

Abstract
The type I keratin 17 (K17) shows a peculiar localization in human epithelial appendages including hair follicles, which undergo a growth cycle throughout adult life. Additionally K17 is induced, along with K6 and K16, early after acute injury to human skin. To gain further insights into its potential function(s), we cloned the mouse K17 gene and investigated its expression during skin development. Synthesis of K17 protein first occurs in a subset of epithelial cells within the single-layered, undifferentiated ectoderm of embryonic day 10.5 mouse fetuses. In the ensuing 48 h, K17-expressing cells give rise to placodes, the precursors of ectoderm-derived appendages (hair, glands, and tooth), and to periderm. During early development, there is a spatial correspondence in the distribution of K17 and that of lymphoid-enhancer factor (lef-1), a DNA-bending protein involved in inductive epithelial–mesenchymal interactions. We demonstrate that ectopic lef-1 expression induces K17 protein in the skin of adult transgenic mice. The pattern of K17 gene expression during development has direct implications for the morphogenesis of skin epithelia, and points to the existence of a molecular relationship between development and wound repair.
Keywords: skin, hair, development, wound repair, keratin
In spite of a recent revolution in the discovery of novel genes, the most reliable criterium to define the epithelial nature of a cell remains keratin gene expression. The pattern of keratin protein synthesis provides a useful tool for studying progression through complex programs of epithelial differentiation and cycles of epithelial self-renewal (Moll et al., 1982; O'Guin et al., 1990; Byrne et al., 1994). Keratins belong the superfamily of intermediate filament (IF)1 proteins. There are in excess of 35 keratin genes, and these can be partitioned into two distinct sequence types, I and II, based their genomic structure and nucleotide sequence homology (Fuchs and Weber, 1994). Most keratin genes, including the type II K1–K8 and the type I K9–K20, are expressed in soft epithelia (Moll et al., 1982; O'Guin et al., 1990). In addition, a group of seven type I (Ha1–Ha7) and six type II (Hb1–Hb6) keratin genes are expressed in hard epithelia such as hair and nail (Rogers et al., 1997; Winter et al., 1997). Epithelial cells coordinately regulate the expression of at least one type I and one type II gene, reflecting the requirement for both types of keratin proteins in a 1:1 molar ratio to sustain the assembly of 10-nm filaments (Coulombe, 1993). Many type I and type II keratin genes are de facto regulated in a pairwise fashion and in an epithelial tissue type- and differentiation-specific manner, even though any combination of type I and type II proteins can produce a filamentous polymer in vitro (Hatzfeld and Franke, 1985). The notions of sequence diversity, pairwise regulation, and differentiation-specific expression of keratin genes have largely been conserved throughout evolution, suggesting the existence of a functional link between keratin proteins and the diversity of structure and function of epithelial tissues.
Our laboratory is interested in the regulation and function of three keratin genes, the type II K6 and the type I K16 and K17, which cannot be readily associated with a well-defined epithelial context (O'Guin et al., 1990). These genes show an analogous pattern of expression in human skin that includes a constitutive expression in the hair follicle, sporadic expression in the thick epidermis of palm and sole skin, but no expression in interfollicular epidermis (Moll et al., 1983; Troyanovsky et al., 1989). K17 is also expressed in the myoepithelium surrounding secretory epithelial cells in many glandular tissues (Troyanovsky et al., 1989). Moreover, K6, K16, and K17 are induced under conditions associated with enhanced proliferation and abnormal differentiation, such as viral infection, inflammation, psoriasis, carcinoma, and after acute injury to various stratified epithelia (Weiss et al., 1984; Stoler et al., 1988; Jiang et al., 1994; Leigh et al., 1995; Paladini et al., 1996; McGowan and Coulombe, 1998). Although its functional significance has not yet been established, the accumulation of K6, K16, and K17 occurs at the expense of the keratins normally expressed under basal conditions and correlates with major changes in epithelial cytoarchitecture (Paladini et al., 1996; Coulombe, 1997; McGowan and Coulombe, 1998).
In comparison with the other wound-induced keratins, K6 and K16, relatively little is known about the significance of K17 gene expression and the properties and function(s) of K17 protein. To gain some insight into this question we wanted to examine K17 expression in a context potentially related to wound repair, epidermal development. Whereas one functional human 17 gene and two pseudogenes have been characterized (Troyanovsky et al., 1992), only a partial cDNA is available for mouse K17 (Knapp et al., 1987). To facilitate our studies we cloned the full-length gene for mouse K17 and examined its expression by producing a high-titer monospecific antiserum. We discovered that K17 is expressed at an early stage of skin morphogenesis according to a complex spatial pattern, and that the transcription lymphoid-enhancer factor (lef-1) is likely to play a role in its regulation. Our data led us to propose a revised model for skin morphogenesis, and suggest the existence of a molecular relationship between development and the wound repair response in adult skin epithelia.
Materials and Methods
Materials
Materials were obtained from the following sources: 129SvJ mouse genomic library packaged into Lambda Fix II and pBluescript vector from Stratagene (La Jolla, CA); restriction endonucleases and DNA modifying enzymes from New England Biolabs (Beverly, MA); Superscript II reverse transcriptase from Life Technologies (Grand Island, NY); Nytran membranes from Schleicher & Schuell (Keene, NH); Hybond-N filters from Amersham Corp. (Arlington Heights, IL); phage and plasmid DNA purification kits from QIAGEN Corp. (Santa Clarita, CA); LA-Taq DNA polymerase from Panvera Corp. (Madison, WI); alkaline-phosphatase antibody detection kit from Bio-Rad Laboratories (Hercules, CA). All other chemicals were reagent grade and were typically obtained from Sigma Chemical Co. (St. Louis, MO).
Animal Protocols
All studies involving animals were reviewed by the Johns Hopkins University Animal Use and Care Committee (Baltimore, MD). For studies involving experimental injury adult mice were anesthetized with avertin and their backs were epilated with Nair cream (Carter-Wallace Inc., New York, NY). A surgical area was disinfected and full thickness skin wounds were made with a 4-mm punch (AcuPunch; Acuderm Inc., Ft. Lauderdale, FL). The injured area was cleaned and covered with anti-bacterial ointment until the time of killing. For developmental studies, mouse embryos were isolated from timed pregnancies of B6C3F1/J mice (Jackson Laboratory, Bar Harbor, ME). Individual matings were initiated at 6:00 pm and monitored the next morning at 8:00 am. At this time the males were removed and the females were examined for the appearance of a vaginal plug. If copulatory plugs were detected, the date was recorded and the pregnancy was timed using 12 noon as embryonic day (e) = 0.5 d (Kaufman, 1992). On days 8, 9, 10, 12, 14, 16, and 18 at 12:00 noon, the embryos were isolated and fixed in either Bouin's fixative for subsequent embedding in paraffin or frozen in OCT Compound (Tissue-Tek; Sakura Finetek, Torrance, CA) for preparation of cryosections. Additional tissues from lef-1 overexpressing and lef-1–null mice were obtained from E. Fuchs (University of Chicago, Chicago, IL) (Zhou et al., 1995) and R. Grosschedl (University of California, San Francisco, CA) (van Genderen et al., 1994), respectively.
RNA Isolation
RNA samples were obtained from control and experimentally wounded mouse back skin using guanidine thioisocyanate (GTC)-phenol extraction method (Bessho et al., 1993). The wounded tissue samples were extracted 72 h after experimental injury to enhance the abundance of wound-induced keratin gene mRNAs.
Immunological Analyses
For immunoblot analyses, known quantities of recombinant human keratins were electrophoresed, transferred to nitrocellulose, and the blots were incubated with primary antisera diluted in blocking buffer (Tris-buffered saline with 0.5% Tween 20 and 5% powdered milk). Bound primary antibodies were revealed by alkaline phosphatase–conjugated secondary antibodies as recommended by the manufacturer (Bio-Rad Laboratories). The primary antisera for K17 and K6 were prepared as described below. Immunohistochemical analyses were performed on 5-μm sections prepared from either paraffin-embedded or fresh-frozen tissues. The sections were first reacted with the primary antisera and then revealed with either a peroxidase-based reaction (Kirkegaard and Perry Laboratories, Gaithersburg, MD) or by indirect immunofluorescence (Jackson Immunological Reagents, West Grove, PA). In addition to the antisera mentioned above, other primary antisera used in these studies were: a guinea pig anti-K8/18 purchased from ARP laboratories (Belmont, MA); a mouse monoclonal anti-K10 (clone K8.60) from Sigma Chemical Co.; a mouse monoclonal anti-K14 (LL001) donated by I. Leigh (London Hospital Medical College, London, UK) (Purkis et al., 1990); and a rabbit anti–lef-1 donated by R. Grosschedl (van Genderen et al., 1994). Double-immunofluorescence staining using primary antibodies from the same species (rabbit anti-K17 and rabbit anti–lef-1) was performed as described (Lewis-Carl et al., 1993).
Screening the Mouse Genomic Library
Approximately 1.5 d × 106 phage clones were screened with [α-32P]dCTP radiolabeled probes. The initial round of screening was performed using two probes. The first probe was derived from the rod portion of the published human keratin 16 cDNA (Paladini et al., 1995). This domain is highly conserved among the type I keratin sequences, particularly K14, K16, and K17. The second probe was generated by reverse transcription (RT)-PCR using RNA isolated from mouse back skin 72 h after experimental wounding as described (Takahashi et al., 1998). The oligonucleotides were based on the partial mouse K17 cDNA sequence published by Knapp and colleagues (Knapp et al., 1987) and amplified a 700-bp fragment containing the COOH terminus and 3′ untranslated regions of this mRNA. The strategy was to use the first probe as a means of identifying all the type I keratin genes contained in the library and then apply the K17-specific probe on a duplicate set of filters to identify the K17 containing phages. Hybridization and washes were carried out as suggested by the manufacturer (Stratagene). Positive colonies were isolated by repeated plaque purification as described (Sambrook et al., 1989). Phage DNA was extracted and analyzed by restriction enzyme digestion and Southern blotting. DNA fragments expected to contain expressed regions of the K17 gene were subcloned in pBluescript KS+ and sequenced. DNA sequencing and reconstruction of mouse keratin 17 gene locus was performed with the assistance of the Johns Hopkins University DNA Sequencing Core Facility (Baltimore, MD).
Mouse K17 cDNA Clone
A cDNA clone containing the entire coding sequence of K17 was obtained via RT-PCR with oligonucleotides derived from the SvJ129 genomic sequence using RNA from experimentally wounded back skin of a SvJ129 mouse. The primers used were: forward primer, 5′-ACGTGGATCCCGCTGCCACCATGACCACCA-3′; and reverse primer, 5′-GCTGTAGCAGGAGGGTGATGCCGGAGCGGA-3′.
The PCR conditions applied were 94°C for 1 min (denaturation); 55°C for 1 min (annealing), and 72°C for 2 min (elongation) for 25 cycles. The 1.3-kb product obtained (as expected) was subcloned in pGEM-T (Promega Corp., Madison, WI) and subjected to DNA sequence analysis as described above. Sense and anti-sense probes corresponding to the 3′-untranslated region-specific probe were also generated and used by Carroll et al. (1997) for in situ analysis of K17 mRNA expression.
Antibody Production and Characterization
Synthetic peptides (see below) were conjugated to maleimide-activated keyhole limpet hemocyanin carrier as described by the manufacturer (Pierce Chemical Co., Rockford, IL) and used for the production of monospecic anti-keratin polyclonal antisera in rabbits according to standard procedures (Covance Research Products, Denver, PA). A 12-mer peptide, NH2-C-S-S-R-E-Q-V-H-Q-T-T-R-COOH, was used for the production of an antiserum to K17 (anti-K17), and a 9-mer peptide, NH2-C-S-S-T-I-K-Y-T-T-COOH, was used for the production of an antiserum that recognizes all K6 isoforms (anti-K6gen; Takahashi et al., 1998). Bleeds were tested with the immunoblot procedures described above using recombinant keratin proteins prepared as described (Takahashi et al., 1994; Wawersik et al., 1997).
Results
Isolation and Characterization of Genomic and cDNA Clones for Mouse K17
We screened 106 clones from a 129 SvJ mouse genomic library (average insert size: 15–20 kb). An initial pool of 31 phage clones was established in which two clones (designated 16-9 and 16-61) were found to hybridize strongly with a K17-specific probe. Further analyses on these two clones revealed that they contain overlapping inserts that included the 3′ portion of a type I keratin gene whose coding and 3′ noncoding sequences were identical to the previously reported partial K17 cDNA (Knapp et al., 1987). Genomic walking yielded a third phage, designated 16-8, which contained the 5′ portion of this gene. DNA sequence analyses revealed the presence of a 925 nucleotide overlap between the genomic inserts contained in phages 16-8 and 16-9. The contiguity of these two inserts in 129 SvJ mouse genomic DNA was confirmed by a PCR experiment (data not shown).
The nucleotide sequence of all exons and intron–exon junctions, 5′ upstream and 3′ noncoding sequences was determined for the candidate K17 genomic clone (see Fig. 2). The boundaries of the functional elements in the gene isolated were established via comparison with the previously cloned human K17 gene (Troyanovsky et al., 1992) and a near full-length mouse K17 cDNA clone (see below). The candidate mouse K17 gene contains eight exons interrupted by seven introns (1–7). The sequence of the exon– intron boundaries (Fig. 2) conforms to the consensus splicing signals (Shapiro and Senapathy, 1987). All introns occur within the protein coding sequence (Figs. 1 and 2) at positions that are perfectly conserved in all type I keratin genes, including human K17 (Troyanovsky et al., 1992).
Figure 2.

Nucleotide sequence of exons and flanking regions for the mouse K17 gene. Sequences corresponding to protein coding strand for exons 1–8 are shown in uppercase letter, and the predicted amino acid encoded (one-letter code) is indicated directly above. Amino acids are numbered at right starting with the initial methionine. The 5′ boundary of exon 1 has not been determined. Sequences corresponding to intron boundaries are shown as lowercase letters. In the 5′ upstream sequence, the canonical TATA sequence (−95 bp from the ATG translation start) is underlined, as are potential binding sites for known transcription factors (Boulikas, 1994). In the 3′ sequence, the canonical polyA signal sequence (AATAAA) is underlined. This sequence can be accessed at GenBank (GenBank/EMBL/DDBJ accession number applied for).
Figure 1.

Relationship between genomic and protein domain structure for mouse K17. From the two overlapping phage clones designated 16-8 and 16-9 shown in A, a full-length type I keratin gene was reconstructed as shown in B. DNA sequencing showed that this gene contains an open reading frame identical to that of a near full-length (this study) and partial (Knapp et al., 1987) cDNA clones for mouse K17, and highly related to a genomic clone for human K17 (Troyanovsky et al., 1992). The structure of the mouse K17 gene is identical to that of the human orthologous gene and all other type I keratin genes (Troyanovsky et al., 1992), in that it contains eight exons (black boxes in B) and seven intervening introns comprised within ∼5 kilobases of sequence. (C) K17 is predicted to show the tripartite domain structure common to all IF proteins with a nonhelical (HEAD) domain at the NH2 terminus, a central domain featuring extended regions of heptad repeats (1A, 1B, 2A, and 2B), and another nonhelical (TAIL) domain at the COOH terminus. There is no obvious relationship beween the genomic and protein domain structures.
Oligonucleotide primers spanning the putative translation start codon (forward) and 3′ non-translated region (reverse) in the candidate K17 gene were used to obtain an ∼1.3-kb cDNA clone by RT-PCR. The nucleotide sequence of the cDNA clone is identical to the open reading frame contained within exons 1–8 in the candidate K17 genomic sequence and to the previously reported mouse partial K17 cDNA (data not shown) (Fig. 2). Together with the distribution of the corresponding keratin protein observed via immunohistochemistry (see below), this led us to conclude that the gene and cDNA that we have isolated encode mouse keratin 17.
An alignment of the cDNA and genomic sequences is shown diagramatically in Fig. 1. As for other keratin genes there is no obvious correspondence between the placement of introns and the predicted protein domains. The 5′ leader, the entire head domain and the 1A subdomain of the rod are encoded by exon 1, the largest of the eight exons (Table I). Exons 2–6 code for nearly all the remaining portions of the postulated α-helical rod domain including linkers, the exception being the last residue of subdomain 2B at the COOH terminus. The very short exon 7 codes for this last residue and the first eight amino acids of the nonhelical tail domain. Finally, the remainder of the tail domain (31 amino acids) and the entire 3′ nontranslated region are encoded by exon 8.
Table I.
Comparing the Mouse and Human K17 Genes
| Mouse K17 | Human K17 | |||
|---|---|---|---|---|
| Total length of the gene | 4,852 bp | 5,146 bp | ||
| Length of coding sequence | 433 aa | 432 aa | ||
| Size of exons/introns (in base pairs) | ||||
| Exon 1 | 432 | 432 | ||
| Intron 1 | 756 | 1,045 | ||
| Exon 2 | 83 | 83 | ||
| Intron 2 | 365 | 438 | ||
| Exon 3 | 157 | 157 | ||
| Intron 3 | 612 | 600 | ||
| Exon 4 | 162 | 162 | ||
| Intron 4 | 538 | 501 | ||
| Exon 5 | 126 | 126 | ||
| Intron 5 | 89 | 86 | ||
| Exon 6 | 221 | 221 | ||
| Intron 6 | 87 | 100 | ||
| Exon 7 | 26 | 23 | ||
| Intron 7 | 823 | 847 | ||
| Exon 8 | 278 | 291 | ||
| Predicted protein size | 48,159 D | 48,103 D | ||
| Observed protein size (SDS-PAGE) | ∼48 kD | ∼48 kD | ||
| Chromosomal mapping | 11 | 17q12-q21 | ||
| Identity of upstream gene | K17-like | K17 pseudogene | ||
| Distance to upstream gene | 2 kb | 2 kb | ||
| Downstream gene | K16 | K16 | ||
| Distance to downstream gene | — | 5.0 kb |
Inspection of the 5′ upstream sequence from our cloned sequence revealed the presence of a TATAA motif located 95 bases upstream of a potential translation start site with the consensus sequence defined by Kozak (1987) (Fig. 2). Inspection of the proximal 5′ upstream sequence revealed the presence of consensus binding sites for several known transcription factors (Fig. 2). These include AP-2, which has been shown to concentrate at sites of future hair follicle formation during epidermal development (Byrne et al., 1994), and several NF-IL-6(C/EBP-β) and GAS sites, which are involved in the upregulation gene expression during inflammation and viral infection as previously discussed for the human K17 gene (Proby et al., 1993; Jiang et al., 1994; Troyanovsky and Leube, 1994; Bonnekoh et al., 1995; Vogel et al., 1995; Komine et al., 1996). A detailed analysis of the complete K17 promoter sequence will be reported elsewhere.
The Human and Mouse K17 Gene and Predicted Protein Sequences Are Highly Conserved
Table I compares various functional elements from the mouse (this study) and human K17 gene (Troyanovsky et al., 1992). At ∼4.8 kb in total length, the mouse gene is slightly shorter than its human orthologue, due primarily to shorter introns 1 and 2. Whereas exon 8 is shorter in mouse at the level of the 3′ noncoding portion, exons 1–7 are of identical size between mouse and human (Table I). As a result, the K17 protein is predicted to be of similar size in human and mouse (see below). As illustrated in Fig. 1, the mouse K17 gene is flanked by a K17-like gene on its 5′ side and the K16 gene on the 3′ side. A characterization of the latter will be reported elsewhere (McGowan, K.M., K.M. Hess, and P.A. Coulombe, manuscript in preparation). Of note, Troyanovsky et al. (1992) showed that the functional human K17 gene is flanked by a K17 pseudogene on its 5′ side and the functional K16 gene on its 3′ side, all of which reside within the cluster of type I keratin genes on chromosome 17q. It appears likely that these keratin genes reside on chromosome 11q in mouse, since it is syntenic with human chromosome 17q and contains a cluster of type I genes (Nadeau et al., 1989).
An alignment of the predicted amino acid sequences of mouse and human K17, as well as human K14 and K16, is shown on Fig. 3. The mouse sequence (433 amino acids) is longer than human K17 by one amino acid (Table I) due to the insertion of a Pro residue at position 400 within the tail domain (Fig. 3). The primary sequence of the head, rod, and tail domains of mouse and human K17 show 88% (73 out of 83 residues), 96% (302 of 312 residues), and 97% identity (37 of 38 residues), respectively. Most of the amino acid substitutions between mouse and human K17 are conservative in nature, and four of them render mouse K17 identical to human K14 (see residues marked by # in Fig. 4). This exceptionally high degree of sequence relatedness between mouse and human K17 suggests, when interpreted in light of their highly conserved regulation (see below), that a K17 ancestral gene was present before the divergence of these species during evolution, and that the function(s) of the protein is conserved.
Figure 3.
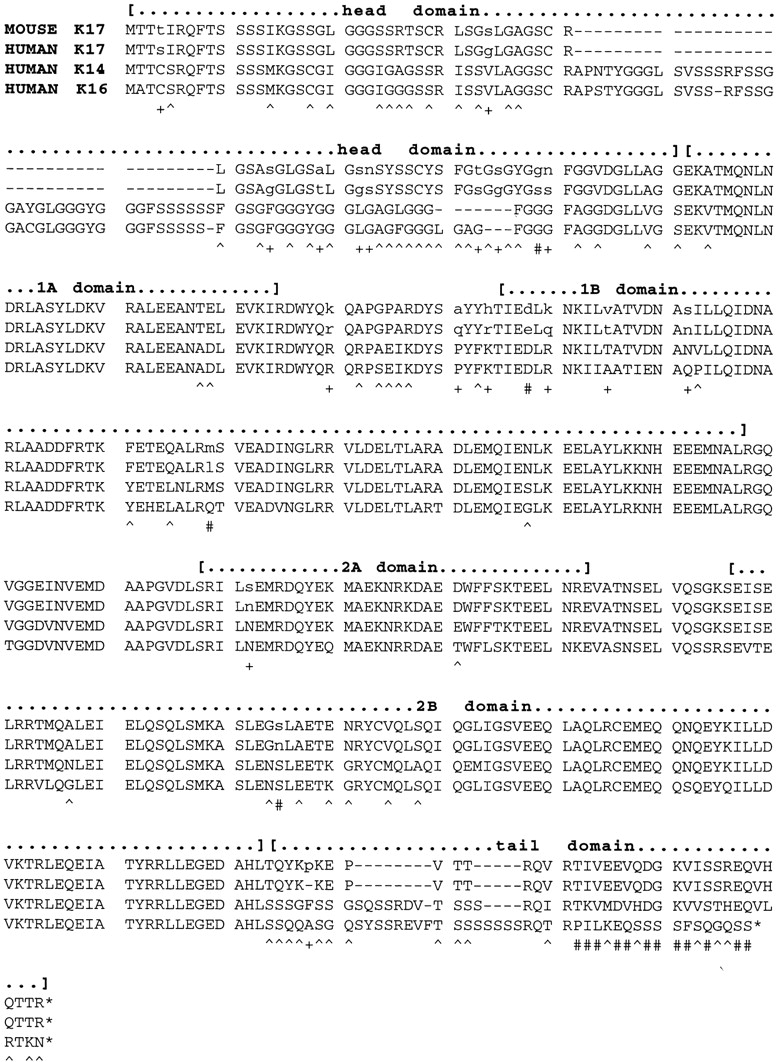
Alignment of the predicted amino acid sequences for mouse K17 (this study) and human K17 (Troyanovsky et al., 1992), K14 (Marchuk et al., 1984), and K16 (Paladini et al., 1995). This alignment was produced using the DNASIS v.3.5 software (Hitachi Software Engineering Inc., Tokyo, Japan), using default parameters. The boundaries of the major domains shared by all IF proteins (Fuchs and Weber, 1994) are indicated with brackets: a nonhelical head domain at the NH2 terminus; the α-helical subdomains 1A, 1B, 2A, and 2B characteristic of the central rod domain; and the nonhelical tail domain at the COOH terminus. Asterisks, nonsense stop codons. Underneath the aligned sequences, the symbols + and # identify residues that are different between mouse and human K17; #, the subset for which mouse K17 is identical to either K14 or K16; ^, residues that are conserved between mouse and human K17 but different in both K14 and K16.
Figure 4.

Validation of the specificity of a rabbit antiserum raised against a conjugated 12-mer peptide corresponding to the COOH terminus of mouse and human K17. (A) 50 ng of FPLC-purified human recombinant K14 (lane 1), K16 (lane 2), and K17 (lane 3) were prepared as described (Wawersik et al., 1997), electrophoresed in triplicate, and transferred on nitrocellulose membranes. These were reacted with the monoclonal antibody LL001 directed against K14 (Purkis et al., 1990), a rabbit polyclonal antiserum directed against K16 (Takahashi et al., 1994), and the newly produced antiserum against K17 (this study). (B) Immunoblot analysis of electrophoresed total skin proteins (20 μg) prepared from the back of an adult mouse (lane 2). A single band comigrating with purified human recombinant K17 (25 ng; lane 1), with an apparent M r of 48 kD, reacts with the polyclonal anti-K17 peptide antiserum made for this study.
As pointed out for the human sequences (Troyanovsky et al., 1992), mouse K17 is more related to K14 (73% identity) than to K16 (66% identity), due primarily to a better conservation of the tail domains in K17 and K14 (Fig. 3). In addition, the central rod domain of mouse K17 is more related to that of human K14 (88% identity) than to human K16 (81% identity). We recently showed that unlike K16, human K14 and K17 are capable of forming very stable heterotetramers with a variety of type II keratin partners, including K5, K6, and K8 (Wawersik et al., 1997).
Expression of K17 in Intact and Wounded Adult Mouse Trunk Skin
The commercially available antiserum against K17 (monoclonal; Sigma Chemical Co.) has a low titer and does not react with mouse K17 (our unpublished data). This prompted us to produce a polyclonal antiserum in rabbits using a hapten-conjugated 11-mer peptide that is perfectly conserved at the COOH terminus of human and mouse K17, and which is distinct from the related K14 sequence (Fig. 3). When used for Western blotting at a high dilution (1:10,000), the rabbit antiserum does not react with K14 and K16, but reacts strongly with purified recombinant human K17 (Fig. 4 A). In total insoluble proteins extracted from mouse trunk skin, the rabbit antiserum recognizes a single antigen with an apparent M r of 48 kD (Fig. 4 B) which comigrates with human K17 under standard gel buffer conditions (Fig. 4). This reactivity is not seen with the preimmune serum from the same rabbit, and can be successfully competed out with the free peptide used in the production of the antiserum (data not shown). These data establish that we produced a high-titer antiserum that is monospecific for K17, and confirms that the mouse and human K17 proteins do not differ significantly in their apparent M r by conventional gel electrophoresis.
When applied on sections of adult mouse trunk skin (Fig. 5), the K17 antiserum reacts strongly with hair follicles (Fig. 5, A and B) and sweat glands (data not shown), but not with normal interfollicular epidermis. In hair follicles, the entire outer root sheat (ORS) is immunostained, whether the follicle is actively growing (anagen phase; Fig. 5, A and B) or resting (data not shown). Most, if not all, ORS keratinocytes are positively stained for K17, unlike K6, whose protein distribution is restricted to the innermost ORS layers (Fig. 5, compare panels C, D, and E). Although the difference in K6 and K17 distribution is best seen in the context of larger hair follicles, such as those presented in Fig. 5, it applies for all hair types (see Fig. 11, showing longitudinally sectioned pelage hair). Similar findings were obtained in human skin samples (data not shown).
Figure 5.
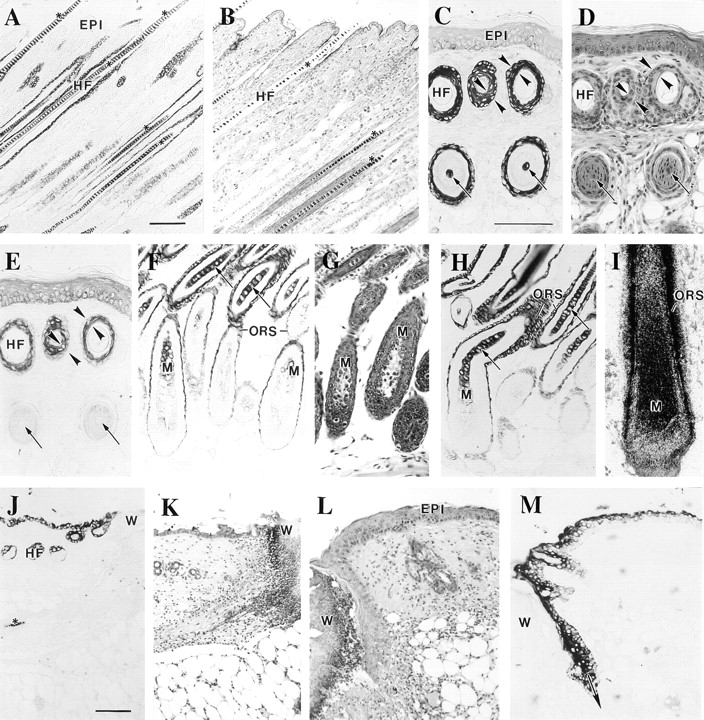
Immunolocalization of K17 protein in sections prepared from adult mouse skin. Tissues were fixed, paraffin embedded, and 5-μm sections were processed for hematoxylin-eosin staining, for K17 of K6 immunohistochemistry using a peroxidase-based detection method, or for in situ hybridization as described (Carroll et al., 1997). A (K17 stain) and B (H & E), consecutive cross sections of intact trunk skin showing anagen phase hair follicles (HF). C (K17 stain), D (H & E), and E (K6 stain) represent consecutive cross sections of adult tail skin where on can observe profiles of hair follicles at two different levels. In contrast to K6, K17 immunostaining occurs in both levels, indicating a more extensive distribution along the longitudinal axis of the follicle. Furthermore, K17 staining is present in all the ORS layers whereas K6 is polarized to the innermost layers, as indicated by the opposing arrowheads in C, D, and E. Finally, the arrows in these frames point to the occurrence of K17 staining in the hair shaft in the lower segment of the follicle. F (K17 stain), G (H & E), and H (K17 stain) document the occurrence of K17 immunoreactivity in the matrix epithelial cells (M) and early differentiating trichocytes (arrows). This staining is clearly distinct from that specific to the ORS compartment. (I) Localization of K17 mRNA by in situ hybridization using a 3′ noncoding probe. In this large vibrissae follicle, a strong hybridization occurs for both the hair matrix (M) and hair epithelial cells as well as the ORS. J (K17 stain), K, L (H & E), and M (K17 stain) illustrate the induction of K17 immunoreactivity in wound edge epidermis at 8 h (J and K) and 48 h (L and M) after injury. W, wound site. Bars: (A and B) 100 μm; (C–I) 100 μm; (J–M) 100 μm.
Figure 11.
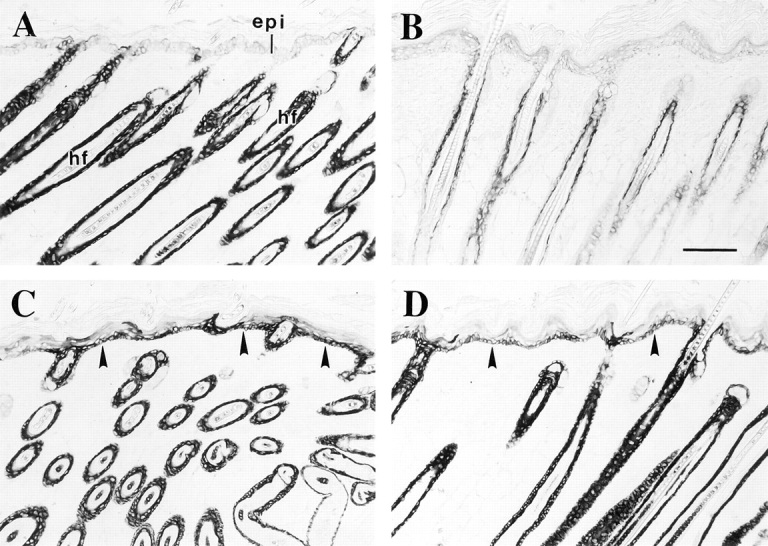
Immunolocalization of keratins in the skin of transgenic mice that ectopically express lef-1 in the basal layer of epidermis. Skin tissue samples were fixed in Bouin's, paraffin embedded, and then sections were immunostained for either K17 or K6 using a peroxidase-based detection method. (A) Trunk skin from a 11-d-old nontransgenic littermate control stained for K17. As in Fig. 5, the hair follicle (hf) ORS is strongly immunostained, whereas the interfollicular epidermis (epi) is negative. (B–D) Trunk skin from a 11-d-old transgenic animal stained for K6 (B) and K17 (C and D). Consistent with the normal histology (data not shown), K6 protein occurs in the innermost layers of the hair follicle ORS, whereas the epidermis is negative. In contrast, the basal layer of transgenic epidermis shows a strong signal for K17 (arrowheads in C and D), as does the hair follicle ORS. Arrowhead, dermo-epidermal interface. Bar, 100 μm.
Close examination of the hair bulb area revealed the consistent presence of faint immunostaining for K17 in the matrix cells and stronger staining in the early differentiating hair epithelial cells (trichocytes) (Fig. 5, F–H). The signal is lost in the upper segments of the hair (data not shown), owing probably to epitope masking. Such immunostaining for K17 occurs in all types of hair follicles, and its specificity is supported by control experiments involving the use of preimmune serum or the addition of the peptide immunogen to the anti-K17 serum (data not shown). Reexamination of in situ hybridization data (Carroll et al., 1997), which made use of a probe corresponding to the 3′ portion of the K17 mRNA, revealed the presence of K17 transcripts in the early differentiating trichocytes of vibrissae (Fig. 5 I) and pelage hair follicles (data not shown). A similar outcome was observed in a previous report by Panteleyev et al. (1997), but these authors attributed their results to the presence of melanin granules. We have observed this same pattern of immunostaining using samples obtained from unpigmented mice. Furthermore, immunoreactivity for K17 can be observed in Western blots analysis of protein extracts prepared from mouse hair clippings (the lack of K14 in such extracts confirms the absence of ORS contamination; data not shown). Finally, we can detect reporter transgene activity in trichocytes in the context of studies involving the 5′ upstream sequence of the mouse K17 gene (McGowan, K.M., and P.A. Coulombe, unpublished observations). Our ability to detect K17 protein in trichocytes, an occurrence that has never been reported before, may be primarily the result of the high titer and specificity displayed by our polyclonal antiserum.
We previously showed, using the commercially available monoclonal antibody (Sigma Chemical Co.), that K17 is induced at the wound edge starting 12 h after acute injury to human epidermis (Paladini et al., 1996). Likewise, Knapp et al. (1987) showed that a mouse type I keratin mRNA, which later on was identified as K17 (see Troyanovsky et al., 1992; Schweizer, 1993), is induced in skin wounds. Application of our novel antiserum to sections of adult mouse wounded skin showed that K17 is already induced by 8 h after acute injury (Fig. 5, E and F). In older wounds, K17 synthesis extends distally from the cut edge of the wound and is particularly strong in the migrating epidermal tissue, where it occurs in all epidermal layers (Fig. 5, G and H; 48 h after injury). The epidermal staining for K17 eventually disappears after wound closure (data not shown). These data establish that, as was found to be the case for K6 and K16 in human skin (Paladini et al., 1996), K17 is an early marker of the process of keratinocyte activation after injury and is expressed in the migrating epithelium.
Onset of K17 Expression Occurs Early in Developing Mouse Skin
Until ∼e9.5 d, the single-layered ectoderm covering the surface of the embryo expresses the keratins 8 and 18 typical of simple epithelia (Jackson et al., 1981). The earliest molecular indices of epidermal gene expression, starting at ∼e9.5 d, occur in the form of the transcription factor AP2 and its target genes K5 and K14. Byrne et al. (1994) showed that onset of expression of these genes takes place according to a complex spatiotemporal pattern that does not correlate with the initial stratification of the ectoderm. In agreement with previous reports (Jackson et al., 1981; Thorey et al., 1993), we find that e9.5-d ectoderm stains uniformly with an antiserum directed against K8/K18 but sporadically with antisera directed against K14 (data not shown). At e10.5 d, expression of K14 has generalized to a significant portion of the surface ectoderm. These data establish the validity of our antibody tools. We performed triple-staining studies that include the DNA stain Hoechst to analyze the context in which K17 expression is initiated in the early stages of skin morphogenesis. These studies made use of saggital sections of whole e12.5-d embryos, as these feature areas with single layered ectoderm (e.g., upper dorsum), two-layered ectoderm (e.g., lower dorsum), and stratified epithelium (e.g., ventral surface).
We find that induction of K17 expression takes place in cells that express both K8–K18 and K14. The induction is independent of stratification of the epithelium, as it is seen in single-layered ectoderm (Fig. 6, A–F), in the bottom layer of two-layered epithelium (Fig. 6, G and H), in the top layer of the stratified epithelium on the ventral surface of the embryo (Fig. 6, I and J). In favorable sections, the filamentous nature of the K17 staining can clearly be seen at higher magnification (Fig. 6 K). As previously reported (Greer and Roop, 1991; Byrne et al., 1994), no expression of K10 can be detected in sections prepared from e12.5-d embryos (data not shown). These data establish that K17 protein synthesis occurs shortly after the onset of K14 expression, before the onset of epithelial differentiation-specific gene expression.
Figure 6.

Distribution of K17 protein an early stage of mouse skin development. Triple- immunofluorescence staining was performed on sections prepared from fresh-frozen e12.5-d mouse embryos to assess the context in which K17 expression first occurs in the embryonic ectoderm. (A–C) Portion of the upper dorsum showing single-layer ectoderm as determined by the vital Hoechst stain for DNA (C). A single cell is seen to express K17 (arrowhead in A), whereas K14 expression is uniformly established at that stage (B). (D–F) Another example of single-layer ectoderm (Hoechst stain in F). Two adjacent cells are seen to express K17 (arrowhead in D), whereas uniform K8–K18 expression is maintained at that stage (E). (G and H) Portion of the lower dorsum showing two-layered ectoderm. In this example, K17 induction occurs in the bottom layer of the ectoderm (arrowhead in G), which is otherwise uniformly stained for K14 (H). (J and K) Portion of the ventral surface showing three-layered ectoderm. In this case, K17 induction occurs in the top layer of the ectoderm (arrowhead in I), whereas again the staining for K14 is uniform (J). (K) At higher magnification, the filamentous nature of the anti-K17 staining is readily apparent in individual epithelial cells. In all panels, the arrow depicts the interface between the surface epithelium and the underlying mesenchyme. Bars: (A–J) 20 μm; (K) 10 μm.
Onset of K17 Expression Reflects the Adoption of a Nonepidermal Cell Fate in Embryonic Skin
As mentioned above, we observe very patchy expression of the K17 gene in the ectoderm of the upper dorsum region of e10.5-d embryos (Fig. 7 A). Examination of sections prepared from several embryos suggest a periodicity in the distribution of K17, in that clusters of one to three positively stained cells occur at regularly spaced intervals along the dorsal surface of the embryo. This staining pattern is more firmly established at e12.5 d (Fig. 7 B). Between e12.5 and e14.5 d, the two-layer ectoderm further stratifies to give rise to embryonic epidermis over most of the embryo surface, whereas the first signs of hair follicle induction occur in the form of an invagination of small groups of epithelial cells into the underlying mesenchyme. We find that the placodes and the proximal epithelial tissue are both strongly immunostained for K17 (Fig. 7, C, D, and E show various stages of placode formation in e14.5-d epidermis). Continued downward expansion of the placodes gives rise to primary hair germs, starting at e14.5 d and well established by e16.5 d (Mann, 1962; Hardy and Vielkind, 1996). The strong expression of K17 persists in the primary hair germs at e14.5 d (Fig. 7 F) and e16.5 d (Fig. 7 G), and examination of sections from several embryos shows that the majority of the epithelial cells in primary hair germs are K17 positive. Collectively, our data suggest that the precursors of pelage and vibrissae hair follicles arise from small clusters of K17–positive ectoderm that first appear in the undifferentiated ectoderm of e10.5 and e12.5 mouse embryos.
Figure 7.

Distribution of K17 protein during skin development. Sections were prepared from fresh-frozen e10.5-, e12.5-, e14.5-, and e16.5-d mouse embryos and from newborn mice and immunostained for K17 using a peroxidase-based detection method. Examples were chosen so as to represent the continuum of events characterizing skin morphogenesis. K17 is first induced in the single layer ectoderm in e10.5-d embryos (arrowheads in A). At e12.5 d, small clusters of K17-positive cells occur at periodic intervals in the cross sectioned ectoderm (arrowheads in B). These clusters subsequently evolve into placodes and then primary hair germs, as suggested by the pattern of K17-staining in frames C–G (open arrowheads). At e14.5 d, the periderm (p) is also distinctly stained for K17 (C–E). Moreover, beginning at e14.5 d (F) and firmly established by e16.5 d (bracket in G), the basal layer of embryonic epidermis expresses K17. Between e16.5 d and birth (H), the staining for K17 in the basal layer as well as the periderm is gradually lost. At birth, the skin features an adult pattern of K17 expression, in that the latter is largely restricted to the hair follicles. In all frames, the small arrow depicts the interface beween the surface epithelium and the underlying mesenchyme. See text for further explanation. Bars: (A–G) 50 μm; (H) 82 μm.
Another ectodermal cell type that is uniformly and consistently immunopositive when stained with anti-K17 is the periderm. As is the case of K6 (see below), the periderm staining begins at e14.5 d (Fig. 7, C–F) and extends (Fig. 7 G, e16.5-d sample) until this layer is lost from the surface of the embryos shortly before birth (Fisher, 1994).
The embryonic epidermis is mostly negative for K17, with the exception of a transient expression in the basal layer starting at e14.5 (Fig. 7 F) and peaking at e16.5 d (Fig. 7 G). This finding is rather intriguing, as differentiation specific gene expression is already ongoing in the embryonic epidermis during this period (data not shown) (Dale et al., 1985; Greer and Roop, 1991; Byrne et al., 1994). This expression has already begun to recede towards developing hair follicles by e18.5 d and is further restricted to the neck of hair follicles openings in dorsal trunk skin of newborn mice (Fig. 7 H).
K17 Expression Is Partially Uncoupled from That of K6 in Embryonic Skin
We examined the expression of K6 in our collection of embryo specimens using a newly produced rabbit polyclonal antiserum (refer to Materials and Methods). This type II keratin is coregulated with K17 in wound edge epidermis after injury to human and mouse adult skin (Paladini et al., 1996; Takahashi et al., 1997, 1998), and is presumed to be the type II pairing partner for K17 in several stratified epithelia (e.g., O'Guin et al., 1990). In e12.5-d embryos, a strong staining for K6 protein occurs in the oral mucosa whereas the surface ectoderm is negative (data not shown). In e14.5-d embryos, K6 immunoreactivity occurs the innermost aspect of the developing vibrissae follicles (Fig. 8 A) and in the periderm layer, which is uniformly stained (Fig. 8, A and B). Similarly to K17, the periderm staining for K6 persists (Fig. 8 C) until the loss of this layer before birth (data not shown). In contrast to K17, however, we detect K6 immunoreactivity at a relatively late stage of pelage hair follicle development (e16.5 d), where it is restricted to a subset of epithelial cells (Fig. 8 C). This pattern is maintained in e18.5-d embryos and newborn mouse skin (data not shown). Consistent with previous reports situating the onset of hair-specific keratin gene expression at e14.5–e15.5 d (Zhou et al., 1995), onset of K6 expression appears to coincide with the beginning of differentiation in developing hair follicles. These data establish that with the notable exception of the entire periderm and a small subset of hair epithelial cells (see Discussion), K17 expression is largely uncoupled from that of K6 in developing skin.
Figure 8.

Distribution of K6 isoform proteins during skin development. Sections were prepared from fresh-frozen mouse embryos and immunostained for K6 protein using a peroxidase-based detection method. K6 staining is first detected in e14.5-d embryos, where it is restricted to a subset of cells within the vibrissae (vib in A) and the periderm (p; shown are whisker pads in A and trunk skin in B). At e16.5 d, K6 staining is detected in a small subset of epithelial cells within primary hair germs (arrows in C), coinciding with the onset of differentiation-specific gene expression (see text). Solid arrowhead, interface between the surface epithelium and the underlying mesenchyme. Bar, 100 μm.
Expression of K17 Coincides with That of the Transcription Factor lef-1 during Skin Development
The epithelial placodes that serve as precursors of hair follicles and other appendages form directly above condensates of specialized mesenchymal cells with inductive properties (Sakakura et al., 1976; Kollar et al., 1983; Hardy, 1992; Jowett et al., 1993). Lef-1, a protein that belongs to the HMG box family of transcription factors (Clevers and Grosscheld, 1996), is known to play a critical role in the epithelial–mesenchymal interactions that underlie appendageal morphogenesis. Lef-1 is expressed in the cell condensates that form at sites of future hair follicle formation (e.g., Zhou et al., 1995; Kratochwil, 1996), and mice harboring a null mutation in their lef-1 gene show a significantly reduced number of hair follicles, a failure to form teeth and mammary glands, and die shortly after birth (van Genderen et al., 1994). Given this, we decided to relate the expression of K17 to that of lef-1 in our collection of mouse embryo sections.
Immunostainings for lef-1 performed on frozen sections prepared from e12.5-, e14.5-, and e16.5-d mouse embryos confirmed the elegant whole mount in situ hybridization findings reported by Zhou et al. (1995). The lef-1 protein staining is predominantly nuclear, as expected for a transcription factor, and is initially manifested in and around epithelial placodes (Fig. 9, A and B; e14.5-d embryo dorsum). The presence of lef-1 in both the epithelial cells forming the placode and in proximal mesenchymal cells is consistent with its role in epithelial–mesenchymal interactions (Kratochwil et al., 1996). These sites of epithelial– mesenchymal interactions, as identified by lef-1 protein stainings, also display a strong signal for K17 (Fig. 9, A and B). A survey of samples obtained from dual-staining experiments of e14.5-d embryos show that the spatial correspondence between the signals for K17 and lef-1 proteins is striking. This is well illustrated in the context of double-staining experiments (hair placode shown in Fig. 9, C and D) and staining of consecutive frozen sections (vibrissae shown in Fig. 9, E and F). As hair placodes evolve into primary hair germs, e.g., in e16.5-d embryos, the spatial correspondence between the two signals persists only at the tip region near the mesenchyme (data not shown). In addition, the basal layer of e16.5-d interfollicular epidermis is clearly positive for both K17 and lef-1 proteins (Fig. 9, B and C). In contrast to K17, however, we could not detect a convincing signal for lef-1 in the periderm layer at any stage of its formation. In newborn skin, the signal for lef-1 is mostly restricted to the actively growing (bulb) segment of the follicles, as previously reported (Zhou et al., 1995), whereas K17 is distributed throughout the outer root sheath and occurs in proximal epidermis as well (refer to Fig. 7 H). Thus, it would appear that lef-1 protein is not synthesized in differentiating epithelial cells of embryonic mouse skin.
Figure 9.
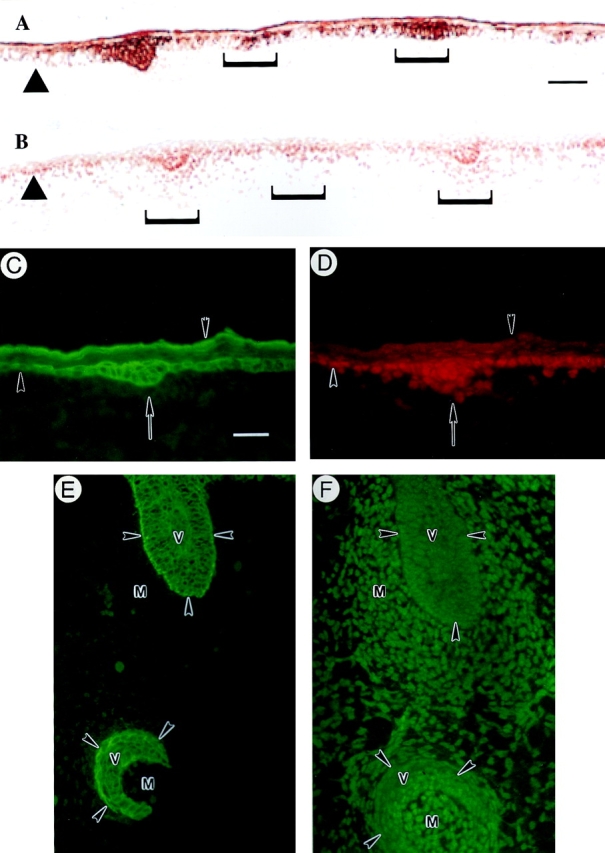
Colocalization of K17 and lef-1 during skin development. Sections were prepared from fresh-frozen mouse embryos at specific developmental stages and immunostained for K17 and for the transcription factor lef-1, either on consecutive (A and B and E and F) or the same sections (C and D, double immunofluorescence). (A and B) Upper dorsum of a e12.5-d embryo stained for K17 (A) and lef-1 (B). Brackets denote placodes at various stages of their formation resulting from epithelio–mesenchymal interactions. A strong, epithelium-restricted staining for K17 is seen in these placodes, while the signal for lef-1 is confined to the nucleus in both the epithelial cells and the proximal mesenchymal cells. (C and D) Upper dorsum of a e12.5-d embryo subjected to double immunofluorescence for K17 (C) and lef-1 (D). K17 is expressed strongly in the placode epithelial cells denoted by an arrow and in the periderm (inverted arrowhead), and sporadically in the embryonic basal layer. Although excluded from the periderm, a strong nuclear staining for lef-1 is detected in the placode epithelial cells and in the mesenchymal cells proximal to it. In addition, embryonic basal cells that express K17 strongly are also immunopositive for lef-1. The upward arrowheads in A–D depict the interface between the epithelium and the underlying mesenchyme. (E and F) Consecutive sections through vibrissae follicles (v) from an e14.5-d embryo subjected to immunofluorescence for K17 (E) and lef-1 (F). Although K17 immunoreactivity is restricted to the epithelial cells, the signal for lef-1 is particularly strong in the nucleus of mesenchymal cells (m) surrounding the epithelium. Arrowheads, interface between the epithelium and the mesenchyme. Bars: (A and B) 70 μm; (C–F) 20 μm.
Keratin 17 is expressed in several mature epithelia whose development is known to depend on epithelial– mesenchymal interactions mediated by lef-1. These include vibrissae, salivary glands, tooth, and thymus (Fig. 10, A–D, respectively). To further probe the relationship between K17 and lef-1 expression, we examined these tissues in e14.5-d mouse embryos. Again, we detected K17 immunoreactivity at an early step in the morphogenesis of these tissues (Fig. 10, A′–D′). Collectively, these findings establish that the K17 gene is expressed in a number of developing tissues whose formation partly depend upon lef-1.
Figure 10.
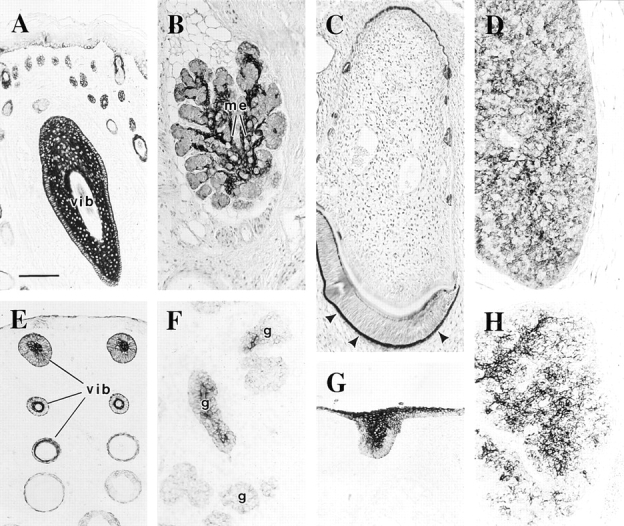
Distribution of K17 protein in adult- and embryonic-stage epithelia known to develop in a lef-1–dependent manner. Sections were prepared from either paraffin-embedded (A–D) or fresh-frozen e14.5-d whole embryos (E–H), and immunostained for K17 using a peroxidase-based detection method. A (adult) and E (embryo), whisker pads; vib, a tangentially sectioned vibrissae follicle in A. B (adult) and F (embryo), submaxillary gland; me, myoepithelium that envelops the glandular epithelial tissue. C (adult) and G (embryo), tooth. Arrowhead, enamel organ. D (adult) and H (embryo), thymus. Bar, 100 μm.
Ectopic Expression of lef-1 Correlates with K17 Induction in the Epidermis of Adult Transgenic Mice
The intriguing correspondence between the timing and location of lef-1 protein synthesis, a transcription factor allegedly involved in the regulation of hair-specific keratin genes, and K17, a component of the pilosebaceous apparatus, raises the possibility that the former may contribute to regulate the latter during early skin morphogenesis. In support of this possibility, inspection of the 5′ upstream sequence of the mouse K17 gene reveals the presence, in three distinct locations, of a nucleotide sequence motif, 5′-CTTTGWW-3′, which fits perfectly the consensus DNA binding site for lef-1 (Powell et al., 1991; Clevers and Grosscheld, 1996). A perfect consensus binding site for lef-1 occurs in the third intron of the human K17 gene (GenBank/EMBL/DDBJ accession number Z19574) (Troyanovsky et al., 1992). Such lef-1 consensus binding sites occur in the 5′ upstream regulatory region of all the hair follicle-specific genes examined so far, and can mediate the transactivation of a reporter gene in a lef-1–dependent manner in an heterologous expression assay ex vivo (Zhou et al., 1995). Based on these findings we examined the expression of K17 in two mouse models in which lef-1 expression has been genetically manipulated: mice that ectopically overexpress lef-1 in epidermis via a keratin 14 gene promoter (Zhou et al., 1995), and mice harboring a null mutation for lef-1 (van Genderen et al., 1994).
Transgenic mice that express lef-1 under the control of the K14 gene promoter show the presence of lef-1 in the basal layer of epidermis, oral mucosa, and a few other stratified epithelia. These mice feature an abnormal patterning of hair follicles over their trunk, and consistent with the postulated role for lef-1 in appendageal morphogenesis (van Genderen et al., 1994), they show ectopic appendages in the oral mucosa (Zhou et al., 1995). Paraffin-embedded skin samples from 11-d-old transgenic pups and controls were provided by E. Fuchs. The epidermis of these mice display a normal morphology and distribution of key markers of terminal differentiation, such as K10 and filaggrin (data not shown) (Zhou et al., 1995). Additionally, the interfollicular epidermis does not stain positively for K6 (Fig. 11 B), a marker for abnormal proliferation that is coregulated with K17 after various acute challenges to the skin (Takahashi et al., 1997; 1998). Yet, unlike the situation in control (Fig. 11 A), a significant fraction of basal cells in interfollicular epidermis express K17 in lef-1 transgenic tissue (Fig. 11, C and D). The specificity of this induction and the absence of any other detectable changes in the properties of progenitor basal keratinocytes in the mature epidermis of [K14 promoter] lef-1 mice supports the possibility that lef-1 may be involved in aspects of K17 expression at an early stage of appendageal morphogenesis.
Transgenic mice that harbor a null mutation in the lef-1 gene feature a significantly reduced number of hair follicles that are reportedly shorter and delayed in their maturation (van Genderen et al., 1994). We found that the hair follicle profiles present show positive immunostaining for K17 in the ORS, however these samples lacked hair shafts (data not shown). Whether these follicles form as a result of functional redundancy with other related transcription factors in lef-1–null mice, or whether lef-1 preferentially impacts on the formation of a specific type of hair follicle, remains unclear at present. From this we conclude that lef-1 expression is not essential for the morphogenesis of all pelage hairs or for the expression of K17 in the ORS.
Discussion
K17 and the Onset of Epithelial Differentiation during Skin Development
Essential to understanding the mechanisms that underlie the developmental choices that occur during mammalian organogenesis is the identification of the stages at which commitment to various cell lineages and subsequent differentiation occur. The embryonic epidermis represents an excellent system for such studies as it differentiates and expands from a single-layered embryonic ectoderm to form a stratified squamous epithelium. Concomitantly, cells from within this tissue undergo separate programs of differentiation to form epidermally derived appendages like hair, nail, teeth, and glands. The monitoring of epidermal and hair-specific keratin gene expression has significantly contributed to our current understanding of these processes (Dale et al., 1985; Kopan and Fuchs, 1989; Greer and Roop, 1991; Byrne et al., 1994). We surmised that given their constitutive expression in various appendages and their rapid induction after acute injury, the nonepidermal keratin genes K6 and K17 may provide additional insights into skin development. We found that expression of K17 is initiated in the undifferentiated ectoderm at the single- or two-layer stage, starting in e10.5-d mouse embryos. The spatiotemporal pattern of K6 expression is different, and coincides with the onset of expression of several differentiation-specific markers at a later time (see below). Our data point to the intriguing possibility that onset of K17 expression in the embryonic ectoderm reflects a commitment towards distinct epithelial lineages, and prompt us to propose a revised model for skin development as shown in Fig. 12.
Figure 12.

A model for early skin morphogenesis. This model relates the induction of K17 expression in developing mouse skin to other molecular events as described by Byrne et al. (1994). For the sake of simplicity, this model focuses on the events involving keratinocytes and ignores the other cell types. Shading, presence of K17 protein. (Phase 1) The onset of K5–K14 expression in the single-layer embryonic ectoderm, beginning at ∼e9.5 d, reflects a commitment towards the formation of a complex epithelium. Induction of K5–K14 occurs concomitant with, but independently of, stratification to a two-layered epithelium. (Phase 2) The onset of K17 expression, starting at ∼e10.5 d, reflects the determination of the major lineages in skin epithelia. Specifically, we postulate that K17 is induced in the subset of K8–K18, K5–K14 expressing epithelial cells that are responding to the mesenchymal cues that induce appendage formation, and in cells that will give rise to periderm. As is the case for K5–K14 approximately a day earlier, onset of K17 expression may occur in single-layer ectoderm or alternatively, in either layer of a two-layer-thick epithelium, depending on the region of the body. (Phase 3) Cell expansion and onset of epithelial polarization. Between e12.5 and e14.5 d, the epithelial sheet thickens considerably and clusters of K17-expressing epithelial cells extend into the underlying mesenchyme at sites of strong lef-1 transcription factor expression. Consistent with the lack of differentiation-specific gene expression, we propose that this time window corresponds primarily to a cell expansion phase. (Phase 4) Onset of cell type-specific differentiation. Starting at e14.5 d as previously reported, terminal differentiation is initiated within the various lineages as manifested through K1–K10 expression in embryonic epidermis and hair-specific keratin gene expression in hair germs. The periderm is destined to be lost via shedding beginning at e16.5 d.
In this model, the morphogenesis of the major types of skin epithelia is segmented into four distinct yet logically interconnected phases, which can be described as follows:
Phase 1. Commitment to Form a Complex Epithelium.
Up to ∼e9.5 d, the single-layered ectoderm covering the embryo is undifferentiated and expresses the keratins characteristic of simple epithelia, K8, and K18 (Jackson et al., 1981; Thorey et al., 1993). We propose that the onset of transcription factor AP-2 expression, soon followed by that of its target genes K5 and K14 (Byrne et al., 1994), reflects a commitment towards the formation of a complex epithelium. Of note, K5 and K14 (along with variable levels of K15) are the only keratins that are expressed in all the complex (soft) epithelia in the adult, where they typically occur in the least differentiated progenitor cells (Moll et al., 1982; O'Guin et al., 1990).
Phase 2. Definition of Major Epithelial Lineages.
We propose that onset of K17 expression in a subpopulation of the K5–K14- and K8–K18-expressing embryonic ectodermal cells, which occurs in the e10.5 –e12.5-d time window, signifies a commitment to form a nonepidermal tissue such as a hair follicle, a gland, or the periderm. This commitment is the result of a set of specific molecular instructions (e.g., lef-1–mediated epithelio–mesenchymal interactions) that do not take place in epidermal precursors, which represents the default pathway (Kopan and Fuchs, 1989). We have demonstrated that onset of K17 expression may occur within the single-layer embryonic ectoderm, or in the lower or upper layer of the two-layer ectoderm (depending on the region of the embryo), in cells that are mitotically active (this study; Byrne et al., 1994). These data extend the work of Byrne and colleagues (1994), who observed that the initial stratification of the ectoderm does not correspond to a polarization or differentiation event. Moreover, our temporal assignment for this phase is in complete agreement with the results obtained by Sanes and coworkers (1986), who used retrovirally marked cells to determine the genealogy of the various cell types in several mouse tissues. Their study showed that commitment to peridermal and follicular cell fates occurs at ∼e11 d, but that before this all ectodermal cells maintain pluripotentiality.
Phase 3. Cellular Expansion.
For a period of ∼48 hours that extends from the onset of K17 gene expression to that of differentiation-specific genes, there is a significant increase in epithelial cell number without molecular indices of terminal differentiation (see below). We propose that this period corresponds to an expansion of relatively undifferentiated cell pools within each of the major epithelial lineages. Obviously, additional studies are required to examine gene expression during this poorly-defined phase.
Phase 4. Onset of Lineage-specific Epithelial Differentiation.
Several differentiation-related genes are first expressed starting at e13.5–e15.5 d, such as K1–K10 (this study; Byrne et al., 1994), filaggrin, and loricrin (Byrne et al., 1994; Greer and Roop, 1991) in epidermis, and hard keratin genes (Byrne et al., 1994) as well as K6 (this study) in hair follicles. In all these instances, expression of the marker occurs in a subset of cells within the maturing epithelium, reflecting a polarization in the developing tissue. Consequently, this phase represents the last one in our model for the morphogenesis of skin epithelia, as it corresponds to the onset of terminal differentiation.
Although the four steps described occur sequentially for any individual element (e.g., a single hair follicle), it must be realized, as alluded to in the results section, that they are not synchronized over the entire skin tissue, since development proceeds according to spatial and temporal gradients.
K17 Is a Candidate Target Gene for Regulation by Lef-1
Considerable progress has been made towards identifying the mechanisms and molecules that mediate the reciprocal, inductive interactions between mesenchymal and epithelial tissues that play an integral role in hair follicle morphogenesis. One such molecule is the lymphoid enhancer factor, lef-1 (van Genderen et al., 1994; Zhou et al., 1995; Clevers and Grosschedl, 1996). Lef-1 is a transcription factor capable of bending DNA, a property that is thought to influence gene transcription by facilitating the association of other transcription factors (for review see Clevers and Grosschedl, 1996). Targeted inactivation of the lef-1 gene produced major alterations in epithelial tissues that share a dependency upon epithelial–mesenchymal interactions for proper morphogenesis (van Genderen et al., 1994). The spatiotemporal pattern of K17 expression during development suggest strongly that this keratin gene may be regulated by signals generated in the context of such epithelio–mesenchymal interactions, possibly lef-1.
We presented several lines of evidence that support this notion. First, we have shown a temporal and spatial correspondence between K17 and lef-1 protein synthesis in hair placodes. Second, we have presented data depicting K17 protein synthesis during the formation of other lef-1 target organs. Third, we provide convincing evidence that K17 is expressed in precursor matrix epithelial cells and early differentiating trichocytes of fully formed hair follicles. These cells give rise to the structures of the hair, and the programs of gene expression associated with these include several hard keratin genes postulated to be directly regulated by lef-1. To our knowledge, K17 represents the first instance of a keratin gene expressed in both hard and soft epithelial tissues. Fourth, there are three consensus binding sites for lef-1 in the mouse K17 gene, and at least one in the human K17 gene (the full 5′ upstream sequence of this gene is not yet available). These consensus sites are identical to those found in hair keratin genes (Powell et al., 1991, 1992), where they have been found to mediate a direct transactivation (Zhou et al., 1995). Fifth and finally, we found that K17 protein is synthesized in the epidermal basal layer of transgenic mice that ectopically express lef-1 under the control of the K14 promoter.
The connection between K17 and lef-1 expression is important when one considers the delay between the expression of lef-1, which begins at ∼e11 d (Zhou et al., 1995; Kratochwil et al., 1996), and the subsequent induction of hair keratin genes at ∼e14.5–15.5 d. The potential relationship between lef-1 expression and K17 upregulation could provide a critical molecular link between the expression of this transcription factor and the actual commitment to undergo appendageal organogenesis. The location of binding sites for lef-1 is different in the K17 and the hair-specific keratin genes (data not shown). This may represent an important determinant for the manner with which lef-1, being an architectural transcription factor, impacts on the expression of these keratin genes during development. Studies are in progress to test whether lef-1 can directly transactivate the K17 gene promoter.
K17, Wound Healing, and Epidermal Development
Together with our previous work (Paladini et al., 1996; Takahashi et al., 1997, 1998), the studies reported here establish that K6, K16, and K17 proteins are all induced in epidermis at the proximal edge of the wound within hours after acute injury to mouse and human skin. The K17 specific antisera used for this study allows us to modify our early studies, in that it appears that the induction of K6, K16, and K17 genes occurs on the same time scale. At least early on, this induction occurs in suprabasal keratinocytes, i.e., in cells that have already begun to differentiate. Thereafter, expression of K6, K16, and K17 spreads distally from the cut edge of the wound, is maintained in keratinocytes that are migrating into the wound site, and ceases upon the completion of reepithelialization. The significance of this phenomenon, and the precise contribution of K6, K16, and K17, remain to be ascertained. Schermer and colleagues (1989) proposed the interesting idea that this induction may reflect a switch to an alternative, possibly more primitive mode of epithelial differentiation. From previous work we postulated that these keratins, K16 in particular, may contribute to promote the cytoarchitectural changes that are typical of keratinocyte activation (Paladini et al., 1997; McGowan et al., 1998). In this regard, the data we report here have an impact at two distinct levels. First, the notion that K17 is synthesized in soft and hard epithelial cells and that this protein is a major component of regenerating epithelia is an intriguing one. We have preliminary data further supporting this concept that involve studies with the WE6 antigen, which is wound inducible in the surface epithelium of the newt and frogs (Estrada et al., 1993) and corresponds to a type I keratin (Klatt, K., and R. Tassava, personal communication). We detect WE6 immunoreactivity in the early differentiating cells of hard epithelia in the mouse such as hair, nail, and filiform papillae of the tongue (Coulombe, P.A., unpublished observations). Additional studies are required to further explore the notion that K17, and possibly other wound repair-associated keratins, could perform a key function in both soft and hard epithelia under resting conditions. Second, our findings argue that the program of gene expression characteristic of wound-activated adult keratinocyte (for review see Coulombe, 1997) may bear a relationship to that executed by epithelial cells at specific stages of embryonic development, before expression of true differentiation-specific genes. The existence of such a molecular relationship between embryonic development and adult tissue regeneration has already been evidenced in other tissues, liver in particular (Michapoulos, 1997).
The event that most convincingly links embryonic development and wound repair in skin epithelia is the abundant expression of K17 in developing hair placodes. In this case as in adult skin wounds, a group of K17-expressing keratinocytes coordinately expands or migrates into a connective tissue, suggesting that expression of this particular keratin may be a molecular trait of keratinocytes involved in migration or tissue expansion. Additional lines of evidence support this contention. Appendages have long been known to contribute keratinocytes to the resurfacing of wound sites in adult skin, especially in cases involving partial thickness wounds (Clark, 1993), and we find that K17 expression is upregulated in the ORS of hair follicles located proximal to the wound site (data not shown). Moreover, experimental evidence has recently been provided for a similar phenomenon involving epithelial cells originating from eccrine sweat glands (Miller et al., 1998). An exciting possibility is that the cell type involved in this case would be the myoepithelial cells, which express K17 constitutively (see below).
In terms of keratin gene expression profile, however, the periderm appears to better mimic the activated keratinocyte of adult skin wounds. In developing skin, indeed, uniform and abundant co-expression of K6 and K17 occurs only in the periderm. The function of the periderm remains a mystery (Fisher, 1994). Periderm has been suggested to emanate from sites of epithelio–mesenchymal interactions (M'Boneko and Merker, 1988). The morphological changes taking place as it emerges (M'Boneko and Merker, 1988) bear a striking resemblance to activated keratinocytes at the edge of adult skin wounds (Paladini et al., 1996). An open question is whether K17 induction in the periderm is a byproduct of the inductive interactions that lead to appendage formation or whether it represents a critical event in the morphogenesis of this poorly understood cell type. The degree to which the molecular relationship between periderm and the wound-activated adult skin keratinocyte extends beyond keratin gene expression deserves further investigation.
Other than hair placodes and periderm, K17 is expressed transiently but uniformly in the basal layer of embryonic epidermis ∼e16.5 d. By that time, the developing epidermis already features a mature character as judged by various markers of adult epidermis (Fuchs and Byrne, 1994). This aspect of embryonic K17 expression is of significance, as it provides further support to the notion that K17 expression in epidermis is compatible with mitosis and not a manifestation of advanced differentiation. This finding raises several questions, however, including the fate adopted by these embryonic basal cells, and the mechanism(s) responsible for spatially restricting the expression of this gene to the postmitotic layers in adult epidermis subjected to injury and other forms of challenge (as discussed above).
At another level, our findings may have significant implications with regard to a recent study of perfect wound healing in K8-null embryos (Brock et al., 1996). These studies concluded, on the basis that reepithelialization is not altered in K8-null embryos, that keratin IFs are not an integral part of the fetal wound repair process. We would assert that one must look beyond the K8–K18 network, as these filaments may not play a substantial role once the skin-specific keratins have begun to accumulate. It may be possible that the keratins already present at e11.5 d, which at least include K5, K14, and K17, are sufficient to bring about wound closure. We are currently exploring the possibility that the wound repair keratins K6, K16, and K17 may be expressed during reepithelialization of fetal wounds.
Speculation on the Function of K17
A fundamental question in the field of keratin filament biology is why there exists so many homologous proteins. By virtue of its primary sequence, K17 belongs to a subgroup of type I keratin genes that includes K14 and K16. A comparison of their amino acid sequences reveals that individual substitutions tend to be grouped within defined segments of these proteins. Thus, the NH2-terminal head domains of K14 and K16 are clearly more related to one another than either is to K17. Conversely, homology within the COOH-terminal tail domain is greatest between K14 and K16 for the proximal half segment, while being greatest for K14 and K17 for the distal half segment. It is unclear how these variations result in functional differences between these type I keratins. What is known is that human K16 can not entirely complement the phenotype associate with a K14-null mutation in transgenic mice, a behavior that we could map to the COOH-terminal 105 amino acids (Paladini, R.D., and P.A. Coulombe, unpublished data).
Our findings point to a dual function for K17 that may help to explain the phenotype and ultrastructural pathology characteristic of pachyonychia congenita and steatocystoma multiplex, two clinical disorders that can arise from missense mutations affecting the primary sequence of K17 (McLean et al., 1995; Smith et al., 1997). On the one hand, the occurrence of cytolysis in at least some K17 mutant-expressing epithelia (McLean et al., 1995) is consistent with a structural role akin to that already defined for other keratin proteins. It is possible that the synthesis of K17 protein in the hair matrix and lower portions of the hair shaft impacts the formation of this structure and would explain the presence of twisted hairs (pili torti) in PC patients. On the other hand, our description of the developmental expression of K17, especially during odontogenesis and gland formation, may explain the linkage between K17 mutation the occurrence of developmental anomalies such as those characterizing the eyebrow, sweat glands, and teeth (Munro et al., 1994; McLean et al., 1995). This would render K17 unique among the keratin genes so far linked to inherited diseases by suggesting a function for this protein during the morphogenesis of these tissues.
The context and subcellular distribution of a given protein can also convey hints about its function. Many of the epithelial cell types expressing K17, whether normal or activated, show contractile or motile activity. An example of this are the myoepithelial cells of glandular tissues, whose cytoplasm displays arrays of actin and myosin filaments organized in a sarcomere-like fashion (Raubenheimer, 1987; Redman, 1994). Contraction of these star-shaped myoepithelial cells, whose long cytoplasmic projections envelop the lumenal tissue, is thought to supply the force needed for propelling the glandular contents into the ducts (Raubenheimer, 1987; Redman, 1994). Interestingly, as stated for the periderm epithelium above, myoepithelial cells resemble activated keratinocytes proximal to the wound edge (Paladini et al., 1996) in many respects. Thus it would appear K17-expressing epithelial cells often feature a prominent actin-based molecular machinery. Although this relationship remains to be established, recent work would suggest that such cross-talk between filament networks, as mediated by linker proteins like plectin (Wiche, 1989) and BPAG (Yang et al., 1996), is not only possible but essential (Bousquet and Coulombe, 1996). This raises the possibility that K17 containing intermediate filament networks possess unique properties that render them capable of making such connections.
Acknowledgments
We are grateful to members of the Coulombe laboratory for their advice and support, and wish to extend a very special thank you to E. Fuchs and R. Grosscheld for their generous gift of transgenic mouse tissues and antibodies; to J. Carroll (Genetics Institute, Andover, MA), for sharing in situ hybridization data; to R. Tassava (Ohio State University, Columbus, OH) and K. Klatt (Denison University, Granville, OH) for helpful discussions; and G. Rogers (University of Adelaide, Adelaide, Australia), for advice on hair protein extraction. We also thank I. Leigh (Imperial Cancer Research Foundation, London, UK) for providing the LL001 antibody.
Abbreviations used in this paper
- e
embryonic day
- IF
intermediate filament
- K
keratin
- ORS
outer root sheat
- RT
reverse transcriptase
Footnotes
P.A. Coulombe was the recipient of Junior Faculty Research Award from the American Cancer Society (A-76019). K. McGowan was supported by NRSA fellowship (CA-67513) from the National Cancer Institute. This work was supported by a National Institutes of Health grant to P.A. Coulombe (AR-44232).
Address all correspondence to P.A. Coulombe, Department of Biological Chemistry, The Johns Hopkins University School of Medicine, 725 N. Wolfe St., Baltimore, MD 21205. Tel.: (410) 614-0510. Fax: (410) 955-5759. E-mail: coulombe@jhmi.edu
References
- Bessho Y, Nakanishi S, Nawa H. Glutamate receptor agonists enhance the expression of BDNF mRNA in cultured cerebellar granule cells. Mol Brain Res. 1993;18:201–208. doi: 10.1016/0169-328x(93)90190-z. [DOI] [PubMed] [Google Scholar]
- Bonnekoh B, Huerkamp C, Wevers A, Geisel J, Sebok B, Bange FC, Greenhalgh DA, Bottger EC, Krieg T, Mahrle G. Up-regulation of keratin 17 expression in human HaCaT keratinocytes by interferon-gamma. J Invest Dermatol. 1995;104:58–61. doi: 10.1111/1523-1747.ep12613492. [DOI] [PubMed] [Google Scholar]
- Boulikas T. A compilation and classification of DNA binding sites for protein transcription factors from vertebrates. Crit Rev Eukaryot Gene Expr. 1994;4:117–322. doi: 10.1615/critreveukargeneexpr.v4.i2-3.10. [DOI] [PubMed] [Google Scholar]
- Bousquet O, Coulombe PA. Cytoskeleton: missing links found? . Curr Biol. 1996;6:1563–1566. doi: 10.1016/s0960-9822(02)70772-0. [DOI] [PubMed] [Google Scholar]
- Brock J, McCluskey J, Baribault H, Martin P. Perfect wound healing in the keratin 8 deficient mouse embryo. Cell Motil Cytoskeleton. 1996;35:358–366. doi: 10.1002/(SICI)1097-0169(1996)35:4<358::AID-CM7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Byrne C, Tainsky M, Fuchs E. Programming gene expression in developing epidermis. Development (Camb) 1994;120:2369–2383. doi: 10.1242/dev.120.9.2369. [DOI] [PubMed] [Google Scholar]
- Carroll JM, Crompton T, Teery JP, Watt FM. Transgenic mice expressing IFN-gamma in the epidermis have eczema, hair hypopigmentation, and hair loss. J Invest Dermatol. 1997;108:412–422. doi: 10.1111/1523-1747.ep12289702. [DOI] [PubMed] [Google Scholar]
- Clark, R.A.F. 1993. Mechanisms of Cutaneous Wound Repair. Vol. I, T.B. Fitzpatrick, A.Z. Eisen, K. Wolff, I.M. Freedberg, and M.D. Austen, editors. McGraw-Hill, New York.
- Clevers HC, Grosschedl R. Transcriptional control of lymphoid development: lessons from gene targeting. Immunol Today. 1996;17:336–343. doi: 10.1016/0167-5699(96)10019-0. [DOI] [PubMed] [Google Scholar]
- Coulombe PA. The cellular and molecular biology of keratins: beginning a new era. Curr Opin Cell Biol. 1993;5:17–29. doi: 10.1016/s0955-0674(05)80004-3. [DOI] [PubMed] [Google Scholar]
- Coulombe PA. Towards a molecular definition of keratinocyte activation after acute injury to stratified epithelia. Biochem Biophys Res Comm. 1997;236:231–238. doi: 10.1006/bbrc.1997.6945. [DOI] [PubMed] [Google Scholar]
- Dale BA, Holbrook KA, Kimball JR, Hoff M, Sun TT. Expression of epidermal keratins and filaggrin during human fetal skin development. J Cell Biol. 1985;101:1257–1269. doi: 10.1083/jcb.101.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada, C.M., C.D. Park, M. Castilla, and R.A. Tassava. 1993. Monoclonal antibody WE6 identifies an antigen that is up-regulated in the wound epithelium of newts and frogs. In Limb Development and Regeneration. Wiley-Liss Inc., New York. 272–282. [PubMed]
- Fisher, C. 1994. The cellular basis of development and differentiation in mammalian keratinizing epithelia. In The Keratinocyte Handbook. I.M. Leigh, B. Lane, and F. Watt, editors. Cambridge University Press, Cambridge, UK. 131–150.
- Fuchs E, Weber K. Intermediate Filaments: Structure, Dynamics, Function, and Disease. Annu Rev Biochem. 1994;63:345–382. doi: 10.1146/annurev.bi.63.070194.002021. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Byrne C. The epidermis: rising to the surface. Curr Opin Genet Dev. 1994;4:725–736. doi: 10.1016/0959-437x(94)90140-x. [DOI] [PubMed] [Google Scholar]
- Greer J, Roop D. Loricrin, a major keratinocyte cell envelope protein, is expressed late in development. J Invest Dermatol. 1991;96:553–560. [Google Scholar]
- Gurdon JB. The generation of diversity and pattern in animal development. Cell. 1992;68:185–199. doi: 10.1016/0092-8674(92)90465-o. [DOI] [PubMed] [Google Scholar]
- Hardy MH, Vielkind U. Changing patterns of cell adhesion molecules during mouse pelage hair follicle development. I. Follicle morphogenesis in wilt-type mice. Acta Anat. 1996;157:169–182. doi: 10.1159/000147879. [DOI] [PubMed] [Google Scholar]
- Hardy MH. The secret life of the hair follicle. Trends Genet. 1992;8:55–61. doi: 10.1016/0168-9525(92)90350-d. [DOI] [PubMed] [Google Scholar]
- Hatzfeld M, Franke WW. Pair formation and promiscuity of cytokeratins: formation in vitro of heterotypic complexes and intermediate-sized filaments by homologous and heterologous recombinations of purified polypeptides. J Cell Biol. 1985;101:1826–1841. doi: 10.1083/jcb.101.5.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson BW, Grund C, Winter S, Franke WW, Illmensee K. Formation of cytoskeletal elements during mouse embryogenesis II. Epithelial differentiation and intermediate filaments in early post-implantation embryos. Differentiation. 1981;20:203–216. doi: 10.1111/j.1432-0436.1981.tb01177.x. [DOI] [PubMed] [Google Scholar]
- Jiang CK, Flanagan S, Ohtsuki M, Shuai K, Freedberg IM, Blumenberg M. Disease-activated transcription factor: allergic reactions in human skin cause nuclear translocation of STAT-91 and induce synthesis of keratin K17. Mol Cell Biol. 1994;14:4759–4769. doi: 10.1128/mcb.14.7.4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowett AK, Vajno S, Ferguson MWJ, Sharpe PI, Thesleff I. Epithelial-mesenchymal interactions are required for msx-1 and msx-2 gene expression in the developing murine molar tooth. Development (Camb) 1993;117:461–470. doi: 10.1242/dev.117.2.461. [DOI] [PubMed] [Google Scholar]
- Kaufman, M.H. 1992. The Atlas of Mouse Development. Academic Press, New York. 1–16.
- Knapp B, Rentrop M, Schweizer J, Winter H. Three cDNA sequences of mouse type I keratins. Cellular localization of the mRNAs in normal and hyperproliferative tissues. J Biol Chem. 1987;262:938–945. [PubMed] [Google Scholar]
- Kollar, E.J. 1983. Epithelial–mesenchymal interactions in the mammalian integument: Tooth development as a model instructive induction. In Epithelial-Mesenchymal Interactions in Development. R.H. Sawyer and J.F. Fallon, editors. Praeger Press, New York. 27–50.
- Komine M, Freedberg IM, Blumenberg M. Regulation of epidermal expression of keratin K17 in inflammatory skin diseases. J Invest Dermatol. 1996;107:569–575. doi: 10.1111/1523-1747.ep12582820. [DOI] [PubMed] [Google Scholar]
- Kopan R, Fuchs E. A new look into an old problem: Keratins as tools to investigate determination, morphogenesis, and differentiation in skin. Genes Dev. 1989;3:1–15. doi: 10.1101/gad.3.1.1. [DOI] [PubMed] [Google Scholar]
- Kozak M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochwil K, Dull M, Farinas I, Galceran J, Grosscheld R. Lef1 expression is activated by BMP-4 and regulates inductive tissue interactions in tooth and hair development. Cell. 1996;10:1382–1394. doi: 10.1101/gad.10.11.1382. [DOI] [PubMed] [Google Scholar]
- Leigh IM, Navsaria H, Purkis PE, McKay IA, Bowden PE, Riddle PN. Keratins (K16 and K17) as markers of keratinocyte hyperproliferation in psoriasis in vivo and in vitro. Br J Dermatol. 1995;133:501–511. doi: 10.1111/j.1365-2133.1995.tb02696.x. [DOI] [PubMed] [Google Scholar]
- Lewis-Carl SA, Gillette-Ferguson I, Ferguson DG. An indirect immuno-fluorescecnce procedure for staining the same cryosection with two mouse monoclonal antibodies. J Histochem Cytochem. 1993;41:1273–1278. doi: 10.1177/41.8.7687266. [DOI] [PubMed] [Google Scholar]
- M'Boneko V, Merker H-J. Development and morphology of the periderm of mouse embryos (Days 9-12 of gestation) Acta Anat. 1988;133:325–336. doi: 10.1159/000146662. [DOI] [PubMed] [Google Scholar]
- Mann SJ. Prenatal formation of hair follicle types. Anat Rec. 1962;144:135–142. [Google Scholar]
- Marchuk D, McCrohon S, Fuchs E. Remarkable conservation of structure among intermediate filament genes. Cell. 1984;39:491–498. doi: 10.1016/0092-8674(84)90456-2. [DOI] [PubMed] [Google Scholar]
- McGowan, K., and P.A. Coulombe. 1998. The wound repair-associated keratins K6, K16 and K17: Insights into the role of intermediate filaments in specifying keratinocyte cytoarchitecture. In Subcellular Biochemistry: Intermediate Filaments. J.R. Harris and H. Herrmann, editors. Plenum Publishing Co., London, UK. In press. [PubMed]
- McLean WHI, Lane EB. Intermediate filaments in diseases. Curr Opin Cell Biol. 1995;7:118–125. doi: 10.1016/0955-0674(95)80053-0. [DOI] [PubMed] [Google Scholar]
- McLean WHI, Rugg EL, Lunny DP, Morley SM, Lane EB, Swensson O, Dopping-Hepenstal PJC, Griffiths WAD, Eady RAJ, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet. 1995;9:273–278. doi: 10.1038/ng0395-273. [DOI] [PubMed] [Google Scholar]
- Michapoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- Miller S, Burke EM, Rader MD, Coulombe PA, Lavker RM. Re-epithelialization of porcine skin wounds by the sweat apparatus. J Invest Dermatol. 1998;110:13–19. doi: 10.1046/j.1523-1747.1998.00087.x. [DOI] [PubMed] [Google Scholar]
- Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;3:11–24. doi: 10.1016/0092-8674(82)90400-7. [DOI] [PubMed] [Google Scholar]
- Moll R, Krepler R, Franke WW. Complex cytokeratin polypeptide patterns observed in certain human carcinomas. Differentiation. 1983;23:256–269. doi: 10.1111/j.1432-0436.1982.tb01291.x. [DOI] [PubMed] [Google Scholar]
- Munro CS, Carter S, Bryce S, Hall M, Rees JL, Kunkeler L, Stephenson A, Strachan T. A gene for pachyonychia congenita is closely linked to the keratin gene cluster on 17q12-q21. J Med Genet. 1994;30:675–678. doi: 10.1136/jmg.31.9.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau JH, Berger FG, Cox DR, Crosby JL, Davisson MT, Ferrara D, Fuchs E, Hart C, Hunihan L, Lalley PA, et al. A family of type I keratin genes and the homeobox-2 gene complex are closely linked to the rex locus on mouse chromosome 11. Genomics. 1989;5:454–462. doi: 10.1016/0888-7543(89)90009-8. [DOI] [PubMed] [Google Scholar]
- O'Guin, W.M., A. Schermer, M. Lynch, and T.T. Sun. 1990. Differentiation-specific expression of keratin pairs. In Cellular and Molecular Biology of Intermediate Filaments. R.D. Goldman and P.M. Steinert, editors. Plenum Publishing Co., New York. 301–334.
- Osborn M, Weber K. Tumor diagnosis by intermediate filament typing: A novel tool for surgical pathology. Lab Invest. 1983;48:372–393. [PubMed] [Google Scholar]
- Paladini RD, Takahashi K, Gant TM, Coulombe PA. cDNA cloning and bacterial expression of the human type I keratin 16. Biochem Biophys Res Comm. 1995;215:517–523. doi: 10.1006/bbrc.1995.2495. [DOI] [PubMed] [Google Scholar]
- Paladini RD, Takahashi K, Bravo NS, Coulombe PA. Onset of re-epithelialization after skin injury correlates with a reorganization of keratin filaments in wound edge keratinocytes: defining a potential role for keratin 16. J Cell Biol. 1996;132:381–397. doi: 10.1083/jcb.132.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panteleyev AA, Paus R, Wanner R, Nurnberg W, Eichmuller S, Thiel R, Zhang J, Henz BM, Rosenbach T. Keratin 17 gene expression during the murine hair cycle. J Invest Dermatol. 1997;108:324–329. doi: 10.1111/1523-1747.ep12286476. [DOI] [PubMed] [Google Scholar]
- Powell B, Crocker L, Rogers G. Hair follicle differentiation: expression, structure and evolutionary conservation of the hair type II keratin intermediate filament gene family. Development (Camb) 1992;114:417–433. doi: 10.1242/dev.114.2.417. [DOI] [PubMed] [Google Scholar]
- Powell, B.C., and G.E. Rogers. 1997. The role of keratin proteins and their genes in the growth, structure and properties of hair. In Formation and Structure of Human Hairs, P. Jollès, H. Zahn, and H. Höcker, editors. Birkhaüser-Verlag, Basel, Switzerland. 59–148. [DOI] [PubMed]
- Powell BC, Nesci A, Rogers GE. Regulation of keratin gene expression in hair follicle differentiation. Ann NY Acad Sci. 1991;642:1–20. doi: 10.1111/j.1749-6632.1991.tb24376.x. [DOI] [PubMed] [Google Scholar]
- Proby CM, Churchill L, Purkis PE, Glover MT, Sexton CJ, Leigh IM. Keratin 17 expression as a marker for epithelial transformation in viral warts. Am J Pathol. 1993;143:1667–1678. [PMC free article] [PubMed] [Google Scholar]
- Purkis PE, Steel JB, Mackenzie IC, Nathrath WB, Leigh IM, Lane EB. Antibody markers of basal cells in complex epithelia. J Cell Sci. 1990;97:39–50. doi: 10.1242/jcs.97.1.39. [DOI] [PubMed] [Google Scholar]
- Raubenheimer EJ. The myoepithelial cell: Embryology, function, and proliferative aspects. CRC Crit Rev Clin Lab Sci. 1987;25:161–193. doi: 10.3109/10408368709105881. [DOI] [PubMed] [Google Scholar]
- Redman RS. Myoepithelium of salivary glands. Micr Res Tech. 1994;27:25–45. doi: 10.1002/jemt.1070270103. [DOI] [PubMed] [Google Scholar]
- Rogers MA, Langbein L, Praetzel S, Moll I, Krieg T, Winter H, Schweizer J. Sequences and differential expression of three novel human type-II hair keratins. Differentiation. 1997;61:187–194. doi: 10.1046/j.1432-0436.1997.6130187.x. [DOI] [PubMed] [Google Scholar]
- Sakakura T, Nishizuka Y, Dawe CJ. Mesenchyme-dependent morphogenesis and epithelial-specific cytodifferentiation in mouse mammary gland. Science. 1976;194:1439–1441. doi: 10.1126/science.827022. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 546 pp.
- Sanes JR, Rubenstein JLR, Nicolas J-F. Use of a recombinant retrovirus to study post-implantation cell lineage in mouse embryos. EMBO (Eur Mol Biol Organ) J. 1986;5:3133–3142. doi: 10.1002/j.1460-2075.1986.tb04620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermer A, Jester JV, Hardy C, Milano D, Sun TT. Transient synthesis of K6 and K16 keratins in regenerating rabbit corneal epithelium: keratin markers for an alternative pathway of keratinocyte differentiation. Differentiation. 1989;42:103–110. doi: 10.1111/j.1432-0436.1989.tb00611.x. [DOI] [PubMed] [Google Scholar]
- Schweizer, J. 1993. Murine epidermal keratins. In Molecular Biology of the Skin: The Keratinocyte. M. Darmon and M. Blumenberg, editors. Academic Press, Inc., San Diego, CA. 33–78.
- Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987;15:7155–7174. doi: 10.1093/nar/15.17.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FJ, Corden LD, Rugg EL, Ratnavel R, Leigh IM, Moss C, Tidman MJ, Hohl D, Huber M, Kunkeler L, et al. Missense mutations in keratin 17 cause either pachyonychia congenita type 2 or a phenotype resembling steatocystoma multiplex. J Invest Dermatol. 1997;108:220–223. doi: 10.1111/1523-1747.ep12335315. [DOI] [PubMed] [Google Scholar]
- Stoler A, Kopan R, Duvic M, Fuchs E. Use of monospecific antisera and cRNA probes to localize the major changes in keratin expression during normal and abnormal epidermal differentiation. J Cell Biol. 1988;107:427–446. doi: 10.1083/jcb.107.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Coulombe PA. Defining a region of the human keratin 6a gene that confers inducible expression in stratified epithelia of transgenic mice. J Biol Chem. 1997;272:11979–11985. doi: 10.1074/jbc.272.18.11979. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Folmer J, Coulombe PA. Increased expression of keratin 16 causes anomalies in cytoarchitecture and keratinization in transgenic mouse skin. J Cell Biol. 1994;127:505–520. doi: 10.1083/jcb.127.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yan B, Yamanishi K, Imamura S, Coulombe PA. The two functional type II keratin 6 genes of mouse show a differential regulation and evolved independently from their human orthologs. Genomics. 1998;53:170–183. doi: 10.1006/geno.1998.5476. [DOI] [PubMed] [Google Scholar]
- Thorey IS, Meneses JJ, Neznanov N, Kulesh DA, Pedersen RA, Oshima RG. Embryonic expression of human keratin 18 and K18-beta-galactosidase fusion genes in transgenic mice. Dev Biol. 1993;160:519–534. doi: 10.1006/dbio.1993.1326. [DOI] [PubMed] [Google Scholar]
- Troyanovsky SM, Guelstein VI, Tchipysheva TA, Krutovskikh VA, Bannikov GA. Patterns of expression of keratin 17 in human epithelia: dependency on cell position. J Cell Sci. 1989;93:419–426. doi: 10.1242/jcs.93.3.419. [DOI] [PubMed] [Google Scholar]
- Troyanovsky SM, Leube RE. Activation of the silent human cytokeratin 17 pseudogene-promoter region by cryptic enhancer elements of the cytokeratin 17 gene. Eur J Biochem. 1994;225:61–69. doi: 10.1111/j.1432-1033.1994.00061.x. [DOI] [PubMed] [Google Scholar]
- Troyanovsky SM, Leube RE, Franke WW. Characterization of the human gene encoding cytokeratin 17 and its expression pattern. Eur J Cell Biol. 1992;59:127–137. [PubMed] [Google Scholar]
- van Genderen C, Okamura RM, Farinas I, Quo R-G, Parslow TG, Bruhn L, Grosscheld R. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in Lef-1-deficient mice. Genes Dev. 1994;8:2691–2703. doi: 10.1101/gad.8.22.2691. [DOI] [PubMed] [Google Scholar]
- Vogel U, Denecke B, Troyanovsky SM, Leube RE, Bottger EC. Transcriptional activation of psoriasis-associated cytokeratin K17 by interferon-gamma. Analysis of gamma-interferon activation sites. Eur J Biochem. 1995;227:143–149. doi: 10.1111/j.1432-1033.1995.tb20370.x. [DOI] [PubMed] [Google Scholar]
- Wawersik M, Paladini RD, Noensie E, Coulombe PA. A proline residue in the α-helical rod domain of type I keratin 16 destabilizes keratin heterotetramers and influences incorporation into filaments. J Biol Chem. 1997;272:32557–32565. doi: 10.1074/jbc.272.51.32557. [DOI] [PubMed] [Google Scholar]
- Weiss RA, Eichner R, Sun TT. Monoclonal antibody analysis of keratin expression in epidermal diseases: a 48- and 56-kd keratin as molecular markers for hyperproliferative keratinocytes. J Cell Biol. 1984;98:1397–1406. doi: 10.1083/jcb.98.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiche G. Plectin: general overview and appraisal of its potential role as a subunit protein of the cytomatrix. Crit Rev Biochem Mol Biol. 1989;24:41–67. doi: 10.3109/10409238909082551. [DOI] [PubMed] [Google Scholar]
- Winter H, Rogers MA, Langbein L, Stevens HP, Leigh IM, Labreze C, Roul S, Taieb A, Krieg T, Schweizer J. Mutations in the hair cortex keratin hHb6 cause the inherited hair disease monilethrix. Nat Genet. 1997;16:372–374. doi: 10.1038/ng0897-372. [DOI] [PubMed] [Google Scholar]
- Yang Y, Dowling J, Yu QC, Kouklis P, Cleveland DW, Fuchs E. An essential cytoskeletal linker protein connecting actin microfilaments to intermediate filaments. Cell. 1996;86:655–665. doi: 10.1016/s0092-8674(00)80138-5. [DOI] [PubMed] [Google Scholar]
- Zhou P, Byrne C, Jacobs J, Fuchs E. Lymphoid enhancer factor 1 directs hair follicle patterning and epithelial cell fate. Genes Dev. 1995;9:700–713. doi: 10.1101/gad.9.6.700. [DOI] [PubMed] [Google Scholar]