

Abstract
We previously hypothesized that the type I keratin 16 (K16) plays a role in the process of keratinocyte activation that occurs in response to skin injury (Paladini, R.D., K. Takahashi, N.S. Bravo, and P.A. Coulombe. 1996. J. Cell Biol. 132:381–397). To further examine its properties in vivo, the human K16 cDNA was constitutively expressed in the progenitor basal layer of transgenic mouse skin using the K14 gene promoter. Mice that express approximately as much K16 protein as endogenous K14 display a dramatic postnatal phenotype that consists of skin that is hyperkeratotic, scaly, and essentially devoid of fur. Histologically, the epidermis is thickened because of hyperproliferation of transgenic basal cells, whereas the hair follicles are decreased in number, poorly developed, and hypoproliferative. Microscopically, the transgenic keratinocytes are hypertrophic and feature an altered keratin filament network and decreased cell–cell adhesion. The phenotype normalizes at ∼5 wk after birth. In contrast, control mice expressing a K16-K14 chimeric protein to comparable levels are normal. The character and temporal evolution of the phenotype in the K16 transgenic mice are reminiscent of the activated EGF receptor– mediated signaling pathway in skin. In fact, tyrosine phosphorylation of the EGF receptor is increased in the newborn skin of K16 transgenic mice. We conclude that expression of K16 can significantly alter the response of skin keratinocytes to signaling cues, a distinctive property likely resulting from its unique COOH-terminal tail domain.
Keywords: skin, keratin, adhesion, wound repair, transgenic mouse
Full thickness injury to the epidermis, a stratified squamous epithelium, induces dramatic morphological changes in suprabasal keratinocytes located at the edges of the wound. Before beginning migration into the wound site at 16–24 h after injury in the form of a stratified epithelial sheet (3, 12), suprabasal cells at the wound edge undergo a process termed keratinocyte activation (14). During this activation phase, changes in gene expression contribute to major morphological alterations in the cytoarchitecture of the keratinocytes. These include cellular hypertrophy, the fragmentation of keratin filaments and their relocation adjacent to the nucleus, and changes in the number and structure of desmosomes that correlate with an increase in space between keratinocytes (12, 69). As a result, the process of terminal differentiation in these cells is perturbed.
One of the hallmarks of the epidermis is the programmed expression of keratin genes that is determined by the location and function of the keratinocyte within the epidermis (21). Basal cells, which are the progenitor, dividing cells of the epidermis, express keratin K5 (type II), keratin K14 (type I) (66), and low amounts of keratin K15 (type I) (48). By exiting the cell cycle and migrating upward, basal cells begin the terminal differentiation process that results in the production of squames. A distinctive molecular change concomitant with the onset of differentiation is the switch from the expression of K5 and K14 to the expression of the type II keratin K1 and the type I keratin K10 (22). A structural function for keratins in the epidermis has been clearly defined by several transgenic mouse studies in which either the introduction of dominant-negative mutant proteins (17, 23, 70, 73, 87) or the absence of the protein (48) resulted in an epidermis that was susceptible to blistering. The location of the blistering within the epidermis is determined by where the genetic alteration has occurred. Mutations in the keratin genes for K1, K5, K2e, K9, K10, and K14 have been found to cause several human genodermatoses such as epidermolysis bullosa simplex, epidermolytic hyperkeratosis, and palmoplantar keratoderma (15, 39, 56).
The type II keratin K6 and the type I keratins K16 and K17 are constitutively expressed at low levels in a number of stratified epithelium including palmar and plantar epidermis, tongue, oral mucosa, and the outer root sheath of the hair follicle (55, 60, 68). These keratins are not present in normal interfollicular epidermis but their expression is rapidly induced in suprabasal cells located at the edges of wounds. In addition to this induction, K6, K16, and K17 are also expressed in stratified epithelia featuring hyperproliferation or abnormal differentiation, including psoriasis and cancer (55, 79, 90). Keratinocytes from these abnormal conditions feature some of the cytoarchitectural changes that are observed in wound edge keratinocytes including hypertrophy, filament abnormalities, and reductions in cell–cell adhesion (82). These data suggest a relationship between the expression of these keratins and differences in keratinocyte differentiation and cytoarchitecture.
A remarkable feature of K6, K16, and K17 is the high degree of sequence identity that they share with their basal keratin counterparts (K5 and K14). For example, along the tripartite head–rod–tail domain structure of keratins, K16 and K14 are 91%–86%–38% identical in their primary sequence (55, 85). The question arises as to why a keratinocyte under certain conditions would synthesize two new keratins that are very similar to the keratins that are already present. One simple possibility is that the function that these keratins are performing is not strictly structural. There is ample evidence that this may be true for keratin 16.
For example, a dominant-negative mutation in K16 that has been reported in a case of Jadassohn-Lewandowsky Pachyonychia Congenita results in filament aggregation near the nucleus and a decrease in the number of desmosomes (57). However, cytolysis does not occur in these keratinocytes, which is in stark contrast to the fact that comparable mutations in the other epidermal keratins (K1, K2e, K5, K9, K10, and K14) do result in cytolysis (15, 56). In addition, the tissue-specific overexpression of wild-type human K16 in transgenic mouse epidermis resulted in aberrant keratinization and hyperproliferation (82). These changes were also accompanied by keratin filament reorganization and a decrease in desmosomes at the cell surface. Finally, biochemical and cell culture experiments revealed that K16, unlike K5, K6, and K14 promoted the formation of short filaments that preferentially localized near the nucleus of cells (69). These data have led to the hypothesis that K16 may play a role in promoting many of the cytoarchitectural changes that occur in suprabasal keratinocytes in response to wound healing. This is quite distinct from the structural role that the other epidermal keratins perform in maintaining keratinocyte integrity and begins to address the issue of why epidermal function and homeostasis might require so many different keratin genes.
The ability to get answers to these pressing questions has been facilitated by a mouse study that clearly defined the structural function of the highly related keratin 14 by gene inactivation (48). Basal cells lacking a keratin filament network because of the absence of K14 suffered extensive cytolysis resulting from the inability of these cells to withstand mechanical trauma, resulting in an EBS-like phenotype. This severe phenotype results in the death of the K14 null mice, generally ∼2 d after birth. Our goal has been to target the expression of human K16 to the basal layer of these mice as a test to determine if it can functionally complement the lethal phenotype of these mice. The results of these experiments will provide in vivo information directly comparing the functional properties of K14 and K16.
As a first step in creating the keratin replacement mice, transgenic mice were generated that targeted the expression of K16 to the basal layer of the epidermis. As a control, mice expressing a K16-C14 chimeric protein containing the head and rod domains of K16 and the COOH-terminal tail domain of K14 were also generated. The K16 transgenic mice displayed a strong skin phenotype including epidermal hyperproliferation and abnormalities in hair follicle morphogenesis and hair growth. Multiple abnormalities in keratin filament organization and cell–cell adhesion were also observed further correlating the expression of K16 with changes in keratinocyte cytoarchitecture. The phenotype was quite distinct at the level of the organism from the previous study (82) suggesting that the context of K16 expression may be very important in determining its effect. None of these phenotypic changes were observed in the control animals allowing for implication of the COOH-terminal tail domain of K16 as being responsible for its functional properties.
Materials and Methods
Generation and Screening of Transgenic Mice
All protocols involving mice were approved by the Johns Hopkins University Animal Care and Use Committee (Baltimore, MD). Transgenic mice were generated by pronuclear injection using established protocols (37). The plasmids pET-K16 (69) and pET-K16-C14 (89) were modified to include a BamHI restriction site 5′ of the ATG start codon. A BamHI digest of each plasmid generated a 1.4-kb piece of DNA encoding the complete cDNA for each construct. This was subcloned into the BamHI site of the modified K14 cassette expression vector (75). Clones of the correct orientation were digested with HindIII and EcoRI to generate a 6-kb piece of DNA containing 2 kb of human K14 promoter sequence, the rabbit β-globin intron, the cDNA of interest, and 0.6 kb of human K14 polyA sequence. Purified DNA was microinjected into fertilized B6C3 F2 mouse embryos. At 3 wk of age, potential founders were anesthetized to obtain a small piece of tail. Genomic DNA was isolated, digested with EcoRV, subjected to 0.8% agarose gel electrophoresis, and then transferred to a nylon membrane (NEN Life Science Products, Boston MA). Membranes were prehybridized for 30 min at 65°C in hybridization buffer (50 mM Tris, pH 7.4, 1 M NaCl, 1% SDS, and 10% PEG 8000). Hybridization was carried out overnight at 65°C. Blots were washed for 20 min at 65°C four times with 0.1× SSC, 0.5% SDS. The 1.4-kb BamHI fragments from plasmids pET-K16 and pET-K16-C14 were used as probes to detect the presence of the transgene by Southern blotting. These probes are specific for transgenic animals as there is no reaction with non-transgenic mouse DNA. All subsequent generations of mice were screened identically.
Determination of Transgene Protein Expression in the Epidermis
To determine the amount of transgene protein that is expressed in the epidermis, urea soluble proteins were isolated from dorsal skin. A 1-cm2 piece of dorsal skin was isolated from killed animals and quick frozen in liquid nitrogen. The frozen skin was crushed in a metal mortar and pestle kept cold with dry ice. The fragmented pieces were homogenized in 1 ml of buffer A (6.5 M urea, 50 mM Tris, DTT, PMSF, EDTA) for 1 min using a PT 1200 Polytron (Kinematica AG, Littau, Switzerland) on ice. The homogenates were incubated at 37°C for 30 min. Supernatants were isolated and the protein concentration was determined using the Bradford assay (6) with reagents purchased from Bio-Rad Laboratories (Hercules, CA). Equivalent amounts of isolated proteins (20 μg) were resolved via 8% SDS-PAGE and then transferred to nitrocellulose. In addition, varying amounts of human recombinant K16, K14, or K16-C14 proteins were electrophoresed and transferred in tandem to determine the amount of transgene expressed. Western blotting was performed using the alkaline phosphatase method (Bio-Rad Laboratories). Human K16 was detected using the previously described rabbit polyclonal anti–human K16 antibody (82) or with K8.12 (anti-K13, K15, and K16; Sigma Chemical Co., St. Louis, MO). Mouse and human K14 and human K16-C14 were detected with the mouse mAb LL001 (71) that recognizes an identical sequence shared between the human and mouse K14 tails. Western blots were scanned using NIH Image 1.59 software (developed at the U.S. National Institutes of Health and available on the world wide web via http://rsb.info.nih.gov/nih-image/). The approximate amount of transgene protein made was calculated from standard curves established with the purified recombinant keratins. The quantitation analysis was done three times for each of the transgenic lines.
Culture and Analysis of Primary Mouse Keratinocytes
Primary cultures of skin keratinocytes were established essentially as described (74). Newborn mice were killed by decapitation and their limbs were removed. The tails were used to isolate DNA for determining genotype. Mice were washed with 70% ethanol, 10% Proviodine, and then 70% ethanol. The skin was removed from the trunk, and washed three times in sterile PBS–fungizone. The skin was floated (epidermis upward) overnight in a 10-cm petri dish containing a solution of 0.25% trypsin and 40 μg/ml DNase in PBS at 4°C. The next day the epidermis was separated from the dermis and transferred to a small petri dish containing mouse keratinocyte medium where it was gently scraped with curved forceps to isolate keratinocytes. The cells were pelleted and resuspended in mouse keratinocyte medium. The suspension was layered on a lympholyte M gradient (GIBCO-BRL, Gaithersburg, MD) and centrifuged for 30 min at 500 rpm and 4°C. The living keratinocytes located at the interface of the two media were isolated, centrifuged, and then resuspended. The cells were counted and plated at a density of 2 × 105 cells/cm2 on glass cover slips. 3–4 d after plating, the primary keratinocytes were processed for immunofluorescence. The coverslips were washed three times in PBS and fixed for 15 min with 100% methanol at −20°C. After three washes with PBS, coverslips were blocked with 5% normal goat serum in PBS. Primary antibodies were diluted in blocking buffer and incubated for 45 min at room temperature. FITC-conjugated goat anti–mouse and anti–rabbit, rhodamine-conjugated goat anti–mouse and –rabbit, and FITC-conjugated goat anti–guinea pig secondary antibodies were used to detect bound primary antibodies. Coverslips were mounted onto slides and analyzed via fluorescence microscopy. Primary antibodies used were mouse mAbs LL001(anti-K14) (71) LL025 (anti-K16) (43), K8.60 (anti-K10; Sigma Chemical Co.), K8.12 (anti-K16; Sigma Chemical Co.) and rabbit polyclonals No. 1275 (anti-K16) (82), K6 general (anti-K6) (see Results), K17 (anti-K17) (see Results), and No. 5054 (anti-K5 and -K6) (44).
Morphological Analyses
Mouse tissues were fixed in Bouin's overnight at 4°C. The fixed tissues were embedded in paraffin and 5-μm sections were stained with hematoxylin and eosin or immunostained using the HRP procedure as per the manufacturer's protocol (Kirkegaard & Perry Labs, Gaithersburg, MD). In addition, freshly isolated dorsal and ventral skin was washed in 4°C PBS and quick frozen in liquid nitrogen using OCT (Sakura Finetek, Torrance, CA) as the freezing medium. 8-μm sections were cut, placed onto superfrost slides (VWR Scientific Products, West Chester, PA), and immediately stored at −20°C until used. Unfixed sections were air dried for 2 min, and then hydrated in PBS. Sections were analyzed either using the HRP protocol described above or by using indirect immunofluorescence. For electron microscopy, ventral skin tissues were fixed in 0.1 M sodium cacodylate and 2.5% glutaraldehyde, post-fixed in 1% osmium tetroxide, and then embedded in LX112 epoxy-resin (Ladd Research Industries Inc., Burlington, VT). Ultrathin sections (50–70 nm) were placed on copper grids, counterstained with uranyl acetate and lead citrate, and then visualized using a transmission electron microscope (EM10A; Carl Zeiss Inc., Thornwood, NY) operated at 60 kV.
Immunoblot Analysis of the EGF Receptor
Newborn mice were killed and their skin was isolated and quick frozen in liquid nitrogen. The skin was crushed as described above and homogenized in 2 ml of lysis buffer (50 mM Tris, pH 7.4, 1% sodium deoxycholate, 1% Triton X-100, .1% SDS, 150 mM NaCl, .5 mM EDTA, 1 mM EGTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM PMSF, 20 μm NaVO4, and 10 mM NaF) for 1 min using the polytron. The lysates were centrifuged and the supernatants were isolated and the protein concentration was determined as described above. Equivalent amounts of protein (∼30 μg) were resolved via SDS-PAGE (8%) and electrophoresed onto Nitrocellulose. The blots were incubated with an anti-EGF receptor (EGFR)1 or an anti-phosphotyrosine antibody (both from Upstate Biotechnology Inc., Lake Placid, NY) followed by incubation with HRP-conjugated secondary antibodies (Jackson Immunoresearch, West Grove, PA). The signal was detected using ECL reagents from Amersham Life Sciences (Arlington Heights, IL) and autoradiography film from Kodak (Rochester, NY). The film was scanned using NIH image 1.59 as described above. As a control for EGFR tyrosine phosphorylation, 200 μl of 10 μg/ml EGF (Sigma Chemical Co.) was subcutaneously injected into a newborn mouse 10 min before the mouse was killed.
Results
Transgenic Mice Expressing Human K16 Display a Skin Phenotype
As the first step in creating mice that express human K16 in the absence of K14, transgenic mice were generated to express either the K16 or K16-C14 cDNA in the basal layer of stratified squamous epithelia. The cDNAs were subcloned into a modified version of the K14 expression cassette (88, 75) as shown in Fig. 1 A. The two constructs were transiently transfected in PTK2 cells and in both cases the transgene was expressed and incorporated into the endogenous keratin network as detected by indirect immunofluorescence (data not shown). The K16-C14 chimeric cDNA encodes the head and rod of K16 fused to the tail of K14. The two keratins share 91% amino acid sequence identity in their head domains and 86% identity in their rod domains. The identity is only 38% in the tail domain. As previously shown (69), K16, when transiently expressed in PtK2 cells, can cause the reorganization of the endogenous keratin filament network whereas K14 doesn't have the same ability. When the chimera was transfected into PtK2 cells, the results were similar to those obtained with K14 (data not shown), suggesting that the tail domains are responsible for the differences seen between the two keratins. Therefore, the chimeric construct was chosen as a control and was predicted to behave in a K14-like fashion.
Figure 1.
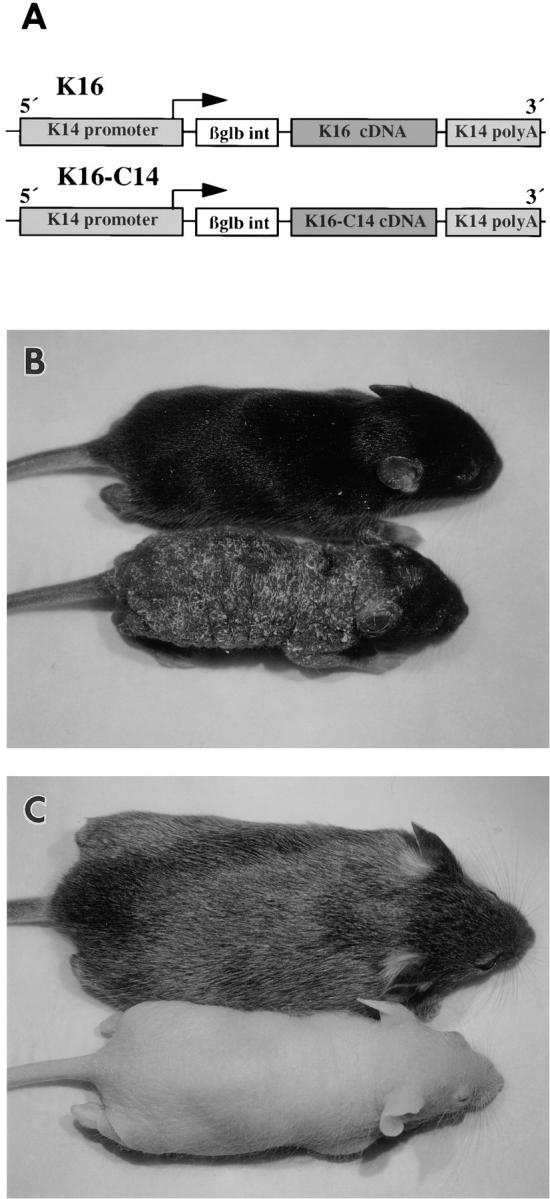
Generation of transgenic mice. (A) Schematic representation of the DNA constructs used. The human K16 and K16-C14 cDNAs were subcloned into a modified version of the K14 expression cassette (75). The cassette features 0.6 kb of human K14 polyA sequence, the rabbit β-globin intron, and 2 kb of human K14 promoter sequence (88). Arrow, direction and initiation site of transcription. (B) Phenotype of the K16 transgenic mice at 9 d after birth (No. 21 line). Wild-type (top) and homozygous transgenic (bottom) littermates are pictured. The smaller phenotypic mouse shows an absence of fur coat and a flaky, wrinkled, and thickened skin surface. Also, the ears have not yet completely erupted. (C) Phenotype at 3 wk (No. 10 line). Wild-type (top) and homozygous transgenic (bottom) littermates are pictured. The first hair cycle in mice is complete at ∼3 wk (5, 20) and the phenotypic littermate is essentially lacking the first hair coat. The epidermis, however, is no longer flaky and wrinkled in appearance. In contrast, the K16-C14 transgenic mice have a wild-type appearance (data not shown).
Four K16 transgenic founder mice and three K16-C14 chimera transgenic founder mice were obtained. Each of the lines has been found to have a single transgene insertion site. All seven founders were able to breed successfully and each line was bred to homozygosity at the insertion locus. Beginning at ∼5 d after birth, the K16 transgenic mice develop a severe phenotype easily discernible by eye (Fig. 1 B). This phenotype occurs in three of the K16 transgenic lines. Phenotypic mice show a thickened and flaky skin over the entire surface of their body. The normal emergence of hair at ∼6–7 d after birth does not occur. The emergence of the ears is also delayed in the phenotypic mice. In two of the phenotypic K16 lines blistering occurs in a restricted area located on the underside of the front limbs. By ∼3 wk, the skin of the phenotypic mice has a more normal appearance (Fig. 1 C) except for the obvious lack of hair. The phenotypic mice are generally smaller than non-phenotypic littermates. At ∼5 wk, the phenotype normalizes and hair growth begins. The resultant hairs, however, are shorter and fewer in number compared with non-phenotypic littermates (data not shown). Southern blotting of DNA from phenotypic and non-phenotypic littermates revealed that phenotypic mice were homozygous for the presence of the transgene while non-phenotypic mice were either heterozygous or negative for the transgene. Mice from the three control K16-C14 chimera lines exhibited no obvious phenotype at any age even when homozygous for the transgene.
The Amount of Transgene Protein Expression in the Epidermis Determines the Phenotype
The fact that only mice homozygous for the transgene were phenotypic suggested that the amount of transgene being expressed was determining the phenotype. To examine this, the amount of transgene expressed in back skin from non-transgenic, heterozygous, and homozygous littermates was determined. Total urea-extractable proteins from back skin were analyzed by SDS-PAGE and Western blotting. Using known amounts of human recombinant keratins 14 and 16, a standard curve was established using densitometry and the amount of transgene expressed was quantitated with respect to endogenous levels of mouse K14.
In Fig. 2 A the results are shown for the phenotypic K16 transgenic line Nos. 10, 21, and 6 and in Fig. 2 B are the results for the K16-C14 chimera line Nos. A2, B1, and C1. Rabbit polyclonal antibody No. 1275, which specifically reacts with the tail of K16 (human and mouse; see references 18, 55) was used to detect the K16 transgene. Mouse mAb LL001 (71), which recognizes a tail epitope that is identical between human and mouse K14 was used to detect mouse K14 and the human K16-C14 transgene. The amount of mouse K14 in the extracts was equivalent regardless of the genotype, phenotype, or transgene line from which they were derived as determined by densitometry of the K14 blot (Fig. 2 A). From this result, mouse K14 was used as an internal reference to allow for quantitation of transgene protein.
Figure 2.



Determination of the level of transgene expression in the epidermis. Urea-soluble protein extracts were obtained from the epidermides of 7-d-old control, heterozygous, and homozygous littermates from the various transgene lines. Equivalent amounts of total urea-extractable protein (20 μg) were electrophoresed via SDS-PAGE and transferred to nitrocellulose for subsequent Western blot analysis. (A) Western blot analysis of the K16 transgenics. The indicated amounts of purified human recombinant K14 and K16 were used to establish a standard curve for densitometric analysis. The LL001 antibody (71) was used to detect K14 and the 1275 antibody (82) to detect K16. (B) Western blot analysis of the K16-C14 chimera transgenics. The LL001 antibody was used to detect both mouse K14 and the K16-C14 transgene protein. Their position of migration of each protein is indicated by the arrows on the right. Purified recombinant K16-C14 was used to determine the migration position of the transgene product. (C) The three No. 21 line samples were subjected to Western blot analysis using the K8.12 antibody (this antibody is known to react with at least K13, K15, and K16). No bands of molecular weight ⪝40 kD were detected in any of the three samples. Note that a minor degradation product of the purified K16 (see D) reacts with the antibody. The lower arrow indicates the bottom of the gel. (D) The same samples were subjected to SDS-PAGE and stained with Coomassie blue. The are no significant protein products below the type I keratin cluster (⪝40 kD) and no significant differences in the total amount of keratin proteins among the three samples.
Extracts from control mice from either of the three K16 lines showed little or no reactivity when probed with the 1275 antibody (Fig. 2 A). (The faint reactivity could be due to endogenous mouse K16 that has yet to be fully characterized; see reference 55.) A strong signal was detected in each of the three heterozygous samples. Line No. 10 expresses the K16 transgene at a level of ∼30% that of mouse K14. Line No. 6 expresses at ∼40%. Line No. 21 expresses an amount intermediate between the Nos. 10 and 6 lines. The levels approximately doubled when the values for the homozygous mice were determined. K16 transgenic line No. 13, which does not display a skin phenotype, was found to express the transgene at approximately half the level of the No. 10 line (data not shown). Therefore, expression of the K16 transgene to at least 60% the level of endogenous mouse K14 is sufficient to cause development of a skin phenotype.
The presence of minor amounts of faster migrating protein that reacted with the 1275 antibody in the homozygous samples suggested that the transgene was partially degraded and that there might be other proteolytic fragments not recognized by the antibody. To investigate this possibility the samples were subjected to western analysis using an antibody that recognizes a rod epitope present in K16 (K8.12). No prominent bands of molecular weight ⪝40 kD were observed in either the wild-type, heterozygous, or homozygous K16 transgenic samples (Fig. 2 C). In addition, when the same samples were subjected to SDS-PAGE and stained with Coomassie blue the vast majority of proteins were either full-length type I or type II keratins (Fig. 2 D). There were no prominent protein bands having a molecular weight ⪝40 kD in any of the three samples. These data argue that there is very little stable degradation product(s) derived from the transgenic protein and that it is the full-length K16 that leads to development of a phenotype.
The Western blot analysis of the three chimera lines is shown in Fig. 2 B. Line No. A2 did not express the transgene. The other two lines did but only line No. B1 expressed at levels equivalent to the K16 phenotypic lines. In the heterozygous state the level was ∼50% and in the homozygous state it was ∼70%. Mice homozygous in the No. B1 line, however, have no obvious skin phenotype despite expressing the transgene at a level that does cause a phenotype in the K16 transgenics. This supports that idea that the K16-C14 chimera, functionally, is similar to K14.
We previously speculated that K16 protein may turnover faster because of its decreased ability to form stable heterotetramers with a variety of type II keratin partners (89). Consistent with this, the mRNA levels for the K16 transgene are higher than anticipated based on the steady-state protein levels measured in transgenic mice (data not shown). Moreover when the K16 and K16-C14 transgenes are bred into the K14 null background, the amount of transgenic protein made is increased (Paladini, R., and P.A. Coulombe, unpublished data). Both transgenic proteins bear the 188Pro residue that acts as a tetramer destabilization determinant (89). Additional studies on these transgenic mouse lines will provide insight into the potential posttranslational mechanisms that may regulate K16 function.
Phenotypic Skin Features Acanthotic Epidermis and a Reduction of Hair Follicles
To examine the histological changes in the phenotypic mice, trunk skin from non-transgenic, heterozygous, and homozygous 7-d-old littermates from a K16 phenotypic line was obtained and examined by light microscopy (Fig. 3, D, G, and J, respectively). Trunk skin was also obtained from a 7-d-old homozygous mouse from the No. B1 chimera control line (Fig. 3 A). The epidermis from the K16 transgenic homozygote was significantly thickened when compared with control epidermis (compare Fig. 3, J and A; D and G). This acanthosis involves an expansion of both the spinous and granular layers. Many of the cells in the spinous layer are hypertrophic (see EM data below). In addition, the stratum corneum of the epidermis is also thickened (hyperkeratosis). As previously mentioned, blistering occurs in two of the K16 transgenic lines. The blistering occurred in the suprabasal layers of the epidermis (Fig. 3 C) and was extensive along the ventral surface around the limbs of the animals. In addition, ventral skin from phenotypic mice featured areas of parakeratosis (Fig. 3 F) and the presence of Munro microabcesses, a common feature of psoriasis (11). In both the blistered and parakeratotic epidermis, there was the presence of a large dermal infiltrate suggesting an inflammatory response. The other major morphological aberration of the phenotypic skin was a marked reduction in the number of hair follicles. Many of the remaining follicles were misoriented and did not extend deeply into the hypodermis (Fig. 3 J) suggesting that there might a partial impairment of follicle morphogenesis. None of these differences were seen in either the control or heterozygote littermates or in the homozygote chimera control.
Figure 3.
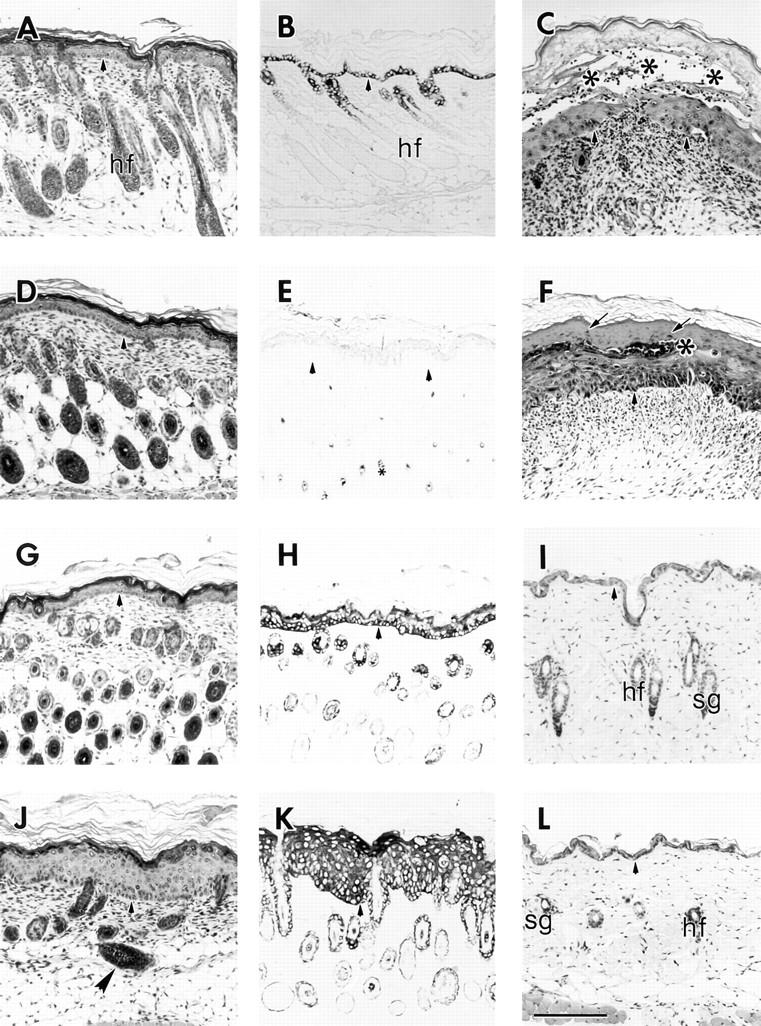
Light microscopy of the skin of control and transgenic mice. 5-μm paraffin sections from mice were counterstained with hematoxylin and eosin (H & E) or were subjected to immunostaining using the HRP procedure (No. 6 line). (A, D, G, and J) H & E–stained section of trunk skin from 7 d mice are shown. The epidermides of a homozygote chimera (A), a control wild-type (D) and a K16 heterozygote (G) have a comparable thickness and similar hair follicle profiles. The epidermis of the K16 homozygote (J) is significantly thickened compared with the others and there is a reduction in the number of hair follicles. Some of the follicles are also incorrectly oriented (large arrowhead). (B, E, H, and K) Immunohistochemical detection of the transgene in skin section from 7 d chimera homozygote (B), control wild-type (E), K16 heterozygote (H), and K16 homozygote (K). The LL001 antibody was used to detect the chimera transgene (B) and the 1275 antibody to detect the K16 transgene (E, H, K). The chimera transgene expression is correctly restricted to the outer root sheath of hair follicles and the basal layer of the epidermis. Control skin (E) shows no expression of human K16 (small asterisk, melanin granules in hair follicle profiles) while the K16 heterozygote (H) features the correct regulation of the transgene. The K16 homozygote (K), however, shows K16 expression throughout all layers of the epidermis. (C) H & E staining of 7 d ventral skin from a K16 homozygote that features blistering. Note the large blister (asterisks) that occurs within the suprabasal layers of the epidermis. (F) H & E of ventral skin from the same mouse that features parakeratosis. The arrows indicate parakeratotic nuclei and the asterisk denotes a Munro microabcess, a common feature of psoriasis. C and F also illustrate the presence of a large dermal infiltrate suggesting an inflammatory response. (I and L) H & E staining of 41 d dorsal skin from a wild-type control (I) and K16 homozygote (L), (No. 10 line). Note that at this age the epidermides of the two mice are of comparable thickness. hf, hair follicle; sg, sebaceous gland. Arrowheads, indicate the dermal–epidermal junction. Bar, 100 μm.
The distribution of the transgene product in these mice was determined by performing immunohistochemical analysis. Control mice had no staining for the transgene in the epidermis (Fig. 3 E) but occasionally displayed some faint reactivity in the outer root sheath of hair follicles (presumably, mouse K16; data not shown). The presence of the transgene product in K16 heterozygous mice was largely confined to the outer root sheath of hair follicles and the basal layer of the epidermis, confirming the proper expression of the transgene in a K14-like fashion (Fig. 3 H; 88). However, in the homozygous K16 transgenic skin strong and uniform staining was present throughout all the living layers of the epidermis (Fig. 3 K). The thickness of the epidermis coupled with this pattern of expression was suggestive of a hyperproliferative epidermis (69). The chimeric transgene was properly expressed in the outer root sheath of hair follicles and in the basal layer of the epidermis (Fig. 3 B). Since the antibody used to detect the chimeric protein recognizes the tail domain of K14, it is not possible to distinguish the chimeric protein from the endogenous mouse K14. The protein is present, however, as determined from the Western blot data (see Fig. 2 B). K16 expression in the chimera homozygotes is restricted to the outer root sheath (ORS) of hair follicles (data not shown).
As mentioned earlier, the phenotype of the K16 mice begins to normalize at ∼5 wk after birth. That is, the mice begin growing hair and their skin becomes increasingly normal. Dorsal skin from a 6-wk-old non-transgenic mouse and a homozygous K16 transgenic littermate was processed for light microscopic analysis to confirm these observations. The epidermides of both the control (Fig. 3 I) and the homozygote (Fig. 3 L) appeared similar with respect to thickness and general morphology.
Hair Morphology of the Phenotypic Mice Is Abnormal
Dorsal hairs from 41-d-old wild-type, heterozygous, and homozygous littermates were obtained and examined using light microscopy. Hairs from the homozygous mouse were shorter and the distal ends were often curved and shaped like a sickle (Fig. 4 A). Approximately 80% of the hairs from the homozygous mouse featured this abnormal shape. This same morphology was present in dorsal hair examined from older (6 mo) homozygous mice although the frequency was slightly lower (∼60%). Hair from wild-type (Fig. 4 B), and heterozygous littermates (data not shown) were long and featured straight distal ends. Essentially all of the hairs examined (98%) featured this morphology. The morphology of the phenotypic hairs were very similar to those obtained by Moore et al. (61) in their study in which they treated newborn mice with subcutaneous injections of EGF. Hairs from the first coat of these mice were reduced in diameter, shorter, and featured curvature of the distal ends.
Figure 4.

Light microscopy of hair from control and transgenic mice (No. 10 line). Dorsal hairs from a 41-d-old wild-type and homozygous transgenic litter mate were placed on slides, coverslipped, and examined using light microscopy. Control hairs (B) are long and straight whereas the hairs from the homozygous littermate (A) are shorter and feature distal ends that are curved and sickle shaped. Bar, 100 μm.
Phenotypic Epidermis Is Hyperproliferative and Features Aberrations in Terminal Differentiation
To examine the effects of K16 expression in the basal layer of the epidermis on differentiation, immunohistochemistry was performed. K14 was examined as a marker for basal cells, K10 was used as a marker for early terminal differentiation, and filaggrin was used as a marker for late differentiation. Wild-type, heterozygous, and homozygous K16 transgenic littermates were examined along with homozygotes from the No. B1 chimera line. Data for the heterozygote K16 transgenics is not shown as the results are identical to those obtained with wild-type mice.
K14, which is expressed in the basal layer of stratified epithelia, including the outer root sheath of hair follicles (16) was restricted to that location in both the chimera homozygote and the control sample (Fig. 5, A and B). In contrast, K14 was reduced in the basal layer and extended strongly in the suprabasal layers of the phenotypic sample (Fig. 5 C). This pattern of K14 localization is reminiscent of both hyperproliferative and wounded epidermis (17, 69). The expression of K10, an early marker of epidermal keratinocyte differentiation (21, 68), was detected in the suprabasal layers of all three samples (Fig. 5, D–F) with the only difference being that the area of K10 expression was thickened in the phenotypic sample due to the increased number of suprabasal layers. In contrast to the seemingly normal expression of K10, filaggrin expression in the phenotypic sample was abnormal (Fig. 5 I). Areas of the epidermis featured an expansion of filaggrin expression (compare to the thin layer of granular staining seen in the homozygous chimera and control samples; Fig. 5, G and H). In addition, there are some areas of the epidermis where the signal appeared decreased or even absent. These data suggest that there are deviations in the program of terminal differentiation executed in the phenotypic epidermis.
Figure 5.

Immunolocalization of epidermal differentiation markers in the skin. 5-μm paraffin sections were stained using the HRP procedure (No. 21 line). (A–C) Expression of K14 in the chimera homozygote (A), wild-type control (B), and K16 homozygote (C). K14 is properly expressed in the basal layer of the epidermis and the outer root sheath in the two controls. However, in the K16 homozygote (C), the expression is now detected primarily suprabasally. (D–F) Expression of K10 in the chimera homozygote (D), wild-type control (E), and K16 homozygote (F). K10 is properly localized to the suprabasal layers of the epidermis in all three samples. Expansion of K10 expression in F is due to the increased thickness of the suprabasal layers. (G–I) Filaggrin expression in the chimera homozygote (G), wild-type control (H), and K16 homozygote (I). Filaggrin is expressed in the granular layer of the epidermis in both G and H. In the K16 homozygote (I) some areas of agranulosis are observed (large asterisk) along with ectopic spinous layer expression (arrows). Arrowheads, dermal–epidermal junction. hf, hair follicle. Small asterisks, melanin granules in hair follicle profiles. Bar, 100 μm.
To determine whether or not the epidermis of the phenotypic mice was hyperproliferative, mice were injected with BrdU 2 h before they were killed. An anti-BrdU antibody was used to detect cells that were in S phase at the time of sacrifice. The K16 transgenic phenotypic epidermis had many more BrdU-positive nuclei compared with either of the two controls (compare Fig. 6, C with A and B). Quantitatively, the phenotypic epidermis had an approximately threefold higher mitotic index than either of the two controls. The opposite effect was observed in the hair follicles. The control mouse follicles had many nuclei that were BrdU positive, whereas follicles from the K16 homozygote showed fewer positive nuclei. These data indicate that the epidermis is hyperproliferative whereas the hair follicles are hypoproliferative. At ∼3 wk, when the first hair cycle is completed (5, 20) control epidermis and hair follicles have very few BrdU-positive nuclei (Fig. 6 D). In striking contrast, there are many BrdU-positive nuclei in the hair follicles of the phenotypic mice (Fig. 6 E). This data suggests that the first hair cycle in the phenotypic mice is dramatically delayed.
Figure 6.
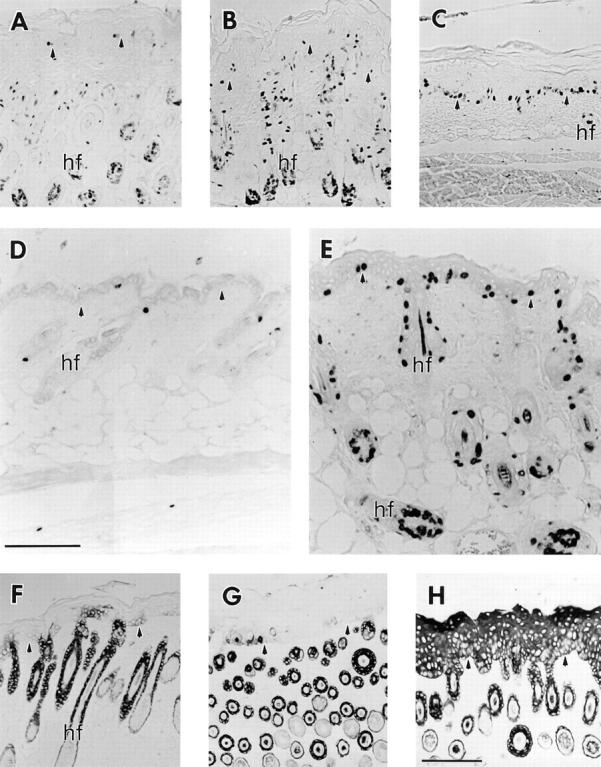
Immunolocalization of hyperproliferation markers in the skin. Mice were injected with BrdU 2 h before sacrifice, and samples from dorsal trunk skin were paraffin-embedded, sectioned, and immunostained using the HRP procedure (No. 6 line). (A–C) Trunk skin from a 7 d chimera homozygote (A), a wild-type control (B), and a K16 homozygote (C) were stained with an anti-BrdU antibody. The two controls (A and B) exhibit low labeling in the epidermis while the hair follicles are highly labeled. In the K16 homozygote (C), however, the follicles show little mitotic activity while the epidermis is highly labeled. (D and E) Skin from 21-d-old wild-type control (D) and a K16 homozygote (E) stained with the anti-BrdU antibody. Note that the control shows very little labeling in both the epidermis and the telogen stage hair follicles. In contrast, phenotypic epidermis still features high mitotic activity and, in addition, the anagen stage hair follicles are now highly labeled. (F–H) Trunk skin from a 7-d-old chimera homozygote (F), a wild-type control (G), and a K16 homozygote (H) were stained with an anti-K17 antibody. K17 expression was restricted to the outer root sheath of hair follicles in the two controls (F and G). In the K16 homozygote, however, K17 expression was detected in all layers of the epidermis. Arrowheads, the dermal–epidermal junction. hf, hair follicle. Bar, 100 μm.
In addition to examining the mitotic activity in skin epithelia, immunohistochemistry was performed to detect K6 and K17. In addition to being rapidly induced in wound edge keratinocytes after injury to the epidermis (69), these keratins are commonly associated with hyperproliferation or altered differentiation in skin (79, 81, 90). K17, which is normally expressed in the outer root sheath of the hair follicle (55, 84), was detectable in all layers of the epidermis from the K16 homozygote (Fig. 6 H). This was in stark contrast to the normal K17 expression pattern that was observed for the two controls (Fig. 6, F and G). K6 was detected in the inner layer of the outer root sheath of hair follicles in both of the controls (data not shown). K6 could not be detected in the interfollicular epidermis. Phenotypic epidermis, on the other hand, featured the expression of K6 in the suprabasal layers (data not shown). The detection of mouse K16, which is presumably also induced in the hyperproliferative epidermis is not possible due to probable cross reactivity of the 1275 antibody (see Fig. 3 K).
Basal Keratinocytes of the Phenotypic Epidermis Feature Multiple Cytoarchitectural Anomalies
To finely characterize the cytoarchitectural changes in the keratinocytes of the phenotypic epidermis, ventral skin from 6-d-old littermates was examined using transmission electron microscopy. The control epidermis appears normal at low magnification (Fig. 7 A) All of the layers of the epidermis, from the basal cells to the stratum corneum, are clearly observed along with the distinctive features of each layer.
Figure 7.

Transmission electron microscopy of phenotypic transgenic mouse epidermis (No. 6 line). Ventral skin was isolated from 6-d-old wild-type control and K16 homozygous littermates and processed for electron microscopy. (A and C) Low magnification of epidermis. While all layers of the epidermis can be visualized in the control (A), only the basal and spinous layers can be seen in the phenotypic epidermis (C), underscoring the dramatic difference in thickness between the two. (B and D) Basal cells shown at higher magnification. (B) Control mouse epidermis. (D) Phenotypic K16 transgenic mouse epidermis shown at the same magnification. Note the elongated shape of the basal cells, the bundling of keratin filaments (kf), cytoplasmic areas devoid of keratin filaments (large asterisks), and the numerous gaps between cells (small asterisks). The inset shows electron dense inclusions in mitochondria (short arrows) near the nucleus of a suprabasal cell. Large arrowheads, the basal lamina. Small arrowheads, desmosomes. ba, basal layer; sp, stratum spinosum; sg, stratum granulosum; sc, stratum corneum; Nu, nucleus; m, mitochondria; kf, keratin filaments. Bars: (A and C) 10 μm; (B and D) 2 μm.
The normal cytoarchitectural features of control basal cells are clearly observed under higher magnification (Fig. 7 B). The basal cells have a rounded shape and are tightly packed together as there is no observable intercellular space. The keratin filaments are loosely packed together and are well distributed throughout the cell. There are multiple hemidesmosomal as well as desmosomal attachments.
The large difference in thickness between control and phenotypic epidermis is accentuated when observing phenotypic epidermis at the low magnification (Fig. 7 C). Whereas the entirety of the control epidermis was visible at this magnification (Fig. 7 A), only the basal and spinous layers could be seen in the phenotypic epidermis. The basal cells have an elongated shape and many gaps can be seen between cells at both the basal and suprabasal layers. The suprabasal cells have not reoriented their axis upon leaving the basal layer. In addition, some suprabasal cells featured bi-lobed nuclei, which has been observed in epidermolytic hyperkeratosis (23).
The cytoarchitectural differences are much more apparent when observed at the higher magnification (Fig. 7 D). The basal cells are very elongated, as if they were being pulled from above. Hemidesmosomal contacts appear normal although the basement membrane of the phenotypic skin often had an undulating characteristic. The filaments in the basal cells are packed into very electron-dense bundles. As a result of this, many areas of the cytoplasm in basal cells appear devoid of keratin filaments (Fig. 7 D, asterisks). The many gaps between basal cells are obvious and the number of desmosomes appear to be reduced. Another distinctive feature of the phenotypic epidermis was the presence of electron-dense inclusions in the mitochondria of suprabasal cells (Fig. 7 D, inset). These were also observed in basal cells although to a lesser extent. The presence of these mitochondrial inclusions has been previously described (82).
Transgenic Epidermis Has Multiple Defects in Cell–Cell Adhesion
The electron microscopy data indicated that there were large gaps between basal cells in the epidermis. In addition, the number of desmosomes was reduced providing a possible explanation for the decrease in cell adhesion. To better define the changes in cell–cell adhesion, immunofluorescence of frozen tissue sections was performed to examine the distribution of molecules involved in both actin and keratin-associated adhesion complexes.
Desmoplakin was examined as a marker of desmosomal adhesion. Detection was primarily at the surface of basal cells in the areas of cell–cell contact (Fig. 8 A). The intensity of staining increased on the surface of suprabasal cells coinciding with the maturation of desmosomes as keratinocytes differentiate (91). Cell–cell staining in the majority of basal and suprabasal cells from the phenotypic epidermis was reduced (Fig. 8 B) as would be expected from the EM data.
Figure 8.

Immunolocalization of cell adhesion molecules in the epidermis (No. 10 line). 8-μm frozen sections of skin from control (A, C, E, G, I, and K) and K16 homozygote littermates (B, D, F, H, J, and L) were used for the localization of cell adhesion molecules by indirect immunofluorescence. For the α6 integrin the HRP method was used. Sections were stained for Desmoplakin (A and B), E-cadherin (C and D), β-catenin (E and F), Connexin 26 (G and H), α3 integrin (I and J), and α6 integrin (K and L). Desmoplakin staining in the homozygote was not only reduced at regions of cell– cell contact but was also prevalent in the cytoplasm of cells (asterisks, compare B and A). E-Cadherin was absent from the basal layer of the homozygote (asterisks in D) and was more disorganized suprabasally compared with control (C). The results of the β-catenin staining were similar to those obtained with desmoplakin with the additional fact that staining seemed reduced in the basal layer of the homozygote (asterisks, compare F and E). Connexin 26, which is normally expressed in the hair follicle (arrow) and not the epidermis (G) in control skin, was dramatically induced suprabasally in the homozygote (H). Expression of α3 integrin extended suprabasally (asterisks) in homozygous epidermis (J) as opposed to its normal basal expression in control (I). α6 integrin expression appeared comparable between homozygous (L) and control (K) epidermis with the possible exception of increased lateral and apical staining of basal cells in the homozygote. Arrowheads, the dermal–epidermal junction. hf, hair follicle. Bars: (A–J) 50 μm; (K and L) 50 μm.
To examine adherens junctions, an antibody was used to detect E-cadherin. Normal epidermis featured E-cadherin expression in all layers of the epidermis at sites of cell–cell contact (Fig. 8 C; see reference 67). The expression of E-cadherin was essentially absent in the basal cells of phenotypic epidermis (Fig. 8 D). In addition, the organization of E-cadherin in suprabasal cells was disturbed as evidenced by its decreased localization at areas of cell–cell contact. It is interesting to note that in human skin diseases such as pemphigus and Darier's the expression of E-cadherin is markedly reduced on the surface of acantholytic cells (25).
This mislocalization and absence of E-cadherin suggested that other molecules involved in actin-based cell– cell adhesion might be perturbed. To test this the localization of β-catenin, a cytoplasmic protein involved in stabilizing adherens junctions, was determined. β-Catenin was detected in all layers of the epidermis at areas of cell–cell contact from control skin (Fig. 8 E). The localization of β-catenin in phenotypic epidermis was similar to that of E-cadherin (Fig. 8 F), in that it was reduced in some areas of the basal layer and was generally reduced at cell–cell interfaces in all layers of the phenotypic epidermis. β-Catenin was not detected in the nuclei of any cells in the epidermis. Similar results were obtained for plakoglobin, a cytoplasmic molecule that is part of adherens junctions and desmosomes (19) (data not shown), with the only difference being that it was also present in basal cells presumably because of stabilizing interactions with components of desmosomes.
Gap junctions, a type of cell–cell adhesion complex that metabolically couples adjacent cells, were examined in control and phenotypic skin. Connexin 26 is normally expressed in the outer root sheath of the hair follicle but not in the epidermis (76). However, wounding of the epidermis induces rapid expression of connexin 26 in suprabasal cells near the wound edge (26). Control skin featured connexin 26 expression only in the hair follicles (Fig. 8 G). Phenotypic epidermis, however, featured the strong expression of connexin 26 in all suprabasal layers of the epidermis (Fig. 8 H). This data suggests that metabolic coupling between keratinocytes may also be modified in the phenotypic epidermis.
Keratin- and actin-based cell–matrix adhesion was examined by localizing the α6 and α3 integrin subunits on frozen skin sections. The α6 integrin subunit, which is part of the hemidesmosome structure that attaches to keratin filaments and the basal lamina, is restricted to the basal and, less frequently, lateral sides of basal keratinocytes (10). This was the localization observed in epidermis from both the control and phenotypic animals (Fig. 8, K and L). The α3 integrin, a key component of the bridge between the basal lamina and actin filaments, was localized to the basal, lateral, and apical sides of basal cells in control epidermis (Fig. 8 I; see reference 10). This localization was similar in phenotypic epidermis except for certain areas where the expression extended suprabasally (Fig. 8 J).
Keratin Filaments Are Reorganized in Primary Keratinocyte Cultures from K16 Transgenic Mice
To visualize the entire keratin filament network in transgenic basal keratinocytes, newborn mice were killed and isolated keratinocytes were placed in culture and processed for immunofluorescence using a variety of antibodies against different keratins. When cultured cells from non-transgenic mice were stained to detect K16, ∼10% of the cells were positive for a keratin presumed to be mouse K16 (Fig. 9 A). When double stained with an antibody that recognizes the endogenous K14, the two immunofluorescence signals were found to coalign (Fig. 9 D). All cells from heterozygous transgenic mice were positive for the human K16 transgene (Fig. 9 B). The transgene colocalized with the endogenous keratin network as determined by double staining with an antibody to K14 (Fig. 9 E). No abnormalities were observed in the organization of the keratin filaments within these cells. Other keratins analyzed including K5, K6, and K17 were found to colocalize with the K16 transgene product (data not shown). No K10 staining was observed suggesting that the cells in culture are basal-like (data not shown). In contrast to wild type and heterozygotes, a subset of the homozygote keratinocytes when stained for the K16 transgene featured a severely disrupted keratin network in which the filaments had relocalized in a perinuclear fashion (Fig. 9 C). This was very reminiscent of the results obtained when K16 was transfected into Ptk2 cells (69) and when K16 was overexpressed in the suprabasal cells of trangenic mice (82). The endogenous network also featured this relocalization as determined by staining for K14 (Fig. 9 F). The type II keratins K5 and K6 also colocalized to these reorganized filaments (data not shown). The fact that this relocalization is only seen in the homozygous cells strongly argues that a certain amount of K16 is needed with respect to the other keratins to cause this effect.
Figure 9.
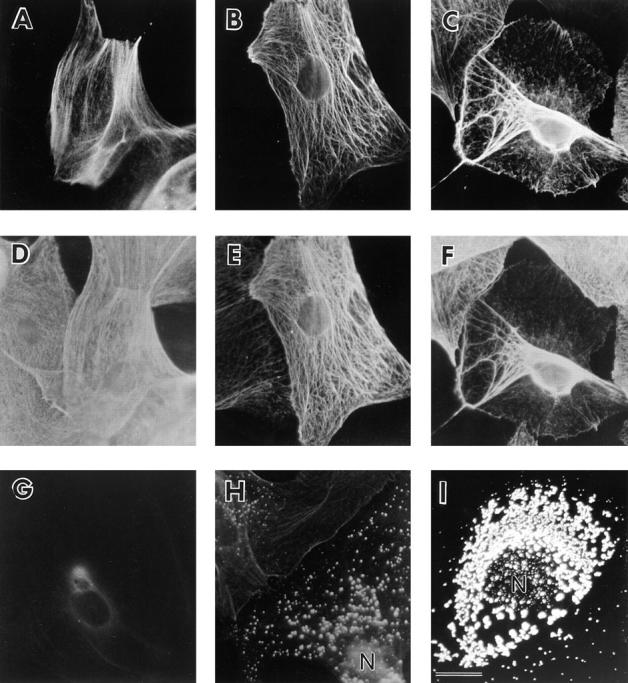
Immunofluorescent analysis of keratins in cultured primary mouse keratinocytes (No. 10 line). Keratinocytes were isolated from the epidermides of newborn control, K16 heterozygote, and K16 homozygote littermates, cultured, and processed for indirect, double-immunofluorescence to analyze the keratin filament networks. (A–C) Keratinocytes from wild-type control (A), heterozygote (B), and homozygote littermates (C) were stained with the 1275 antibody to detect K16. (D–F) These same cells were double stained with the LL001 antibody to detect K14. A low percentage of wild-type cells express a keratin that is recognized by the 1275 antibody (A). Transgene expression in the heterozygote sample was detectable in all cells (B) and colocalized with the endogenous network (E). A subset of homozygous cells (C) featured a network in which the bulk of keratin filaments were located near the nucleus. This included the endogenous keratins (F). (G–I) Wild-type (G), heterozygote (H), and homozygote (I) keratinocytes were stained with K8.12 antibody, which recognizes K16 in an aggregated form (82). A small subset of heterozygote cells (H) featured punctate staining distributed throughout the cytoplasm. In contrast, many homozygous cells as shown in I feature a much higher density of punctate staining near the nucleus. N, nucleus. Bar, 25 μm.
A possible mechanism to explain the relocalization of keratin filaments was provided by data using an antibody to K16 that preferentially recognizes an aggregated, non-filamentous form of the protein in cultured epithelial cells (82). Whereas wild-type keratinocytes were negative using this antibody (Fig. 9 G), a subset of keratinocytes from heterozygous K16 mice showed a punctate pattern of staining with this antibody (Fig. 9 H). These aggregates were distributed throughout the cytoplasm and were not preferentially located near the nucleus. The keratin filament networks of these cells appeared normal when stained with the K14 antibody (data not shown). The percentage of cells with the punctate staining pattern was significantly higher in keratinocytes derived from homozygous K16 mice. In addition, a subset of cells showed a much higher density of staining near the nucleus, with a concomitant decrease in cytoplasmic staining (Fig. 9 I). When these were double stained for K14, the keratin network was found to be reorganized around the nucleus (data not shown). These data suggest a relocalization mechanism in which the first step is K16 integration throughout the keratin filament network of the cell. At certain areas (perhaps resulting from high K16 amounts), the filaments form micro-aggregates. These micro-aggregates may then subsequently relocalize to a perinuclear location.
Tyrosine Phosphorylation of the EGFR in K16 Transgenic Mice
The general morphology of the skin and the temporal nature of the phenotype in the K16 transgenic mice suggested that an activated EGF receptor might be involved in generating the phenotype (see Discussion for details). To test this, skin from newborn wild-type, heterozygous, and homozygous littermates was obtained and the amount and the tyrosine phosphorylation status of the receptor was determined by Western blot analysis. EGF was subcutaneously injected into a control littermate to generate tyrosine-phosphorylated receptor to serve as a reference (Fig. 10, control + EGF).
Figure 10.

Determination of EGFR levels and tyrosine phosphorylation in the K16 transgenic mice (No. 6 line). Equivalent amounts (∼30 μg) of newborn skin lysates from control, heterozygous, and homozygous K16 transgenic littermates were electrophoresed via SDS-PAGE and transferred to nitrocellulose for Western blot analysis (in duplicate). Top blot, the four samples were probed with an antibody specific for the EGFR. Approximately equivalent amounts of the receptor were present in each of the samples as determined by densitometry. Bottom blot, the samples were probed with an antibody specific for phosphotyrosine. The control + EGF sample featured a strong band migrating at the same apparent molecular weight as the EGFR. The control and heterozygous samples featured faint reactivity. The homozygous sample is increased twofold with respect to the non-stimulated controls.
When the lysates were probed with an anti-EGFR antibody, approximately equivalent amounts of the ∼170-kD receptor was detected in each of the samples as determined by densitometry (Fig. 10, top blot). When the control + EGF sample was probed with an anti-PTyr antibody a large band of the same apparent molecular weight was present suggesting that the EGF receptor was highly tyrosine phosphorylated in response to the EGF treatment (Fig. 10, bottom blot). In contrast, control and heterozygous samples featured faint reactivity of the receptor. The homozygous sample, however, was increased twofold with respect to the unstimulated controls as determined by densitometry. It is relevant to note that the newborn skin of the K16 transgenic homozygous mice has a normal morphology with respect to control skin. However, 6-d-old phenotypic skin did not show increased EGFR tyrosine phosphorylation when compared with control skin (data not shown). These data suggest that the activation of the EGFR may be the result of K16 expression in basal cells and that this activation may contribute to the observed phenotype.
Discussion
The Ectopic Expression of K16 in Transgenic Mouse Skin Delays Skin Development
The expression of K16 protein to a certain level with respect to the endogenous type I keratins in basal cells resulted in multiple changes in both the development, differentiation, and maturation of transgenic mouse skin. The thickened and hyperproliferative epidermis of the phenotypic mice is first observed 2 d after birth (data not shown). During the first week of postnatal growth, the phenotypic epidermis features the aberrant expression of multiple markers of keratinocyte localization and differentiation. In addition, the hair follicles are hypoproliferative and reduced in number compared with control animals. Furthermore, many of the follicles are grossly disoriented and misaligned in the hypodermis. By the end of the first hair cycle in mice at ∼3 wk of age (5, 20), the phenotypic mice are essentially hairless. However, as opposed to control mice, at day 21 the phenotypic hair follicles are now proliferative and in the anagen phase.
Beginning at ∼4–5 wk after birth the phenotypic mice begin to grow hair and their epidermis becomes thinner. There is an apparent normalization of the skin phenotype by this time. However, by the end of the second hair cycle at ∼6 wk (5, 20), hair follicles from phenotypic mice are still in the anagen phase as opposed to control follicles that are in the resting phase (data not shown). Although shorter, curved hairs are being produced, the staging of the follicles is still aberrant. In short, the normal pattern of embryonic and postnatal development is severely disrupted in transgenic skin until ∼5 wk after birth. Whereas other epidermal keratins have been ectopically expressed in different epithelia and have impaired both the structure and function of the targeted tissue, they had no apparent effect on the development of the transgenic epithelia (e.g., references 1, 4). This is the first example of a keratin being able to influence the morphogenesis of a complex epithelium. It appears likely that the use of the K14 promoter, which is active in the progenitor cells of the skin starting at a very early stage of development (9), is a major factor that contributes to the phenotype. This is in contrast to our previous study involving increased expression of K16 under the control of its own promoter (82) in which there was no impairment of skin maturation.
Why does the expression of wild-type keratin 16, but not the K16-C14 chimera, in basal keratinocytes lead to a phenotype that is temporally regulated? Clearly, transgenic basal keratinocytes expressing K16 are responding differently to the signals that participate in the normal development of newborn skin. It would follow then that signal transduction in the transgenic basal keratinocytes may deviate from that of the normal basal keratinocytes. These differences could lie in the sensing, interpreting, and execution of signals or it could lie in the actual composition of molecules involved. The exact details of these possible differences are of great interest as they could reveal the molecular mechanisms by which keratinocytes may be able to reprogram themselves as a function of keratin gene expression. While there may appear to be a countless number of possibilities, there is ample evidence to suggest that the EGF receptor pathway may be involved in generating the phenotype seen in the K16 transgenic mice.
The EGF Receptor and Skin Development
EGF was originally described as a protein that could promote proliferation and keratinization of the epidermis while inhibiting hair growth when administered to newborn mice (13, 46). In studies that extended these observations, the administration of EGF to newborn mice resulted in thinner, curved hairs that grew slower (61). Furthermore, EGF treatment delayed the development of the hair follicles during the growth of the first coat while eliciting hyperproliferation in the epidermis (61, 62). The effects of the treatments diminished with the increasing age of the mice.
The EGFR is expressed in basal cells of the epidermis as well as in the outer root sheath of hair follicles (28, 29, 32). Expression levels in the epidermis are highest at birth and by 4 wk have been reduced ∼10-fold (29). Interestingly, basal cells overlying dermal condensates (placodes) that mark the first stage of hair follicle development are devoid of EGFR (65). Furthermore, EGFR staining is essentially absent in hair follicles until ∼5 d after birth (32, 65).
Recently, many genetically manipulated mice have been created that have further delineated the role of the EGFR and its ligands in the development of skin. When TGFα, an autocrine growth factor of epidermal cells, was overexpressed in basal keratinocytes and the outer root sheath of hair follicles, the resulting mice had a scaly and thickened epidermis that was hyperproliferative (86). Hair growth was stunted and hair follicles were misaligned. After 5 wk, the scaliness disappeared and hair growth was partially restored. In contrast, when the TGFα gene was inactivated by homologous recombination, the mice had wavy hair and misaligned hair follicles (50, 51). The epidermis, however, was unaffected and increasing age reduced the severity of the hair phenotype.
Inactivation of the EGFR by homologous recombination (59, 78, 83) and the targeting of a dominant-negative EGFR to the basal layer and outer root sheath of the epidermis (63) have further emphasized the role of the receptor in hair development. In each of these studies hair follicles were severely misoriented and crowded and hair growth was significantly delayed. Although the epidermis of the dominant-negative EGFR mouse was hyperproliferative (63), the epidermides from the EGFR-deficient mice were thinner and hypoproliferative (59, 78, 83).
Collectively, these in vivo data suggest a prominent role for the EGFR in the development of the epidermis and the hair follicles. More specifically, they point to the EGFR as having the role of promoting proliferation in the epidermis while delaying the differentiation of hair follicles in normal skin (32, 62). When comparing this body of data with the phenotype of the K16 transgenic mice, the parallels are striking. The distinguishing features of the K16 transgenic mice including the hyperproliferative epidermis, the problems of follicle orientation and morphogenesis, and the delay in hair growth as well as the shape of the mature hairs strongly argue that an activation of the EGFR pathway may be involved in generating the phenotype.
In newborn homozygous mice, the amount of the EGFR in the skin was comparable to the control littermates. However, the tyrosine phosphorylation of the receptor was twofold that of the controls. When the same analysis was performed on 6-d-old phenotypic mice and control littermates there was no difference in either the levels or the tyrosine phosphorylation status of the receptor. Phenotypic changes in the skin of homozygous mice are first noticeable in 2-d-old mice while newborn skin appears normal. Based on these data, activation of the EGFR precedes the phenotypic changes observed in the skin. The next step will be to define the molecular mechanism by which K16-expressing basal keratinocytes activate the EGFR signaling pathway. This may be difficult to determine as the EGFR signaling pathway is quite complex (34). The second messenger molecules that participate in the pathway are also shared by other tyrosine kinase receptors and the cytokine receptors (34). Further complicating matters is the presence of multiple ligands in the skin for the EGFR (78). Whatever the possible mechanism may be, its definition will be very interesting as the EGFR is upregulated in wound edge keratinocytes (80) and is expected to play a prominent role in the wound healing process (7, 8, 64, 77).
Parallels and Relevance to Wound Healing
When the skin is wounded, suprabasal keratinocytes located at the edges of the wound undergo a process called keratinocyte activation (14, 30). Essentially, these keratinocytes do not proceed along the normal differentiation pathway but instead are directed to participate in the re-epithelialization of the wound. One of the earliest detectable events to occur is the expression of K16 protein in these keratinocytes at 4–6 h after injury (69). In addition, K6 and K17 are also induced while the synthesis of the differentiation specific keratins, K1 and K10, is reduced (52, 55, 69). As the activation process occurs, the keratin filaments are redistributed behind the nucleus as the cell attains a migratory morphology. As this happens, multiple changes occur, which result in the dramatic decrease in cell adhesion among the keratinocytes. The number of desmosomal attachments between cells decreases (12, 69). In addition, the surface levels of E-cadherin are reduced (31) and the expression of connexin 26 is induced (26). Furthermore, there is the induction of the α5β1 integrin and the suprabasal localization of the endogenous integrin subunits (36). Together these molecular changes result in hypertrophic suprabasal keratinocytes that are loosened from each other and are now able to migrate into the wound site.
Many of these same changes are observed in the phenotypic transgenic basal keratinocytes. K6 and K17 were expressed. The EM data revealed that the hypertrophic keratinocytes had fewer desmosomes, bundled filaments, and increased space between them. E-Cadherin was essentially absent in the basal layer and Connexin 26 was induced suprabasally. The α3 integrin was also found in the suprabasal layers. It is interesting to note that the expression of K16 in the basal layer, as opposed to its normal expression suprabasally, also results in keratinocytes that are quite similar to activated suprabasal keratinocytes. This would imply that K16 expression, along with K6 and K17 has the potential to drive a keratinocyte into the activated state. In this case, the result is a hyperproliferative epidermis where the balance between proliferation and differentiation has been disturbed.
The fact that K16 expression can cause a relocation of keratin filaments from the periphery to near the nucleus of keratinocytes provides a potential explanation for some of the changes in cell–cell adhesion. The type II epidermal keratins K5 and K6 both colocalize with K16 during this process. In addition desmoplakin, a key component of desmosomes, also localizes to the redistributed filaments (data not shown). K5 and K6 both have the ability to interact with the tail domain of desmoplakin whereas the type I epidermal keratins do not (41, 58). By interacting with K5 and K6 in the context of filaments, K16 may indirectly affect the localization of desmoplakin and the stability of desmosomes.
The assembly of desmosomes is dependent on the prior formation of adherens junctions (2, 27, 47). The fact that E-cadherin is dramatically decreased in the basal layer of the phenotypic epidermis suggests that this may also contribute to the decreased number of desmosomes. It is interesting to note that E-cadherin and the EGFR colocalize in epithelial cells (24). Furthermore, the EGFR associates with the cadherin–catenin complex and phosphorylates β-catenin and plakoglobin when stimulated by EGF (38). It could be that some of the aforementioned events are required for keratinocyte activation.
Role of K16's Tail Domain
The fact that K16 but not the K16-C14 chimeric protein causes dramatic changes to occur in keratinocytes when expressed at comparable levels implicates the last ∼105 amino acids of K16 as being the sequence responsible for generating the phenotype. The first ∼60 amino acids are located in the 2B rod domain. Of these ∼60 amino acids, there are only five differences between K14 and K16. Three of these five are conservative substitutions. The remaining ∼45 amino acids constitute the tail domain. The sequence identity remains high along the first half of the tail but drops dramatically along the second half of the tail domain. There are many serines in this region of K16 and the DGK(L/V)VS(T/E) motif, which is conserved in several type I sequences from diverse species is absent (45 and references therein).
The exact role of the tail domains of intermediate filaments is still a subject of debate. Many studies have pointed to the tail domain as being important in regulating the lateral aggregation of 10-nm filaments (33, 35, 40). Other studies have implicated the tail domain as being required for the stabilization of filaments and for the proper organization of intermediate filament networks in vivo (49, 53, 92). In addition, the absence of the tail domain of Xenopus vimentin resulted in the nuclear accumulation of the protein (72). The tail domains are also substrates for posttranslational modifications (42) and are prime candidates for possible interactions with other cellular proteins.
The fact that keratin filaments of the transgenic basal keratinocytes are thickly bundled and often show a perinuclear location argue strongly that it is the tail domain of K16 that is responsible for these changes in cellular localization and lateral packing. The mechanism by which this may occur is an active area of our investigations. As mentioned above, the lack of the DGK(L/V)VS(T/E) sequence and the presence of multiple serines may be important clues that could help in leading to a molecular understanding of these events.
Our results strongly suggest that a certain amount of K16 needs to be expressed to cause dramatic changes in keratinocytes. Furthermore, the data argue that it is the tail domain of K16 that causes these changes. The results also suggest that the location of K16 expression in the epidermis is very important in determining the subsequent phenotypic changes since a different phenotype was observed in transgenic mice overexpressing human K16 off of its own promoter (81). This argues that the effects of K16 on keratinocytes may be highly dependent on the specific keratin composition and the resulting filament network of the keratinocyte. Finally, our results provide further support for the idea that a primary function of K16 is to modulate the pre-existing keratin filament network of keratinocytes in addition to providing structural support. The replacement mice that we are generating in which K16 is expressed in the absence of K14 in basal keratinocytes will provide answers to many of these questions.
Footnotes
Our thanks to Ms. S. Brust and Ms. A. Chen (Johns Hopkins University, Transgenic Core Facility) for the production of transgenic mice. Special thanks are due to Dr. E. Fuchs and Dr. T. Tanaka for providing the K14 expression cassette. We also thank Dr. M. Takaechi (E-cadherin), Dr. R. Hynes (α3 and β1 integrins), Dr. F. Giancotti (α6 and β4 integrins), Dr. D. Paul (connexin 26), Dr. B. Dale (filaggrin), Dr. I. Leigh (keratins 14 and 16), Dr. K. Green (desmoplakin), and Dr. E. Fuchs (keratin 5) for providing antibodies. Dr. L. Hansen provided valuable help for the EGFR Western experiments. Thanks also to Dr. J. Porter and Dr. K. McGowan for their help.
P.A. Coulombe is the recipient of a Junior Faculty Research Award from the American Cancer Society. This work was supported by National Institutes of Health grant AR44232.
Address all correspondence to Pierre A. Coulombe, Ph.D., Dept. of Biological Chemistry, Johns Hopkins University School of Medicine, 725 N. Wolfe Street, Baltimore, MD 21205. Tel.: (410) 614-0510. Fax: (410) 955-5759. E-mail: coulombe@jhu.edu
1. Abbreviation used in this paper: EGFR, EGF receptor.
References
- 1.Albers KM, Davis FE, Perrone TN, Lee EY, Liu Y, Vore M. Expression of an epidermal keratin protein in liver of transgenic mice causes structural and functional abnormalities. J Cell Biol. 1995;128:157–169. doi: 10.1083/jcb.128.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amagai M, Fujimori T, Masunaga T, Shimizu H, Nishikawa T, Shimizu N, Takeichi M, Hashimoto T. Delayed assembly of desmosomes in keratinocytes with disrupted classic-cadherin-mediated cell adhesion by a dominant negative mutant. J Invest Dermatol. 1995;104:27–32. doi: 10.1111/1523-1747.ep12613462. [DOI] [PubMed] [Google Scholar]
- 3.Bereiter-Hahn, J. 1986. Epidermal cell migration and wound repair. In Biology of the Integument, Vol. 2: Vertebrates. J. Bereiter-Hahn, A.G. Matoltsy, and K.S. Richards, editors. Springer-Verlag, Berlin. 443–471.
- 4.Blessing M, Ruther U, Franke WW. Ectopic synthesis of epidermal cytokeratins in pancreatic islet cells of transgenic mice interferes with cytoskeletal order and insulin production. J Cell Biol. 1993;120:743–755. doi: 10.1083/jcb.120.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borum K. Hair patterns and hair succession in the albino mouse. Acta Pathol Microbiol Scand. 1954;34:521–541. doi: 10.1111/j.1699-0463.1954.tb00299.x. [DOI] [PubMed] [Google Scholar]
- 6.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 7.Brown GL, Curtsinger LD, Brightwell JR, Ackerman DM, Tobin GR, Polk HC, Jr, George-Nascimento C, Valenzuela P, Schultz GS. Enhancement of epidermal regeneration by biosynthetic epidermal growth factor. J Exp Med. 1986;163:1319–1324. doi: 10.1084/jem.163.5.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown GL, Nanney LB, Griffen J, Cramer AB, Yancey JM, Curtsinger LJD, Holtzin L, Schultz GS, Jurkiewicz MJ, Lynch JB. Enhancement of wound healing by topical treatment with epidermal growth factor. N Engl J Med. 1989;321:76–79. doi: 10.1056/NEJM198907133210203. [DOI] [PubMed] [Google Scholar]
- 9.Byrne C, Tainsky M, Fuchs E. Programming gene expression in developing epidermis. Development (Camb) 1994;120:2369–2383. doi: 10.1242/dev.120.9.2369. [DOI] [PubMed] [Google Scholar]
- 10.Carter WG, Kaur P, Gil SG, Gahr PJ, Wayner EA. Distinct functions for integrins α3 β1 in focal adhesions and α6 β4/bullous pemphigoid antigen in a new stable anchoring contact (SAC) of keratinocytes: Relation to hemidesmosomes. J Cell Biol. 1990;111:3141–3154. doi: 10.1083/jcb.111.6.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christophers, E., and W. Sterry. 1993. Psoriasis. In Dermatology in General Medicine. Vol. I. T.B. Fitzpatrick, A.Z. Eisen, K. Wolff, I.M. Freedberg, and K.F. Austen, editors. McGraw-Hill Inc., New York. 489–515.
- 12.Clark, R.A.F. 1993. Mechanisms of cutaneous wound repair. In Dermatology in General Medicine. Vol. I. T.B. Fitzpatrick, A.Z. Eisen, K. Wolff, I.M. Freedberg, and K.F. Austen, editors. McGraw-Hill, New York. 473– 486.
- 13.Cohen S, Elliot GA. The stimulation of epidermal keratinization by a protein isolated from the submaxillary gland of the mouse. J Ivest Dermatol. 1963;40:1–5. doi: 10.1038/jid.1963.1. [DOI] [PubMed] [Google Scholar]
- 14.Coulombe PA. Towards a molecular definition of keratinocyte activation after acute injury to stratified epithelia. Biochem Biophys Res Comm. 1997;236:231–238. doi: 10.1006/bbrc.1997.6945. [DOI] [PubMed] [Google Scholar]
- 15.Coulombe, P.A., and E. Fuchs. 1994. Molecular mechanisms of keratin gene disorders and other bullous diseases of the skin. In Molecular Mechanisms in Epithelial Cell Junctions: From Development to Disease. S. Citi, editor. R.G. Landes Co., Austin, TX. 259–285.
- 16.Coulombe PA, Kopan R, Fuchs E. Expression of keratin K14 in the epidermis and hair follicle: insights into complex programs of differentiation. J Cell Biol. 1989;109:2295–2312. doi: 10.1083/jcb.109.5.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coulombe PA, Hutton ME, Vassar R, Fuchs E. A function for keratins and a common thread among different types of epidermolysis bullosa simplex diseases. J Cell Biol. 1991;115:1661–1674. doi: 10.1083/jcb.115.6.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coulombe PA, Bravo NS, Paladini RD, Nguyen D, Takahashi K. Overexpression of human keratin 16 produces a distinct phenotype in transgenic mouse skin. Biochem Cell Biol. 1995;73:611–618. doi: 10.1139/o95-067. [DOI] [PubMed] [Google Scholar]
- 19.Cowin P, Kapprell HP, Franke WW, Tamkun J, Hynes RO. Plakoglobin: a protein common to different kinds of intercellular adhering junctions. Cell. 1986;46:1063–1073. doi: 10.1016/0092-8674(86)90706-3. [DOI] [PubMed] [Google Scholar]
- 20.Dry F. Coat of the mouse (mus musculus) J Genet. 1926;16:287–340. [Google Scholar]
- 21.Fuchs E. Epidermal differentiation and keratin gene expression. J Cell Sci Suppl. 1993;17:197–208. doi: 10.1242/jcs.1993.supplement_17.28. [DOI] [PubMed] [Google Scholar]
- 22.Fuchs E, Green H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell. 1980;19:1033–1042. doi: 10.1016/0092-8674(80)90094-x. [DOI] [PubMed] [Google Scholar]
- 23.Fuchs E, Esteves RA, Coulombe PA. Transgenic mice expressing a mutant keratin 10 gene reveal the likely genetic basis for epidermolytic hyperkeratosis. Proc Natl Acad Sci USA. 1992;89:6906–6910. doi: 10.1073/pnas.89.15.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukuyama R, Shimizu N. Detection of epidermal growth factor receptors and E-cadherins in the basolateral membrane of A431 cells by laser scanning fluorescence microscopy. Jpn J Cancer Res. 1991;82:8–11. doi: 10.1111/j.1349-7006.1991.tb01737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furukawa F, Takigawa M, Matsuyoshi N, Shirahama S, Wakita H, Fujita M, Horiguchi Y, Imamura S. Cadherins in cutaneous biology. J Dermatol. 1994;21:802–813. doi: 10.1111/j.1346-8138.1994.tb03294.x. [DOI] [PubMed] [Google Scholar]
- 26.Goliger JA, Paul DL. Wounding alters epidermal connexin expression and gap junction-mediated intercellular communication. Mol Biol Cell. 1995;6:1491–1501. doi: 10.1091/mbc.6.11.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green KJ, Geiger B, Jones JC, Talian JC, Goldman RD. The relationship between intermediate filaments and microfilaments before and during the formation of desmosomes and adherens-type junctions in mouse epidermal keratinocytes. J Cell Biol. 1987;104:1389–1402. doi: 10.1083/jcb.104.5.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green MR, Couchman JR. Distribution of epidermal growth factor receptors in rat tissues during embryonic skin development, hair formation, and the adult hair growth cycle. J Invest Dermatol. 1984;83:118–123. doi: 10.1111/1523-1747.ep12263298. [DOI] [PubMed] [Google Scholar]
- 29.Green MR, Basketter DA, Couchman JR, Rees DA. Distribution and number of epidermal growth factor receptors in skin is related to epithelial cell growth. Dev Biol. 1983;100:506–512. doi: 10.1016/0012-1606(83)90243-9. [DOI] [PubMed] [Google Scholar]
- 30.Grinnell F. Wound repair, keratinocyte activation and integrin modulation. J Cell Sci. 1992;101:1–5. doi: 10.1242/jcs.101.1.1. [DOI] [PubMed] [Google Scholar]
- 31.Grzesiak JJ, Pierschbacher MD. Changes in the concentrations of extracellular Mg++ and Ca++down-regulate E-cadherin and up-regulate α2 β1 integrin function, activating keratinocyte migration on type I collagen. J Invest Dermatol. 1995;104:768–774. doi: 10.1111/1523-1747.ep12606983. [DOI] [PubMed] [Google Scholar]
- 32.Hansen LA, Alexander N, Hogan ME, Sundberg JP, Dlugosz A, Threadgill DW, Magnuson T, Yuspa SH. Genetically null mice reveal a central role for epidermal growth factor receptor in the differentiation of the hair follicle and normal hair development. Am J Pathol. 1997;150:1959–1975. [PMC free article] [PubMed] [Google Scholar]
- 33.Heins S, Wong PC, Muller S, Goldie K, Cleveland DW, Aebi U. The rod domain of NF-L determines neurofilament architecture, whereas the end domains specify filament assembly and network formation. J Cell Biol. 1993;123:1517–1533. doi: 10.1083/jcb.123.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heldin C-H. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–224. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- 35.Herrmann H, Haner M, Brettel M, Muller SA, Goldie KN, Fedtke B, Lustig A, Franke WW, Aebi U. Structure and assembly properties of the intermediate filament protein vimentin: The role of its head, rod and tail domains. J Mol Biol. 1996;264:933–953. doi: 10.1006/jmbi.1996.0688. [DOI] [PubMed] [Google Scholar]
- 36.Hertle MD, Kubler MD, Leigh IM, Watt FM. Aberrant integrin expression during epidermal wound healing and in psoriatic epidermis. J Clin Invest. 1992;89:1892–1901. doi: 10.1172/JCI115794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hogan, B., R. Beddington, F. Costantini, and E. Lacy. 1994. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 497 pp.
- 38.Hoschuetzky H, Aberle H, Kemler R. β-Catenin mediates the interaction of the cadherin–catenin complex with epidermal growth factor receptor. J Cell Biol. 1994;127:1375–1380. doi: 10.1083/jcb.127.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korge BP, Krieg T. The molecular basis for inherited bullous diseases. J Mol Med. 1996;74:59–70. doi: 10.1007/BF00196781. [DOI] [PubMed] [Google Scholar]
- 40.Kouklis PD, Hatzfeld M, Brunkener M, Weber K, Georgatos SD. In vitro assembly properties of vimentin mutagenized at the β-site tail motif. J Cell Sci. 1993;129:1049–1060. doi: 10.1242/jcs.106.3.919. [DOI] [PubMed] [Google Scholar]
- 41.Kouklis PD, Hutton E, Fuchs E. Making a connection: Direct binding between keratin intermediate filaments and desmosomal proteins. J Cell Biol. 1994;127:1049–1060. doi: 10.1083/jcb.127.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ku N-O, Liao J, Chou C-F, Omary MB. Implications of intermediate filament phosphorylation. Cancer Metastasis Rev. 1996;15:429–444. doi: 10.1007/BF00054011. [DOI] [PubMed] [Google Scholar]
- 43.Leigh IM, Navsaria H, Purkis PE, McKay IA, Bowden PE, Riddle PN. Keratins (K16 and K17) as markers of keratinocyte hyperproliferation in psoriasis in vivo and in vitro. Br J Dermatol. 1995;133:501–511. doi: 10.1111/j.1365-2133.1995.tb02696.x. [DOI] [PubMed] [Google Scholar]
- 44.Lersch R, Stellmach V, Stocks C, Giudice G, Fuchs E. Isolation, sequence, and expression of a human keratin K5 gene: transcriptional regulation of keratins and insights into pairwise control. Mol Cell Biol. 1989;9:3685–3697. doi: 10.1128/mcb.9.9.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leube RE, Bader BL, Bosch FX, Zimbelmann R, Achtstaetter T, Franke WW. Molecular characterization and expression of the stratification-related cytokeratins 4 and 15. J Cell Biol. 1988;106:1249–1261. doi: 10.1083/jcb.106.4.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levi-Montalcini R, Cohen S. Effects of the extract of the mouse submaxillary salivary glands on the sympathetic system of mammals. Ann NY Acad Sci. 1960;85:324–341. doi: 10.1111/j.1749-6632.1960.tb49963.x. [DOI] [PubMed] [Google Scholar]
- 47.Lewis JE, Wahl JKR, Sass KM, Jensen PJ, Johnson KR, Wheelock MJ. Cross-talk between adherens junctions and desmosomes depends on plakoglobin. J Cell Biol. 1997;136:919–934. doi: 10.1083/jcb.136.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lloyd C, Yu QC, Cheng J, Turksen K, Degenstein L, Hutton E, Fuchs E. The basal keratin network of stratified squamous epithelia: Defining K15 function in the absence of K14. J Cell Biol. 1995;129:1329–1344. doi: 10.1083/jcb.129.5.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu X, Lane EB. Retrovirus-mediated transgenic keratin expression in cultured fibroblasts: specific domain functions in keratin stabilization and filament formation. Cell. 1990;62:681–696. doi: 10.1016/0092-8674(90)90114-t. [DOI] [PubMed] [Google Scholar]
- 50.Luetteke NC, Qiu TH, Peiffer RL, Oliver P, Smithies O, Lee DC. TGF α deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell. 1993;73:263–278. doi: 10.1016/0092-8674(93)90228-i. [DOI] [PubMed] [Google Scholar]
- 51.Mann GB, Fowler KJ, Gabriel A, Nice EC, Williams RL, Dunn AR. Mice with a null mutation of the TGF alpha gene have abnormal skin architecture, wavy hair, and curly whiskers and often develop corneal inflammation. Cell. 1993;73:249–61. doi: 10.1016/0092-8674(93)90227-h. [DOI] [PubMed] [Google Scholar]
- 52.Mansbridge JN, Knapp AM. Changes in keratinocyte maturation during wound healing. J Invest Dermatol. 1987;89:253–263. doi: 10.1111/1523-1747.ep12471216. [DOI] [PubMed] [Google Scholar]
- 53.McCormick MB, Kouklis P, Syder A, Fuchs E. The roles of the rod end and the tail in vimentin IF assembly and IF network formation. J Cell Biol. 1993;122:395–407. doi: 10.1083/jcb.122.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deleted in proof.
- 55.McGowan K, Coulombe PA. The wound repair-associated keratins K6, K16 and K17: Insights into the role of intermediate filaments in specifying keratinocyte cytoarchitecture. Subcell Biochem. 1998;31:173–204. [PubMed] [Google Scholar]
- 56.McLean WHI, Lane EB. Intermediate filaments in diseases. Curr Opin Cell Biol. 1995;7:118–125. doi: 10.1016/0955-0674(95)80053-0. [DOI] [PubMed] [Google Scholar]
- 57.McLean WHI, Rugg EL, Lunny DP, Morley SM, Lane EB, Swensson O, Dopping-Hepenstal PJC, Griffiths WAD, Eady RAJ, Higgins C, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet. 1995;9:273–278. doi: 10.1038/ng0395-273. [DOI] [PubMed] [Google Scholar]
- 58.Meng JJ, Bornslaeger EA, Green KJ, Steinert PM, Ip W. Two-hybrid analysis reveals fundamental differences in direct interactions between desmoplakin and cell type-specific intermediate filaments. J Biol Chem. 1997;272:21495–21503. doi: 10.1074/jbc.272.34.21495. [DOI] [PubMed] [Google Scholar]
- 59.Miettinen PJ, Berger JE, Meneses J, Phung Y, Pederssen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking the epidermal growth factor receptor. Nature. 1995;376:337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- 60.Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31:11–24. doi: 10.1016/0092-8674(82)90400-7. [DOI] [PubMed] [Google Scholar]
- 61.Moore GP, Panaretto BA, Robertson D. Effects of epidermal growth factor on hair growth in the mouse. J Endocrinol. 1981;88:293–299. doi: 10.1677/joe.0.0880293. [DOI] [PubMed] [Google Scholar]
- 62.Moore GPM, Panaretto BA, Robertson D. Epidermal growth factor delays the development of the epidermis and hair follicles of mice during growth of the first coat. Anat Rec. 1983;205:47–55. doi: 10.1002/ar.1092050107. [DOI] [PubMed] [Google Scholar]
- 63.Murillas R, Larcher F, Conti CJ, Santos M, Ullrich A, Jorcano JL. Expression of a dominant negative mutant of epidermal growth factor receptor in the epidermis of transgenic mice elicits striking alterations in hair follicle development and skin structure. EMBO (Eur Mol Biol Organ) J. 1995;14:5216–5223. doi: 10.1002/j.1460-2075.1995.tb00206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nanney LB. Epidermal and dermal effects of epidermal growth factor during wound repair. J Invest Dermatol. 1990;94:624–629. doi: 10.1111/1523-1747.ep12876204. [DOI] [PubMed] [Google Scholar]
- 65.Nanney LB, Magid M, Stoscheck CM, King LE., Jr Comparison of epidermal growth factor binding and receptor distribution in normal human epidermis and epidermal appendages. J Invest Dermatol. 1984;83:385–393. doi: 10.1111/1523-1747.ep12264708. [DOI] [PubMed] [Google Scholar]
- 66.Nelson WG, Sun TT. The 50- and 58-kdalton keratin classes as molecular markers for stratified squamous epithelia: Cell culture studies. J Cell Biol. 1983;97:244–251. doi: 10.1083/jcb.97.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nose A, Takeichi M. A novel cadherin cell adhesion molecule: Its expression patterns associated with implantation and organogenesis of mouse embryos. J Cell Biol. 1986;103:2649–2658. doi: 10.1083/jcb.103.6.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O'Guin, W.M., A. Schermer, M. Lynch, and T.-T. Sun. 1990. Differentiation-specific expression of keratin pairs. In Cellular and Molecular Biology of Intermediate Filaments. R.D. Goldman, P.M. Steinert, editors. Plenum Publishing Corp., London. 301–334.
- 69.Paladini RD, Takahashi K, Bravo NS, Coulombe PA. Onset of re-epithelialization after skin injury correlates with a reorganization of keratin filaments in wound edge keratinocytes: Defining a potential role for K16. J Cell Biol. 1996;132:381–397. doi: 10.1083/jcb.132.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Porter RM, Leitgeb S, Melton DM, Swensson O, Eady RAJ, Magin TM. Gene targeting at the mouse keratin 10 locus: Severe skin fragility and changes in cytokeratin expression in the epidermis. J Cell Biol. 1996;132:925–936. doi: 10.1083/jcb.132.5.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Purkis PE, Steel JB, Mackenzie IC, Nathrath WB, Leigh IM, Lane EB. Antibody markers of basal cells in complex epithelia. J Cell Sci. 1990;97:39–50. doi: 10.1242/jcs.97.1.39. [DOI] [PubMed] [Google Scholar]
- 72.Rogers KR, Eckelt A, Nimmrich V, Janssen KP, Schliwa M, Herrmann H, Franke WW. Truncation mutagenesis of the non-α-helical carboxyterminal tail domain of vimentin reveals contributions to cellular localization but not to filament assembly. Eur J Cell Biol. 1995;66:136–150. [PubMed] [Google Scholar]
- 73.Rothnagel JA, Greenhalgh DA, Wang XJ, Sellheyer K, Bickenbach JR, Dominey AM, Roop DR. Transgenic models of skin diseases. Arch Dermatol. 1993;129:1430–1436. [PubMed] [Google Scholar]
- 74.Rouabhia M, Germain L, Belanger F, Guignard R, Auger FA. Optimization of murine keratinocyte culture for the production of graftable epidermal sheets. J Dermatol. 1992;19:325–334. doi: 10.1111/j.1346-8138.1992.tb03234.x. [DOI] [PubMed] [Google Scholar]
- 75.Saitou M, Sugai S, Tanaka T, Shimouchi K, Fuchs E, Narumiya S, Kakizuka A. Inhibition of skin development by targeted expression of a dominant-negative retinoic acid receptor. Nature. 1995;374:159–162. doi: 10.1038/374159a0. [DOI] [PubMed] [Google Scholar]
- 76.Salomon D, Masgrau E, Vischer S, Ullrich S, Dupont E, Sappino P, Saurat JH, Meda P. Topography of mammalian connexins in human skin. J Invest Dermatol. 1994;103:240–247. doi: 10.1111/1523-1747.ep12393218. [DOI] [PubMed] [Google Scholar]
- 77.Schultz GS, White M, Mitchell R, Brown G, Lynch J, Twardzik DR, Todaro GJ. Epithelial wound healing enhanced by transforming growth factor-alpha and vaccinia growth factor. Science. 1987;235:350–352. doi: 10.1126/science.3492044. [DOI] [PubMed] [Google Scholar]
- 78.Sibilia M, Wagner ER. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science. 1995;269:234–237. doi: 10.1126/science.7618085. [DOI] [PubMed] [Google Scholar]
- 79.Stoler A, Kopan R, Duvic M, Fuchs E. Use of monospecific antisera and cRNA probes to localize the major changes in keratin expression during normal and abnormal epidermal differentiation. J Cell Biol. 1988;107:427–446. doi: 10.1083/jcb.107.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stoschek CM, Nanney LB, King LE., Jr Quantitative determination of EGF-R during epidermal wound healing. J Invest Dermatol. 1992;99:645–649. doi: 10.1111/1523-1747.ep12668143. [DOI] [PubMed] [Google Scholar]
- 81.Takahashi K, Coulombe PA. Defining a region of the human keratin 6a gene that confers inducible expression in stratified epithelia of transgenic mice. J Biol Chem. 1997;272:11979–11985. doi: 10.1074/jbc.272.18.11979. [DOI] [PubMed] [Google Scholar]
- 82.Takahashi K, Folmer J, Coulombe PA. Increased expression of keratin 16 causes anomalies in cytoarchitecture and keratinization in transgenic mouse skin. J Cell Biol. 1994;127:505–520. doi: 10.1083/jcb.127.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Threadgill DW, Dlugosz AA, Hansen LA, Tennebaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, et al. Targeted disruption of mouse EGF receptor: Effect of genetic background on mutant phenotype. Science. 1995;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- 84.Troyanovsky SM, Guelstein VI, Tchipysheva TA, Krutovskikh VA, Bannikov GA. Patterns of expression of keratin 17 in human epithelia: dependency on cell position. J Cell Sci. 1989;93:419–426. doi: 10.1242/jcs.93.3.419. [DOI] [PubMed] [Google Scholar]
- 85.Troyanovsky SM, Leube RE, Franke WW. Characterization of the human gene encoding cytokeratin 17 and its expression pattern. Eur J Cell Biol. 1992;59:127–137. [PubMed] [Google Scholar]
- 86.Vassar R, Fuchs E. Transgenic mice provide new insights into the role of TGF-alpha during epidermal development and differentiation. Genes Dev. 1991;5:714–727. doi: 10.1101/gad.5.5.714. [DOI] [PubMed] [Google Scholar]
- 87.Vassar R, Coulombe PA, Degenstein L, Albers K, Fuchs E. Mutant keratin expression in transgenic mice causes marked abnormalities resembling a human genetic skin disease. Cell. 1991;64:365–380. doi: 10.1016/0092-8674(91)90645-f. [DOI] [PubMed] [Google Scholar]
- 88.Vassar R, Rosenberg M, Ross S, Tyner A, Fuchs E. Tissue-specific and differentiation-specific expression of a human K14 keratin gene in transgenic mice. Proc Natl Acad Sci USA. 1989;86:1563–1567. doi: 10.1073/pnas.86.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wawersik M, Paladini RD, Noensie E, Coulombe PA. A proline residue in the α-helical rod domain of type I keratin 16 destabilizes keratin heterotetramers and influences incorporation into filaments. J Biol Chem. 1997;272:32557–32565. doi: 10.1074/jbc.272.51.32557. [DOI] [PubMed] [Google Scholar]
- 90.Weiss RA, Eichner R, Sun TT. Monoclonal antibody analysis of keratin expression in epidermal diseases: A 48- and 56-kdalton keratin as molecular markers for hyperproliferative keratinocytes. J Cell Biol. 1984;98:1397–1406. doi: 10.1083/jcb.98.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.White FH, Gohari K. Desmosomes in hamster cheek pouch epithelium: their quantitative characterization during epithelial differentiation. J Cell Sci. 1984;66:411–429. doi: 10.1242/jcs.66.1.411. [DOI] [PubMed] [Google Scholar]
- 92.Wilson AK, Coulombe PA, Fuchs E. The roles of K5 and K14 head, tail, and R/K L L E G E domains in keratin filament assembly in vitro. J Cell Biol. 1992;119:401–414. doi: 10.1083/jcb.119.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]