

Abstract
When vertebrate somatic cells are selectively irradiated in the nucleus during late prophase (<30 min before nuclear envelope breakdown) they progress normally through mitosis even if they contain broken chromosomes. However, if early prophase nuclei are similarly irradiated, chromosome condensation is reversed and the cells return to interphase. Thus, the G2 checkpoint that prevents entry into mitosis in response to nuclear damage ceases to function in late prophase. If one nucleus in a cell containing two early prophase nuclei is selectively irradiated, both return to interphase, and prophase cells that have been induced to returned to interphase retain a normal cytoplasmic microtubule complex. Thus, damage to an early prophase nucleus is converted into a signal that not only reverses the nuclear events of prophase, but this signal also enters the cytoplasm where it inhibits e.g., centrosome maturation and the formation of asters. Immunofluorescent analyses reveal that the irradiation-induced reversion of prophase is correlated with the dephosphorylation of histone H1, histone H3, and the MPM2 epitopes. Together, these data reveal that a checkpoint control exists in early but not late prophase in vertebrate cells that, when triggered, reverses the cell cycle by apparently downregulating existing cyclin-dependent kinase (CDK1) activity.
Keywords: checkpoint control, G2/M transition, CDK1, mitosis, radiation
As emphasized by Mazia (39), there are two points of no return during mitosis. One is in late prophase and passage through this point leads to nuclear envelope breakdown (NEB)1 and an irreversible commitment to mitosis. The other is at the metaphase–anaphase (M–A) transition when pathways are activated that trigger chromatid disjunction and exit from mitosis. We now know that both of these transition points (40) are guarded by signal transduction pathways, termed checkpoint controls (16), which inhibit or delay progress through the cell cycle (including mitosis) until the event(s) being monitored are completed or mistakes corrected.
The checkpoint controlling the M–A transition monitors kinetochore attachment to the spindle (for review see 51). This pathway is based on a negative feedback loop in which kinetochores release an inhibitor of anaphase until they are properly attached to spindle microtubules (Mts), and just one unattached kinetochore can delay anaphase for many hours. Within the past few years rapid progress has been made in understanding how this checkpoint works at the molecular level. In part through the activity of Mad (e.g., 7, 37), Bub (e.g., 20, 56) and other gene products (like cdc20; 21, 29), unattached kinetochores appear to inhibit the activity of ubiquitin ligases, known as anaphase-promoting complexes (APCs; for review see 45), that target proteins for proteolysis. Once all the kinetochores are attached and the wait anaphase signal is abrogated, APCs induce anaphase by degrading those proteins responsible for sister chromatid cohesion (e.g., 8, 12, 19, 54). After this process has been initiated, another pathway is then triggered that inactivates the mitotic cyclin-dependent kinase (CDK1) by the ubiquination and subsequent proteolysis of its associated cyclin B (for reviews see 22, 45). The inactivation of CDK1 then allows the cell to escape the mitotic condition and leads to the hallmark features of telophase including chromosome decondensation, nuclear envelope reformation, and cytokinesis.
Like exit from mitosis, entry into mitosis is also subject to checkpoint controls, at least one of which prohibits the G2/M transition in response to DNA damage. In budding or fission yeast, this DNA damage checkpoint pathway is based on RAD, MEC1, and other gene products (for reviews see 11, 60), and it remains functional during mitosis (and can be triggered in mid-anaphase in budding yeast by dicentric chromosomes; e.g., 62). Although relatively poorly understood, mammalian cells also have one or more checkpoint control pathways during G2 that prohibit entry into mitosis in response to DNA damage (for reviews see 25, 42, 43).
Unlike budding yeast, in vertebrates the checkpoint that prevents entry into mitosis in response to DNA damage appears to become nonfunctional very early in the mitotic process. This is suggested from Carlson's (5; for review see 6) early discovery that when grasshopper neuroblasts are irradiated with UV light during early to mid-prophase they return to interphase, but when irradiated during late prophase they continue into and complete mitosis. Subsequent UV (15) and visible spectrum (23, 44) studies, using highly focused microbeams, revealed that this radiation-induced return of prophase cells to interphase is a general feature of animals and that it is due to damage to a nuclear and not cytoplasmic component.
The goal of our study was to better characterize the radiation-induced reversion of prophase in vertebrate somatic cells, which has never been explored in a systematic manner. To this end we show that the checkpoint responsible for this phenomenon becomes nonfunctional in PtK1 cells ∼30 min before NEB. Prophase cells irradiated in the nucleus before this point return to interphase, whereas those irradiated after this point enter and complete mitosis with normal kinetics even if they contain broken chromosomes. Early to mid-prophase cells that have been induced to return to interphase possess a normal interphase cytoplasmic microtubule complex. This suggests that radiation damage to a prophase nucleus is converted into a signal that not only reverses the nuclear events of prophase, but which also enters the cytoplasm and ultimately inhibits the maturation of centrosomes into mitotic centers. That this checkpoint pathway involves the cytoplasm was then confirmed by experiments on cells containing two prophase nuclei. Finally, we determined how specific markers for CDK1 activity, including antibodies against phosphorylated histone H1, histone H3, and MPM2 epitopes change as prophase cells are induced to return to interphase. The results of these studies strongly suggest that the radiation-induced reversion of prophase is correlated with the downregulation of existing CDK1 activity.
Materials and Methods
Cell Culture
Stock cultures of PtK1 (rat kangaroo kidney) cells were grown in a 5% CO2 atmosphere in Ham's F-12 medium supplemented with 10% FCS and antibiotics (e.g., 27). Stock cultures of CHO cells were grown in a 5% CO2 atmosphere in MEM supplemented with 10% FCS and antibiotics. For experiments, cells were trypsinized from stock T flasks and plated on 25-mm2 coverslips in the bottom of plastic Petri dishes. These were then incubated at 37°C until the coverslip cultures became mitotically active. They were then mounted in Rose chambers in L-15 media, pH 7.2, supplemented with 10% FCS and 10 mM Hepes. Newt lung cells were grown from primary explant cultures as described in Rieder and Hard (48).
Microbeam Irradiation
All microbeam irradiations and most of the time-lapse video light microscopy (LM) were conducted on our laser microsurgery workstation (9). After frequency doubling, the 5–7-ns pulses of 532-nm (green) light derived from our Q-switched Nd:YAG laser are focused through a Nikon (Tokyo, Japan) 1.4 NA differential interference contrast (DIC) objective into an Airy disk that approximates a 0.3 × 0.3 × 0.5-μm sausage shape (9). When attenuated to 1 μJ/pulse at the level of the specimen, this microbeam can sever a chromosome in a living PtK1 cell across its short axis (∼2 μm) in <20 pulses (27). The mechanism of this ablative photodecomposition is unknown, but it is clear that damage to the specimen is restricted to the volume contained within the Airy disk (28).
For microbeam experiments, a Rose chamber was placed in a 37°C heating block (50) that was mounted on the stage of the microsurgery workstation. A suitable prophase cell was then located within the chamber using a 60× DIC objective, and its behavior was followed in vivo by video- enhanced time-lapse DIC LM. This shuttered video-enhanced system (9) is based on a Paultek 1000 video camera (Paultek Imaging, Grass Valley, CA), an Image 1 (Universal Imaging, West Chester, PA) image processing system, and a Sony (model LVR 3300; Sony Corp., Tokyo, Japan) video disk recorder. Cells were initially recorded at 1 frame per 8 s which was then increased to 1 frame per second just before irradiation. After irradiation, the framing rate was decreased to 1 frame per 30 s, and the cell was followed until fixation or until it was transferred to another microscope for long-term observations. Under either circumstance, an objective scribe was used to mark the location of the experimental cell on the coverslip before it was removed from the stage of the laser microsurgery microscope.
In one set of experiments, mitotically active coverslip cultures of PtK1 cells were removed from their Petri dishes, drained of excess media, and then placed cell-side up in a box designed to evenly irradiate samples with UV-B light. They were then irradiated for 30 s before being returned to their Petri dishes and incubator.
Long-term Video Light Microscopy
For long term (>6 h) studies, the Rose chamber containing the experimental cell was removed from the microsurgery workstation and placed in a 37°C heating block (50) on the stage of a Nikon Optiphot LM. The cell was then followed using 546-nm (± 20 nm) shuttered light by phase-contrast optics. Images were captured once every 10 min using a Paultek 1000 video camera and were stored on a Sun Sparc 10 workstation running ISEE (Innovisions Corp., Durham, NC). The medium within the Rose chamber was replaced every 24 h.
Immunofluorescence Microscopy
MPM2 (monoclonal; no. 05-368), phosphohistone H1 (rabbit polyclonal; no. 06-597), and phosphohistone H3 (rabbit polyclonal; no. 06-570) antibodies were purchased from Upstate Biotechnology, Inc. (Lake Placid, NY). For MPM2 staining, cells were rinsed in PHEM buffer, fixed in 0.7% glutaraldehyde in PHEM for 15 min, rinsed, and then permeabilized with 0.2% Triton in PBS with 0.1% Tween-20 (PBST). After reducing with NaBH4, the cultures were blocked in 8% BSA in PBST, rinsed, and then stained with the primary antibody at a 1:4,000 dilution for 1 h at 37°C. They were then rinsed and incubated in an FITC-conjugated goat anti– mouse antibody (Sigma Chemical Co., St. Louis, MO) for 30 min at 37°C. After rinsing, the coverslips were stained with Hoechst 33342, rinsed, and then mounted on microscope slides in a 50:50 mixture of PBS and glycerol containing 1 mg/ml p-phenylenediamine.
The procedure used for phosphohistone H3 staining was as follows. Cultures were rinsed in PBS and then fixed in ice-cold (−20°C) methanol for 10 min, rinsed in PBS, and blocked with 8% BSA in PBST. They were then rinsed and incubated in the primary antibody for 2.5–3 h at room temperature, and further processed as described above for MPM2 staining. For phosphohistone H1 staining, the cultures were rinsed in PBS and fixed in 2% paraformaldehyde in PBS for 10 min. They were then postfixed/extracted in ice-cold (−20°C) methanol for 10 min, rinsed in PBS, and then blocked with 8% BSA in PBST. They were then stained as described above for MPM2.
Cells stained for indirect IMF LM were examined and photographed on a Nikon Optiphot (Nikon Corp., Tokyo, Japan) equipped with a Quad Fluor epifluorescence attachment and a 60× 1.4 NA Planapo objective. Optical sections were captured using a Photometrics PXL 1400 (Tuscon, AZ) cooled charge-coupled device, processed, and then stored on a SGI workstation using ISEE software (Innovision Corp.). In all cases the exposure used to record images of experimental cells stained for nuclear epitopes (i.e., phosphorylated histone H1, H3, and MPM2) was set to be the maximum exposure in which the interphase nuclei surrounding the experimental nucleus still remained invisible.
Image Manipulation and Photography
Selected DIC and epifluorescent digital images were exported from Image 1 or ISEE into Adobe Photoshop (Adobe Systems Inc., Mountain View, CA). After contrast manipulation, masking, and montaging, the final plates were printed with a Tektronix Phaser 450 dye sublimation printer (Wilsonville, OR).
Results
The Duration of Prophase in PtK1 Cells
Traditionally defined, the prophase stage of mitosis starts with the first visible signs of chromosome condensation and ends at NEB. For our studies it was important to know the approximate duration of prophase in PtK1 cells at 37°C. In our first attempts to obtain this data we used time-lapse video DIC LM to follow individual interphase cells in a growing culture for 10–12 h in hopes of catching some as they entered and proceeded through prophase (Fig. 1). These records reveal that late G2 PtK1 nuclei contain 1–3 prominent nucleoli and a variable number of small discrete patches of heterochromatin which appear, in 0.2–0.3-μm-thick optical sections, to reside in a relatively unstructured (i.e., optically clear) nuclear matrix (Fig. 1 A). As chromosome condensation is initiated, the nuclear matrix becomes progressively granulated (Fig. 1 B), and over time these granules grow and fuse (Fig. 1 C) to form linear ribbons that are ultimately resolved as individual chromosomes (Fig. 1, D and E).
Figure 1.

(A–F) The G2/ mitosis transition in a PtK1 cell. (A–F) Selected video frames from a time-lapse recording of a G2 cell (A) as it enters (B and C) and proceeds through (D and E) prophase of mitosis. Late G2 nuclei contain 1–3 nucleoli that are suspended in a relatively unstructured nuclear matrix that is also studded with small granules of heterochromatin (A). In optical DIC sections the earliest signs of chromosome condensation are signaled by a rapid increase in the number of small granules within the nuclear matrix (B), which over time grow and fuse to ultimately form recognizable chromosomes (C and D). In this example, NEB (E) occurred ∼70 min after the first visible signs of chromosome condensation, and the cell formed a normal bipolar prometaphase spindle (F). In this and all subsequent figures elapsed time (in min) is shown in the lower left-hand corner of each frame. Bar, 10 μm.
This approach proved impractical for gathering a significant sample size within a reasonable period because in living cells G2 nuclei cannot be distinguished by conventional LM from G1 or S nuclei, and because few of the PtK1 cells in a random population are in the cell cycle. However, with practice it becomes quite easy to distinguish an interphase nucleus (e.g., Fig. 1 A) from one that is in the early stages of chromosome condensation. To estimate how long prophase lasts in PtK1, we therefore followed cells from the earliest clear signs of chromosome condensation until NEB. The results of this study revealed that the average duration of prophase in PtK1 cells is 60 ± 2.5 min (n = 27; range = 45–89 min). This is a minimum average because chromosome condensation started before being clearly definable by video LM (e.g., 46).
Chromosomal Damage in Late Prophase Does Not Affect Cell Cycle Advance
To determine whether chromosomal breakage during late prophase affected cell cycle progression, we damaged the DNA in late prophase cells by selectively irradiating their nuclei through the objective lens with 532-nm laser light. To spread the irradiation across a selected region or area, as is required for cutting a chromosome or other organelle, the cell is slowly moved through the stationary beam by a motorized microscope stage. As a result, that region of the specimen moved through the beam is stitched by the laser pulses, leaving a visible linear pattern of denatured protein (i.e., a sniglet) in the plane of the Airy disk (28).
When cells in late prophase were stitched in the nucleus with up to 300 pulses of green (532-nm) laser light they always progressed into prometaphase (n = 11; Fig. 2). Mitosis in these cells was normal even when they were subsequently found during prometaphase to contain chromosome fragments (Fig. 2 D, arrows). Spindle formation took ∼1 h and, as in nonirradiated controls (49), the cells did not enter anaphase until the last kinetochore attached to the spindle. These observations confirm that by late prophase, vertebrate cells no longer have a mechanism to arrest progress into mitosis in response to extensive damage to the chromatin.
Figure 2.

(A–F) Nuclear irradiations in late prophase that induce chromosome breaks have no effect on progression into and through mitosis. Six selected frames from a time-lapse video DIC record of a PtK1 cell progressing through mitosis after its nucleus was stitched in late prophase (∼20 min before NEB) with 100 pulses of 532-nm laser light. (A) Just before irradiation; (B) just after the nucleus was irradiated near one of its nucleoli. Arrows, the linear sniglet of denatured material (also seen in C) produced by translating the Airy disk within the nucleus. (C) NEB. During spindle formation (D), several chromosome fragments were ejected into the cytoplasm (arrows). After all the chromosomes congressed the cell entered anaphase (E) and the irradiation-induced acentric chromosome fragments were randomly sorted during cytokinesis (F, arrows). Bar, 10 μm.
Chromosomal Damage in Early Prophase Causes a Reversion to Interphase
During the initial stages of this study we found that PtK1 cells in early prophase sometimes, but not always, returned to interphase after they were irradiated randomly in the nucleus with <10 pulses of laser light. During these short (<1-s) irradiations no attempt was made to move the nucleus through the beam and, as a result, the same volume received all the pulses. By using an empirical approach we subsequently determined that early prophase cells always returned to interphase when their nuclei were slowly moved through the beam during a 10-s (100-pulse) irradiation (n = 17; Fig. 3). Although the duration and intensity of this irradiation were well above the threshold needed to induce reversion, we adopted this strategy for the remainder of our studies because it ensured that the early prophase cells we irradiated would return to interphase.
Figure 3.
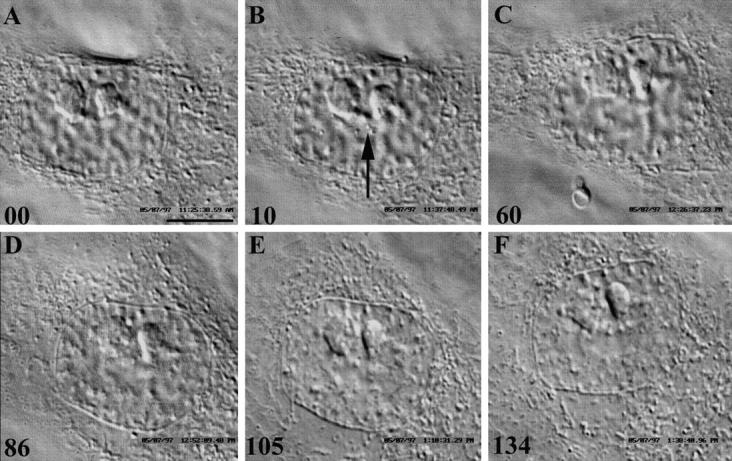
(A–F) Nuclear irradiations during early to mid-prophase inhibit progression into mitosis and induce the cell to return to interphase. Six selected frames from a time-lapse video DIC sequence of a mid-prophase PtK1 cell returning to interphase in response to a 10-s nuclear irradiation with 532-nm laser light. (A) Just before irradiation. Note the two prominent nucleoli and the condensing chromosomes. (B) Just after irradiation. Arrow, clumps of denatured protein formed within the nucleus in response to laser irradiation. Over the next 90–120 min (C–F), the chromosomes progressively decondense and the nucleus returns to an interphase-like state (E and F). Time, in min, is at the lower left-hand corner of each frame. Bar, 10 μm.
Once an early prophase nucleus was irradiated, the condensing chromosomes began to slowly and progressively decondense (Fig. 3, A–F). As a result, over the next 90– 120 min the nucleus returned to what appeared by DIC (and phase-contrast) LM as an interphase-like state (refer to Fig. 1 A and Fig. 3, E and F). We followed six of these cells for long periods and, although they never re-entered mitosis, they were still viable when we terminated the observations 48–72 h later (data not shown). We also fixed three for the indirect immunofluorescent localization of Mts at a time (90–120 min after irradiation) when the cell would have certainly been in prometaphase had it not been irradiated. In all cases, the distribution of Mts in these cells resembled those found in adjacent interphase cells, i.e., they contained an interphase cytoplasmic Mt complex and not the two radial astral arrays of Mts characteristic of prometaphase cells (data not shown).
When we irradiated the cytoplasm adjacent to the nucleus of early prophase cells with 100 pulses from the laser microbeam, the cells invariably progressed normally through prophase and entered prometaphase (n = 5; data not shown). We repeated these nuclear and cytoplasmic irradiation experiments on prophase CHO and newt lung cells and obtained the same results (data not shown). From these experiments we conclude that the irradiation-induced reversion of prophase is due to damage to the nucleus and not the cytoplasm, and that during this process progression of the cytoplasm into the mitotic state is also reversed and/or inhibited.
Approximately 3% of the cells in a growing PtK1 culture contain two nuclei (i.e., they are binucleated) which, as the cell enters mitosis, condense their chromosomes synchronously (53). When we irradiated just one nucleus in a cell containing two early prophase nuclei with 100 pulses from our microbeam, both nuclei always returned to interphase (n = 4; Fig. 4). However, if we irradiated the cytoplasm with same dose, both nuclei always progressed normally into mitosis and the cell formed a multipolar spindle (n = 3; data not shown). These results confirm that the biochemical pathway involved in the reversion process is not fully contained within the nucleus, but that it also involves the cytoplasm.
Figure 4.

(A–C) Selectively irradiating one nucleus in a cell containing two early prophase nuclei induces both to return to interphase. Three selected frames, from a time-lapse video DIC record, of a binucleated PtK1 cell in which both nuclei were in prophase and synchronously condensing their chromosomes. The cell pictured in A is just after one of the nuclei was irradiated with 100 pulses of 532-nm laser light. Note the track of denatured material (arrows). Over the next 2 h (B and C) the chromosomes in both nuclei progressively decondensed (refer to Fig. 3). Bar, 10 μm.
At What Point Do Prophase PtK1 Cells Become Refractory to Nuclear Irradiation?
So far our data confirm that vertebrate somatic cells possess a point of no return during prophase past which the cell becomes refractory to irradiation and thus is committed to enter prometaphase. To estimate when this transition occurs with respect to the duration of prophase, we determined how long cells that had been irradiated in the nucleus, but yet progressed into prometaphase, remained in prophase before NEB (n = 11). We found that the longest time between irradiation and NEB was 34 min, which means that the point of no return occurs near the middle of prophase (which lasts ∼60 min in PtK1 cells; refer to above). This conclusion is consistent with our observation that the number of prometaphase cells in growing PtK1 cultures, that have been irradiated with UV-B light, drops sharply 30 min after the irradiation (data not shown). This sudden decrease in prometaphase cells is due to the fact that those prophase cells that were <30 min away from NEB (and thus past the point of no return) at the time of irradiation progress into prometaphase, whereas those that were in an earlier stage of prophase fail to enter prometaphase and return to interphase.
Prophase-specific Nuclear Antigens Are Lost When Nuclei Are Induced by Irradiation to Return to Interphase
A major problem in studying the progressive biochemical changes that occur in the nucleus as it transits prophase is that many of the fixation protocols used to preserve antigens, including those based on acetone, methanol, and formaldehyde, grossly disturb chromosome morphology. As a result, it is impossible in cultures fixed with these reagents to define with certainty, by phase-contrast or even fluorescence LM, the stage of chromosome condensation at the time of fixation, or sometimes even to distinguish early prophase from interphase cells. To eliminate this problem, we followed individual living cells by time-lapse video LM throughout the experiment until the point of fixation. After processing for immunofluorescence microscopy (IMF) the same cell was then relocated for imaging. Although this approach provides data on only one cell within a culture, it is the only way to clearly demonstrate that a nucleus was in early to mid-prophase at the time of experimentation and/or fixation.
A number of biochemical changes occur in the nucleus as it transits from G2 into prophase. One of the earliest appears to be the mitosis-specific phosphorylation of histone H3 which is thought to be required for chromosome condensation (for review see 18). Prophase is also temporally coordinated with the hyperphosphorylation of the linker histone H1, which is not required for chromosome condensation (for review see 31). Finally, as G2 cells enter mitosis, phosphorylated epitopes recognized by the MPM2 antibody progressively accumulate within the nucleus (10). More than 40 phosphorylated proteins are recognized in mitotic cells by the MPM2 antibody (61) and they include, e.g., DNA topoisomerase II alpha, CDC25C, histone H1, and a H1 kinase (e.g., 32, 34, 55).
Histone H3 Phosphorylation
The antibody used for this part of our study is specific for the phosphorylated (Ser10) form of the amino terminus of histone H3 (18). After fixing cells in methanol it stains chromatin/chromosomes from the earliest stages of condensation in prophase in a wide variety of mammalian cells, and the intensity of the labeling is positively correlated with the degree of chromosome condensation. We found that early prophase nuclei in PtK1 cells, as judged by the degree of chromosome condensation just before fixation, stained intensely with this antibody (Fig. 5, A–C). We irradiated nuclei in 7 cells that were in a similar stage of early prophase with 100 pulses from the laser microbeam, and then fixed and stained these cells as they were decondensing their chromosomes. The nuclei in all of those cells fixed 30 s to 30 min after irradiation still stained relatively intensely for phosphorylated H3 (Fig. 5, D–G). However, those cells that were fixed >90 min after irradiation exhibited only a few very weak patches of stain similar to those of surrounding interphase cells (Fig. 5, H–K). Thus, triggering the nuclear damage checkpoint in early prophase cells leads to the dephosphorylation of histone H3, which was originally phosphorylated during the earliest stages of chromosome condensation.
Figure 5.

The phosphorylated form of histone H3 progressively disappears from prophase nuclei as they return to interphase in response to nuclear irradiations. (A–C) An early prophase control cell that was first photographed alive using DIC optics (A) before fixing and staining with Hoechst (B) and for the indirect immunofluorescent localization of phosphorylated histone H3 (C). Note that the prophase nucleus, but not the surrounding interphase nuclei (B), stains intensely for the presence of phosphorylated histone H3. (D–G and H–K) Two experimental prophase nuclei that were fixed in the process of returning to interphase in response to nuclear irradiations. In these examples, living cells in early prophase (D and H) were irradiated in the nucleus with 100 laser pulses of 532-nm laser light and followed by time-lapse video LM for 30 (E) or 90 (I) min before fixation. After fixation they were stained with Hoechst (F and J, arrows) and for the indirect immunofluorescent localization of phosphorylated histone H3 (G and K). The exposures in C, G, and K were normalized so that other nonmitotic nuclei within the same field (see B, F, and J) were not visible. Note that reverting nuclei contain significant amounts of phosphorylated histone H3 30 min (G) but not 90 min (K) after irradiation. Bars, 10 μm.
Histone H1 Phosphorylation
We used a polyclonal antibody that specifically recognizes phosphorylated forms of H1 in diverse cell types (38). As previously reported for other cells, most interphase nuclei within a PtK1 culture stained weakly with this antibody, but the staining intensity increased dramatically as cells entered mitosis. Relative to the nuclei of interphase cells, the nuclei of early prophase PtK1 cells showed an enhanced staining with this antibody, and within the nucleus the distribution of the staining pattern was not restricted to the condensing chromosomes (Fig. 6, A–C).
Figure 6.

The phosphorylated form of histone H1 progressively disappears from prophase nuclei as they return to interphase in response to nuclear irradiations. Conditions were the same as in Fig. 5 except that the cells were stained for the phosphorylated form of histone H1. Early prophase PtK1 nuclei that have not been irradiated (A and B) contain significant amounts of phosphorylated histone H1 (C). However, similar prophase nuclei that are fixed 30 min after a nuclear irradiation (D–F) contain very little phosphorylated histone H1 (G). The amount of phosphorylated histone H1 seen in prophase nuclei that are fixed 90 min after a nuclear irradiation is minimal and similar to that seen in surrounding nonirradiated interphase cells (K). Bars, 10 μm.
As found for the phosphohistone H3, the distribution of phosphohistone H1 also changed when prophase cells were induced by nuclear irradiation to return to interphase (n = 6). The nuclei in cells fixed 30 min after irradiation still contained more phosphohistone H1 than the surrounding interphase cells (Fig. 6, D–G). However, the nuclear staining in those fixed 90 min after irradiation was indistinguishable from that of surrounding interphase controls (Fig. 6, H–K). Thus, triggering the nuclear damage checkpoint in prophase cells also leads to the dephosphorylation of histone H1 that had been phosphorylated before nuclear irradiation.
MPM2 Antigens
During interphase, the MPM2 antibody stains small discrete patches within the nuclei of interphase PtK1 cells but the intensity of this staining increases dramatically as the cell enters prophase (58; Fig. 7, A–C). Throughout prophase MPM2 antigens are restricted to the nucleus until NEB, at which time they become associated with the chromosomes and the mitotic apparatus (58). We irradiated the nuclei in six early prophase PtK1 cells and then fixed and stained them for the presence of MPM2 epitopes at various times after irradiation. Relative to the surrounding interphase cells, the nuclei in those cells fixed 30 min after irradiation still exhibited a significant amount of MPM2 staining (Fig. 7, D–G). However, the nuclei in those cells fixed 90 min after irradiation exhibited just a few punctuate and weak regions of stain similar to those of interphase cells (Fig. 7, H–K). Thus, the phosphoepitopes that are present in early prophase nuclei that are recognized by the MPM2 antibody are progressively dephosphorylated when the nucleus is induced to return to interphase by laser irradiation.
Figure 7.

The mitosis-associated phosphorylated epitopes recognized by the MPM2 antibody disappear from prophase nuclei as they return to interphase in response to nuclear irradiations. Conditions were the same as in Fig. 5 except that these cells were stained with a monoclonal antibody to MPM2 antigens. Early prophase (A and B, arrow) PtK1 nuclei contain appreciably more MPM2 epitopes than adjacent interphase nuclei (C). By contrast, similar prophase nuclei fixed 30 min after a nuclear irradiation (D–F) contain significantly fewer MPM2 epitopes (G), whereas those fixed after 90 min (H–J) largely lack these epitopes (K). As evident from the Hoechst image (F), the bright MPM2-positive cell in G is in metaphase of mitosis. Bars, 10 μm.
Discussion
Our study confirms that a cell cycle checkpoint exists in vertebrate somatic cells during prophase that prohibits entry into prometaphase when the nucleus is damaged, and this checkpoint pathway becomes nonfunctional ∼30 min before NEB in PtK1, which is well after the prophase stage of mitosis has begun. Although we assume that this checkpoint is triggered by DNA damage, we cannot exclude the possibility that it monitors damage to other nuclear component(s). However, regardless of what is monitored, when activated it reverses the process of chromosome condensation and inhibits NEB. To date this is the last checkpoint described before the cell becomes committed to enter mitosis. During the remainder of our discussion we will refer to this checkpoint as the one that regulates entry into prometaphase.
Prophase PtK1 cells that are induced to undergo a reversion by our protocol remain viable but do not reenter mitosis during the next 72 h. This likely reflects the fact that our protocol (100 pulses in the nucleus) produces substantial chromatin damage that cannot be repaired in a reasonable period. In support of this conclusion, Carlson (6) found that when mammalian cells in early prophase are exposed to different doses of X-rays, they revert to interphase, but then ultimately reenter and complete a normal mitosis, and that those more heavily irradiated take many more hours to do so. Similar results were also reported by Gaulden and Perry (15) who studied the effects of nuclear UV microbeam irradiations in grasshopper neuroblasts. Thus, the ability of cells to reenter mitosis after an irradiation-induced reversion from prophase is dose dependent, and the (high) doses we use to ensure reversion prohibit reentry into mitosis.
Triggering the Checkpoint Regulating Entry into Prometaphase Likely Downregulates Existing CDK1 Activity
The mitotic condition is induced by activating the Cdc2/cyclin B (CDK1) kinase (for reviews see 22, 30). Unfortunately, the degree of synchronization required to conduct direct biochemical assays for CDK1 activity on populations of prophase cells that have been induced to revert to interphase cannot be obtained. However, since the phosphorylation of histone H1 (e.g., 35, 57) and MPM2 antigens (e.g., 32, 33) occurs directly or indirectly through CDK1 activity, the phosphorylation of these substrates (especially H1) is routinely used to assay CDK1 activity (for reviews see 41, 52). We found that once they have been phosphorylated in response to CDK1, histone H1, histone H3, and the MPM2 epitopes become dephosphorylated (or destroyed) when prophase nuclei are induced to return to interphase. It is possible that the dephosphorylation of these antigens is due, e.g., to an irradiation-induced enhanced activity of unknown phosphatases that are regulated independently of CDK1 activity. However, there is ample evidence that radiation during G2 prohibits the activation of CDK1, and thus entry into mitosis, by either inhibiting the stimulatory function of the Cdc25C phosphatase on CDK1 activity (3, 47) and/or by up-regulating CDK1 inhibitory kinases (Wee1 or Mik1; for reviews see 4, 24, 26, 36, 43). Thus, the most straightforward interpretation of our data is that triggering the checkpoint regulating entry into prometaphase leads to the downregulation of existing CDK1 activity. If true, then the cell possesses a pathway during early but not late prophase that allows it to downregulate active CDK1, and the presence of such a pathway provides a mechanism for reversing the prophase portion of the cell cycle. It is also possible that degradation of this checkpoint in late prophase occurs after CDK1 activation, via the Cdc2/Cdc25 autocatalytic feedback loop (for review see 43), has progressed to a point where it cannot be overridden by the existing checkpoint pathway.
Once the Checkpoint Guarding Entry into Prometaphase Is Abrogated the Cell Does Not Normally Inactivate Its CDK1 Until Conditions Are Met to Trigger the M–A Transition
As we have shown the checkpoint guarding entry into prometaphase in animal somatic cells becomes nonfunctional in mid- to late prophase, after which point the cell no longer has the ability to inactivate CDK1 (i.e., escape the mitotic condition) in response to DNA damage. Indeed, once prophase has ended (at NEB), the cell cannot normally inactivate its CDK1 until those requirements are met for passage through the checkpoint controlling the M–A transition. At this point CDK1 is then inactivated by the ubiquitin-mediated destruction of its associated cyclin B (refer to Introduction) and the cell exits mitosis. These observations raise the possibility that degradation of the checkpoint pathway guarding entry into prometaphase, which appears to coincide with the loss of the cell's ability to inactivate its CDK1 in response to DNA damage, occurs concurrent with the activation of the kinetochore attachment checkpoint. This latter control prohibits the premature inhibition of CDK1 activity, and thus exit from mitosis, until all the chromosomes (kinetochores) are properly attached to the spindle (refer to Introduction). In this regard it is noteworthy that the MAD2 (7) and BUB1 proteins (56), which are important components of the kinetochore attachment checkpoint, are both located in the nucleus of late G2 cells, and that BUB1 is associated with the kinetochores of condensing chromosomes.
The Checkpoint Pathway Guarding Entry into Prometaphase Involves the Cytoplasm
The Mt complex of early prophase cells is replaced, before or sometimes just after NEB (1), with two radial arrays of Mts that focus on the replicated centrosomes. This reorganization of Mts at the G2/M transition correlates with a change in the behavior of Mt plus ends (59) and with the hyperphosphorylation of centrosomal components (58), both of which are though to be mediated by CDK1. In this regard, centrosome maturation has been reported to coincide with the cdc25B-dependent activation of centrosome-associated CDK1 (2, 13).
Radiation damage to G2 cells prohibits progression into mitosis by ultimately inhibiting the activation of CDK1, perhaps thought its affect on Cdc25 phosphatase (e.g., 3, 14, 47). Without exception, the methods used in these studies involved irradiating populations of whole cells, and Cdc25 is found in both the cytoplasm and in the nucleus. As a result, the influence on the cytoplasm of biochemical changes induced within the nucleus after DNA damage is masked in these studies. By contrast, with our methods we could selectively irradiate only the nucleus or cytoplasm. Mitosis proceeds normally when the cytoplasm of prophase cells is irradiated with doses that induce a reversion when targeted to the nucleus. However, similar nuclear irradiations not only induce chromosome decondensation, but it also inhibit progression of the cytoplasm into the mitotic state as evidenced, e.g., by the fact that the centrosomes fail to ultimately form asters, which they would normally do at the end of prophase if the nucleus had not been irradiated. In addition, irradiating one nucleus in a cell containing two prophase nuclei induces both to return to interphase (refer to Fig. 4). Thus, damage to a prophase nucleus is converted into a signal that enters the cytoplasm and spreads throughout the cell to inhibit progression of the cytoplasm into the mitotic state (and also progression of an adjacent nonirradiated nucleus). The molecular mechanisms behind this signaling pathway remain to be determined. Since cdc25 has been implicated in activating both nuclear and cytoplasmic CDK1, it is possible that triggering this nuclear damage checkpoint inhibits nuclear cdc25 activity, and that this same inhibitory signal spreads to the cytoplasm to delay centrosome maturation by a similar mechanism.
Acknowledgments
The authors thank A. Khodjakov for his stimulating discussions and suggestions throughout this study; C. Hughes for her tireless cell culturing assistance and for helping with the UV radiation and IMF experiments; C. Moorhead for her assistance with determining the average minimum duration of prophase in PtK1 cells; and M. Savoian (all four from Wadsworth Center, Albany, NY), G. Sluder (University of Massachusetts Medical Center, Worcester, MA), and H. Piwnica-Worms (Washington University, St. Louis, MO) for helpful discussion and advice.
Abbreviations used in this paper
- CDK1
cyclin-dependent kinase 1
- DIC
differential interference contrast
- IMF
immunofluorescent
- LM
light microscopy
- M–A
metaphase–anaphase
- Mts
microtubules
- NEB
nuclear envelope breakdown
Footnotes
This work was supported, in part, by a grant from the National Institutes of Health (NIH) General Medical Sciences (R01 40198) to C.L. Rieder, and by a grant from NIH/National Center for Research Resources (P4101219) which supports the Wadsworth Center's Biological Microscopy and Image Reconstruction facility as a National Biotechnological Resource.
Address all correspondence to Conly L. Rieder, Division of Molecular Medicine, Wadsworth Center, P.O. Box 509, Albany, New York 12201-0509. Tel.: (518) 474-6774. Fax: (518) 486-4901. E-mail: rieder@wadsworth.org
References
- 1.Aubin JE, Osborn M, Weber K. Variations in the distribution and migration of centriole duplexes in mitotic PtK2cells studied by immunofluorescence microscopy. J Cell Sci. 1980;43:177–194. doi: 10.1242/jcs.43.1.177. [DOI] [PubMed] [Google Scholar]
- 2.Bailly E, Pines J, Hunter T, Bornens M. Cytoplasmic accumulation of cyclin B1 in human cells: association with a detergent-resistant compartment and with the centrosome. J Cell Sci. 1992;101:529–545. doi: 10.1242/jcs.101.3.529. [DOI] [PubMed] [Google Scholar]
- 3.Barth H, Hoffmann I, Kinzel V. Radiation with 1 Gy prevents the activation of the mitotic induces mitosis-promoting factor (MPF) and cdc25-c in HeLa cells. Cancer Res. 1996;56:2268–2272. [PubMed] [Google Scholar]
- 4.Blasina A, Paegle ES, McGowan CH. The role of inhibitory phosphorylation of CDC2 following DNA replication block and radiation-induced damage in human cells. Mol Biol Cell. 1997;8:1013–1023. doi: 10.1091/mbc.8.6.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlson JG. Effects of radiation on mitosis. J Cell Comp Physiol. 1950;35:89–101. doi: 10.1002/jcp.1030350407. [DOI] [PubMed] [Google Scholar]
- 6.Carlson JG. X-ray induced prophase delay and reversion of selected cells in certain avian and mammalian tissues in culture. Radiation Res. 1969;37:15–30. [PubMed] [Google Scholar]
- 7.Chen RH, Waters JC, Salmon ED, Murray AW. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science. 1996;274:242–246. doi: 10.1126/science.274.5285.242. [DOI] [PubMed] [Google Scholar]
- 8.Cohen-Fix O, Peters JM, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiaeis controlled by the APVC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996;10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- 9.Cole RW, Khodjakov A, Wright WH, Rieder CL. A differential interference contrast-based light microscopic system for laser microsurgery and optical trapping of selected chromosomes during mitosis in vivo. J Microsc Soc Amer. 1995;1:203–215. [Google Scholar]
- 10.Davis FM, Taso TY, Fowler SK, Rao PN. Monoclonal antibodies to mitotic cells. Proc Natl Acad Sci USA. 1983;80:2926–2930. doi: 10.1073/pnas.80.10.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feilotter H, Lingner C, Rowley R, Young PG. Regulation of the G2-mitosis transition. Biochem Cell Biol. 1992;70:954–971. doi: 10.1139/o92-140. [DOI] [PubMed] [Google Scholar]
- 12.Funabiki H, Yamano H, Kumada K, Nagao K, Hunt T, Yanagida M. Cut2 proteolysis required for sister-chromatid separation in fission yeast. Nature. 1996;381:438–441. doi: 10.1038/381438a0. [DOI] [PubMed] [Google Scholar]
- 13.Gabrielli BG, DeSouza CPC, Tonks ID, Clark JM, Hayward NK, Ellem KAO. Cytoplasmic accumulation of cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J Cell Sci. 1996;109:1081–1093. doi: 10.1242/jcs.109.5.1081. [DOI] [PubMed] [Google Scholar]
- 14.Gabrielli BG, Clark JM, McCormak AK, Ellem KAO. Ultraviolet light-induced G2 phase cell cycle checkpoint blocks cdc25-dependent progression into mitosis. Oncogene. 1997;15:749–758. doi: 10.1038/sj.onc.1201254. [DOI] [PubMed] [Google Scholar]
- 15.Gaulden ME, Perry RP. Influence of the nucleolus on mitosis as revealed by ultraviolet microbeam irradiation. Proc Natl Acad Sci USA. 1958;44:553–559. doi: 10.1073/pnas.44.6.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartwell LH, Weinert TA. Checkpoints: Controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 17.Heald R, McLoughlin M, McKeon F. Human Wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell. 1993;74:463–474. doi: 10.1016/0092-8674(93)80048-j. [DOI] [PubMed] [Google Scholar]
- 18.Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 19.Holloway SL. Sister chromatid separation in vivo and in vitro. Curr Opin Genet Dev. 1995;5:243–248. doi: 10.1016/0959-437x(95)80015-8. [DOI] [PubMed] [Google Scholar]
- 20.Hoyt MA, Totis L, Roberts BT. S. cerevisiaegenes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–517. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- 21.Hwang LH, Lau FF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW. Budding yeast Cdc20: A target of the spindle checkpoint. Science. 1998;279:1041–1044. doi: 10.1126/science.279.5353.1041. [DOI] [PubMed] [Google Scholar]
- 22.Jackman MR, Pines JN. Cyclins and the G2/M transition. Cancer Surveys. 1997;29:47–73. [PubMed] [Google Scholar]
- 23.Jiang R, Liang H. Mitotic reversion in prophase PtK1cells induced by argon laser microirradiation. Cell Biophys. 1989;14:271–282. doi: 10.1007/BF02797273. [DOI] [PubMed] [Google Scholar]
- 24.Jin P, Gu Y, Morgan DO. Role of inhibitory CDC2 phosphorylation in radiation-induced G2 arrest in human cells. J Cell Biol. 1996;134:963–970. doi: 10.1083/jcb.134.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufmann WK, Paules RS. DNA damage and cell cycle checkpoints. FASEB (Fed Am Soc Exp Biol) J. 1996;10:238–247. doi: 10.1096/fasebj.10.2.8641557. [DOI] [PubMed] [Google Scholar]
- 26.Kharbanda S, Saleem A, Datta R, Yuan Z-M, Weichselbaum R, Kufe D. Ionizing radiation induces rapid tyrosine phosphorylation of P34cdc2 . Cancer Res. 1994;54:1412–1414. [PubMed] [Google Scholar]
- 27.Khodjakov A, Cole RW, McEwen BF, Buttle K, Rieder CL. Chromosome fragments possessing only one kinetochore can congress to the spindle equator in PtK1cells. J Cell Biol. 1997a;136:229–241. doi: 10.1083/jcb.136.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khodjakov A, Cole RW, Rieder CL. A synergy of technologies: combining laser microsurgery with green fluorescent protein (GFP)- tagging. Cell Motil Cytoskel. 1997b;38:311–317. doi: 10.1002/(SICI)1097-0169(1997)38:4<311::AID-CM1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 29.Kim SH, Lin DP, Matsumoto S, Kitaxono A, Matsumoto T. Fission yeast Slp1: an effector of the Mad2-dependent spindle checkpoint. Science. 1998;279:1045–1047. doi: 10.1126/science.279.5353.1045. [DOI] [PubMed] [Google Scholar]
- 30.King RW, Deshaies RJ, Peters J-M, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–1659. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- 31.Koshland K, Strunnikov A. Mitotic chromosome condensation. Annu Rev Cell Dev Biol. 1996;12:305–333. doi: 10.1146/annurev.cellbio.12.1.305. [DOI] [PubMed] [Google Scholar]
- 32.Kuang J, Penkala JE, Wright DA, Saunders GF, Rao PN. A novel M phase-specific H1 kinase recognized by the mitosis-specific monoclonal antibody MPM-2. Dev Biol. 1991;144:54–64. doi: 10.1016/0012-1606(91)90478-l. [DOI] [PubMed] [Google Scholar]
- 33.Kuang J, Ashorn CL. At least two kinases phosphorylate the MPM-2 epitope during Xenopusoocyte maturation. J Cell Biol. 1993;123:859–868. doi: 10.1083/jcb.123.4.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuang J, Ashorn CL, Gonzalez-Kuyvenhoven M, Penkala JE. Cdc25 is one of the MPM-2 antigens involved in the activation of maturation-promoting factor. Mol Biol Cell. 1994;5:135–145. doi: 10.1091/mbc.5.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langan TA, Gautier J, Lohka M, Hollingsworth R, Moreno S, Nurse P, Maller J, Sclafani RA. Mammalian growth-associated H1 histone kinase: a homologue of cdc2/cdc28 protein kinases controlling mitotic entry in yeast and frog cells. Mol Cell Biol. 1989;9:3860–3868. doi: 10.1128/mcb.9.9.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lew DJ, Kornbluth S. Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Curr Opin Cell Biol. 1996;8:795–804. doi: 10.1016/s0955-0674(96)80080-9. [DOI] [PubMed] [Google Scholar]
- 37.Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–531. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- 38.Lu MJ, Dadd CA, Mizzen CA, Perry CA, McLachlan DR, Annunziato AT, Allis CD. Generation and characterization of novel antibodies highly selective for phosphorylated linker histone H1 in Tetrahymena and HeLa cells. Chromosoma. 1994;103:111–121. doi: 10.1007/BF00352320. [DOI] [PubMed] [Google Scholar]
- 39.Mazia, D. 1961. Mitosis and the Physiology of Cell Division. In The Cell. Vol. 3. J. Brachet and A.E. Mirsky, editors. Academic Press, New York. 77–412.
- 40.Mitchison, J.M. 1971. The Biology of the Cell Cycle. Cambridge University Press, London, UK. 313 pp.
- 41.Nigg EA. The substrates of the cdc2 kinase. Sem Cell Biol. 1991;2:261–270. [PubMed] [Google Scholar]
- 42.Nishimoto T, Uzawa S, Schlegel R. Mitotic checkpoints. Curr Biol. 1992;4:174–179. doi: 10.1016/0955-0674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 43.O'Connor PM. Mammalian G1 and G2 phase checkpoints. Cancer Surveys. 1997;29:151–182. [PubMed] [Google Scholar]
- 44.Ohnuki Y, Rlounds DE, Olson RS, Berns MW. Laser microbeam irradiation of the juxtanucleolar region of prophase nucleolar chromosomes. Exp Cell Res. 1972;71:132–144. doi: 10.1016/0014-4827(72)90271-6. [DOI] [PubMed] [Google Scholar]
- 45.Page AM, Hieter P. The anaphase promoting complex. Cancer Surveys. 1997;29:133–150. [PubMed] [Google Scholar]
- 46.Pederson T. Chromatin structure and the cell cycle. Proc Natl Acad Sci USA. 1972;69:2224–2228. doi: 10.1073/pnas.69.8.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng C-Y, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 48.Rieder CL, Hard R. Newt lung epithelial cells: cultivation, use, and advantages for biomedical research. Int Rev Cytol. 1990;122:153–220. doi: 10.1016/s0074-7696(08)61208-5. [DOI] [PubMed] [Google Scholar]
- 49.Rieder CL, Schultz A, Cole R, Sluder G. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol. 1994;127:1301–1310. doi: 10.1083/jcb.127.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rieder CL, Cole RW. Perfusion chambers for high-resolution video light microscopic studies of vertebrate cell monolayers: some considerations and a design. Methods Cell Biol. 1998;56:253–277. doi: 10.1016/s0091-679x(08)60430-6. [DOI] [PubMed] [Google Scholar]
- 51.Rieder CL, Salmon ED. The vertebrate cell kinetochore and its roles during mitosis. Trends Cell Biol. 1998;8:310–318. doi: 10.1016/s0962-8924(98)01299-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sluder G, Thompson EA, Rieder CL, Miller FJ. Nuclear envelope breakdown is under nuclear not cytoplasmic control in sea urchin zygotes. J Cell Biol. 1995;129:1447–1458. doi: 10.1083/jcb.129.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sluder G, Thompson EA, Miller FJ, Hayes J, Rieder CL. The checkpoint control for anaphase onset does not monitor excess numbers of spindle poles or bipolar spindle symmetry. J Cell Sci. 1997;110:421–429. doi: 10.1242/jcs.110.4.421. [DOI] [PubMed] [Google Scholar]
- 54.Straight AF, Belmont AS, Robinett CC, Murray AW. GFP tagging of budding yeast chromosomes reveals that protein-protein interactions can mediate sister chromatid cohesion. Curr Biol. 1996;6:1599–1608. doi: 10.1016/s0960-9822(02)70783-5. [DOI] [PubMed] [Google Scholar]
- 55.Taagepera S, Rao PN, Drake FH, Gorbsky GJ. DNA topoisomerase II alpha is the major chromosome protein recognized by the mitotic phosphoprotein antibody MPM-2. Proc Natl Acad Sci USA. 1993;90:8407–8411. doi: 10.1073/pnas.90.18.8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taylor SS, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89:727–735. doi: 10.1016/s0092-8674(00)80255-x. [DOI] [PubMed] [Google Scholar]
- 57.Th'ng JPH, Wright PS, Hamaguchi J, Lee MG, Norbury CJ, Nurse P, Bradbury EM. The FT10 cell line is a mouse G2 phase mutant with a temperature-sensitive CDC2 gene product. Cell. 1990;63:313–324. doi: 10.1016/0092-8674(90)90164-a. [DOI] [PubMed] [Google Scholar]
- 58.Vandre DD, Davis FM, Rao PN, Borisy GG. Phosphoproteins are components of mitotic microtubule organizing centers. Proc Natl Acad Sci USA. 1984;81:4439–4443. doi: 10.1073/pnas.81.14.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verde F, Dogterom M, Stelzer E, Karsenti E, Leibler S. Control of microtubule dynamics and length by cyclin A- and cyclin B-dependent kinases in Xenopusegg extracts. J Cell Biol. 1992;118:1097–1108. doi: 10.1083/jcb.118.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weinert T. Yeast checkpoint controls and relevance to cancer. Cancer Surveys. 1997;29:109–132. [PubMed] [Google Scholar]
- 61.Westendorf JM, Rao PN, Gerace L. Cloning of cDNAs for M-phase phosphoproteins recognized by the MPM2 monoclonal antibody and determination of the phosphorylated epitope. Proc Natl Acad Sci USA. 1994;91:714–718. doi: 10.1073/pnas.91.2.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang SS, Yeh E, Salmon ED, Bloom K. Identification of a mid-anaphase checkpoint in budding yeast. J Cell Biol. 1997;136:345–354. doi: 10.1083/jcb.136.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]