody detected an additional 49-kD band in the transgene skin extract (Fig. 1 A) that was not present in the wild-type skin extract (Fig. 1 B). The 52-kD band corresponding to endogenous K14 was detected in both transgenic and wild-type skin extracts. Densitometry scanning revealed that in mice heterozygous for the K16-K14 transgene locus, the transgene product was present at 17% ± 3% the level of K14 in the lower expressing line (shown) and at ∼50% the level of K14 in the higher expressing line (data not shown). These mice were then bred to the K14 null animals to generate mice with a basal epidermal layer null for K14 and positive for the K16-14 transgene. Animals were genotyped by Southern blot analysis.
Mice from both K16-K14 transgenic/K14 null lines appeared normal at birth and exhibited no blistering over their body or paws. Animals remained blister-free as they became adults. A more detailed analysis of these mice will be described elsewhere. Histological analysis revealed no signs of epidermal abnormalities, even in the palmar region of the paws, subjected naturally to substantial mechanical stress (Fig. 2 A). In palmar areas and elsewhere throughout the K16-14 replacement skin, an ordered program of differentiation was observed, with mitotically active basal cells in the inner layer, spinous cells, granular layer cells, and stratum corneum (Fig. 2, A and B). This was in marked contrast to K14 null skin, which displayed gross blistering within 2 d of birth (Lloyd et al., 1995). Blistering was very bad in the palmar regions of K14 null skin where rete ridges are typically seen (not shown), and was prominent even in areas with hair follicles (Fig. 2 C). At the histological level, clear cytoplasm and intraepidermal rupturing was seen within the basal layer (Fig. 2 C; Lloyd et al., 1995). These findings demonstrate that in the presence of K16-14, the skin blistering phenotype typical of neonatal K14 null mice was rescued.
Figure 2.

The cytolysis in the basal layer of K14 null epidermis is missing when these cells express the K16-14 transgene. Semithin (0.75 μm) sections of neonatal paw skin biopsies from (A and B) K14 null/K16-14 transgenic and (C) K14 null mice. Skins were embedded in Epon and stained with Toluidine blue. Section in A is from central paw, showing that even in areas rich in rete ridges, the K16-14–expressing, K14 null paw skin appears indistinguishable from wild type. Basal cells from the equivalent region of K14 null skin were fully cytolyzed, leading to complete separation of the upper epidermis (not shown). Areas of paw skin from areas where hair follicles were still sparse, but where rete ridges were no longer present were still normal in the replacement skin (B), but partially blistered in the K14 null skin (C). Bar, 30 μm.
To investigate whether there might be more subtle aberrations in the basal cell cytoarchitecture of K16-14 transgenic/K14 null replacement epidermis, we examined the skin by electron microscopy. As shown in Fig. 3, replacement paw skin basal cells showed abundant keratin filaments (A), not seen in the K14 null cells (B). These filaments associated with desmosomes and with hemidesmosomes, as expected for wild-type keratin networks. Furthermore, no signs of microscopic blistering were visible in these samples. Overall, the macroscopic and microscopic appearance of the K16-14 skin was similar to that of K14 ± or wild-type skin (Fig. 3 C).
Figure 3.

K14 null mice expressing the K16-14 transgene have basal epidermal cells that are rich in keratin filament bundles and similar to wild-type basal cells. Paw skin samples were taken from neonatal age-matched animals that were either K16-14 transgenic/K14 null, K14 null, or wild type. Skins were processed for electron microscopy. (A) Basal cell of K16-14 paw skin in area of rete ridge formation. Basal cells show bundles of keratin filaments (Kf) in cytoplasm. No signs of microblistering or basal cell cytolysis were evident here, or elsewhere throughout the skin. (B) Basal layer of K14 null mouse skin. Note that keratin filament bundles are largely absent. Note also the presence of basal cell cytolysis (asterisks). (C) Basal layer from wild-type mouse skin. Note presence of bundles of keratin filaments (Kf). Nu, nucleus; BL, basal lamina; Mi, mitochondria; De, desmosome; Hd, hemidesmosome. Bar, 0.5 μm.
Generating K18 Transgenic Mice and Quantitating the Level of Transgene Expression
To test the functional significance of the multiplicity of keratin sequences, we next focused our attention on replacing the major epidermal type I keratin K14 with the major simple epithelial type I keratin K18. These keratins share only 48% sequence identity at the amino acid level, and are the most distantly related among the type I keratins. Fig. 4 illustrates the K14 promoter–K18 transgene that was injected into fertilized mouse eggs to engineer transgenic mice. In this case, we did not use any epitope tags or manipulations of K18, which might complicate interpretation of the rescue. Mice that test positive by Southern blot analysis for the K14 promoter–driven K18 transgene were then bred to the F1 generation.
Figure 4.
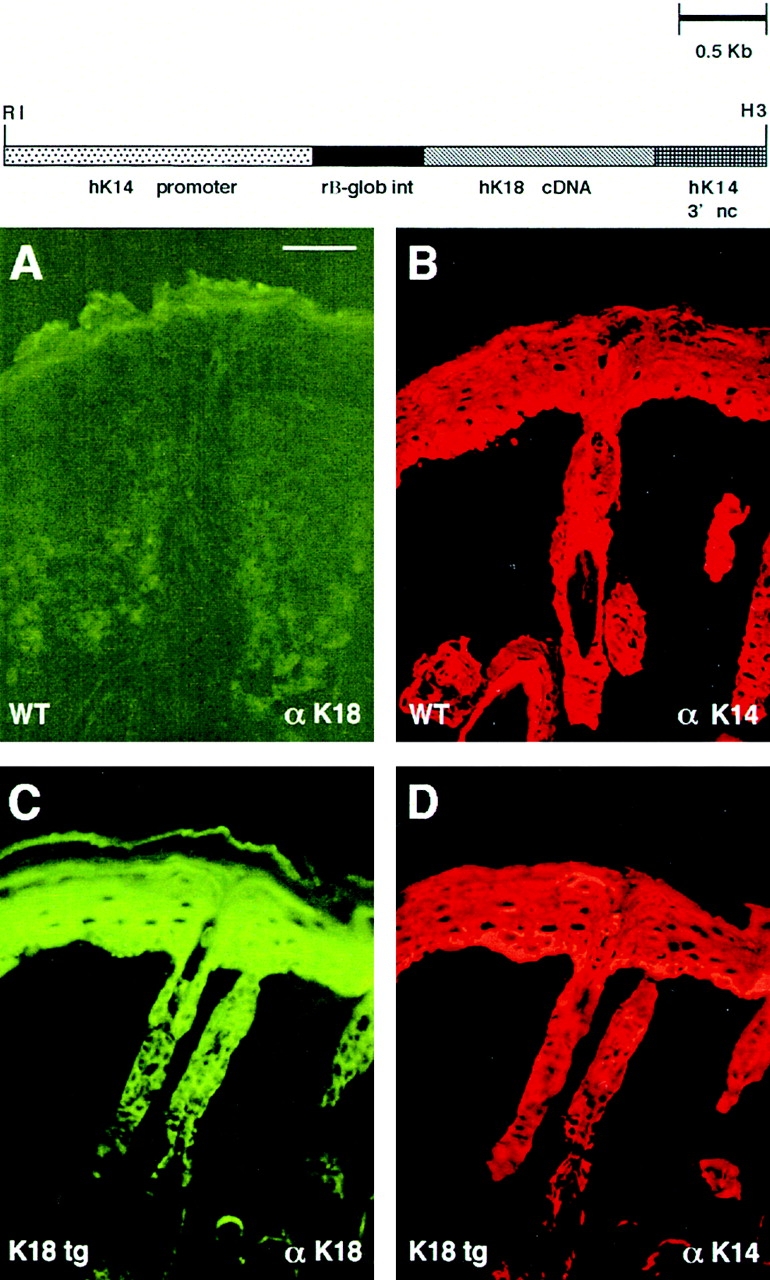
K18 replacement vector and transgene expression in mouse skin. (Top) Schematic depicting the K18 replacement vector. The backbone of the vector is as described in Fig. 1. The full-length human K18 cDNA (Oshima et al., 1986) was introduced into the BamHI site of the vector. Frozen, methanol-fixed sections of back skins (10 μm) of neonatal K18 transgenic mice (tg) on a wild-type K14 background were subjected to double-immunofluorescence microscopy using antibodies against K18 (FITC-conjugated secondary) and K14 (Texas red–conjugated secondary), respectively (see Materials and Methods). A and B, wild-type skin; C and D, K18 transgenic skin. Bar, 40 μm.
We first demonstrated that the αK18 mAb used for our studies did not cross-react with endogenous skin keratins. As judged by indirect immunofluorescence of frozen tissue from normal mice, no αK18 staining was observed (Fig. 4, A and B). In contrast to control skin, tissue from several K18 transgenic mouse lines displayed strong staining with αK18, and this paralleled the staining pattern observed with αK14 (example shown in Fig. 4, C and D, respectively). Both αK18 and αK14 staining often extended beyond the basal and into the suprabasal layers, reflective of the ability of epidermal keratins to persist long after their synthesis (Fuchs and Green, 1980; Roop et al., 1987). Overall, these data demonstrated that K18 was faithfully expressed in a pattern identical to that of K14.
To examine the behavior of K18 protein in basal epidermal keratinocytes, we conducted two-dimensional gel electrophoresis on keratins extracted from K18 transgenic mouse skin. Coomassie blue staining revealed two to three spots, not present in control skin samples, that migrated at the molecular size (∼44 kD) and pKi (5.5) expected for K18 (Fig. 5 A). The identity of these spots was confirmed by αK18 immunoblot analysis (Fig. 5 B). The size and pKi of K18 in transgenic mouse epidermis was comparable to that observed in simple epithelia (Moll et al., 1982), suggesting that its expression in epidermis did not result in any substantial processing or modifications of the protein.
Figure 5.

Polyacrylamide gel analysis of IF proteins. IF proteins were extracted from the skins of neonatal transgenic mice expressing the K18 transgene on a wild-type (A and B) or K14 null (C) background. A–C, duplicate gels were either stained with Coomassie blue (A and C) or transferred to nitrocellulose paper for immunoblot analysis with K18 (B) or K14 (not shown) antibodies. Shown are the type I keratins. K10 is far more abundant than K14 (spot below K10 in A). Note that the K14 spot, confirmed by immunoblot analysis, is missing in C. (Note: we do not know the identity of the very acidic spot in C; it does not migrate at a pKi or size known to keratins, and it was not consistently obtained in our 0.6 M KCl–insoluble extracts.) (D and E) Duplicate samples of IF proteins from wild-type (wt) and K18 transgenic (Tg) mouse skins and dilutions of purified recombinant human K18 or K14 were subjected to one-dimensional gel electrophoresis and immunoblot analysis using αK18 or αK14 antibodies. Loadings were: wt, 4 μg extract; Tg, 2 μg and 4 μg extract, respectively. Recombinant protein loadings are as indicated in nanograms.
Heterozygous K18 positive mice were mated with K14 heterozygous mice, and eventually bred further to produce K18 positive/K14 null animals. As shown in Fig. 5 C, the keratin extracts from the skins of these animals lacked the K14 spots, but still contained K18 spots of approximately the same isoelectric points, size, and levels as those seen in the transgenic skin extracts.
To assess the levels of K18 transgene expression relative to endogenous K14, we had to modify the approach we had used for the K16-14 protein (see Fig. 1 B), since K18 and K14 did not share a common antibody epitope. In the first approach, we subjected the Coomassie-stained, two-dimensional gels of transgenic samples to densitometry scanning, and compared directly the relative density of the K18 versus K14 spots. Using this method, transgene expression ranged from 5% to 45% of the wild-type K14 levels depending upon the mouse. The example shown in Fig. 5 A is from the transgenic line used for this and subsequent studies, which on a background of two K14 alleles is 35% ± 5%. In the second approach, we used one dimensional PAGE of IF extracts from this line, followed by immunoblot analysis with antibodies against K18 and K14, respectively. In this case, we used varying dilutions of purified recombinant human K18 and K14 as standards, so that we could determine the approximate number of nanograms of K18 and K14 in the transgenic skin extracts (Fig. 5, D and E). Two different anti-K18 antibodies, multiple gels, and multiple exposures were used in the quantitation, which again provided an estimate that the sample contained K18 at levels that were 35% ± 5% the levels of K14.
To further explore the K18 protein on a K14 null background, we conducted immunoblot analysis, this time using one-dimensional gel electrophoresis that enabled us to examine and compare more samples. As shown in Fig. 6, the immunoblot data verified the presence of K18 and the absence of K14 in the skin of some offspring from heterozygous matings. Moreover, the K18 isolated from these skins was found in the insoluble cytoskeletal fraction, along with the other keratins, consistent with the notion that it existed as part of the keratin network. The levels of K18 were at least comparable to, if not higher than, those in the K18 transgenic mice.
Figure 6.

Immunoblot analyses of skin IF proteins from wild-type, K14 null, K18 transgenic, and K18 rescue mice. Triton X-100– insoluble and –soluble protein extracts were prepared from back skins of 1–2-d-old mice, resolved by electrophoresis through 10% SDS–polyacrylamide gels (Wu et al., 1982) and transferred to nitrocellulose paper. The blot was then sequentially hybridized with antibodies against K18, K14, and K5. After each hybridization, bound antibody was visualized by chemiluminescence (Amersham Corporation), and the blot was then stripped to remove the bound antibody before proceeding with the next antibody. Extracts are as indicated: WT, wild-type; KO, K14 null; K18 tg, K18 transgenic; and K18 res, two different lines of K18 rescue. Note that both transgene protein and endogenous keratins reside in the insoluble fraction. Molecular mass standards at right in kD.
Immunofluorescence of skin sections from the K18 transgenic/K14 null mice revealed a pattern of αK18 antibody staining that was similar to what we saw in the transgenic mouse skin; in this case, however, K14 was absent (Fig. 7, A and B, respectively). This K18 was strictly due to transgene expression and not to induction of endogenous K18, since no αK18 staining was detected in K14 null skin (not shown). The pattern of expression of K18 paralleled that of K5, the normal partner of K14 (Fig. 7, C and D). The exchange that we genetically engineered between K18 and K14 did not result in the induction within the basal layer of K6 and K16, the keratin pair known to be expressed normally in the outer root sheath of the hair follicle (see Fig. 7 E) and induced upon wound healing and a variety of hyperproliferative disorders in the skin (Sun et al., 1984; Mansbridge and Knapp, 1987; Paladini et al., 1996). Thus, the genetic exchange appeared to occur in the absence of other alterations in keratin expression.
Figure 7.
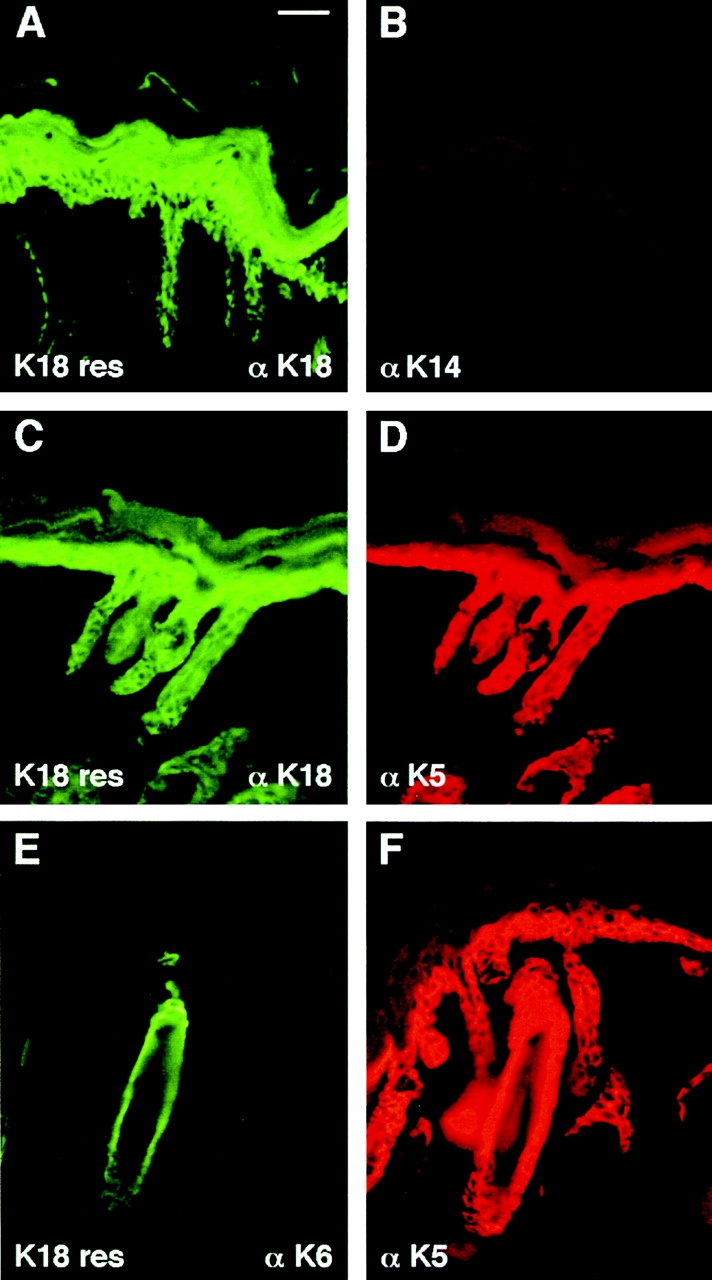
K18 expression is still maintained and does not induce K6 when K18 transgenic mice are bred onto a K14 null background. Neonatal back skins and paw skins of control (not shown) and K18+/K14 null mice (K18 res) were frozen and methanol-fixed, sectioned (10 μm), and then stained with antibodies against the keratins indicated. (Note: depending upon the fixation/processing conditions, antibody staining of K18 sometimes extended into the suprabasal layers. This happened inconsistently in control as well as transgenic skin, and in all cases, was always paralleled by an identical staining pattern with αK5.) Note the wild-type staining pattern of αK6 in the outer root sheath of hair follicles; suprabasal epidermal induction of K6, a typical marker of hyperproliferative disorders did not occur in K18 rescue skin. Bar, 40 μm.
Expression of the K18 Transgene on a K14 Null Background Restores Back Skin Morphology, but Not Paw Blistering
When bred on either a K14 wild-type background or a K14 +/− heterozygous background, our K18 transgenic mice appeared phenotypically and histologically indistinguishable from the wild type. These animals lived to adulthood, showed no loss in viability, and developed no signs of aberrations in their skin or hair coat. This was important, since there are a number of cases where ectopic expression of a keratin can induce phenotypic changes (Powell and Rogers, 1990; Blessing et al., 1993; Paladini and Coulombe, 1998).
Even when bred to produce K18+/K14 null mice, certain features of the animals were typical of wild-type mice. Most notably, the back skin morphology of K18+/K14 null mice was similar to that of wild-type back skin, exhibiting no signs of basal cell cytolysis or of alterations in terminal differentiation (Fig. 8 A). Within 2–3 d after birth, however, the paws of these mice began to display signs of EBS, including signs of basal cell degeneration within the cytoplasm of basal cells (Fig. 8 B). In most regions of the ventral paw, basal epidermal cells showed gross signs of cytolysis (Fig. 8 C), and often the basal layer was completely degenerated, leading to detachment of the epidermis, and only remnants of basal cells attached to the blister floor (Fig. 8 D). Typical of EBS, suprabasal layers remained intact, reflective of expression of K1 and K10, concomitant with downregulation of K5 and K14 in these layers (Fuchs and Green, 1980; Roop et al., 1987). However, in both severely cytolyzed and in detached regions, the overall morphology of the epidermis was distorted, suggesting that significant perturbations had occurred in the biochemistry of these cells, presumably beginning before the time at which they exited the basal layer.
Figure 8.
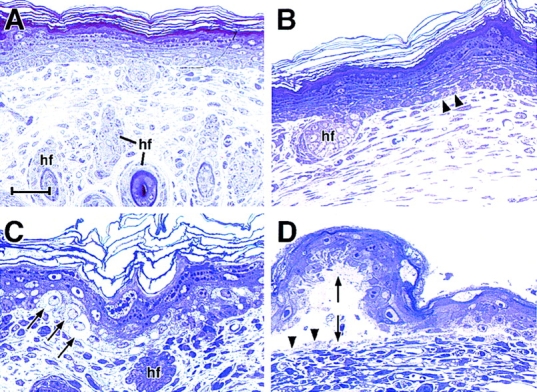
K18 expression does not rescue the blistered paw phenotype of K14 null mice. Neonatal back skins and paw skins from K18+/K14+, K18+/K14−, and wild-type littermates were embedded in Epon, sectioned (0.75 μm), and then stained with Toluidine blue. Sections shown are from K18 transgenic skin on a K14 null background. (A) back skin, showing no obvious abnormalities; (B) paw skin, depicting early signs of basal cell degeneration (arrowheads); (C) paw skin showing clear signs of basal cell cytolysis (arrows); (D) paw skin showing blister resulting from completely degenerated basal epidermal layer. Double arrow, blister; arrowheads in D, fragments of basal cells left on the blister floor, indicative of basal cell rupturing. Bars: (A and B) 40 μm; (C and D) 20 μm.
The phenotype of these mice was sufficiently severe that they died within several days after birth unless given special care. While we did not analyze internal complications in detail, the mice also suffered from degeneration of the tongue and oral epithelia, places where the K14 promoter is known to be active. Taken together, the expression of K18 seemed to restore some, but clearly not all, of the defects caused by removal of K14 expression in mice.
Restoration of the 10-nm Filament Network in Basal Cells of K18+/K14 Null Back Skin, but Not Paw Skin
To evaluate the effects of K18 expression on cytoskeletal architecture, we first examined the basal keratinocytes in the paw and back skin of K18 transgenic mice on a wild-type background. At the ultrastructural level, filament bundles within K18 basal epidermal cells appeared indistinguishable from those in wild-type basal epidermal cells, and no signs of keratin clumps or other perturbations were detected (Fig. 9 A). Desmosomes and hemidesmosomes appeared normal, and both were surrounded by densely staining keratin, as expected for the comparable wild-type structures. Basal keratinocytes cultured from the skin of K18 transgenic mice also displayed a seemingly normal keratin network, which colabeled with antibodies against K18, K5, and K14 (data not shown). Taken together, these findings were in agreement with similar in vitro findings by Lu and Lane (1990), who demonstrated that K18 is able to integrate into an epidermal keratin network without perturbation. The result was also consistent with the normal appearance of the K18 transgenic mice.
Figure 9.
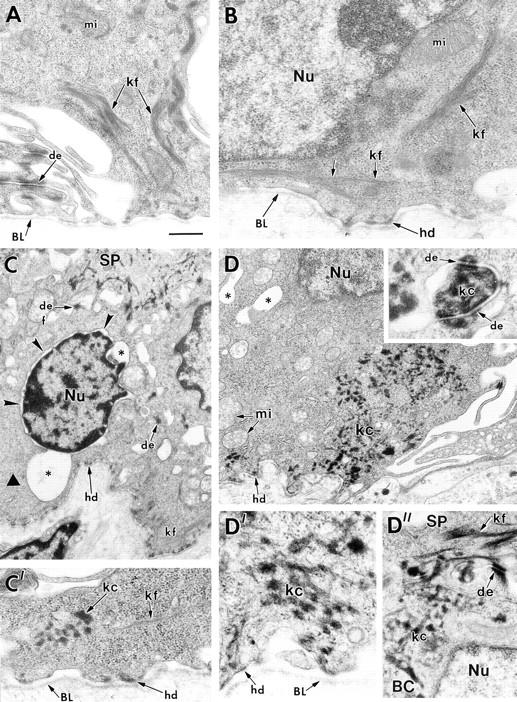
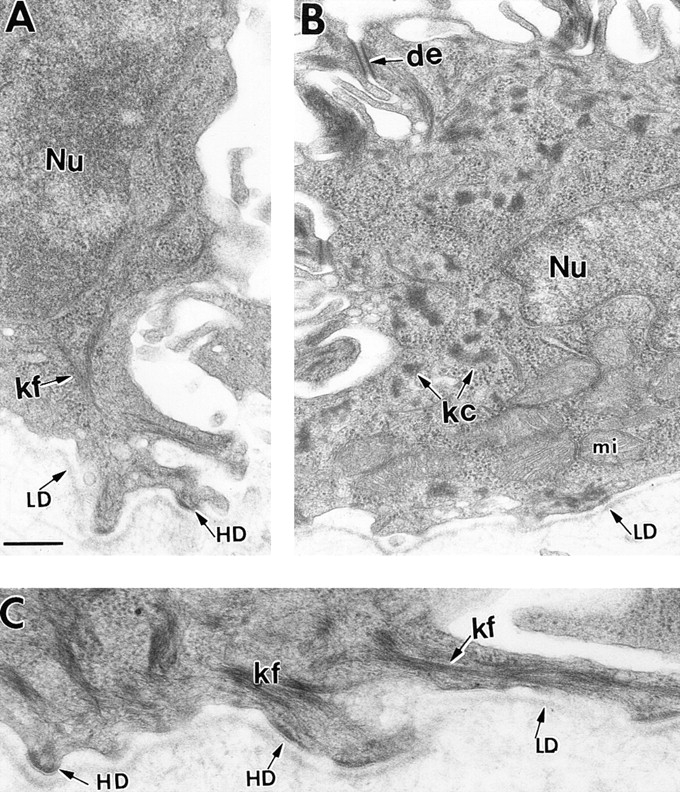
Ultrastructure of basal cells from the back skins and paw skins of K18 transgenic mice bred on either a wild-type or a K14 null background. Skins of 1–2-d-old K18 transgenic mice on either a wild-type or K14 null background were processed for electron microscopy as described in the Materials and Methods. (A) basal cells from K18 transgenic/K14 wild-type back skin, depicting normal keratin filament bundles (kf) and desmosomes (de). Paw skin showed similar morphology. (B– D) basal cells from K18 transgenic/K14 null back skin (B and C) or paw skin (D). The majority of basal cells in back skin displayed normal morphology and keratin filament bundles, similar to that seen in B. An occasional basal cell from back skin exhibited signs of cytolysis (asterisks and small arrowheads), with some regions of the cytoplasm devoid of keratin filaments (large arrowhead in C) and other regions showing some small aggregates or clumps of keratin (C′, kc). Many cells from paw skin displayed prominent clumping of keratin both in the cytoplasm and associated with the desmosomes (D and inset to D, respectively). D′ and D″ show higher magnification to visualize these clumps of keratin in more detail. Note that spinous cells (SP) contained a largely normal keratin network, reflective of the induction of K1 and K10 in these layers. BL, basal lamina; mi, mitochondria; hd, hemidesmosome; Nu, nucleus; BC, basal cell. Bar in A: (A) 0.4 μm; (B, C′, D′, D″) 0.3 μm; (C) 0.9 μm; (D) 0.8 μm; (inset to D) 0.1 μm.
We next examined the cytoskeletal architecture of the skin of K18 positive/K14 null mice. A number of the back skin basal cells of these mice displayed seemingly normal cytoskeletal networks, with bundles of cytoplasmic keratin filaments connecting to hemidesmosomes and desmosomes (Fig. 9 B). This was in contrast to the back skin of K14 null mice, but it was similar to that seen in the K16-14 rescue mice (Fig. 3). In these regions of the skin, no signs of microblisters were detected, and the basal cells appeared generally healthy.
Whereas the majority of back skin basal cells from K18 transgenic/K14 null mice appeared ultrastructurally normal, an occasional cell exhibited signs of degeneration (Fig. 9 C, asterisk). Some vacuolization was also seen, a feature also frequently found in Weber-Cockayne EBS, the mildest form of human EBS (for review see Anton-Lamprecht, 1994; Fuchs and Cleveland, 1998). A few bundles of keratin filaments were still detected in these cells, but regions of the cytoplasm were devoid of such filaments (Fig. 9 C, large arrowhead). In a few regions of the cytoplasm of these cells, a mixture of filaments and aggregates of keratin-like material were detected (Fig. 9 C′). Clumps of keratin were not seen in the K14 null skin (Lloyd et al., 1995), but are characteristic of dominant-negative acting keratin mutants in mice and in humans (Vassar et al., 1991; Anton-Lamprecht, 1994).
Abnormalities in keratin networks were significantly more prominent in the basal cells of the paw skin of K18 positive/K14 null mouse skin, i.e., in regions where overt skin blistering was also readily detected (Fig. 9 D). In many of these cells, large areas of cytoplasm were devoid of keratin, while other areas were densely packed with clumps or aggregates of keratin-like material. These clumps often associated with desmosomes (Fig. 9 D, inset), and at higher magnification, they appeared as aggregates of short filamentous-like structures (D′). In Fig. 9 D″, depicting the junction between a basal cell (BC) and a spinous cell (SP), note that the spinous cells contained an array of keratin filaments; this is again consistent with the switch to expression of K1 and K10 in terminally differentiating layers.
Immunoelectron microscopy confirmed that the clumps of material in the basal cells were keratin, and that they labeled not only with antibodies against K18, but also with antibodies against K5 (Fig. 10). Since these clumps were not detected in K14 null mice, we conclude that the clumps represent a new structure produced from a combination of K18 and K5, or possibly other cytoplasmic proteins.
Figure 10.
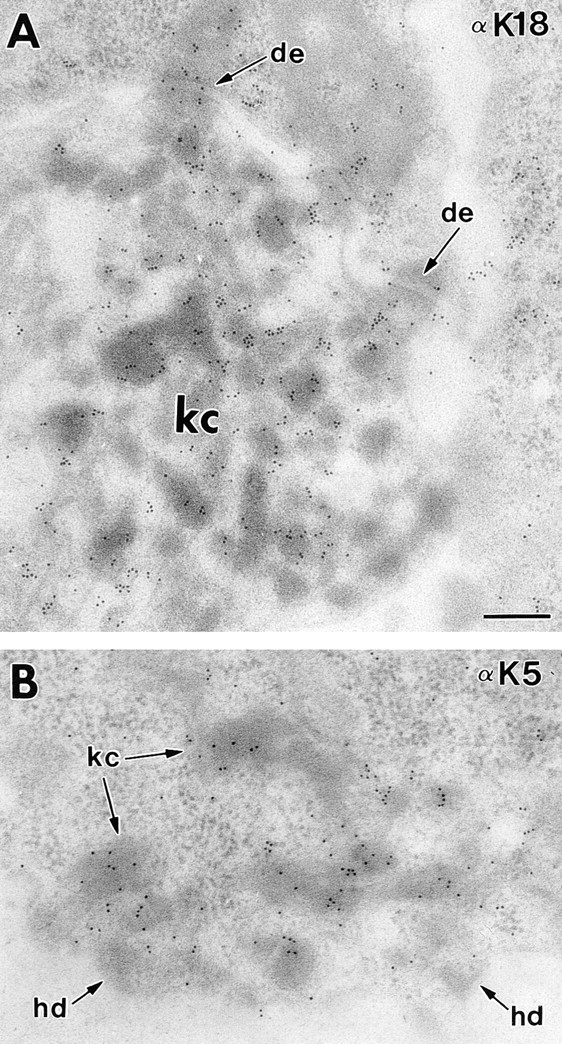
Keratin clumps in K18 transgenic/K14 null basal cells contain a mixture of K5 and K18. Paw skin from K18 transgenic/ K14 null animals was embedded in Lowicryl and subjected to 30-nm gold labeling with antibodies against either K18 (A) or K5 (B) as described previously (Coulombe et al., 1989). de, desmosome; hd, hemidesmosome; kc, keratin clumps. Bar, 0.2 μm.
The Keratin Network in K18 Replacement Skin Cannot Withstand Mechanical Trauma
A priori, the detection of keratin clumps in the K18 replacement skin could imply that the assembly process is defective, or that there is an excess of K5 relative to K18. Either case might result in the accumulation of partially polymerized keratin material. Alternatively, it could be an indication that the resulting keratin network formed by K18/K5 filaments is unstable, and collapses partially upon mechanical stress. To distinguish between these possibilities, we conducted an experiment to see how the basal epidermal layer would perform under mechanical stress.
Four 2-d-old K14 null/K18+ replacement mice and two control littermates were subjected to mild rubbing of the back skin, which was then processed for ultrastructural analysis immediately thereafer (within 10 min). In each case, only the left side of the middle lower back was rubbed (12 times with a cotton-tipped applicator); the right side of each mouse was not rubbed, and was used as a control for each mouse.
Some basal epidermal cells of rubbed, K18 transgenic/ K14 null skin survived the rubbing, as judged by the presence of a seemingly normal keratin filament network and generally healthy cytoplasm (Fig. 11 A). However, a number of cells displayed clumps or aggregates of keratin material (Fig. 11 B). The aberrancies in keratin networks were similar, but more extensive than those seen in occasional back skin cells of unrubbed K18 replacement skin and in many paw skin cells. As expected, we did not observe signs of cell cytolysis, a delayed response which in human EBS occurs subsequent to alterations in cytoskeletal architecture (for review see Anton-Lamprecht, 1994).
Figure 11.
In K18 transgenic/K14 null basal cells, the morphological deviations from a wild-type keratin network become more pronounced upon mechanical stress. Two separate litters of neonatal mice were used for these studies. For each litter, two K18 transgenic/K14 null and one wild-type littermates were subjected to lateral rubbing of the skin on one side of the back, and no rubbing on the other side. After the rubbing experiment, skins from each side of the back of each animal were processed for conventional electron microscopy. Skin samples were analyzed for alterations in the morphology of the basal keratin network. Many of the basal cells maintained a proper keratin network (A). Approximately 20% of the basal cells of K18 transgenic/K14 null back skin showed signs of keratin clumping on the rubbed side (B). The majority of the basal cells from unrubbed back skin displayed keratin filaments typical of wild-type skin (not shown). Wild-type skin showed abundant keratin filament bundles and no keratin clumps irrespective of whether the skin was rubbed or not (C). Bar in A: (A and B) 0.4 μm; (C) 0.3 μm.
In contrast to the basal cells from rubbed back skin of the K18+/K14 null mice, cells from similarly rubbed back skin of control mice appeared uniformly healthy, with little or no perturbations in overall morphology (Fig. 11 C). Taken together, our findings suggest that the perturbations detected in basal cells of K18 replacement skin are accentuated by mechanical stress.
When Combined in the Presence of 6 M Urea, K5 and K18 Associate in a 1:1 Ratio to Form a Complex That Then Assembles into Filaments Efficiently In Vitro
Our in vivo studies suggested that K5 and K18 can assemble into keratin filaments. To test this hypothesis in vitro and analyze this interaction in more detail, we first demonstrated that K5 and K18 can form stable heterotypic complexes under 6 M urea buffer conditions. We showed this by anion exchange chromatography (not shown) and by chemical cross-linking (Fig. 12 A). Cross-linking using BS3 showed that the major K5-K18 and K5-K14 species formed under these conditions was a 240-kD product, with small amounts of an ∼130-kD product (Fig. 12 A). As shown previously (Coulombe and Fuchs, 1990), these species correspond to covalently cross-linked heterotetramers and heterodimers of keratins, respectively. When the urea concentration was raised to 8 M under otherwise identical conditions, the K5-K18 tetramers were destabilized to a greater extent than K5-K14 ones (data not shown). Taken together, these data show that K18 can readily form heterotetramers in combination with K5, although these are slightly less stable than those formed from K5-K14.
Figure 12.

Assembly of K18 and K5 into 10-nm keratin filaments. (A and B) Pure preparations of K5, K18, and K14 in 6 M urea buffers were obtained by FPLC chromatography, as described by Coulombe and Fuchs (1990). Keratin-rich fractions were pooled, and the protein concentration measured using an assay kit (Bio-Rad Laboratories). Equimolar mixtures of K5/K18 or K5/K14 were subjected to anion-exchange chromatography to purify the heteromeric complexes. Complexes were confirmed by chemical cross-linking with BS3 as described previously (Wawersik et al., 1997). Samples of unlinked (−) and cross-linked (+) proteins were subjected to SDS-PAGE, and the gel was stained with Coomassie blue to visualize the protein. D, dimer; T, tetramer. (B and C) Complexes of either K5/K18 (B) or K5/K14 (C) were subjected to in vitro filament assembly as described in Materials and Methods. Filaments were visualized under a Philips CM10 electron microscope. Bar, 100 nm.
When dialyzed under standard epidermal keratin assembly buffer, K5-K18 heterotypic complexes polymerized efficiently (90%) into 10-nm filaments that are similar to those formed by K5-K14 (>95% efficiency) (Fig. 12, compare B and C, respectively). Under in vitro assembly conditions, it was not possible to distinguish reliably those filaments formed from K5-K18 versus those formed from K5-K14. Taken together with our in vivo data, these results provide further evidence that K18 is an effective partner for K5, and that the failure of K18 to fully rescue the EBS phenotype resides in the physical properties of the filament network, rather than in K18's ability to assemble into a 10-nm filament network.
Discussion
Previously, we showed that K14 null animals exhibit blistering upon physical stress, a feature that is due to mechanically induced degeneration in their epidermal basal layer (Lloyd et al., 1995). This study provided compelling evidence that the epidermal keratin network functions to impart mechanical integrity to keratinocytes. Stress-induced blistering of the basal epidermal layer is the hallmark of EBS in humans, and it is known that genetic alterations in the K5 and K14 network are responsible for this disorder (Bonifas et al., 1991; Coulombe et al., 1991; Lane et al., 1992). Although human EBS typically involves keratin filament–disrupting mutations in the K5 or K14 genes and is inherited in an autosomal dominant fashion, rare cases of recessive homozygous mutations have been described that show a dramatic reduction in keratin filaments within the basal layer of the skin (Chan et al., 1994; Rugg et al., 1994). True K14 null mutations have been engineered in mice, and these mice exhibit an EBS phenotype (Lloyd et al., 1995).
Our previous studies on K14 null mice demonstrated that in the absence of K14, there is no dramatic upregulation of other type I keratins to provide compensation for the loss of this keratin (Lloyd et al., 1995). In the absence of a partner, the basal type II keratin K5 was still present, but unable to form a keratin filament network. This was consistent with predictions made from in vitro filament assembly studies arguing that K5 and K14 form obligatory heterodimers (Coulombe and Fuchs, 1990; see also Hatzfeld and Weber, 1990; Steinert, 1990). In the absence of K14, there is a very sparse keratin filament network made in the basal layer, resulting from the presence of very low levels of K15 in postnatal mouse skin (Lloyd et al., 1995). However, this sparse network was not able to sustain the mechanical stress normally handled by the more robust bundles of K5-K14 filaments.
To test the possibility that a non-epidermal type I keratin might be able to restore mechanical stability to the basal epidermal keratin network, we chose K18 as our replacement for K14. Indeed, K8/K18 are the major keratins of simple epithelial tissues just as K5 and K14 are the key keratins of stratified epithelia (Moll et al., 1982). Moreover, in contrast to most keratins, these two keratin pairs are often expressed in the absence of any other keratins, establishing that in their respective tissue, they can on their own form extensive keratin networks. Since the dividing populations of simple epithelia and epidermis are so dramatically different in their morphology and in their physical properties, we felt that this replacement would provide a major test of whether there are fundamental differences in the two respective keratin networks.
On a wild-type background, our K18 transgenic mice displayed no overt phenotype and showed no loss of viability with age. This was also true for K18 transgenic mice on a heterozygous K14 background, where the levels of K18 approached those of K14. We have cultured the transgenic basal epidermal cells, and they too behaved normally and exhibited a normal keratin network that labeled with antibodies against endogenous basal keratins or K18 (Hutton, E., and E. Fuchs, unpublished observations). Thus, both in vivo and in vitro, K18 seemed to integrate into the endogenous K5-K14 keratin network without any noticeable perturbations. Moreover, in vivo, a basal keratin network composed of K5, K14, and K18 seemed able to withstand the natural environmental stresses in a fashion analogous to the K14-K5 network. This was the case irrespective of whether the mice contained one or two of their K14 alleles.
Overall, the behavior of the transgenic K18 mice seemed to be markedly different from that of transgenic K16 mice generated using the same K14 basal epidermal promoter: in that case, expression of K16 at >60% the levels of endogenous K14 resulted in skin that was hyperkeratotic, scaly and devoid of fur (Paladini and Coulombe, 1998). It could be that the differences in K18 versus K16 transgenic phenotypes stem from the higher level of expression (or increased stability) of K16 than K18. However, it seems more likely that these differences stem from inherent differences in keratins, particularly since the K16-14 hybrid protein behaved normally when expressed in mice at comparable levels to K16 (Paladini and Coulombe, 1998). Taken together, even at the transgenic level, there appear to be differences in the behavior of type I keratins expressed under the control of a basal epidermal keratin promoter.
Even on a K14 null background, transgenic K18 still functioned quite well in back skin, where it coassembled with K5 to form a network of keratin filaments in the basal epidermal layer. That K5 and K18 can form bona fide 10-nm filaments in the absence of K14 was also confirmed from our in vitro studies. These in vitro–assembled K18/ K5 filaments were similar in structure, appearance, and concentration to the epidermal counterparts. Taken together, these findings indicate that K18 is a suitable partner for K5 in keratin filament assembly.
The keratin filaments formed by K5 and K18 in vivo associated both with hemidesmosomes and with desmosomes in the basal epidermal layer. While the molecular details of the interaction between hemidesmosomes and IFs have not been fully elucidated, in vitro studies suggest that the interaction between desmosomes and keratin filaments occurs through binding of the desmoplakin tail segment to the head domain of type II epidermal keratins (Kouklis et al., 1994; Kowalczyk et al., 1997). In the K18 positive/K14 null mice, the K5 head domain is presumably still available to associate with desmoplakin, and this could explain why we did not observe any perturbation in keratin filament attachments to cell junctions. Interestingly, the interaction between K8 and desmoplakin seems to be much weaker than that with K5 (Kouklis et al., 1994), leading us to wonder whether desmosomal interactions might have been compromised if we had exchanged the type II rather than the type I keratins. The answer to this question must await further replacement studies.
Despite the ability of K5 and K18 to assemble into filaments and to make a keratin filament network in basal epidermal cells, K18 did not fully compensate for the loss of K14, even at the highest levels that we were able to achieve in the present study. These mice still died within several days after birth, presumably because of both internal and external complications (see Lloyd et al., 1995 for a description of internal abnormalities in the K14 null mice). Within the skin, the severity in blistering and perturbations in the keratin network seemed to correlate with degree of mechanical stress, as judged by the fact that the pathology was most prominent in paw skin, a region of natural stress, and it was exacerbated in response to physical rubbing of back skin. Taken together, these observations suggest a model whereby the K18/K5 network assembles, but is unable to withstand mechanical stress, leading to a collapse of the network and skin blistering. In the future, cytoskeletal tension measurements might be used to address this hypothesis directly.
The current limitations of keratinocyte promoter activity and/or K18 turnover rates precluded our examining the consequences of expressing higher levels of K18, and thus we cannot say whether comparable to wild-type K14 levels of K18 might better compensate for the loss of K14 in our mice. This said, mice and humans harboring only a single functional K14 allele behave normally (Chan et al., 1994; Rugg et al., 1994; Lloyd et al., 1995), and in the present study, K14 null mice expressing the K16-14 transgene survived to adulthood and showed no overt signs of skin blistering. Thus, while gross differences in filament density can certainly influence basal epidermal cell survival (Chan et al., 1994; Rugg et al., 1994; Lloyd et al., 1995), more subtle differences in filament density may be less important than the particular keratins involved in conferring mechanical integrity to a tissue.
Our data lend support to the notion that the specific keratins composing an IF network in vivo are central to cytoskeletal architecture and design in cells (for recent related papers on this subject, see Paladini et al., 1996; Paladini and Coulombe, 1998). Before our study, a number of differences had already been recognized between K8/K18 and K5/K14. For instance, the physical properties of K8/ K18 heterodimers and filaments assembled from them differ markedly from K5/K14 heterodimers and their corresponding filaments (Franke et al., 1983; Hatzfeld and Franke, 1985; Coulombe and Fuchs, 1990; Hatzfeld and Weber, 1990; Hoffmann and Franke, 1997). Moreover, in vivo, K8/K18 networks can reorganize during mitosis in some cells, whereas K5/K14 networks do not, suggesting the possibility that K8/K18 networks may be more dynamic than K5/K14 networks (Franke et al., 1983). Dramatic sequence and size differences in the head and tail segments and in the posttranslational modifications of keratins are likely to account for at least a part of these dramatic variations in keratin networks (Lu and Lane, 1990; Liao et al., 1995 and references therein; for review see Fuchs and Weber, 1994). It is tempting to speculate that K18 cannot fully compensate for K14 because it creates a more dynamic keratin network that is not entirely compatible with the structural requirements imposed upon a tissue like the epidermis.
The seemingly less stable aspects of the K5/K18 network could also have arisen from genetically forcing an undesirable partner upon K5. That keratins have distinct preferences for their partners has been recently inferred from K18 knockout studies, where when left without its normal partner, K8 can successfully compete for K19, normally the partner of K7 (Magin et al., 1998). To assess whether K8/K18 can replace K5/K14 must await further replacement studies.
In summary, our study suggests that the multiplicity of keratin sequences is not simply an evolutionary quirk, but that these proteins have diverged to perform specific functions in higher eukaryotic epithelia. Whereas keratin networks are a universal feature of epithelia, their density, composition, and organization vary dramatically and seem to be tailored to the varied shapes and structural requirements of individual epithelial cells.
Acknowledgments
This work was supported by grants from the National Institutes of Health (AR27883, to E. Fuchs; AR44232 to P. Coulombe). E. Fuchs is a Howard Hughes Medical Institute investigator.
Abbreviations used in this paper
- BS3
bis(sulfosuccinimidyl)suberate
- EBS
epidermis bullosa simplex
- IF
intermediate filament
Footnotes
A special thank you goes to L. Degenstein for her help in transgenic mice aspects of this work, and in conducting the mouse skin rubbing experiments. We thank D. Dugger for technical assistance in transgenic mouse engineering; G. Strasser and D. Lourim for technical assistance in some phases of the cell and molecular biology; Dr. C. Bauer for his expert assistance in electron microscopy and in assisting with some of the photography and figure preparations; E. Smith and C. Wellek for their help with the computer-assisted art work; J. Fradette (University Laval, Quebec, Canada) for providing the chemical cross-linking data; Dr. M.B. Omary (Stanford University, Palo Alto, CA) for providing the pET-K18 bacterial expression clone; and Dr. D. Roop (Baylor University School of Medicine) for his gift of anti-K6 antiserum.
Address all correspondence to Elaine Fuchs, Howard Hughes Medical Institute, Department of Molecular Genetics and Cell Biology, The University of Chicago, 5841 S. Maryland Avenue, Room N314, MC1028, Chicago, IL 60637. Tel.: (773) 702-1347. Fax: (773) 702-0141.
References
- Allen E, Yu Q-C, Fuchs E. Abnormalities in desmosomes, proliferation and differentiation in the epidermis of mice expressing a mutant desmosomal cadherin. J Cell Biol. 1996;133:1367–1382. doi: 10.1083/jcb.133.6.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton-Lamprecht I. Ultrastructural identification of basic abnormalities as clues to genetic disorders of the epidermis. J Invest Dermatol. 1994;103:65–125. doi: 10.1111/1523-1747.ep12398887. [DOI] [PubMed] [Google Scholar]
- Baribault H, Price J, Miyai K, Oshima RG. Mid-gestational lethality in mice lacking keratin 8. Genes Dev. 1993;7:1191–1201. doi: 10.1101/gad.7.7a.1191. [DOI] [PubMed] [Google Scholar]
- Baribault H, Penner J, Iozzo RV, Wilson-Heiner M. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 1994;8:2964–2973. doi: 10.1101/gad.8.24.2964. [DOI] [PubMed] [Google Scholar]
- Blessing M, Ruther U, Franke W. Ectopic synthesis of epidermal cytokeratins in pancreatic islet cells of transgenic mice interferes with cytoskeletal order and insulin production. J Cell Biol. 1993;120:743–755. doi: 10.1083/jcb.120.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifas JM, Rothman AL, Epstein EH. Epidermolysis bullosa simplex: evidence in two families for keratin gene abnormalities. Science. 1991;254:1202–1205. doi: 10.1126/science.1720261. [DOI] [PubMed] [Google Scholar]
- Byrne C, Tainsky M, Fuchs E. Programming gene expression in developing epidermis. Development (Camb) 1994;120:2369–2383. doi: 10.1242/dev.120.9.2369. [DOI] [PubMed] [Google Scholar]
- Chan Y-M, Anton-Lamprecht I, Yu Q-C, Jackel A, Zabel B, Ernst J-P, Fuchs E. A human keratin 14 “knockout”: the absence of K14 leads to severe epidermolysis bullosa simplex and a function for an intermediate filament protein. Genes Dev. 1994;8:2574–2587. doi: 10.1101/gad.8.21.2574. [DOI] [PubMed] [Google Scholar]
- Cheng J, Syder AJ, Yu Q-C, Letai A, Paller AS, Fuchs E. The genetic basis of epidermolytic hyperkeratosis: a disorder of differentiation-specific epidermal keratin genes. Cell. 1992;70:811–819. doi: 10.1016/0092-8674(92)90314-3. [DOI] [PubMed] [Google Scholar]
- Chipev CC, Korge BP, Markova N, Bale SJ, DiGiovanna JJ, Compton JG, Steinert PM. A leucine→ proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell. 1992;70:821–828. doi: 10.1016/0092-8674(92)90315-4. [DOI] [PubMed] [Google Scholar]
- Coulombe P, Fuchs E. Elucidating the early stages of keratin filament assembly. J Cell Biol. 1990;111:153–169. doi: 10.1083/jcb.111.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe PA, Kopan R, Fuchs E. Expression of keratin K14 in the epidermis and hair follicle: Insights into complex programs of differentiation. J Cell Biol. 1989;109:2295–2312. doi: 10.1083/jcb.109.5.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe PA, Hutton ME, Letai A, Hebert A, Paller AS, Fuchs E. Point mutations in human keratin 14 genes of epidermolysis bullosa simplex patients: genetic and functional analyses. Cell. 1991;66:1301–1311. doi: 10.1016/0092-8674(91)90051-y. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick, T.B., A.Z. Eisen, K. Wolff, I.M. Freedberg, and K.F. Austen. 1993. Dermatology in general medicine. Vol. I and II. McGraw-Hill, Inc., New York. 2979 pp.
- Franke WW, Schiller DL, Hatzfeld M, Winter S. Protein complexes of intermediate-sized filaments: melting of cytokeratin complexes in urea reveals different polypeptide separation characteristics. Proc Natl Acad Sci USA. 1983;80:7113–7117. doi: 10.1073/pnas.80.23.7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E, Cleveland D. A structural scaffolding of intermediate filaments in health and disease. Science. 1998;279:514–519. doi: 10.1126/science.279.5350.514. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Green H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell. 1980;19:1033–1042. doi: 10.1016/0092-8674(80)90094-x. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Weber K. Intermediate filaments: structure, dynamics, function, and disease. Ann Rev Biochem. 1994;63:345–382. doi: 10.1146/annurev.bi.63.070194.002021. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Esteves RA, Coulombe PA. Transgenic mice expressing a mutant keratin 10 gene reveal the likely genetic basis for epidermolytic hyperkeratosis. Proc Natl Acad Sci USA. 1992;89:6906–6910. doi: 10.1073/pnas.89.15.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzfeld M, Franke WW. Pair formation and promiscuity of cytokeratins: formation in vitro of heterotypic complexes and intermediate-sized filaments by homologous and heterologous recombinations of purified polypeptides. J Cell Biol. 1985;101:1826–1841. doi: 10.1083/jcb.101.5.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzfeld M, Weber K. The coiled coil of in vitro assembled keratin filaments is a heterodimer of type I and II keratins: Use of site-specific mutagenesis and recombinant protein expression. J Cell Biol. 1990;110:1199–1210. doi: 10.1083/jcb.110.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann I, Franke WW. Heterotypic interactions and filament assembly of type I and type II cytokeratins in vitro: viscometry and determinations of relative affinities. Eur J Cell Biol. 1997;72:122–132. [PubMed] [Google Scholar]
- Kouklis P, Hutton E, Fuchs E. Making the connection: Keratin intermediate filaments and desmosomes proteins. J Cell Biol. 1994;127:1049–1060. doi: 10.1083/jcb.127.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk AP, Bornslaeger EA, Borgwardt JE, Palka HL, Dhaliwal AS, Corcoran CM, Denning MF, Green KJ. The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin–plakoglobin complexes. J Cell Biol. 1997;139:773–784. doi: 10.1083/jcb.139.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N, Omary MB. Identification of the major physiologic phosphorylation site of human keratin 18: Potential kinases and a role in filament reorganization. J Cell Biol. 1994;127:161–171. doi: 10.1083/jcb.127.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N, Michie S, Oshima RG, Omary MB. Chronic Hepatitis, hepatocyte fragility, and increased soluble phosphoglycokeratins in transgenic mice expressing a keratin 18 conserved arginine mutant. J Cell Biol. 1995;131:1303–1314. doi: 10.1083/jcb.131.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N, Michie SA, Soetikno RM, Resurreccion EZ, Broome RL, Oshima RG, Omary MB. Susceptibility to hepatotoxicity in transgenic mice that express a dominant-negative human keratin 18 mutant. J Clin Invest. 1996;98:1034–1046. doi: 10.1172/JCI118864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku NO, Wright TL, Terrault NA, Gish R, Omary MB. Mutation of human keratin 18 in association with cryptogenic cirrhosis. J Clin Invest. 1997;99:19–23. doi: 10.1172/JCI119127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane EB, Rugg EL, Navsaria H, Leigh IM, Heagerty AHM, Ishida-Yamamoto A, Eady RAJ. A mutation in the conserved helix termination peptide of keratin 5 in hereditary skin blistering. Nature. 1992;356:244–246. doi: 10.1038/356244a0. [DOI] [PubMed] [Google Scholar]
- Lersch R, Stellmach V, Stocks C, Giudice G, Fuchs E. Isolation, sequence, and expression of a human keratin K5 gene: transcriptional regulation of keratins and insights into pairwise control. Mol Cell Biol. 1989;9:3685–3697. doi: 10.1128/mcb.9.9.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Coulombe PA, McCormick MB, Yu Q-C, Hutton E, Fuchs E. Disease severity correlates with position of keratin point mutations in patients with epidermolysis bullosa simplex. Proc Natl Acad Sci USA. 1993;90:3197–3201. doi: 10.1073/pnas.90.8.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leube RE, Bader BL, Bosch FX, Zimbelmann R, Achtstaetter T, Franke WW. Molecular characterization and expression of the stratification-related cytokeratin 4 and 15. J Cell Biol. 1988;106:1249–1261. doi: 10.1083/jcb.106.4.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Lowthert LA, Ku N, Fernandez R, Omary MB. Dynamics of human keratin 18 phosphorylation: Polarized distribution of phosphorylated keratins in simple epithelial tissues. J Cell Biol. 1995;131:1291–1301. doi: 10.1083/jcb.131.5.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd C, Yu Q-C, Cheng J, Turksen K, Degenstein L, Hutton E, Fuchs E. The basal keratin network of stratified squamous epithelia: Defining K15 function in the absence of K14. J Cell Biol. 1995;129:1329–1344. doi: 10.1083/jcb.129.5.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loranger A, Duclos S, Grenier A, Price J, Wilson-Heiner M, Baribault H, Marceau N. Simple epithelium keratins are required for maintenance of hepatocyte integrity. Am J Pathol. 1997;151:1673–1683. [PMC free article] [PubMed] [Google Scholar]
- Lu X, Lane EB. Retrovirus-mediated transgenic keratin expression in cultured fibroblasts: specific domain functions in keratin stabilization and filament formation. Cell. 1990;62:681–696. doi: 10.1016/0092-8674(90)90114-t. [DOI] [PubMed] [Google Scholar]
- Magin TM, Schroder R, Leitgeb S, Wanninger F, Zatloukal K, Grund C, Melton DW. Lessons from keratin 18 knockout mice: Formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8 positive aggregates. J Cell Biol. 1998;140:1441–1451. doi: 10.1083/jcb.140.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansbridge JN, Knapp AM. Changes in keratinocyte maturation during wound healing. J Invest Dermatol. 1987;89:253–262. doi: 10.1111/1523-1747.ep12471216. [DOI] [PubMed] [Google Scholar]
- Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors, and cultured cells. Cell. 1982;31:11–24. doi: 10.1016/0092-8674(82)90400-7. [DOI] [PubMed] [Google Scholar]
- Nelson W, Sun T-T. The 50- and 58-kdalton keratin classes as molecular markers for stratified squamous epithelia: Cell culture studies. J Cell Biol. 1983;97:244–251. doi: 10.1083/jcb.97.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima RG, Millan JL, Cecena G. Comparison of mouse and human keratin 18: a component of intermediate filaments expressed prior to implantation. Differentiation. 1986;33:61–68. doi: 10.1111/j.1432-0436.1986.tb00411.x. [DOI] [PubMed] [Google Scholar]
- Paladini RD, Coulombe PA. Directed expression of keratin 16 to the progenitor basal cells of transgenic mouse skin delays skin maturation. J Cell Biol. 1998;142:1035–1051. doi: 10.1083/jcb.142.4.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini RD, Takahashi K, Bravo NS, Coulombe PA. Onset of reepithelialization after skin injury correlates with a reorganization of keratin filaments in wound edge keratinocytes: Defining a potential role for keratin 16. J Cell Biol. 1996;132:381–397. doi: 10.1083/jcb.132.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter RM, Leitgeb S, Melton DW, Swensson O, Eady RA, Magin TM. Gene targeting at the mouse cytokeratin 10 locus: Severe skin fragility and changes of cytokeratin expression in the epidermis. J Cell Biol. 1996;132:925–936. doi: 10.1083/jcb.132.5.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell BC, Rogers GE. Cyclic hairloss and regrowth in transgenic mice overexpressing an intermediate filament gene. EMBO (Eur Mol Biol Organ) J. 1990;9:1485–1493. doi: 10.1002/j.1460-2075.1990.tb08266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roop DR, Huitfeldt H, Kilkenny A, Yuspa SH. Regulated expression of differentiation-associated keratins in cultured epidermal cells detected by monospecific antibodies to unique peptides of mouse epidermal keratins. Differentiation. 1987;35:143–150. doi: 10.1111/j.1432-0436.1987.tb00162.x. [DOI] [PubMed] [Google Scholar]
- Rothnagel JA, Dominey AM, Dempsey LD, Longley MA, Greenhalgh DA, Gagne TA, Huber M, Frenk E, Hohl D, Roop DR. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science. 1992;257:1128–1130. doi: 10.1126/science.257.5073.1128. [DOI] [PubMed] [Google Scholar]
- Rugg EL, McLean WHI, Lane EB, Pitera R, McMillan JR, Dopping-Hepenstal PJC, Navsaria HA, Leigh IM, Eady RAJ. A functional “knockout” of human keratin 14. Genes Dev. 1994;8:2563–2573. doi: 10.1101/gad.8.21.2563. [DOI] [PubMed] [Google Scholar]
- Steinert PM. The two-chain coiled-coil molecular of native epidermal keratin intermediate filaments is a type I-type II heterodimer. J Biol Chem. 1990;265:8766–8774. [PubMed] [Google Scholar]
- Stoler A, Kopan R, Duvic M, Fuchs E. The use of monospecific antibodies and cRNA probes reveals abnormal pathways of terminal differentiation in human epidermal diseases. J Cell Biol. 1988;107:427–446. doi: 10.1083/jcb.107.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, T.-T., R. Eichner, A. Schermer, D. Cooper, W.G. Nelson, and R.A. Weiss. 1984. The transformed phenotype. In The Cancer Cell. Vol. 1. A. Levine, W. Topp, G. van de Woude, and J.D. Watson, editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York. 169–176.
- Vassar R, Rosenberg M, Ross S, Tyner A, Fuchs E. Tissue-specific and differentiation-specific expression of a human K14 keratin gene in transgenic mice. Proc Natl Acad Sci USA. 1989;86:1563–1567. doi: 10.1073/pnas.86.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Coulombe PA, Degenstein L, Albers K, Fuchs E. Mutant keratin expression in transgenic mice causes marked abnormalities resembling a human genetic skin disease. Cell. 1991;64:365–380. doi: 10.1016/0092-8674(91)90645-f. [DOI] [PubMed] [Google Scholar]
- Warhol MJ, Lucocq JM, Carlemalm E, Roth J. Ultrastructural localization of keratin proteins in human skin using low-temperature embedding and the protein A-gold technique. J Invest Dermatol. 1985;84:69–72. doi: 10.1111/1523-1747.ep12274837. [DOI] [PubMed] [Google Scholar]
- Wawersik M, Paladini RD, Noensie E, Coulombe PA. A proline residue in the alpha-helical rod domain of type I keratin 16 destabilizes keratin heterotetramers. J Biol Chem. 1997;272:32557–32565. doi: 10.1074/jbc.272.51.32557. [DOI] [PubMed] [Google Scholar]
- Wu Y-J, Parker LM, Binder NE, Beckett MA, Sinard JH, Griffiths CT, Rheinwald JG. The mesothelial keratins: a new family of cytoskeletal proteins identified in cultured mesothelial cells. Cell. 1982;31:693–703. doi: 10.1016/0092-8674(82)90324-5. [DOI] [PubMed] [Google Scholar]