

Abstract
Expression of most RNA polymerase II transcripts requires the coordinated execution of transcription, splicing, and 3′ processing. We have previously shown that upon transcriptional activation of a gene in vivo, pre-mRNA splicing factors are recruited from nuclear speckles, in which they are concentrated, to sites of transcription (Misteli, T., J.F. Cáceres, and D.L. Spector. 1997. Nature. 387:523–527). This recruitment process appears to spatially coordinate transcription and pre-mRNA splicing within the cell nucleus. Here we have investigated the molecular basis for recruitment by analyzing the recruitment properties of mutant splicing factors. We show that multiple protein domains are required for efficient recruitment of SR proteins from nuclear speckles to nascent RNA. The two types of modular domains found in the splicing factor SF2/ ASF exert distinct functions in this process. In living cells, the RS domain functions in the dissociation of the protein from speckles, and phosphorylation of serine residues in the RS domain is a prerequisite for this event. The RNA binding domains play a role in the association of splicing factors with the target RNA. These observations identify a novel in vivo role for the RS domain of SR proteins and suggest a model in which protein phosphorylation is instrumental for the recruitment of these proteins to active sites of transcription in vivo.
Keywords: nucleus, phosphorylation, pre-mRNA splicing, recruitment, transcription
Efficient gene expression in vivo requires the coordinated execution of multiple nuclear processes, including transcription and pre-mRNA splicing. Upon transcription, heterogeneous nuclear ribonucleoproteins and pre-mRNA splicing factors rapidly associate with the elongating transcript. Excision of introns is then catalyzed by the assembled spliceosome and in many cases occurs cotranscriptionally (Sass and Pederson, 1984; Beyer and Osheim, 1988; Matunis et al., 1993; Kiseleva et al., 1994). Biochemical evidence for tight coupling of transcription and pre-mRNA splicing includes the physical interaction of the COOH-terminal domain (CTD)1 of the large subunit of RNA polymerase II (pol II) with pre-mRNA splicing factors and the finding that splicing is severely inhibited in cells that contain a form of the large subunit of RNA pol II with a reduced CTD (Mortillaro et al., 1996; Vincent et al., 1996; Yuryev et al., 1996; Corden and Patturajan, 1997; Kim et al., 1997; McCracken et al., 1997). Morphological evidence for spatial and temporal coupling of the two processes is provided by electron and fluorescence microscopy studies, which have demonstrated cotranscriptional splicing intermediates and the presence of spliceosomes on nascent transcripts (Beyer and Osheim, 1988; Báuren and Wieslander, 1994; Kiseleva et al., 1994).
Virtually all proteins involved in pre-mRNA splicing are enriched in numerous nuclear compartments, or speckles, in addition to their diffuse distribution throughout the nucleoplasm (for review see Spector, 1993). Some splicing factors are also found in the coiled body, a nuclear body of unknown function (Lamond and Carmo-Fonseca, 1993). Morphological examination of speckles by electron microscopy reveals that they consist of two distinct structures: granules 20–25 nm in diameter that are clustered in the interchromatin space and hence are termed interchromatin granule clusters (IGCs), and fibrils ∼5 nm in diameter, termed perichromatin fibrils (PFs), often extend from the periphery of IGCs and are also found dispersed throughout the nucleoplasm (Fakan, 1994). Nucleotide incorporation experiments and gene mapping studies showed that sites of active transcription are found dispersed throughout the nuclear volume and that the majority of actively transcribing genes are found within PFs and are largely excluded from IGCs (Jackson et al., 1993, 1998; Wansink et al., 1993; Fakan, 1994; Xing et al., 1995). This is supported by fluorescence microscopy data, which have placed the position of numerous genes at the periphery of or between speckles (Huang and Spector, 1991; Xing et al., 1995; Dirks et al., 1997). These observations indicate that IGCs are not sites of active transcription and thus not sites of cotranscriptional splicing. It has been proposed that IGCs are storage/reassembly sites for pre-mRNA splicing factors and that splicing factors are recruited from these compartments to sites of active transcription (Jiménez-García and Spector, 1993; for review see Misteli and Spector, 1998). Recent observations using time-lapse experiments in living cells support this model. Upon activation of a gene, pre-mRNA splicing factors were seen to be released from speckles and to be redistributed to sites of active transcription in a recruitment process (Misteli et al., 1997). This recruitment process might ensure the sufficient supply of splicing factors to sites of active transcription and thus serve as a means to spatially coordinate transcription and pre-mRNA splicing in vivo (Jiménez-García and Spector, 1993; Huang and Spector, 1996; Neugebauer and Roth, 1997; for review see Misteli and Spector, 1998). The molecular basis of splicing factor recruitment to nascent RNAs has been unknown.
One prominent component of nuclear speckles is the family of SR proteins (for reviews see Fu, 1995; Manley and Tacke, 1996). SR proteins are essential for constitutive splicing in vitro and can influence alternative splice site selection in a concentration-dependent manner (for review see Horowitz and Krainer, 1994). Classical SR proteins contain a COOH-terminal domain rich in ser/arg-dipeptides (RS domain) and either one or two RNA-recognition motifs (RRMs). In addition, a number of SR-like proteins have been described that contain at least one RS domain but differ from classical SR proteins in their overall domain structure (for review see Fu, 1995). The RS domain has been suggested to contain a targeting signal to mediate localization of these proteins to speckles. In the Drosophila protein Tra2, the RS domain, and specifically a short peptide sequence within the domain, is necessary and sufficient for the localization of the protein to speckles (Li and Bingham, 1991; Hedley et al., 1995). In mammalian SR proteins, the RS domain contributes to their localization but is not always necessary, and only in some cases (for example SRp20) is it sufficient for proper subnuclear localization (Cáceres et al., 1997; Gama-Carvalho et al., 1997). SR proteins are phosphoproteins, and several kinases that specifically phosphorylate serine residues in the RS domain and a protein phosphatase 1 (PP1) activity that dephosphorylates SR proteins have recently been identified (Meyrand et al., 1993; Gui et al., 1994; Mermoud et al., 1994; Colwill et al., 1996b ; Misteli and Spector, 1996; Rossi et al., 1996; Duncan et al., 1998; Kuroyanagi et al., 1998; Wang et al., 1998; for review see Misteli and Spector 1998).
Here we have investigated the molecular mechanism by which SR proteins are recruited from nuclear speckles to active sites of transcription in living cells upon activation of a nearby gene. We find that all modular domains of SR proteins are involved in the efficient recruitment and that different types of domains exert distinct functions in the recruitment process. Specifically, we show that phosphorylation of serine residues in the RS domain is required for the efficient recruitment of SR proteins to a site of active transcription in vivo.
Materials and Methods
Epitope-tagged Expression Plasmids
SF2/ASF, SC35, SRp20, and SRp40 deletion mutants in the pCG-T7 expression vector have been described (Cáceres et al., 1997). RG and GS replacement mutants in the same vector have been described (Cáceres and Krainer, 1993). GFP-SF2/ASF-ΔRS was generated by cutting GFP-SF2/ ASF with ApaI and religation. GFP-SF2/ASF-RG and -GS were generated by replacing a SacII-BamHI fragment in GFP-SF2/ASF-WT with the corresponding fragment from pCG-T7-SF2/ASF-RG or -GS mutants, respectively.
Cell Lines, Transfections, and Western Blotting
HeLa cells stably expressing the rat homeobox genepem were generated by cotransfection of pPem-89 containing the tetracycline promoter followed by the genomic pem sequence including the 3′ UTR (Maiti et al., 1996), the plasmid EV-124 containing the tetracycline transactivator, and the neomycin gene for selection (Gossen and Bujard, 1992). HeLa cell clones that exhibited highly repressible expression of pem RNA by addition of 1 μg/ml tetracycline were selected in 1 mg/ml G418, and colonies were screened by Northern blot analysis. Pem-HeLa cells were routinely grown in DME (GIBCO BRL, Gaithersburg, MD) supplemented with 10% FCS (Hyclone Labs, Logan, UT) in the presence of 0.4 mg/ml G418 (GIBCO BRL) and transfected as described in Huang and Spector (1996). SR protein mutants and a β-tropomyosin minigene (Helfman et al., 1988) were transfected by electroporation of 7 μg DNA and 13 μg of salmon sperm DNA at 240 V using a Bio-Rad gene pulser (Hercules, CA). For Western blotting, cell lysates were prepared at 14 h posttransfection and blotted as described in Misteli and Spector (1996). Monoclonal mouse anti–SF2/ASF antibody recognizing RRM1 of SF2/ASF (Hanamura et al., 1998) was used for Western blotting at 1:5. For analysis of the phosphorylation state of proteins, lysates were treated for 30 min at 37°C with 500 U/ml alkaline phosphatase (New England Biolabs, Beverly, MA).
In Situ Hybridization and Indirect Immunofluorescence
HeLa cells carrying the stably integrated rat pem gene were fixed at room temperature in freshly made 2% paraformaldehyde, 0.5% Triton in PBS for 5 min, followed by fixation in 2% paraformaldehyde in PBS for 15 min (Spector et al., 1998). For detection of target DNA, coverslips were washed twice in PBS for 5 min and incubated for 90 min at 37°C in 50 μl of 100 μg/ml RNAse A (Boehringer Mannheim, Indianapolis, IN) in PBS on a glass slide. Coverslips were washed three times in PBS for 5 min each, followed by a rinse in 2× SSC and two 10 min washes in 2× SSC at room temperature. Immediately before addition of the hybridization cocktail, cells were denatured in 70% formamide, 2× SSC for 7 min at 78°C. For detection of RNA, the RNAse step and the denaturation step were omitted. Nick translated probe representing the entire integrated pem sequence, generated as described in Langer et al. (1981) in 2× SSC, 10% dextran sulfate, and 1 mg/ml yeast tRNA, was added in a total volume of 20 μl to the coverslip, sealed with rubber cement, and incubated overnight at 37°C. Coverslips were washed in 2× SSC three times for 15 min at room temperature and once for 10 min in 4× SSC at room temperature. The probe was detected by incubation for 60 min at room temperature with avidin-FITC (2.5 μg/ml) in 4× SSC. Coverslips were then washed four times for 15 min in 4× SSC. After detection of the probe, cells were washed twice in PBS for 5 min each, and indirect immunofluorescence was performed as described in Misteli and Spector (1996) without additional fixation or permeabilization steps. Anti-T7 monoclonal antibody (Novagen, Madison, WI) was used at 1:400 in PBS, anti-SC35 antibody (Fu and Maniatis, 1990) at 1:800, anti-SF2/ASF antibody (Hanamura et al., 1998) at 1:50. Fluorescently labeled secondary antibodies and avidin-FITC were from Cappel (Durham, NC).
Microscopy
Images were acquired either on a Nikon FXA microscope (Melville, NY) equipped with a Photometrics SenSys cooled CCD camera (1,320 × 1,035 array; 6.7-μm pixel size) using Oncor Image 2.0.5 (Oncor, Gaithersburg, MD) or on a Zeiss LSM410 confocal laser scanning microscope (Thornwood, NY) using simultaneous scans to avoid shift between the two optical channels. Time-lapse microscopy of living cells was performed as described (Misteli et al., 1997). In brief, cells at 14 h after transfection were grown in a FCS2 live-cell microscopy chamber (Bioptechs, Butler, PA) at 37°C and supplied with fresh DMEM medium free of phenol red (GIBCO BRL, Gaithersburg, MD), supplemented with 10% FCS. The FCS2 chamber was mounted on a Zeiss Axiovert 405M inverted fluorescence microscope equipped with a Photometrics Nu200 cooled CCD camera (1,320 × 1,035 array; 6.7-μm pixel size). For routine observation, a 100×/N.A. 1.3 oil immersion lens was used. Images were acquired using Oncor Image 2.0.5 software. Exposure times were 0.2–0.8 s and a GFP filter (Chroma Technology, Brattleboro, VT) was used. Images were pseudocolored using the standard Oncor Image 2.0.5 256-level pseudocolor look-up table. The spectrum of pseudocolors represents the intensities of the fluorescence signal measured for each pixel (gray level 0 in blue, gray level 255 in red), and speckles appear in red. Line profiles were obtained from unprocessed images with no saturated pixels on a 256–grey value scale using Oncor Image 2.0.5.
Recruitment Assay in Living Cells
BK virus–transformed hamster fibrosarcoma BKT-1B cells (Moens et al., 1990) were grown and transcription induced as described (Misteli et al., 1997). In situ hybridization using nick translated biotinylated probes for the full-length BK virus DNA was performed as described (Misteli et al., 1997). Images of a particular series were accurately aligned using morphological markers such as shape of nuclei, position of nucleoli, and occasionally cytoplasmic morphological features.
Results
Effect of Deletion Mutations on the Recruitment Behavior of SR Proteins In Vivo
To gain insight into the molecular mechanisms of recruitment of pre-mRNA splicing factors, we tested the ability of deletion mutants of SR proteins to be recruited to a newly formed site of transcription in vivo. We used SR protein mutants, which have previously been characterized with respect to their subnuclear localization and their function in alternative splice site selection in vitro and in vivo (Cáceres and Krainer, 1993; Cáceres et al., 1997). HeLa cells carrying a stably integrated copy of the rat homeobox gene, pem, were used as an assay system (Maiti et al., 1996). In this cell line, the pem gene, which contains four spliced introns, is under the control of the inducible tetracycline promoter (Gossen and Bujard, 1992). To assess the recruitment ability of a protein, wild-type or mutant splicing factors tagged with the T7 epitope were expressed for 14 h in the absence of pem transcription. The pem locus was then activated for 4 h, and pem RNA was detected by in situ hybridization. Splicing factors were visualized by virtue of the epitope tag. Recruitment of splicing factors to the induced site of transcription was indicated by the colocalization of the in situ hybridization signal and the immunofluorescence signal.
In the absence of pem expression, the pem locus was not associated with nuclear speckles and was typically found between speckles in greater than 95% of cells (Fig. 1). Upon activation of the pem locus, recruitment of endogenous splicing factors (SF2/ASF, U2-B″, SC35) from speckles to the new site of transcription was readily detected (Fig. 2 A, a–c; data now shown). Recruitment was visualized by the localization of SF2/ASF (Fig. 2 A, a), detected with a specific monoclonal antibody against SF2/ASF (Hanamura et al., 1998), at the site of the pem RNA signal (Fig. 2 A, b). This is in agreement with observations by time-lapse microscopy in living cells that showed that transiently transfected SF2/ASF is efficiently recruited from speckles to an active transcription site upon activation of stably integrated genes (Misteli et al., 1997). Recruitment is a rapid process, and accumulation of SF2/ASF at the site of transcription is detectable within 10–20 min after activation of the target genes. Just like endogenous SF2/ASF, transiently expressed full-length SF2/ASF (Fig. 2 A, d–f) was recruited to the induced site of pem transcription, as indicated by the overlap of the two signals. Consistent with the observation that active genes frequently localize to the periphery of speckles (Huang and Spector, 1991; Xing et al., 1995; Dirks et al., 1997), in both cases the spots containing the RNA and splicing factor signals were typically closely associated with the periphery of a larger speckle (Fig. 2 A, a and d, insets). Identical results were obtained using conventional wide-field fluorescence or confocal microscopy (data not shown).
Figure 1.
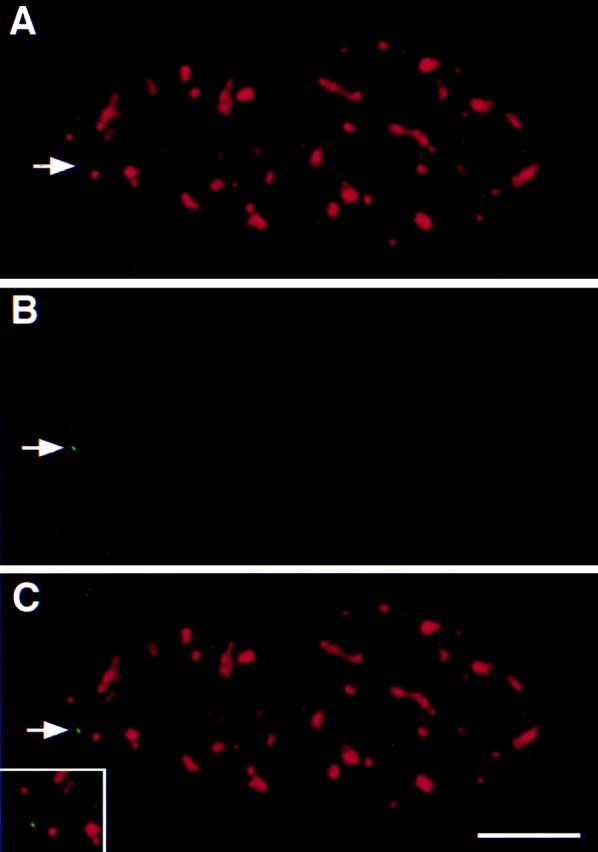
Localization of the transcriptionally inactive pem gene in HeLa cells. HeLa cells carrying the pem gene were grown for 24 h in the presence of 1 μg/ml tetracycline and fixed. The RNA was digested with RNAse, cells were denatured, and the pem gene was localized by in situ hybridization (B) and splicing factor SC35 localized by indirect immunofluorescence (A). In more than 95% of cells, splicing factor SC35 was found not to be associated with the transcriptionally inactive pem locus (C). The position of the pem locus is indicated by an arrow. Bar, 5 μm.
Figure 2.


(A) Recruitment of SF2/ASF mutants to transcription sites. Endogenous SF2/ASF (a–c), full-length SF2/ASF (d–f), or SF2/ASF mutants lacking RRM1 (g–i), RRM2 (j–l), or the RS domain (m–o) were transfected into pem-HeLa cells in the presence of tetracycline to inhibit pem expression. 14 h after transfection, tetracycline was removed to induce pem transcription for 4 h. Cells were fixed, and the SR protein was detected using an anti-SF2/ASF antibody (a, red) or an anti–T7 epitope antibody (d, g, j, and m, red). Pem RNA was detected by in situ hybridization (b, e, h, k, and n, green). Endogenous SF2/ASF and full-length SF2/ ASF were recruited to and accumulate at the site of pem transcription. Deletion of any single domain prevented recruitment. Arrows and arrowheads indicate the sites of pem transcription. (B) A test line was drawn so as to include a splicing factor compartment and the site of pem RNA. The relative fluorescence intensity along the line for both the splicing factor signal (red) and the pem RNA signal (green) was measured in arbitrary units. The fluorescence intensity peaks for endogenous and wild-type SF2/ASF protein coincided with the peak for the pem RNA, indicating recruitment of splicing factor to the site of pem RNA. For mutants of SF2/ASF, the two peaks were separated, indicating the absence of the splicing factor mutants from the site of pem RNA. Bar, 5 μm.
We tested the effect of deletion of either the RS domain or one of the two RRMs of SF2/ASF on the recruitment of the protein to the pem RNA. Deletion of either RRM1 (Fig. 2 A, g–i), RRM2 (Fig. 2 A, j–l), or the RS domain (Fig. 2 A, m–o) prevented accumulation of the mutant protein at the site of induced pem transcription. The pem RNA was now typically found as a distinct spot from speckles and was free of accumulated mutant splicing factor (Fig. 2 A, g, j, and m, insets). The presence or absence of splicing factors at the site of pem RNA was verified by fluorescence intensity measurements along a test-line through the site of pem RNA accumulation and a nearby speckle (Fig. 2 B). In the case of endogenous SF2/ASF and transiently expressed wild-type SF2/ASF, the peaks of the two signals coincided. In contrast, for the deletion mutants the peaks for the pem RNA signal and the mutant splicing factor signal were clearly separated (Fig. 2 B). Quantitation of the number of cells that showed colocalization of splicing factor and the pem RNA demonstrated that endogenous SF2/ASF and wild-type SF2/ASF accumulated in more than 90% of cells at the site of pem RNA, whereas the deletion mutants accumulated in ∼30% of cells with pem RNA (Fig. 3). This latter number most likely reflects the random association of the pem transcription site with speckles in the mammalian cell nucleus.
Figure 3.

Quantitative analysis of recruitment. Cells were scored for accumulation of the wild-type or mutant SR proteins at the site of pem transcription, as detected by in situ hybridization and indirect immunofluorescence. The percentage of cells (n = 200 from 3 experiments) showing accumulation of splicing factor at the site of transcription is shown ±SD. Endogenous and wild-type SR proteins were efficiently recruited to the site of transcription in more than 90% of cells, whereas deletion mutants of SF2/ASF, SRp40, SC35, or SRp20 were typically found at the site of pem RNA in ∼30% of cells (see Results).
The RS domains of various members of the SR protein family all contain multiple arg/ser-dipeptides but are significantly distinct from each other at the amino acid level (for review see Fu, 1995). We tested whether the RS domain of several SR proteins was required for their recruitment. As for SF2/ASF, deletion of the RS domain in SRp40, SRp20, or SC35 prevented the mutant proteins from accumulating at transcription sites (Fig. 3). Furthermore, none of the single RRMs or RS domains was sufficient for recruitment (Fig. 3 and data not shown), indicating that the requirement for recruitment was not strictly amino acid sequence specific, but might be caused by the charge distribution within the domain. The lack of recruitment of all mutants tested was not due to the impaired function of the proteins since they have been shown to be functionally active in alternative splice site selection in vivo (Cáceres et al., 1997).
Evidence for Distinct Roles for the RS Domain and the RRM in the Recruitment Process
Evidence for distinct functions for the RS domain versus the RRMs of SF2/ASF was obtained in an assay in which recruitment of SR proteins to a transiently expressed intron-containing β-tropomyosin (β-TM) minigene was measured. This assay was previously used to demonstrate that recruitment is dependent on the presence of an intron in the target RNA (Huang and Spector, 1996). Similar to the situation during intermediate stages of adenoviral infection, transient overexpression of the β-TM minigene produces large amounts of pre-mRNA, and as a consequence, the majority of endogenous splicing factors dissociate from speckles and associate with sites of β-TM transcription, resulting in the depletion of splicing factors from native speckles (Jiménez-García and Spector, 1993; Pombo et al., 1994; Huang and Spector, 1996; Gama-Carvalho et al., 1997). Under these conditions, transiently expressed full-length SF2/ASF (Fig. 4 A, red) was strongly recruited to active sites of β-TM expression (Fig. 4 A, green). In contrast, deletion of either the first or second RRM in SF2/ASF resulted in the diffuse distribution of the mutant splicing factor throughout the cell nucleus (Fig. 4 B, red) and prevented significant accumulation of the mutant protein at sites of β-TM transcription (Fig. 4 B, green). This was most likely due to the weakened interaction of SF2/ASF with β-TM RNA in the absence of either one of its two RRMs (Cáceres and Krainer, 1993; Wu and Maniatis, 1993; Zuo and Manley, 1993). Note that the yellow signal in the overlay panel is caused by the diffuse distribution of the mutant protein and does not represent specific accumulation at transcription sites. In contrast, SF2/ASF lacking the RS domain was largely retained in speckles that were frequently (∼60%) observed to be distinct from sites of β-TM-transcription (Fig. 4 C, arrows). The presence of speckles distinct from β-TM transcription sites indicates that this mutant was less efficiently displaced from endogenous speckles. These observations suggest that in the absence of either one of the RRMs, the mutant protein was released from speckles but could not efficiently bind the target RNA, whereas in the absence of the RS domain, the mutant protein was not efficiently released. Thus, we propose that the RS domain and the RRMs play distinct roles in the recruitment process in vivo.
Figure 4.

Distinct roles in the recruitment process for RRMs and the RS domain. HeLa cells were cotransfected with a β-TM minigene and the indicated SF2/ASF construct. 14 h after transfection, SF2/ASF was detected by immunofluorescence using an anti–T7 epitope antibody (red), and β-TM RNA was visualized by in situ hybridization (green). (A) Full-length SF2/ASF was efficiently recruited to sites of β-TM transcription. (B) Deletion of the first RRM prevented recruitment and resulted in the diffuse distribution of SF2/ASF-ΔRRM1. Identical results were obtained for SF2/ASF lacking RRM2 (data not shown). (C) Deletion of the RS domain resulted in the retention of SF2/ASF-ΔRS in native speckles at sites distinct from β-TM transcription sites. Bar, 5 μm.
The RS Domain Is Required for Recruitment in Living Cells
To further analyze the role of the RS domain in vivo, we tested the effect of deletion of the RS domain on recruitment of SF2/ASF in living cells using a previously characterized time-lapse microscopy assay (Misteli et al., 1997). BKT-1B cells carry stably integrated cAMP-inducible early genes of BK virus (Moens et al., 1990), and recruitment to the induced transcription site of BK early genes can be monitored in individual living cells using SF2/ASF fused to the green fluorescent protein (GFP) (Misteli et al., 1997). As previously demonstrated, upon stimulation of BKV early genes, GFP-SF2/ASF was recruited from speckles and accumulated at the site of transcription (Fig. 5, top, arrow) (Misteli et al., 1997). The accumulation is detected as an area of higher GFP-SF2/ASF concentration (Fig. 5, red; see figure legend) in the form of a peripheral extension protruding from a nearby speckle (Misteli et al., 1997). The extension typically forms gradually upon addition of cAMP after an initial lag period of about 10 min and peaks at the time of maximal gene expression, in the case of immediate early genes of BK virus at ∼60 min (Moens et al., 1990; Misteli et al., 1997). In contrast, in the absence of the RS domain, SF2/ASF was not recruited to the site of transcription (Fig. 5, bottom, arrow). No peripheral extension could be seen at any time during the induction time of immediate early genes of BK virus, and no accumulation of the splicing factor mutant was detected at the site of transcription (Fig. 5, bottom). This observation shows that the RS domain is required for the efficient recruitment of splicing factors to transcription sites in living cells.
Figure 5.

The RS domain of SF2/ ASF is required for recruitment in living cells. Transcription of BK virus was triggered by addition of 50 μg/ml cAMP to the growth medium, and splicing factor distribution was imaged at the indicated times after induction of transcription. After acquisition of the final image, the cell was fixed, and the position of the induced RNA was detected by fluorescence in situ hybridization. (Top) Full-length SF2/ASF formed a peripheral extension from an existing speckle (red), and SF2/ASF accumulated at the site of BK early gene transcription (arrow). (Bottom) Deletion of the RS domain prevented dissociation of the splicing factor and its accumulation at the site of transcription. Pseudocolored images are shown. Speckles appear in red against a yellow/green background representing the nucleoplasmic pool of the splicing factor. The spectrum of pseudocolors represents the intensities of the fluorescence signal measured for each pixel (blue lowest, red brightest). Arrow, position of the RNA signal. The time after induction of gene expression is indicated in minutes. Bar, 1.5 μm.
Phosphorylation of Serine Residues in the RS Domain Is Required for Efficient Recruitment
A hallmark of the RS domain is its significant posttranslational phosphorylation on serine residues (Roth et al., 1991; Gui et al., 1994; Colwill et al., 1996b ). To test whether phosphorylation of the RS domain is involved in recruitment to transcription sites, we used mutants of SF2/ ASF in which the RS-dipeptides in the RS domain had been replaced by either RG- or GS-dipeptides (Fig. 6 A) (Cáceres and Krainer, 1993). Western blot analysis of total cell lysates showed that wild-type SF2/ASF was efficiently phosphorylated in vivo in BHK cells as judged by a shift in mobility after alkaline phosphatase treatment (Fig. 6 B). This is in agreement with the recent finding that in vivo endogenous SF2/ASF is predominantly found in a highly phosphorylated form (Hanamura et al., 1998). A similar shift was observed for SF2/ASF-GS, demonstrating that SF2/ASF-GS was efficiently phosphorylated in vivo (Fig. 6 B). In contrast, deletion of the RS domain prevented all detectable phosphorylation of the protein (Fig. 6 B) as has been predicted by phospho-epitope mapping studies which demonstrated that all phosphorylation sites in SF2/ASF map to the RS domain (Colwill et al., 1996b ). Similarly, the RG mutant was not phosphorylated in BHK cells and migrated identically in the presence or absence of alkaline phosphatase treatment (Fig. 6 B).
Figure 6.


Phosphorylation of serine residues in the RS domain is required for recruitment. (A) Amino acid sequence of the RS domain of SF2/ASF. The RS domain of SF2/ASF was either deleted or the RS-dipeptides substituted with either RG- or GS-dipeptides. (B) Immunoblot analysis of the phosphorylation state of SF2/ASF in vivo. The indicated T7-tagged constructs were transfected into BHK cells, and total cell lysates were probed by Western blotting with (+) or without (−) alkaline phosphatase digestion. Full-length SF2/ASF and SF2/ASF-GS were phosphorylated in vivo, and SF2/ASF-ΔRS and SF2/ASF-RG were not phosphorylated in vivo.
The ability of these mutants to be recruited to nascent RNA was then assessed in living cells using the BKT system. The recruitment ability of the mutant proteins in living cells correlated with the phosphorylation state of the RS domain (Fig. 7). SF2/ASF-GS, like SF2/ASF-WT, was efficiently recruited to sites of transcription (Fig. 7) as shown by the formation of a peripheral extension from a nearby speckle and the accumulation of the mutant protein at the site of transcription. The time course of accumulation of the mutant protein was indistinguishable from the wild-type protein. In contrast, the RG mutant was retained in speckles after stimulation of transcription of BK early genes (Fig. 7, bottom) and was not recruited to the induced genes. This behavior was identical to the ΔRS mutant (see Fig. 5), and no accumulation of splicing factors was observed at any time. These observations demonstrate that serine phosphorylation of the RS domain is required for the recruitment of SR proteins from nuclear speckles to transcription sites in vivo.
Figure 7.

Phosphorylation of serine residues in the RS domain is required for recruitment in living cells. The recruitment assay was performed as described in Fig. 5. (Top) The phosphorylated SF2/ASF-GS mutant was recruited to the newly formed site of transcription (arrow). (Bottom) The unphosphorylated SF2/ASF-RG mutant was retained in nuclear speckles and did not accumulate at the transcription site (arrow). Time after induction of gene expression is indicated in minutes. Images are pseudocolored; speckles appear in red against a green/ yellow background representing the nucleoplasmic pool of the splicing factor. Arrow, position of the RNA signal. Bar, 1.5 μm.
Discussion
We have probed the molecular mechanism by which pre-mRNA splicing factors are recruited from nuclear speckles to nascent RNAs in vivo. In a stable, inducible gene expression system, deletion of any single modular domain in four SR proteins tested prevented efficient recruitment to the nascent RNA. Furthermore, in a transient transfection assay in which large amounts of target RNA are produced, we found that the different types of domains play distinct roles in recruitment. Under these conditions, deletion of a single RRM in SF2/ASF resulted in the diffuse distribution of the mutant protein in the cell nucleus in the absence of any accumulation at sites of transcription. In contrast, deletion of the RS domain resulted in a protein that was largely retained in nuclear speckles when challenged with high levels of β-TM RNA. The simplest interpretation of these observations is that recruitment of SR proteins from nuclear speckles to a site of transcription is a process involving at least two steps, namely the release of splicing factors from speckles, mediated by the RS domain, followed by their association with the target RNA, mediated by the RRMs. The role of the RRMs in associating with the target RNA is consistent with the well-documented role of the RRMs in binding to RNA (for review see Fu, 1995). The role of the RS domain in the release from speckles is consistent with the observed effect of deletion of the RS domain on subnuclear distribution of RS domain–containing proteins. In SRp20 and SC35, both of which contain only one RRM, the RS domain determines the subnuclear localization (Cáceres et al., 1997). The RS domain is necessary for localization to speckles and is sufficient to target a cytoplasmic reporter protein to nuclear speckles (Cáceres et al., 1997). Similarly, in the SR-like Drosophila proteins Tra and SWAP, the RS domain is sufficient for speckled localization (Li and Bingham, 1991; Hedley et al., 1995). These data argue that the RS domain can control the association/dissociation properties of SR proteins with speckles. In support of this idea, we have observed that the RS domain of SRp20 is sufficient for the release of a reporter protein from speckles upon transient expression of β-TM RNA (Misteli, T., and D.L. Spector, unpublished observation). In SF2/ASF, and also SRp40, association of the protein with nuclear speckles is not dependent on the RS domain alone since deletion of the RS domain only reduces the level of the protein in speckles but does not completely prevent its association (Cáceres et al., 1997). It is likely that for proteins that contain multiple RRMs, such as SF2/ASF, SRp40, or U2AF65, the speckle localization is additionally mediated by protein–protein or protein–RNA interactions occurring through the RRMs, possibly via snRNPs or other RS domain–containing proteins (Cáceres et al., 1997; Gama-Carvalho et al., 1997). An example of such an indirect localization is the Tra protein lacking its RS domain. This mutant localizes to speckles provided that it can interact with RS domain–containing proteins such as Tra2, but deletion of the interacting domain displaces the protein from speckles (Hedley et al., 1995). Thus, the dissociation and association of RS domain–containing proteins might be mediated by two distinct mechanisms (see also Cáceres et al., 1997). Our observations suggest that the RS domain functions in recruitment in the context of SR proteins. However, the recruitment function might be a more general feature of the RS domain in the context of other proteins as well. For example, in U2AF65, an SR-like protein not belonging to the classical SR family of proteins, the NH2-terminal RS domain is required for recruitment to adenoviral transcripts, while the RRMs are not required for this event (Gama-Carvalho et al., 1997).
A trivial explanation for our observations would be that the recruitment properties of the proteins are the consequence of their functional inactivation as a result of the deletions. This is unlikely, since no correlation between function of the protein and recruitment behavior has been observed. In vivo and in vitro, wild-type SF2/ASF strongly activates the use of the 13-S 5′ splice site in the adenoviral E1A transcript in a concentration-dependent manner (Cáceres and Krainer, 1993; Zuo and Manley, 1993; Cáceres et al., 1997). The same is true for SF2/ASF lacking the RS domain or lacking the first RRM. However, in our recruitment assay the wild-type protein was efficiently recruited, while a mutant lacking the RS domain was not, suggesting that the recruitment behavior does not simply reflect the functional properties of the protein. It is not clear at present how a mutant protein, such as SF2/ASF-ΔRS, is able to influence alternative splice site selection, although it is apparently not recruited to the target RNA. One possibility is that a steady-state level of protein below our detection limit is sufficient to affect splice site selection. Alternatively, it is possible that overexpression of the mutant protein affects the choice of splice sites indirectly.
Because the majority of active transcription sites are localized at the periphery of or outside of speckles (Wansink et al., 1993; Zhang et al., 1994; Xing et al., 1995; Neugebauer and Roth, 1997), dissociation of splicing factors from speckles is a requirement for efficient pre-mRNA splicing in vivo. Our results from living cell experiments extend observations that imply a function for phosphorylation in the dissociation of SR proteins from nuclear speckles. Several lines of evidence now point to a role of phosphorylation of serine residues in the RS domain as a switch for the dissociation of splicing factors from nuclear speckles. First, we find that in living BHK cells, SF2/ASF molecules lacking either the RS domain or the serine phosphorylation sites in the RS domain were not observed to dissociate from speckles upon transcriptional activation of a nearby gene in contrast to the wild-type protein (Fig. 7) (Misteli et al., 1997). Second, we observe that in the absence of the RS domain, SF2/ASF was retained in nuclear speckles distinct from sites of β-TM transcription, whereas deletion of either RRM resulted in the diffuse nuclear distribution of the mutant protein (Fig. 4). Third, in living interphase cells, the dynamic movements of splicing factors at the periphery of speckles have been interpreted to represent the continuous movement of splicing factors from speckles to nearby active genes as factors are being recruited (Misteli et al., 1997). In agreement with a role of phosphorylation in the release of factors, these movements cease upon addition of protein kinase inhibitors (Misteli et al., 1997). Fourth, several kinases, SRPK-1, -2 and Clk-1, -2, -3, and topoisomerase I, which specifically phosphorylate SR proteins, have recently been identified (Gui et al., 1994; Colwill et al., 1996b ; Rossi et al., 1996; Duncan et al., 1997; Kuroyanagi et al., 1998; Wang et al., 1998). Overexpression of SRPKs or Clks results in the redistribution of splicing factors from a speckled pattern to a diffuse nuclear pattern and an increase in the solubility of splicing factors. Fifth, a ser/thr phosphatase I activity has been identified based on its ability to prevent the association and accumulation of splicing factors in enlarged speckles upon inhibition of transcription, suggesting that dephosphorylation of SR proteins facilitates their association with this nuclear compartment (Misteli and Spector, 1996). Finally, our in vivo results also correlate well with in vitro data on the effect of phosphorylation on splicing factor function. In vitro analyses demonstrate that phosphorylation of SR proteins is a prerequisite for their efficient incorporation into spliceosomes and for their proper functioning in the splicing process (Tazi et al., 1993; Mermoud et al., 1994; Cao et al., 1997; Xiao and Manley, 1997). Phosphorylation of SR proteins would thus be expected to occur before the association of SR protein with the nascent RNA, which is thought to occur outside of nuclear speckles.
Control of the release of splicing factors from nuclear speckles might be important to ensure the sufficient supply of splicing factors to transcription sites. In addition, it might also play a role in regulation of alternative splice site selection. In vitro and in vivo, the choice of splice site is determined by the ratio of multiple splicing factors (for review see Horowitz and Krainer, 1994). Controlling the release of factors from nuclear compartments might be a way to modify the relative concentrations of multiple factors in the nucleoplasm and thus modulate the choice of splice site. In support of this interpretation, overexpression of Clk kinases causes a switch of splice sites in several RNA templates in vivo (Duncan et al., 1997, 1998). The recently identified SR protein kinases are good candidates to mediate the release of SR proteins from speckles. However, the large number of serine residues in the RS domain and the overlapping substrate specificities of the identified kinases makes it difficult to determine which serine residues, or combinations thereof, are important for the proper function and to address the precise roles of the kinases in vivo (Colwill et al., 1996a ; Labourier et al., 1998; Wang et al., 1998).
These observations can be summarized in a working model for the molecular mechanism of splicing factor recruitment (Misteli and Spector, 1997). In this scenario, splicing factors reside in nuclear speckles, predominantly IGCs, and phosphorylation of serine residues in the RS domain by SR protein kinases has two effects: (a) splicing factors are displaced from speckles either individually or complexed with other factors of the RNA-processing machinery (Gui et al., 1994; Colwill et al., 1996b ; Duncan et al., 1998; Wang et al., 1998) and (b) splicing factors become “active,” i.e., they can be incorporated into the spliceosome (Mermoud et al., 1992, 1994; Cao et al., 1997). During or after completion of the splicing reaction, which takes place in PFs outside of IGCs, the proteins are dephosphorylated (Mermoud et al., 1992, 1994; Misteli and Spector, 1996), allowing them to return to the IGCs, where a new round of the phosphorylation cycle starts (see Misteli and Spector, 1998). We suggest that this arrangement provides a means to control the supply of active splicing factors at sites of transcription and, thus, is instrumental in the spatial coordination of transcription and pre-mRNA splicing within the mammalian cell nucleus.
Acknowledgments
We thank members of the Krainer and Spector laboratories for discussion and critical reading of the manuscript.
Abbreviations used in this paper
- β-TM
β-tropomyosin
- BKV
BK virus
- CTD
COOH-terminal domain
- GFP
green fluorescent protein
- IGC
interchromatin granule cluster
- PF
perichromatin fibril
- Pol II
RNA polymerase II
- PP1
protein phosphatase 1
- RRM
RNA recognition motif
Footnotes
T. Misteli dedicates this paper to the memory of Thomas Kreis.
T. Misteli was supported by the Human Frontiers Science Program and the Roche Research Foundation. A.R. Krainer was supported by National Cancer Institute grant CA13106, M.F. Wilkinson was supported by National Institutes of Health (NIH) grant GM39586 and National Science Foundation grant MCB-9307963, and D.L. Spector was supported by NIH grant GM42694.
Address all correspondence to D.L. Spector, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724. Tel.: (516) 367-8456. Fax: (516) 367-8876. E-mail: spector@cshl.org
References
- Báuren G, Wieslander L. Splicing of Balbiani ring 1 gene pre-mRNA occurs simultaneously with transcription. Cell. 1994;76:183–192. doi: 10.1016/0092-8674(94)90182-1. [DOI] [PubMed] [Google Scholar]
- Beyer AL, Osheim YN. Splice site selection, rate of splicing, and alternative splicing on nascent transcripts. Genes Dev. 1988;2:754–765. doi: 10.1101/gad.2.6.754. [DOI] [PubMed] [Google Scholar]
- Cáceres JF, Krainer AR. Functional analysis of pre-mRNA splicing factor SF2/ASF structural domains. EMBO (Eur Mol Biol Organ) J. 1993;12:4715–4726. doi: 10.1002/j.1460-2075.1993.tb06160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres JF, Misteli T, Screaton G, Spector DL, Krainer AR. Role of the modular domains of SR-proteins in subnuclear localization and alternative splicing specificity. J Cell Biol. 1997;138:225–238. doi: 10.1083/jcb.138.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Jamison SF, Garcia-Blanco MA. Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. RNA. 1997;3:1456–1467. [PMC free article] [PubMed] [Google Scholar]
- Colwill K, Feng LL, Yeakley JM, Gish GD, Cáceres JF, Pawson T, Fu X-D. SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. J Biol Chem. 1996a;271:24569–24575. doi: 10.1074/jbc.271.40.24569. [DOI] [PubMed] [Google Scholar]
- Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. The Clk/Sty protein kinase phosphorylates splicing factors and regulates their intranuclear distribution. EMBO (Eur Mol Biol Organ) J. 1996b;15:265–275. [PMC free article] [PubMed] [Google Scholar]
- Corden JL, Patturajan M. A CTD function linking transcription to splicing. Trends Biochem Sci. 1997;22:413–416. doi: 10.1016/s0968-0004(97)01125-0. [DOI] [PubMed] [Google Scholar]
- Dirks RW, de Pauw ESD, Raap AK. Splicing factors associate with nuclear HCMV-IE transcripts after transcriptional activation of the gene, but dissociate upon transcription inhibition: evidence for a dynamic organization of splicing factors. J Cell Sci. 1997;110:505–513. doi: 10.1242/jcs.110.4.515. [DOI] [PubMed] [Google Scholar]
- Duncan PI, Stojdl DF, Marius RM, Bell JC. In vivo regulation of alternative pre-mRNA splicing by the Clk1 protein kinase. Mol Cell Biol. 1997;17:5996–6001. doi: 10.1128/mcb.17.10.5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan PI, Stojdl DF, Marius RM, Scheit KH, Bell JC. The Clk2 and Clk3 dual-specificity protein kinases regulate the intranuclear distribution of SR proteins and influence pre-mRNA splicing. Exp Cell Res. 1998;241:300–308. doi: 10.1006/excr.1998.4083. [DOI] [PubMed] [Google Scholar]
- Fakan S. Perichromatin fibrils are in situ forms of nascent transcripts. Trends Cell Biol. 1994;4:86–90. doi: 10.1016/0962-8924(94)90180-5. [DOI] [PubMed] [Google Scholar]
- Fu X-D. The superfamily of arginine/serine-rich splicing factors. RNA. 1995;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- Fu X-D, Maniatis T. Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature. 1990;343:437–441. doi: 10.1038/343437a0. [DOI] [PubMed] [Google Scholar]
- Gama-Carvalho M, Krauss R, Chiang L, Valcarcel J, Green M, Carmo-Fonseca M. Targeting of U2AF65 to sites of active splicing in the nucleus. J Cell Biol. 1997;137:975–987. doi: 10.1083/jcb.137.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui JF, Lane WS, Fu X-D. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994;369:678–682. doi: 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- Hanamura A, Cáceres JF, Mayeda A, Franza BR, Krainer A R. Regulated tissue-specific expression of antagonistic pre-mRNA splicing factors. RNA. 1998;4:430–444. [PMC free article] [PubMed] [Google Scholar]
- Hedley ML, Amrein H, Maniatis T. An amino acid sequence motif sufficient for subnuclear localization of an arginine/serine rich splicing factor. Proc Natl Acad Sci USA. 1995;92:11524–11528. doi: 10.1073/pnas.92.25.11524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfman DM, Ricci WM, Finn LA. Alternative splicing of tropomyosin pre-mRNAs in vitro and in vivo. Genes Dev. 1988;2:1627–1638. doi: 10.1101/gad.2.12a.1627. [DOI] [PubMed] [Google Scholar]
- Horowitz DS, Krainer AR. Mechanisms for selecting 5′ splice sites in mammalian pre-mRNA splicing. Trends Genet. 1994;10:100–106. doi: 10.1016/0168-9525(94)90233-x. [DOI] [PubMed] [Google Scholar]
- Huang S, Spector DL. Nascent pre-mRNA transcripts are associated with nuclear regions enriched in splicing factors. Genes Dev. 1991;5:2288–2302. doi: 10.1101/gad.5.12a.2288. [DOI] [PubMed] [Google Scholar]
- Huang S, Spector DL. Intron-dependent recruitment of pre-mRNA splicing factors to sites of transcription. J Cell Biol. 1996;131:719–732. doi: 10.1083/jcb.133.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Hassan AB, Errington RJ, Cook PR. Visualization of focal sites of transcription within human nuclei. EMBO (Eur Mol Biol Organ) J. 1993;12:1059–1065. doi: 10.1002/j.1460-2075.1993.tb05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Iborra FJ, Manders EM, Cook PR. Numbers and organization of RNA polymerases, nascent transcripts, and transcription units in HeLa nuclei. Mol Biol Cell. 1998;9:1523–1536. doi: 10.1091/mbc.9.6.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-García LF, Spector DL. In vivo evidence that transcription and splicing are coordinated by a recruiting mechanism. Cell. 1993;73:47–59. doi: 10.1016/0092-8674(93)90159-n. [DOI] [PubMed] [Google Scholar]
- Kim E, Du L, Bregman DB, Warren SL. Splicing factors associate with hyperphosphorylated RNA polymerase II in the absence of pre-mRNA. J Cell Biol. 1997;136:19–28. doi: 10.1083/jcb.136.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiseleva E, Wurtz T, Visa N, Daneholt B. Assembly and disassembly of spliceosomes along a specific pre-messenger RNP fiber. EMBO (Eur Mol Biol Organ) J. 1994;13:6052–6061. doi: 10.1002/j.1460-2075.1994.tb06952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroyanagi N, Onogi H, Wakabayashi T, Hagiwara M. Novel SR-protein-specific kinase, SRPK2, disassembles nuclear speckles. Biochem Biophys Res Commun. 1998;14:357–364. doi: 10.1006/bbrc.1997.7913. [DOI] [PubMed] [Google Scholar]
- Labourier E, Rossi F, Gallouzi I, Allemand E, Divita G, Tazi J. Interaction between the N-terminal domain of human DNA topoisomerase I and the arginine-serine domain of its substrate determines phosphorylation of SF2/ASF splicing factor. Nucleic Acids Res. 1998;26:2955–2962. doi: 10.1093/nar/26.12.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamond AL, Carmo-Fonseca M. The coiled body. Trends Cell Biol. 1993;3:198–204. doi: 10.1016/0962-8924(93)90214-l. [DOI] [PubMed] [Google Scholar]
- Langer PR, Waldrop AA, Ward DC. Enzymatic synthesis of biotin labeled polynucleotides: novel nucleic acid affinity probes. Proc Natl Acad Sci USA. 1981;78:6633–6637. doi: 10.1073/pnas.78.11.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Bingham PM. Arginine/serine-rich domains of the su(wa) and tra RNA processing regulators target proteins to a subnuclear compartment implicated in splicing. Cell. 1991;67:335–342. doi: 10.1016/0092-8674(91)90185-2. [DOI] [PubMed] [Google Scholar]
- Maiti S, Doskow J, Li S, Nhim RP, Lindsey JS, Wilkinson MF. The Pem homeobox gene. Androgen-dependent and -independent promoters and tissue-specific alternative RNA splicing. J Biol Chem. 1996;271:17536–17546. doi: 10.1074/jbc.271.29.17536. [DOI] [PubMed] [Google Scholar]
- Manley JL, Tacke R. SR proteins and splicing control. Genes Dev. 1996;10:1569–1579. doi: 10.1101/gad.10.13.1569. [DOI] [PubMed] [Google Scholar]
- Matunis EL, Matunis MJ, Dreyfuss G. Association of individual hnRNP proteins and snRNPs with nascent transcripts. J Cell Biol. 1993;121:219–228. doi: 10.1083/jcb.121.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson SD, Wickens M, Bentley D. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997;385:357–361. doi: 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]
- Mermoud JE, Cohen P, Lamond AI. Ser/Thr-specific protein phosphatases are required for both catalytic steps of pre-mRNA splicing. Nucleic Acids Res. 1992;20:5263–5269. doi: 10.1093/nar/20.20.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermoud JE, Cohen PTW, Lamond AI. Regulation of mammalian spliceosome assembly by a protein phosphorylation mechanism. EMBO (Eur Mol Biol Organ) J. 1994;13:5679–5688. doi: 10.1002/j.1460-2075.1994.tb06906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyrand SH, Dwen P, Pederson T. Serine/threonine phosphorylation regulates binding of C hnRNP proteins to pre-mRNA. Proc Natl Acad Sci USA. 1993;90:7764–7768. doi: 10.1073/pnas.90.16.7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Spector DL. Serine/threonine phosphatase 1 modulates the subnuclear distribution of pre-mRNA splicing factors. Mol Biol Cell. 1996;7:1559–1572. doi: 10.1091/mbc.7.10.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Spector DL. Protein phosphorylation and the nuclear organization of pre-mRNA splicing. Trends Cell Biol. 1997;7:135–138. doi: 10.1016/S0962-8924(96)20043-1. [DOI] [PubMed] [Google Scholar]
- Misteli T, Spector DL. The cellular organization of gene expression. Curr Opin Cell Biol. 1998;10:322–331. doi: 10.1016/s0955-0674(98)80007-0. [DOI] [PubMed] [Google Scholar]
- Misteli T, Cáceres JF, Spector DL. The dynamics of a pre-mRNA splicing factor in living cells. Nature. 1997;387:523–527. doi: 10.1038/387523a0. [DOI] [PubMed] [Google Scholar]
- Moens U, Sundsfjord A, Flægstad T, Traavik T. BK virus early RNA transcripts in stably transformed cells: enhanced levels induced by dibutyryl cyclic AMP, forskolin and 12-O-tetradecanoylphorbol-13-acetate treatment. Gen Vir. 1990;71:1461–1471. doi: 10.1099/0022-1317-71-7-1461. [DOI] [PubMed] [Google Scholar]
- Mortillaro MJ, Blencowe BJ, Wei X, Nakayasu H, Du L, Warren SL, Sharp PA, Berezny R. A hyperphosphorylated form of the large subunit of RNA polymerase II is associated with splicing complexes and the nuclear matrix. Proc Natl Acad Sci USA. 1996;93:8253–8257. doi: 10.1073/pnas.93.16.8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer KM, Roth MB. Distribution of pre-mRNA splicing factors at sites of RNA polymerase II transcription. Genes Dev. 1997;11:1148–1159. doi: 10.1101/gad.11.9.1148. [DOI] [PubMed] [Google Scholar]
- Pombo A, Ferreira J, Bridge E, Carmo-Fonseca M. Adenovirus replication and transcription sites are spatially separated in the nucleus of infected cells. EMBO (Eur Mol Biol Organ) J. 1994;13:5075–5085. doi: 10.1002/j.1460-2075.1994.tb06837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi F, Labourier E, Forne T, Divita G, Derancourt J, Riou JF, Antoine E, Cathala G, Brunel C, Tazi J. Specific phosphorylation of SR proteins by mammalian DNA topoisomerase I. Nature. 1996;381:80–82. doi: 10.1038/381080a0. [DOI] [PubMed] [Google Scholar]
- Roth MB, Zahler AM, Stolk JA. A conserved family of nuclear phosphoproteins localized to sites of polymerase II transcription. J Cell Biol. 1991;115:587–596. doi: 10.1083/jcb.115.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass H, Pederson T. Transcription-dependent localization of U1 and U2 small nuclear ribonucleoproteins at major sites of gene activity in polytene chromosomes. J Mol Biol. 1984;180:911–926. doi: 10.1016/0022-2836(84)90263-8. [DOI] [PubMed] [Google Scholar]
- Spector DL. Macromolecular domains within the cell nucleus. Annu Rev Cell Biol. 1993;9:265–315. doi: 10.1146/annurev.cb.09.110193.001405. [DOI] [PubMed] [Google Scholar]
- Spector, D.L., R.D. Goldman, and L.A. Leinwand. 1998. Cells: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 105.1– 105.4.
- Tazi J, Kornstadt U, Rossi F, Jeanteur P, Cathala G, Brunel C, Lührmann R. Thiophosphorylation of U1-70K protein inhibits pre-mRNA splicing. Nature. 1993;363:283–286. doi: 10.1038/363283a0. [DOI] [PubMed] [Google Scholar]
- Vincent M, Lauriault P, Dubois MF, Lavoie S, Bensaude O, Chabot B. The nuclear matrix protein p255 is a highly phosphorylated form of RNA polymerase II largest subunit which associates with spliceosomes. Nucleic Acids Res. 1996;24:4649–4652. doi: 10.1093/nar/24.23.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-Y, Lin W, Dyck JA, Yeakley JM, Songyang Z, Cantley LC, Fu X-D. SRPK2: a differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J Cell Biol. 1998;140:737–750. doi: 10.1083/jcb.140.4.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansink DG, Schul W, van der Kraan I, van Steensel B, van Driel R, de Jong L. Fluorescent labelling of nascent RNA reveals transcription by RNA polymerase II in domains scattered throughout the nucleus. J Cell Biol. 1993;122:282–293. doi: 10.1083/jcb.122.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- Xiao SH, Manley JL. Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev. 1997;11:334–344. doi: 10.1101/gad.11.3.334. [DOI] [PubMed] [Google Scholar]
- Xing Y, Johnson CV, Moen PT, McNeil JA, Lawrence JB. Nonrandom gene organization: structural arrangements of specific pre-mRNA transcription and splicing with SC-35 domains. J Cell Biol. 1995;131:1635–1647. doi: 10.1083/jcb.131.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuryev A, Patturajan M, Litingtung Y, Joshi RV, Gentile C, Gebara M, Corden JL. The C-terminal domain of the largest subunit of RNA polymerase II interacts with a novel set of serine/arginine-rich proteins. Proc Natl Acad Sci USA. 1996;93:6975–6980. doi: 10.1073/pnas.93.14.6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Taneja KL, Singer RH, Green MR. Localization of pre-mRNA splicing in mammalian nuclei. Nature. 1994;372:809–812. doi: 10.1038/372809a0. [DOI] [PubMed] [Google Scholar]
- Zuo P, Manley JL. Functional domains of the human splicing factor ASF/SF2. EMBO (Eur Mol Biol Organ) J. 1993;12:4727–4737. doi: 10.1002/j.1460-2075.1993.tb06161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]