

Abstract
In many malignant cells, both the anchorage requirement for survival and the function of the p53 tumor suppressor gene are subverted. These effects are consistent with the hypothesis that survival signals from extracellular matrix (ECM) suppress a p53-regulated cell death pathway. We report that survival signals from fibronectin are transduced by the focal adhesion kinase (FAK). If FAK or the correct ECM is absent, cells enter apoptosis through a p53-dependent pathway activated by protein kinase C λ/ι and cytosolic phospholipase A2. This pathway is suppressible by dominant-negative p53 and Bcl2 but not CrmA. Upon inactivation of p53, cells survive even if they lack matrix signals or FAK. This is the first report that p53 monitors survival signals from ECM/FAK in anchorage- dependent cells.
Keywords: fibronectin, survival, FAK, p53, apoptosis
Cell survival depends on multiple factors, including signals from cell–cell interactions, the extracellular matrix (ECM),1 and soluble survival/growth factors in serum. When these signals are interrupted, or when stressful conditions such as DNA damage occur, normal cells may undergo apoptosis, a physiological form of programmed cell death (Steller, 1995; Thompson, 1995; Chinnaiyan and Dixit, 1996, 1997; Levine, 1997). Programmed cell death contributes to the normal morphogenetic programs of most tissues and organs (Jacobson et al., 1997), and defects in the regulation of apoptosis are closely correlated with developmental abnormalities, as well as with the pathophysiology of autoimmune diseases and cancer (Hunter, 1997).
Signals from the ECM may play a particularly important role in maintaining normal cytoarchitecture and turnover in mature tissues because of the anchorage-dependence for cell growth and survival exhibited by most normal cell types. During metastatic progression, however, cells lose this anchorage dependence and also display altered adhesive and migratory properties, allowing them to metastasize and to survive and grow in inappropriate environments. It is therefore important to understand at the molecular level how signals from ECM suppress cell death, and what apoptotic pathway is triggered in normal cells when these signals are lost.
Apoptosis can be triggered by multiple mechanisms. Specific soluble factors such as Fas ligand or tumor necrosis factor-α (TNF-α) bind their receptors and directly activate a caspase cascade that leads to programmed cell death (Chinnaiyan and Dixit, 1997). There is strong evidence that the tumor suppressor protein p53 mediates apoptosis in response to conditions that generate genomic instability (Ko and Prives, 1996; Levine, 1997). Cells lacking survival signals from soluble factors transduced by their receptors and/or ECM survival signals transduced by integrins also undergo apoptosis (Frisch and Ruoslahti, 1997). Whether there is a role for p53 in stimulating an apoptotic pathway when survival signals from ECM are absent has not been established. However, mutations in p53 are common in many malignant cell types. The fact that such cells are also no longer anchorage dependent for growth and survival is consistent with the hypothesis that survival signals from ECM, transduced by integrins, suppress p53-regulated apoptosis.
Integrins are receptor αβ heterodimers with overlapping specificity toward ECM components (Damsky and Werb, 1992; Hynes, 1992). Integrin-mediated cell–ECM interactions promote the assembly of cytoskeletal and signaling molecule complexes at sites called focal adhesions. These complexes include Src-family members, focal adhesion kinase (FAK), phosphatidylinositol-3′-kinase (PI3K), and phospholipase C (PLC)-γ (Miyamoto et al., 1995a ,b; Plopper et al., 1995). Integrin ligation activates FAK, which in turn interacts directly with other nonreceptor protein tyrosine kinases, adaptor molecules, and cytoskeletal proteins (Schaller et al., 1994; Schlaepfer et al., 1994; Hanks and Polte, 1997; Ilić et al., 1997; Schlaepfer and Hunter, 1998).
Integrin–FAK signaling complexes have been implicated in the regulation of anchorage-dependent cell survival. Thus, Hungerford et al. (1996) showed that cells become apoptotic if they are microinjected with anti-FAK antibodies, or with a peptide corresponding to the portion of the β1 integrin cytoplasmic domain thought to be required for β1–FAK interaction. In another study, Frisch et al. (1996) showed that constitutively activating FAK by tethering it to the plasma membrane protected MDCK cells from apoptosis caused by loss of matrix contact. Furthermore, Xu et al. (1996) reported that attenuation of FAK expression leads to apoptosis in tumor cells.
The mechanism by which the integrin–FAK signaling pathway suppresses apoptosis is still largely unknown, as is the apoptotic pathway triggered when integrin–FAK signals are interrupted. In this study, we tested the hypothesis that survival signals from ECM, transduced by integrins and FAK, suppress a p53-regulated apoptotic pathway. We used multiple strategies to inactivate FAK and p53 in endothelial cells and fibroblasts under conditions in which the cells were anchorage dependent for survival. Our results document that survival signals from ECM are transmitted by FAK in both cell types, and that in the absence of FAK function, a p53-regulated apoptotic pathway is activated through cytosolic phospholipase A2 (cPLA2) and the Ca2+ and diacylglycerol-independent λ/ι isoform of protein kinase C (PKC). This pathway is Bcl2 suppressible and CrmA resistant and can be repressed by overexpression of the p53 COOH-terminal domain. Thus, this pathway is distinct from both the Fas-induced direct death pathway and other known p53-mediated pathways. This is the first report of a role for p53 in monitoring survival signals from ECM and FAK in anchorage-dependent cells.
Materials and Methods
Mice
Previous studies have described the FAK− (Furuta et al., 1995; Ilić et al., 1995 b) and p53− mice (Tsukada et al., 1993). Before these studies, both strains had been backcrossed to obtain mice with defects in both fak and p53. Inheritance of mutated fak and p53 alleles was determined by PCR analysis (Tsukada et al., 1993; Furuta et al., 1995). Mice were bred and maintained at Kumamoto University Medical School (Kumamoto, Japan) and the University of California San Francisco. Their care was in accordance with guidelines of the respective institutions.
Cell Lines
Rabbit synovial fibroblasts (RSF) were isolated as described previously (Huhtala et al., 1995). Primary cultures were expanded up to passage 3 and frozen. Cells were used between passages 4 and 8. Wild-type and FAK-deficient TT2 embryonic stem (ES) cells and FAK-deficient embryonic fibroblasts were described previously (Ilić et al., 1995 a,b).
Endothelial cells were isolated as follows from E8.0-old fak+ or fak− embryos that also carried mutated p53. Embryonic day 8 (E8.0) embryos dissected from implantation sites were mechanically disaggregated, plated on fibronectin-coated dishes (10 μg/ml), and cultured until confluent in DME with 10% heat-inactivated FCS, sodium pyruvate, nonessential amino acids, 10−4 M β-mercaptoethanol, and penicillin/streptomycin. Cells were detached by enzyme-free PBS-based cell dissociation buffer (GIBCO BRL; Life Technologies, Grand Island, NY), washed with cold PBS, and incubated with rotation for 1 h at 4°C with a rat monoclonal MEC13.3 anti-CD31 (platelet–endothelial cell adhesion molecule [PECAM]-1) antibody (gift of Dr. E. Dejana, Mario Negri Institute, Milan, Italy). Cells that bound the anti–PECAM-1 were then separated using anti–rat Ig-coated magnetic beads (Dynal A.S., Oslo, Norway). After five washes in cold 0.5% BSA/PBS, cells were replated. The purification procedure was repeated twice more after cells reached confluence.
Endothelial cells with an intact p53 gene were isolated from embryoid bodies (EB) derived from wild-type and FAK-deficient TT2 ES cells and then transformed by polyoma middle T (pmT) retrovirus (Fennie et al., 1995). To generate EB for endothelial cell isolation, FAK+ and FAK− TT2 ES cells were cultured for 11 d in suspension in serum-containing medium in the absence of leukemia inhibitory factor. The resulting EB were dispersed with vigorous pipetting in a trypsin/dispase/collagenase solution. Cells were passed through cell strainers to remove remaining clumps, washed, and replated. After reaching confluence, endothelial cells were isolated after incubation with anti–PECAM-1 and IgG-coated magnetic beads, as described above, and plated. The psi-Cre/psi-Crip retroviral packaging system transfected with plasmid containing the genome of pmT–antigen/Neor retrovirus was provided by C. Fennie and L. Lasky (Genentech, San Francisco, CA). Viral titer in supernatants used for transformation was ∼105 cfu/ml. Infection of the once-selected endothelial cell cultures was performed for 4 h with viral supernatant and polybrene as an enhancer (Fennie et al., 1995). Most cells continued to proliferate. Endothelial cells were purified through two more cycles using MEC13.3 anti-CD31 (PECAM-1) antibody and anti–rat Ig-coated magnetic beads.
Analysis of Cell Death in EB
Wild-type and FAK-deficient TT2 ES cells were cultured in suspension in the presence of serum-containing medium (DME containing sodium pyruvate, nonessential amino acids, 10−4 M β-mercaptoethanol, penicillin/ streptomycin, and 10% FBS) for 11 d to form EB (Ilić et al., 1996). They were then cultured for an additional 24 h in the same medium in the presence or absence of 10% serum as a source of growth/survival factors. EB were fixed in 3.7% paraformaldehyde for 20 min and then permeabilized with 0.2% Triton X-100/PBS for 2 min. After washing in PBS, they were subjected to terminal deoxynucleotidyl transferase (TdT)–mediated dUTP nick end labeling (TUNEL) with the In situ Cell Death Detection Kit as recommended by the manufacturer (Boehringer Mannheim Corp., Indianapolis, IN). As a control for nonspecific staining, the reaction mixture was used without enzyme. The last washing was done in 10 μg/ml of Hoechst 33342/PBS to visualize nuclei. Samples were mounted using Vectashield (Vector Laboratories, Burlingame, CA). Fluorescein label incorporated into nucleotide polymers was detected with an epifluorescence microscope (model Axiophot; Carl Zeiss, Inc., Thornwood, NY) equipped with proper filters and photographed (Tri-X film; Eastman Kodak Corp., Rochester, NY).
FACS®-based Apoptosis Assay
Endothelial cell lines were cultured for 48 h with serum and for an additional 24 h either with or without serum. The apoptotic population was determined by a flow cytometric assay (Hamel et al., 1996). Experiments were repeated three times. Serum-starved and control cells were resuspended in 1 ml ice-cold PBS containing 2% FCS, 3% enzyme-free PBS-based cell dissociation buffer (GIBCO BRL), and 5 μg/ml propidium iodide (Sigma Chemical Co., St. Louis, MO). Cells were kept on ice until 10–15 min before addition of Hoechst 33342. After equilibration to room temperature, 4 μg/ml Hoechst 33342 was added to the cell suspension. Detection of Hoechst 33342 staining was done after 6 min, using a dual-laser FACStar+ cell sorter (Becton Dickinson, San Jose, CA).
Analysis of Cell Death in Adherent Cells
Apoptosis in adherent endothelial cell lines and RSF was analyzed using either the Annexin v-Biotin Apoptosis Detection Kit (Calbiochem, San Diego, CA) or Hoechst 33342 staining at 10 μg/ml for 10–15 min. The large nuclei of normal cells stained faintly with Hoechst 33342, whereas condensed apoptotic nuclei stained brightly. Simultaneous staining and quantification with both methods produced similar results.
Plasmids
The sequence of FAK-related nonkinase (FRNK) was created by the introduction of an AflII site between nucleotides 2134–2139 of the mouse FAK cDNA, using the primers (5′-GCTCAGCTCAGCACAATCTTAAGGGAGGAGAAGGTGCAGC-3′ and 5′-GCTGCACCTTCTCCTCCCTTAAGATTGTGCTGAGCTGAGC-3′) and following the protocol outlined in the QuikChange Mutagenesis kit (Stratagene, La Jolla, CA). The primers (5′-GATACCTAGCATCTAGCGGTACCATGGCAGCTGCTTATC-3′ and 5′-GATAAGCAGCTGCCATGGTACCGCTAGATGCTAGGTATC-3′) and the QuikChange protocol were used to create a KpnI site next to the translational start of the mouse FAK cDNA cloned into pBluescript. All nucleotide changes were confirmed by DNA sequencing.
To construct green fluorescent protein–focal adhesion targeting (GFP– FAT) fusion proteins, normal and mutant FAK cDNAs were cut with XhoI (at 2628–2633 bp of fak cDNA) and BamHI (after double hemagglutinin [HA] tag) sites. The fragment was inserted between the XhoI and BamHI sites in the multicloning site of the pEGFP-C1 expression vector (CLONTECH Laboratories, Palo Alto, CA). For expressing GFP–FRNK fusion proteins, the pEGFP-C1 vector was also opened with XhoI at the multicloning site, blunted, and then cut with BamHI. FRNK was cut at the newly introduced AflII site and with BamHI located after the double HA tag. The insert was ligated into the multicloning site of pEGFP-C1 expression vectors in a way that preserved the continuity of the reading frame between GFP and FRNK. To generate a GFP–FAK expression vector, FAK cDNA was cut at the new KpnI site in front of the ATG codon and the BamHI site after the HA tag and inserted into the pEGFP-C1 vector at corresponding sites.
To disrupt the paxillin binding site 2, we introduced a stop codon before the critical R1042 residue and generated mutant GFP–FATΔC13. GFP–FATΔC13 was created by self-ligation of blunted ends after BclI digestion of demethylated GFP–FAT expression vector.
T367, S372, T373, and S374 in GSE p53 were changed to alanines using the QuikChange Mutagenesis Kit and primers 5′-CTACCCGAAGGCCAAGAAGGGCCAGGCTGGGGCCCGCCATAAAAAACCA-ATG-3′ and 5′-CATTGGTTTTTTATGGCGGGCCCCAGCCTGGCCCTTCTTGGCCTTCGGGTAG-3′. All nucleotide changes were confirmed by DNA sequencing.
Mouse Bcl2 was cut out from pBluescript II KS (gift from Z. Werb, University of California San Francisco) and inserted into the multicloning site of pCDNA3.1/Hygro expression vector (Invitrogen, Carlsbad, CA).
All blunting and ligation reactions were performed with either blunting or ligation kits (Takara, Japan, imported by PanVera Corporation, Madison, WI). Plasmid DNAs and agarose-embedded fragments were isolated using appropriate QIAGEN kits (QIAGEN, Valencia, CA).
The expression vectors were gifts: E1B 19K from E. White (Rutgers University, Piscataway, NJ) (White and Cipriani, 1990); CrmA from V. Dixit (University of Michigan, Ann Arbor, MI; Genentech) (Tewari et al., 1995); p53-175, p53-273, and GSE p53 mutants from T. Tlsty and V. Ossovskaya (University of California San Francisco); normal and mutated PKCs from J. Moscat (Universidad Autonoma, Canto Blanco, Madrid, Spain) and S. Ohno (Yokohama City University School of Medicine, Yokohama City, Japan); myristylated HA-tagged AKT from Chiron Corp. (Emeryville, CA); and cPLA2 from A. Stewart and C. Leslie (University of Colorado, Denver, CO).
Matrix Survival Assays
Tissue culture plastic wells were coated for 12 h at 4°C with 25 μg/ml of fibronectin, laminin-1, vitronectin, or collagen I (GIBCO BRL). After coating, and just before plating cells, the coated wells were washed with PBS. Cells for survival assays (RSF and FAK+p53+ endothelial cells) were grown to subconfluency and detached from plastic culture dishes with enzyme-free PBS-based cell dissociation buffer (GIBCO BRL). After washing with PBS, cells were plated on matrix-coated wells at about 75% confluency in serum-free DME with glutamine and nonessential amino acids. After 16 or 24 h of culture (RSF and endothelial cells, respectively), apoptotic cells were quantified as described above.
DNA Transfections and Inhibitor Studies
Primary fibroblast cultures were transfected using the Lipofectamine Plus kit (GIBCO BRL) or SuperFect Transfection Reagent (QIAGEN) according to the manufacturers' instructions. We used 0.5 μg (molar quantity) of DNA per cm2 of 75% confluent cells plated for 24 h on fibronectin. If different DNAs were cotransfected, equimolar amounts of DNA were used. The cells were incubated with the transfection mixture for 5 h (Lipofectamine method) or 2 h (SuperFect method). Then the transfection mixture was removed and replaced with serum/antibiotic-free DME supplemented with glutamine, nonessential amino acids, and pyruvate. After 12 h of culture, apoptotic cells were quantified as described above.
All inhibitors used in this study were purchased from Calbiochem (San Diego, CA) and used according to the manufacturer's recommendation in concentrations outlined in figures and figure legends.
Results
Withdrawal of Serum Induces Apoptosis in FAK− EB and Endothelial Cells Despite the Presence of an Endogenous ECM
Survival signals from ECM in cells that are anchorage dependent for survival can be assessed if they are cultured in the absence of serum but provided with a suitable ECM. In a direct test of the role of FAK in communicating survival signals from ECM, we first compared the ability of EB, derived from wild-type or FAK-deficient ES cells, to survive after withdrawal of serum (Fig. 1 A). Wild-type and FAK− ES cells were grown in suspension for 11 d in the presence of serum, during which time cells formed compacted EB surrounded by endoderm. They were then grown for an additional 24 h in either the presence or absence of serum. Then the EB were fixed and stained with the TUNEL in situ cell death detection kit, which recognizes single- and double-stranded DNA breaks occurring at early stages of apoptosis. Very few cells in either the FAK+ or FAK− EB that had been cultured with serum for the entire 12 d period displayed the intense staining characteristic of cells undergoing apoptosis (Fig. 1 A, (+) SERUM). However, a large number of cells in the FAK− EB that had been grown for the final 24 h in the absence of serum displayed strong TUNEL staining (Fig. 1 A, FAK−, (−) SERUM), whereas the number of TUNEL-positive cells in the FAK+ EB cultured in the absence of serum (Fig. 1 A, FAK+, (−) SERUM) was comparable to that in the EB cultured in the presence of serum for the entire 12 d of the experiment. These data suggest that FAK is required for transduction of survival signals in these cultures when serum is withdrawn.
Figure 1.


FAK suppresses a p53-regulated apoptosis pathway in embryoid bodies and endothelial cells. (A) Withdrawal of serum induces apoptosis in FAK− EB generated from wild-type and FAK− TT2 ES cells (Ilić et al., 1995 a). Cells were cultured for 11 d without leukemia inhibitory factor and in the presence of serum to initiate differentiation. They were then cultured for an additional 24 h either with (+), or without (−) serum. The presence of apoptotic cells was assessed by TUNEL staining, and nuclei were visualized with Hoechst as described in the Materials and Methods. (B) p53 deficiency permits survival, despite the absence of FAK, in anchorage-dependent, serum-deprived endothelial cells. Endothelial cells were isolated from FAK+ or FAK− EB or embryos and immortalized by pmT or incorporation of mutated p53, as described in the Materials and Methods. Apoptosis was assessed by a flow cytometric assay: PI, propidium iodide; Hoechst, Hoechst 33342. In each plot, the events represented in red (upper gated area) correspond to the PI/Hoechst ratios characteristic of an apoptotic population, whereas the events represented in green (lower gated area) correspond to nonapoptotic cells (Hamel et al., 1996). Endothelial cell lines were cultured for 48 h in the presence of serum and for an additional 24 h in the presence (+) or absence (−) of serum. All endothelial lines cultured in the presence of serum, as well as both p53-deficient cell lines cultured without serum, displayed low levels of apoptosis. Of the two endothelial cell lines immortalized by pmT, and therefore wild-type for p53, the FAK+ line exhibited a slightly higher level of apoptosis upon serum removal (6.3 vs. 2.1%). However, the FAK− line showed a much greater (7.8-fold) increase in apoptosis upon serum withdrawal (24.1 vs. 3.1%).
To study in greater detail the role of FAK in promoting survival in a specific cell type, we isolated endothelial cell lines from disaggregated FAK+ or FAK− EB, or from E8.0 FAK+ or FAK− embryos, using magnetic beads coated with a rat monoclonal antibody (MEC13.3) against PECAM-1 (CD31). The isolated endothelial cells were either immortal as a consequence of being derived from FAK+ or FAK− E8.0 embryos that also carried mutated p53 or were transformed by introduction of pmT into FAK+ or FAK− endothelial cells isolated from EB. The two latter lines were wild-type for p53. All four lines displayed an endothelial phenotype by the following criteria: (a) uptake of DiI-labeled acetylated low-density lipoprotein (DiI-Ac-LDL; Wang et al., 1992); (b) binding of labeled Griffonia (Bandeiraea) simplicifolia lectin I–isolectin B4 (Laitinen, 1987); and (c) expression of PECAM-1 as examined by FACS® using MEC13.3 anti-CD31 antibody directly conjugated with FITC (data not shown).
The four endothelial lines were cultured for 48 h in the presence of serum, during which time they elaborated an endogenous fibronectin-containing fibrillar matrix (data not shown). The lines were then cultured in either the presence or absence of serum for an additional 24 h to assess the cells' ability to survive with their endogenous ECM as a source of survival signals. The level of apoptosis in cells harvested from each culture was examined with flow cytometry, using an assay based on the more rapid uptake of Hoechst 33342 dye by apoptotic versus normal nuclei (Hamel et al., 1996). All lines grown continuously in the presence of serum showed a low level of apoptosis in this assay (4.3, 1.1, 2.1, and 3.1%: Fig. 1 B, (+) SERUM). When serum was withdrawn, both p53-deficient endothelial lines continued to display a low level of apoptosis (3.7 and 1.5%: Fig. 1 B, (–) SERUM). Cells that were wild type for both p53 and fak had nearly as low a level of apoptosis (6.3%) after serum withdrawal as before (2.1%). However, the cells that were wild-type for p53, but deficient in fak, showed a 7.8-fold increase in apoptosis (24.1%) after serum withdrawal when compared with these same cells cultured continuously in the presence of serum (3.1%). Furthermore, the serum-deprived FAK− cells with intact p53 displayed almost a fourfold increase in apoptosis when compared with serum-deprived cells that were wild type for both genes (Fig. 1 B, (–) SERUM). Extending the time in the absence of serum to 72 h did not increase the proportion of apoptotic cells in the FAK+ cultures (data not shown). These observations suggest strongly that, in the absence of serum factors, p53 triggers increased programmed cell death in endothelial cells that can be suppressed by survival signals from ECM transduced by FAK.
Specific Matrices Signal Cell Survival in the Absence of Serum in Endothelial Cells and Fibroblasts
The experiments described above suggested that a complex endogenous ECM, generated in the presence of serum, could support survival of FAK+ EB and pmT-immortalized FAK+p53+ endothelial cells after serum withdrawal. To determine more precisely which matrix components are critical for supporting survival of the FAK+p53+ endothelial cells, we plated these cells on defined ECM ligands in the absence of serum. We also tested the ability of ECM ligands to support survival of primary RSF, so as to determine the cell type specificity of the effects of these ligands on survival in the absence of serum. In the case of the endothelial cells, fibronectin and laminin-1 were the most effective in promoting survival, with vitronectin also showing substantial survival-promoting activity. Collagen type I was significantly less effective than the other ECM ligands (Fig. 2 A). In the case of RSF, fibronectin was markedly superior to the other three ligands in promoting survival: the apoptotic index of RSF plated on the latter matrices was no better than that observed for tissue culture plastic (Fig. 2 B). Fibronectin was used for subsequent experiments since it supported survival of both the endothelial cells and fibroblasts.
Figure 2.

Specific matrices signal cell survival in the absence of serum in both endothelial cells and fibroblasts. Tissue culture plastic wells were either coated with fibronectin (FN), vitronectin (VN), laminin-1 (LA), or type-I collagen (CO), or not coated (NC), as described in the Materials and Methods. Cells were plated for 16 h (primary rabbit synovial fibroblasts: RSF) or 24 h (EpmTp53+FAK+ endothelial cells) on these matrices in the absence of serum. SE, serum control. Apoptotic cells in the culture were quantified as described in Materials and Methods. Each sample of at least 100 cells was obtained in triplicate in each experiment. Experiments were repeated at least three times. Error bars show SD. (A) FAK+p53+ endothelial cells. (B) RSF.
Interference with FAK Function Triggers Apoptosis of Primary Fibroblasts in Serum-free Medium
To test the roles of FAK and p53 in regulating survival signals from fibronectin in RSF, we sequentially inactivated the functions of endogenous FAK and p53 in these cells. In the case of FAK, we used two COOH-terminal constructs: FRNK and the FAT domain (Fig. 3 A). FRNK is a naturally occurring alternative transcript to FAK that is expressed in some cell types and constitutes the sequence of FAK COOH-terminal to the kinase domain (Richardson and Parsons, 1996). FAT is a region within FAK and FRNK that contains the sequences necessary for targeting FAK to focal adhesion sites (Hildebrand et al., 1993). FRNK and FAT can interfere with FAK localization and signaling (Gilmore and Romer, 1996; Richardson and Parsons, 1996). To determine the importance of focal adhesion localization for the activity of FAT, we also tested a FAT construct in which the COOH-terminal 13 amino acids were deleted so as to abrogate its ability to target focal adhesion sites (FATΔC13). To follow the expression and localization of these constructs as well as full-length FAK in living cells, the cDNA encoding a GFP was fused in-frame to the NH2 terminus of all of these cDNAs (Fig. 3 A and Materials and Methods).
Figure 3.




GFP–FAT acts as a dominant negative for FAK. (A) RSF were transfected with cDNA encoding a fusion protein containing GFP and FAK, FRNK, FAT, or FATΔ13C (FAT with deletion of 13 COOH-terminal amino acids). (B) At 8 h, all fusion proteins except GFP– FATΔ13C were detected both in focal contacts and diffusely in the cytoplasm. (C) Quantification of the apoptotic index at 16 h (percentage of transfected [green] cells containing condensed, fragmenting nuclei, as detected by Hoechst 33342) for GFP, GFP–FAK, GFP– FAT, GFP–FRNK, and GFP– FATΔ13C transfectants. In contrast to the high apoptotic index (75%) after GFP– FAT transfection, transfections with GFP alone or with GFP fused to intact FAK caused no apoptosis above levels observed in cells plated on fibronectin in the absence of serum (∼20%). GFP–FATΔ13C, which was not detected in focal adhesions, was also unable to induce apoptosis to the same degree as GFP–FAT. (D) Apoptosis assessed by annexin binding and Hoechst 33342 staining. Most RSF expressing GFP–FAT, but not GFP alone or GFP-FAK, are rounded, and their Hoechst 33342-stained nuclei are bright and condensed by 16 h after transfection (arrows). These same cells also show annexin-positive staining (red) in the plasma membrane, indicating they are apoptotic (staining described in Materials and Methods). Untransfected and GFP–FAK–transfected cells are spread (Phase) and have normal nuclei (Hoechst).
When living cultures were examined 8 h after transfection of these constructs, GFP–FAK, GFP–FRNK, and GFP–FAT were detected in focal adhesion sites, as well as diffusely, in successfully transfected cells. In contrast, GFP–FATΔC13 was not detected at focal adhesion sites (Fig. 3 B). Subsequent fixation and staining of the GFP– FRNK– and GFP–FAT–transfected cells with an anti-FAK antibody that recognizes the NH2-terminal region of FAK (and therefore not FAT or FRNK) showed that endogenous FAK was partially or completely displaced from focal adhesion sites at this time (data not shown).
Apoptosis was assessed in these cultures 16 h after transfection by adding Hoechst 33342, which reveals nuclear morphology. The percentage of successfully transfected cells (positive for GFP) that also contained condensed/fragmented nuclei was determined as the apoptotic index for each culture (Fig. 3 C). By 16 h after transfection, most cells expressing GFP–FAT were scored as apoptotic (75%). In contrast, GFP–FATΔC13 triggered a much lower level of apoptosis (38%), indicating the significance of localization to focal adhesion sites for the proapoptotic effect of GFP–FAT. The apoptotic indices for cells transfected with GFP alone or GFP–FAK were similar to the low level found for untransfected fibroblasts cultured on fibronectin in the absence of serum (∼25%). Interestingly, cultures transfected with GFP–FRNK had significantly lower apoptotic indices than cells transfected with GFP–FAT (31 vs. 75%), even though both could target focal adhesion sites.
Apoptosis was also assessed in cultures transfected with GFP alone, GFP–FAK, or GFP–FAT by assessing the ability of unfixed cells to bind biotinylated annexin (annexin v-biotin). Annexin v-biotin binds to phosphatidyl serine, which is normally present in the inner leaflet of the plasma membrane but is translocated to the outer leaflet in cells undergoing apoptosis, thereby becoming accessible to the labeled annexin (Martin et al., 1995). The annexin, Hoechst, and phase microscope images of these cultures are presented in Fig. 3 D. Most GFP–FAT–expressing cells showed evidence of karyorhexis and the presence of brightly stained pyknotic chromatin characteristic of cells undergoing programmed cell death. In contrast, most untransfected cells in the same culture, as well as cells transfected with GFP alone or with full-length FAK fused to GFP (GFP–FAK), had large, lightly stained nuclei, which are characteristic of viable cells. The extent of abnormal nuclear morphology revealed by Hoechst staining correlated highly with annexin binding, thereby confirming by a different method the apoptotic phenotype of GFP–FAT, as compared with GFP–FAK– and GFP-transfected cells.
FRNK has been shown to act as a dominant-negative regulator of FAK for at least some of its functions, including cell spreading (Richardson and Parsons, 1996). In our system, however, ∼70% of the cells expressing GFP– FRNK survived, whereas 75% of cells expressing GFP– FAT died (Fig. 3 C) GFP–FRNK was functional in RSF, as determined by its ability to delay spreading of trypsinized and replated GFP–FRNK–expressing RSF (Fig. 4 A) with a time course similar to that reported by Richardson and Parsons (1996). Furthermore, GFP– FRNK and GFP–FAT were expressed at similar levels in transfected cells, as determined by immunoblotting an equal number of FACS®-sorted GFP–FRNK– and GFP– FAT–expressing cells with anti-GFP (Fig. 4 B). These results are consistent with the proposal that FRNK is a negative regulator of some (i.e., spreading), but not all (i.e., anchorage-dependent survival) functions of FAK. Interestingly, trypsinized and replated GFP–FAT–expressing cells were virtually unable to spread in spite of their initial attachment (Fig. 4 A). The time course of GFP–FAT action in adherent RSF that were transfected with GFP– FAT and left undisturbed demonstrates that accumulation of GFP–FAT in focal contacts leads to cell rounding, detachment, and, ultimately, death (Fig. 3, B and D).
Figure 4.

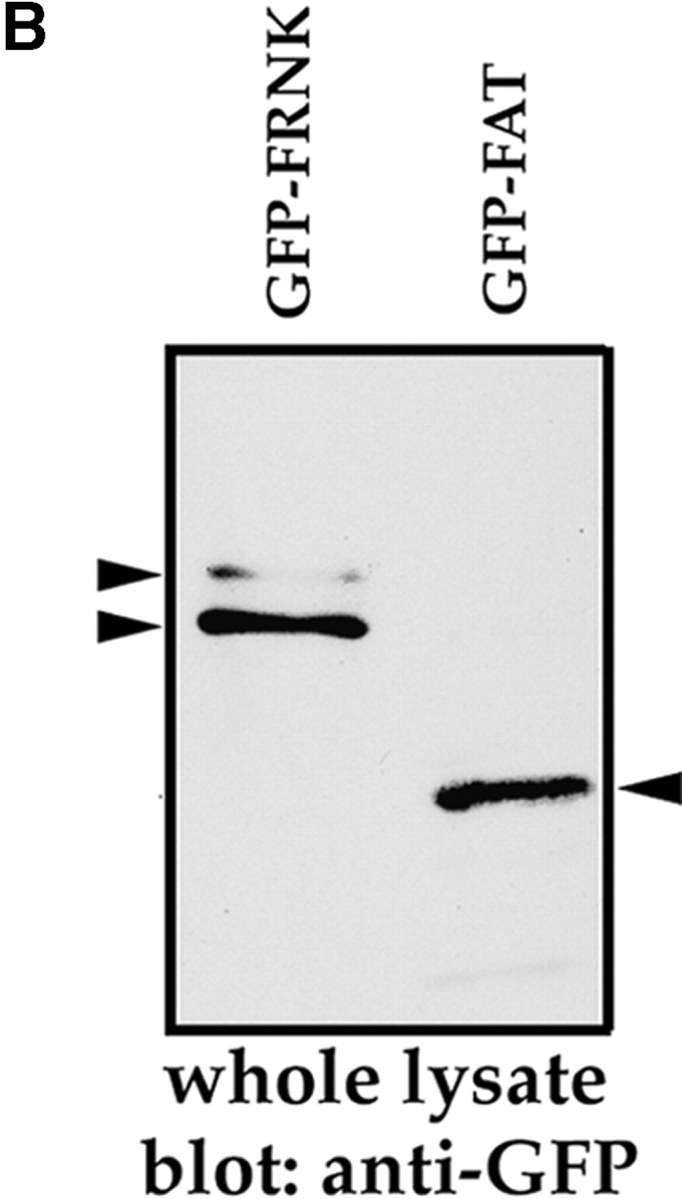
Assessment of GFP–FRNK and GFP–FAT function. (A) Cells expressing GFP–FRNK show delayed spreading. RSF transfected with GFP, GFP–FRNK, or GFP–FAT for 10 h were trypsinized and replated on fibronectin-coated coverslips. 60 min after replating, GFP-transfected cells were completely spread, whereas GFP–FRNK and GFP–FAT transfectants were still rounded. By 2 h, GFP–FRNK–transfected cells had spread, showing focal contact localization of the fusion protein (arrowheads). However, a majority of GFP–FAT–transfected cells remained round, although still attached. (B) Transfected cells express similar amounts of GFP–FRNK and GFP–FAT. Cells expressing GFP–FRNK or GFP–FAT were sorted by FACS® 10 h after transfection. An equal number of sorted cells (5 × 103) of each type were lysed in RIPA buffer, separated on 10% SDS-PAGE, transferred to nitrocellulose, and probed with anti-GFP antibody (Zymed Laboratories, So. San Francisco, CA). Bands were visualized by ECL-Plus detection system (Amersham Corp., Arlington Heights, IL).
Together, the data in Figs. 3 and 4 support the idea that FAT acts as a dominant negative for FAK, interfering with its ability to detect and/or transmit survival signals from fibronectin. In the absence of these FAK-transduced signals, apoptosis was triggered, as was observed for the serum-deprived, p53+FAK− endothelial cells (Fig. 1 B).
Activation of the PI3K/AKT Pathway Does Not Rescue Cells from Death Caused by GFP–FAT
Activated FAK has been shown to bind the p85 regulatory component of PI3K. Furthermore, formation of FAK/p85 complexes and elevated levels of phosphatidylinositol 3,4-diphosphate and 3,4,5-triphosphate (products of PI3K activity), followed by activation of AKT, occur after attachment of cells to ECM (Khwaja et al., 1997). If an anchorage-inducible PI3K/AKT pathway is downstream of FAK in the survival pathway in cells that are cultured under anchorage-dependent conditions, then expression of constitutively active myristylated AKT (Kohn et al., 1998) should rescue cells transfected with GFP–FAT from programmed cell death. We therefore transfected GFP–FAT along with HA-tagged myristylated AKT into RSF cultured on fibronectin in serum-free medium. The AKT construct was correctly localized, as determined by anti-HA tag antibody staining, and was active in rescuing cells from apoptosis induced by overexpression of the FAK-related kinase, PYK2, under the serum-replete culture conditions used by Xiong and Parsons (1997). However, it did not rescue cells dependent on ECM survival signals from apoptosis induced by GFP–FAT (Fig. 5). Furthermore, addition of the stable PI3K inhibitor LY294002 was unable to induce apoptosis of either untransfected cells or cells transfected with GFP alone and cultured in serum-free medium on fibronectin (Fig. 5). These data suggest that the ECM survival pathway downstream of FAK is distinct from the PI3K/AKT survival pathway that is suppressed when PYK2 is overexpressed (Xiong and Parsons, 1997), illustrating the existence of multiple survival pathways in cells.
Figure 5.

The PI3K/AKT pathway cannot rescue the apoptotic response of RSF to GFP–FAT. In RSF cultured on fibronectin without serum, blockade of PI3K activity by LY294002 did not trigger apoptosis, nor did cotransfection of constitutively activated AKT (myr AKT) along with GFP–FAT block apoptosis. This suggests that PI3K/AKT are not downstream mediators of the FAK survival pathway in serum-deprived, anchorage-dependent cells.
Apoptosis Induced by FAK Inactivation in Anchorage-dependent, Serum-deprived Fibroblasts Is Initiated through a p53-regulated Pathway
Turning to the apoptotic pathway triggered by FAK inactivation in anchorage-dependent serum-deprived fibroblasts, we used several approaches to test whether p53 was involved, as we had demonstrated for endothelial cells (Fig. 1 B). In all cases, we determined whether interfering with p53 function or p53 signaling pathways could rescue GFP–FAT–induced apoptosis. The E1B 19K adenoviral protein inactivates the p53 apoptotic pathway triggered by genomic instability (White and Cipriani, 1990; Debbas and White, 1993; Ko and Prives, 1996; Levine, 1997). Cotransfection of the E1B 19K expression vector along with GFP– FAT into serum-deprived RSF plated on fibronectin resulted in a low (background) level of apoptosis (19%; Fig. 6 A), despite the fact that FAK function was inactivated by GFP–FAT. In another approach to interfering with the pathway by which p53 mediates apoptosis (Ko and Prives, 1996; Levine, 1997), Bcl2 was overexpressed in GFP– FAT–transfected RSF. Coexpression of Bcl2 along with GFP–FAT also suppressed apoptosis extensively (35 vs. 82%; Fig. 6 A).
Figure 6.





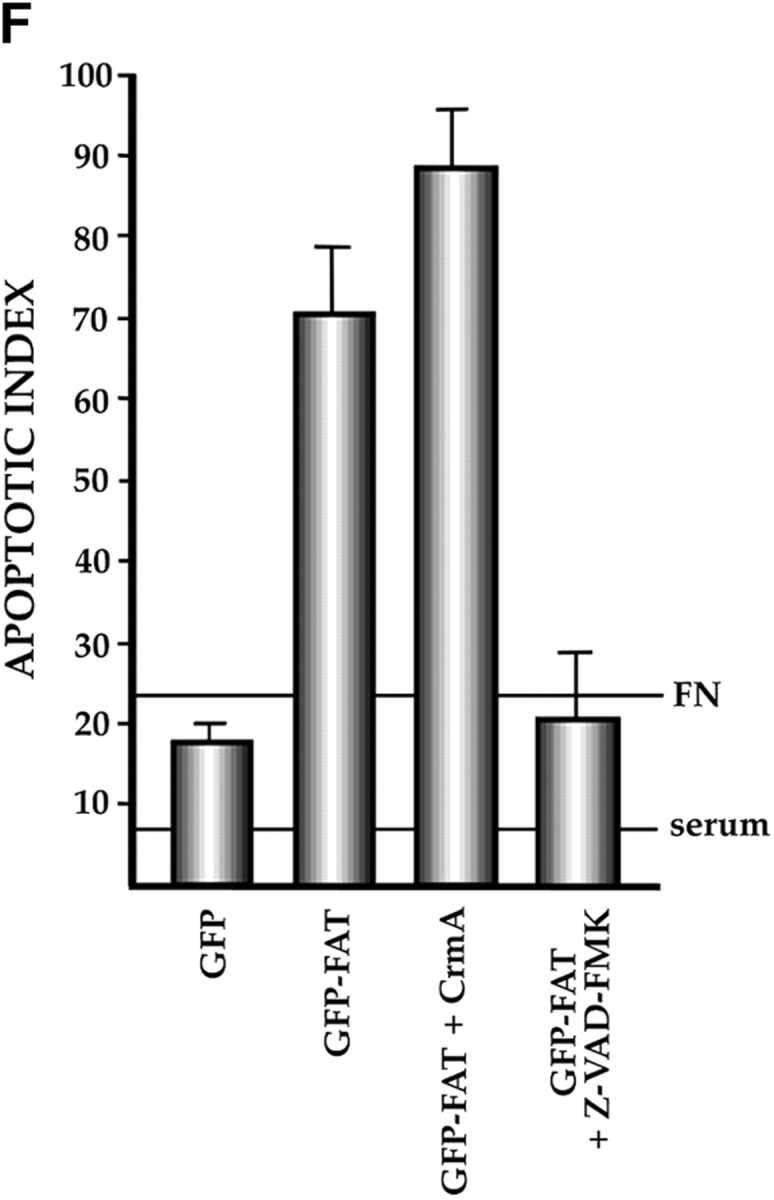
Characterization of the apoptotic pathway triggered in the absence of ECM survival signals in serum-deprived RSF: pharmacological and dominant-negative strategies reveal an apoptotic pathway that is regulated by p53, activated by cPLA2 and PKC λ/ι, and resistant to CrmA. (A) Blocking the function of p53 permits survival of primary RSF despite suppression of FAK function by GFP–FAT. GFP–FAT was cotransfected into fibroblasts plated on fibronectin, with or without either E1B 19K, Bcl2, or a GSE corresponding to the COOH-terminal domain of p53 (C-term p53), which inactivates p53 function (see Materials and Methods). The apoptotic index was high in GFP–FAT–transfected cells, but greatly reduced in cells cotransfected with GFP–FAT and one of the following: E1B 19K, Bcl2, or C-term p53. However, cotransfection of C-term p53, which contained mutated putative PKC phosphorylation sites (mut C-term p53), with GFP–FAT did not rescue cells from apoptosis. (B–D) Apoptosis triggered by inactivation of FAK function in serum-deprived anchorage-dependent RSF requires PKC λ/ι. (B) Inhibitors that block function of all isoforms of PKC (bisindolylmaleimide I and chelerythrine chloride) suppressed GFP–FAT–induced apoptosis, whereas calphostin C, which blocks only the PMA-sensitive isoforms (and therefore not PKC λ/ι), did not. (C) RSF express three isotypes of PKC. To detect PKC isotypes, cells were lysed using RIPA buffer in the presence of proteinase inhibitors. PKCs were immunoprecipitated and detected with anti-PKC α, β, γ, δ, ε, ζ, o, ι, λ, and μ antibodies (Transduction Laboratories, Lexington, KY) after separation in 10% SDS–polyacrylamide gels. Only PKC ε, λ, and ι were detected. + control, PKC isoform standard; − control, precipitation of RSF lysate with nonimmune IgG; anti-PKC, precipitation with antibodies specific for the PKC isoforms. (D). Coexpression of dominant-negative (DN) PKC λ/ι, but not DN PKC ε, blocked GFP–FAT–triggered apoptosis. Cotransfection of the wild-type PKC isoforms with GFP–FAT did not rescue cells from apoptosis. Transfection of wild-type isoforms into GFP-transfected cells did not promote apoptosis. (E) Apoptosis triggered by inactivation of FAK function in serum-deprived anchorage-dependent RSF requires cPLA2. Arachidonic acid induced apoptosis in nontransfected or GFP-transfected RSF. Phospholipases catalyze the release of arachidonic acid from phospholipids. AACOCF3, an inhibitor of cPLA2, but not inhibitors of secretory and Ca2+-independent PLA2 or inhibitors of PLC, rescued cells from apoptosis triggered by GFP–FAT. The panPKC inhibitor bisindolylmaleimide I blocked apoptosis triggered by arachidonic acid, suggesting that cPLA2 is upstream of PKC λ/ι in this pathway. (F) Effects of blocking large and small prodomain caspase functions on apoptosis in GFP–FAT–transfected primary RSF. RSF were cotransfected with GFP–FAT and CrmA or with GFP–FAT alone. At 16 h, the apoptotic index of RSF transfected with GFP–FAT was high whether or not CrmA was also transfected. The small prodomain caspase inhibitor, Z-VAD-FMK, inhibited formation of condensed and fragmented nuclei in serum-deprived GFP–FAT–transfected fibroblasts plated on fibronectin.
To demonstrate the involvement of p53 more directly, we cotransfected (along with GFP–FAT) a genetic suppressor element (GSE) that corresponds to the COOH-terminal domain of p53 (C-term p53, amino acids 312–391 of the rat p53; Ossovskaya et al., 1996). Expression of this construct along with GFP–FAT reduced the apoptotic index significantly (85 vs. 38%: P < 0.01). However, when serines and threonines that are likely phosphorylation sites for PKC (T367, S372, T373, and S374; Milne et al., 1996) were replaced by alanines (mut C-term p53), the construct was no longer able to rescue GFP–FAT–induced apoptosis (Fig. 6 A). This experiment suggests that the C-term p53 GSE functions by a dominant-negative mechanism that interferes with phosphorylation of endogenous p53. Attempts to interfere with p53 function by missense mutations of the DNA binding region at R273 and R175 did not rescue cells from GFP–FAT–induced apoptosis (data not shown). Together, these data point to p53 as a monitor of survival signals transduced by FAK in fibroblasts as well as endothelial cells. The fact that mutating putative PKC phosphorylation sites in the C-term p53 fragment abrogates its ability to interfere with endogenous p53 regulation of apoptosis suggests that this p53-regulated apoptotic pathway is activated by one or more isoforms of PKC when the appropriate ECM or FAK is absent.
Atypical PKC λ/ι and cPLA2 Are Involved in the Pathway That Activates p53-mediated Apoptosis after Interruption of Survival Signals from ECM
Several lines of evidence suggest that a large group of serine/threonine kinases termed PKC are involved in regulating attachment and spreading of cells on ECM (Chun and Jacobson, 1993) and in regulating both cell survival and cell death (Livneh and Fishman, 1997). The many isoforms of PKC have been divided into three groups: classical (α, β, and γ), which are Ca2+- and diacylglycerol-dependent forms; novel (δ, ε, η, and θ), which are Ca++-independent but diacylglycerol-dependent forms; and atypical (ζ and λ/ι), which are Ca2+- and diacylglycerol-independent forms (Livneh and Fishman, 1997). We used several approaches to analyze whether PKC isoforms are engaged either in transduction of survival signals from ECM or in the induction of apoptosis when such signals are withdrawn. In a pharmacological approach, we determined that bisindolylmaleimide I and chelerythrine chloride, inhibitors that affect all PKC isoforms, were able to reduce the apoptotic index significantly in cultures of GFP–FAT–transfected RSF (85 vs. 25–30%) (Fig. 6 B). However, calphostin C, which specifically blocks diacylglycerol-dependent (classical and novel) PKC isoforms, did not suppress GFP–FAT–induced apoptosis. To evaluate these results in terms of the RSF system, we detected expression of only the λ/ι and ε PKC isoforms in these cells (Fig. 6 C). This suggests that for RSF, the atypical PKC isoforms λ/ι are the likely candidates for participating in the apoptotic pathway activated when FAK function is suppressed. Indeed, cotransfection of either GFP or GFP–FAT with dominant-negative constructs of the PKC isoforms detected in RSF confirmed this idea. Thus, dominant-negative λ/ι was able to rescue cells from GFP–FAT– mediated death, whereas dominant-negative PKC ε did not (Fig. 6 D). Neither construct affected survival when cotransfected with GFP alone.
PKC activation by lipid mediators is dependent on release of arachidonic acid from phospholipids. Addition of 100 μM arachidonic acid for 6 h to RSF that had attached and spread on fibronectin in serum-free medium triggered apoptosis in untransfected cells and cells transfected with GFP alone and increased the level of apoptosis triggered by transfection of GFP–FAT (Fig. 6 E). Arachidonic acid is either hydrolytically removed from phospholipids by PLA2 or produced from diacylglycerol, which is released by the action of PLC. Activation of PKC by arachidonic acid can be independent of, or synergistic with, diacylglycerol. Since the λ/ι PKC isoforms implicated above are atypical (Ca2+ and diacylglycerol independent), we would predict that PLA2 rather than PLC would be involved in PKC λ/ι activation in this pathway. Studies with pharmacological inhibitors showed that U-73122, a specific PLC inhibitor, was unable to rescue cells from apoptosis triggered by GFP–FAT transfection of RSF cultured on fibronectin in serum-free medium (Fig. 6 E). However, arachidonyltrifluoremethyl ketone (AACOCF3), a cPLA2 inhibitor, significantly improved the survival rate of GFP– FAT–transfected cells (85 vs. 40%: P < 0.01) (Fig. 6 E).
These studies implicate cPLA2 and PKC λ/ι as participants in activating the p53-regulated apoptotic pathway when ECM survival signals are absent. To determine whether they are part of the same pathway, we tested whether the pan-PKC inhibitor bisindolylmaleimide I could inhibit the high level of apoptosis triggered by addition of arachidonic acid; our results showed that it could (Fig. 6 E). Thus, our data are consistent with the idea that ECM survival signals transduced by FAK in primary fibroblasts suppress a p53-regulated apoptotic pathway activated by cPLA2 and PKC λ/ι.
The p53-regulated Apoptotic Pathway Triggered by Loss of ECM/FAK Signals Is Not Dependent on Large Prodomain Caspases
The activation of a caspase/interleukin 1β-converting enzyme protease cascade has been implicated in both the initiation as well as the execution phases of several apoptotic pathways. The activity of the large prodomain caspases (e.g., caspases 2 and 8) implicated in the initiation of apoptosis by death ligands such as Fas ligand and TNF-α is inhibitable by CrmA, a cowpox virus–derived protein (Tewari et al., 1995; Villa et al., 1997; Wen et al., 1997). If large prodomain caspases are critical for driving apoptosis triggered by the absence of functional FAK in anchorage-dependent, serum-starved RSF, CrmA should block apoptosis in GFP–FAT–expressing cells. However, there was no significant difference in the extent of apoptosis in cultures cotransfected with GFP–FAT plus CrmA versus those transfected with GFP–FAT alone (Fig. 6 F). Thus, in anchorage-dependent, serum-deprived RSF, suppression of FAK function triggers apoptosis through a CrmA-insensitive pathway.
Small prodomain caspases, such as caspase-3, -6, -7, and -9, are thought to be final executors in cell death regardless of the initial signal. Therefore, an inhibitor of these caspases, such as Z-VAD-FMK, should prevent the final stages, although not the initial stages, of programmed cell death triggered by GFP–FAT inactivation of FAK function in anchorage-dependent fibroblasts. Indeed, when cultured on fibronectin in serum-free medium, but in the presence of Z-VAD-FMK, most GFP–FAT–transfected fibroblasts were rounded. However, nuclei were not condensed and fragmented, and therefore these cells were still scored as viable. In contrast, 70–80% of the GFP–FAT– transfected RSF cultured under the same conditions in the absence of Z-VAD-FMK displayed the full apoptotic effect, including fragmented nuclei (Fig. 6 F).
Discussion
Adhesion of many anchorage-dependent cells to ECM is essential for their survival in the absence of trophic factors from serum. If contact with appropriate ECM is denied to such cells, they rapidly undergo apoptosis. After ascertaining that fibronectin was a highly effective survival-promoting ECM ligand for both endothelial cells and primary fibroblasts, we demonstrated four important points in this study related to the transduction of survival signals from ECM in anchorage-dependent cells (Fig. 7): (a) Survival signals from fibronectin are transduced through FAK in both fibroblasts and endothelial cells. (b) The absence of either FAK or an appropriate ECM triggers a p53-regulated apoptosis pathway in serum-deprived endothelial cells and fibroblasts. (c) In anchorage-dependent fibroblasts, this pathway involves activation of cPLA2 and PKC λ/ι, and it is Bcl2 suppressible and CrmA insensitive, which distinguishes it from the direct death pathway triggered by ligation of Fas-family death receptors. (d) The absence of functional p53 enables serum-deprived endothelial cells and fibroblasts to survive despite the absence of FAK and/or an appropriate ECM. This is the first report that survival signals from ECM, transduced by FAK in anchorage-dependent cells, suppress p53-regulated apoptosis.
Figure 7.

Model for the transmission of matrix survival signals. Signals from fibronectin are detected by fibronectin receptors and transduced by FAK. These signals suppress a p53-dependent apoptotic pathway in anchorage-dependent, serum-deprived fibroblasts. When both ECM and serum survival signals are absent, PKC λ/ι is strongly implicated in the activation of p53, while pharmacological evidence also supports a role for cPLA2. FADD, Fas-associated death domain– containing protein; LPDC, large prodomain caspases; SPDC, small prodomain caspases; TRADD, TNF receptor 1–associated death domain protein.
FAK Is Required for Transducing Survival Signals from ECM in Endothelial Cells and Fibroblasts
After ligation of integrins by fibronectin, FAK-containing signaling complexes are formed at sites of adhesion (Miyamoto et al., 1995a ,b; Schlaepfer and Hunter, 1998). Previous work has correlated maintenance of survival with the presence of activated FAK (Frisch et al., 1996; Hungerford et al., 1996). Our studies of endothelial cells derived from FAK+ and FAK− EB and E8.0 embryos tested this correlation directly. They documented conclusively that FAK is required for transducing survival signals from a complex ECM and from fibronectin. Interestingly, both FAK and fibronectin knockout mice have similar phenotypes, showing extensive mesodermal defects and embryonic death at E8–8.5, which is consistent with the hypothesis that these molecules have related functions (George et al., 1993; Furuta et al., 1995).
The importance of FAK in regulating survival of anchorage-dependent cells was confirmed by our studies with primary fibroblasts (RSF). Endogenous FAK function was inactivated by using a dominant-negative construct made of GFP fused to the FAT region of FAK. We chose transfection of the FAT domain as the strategy for interfering with FAK function for two reasons: first, the FAT domain in intact FAK is responsible for targeting it to focal contacts (Hildebrand et al., 1993), and secondly, FAK has been shown to be cleaved by caspase activity at an early stage of Apo-2L–mediated programmed cell death, generating a COOH-terminal fragment similar in size to FAT (Wen et al., 1997; Levkau et al., 1998). The expression of GFP–FAT caused a high level of apoptosis in RSF. The importance of focal contact localization to the proapoptotic effect of GFP–FAT was revealed by our demonstration that expression of a FAT construct that lacked its COOH-terminal 13 amino acids, including a critical R1042 residue (Tachibana et al., 1995), resulted in a significantly reduced apoptotic effect. Interestingly, FRNK, a naturally occurring alternative product of the fak gene, did not trigger a high level of apoptosis when transfected into fibroblasts, even though, like FAT, FRNK lacks kinase activity and can displace FAK from focal contacts (Richardson and Parsons, 1996). Therefore, sequences in FRNK NH2-terminal to FAT must suppress proapoptotic signals in the FAT domain. The ability of these sequences in FRNK to suppress proapoptotic signals in FAT should enable FRNK to be expressed in a regulated fashion and to modulate certain functions of FAK— for instance cell spreading (Richardson and Parsons, 1996)—without killing the cells in which it is expressed.
FAK Suppresses a p53-regulated Apoptotic Pathway
We were able to determine that p53 was involved in the mechanism by which FAK suppresses apoptosis in anchorage-dependent endothelial cells as a consequence of our immortalizing FAK+ and FAK− endothelial cells by two different methods: pmT infection and derivation of endothelial cells from embryos that also carried a mutated p53. Thus, pmT-transformed endothelial cells that were FAK− and p53+ had substantially higher rates of apoptosis after serum withdrawal than either their FAK+p53+ or FAK−p53− counterparts (Fig. 1 B).
Since transformed cells might have altered signaling pathways, especially those that regulate survival, the involvement of p53 in regulating the apoptotic pathway suppressed by survival signals from ECM/FAK was addressed more comprehensively in primary fibroblasts. In the most direct approach, we demonstrated that a truncated COOH-terminal portion of p53 could act in a dominant-negative manner to interfere with the ability of GFP–FAT to trigger apoptosis. This region of p53 contains putative PKC phosphorylation sites, suggesting that phosphorylation by PKC might function to activate or increase the half-life of p53 (Milne et al., 1996). In contrast, mutations in the region of p53 that is required for its DNA binding function did not interfere with GFP–FAT–triggered apoptosis. These data suggest that when FAK function is suppressed, p53 acts directly on downstream components of an apoptotic cascade, rather than acting indirectly by binding DNA and trans-activating target genes. Our data indicating that both E1B 19K and Bcl2 suppress apoptosis when FAK is inactivated also support a role for p53 in this programmed cell death pathway (Debbas and White, 1993; Ko and Prives, 1996; Levine, 1997). Thus, p53 regulates an apoptotic pathway in response to loss of survival signals from ECM and FAK in primary fibroblasts as well as in endothelial cells.
FAK May Function in More Than One Survival Pathway
Two important areas of investigation in this system involve identifying components in the ECM survival pathway that act downstream of FAK, and identifying factors in the apoptotic pathway triggered by the loss of the FAK function that act upstream and downstream of p53. Although the emphasis of the work presented here has been on characterizing the p53-regulated apoptotic pathway, we did ascertain that PI3K and its downstream target AKT were not involved in transmitting survival signals from FAK in serum-deprived, anchorage-dependent fibroblasts. This is in contrast to reports from Khwaja et al. (1997) providing evidence that PI3K/AKT lie downstream of FAK in survival pathways, and from Xiong and Parsons (1997) showing that constitutively activated PI3K or AKT could rescue cells from apoptosis triggered by overexpression of the FAK-related molecule PYK2. These latter results suggested that there might be a balance in some cell types, whereby PI3K/AKT–transmitted survival signals downstream of FAK keep proapoptotic signals from PYK2 in check. The system described in the PYK2 study differed in several respects from that presented here, suggesting that FAK may be involved in more than one survival pathway. First, cells in the PYK2 study were cultured in the presence of serum; therefore, they were not dependent on survival signals from ECM. Since FAK can be activated by receptors for several soluble factors, as well as by ECM receptors, it is likely that a different pathway that involves FAK and PI3K/AKT transmits survival signals from these factors. Secondly, the apoptotic pathway triggered by PYK2 overexpression was suppressible by CrmA and resistant to overexpression of Bcl2, the converse of the pathway reported here. Clearly, additional studies are required to clarify the roles of FAK in transducing survival signals from ECM as well as from soluble factors.
The p53-regulated Apoptotic Pathway Triggered in the Absence of FAK Function Is Activated by cPLA2 and PKC λ/ι and Is Distinct from the Pathway Triggered by Fas-related Death Receptors
To begin to address the mechanisms by which p53 becomes activated when ECM-initiated survival signals are interrupted, we started from the observation that the COOH-terminal region of p53 containing putative PKC phosphorylation sites could act as a dominant negative for the function of endogenous p53, negating its ability to propagate an apoptotic pathway. After documenting the repertoire of PKC isoforms expressed by RSF, we used pharmacological and dominant-negative approaches to determine that PKC λ/ι was required for a successful apoptotic response to the suppression of FAK function by GFP–FAT. This result is consistent with a mechanism by which PKC λ/ι phosphorylates p53 and increases its stability and steady-state level, enabling it to propagate an apoptotic signal. A similar pharmacological approach enabled us to implicate cPLA2, as opposed to PLC, as a potential activator of PKC λ/ι in this pathway. How cPLA2 becomes activated inappropriately in the absence of ECM survival signals remains to be established.
To begin to determine how p53 communicates with the cellular death machinery once it has become activated in response to the loss of ECM/FAK survival signals, we used inhibitors of members of the caspase family suggested to affect early (i.e., initiation) or later (i.e., execution) phases of programmed cell death. CrmA, which blocks the activity of large prodomain caspases that are implicated in the initiation phase of programmed cell death (Villa et al., 1997), did not block p53-regulated apoptosis in RSF triggered either by FAK inactivation or by culture on an inappropriate ECM. This suggests that the commitment to p53-regulated apoptosis, initiated by withdrawal of ECM, is distinct from commitment to the death pathway initiated by Fas ligand or TNF-α (Villa et al., 1997) or by overexpression of the FAK family member PYK2 (Xiong and Parsons, 1997), the apoptotic effects of which are all blocked by CrmA. The CrmA construct we used was active, as indicated by its ability to suppress apoptosis triggered by blockade of soluble growth factor survival signals in serum-dependent cells (data not shown).
Although programmed cell death pathways can be initiated by several different mechanisms, data from many groups suggest that these pathways merge into a final execution phase that is mediated by small prodomain caspases (Chinnaiyan and Dixit, 1996, 1997; Villa et al., 1997). If this is true for the p53-regulated apoptotic pathway described here, the small prodomain caspase inhibitor, Z-VAD-FMK, should block its terminal stages, which are characterized by nuclear fragmentation and cell disintegration. Our data support this idea: in the presence of Z-VAD-FMK, GFP–FAT–transfected RSF were rounded, but at 24 h there was little nuclear fragmentation or cell body disintegration compared with controls. Thus, the CrmA-insensitive, p53-regulated apoptotic pathway that detects the absence of survival signals from ECM in anchorage-dependent RSF requires small prodomain caspases for its execution phase.
In summary (Fig. 7), we have directly documented a role for FAK in transducing survival signals from fibronectin in anchorage-dependent endothelial cells and fibroblasts. By manipulating the expression or function of FAK and/or p53, we show that survival signals from fibronectin, transduced by FAK, suppress a p53-dependent apoptotic pathway in anchorage-dependent cells. Our data suggest that PKC λ/ι and cPLA2 lie upstream of p53 in this pathway. This pathway is suppressible by Bcl2, but not by CrmA, distinguishing it from the direct death pathway triggered by death ligands or PYK2 overexpression.
Acknowledgments
We thank Drs. Steven Hanks, Thomas Parsons, Eileen White, Vishva Dixit, Zena Werb, Thea Tlsty, Valeria Ossovskaya, Jorge Moscat, Shigeo Ohno, Martin Haas, Bert Vogelstein, Allison Stewart, Christina Leslie, Elisabetta Dejana, Christopher Fennie, and Laurence Lasky for providing useful plasmids, antibodies, and reagents. We also thank Drs. Vishva Dixit, Mark Israel, Holly Ingraham, Clifford Lowell, Thea Tlsty, Valeria Ossovskaya, and Olga Genbacev for helpful discussion and comments on the manuscript and Evangeline Leash for expert editorial assistance.
This work was supported by a Biomedical Sciences Grant from the Arthritis Foundation and an American Heart Association Grant-in-Aid to C.H. Damsky, and by a Public Health Service grant R29 CA75240 from the National Cancer Institute to D.D. Schlaepfer. D. Ilić is supported by an American Heart Association Postdoctoral Fellowship. E.A.C. Almeida is supported by National Institutes of Health grant T32-DE07204.
Abbreviations used in this paper
- cPLA2
cytosolic phospholipase A2
- DiI-Ac-LDL
DiI-labeled acetylated low-density lipoprotein
- E
embryonic day
- EB
embryoid bodies
- ECM
extracellular matrix
- ES
embryoid stem cells
- FAK
focal adhesion kinase
- FAT
focal adhesion targeting
- FRNK
FAK-related nonkinase
- GFP
green fluorescent protein
- GSE
genetic suppressor element
- HA
hemagglutinin
- PECAM-1
platelet–endothelial cell adhesion molecule
- PI3K
phosphatidylinositol-3′-kinase
- PKC
protein kinase C
- PLC-γ
phospholipase C-γ
- pmT
polyoma middle T
- RSF
rabbit synovial fibroblasts
- TNF-α
tumor necrosis factor-α
- TUNEL
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling
Footnotes
Duško Ili ć and Eduardo A.C. Almeida contributed equally to this work.
Address all correspondence to Caroline H. Damsky, Ph.D, University of California San Francisco, 513 Parnassus Avenue, HSW-604, San Francisco, CA 94143-0512. Tel.: (415) 476-8922. Fax: (415) 502-7338. E-mail: damsky@cgl.ucsf.edu
References
- Chinnaiyan AM, Dixit VM. The cell-death machine. Curr Biol. 1996;6:555–562. doi: 10.1016/s0960-9822(02)00541-9. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Dixit VM. Portrait of an executioner: the molecular mechanism of FAS/APO-1-induced apoptosis. Semin Immunol. 1997;9:69–76. doi: 10.1006/smim.1996.0055. [DOI] [PubMed] [Google Scholar]
- Chun J-S, Jacobson BS. Requirement for diacylglycerol and protein kinase C in HeLa cell-substratum adhesion and their feedback amplification of arachidonic acid production for optimum cell spreading. Mol Biol Cell. 1993;4:271–281. doi: 10.1091/mbc.4.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsky CH, Werb Z. Signal transduction by adhesion receptors: cooperative processing of extracellular information. Curr Opin Cell Biol. 1992;4:772–781. doi: 10.1016/0955-0674(92)90100-q. [DOI] [PubMed] [Google Scholar]
- Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- Fennie C, Cheng J, Dowbenko D, Young P, Lasky LA. CD34+ endothelial cell lines derived from murine yolk sac induce the proliferation and differentiation of yolk sac CD34+hematopoietic progenitors. Blood. 1995;86:4454–4467. [PubMed] [Google Scholar]
- Frisch SM, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Ruoslahti E, Chan-Hui P-Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y, Ilić D, Kanazawa S, Takeda N, Yamamoto T, Aizawa S. Mesodermal defect in late phase of gastrulation by a targeted mutation of focal adhesion kinase, FAK. Oncogene. 1995;11:1989–1995. [PubMed] [Google Scholar]
- George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development (Camb) 1993;119:1079–1091. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- Gilmore AP, Romer LH. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol Biol Cell. 1996;7:1209–1224. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel W, Dazin P, Israel MA. Adaptation of a simple flow cytometric assay to identify different stages during apoptosis. Cytometry. 1996;25:173–181. doi: 10.1002/(SICI)1097-0320(19961001)25:2<173::AID-CYTO6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Polte TR. Signaling through focal adhesion kinase. Bioessays. 1997;19:137–145. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- Hildebrand JD, Schaller MD, Parsons JT. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions . J Cell Biol. 1993;123:993–1005. doi: 10.1083/jcb.123.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huhtala P, Humphries MJ, McCarthy JB, Tremble PM, Werb Z, Damsky CH. Cooperative signaling by α5β1 and α4β1integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J Cell Biol. 1995;129:867–879. doi: 10.1083/jcb.129.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hungerford JE, Compton MT, Matter ML, Hoffstrom BG, Otey C. Inhibition of pp125FAKin cultured fibroblasts results in apoptosis. J Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. Oncoprotein networks. Cell. 1997;88:333–346. doi: 10.1016/s0092-8674(00)81872-3. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: versatility, modulation and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Ilić D, Furuta Y, Suda T, Atsumi T, Fujimoto J, Ikawa Y, Yamamoto T, Aizawa S. a. Focal adhesion kinase is not essential for in vitro and in vivo differentiation of ES cells. Biochem Biophys Res Commun. 1995;209:300–309. doi: 10.1006/bbrc.1995.1503. [DOI] [PubMed] [Google Scholar]
- Ilić D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Aizawa S. b. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Ilić D, Kanazawa S, Furuta Y, Yamamoto T, Aizawa S. Impairment of mobility in endodermal cells by FAK deficiency. Exp Cell Res. 1996;222:298–303. doi: 10.1006/excr.1996.0038. [DOI] [PubMed] [Google Scholar]
- Ilić D, Damsky CH, Yamamoto T. Focal adhesion kinase: at the crossroads of signal transduction. J Cell Sci. 1997;110:401–407. doi: 10.1242/jcs.110.4.401. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne P, Downward J. Matrix adhesion and ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/AKT cellular survival pathway. EMBO (Eur Mol Biol Organ) J. 1997;16:2783–2794. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Barthel A, Kovacina KS, Boge A, Wallach B, Summers SA, Birnbaum MJ, Scott PH, Lawrence JC, Jr, Richard RA. Construction and characterization of a conditionally active version of the serine/ threonine kinase Akt. J Biol Chem. 1998;273:11937–11943. doi: 10.1074/jbc.273.19.11937. [DOI] [PubMed] [Google Scholar]
- Laitinen L. Griffonia simplicifolialectins bind specifically to endothelial cells and some epithelial cells in mouse tissues. Histochem J. 1987;19:225–234. doi: 10.1007/BF01680633. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Levkau B, Herren B, Koyama H, Ross R, Raines EW. Caspase-mediated cleavage of focal adhesion kinase pp125FAKand disassembly of focal adhesions in human endothelial cell apoptosis. J Exp Med. 1998;187:579–586. doi: 10.1084/jem.187.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livneh E, Fishman DD. Linking protein kinase C to cell-cycle control. Eur J Biochem. 1997;248:1–9. doi: 10.1111/j.1432-1033.1997.t01-4-00001.x. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne DM, McKendrick L, Jardine LJ, Deacon E, Lord JM, Meek DW. Murine p53 is phosphorylated within the PAb421 epitope by protein kinase C in vitro, but not in vivo, even after stimulation with the phorbol ester o-tetradecanoylphorbol 13-acetate. Oncogene. 1996;13:205–211. [PubMed] [Google Scholar]
- Miyamoto S, Akiyama SK, Yamada K. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science. 1995a;267:269–273. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Teramoto H, Coso OA, Gutkind JS, Burbelo PD, Akiyama SK, Yamada KM. Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol. 1995b;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossovskaya VS, Mazo IA, Chernov MV, Chernova OB, Strezoska Z, Kondratov R, Stark GR, Chumakov PM, Gudkov AV. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proc Natl Acad Sci USA. 1996;93:10309–10314. doi: 10.1073/pnas.93.19.10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plopper GE, McNamee HP, Dike LE, Bojanowski K, Ingber DE. Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol Biol Cell. 1995;6:1349–1365. doi: 10.1091/mbc.6.10.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A, Parsons JT. A mechanism for regulation of the adhesion-associated protein tyrosine kinase pp125FAK . Nature. 1996;380:538–540. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src . Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Integrin signalling and tyrosine phosphorylation: just the FAKs? . Trends Cell Biol. 1998;8:151–157. doi: 10.1016/s0962-8924(97)01172-0. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SS, Hunter T, van der Geer P. Integrin-mediated transduction linked to Ras pathway by Grb2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- Tachibana K, Sato K, D'Avirro N, Morimoto C. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK . J Exp Med. 1995;182:1089–1100. doi: 10.1084/jem.182.4.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32b, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Tsukada Y, Tomooka Y, Takai S, Ueda Y, Nishikawa S, Yagi T, Tokunaga T, Takeda N, Suda Y, Abe S, Matsuo I, Ikawa Y, Aizawa S. Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene. 1993;8:3313–3322. [PubMed] [Google Scholar]
- Villa P, Kaufmann SH, Earnshaw WC. Caspases and caspase inhibitors. Trends Biochem Sci. 1997;22:388–393. doi: 10.1016/s0968-0004(97)01107-9. [DOI] [PubMed] [Google Scholar]
- Wang R, Clark R, Bautch V. Embryonic stem cell-derived cystic embryoid bodies form vascular channels: an in vitro model of blood vessel development. Development (Camb) 1992;114:303–316. doi: 10.1242/dev.114.2.303. [DOI] [PubMed] [Google Scholar]
- Wen L-P, Fahrni JA, Troie S, Guan J-L, Orth K, Rosen G. Cleavage of focal adhesion kinase by caspases during apoptosis. J Biol Chem. 1997;272:26056–26061. doi: 10.1074/jbc.272.41.26056. [DOI] [PubMed] [Google Scholar]
- White E, Cipriani R. Role of adenovirus E1B proteins in transformation: altered organization of intermediate filaments in transformed cells that express the 19-kilodalton protein. Mol Cell Biol. 1990;10:120–130. doi: 10.1128/mcb.10.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W-C, Parsons JT. Induction of apoptosis after expression of PYK2, a tyrosine kinase structurally related to focal adhesion kinase. J Cell Biol. 1997;139:529–539. doi: 10.1083/jcb.139.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Owens LV, Sturge GC, Yang X, Liu ET, Craven RJ, Cance WG. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996;7:413–418. [PubMed] [Google Scholar]