

Abstract
Digital imaging of mitochondrial potential in single rat cardiomyocytes revealed transient depolarizations of mitochondria discretely localized within the cell, a phenomenon that we shall call “flicker.” These events were usually highly localized and could be restricted to single mitochondria, but they could also be more widely distributed within the cell. Contractile waves, either spontaneous or in response to depolarization with 50 mM K+, were associated with propagating waves of mitochondrial depolarization, suggesting that propagating calcium waves are associated with mitochondrial calcium uptake and consequent depolarization. Here we demonstrate that the mitochondrial flicker was directly related to the focal release of calcium from sarcoplasmic reticular (SR) calcium stores and consequent uptake of calcium by local mitochondria. Thus, the events were dramatically reduced by (a) depletion of SR calcium stores after long-term incubation in EGTA or thapsigargin (500 nM); (b) buffering intracellular calcium using BAPTA-AM loading; (c) blockade of SR calcium release with ryanodine (30 μM); and (d) blockade of mitochondrial calcium uptake by microinjection of diaminopentane pentammine cobalt (DAPPAC), a novel inhibitor of the mitochondrial calcium uniporter. These observations demonstrate that focal SR calcium release results in calcium microdomains sufficient to promote local mitochondrial calcium uptake, suggesting a tight coupling of calcium signaling between SR release sites and nearby mitochondria.
Keywords: mitochondria, intracellular calcium, mitochondrial potential, sarcoplasmic reticulum, cardiomyocyte
Over recent years, interest in the interplay between mitochondrial function and cellular calcium signaling has grown dramatically, in relation to its impact on cellular calcium signaling (e.g., Jouaville et al., 1995), the regulation of mitochondrial function (Duchen, 1992; Hajnoczky et al., 1995), and the potential role(s) of mitochondria in apoptotic cell death (Kroemer et al., 1997). Several groups have postulated a very close coupling between the release of calcium from intracellular stores and mitochondrial calcium uptake (Rizzuto et al., 1993; Simpson and Russel, 1996), suggesting that mitochondria may be exposed to microdomains of calcium ([Ca2+]i) in which the local calcium concentration far exceeds the average concentration measured throughout the cytosol. These observations suggest that (a) mitochondrial function could be far more responsive to cellular calcium signaling than would be predicted from measurements of global calcium signals, and (b) the uptake by mitochondria of calcium released from nearby endoplasmic or sarcoplasmic reticulum (ER or SR)1 may play a role in shaping cellular calcium signals (e.g., Jouaville et al., 1995).
The improved resolution available with new imaging techniques has also revealed the subcellular functional organization of calcium signaling, demonstrating microdomains of high [Ca2+]i, which reflect focal Ca2+ release from internal Ca2+stores. These apparently elemental events, which may reflect Ca2+release through single Ca2+release channels, have been called calcium sparks in cardiomyocytes (Cheng et al., 1993; Lopez-Lopez et al., 1994) and calcium puffs in oocytes and HeLa cells (Yao et al., 1996; Bootman et al., 1997). Similar local release phenomena have since been described in a number of cell types, and the emerging picture suggests that Ca2+ signaling from internal stores involves a hierarchy of events ranging from highly localized signals that reflect release from single channels, through the release from clustered channels or clustered ER/SR vesicles, and finally to Ca2+ signals that propagate as waves throughout the cell volume (for review see Berridge, 1997). In cardiomyocytes, highly localized and brief sparks have been described, as well as larger focal signals that might reflect the simultaneous occurrence or local propagation of multiple sparks (e.g., Lipp and Niggli, 1994), varying in amplitude, time course, and three-dimensional distribution within the cell.
Mitochondria will take up Ca2+ when exposed to an elevated local cytosolic [Ca2+]i. The functional significance of this uptake pathway has been under debate for many years and has been under considerable scrutiny in a variety of cell types. Mitochondrial respiration generates a large membrane potential (Δψm), which drives oxidative phosphorylation (Mitchell and Moyle, 1967) and which also drives mitochondrial calcium uptake. As mitochondrial calcium uptake operates through an electrogenic uniporter, it causes a depolarization of the mitochondrial potential (for review see Nicholls and Crompton, 1980; see also Duchen, 1992; Loew et al., 1994). While using digital imaging techniques to follow changes in Δψm in rat cardiomyocytes maintained in short-term culture, we have routinely noticed spontaneous transient depolarizations of mitochondria or mitochondrial clusters apparently randomly distributed in space and time through the cell. We therefore proposed that the local transient mitochondrial depolarizations might reflect the mitochondrial responses to microdomains of high [Ca2+]i, and the present study represents our exploration of this hypothesis. It has now become clear that these events are indeed associated with the release of calcium from sarcoplasmic reticular stores and subsequent calcium uptake by neighboring mitochondria. Some of these data have been presented in abstract form (Leyssens, A., M. Anderson, M. Craske, R. Rastoghi, M. Crompton, and M.R. Duchen. 1995. J. Physiol. (Lond.). 123:487P; Duchen, M.R., E. Boitier, D. Jacobson, A. Leyssens, and S. Peuchen. 1997. Biophys J. 72:A294).
Materials and Methods
Preparation of Rat Cardiomyocytes
Isolation Procedure.
Isolated rat cardiomyocytes were prepared as previously described (Leyssens et al., 1996). In brief, Sprague Dawley rats (250 g) were killed by cervical dislocation. The heart was excised, attached to a Langendorff column, and perfused with a Ca2+-free physiological salt solution containing 118 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25 mM NaHCO3, and 11 mM glucose, maintained at 37°C and equilibrated with 95% O2, 5% CO2, pH 7.4. The heart was first rinsed and then treated with 50 μg/100 ml collagenase (type II; Worthington Biochemical Corp., Freehold, NJ) and 30 μg/100 ml hyaluronidase (type I; Sigma Chemical Co., St. Louis, MO) for 10 min. The calcium concentration was then raised in three stages to a final concentration of 0.5 mM. After another 8–10 min, the ventricles were excised, minced, and incubated for a further 30 min with 25 ml of the recycling medium. The resulting material was triturated, filtered through surgical gauze, and then centrifuged (90 s, 30 g). The pellets were washed twice by resuspension and centrifugation and finally resuspended in a Hepes-buffered saline (solution B) containing 118 mM NaCl, 4.8 mM KCl, 25 mM Hepes, 1.2 mM MgSO4, 1 mM CaCl2, 1.2 mM KH2PO4, 11 mM glucose, and 1% BSA.
Preparation of Cardiomyocyte Culture.
The cells were treated for 10 min with 50 μg/ml gentamycin in solution B, centrifuged (90 s, 30 g), and washed three times before resuspension at a density of ∼2 × 105 cells/ml in Medium 199 (Life Technologies, Paisley, Scotland) supplemented with 0.2% BSA (medium A). They were then plated as droplets onto sterile laminin-coated (1 μg/cm2) coverslips, transferred to an incubator at 37°C under an atmosphere of 5% CO2 and 95% air, and allowed to settle. After 3 h, further medium A was added to the Petri dish. A preparation of almost 100% intact (rod-shaped) cells was obtained since only intact cells attach to the coverslip. Medium A was renewed daily, and the cardiomyocytes were kept in culture for up to 4 d. No systematic differences were seen in any of the data presented relating to the time in culture.
Preparation of Neonatal Cardiomyocyte Cultures.
Cardiomyocytes were prepared as previously described by Cumming et al. (1996). In brief, the heart was rapidly removed from neonatal Sprague Dawley pups (<48 h old) after beheading into ice-cold Hepes-buffered oxygenated saline containing (mM): 116 NaCl, 0.8 NaH2PO4, 5.3 KCl, 0.4 MgSO4, and 20 Hepes, pH adjusted to 7.35. Cells were then dispersed by a series of incubations in the same buffer, now containing pancreatin (0.6 mg/ml; GIBCO-BRL, Gaithersburg, MD) and type II collagenase (0.5 mg/ml at ∼260 U/mg; Worthington Biochemical Corp.) at 37°C. After six successive incubations (two for 15 min and four for 10 min), the supernatant was removed, resuspended in 2 ml of FBS (GIBCO-BRL), and pelleted by centrifugation at low speed (800 rpm). The pellets were resuspended in FBS, and the six digests were pooled and then pelleted by low-speed centrifugation before resuspending in an “isolation medium” consisting of 4:1 DME/Medium 199 (GIBCO-BRL) supplemented with 10% horse serum, 5% FBS, and 1% penicillin/streptomycin. To minimize contamination with noncardiomyocytes, cells were preplated in 100-mm dishes for 40 min, and the unattached cells were then resuspended and plated onto sterile glass coverslips coated with laminin (1 μg/cm2) in a droplet of the isolation medium in a humidified incubator overnight. They were then washed and maintained in a medium consisting of 4:1 DME/Medium 199 supplemented with 1% FCS for up to 3 wk.
Experimental Arrangement for Fluorescence Imaging of Single Cardiomyocytes
Cells were incubated with tetra-methyl rhodamine-ethyl ester (TMRE; 3 μM) with the addition of pluronic (0.002%) for 20 min at room temperature. The cells were washed in a physiological saline (solution B, above), and the coverslips were placed into a homemade chamber on the stage of an inverted epifluorescence microscope. Retention of dye was good, and recordings could be made from dishes of cells loaded in this way for several hours without degradation of the signal and without any sign of dye toxicity. Fluorescence was elicited by illumination with a 75-W Xenon arc lamp through 10-nm bandpass filters centered either at 490 or 530 nm. The light intensity was heavily attenuated by neutral density filters because we have found that such attenuation is essential to avoid photodynamic injury to cells when using any mitochondrial potential–sensitive probes. Emitted light at wavelengths longer than 560 nm was projected either to slow scan cooled frame transfer CCD camera (800 × 600 pixel, EEV chip), acquisition rate ∼2 frames per second full frame, binning pixels 3 × 3 (Digital Pixel, Brighton, UK), or to a Hamamatsu 4880 (Hamamatsu, Japan) fast readout cooled interline transfer CCD (480 × 640 pixel chip), acquisition rate ∼14 Hz, binning pixels 4 × 4, controlled from a personal computer using software from Kinetic Imaging (Liverpool, UK). In both cases, the readout was digitized to 12 bits, and data was extracted off-line from the images using appropriate image-processing software (Lucida; Kinetic Imaging, Liverpool, UK).
In addition, a small number of experiments were carried out using a confocal imaging system (model 510; Carl Zeiss, Inc., Thornwood, NY). The imaging of TMRE fluorescence was as described above, but using a 543 HeNe laser line very heavily attenuated (at 1% of laser intensity) with the fluorescence image collected at >560 nm.
Drugs and Solutions
Cells were bathed in a solution containing 118 mM NaCl, 4.8 mM KCl, 25 mM Hepes, 1.2 mM MgSO4, 2 mM CaCl2, 1.2 mM K2HPO4, and 11 mM glucose, pH adjusted to 7.4 with NaOH. All experiments were performed at room temperature. In some experiments, CaCl2 was omitted from the bath solution, and 1 mM EGTA was added with appropriate pH correction. Drugs used included carbonyl cyanide p-trifluoromethoxy-phenylhydrazone (FCCP; 1 μM), thapsigargin (500 nM), and caffeine (10 mM). FCCP was applied locally under pressure ejection from micropipettes. Other compounds were added to the bath solution. In some experiments, cells were loaded with the AM ester of the calcium buffer BAPTA at 5 μM for 30 min. Diaminopentane pentammine cobalt (DAPPAC), an inhibitor of the mitochondrial uniporter (Crompton and Andreeva, 1994), is a nonpermeant agent and was therefore microinjected into the cells (from a micropipette containing 1 mM DAPPAC). Cells were coinjected with 10 μM fura-2–free acid, both as an indicator of successful microinjection and also to provide a control to test the integrity of the sarcoplasmic release pathway, as this is blocked by ruthenium red, the more familiar but less selective inhibitor of the uniporter. The solvent dimethylsulfoxide was used at a maximum concentration of 0.2% and, when tested alone, did not have any effect in the experimental conditions tested. All fluorescent probes and BAPTA-AM were obtained from Molecular Probes (Eugene, OR), and all other drugs were from Sigma Chemical Co. unless specified.
Any direct interaction of calcium and TMRE was excluded using solutions of TMRE in a spectrofluorimeter. Addition of calcium at concentrations from 1 μM to 1 mM failed to have any effect on the excitation and emission spectra of the dye.
Image Processing and Statistical Analysis
To remove nonuniformities in the fluorescence intensity throughout the cell and to resolve transient changes as proportional changes in signal, image series were background subtracted, thresholded, and then ratioed against an image in which each pixel represented a control intensity for the whole series of images. To this end, a virtual image was constructed in which each pixel value was set to the lowest value for that pixel throughout the series because any single image might contain areas of increased intensity, and on the assumption that a minimum signal reflects the greatest mitochondrial polarization (see Results). Evidently, this was only feasible in cells that showed no movement during the course of the experiment.
As a device to represent changing intensities in space and time, we have chosen to present data from image sequences as “line images,” in which each line of the image gives the color-coded fluorescence intensity profile along a chosen line drawn along the length of the cell (see Figs. 1 and 4–8), the post-hoc equivalent of the line scan used for confocal imaging. The images shown were constructed as follows: A line was selected along the long axis of the cell. (Clearly this will exclude some events and will only illustrate events occurring along the chosen line.) The Kinetic Imaging software allows the creation of ASCII files consisting of the pixel values along that line for the full image sequence, typically between 30 and 60 image frames. The ASCII matrices were then read into MatLab (The Mathworks, Inc., Natick, MA), which was used to create the surface plots shown. We have also used as a convention a color look up table that runs from black through red and orange to yellow and white with increasing values for otherwise unprocessed image data, while for images that have been thresholded and ratioed, we have applied a look up table that runs through the spectrum, with blue as the lowest value and red as the highest.
Figure 1.

An increase of TMRE fluorescence signals depolarization of Δψm in a single cardiomyocyte. (a) High-resolution image taken from a cardiomyocyte loaded with TMRE using a confocal imaging system (excitation at 543 nm). The discrete localization of signal to mitochondria and the distribution of mitochondria in bands running along the longitudinal axis of the cell is clear. The asterisks in this and the following panels indicate the position of the nucleus. (b) CCD image of a similar cell. The longitudinal distribution of mitochondria is still evident, despite the loss of resolution. c and d show images of a TMRE-loaded cell before (c) and after (d) application of the uncoupler, FCCP (1 μM). The plot shown in e (a surface plot) and the corresponding “line image” shown in f were obtained from the pixel values extracted from the image series along the line drawn along the axis of the cell as shown in c and d, and they illustrate the evolution of the response to FCCP with time. In response to FCCP, the TMRE signal increased approximately threefold in this cell.
Figure 4.


Characteristics of transient depolarizations of single mitochondria. a, i–iv, shows examples of changes in signal measured over clearly identifiable single mitochondria measured as a function of time. Mitochondria were identified as single objects by thresholding the images, which clearly revealed distinct rod-shaped objects. In each case, the recovery phase of the transient depolarizations was fitted well by a single exponential decay function with time constants indicated. Note particularly the example shown in iv, in which two clearly distinct events were identified, each with a similar time course, suggesting complete repolarization after the first event. Surface plots (b, i and c, i) and their equivalent line images (b, ii and c, ii) are shown; the intensity profile along a line selected to follow the long axis of a single identifiable mitochondrion is shown as a function of time. The band of increased intensity in the middle of the image field is the increased intensity seen over the mitochondrion, and the transient events can clearly be seen to involve a spread of indicator beyond the boundaries of the mitochondrion. Again the example shown in c illustrates two consecutive events occurring within a single mitochondrion with time.
Figure 8.


Transient mitochondrial depolarizations require mitochondrial calcium uptake. (a) After microinjection of a cell with DAPPAC and fura-2–free acid, a challenge with caffeine (10 mM) raised [Ca2+]i and caused a twitch, showing that the SR calcium release mechanism was operational. Nevertheless, the mitochondrial flicker was almost completely suppressed, as shown in the surface and line images shown in c (upper and lower traces, respectively), and compared with a control obtained in the same preparation (b, upper and lower panels). To illustrate the time course of cell shortening, a binary image of the cell was created. The intensity was measured in a region that included the edge of the cell. Since the signal is simply a function of the number of pixels within the region set to unity, cell shortening reduced the signal, illustrating the time course of the twitch.
A statistical analysis of transient events in time and space has been achieved by creating image sequences after processing as described above to yield proportional changes in signal throughout the image with reference to a control image that reflects maximal mitochondrial polarization. The mean signal for the full three-dimensional data set was then computed; if nothing were happening, the mean value should be close to unity. The higher the frequency of transient events, the greater the mean will be. We found that this computed parameter, which we will refer to as the index of variation, accurately followed our subjective impression of the frequency of transient events. Where appropriate, results are presented as mean ± SD. Unless otherwise stated, statistical discriminations were performed with the nonpaired two-tailed Student's t test, and P < 0.05 was regarded as significant. As there was some variability between cultures, experimental test conditions were always compared with controls obtained from the same primary culture. We have also chosen to illustrate a number of examples of the transients to convey the characteristics and range of these events in a number of different cells.
Results
In single intact cardiomyocytes, TMRE partitions between the cytosol and mitochondria with a Nernstian distribution and can be readily seen accumulated within energized mitochondria. In a high-resolution confocal image of a cardiomyocyte loaded with TMRE (Fig. 1 a, see also Fig. 3, a and b), the mitochondria could be clearly identified, running parallel to the longitudinal axis of the cell. In an image taken with our cooled CCD system (Fig. 1 b), individual mitochondria were no longer clearly resolved, but their distribution in bands running along the length of the cell was still clearly identified, and single-stained mitochondria can be resolved at the edge of the cell. The nuclei, effectively mitochondrion-free zones, showed only minimal fluorescence (Fig. 1, c, e, and f). Under the loading conditions used in this study, the intramitochondrial accumulation of TMRE causes autoquenching of fluorescence, much as seen with rhodamine 123 (Emaus et al., 1986; Duchen, 1992; Duchen and Biscoe, 1992; Bunting et al., 1993) and TMRM (Ichas et al., 1997). Thus, TMRE fluorescence in cells routinely increased with a wide range of manipulations expected to depolarize Δψm, thus releasing the dye into the cytosol with effective dequench of the fluorescence. This is illustrated in Fig. 1, c and d, which shows images of a cardiomyocyte before and after application of the uncoupler, FCCP (1 μM). The collapse of Δψm after the addition of the protonophore was signaled by a large increase in TMRE fluorescence. These images were extracted from a series of 50 images, and the sequential change in signal, measured along a line selected along the longitudinal axis of the cell as indicated, is shown in Fig. 1, e and f. This series of images also illustrates how many of the later figures were constructed. In response to FCCP, the signal increased by a mean of 262 ± 18% (SD; n = 17).
Figure 3.
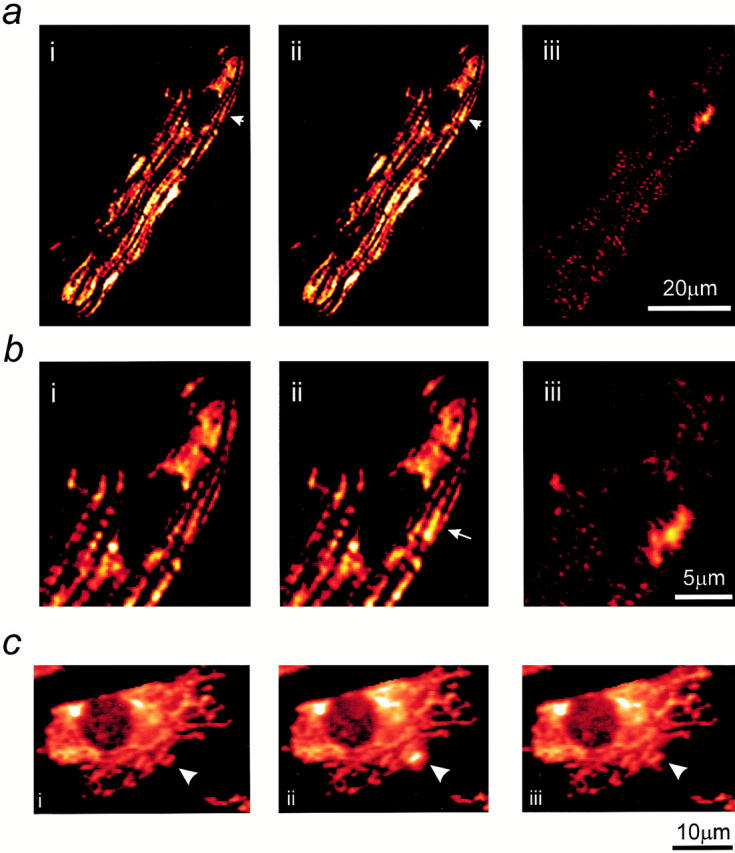
Resolution of transient depolarizations to single mitochondria. In a, a confocal image of an adult myocyte reveals the organization of mitochondria in longitudinal bands. A single mitochondrion showed a transient increase in intensity, and the differential image is shown in iii. b shows an increased magnification of the area of the cell for the three images shown above. In c, a series of images has been extracted from a sequence obtained from the neonatal myocytes in culture to illustrate the occurrence of transient events restricted to single identifiable mitochondria (arrows).
When time series of images of TMRE-loaded myocytes were examined, brief transient changes in signal localized to discrete parts of the cell were invariably seen (total number of cells >200), which we shall refer to as flicker events. Frequently, these later developed into waves of mitochondrial depolarization that sometimes progressed systematically through the cell, culminating in a global depolarization, which was followed rapidly by cell shortening. In the present paper, we will focus on the basis for the brief, transient, localized changes in Δψm, while the basis for later, global changes will be considered in greater detail in a separate study.
Localized Transient Depolarization of Δψm
As an initial experimental protocol, a series of images were acquired in sequence, typically giving an acquisition rate of 1–2 images per second. The transient reversible changes in Δψm localized to small discrete areas of the cells seen in such an image series are illustrated in Fig. 2 a, i and ii (arrows). The events occurred as small increases in signal over a relatively bright baseline signal, with an increase of ∼10–20% for most events. Small events and their distribution were more readily revealed by image processing, in which a running differential of the image series was constructed. In this mode, the preceding image of the series was subtracted from each image, revealing only those pixels in which the signal had changed. Fig. 2 b, i and ii, illustrates the distribution of the flicker events revealed from the differentials of the raw signals illustrated in Fig. 2 a. Individual events could be extremely localized and could involve greater volumes of the cell, and several events could occur simultaneously in different parts of the cell. The changes in signal with time are shown in Fig. 2 c, in which the intensity over small groups of pixels is plotted as a function of time for four different cells. Note the apparent independence of individual transient events in different parts of the cell with time (Fig. 2 c, i). Fig. 2 c, ii, illustrates an isolated brief event to show its occurrence in relation to an otherwise very quiet and stable baseline. The recurrent appearance of brief transients over the same point in the cell (Fig. 2 c, iii) suggests that it is unlikely that the transient events could in fact reflect complete dissipation of potential in single mitochondria (and see below). A further observation that strengthens this view is the relatively small amplitude of the signal compared with that seen with complete dissipation of Δψm (demonstrated after the application of FCCP). This is illustrated in Fig. 2 c, iv, which demonstrates that after a transient depolarization, the signal could still be dramatically increased by the uncoupler.
Figure 2.


Localized transient depolarization of Δψm. a, i and ii, shows a selection of images extracted from image series obtained from two cells over a period of ∼60 s and illustrates the transient localized depolarizations of mitochondria or groups of mitochondria throughout the cell with time. Each arrowhead points to a region of the image that shows a marked transient increase in intensity, and the times of the individual images are indicated. In b, i and ii, the differential images corresponding to those selected in a, i and ii, are shown, revealing the distribution over which the signal has changed between image frames. The black calibration bars to the right of each group of images represent 20 μm. The traces below (c) illustrate the change in intensity with time for events identified in each of four cells. In each case, an area or areas of just a few pixels were selected to limit the collection of signal to a small volume of the cell. In c, i, three widely separated areas within a cell were chosen, over which single events occurred during the sampling period. The record in ii shows a single brief event highly localized to a small part of a cell superimposed on a very quiet baseline. In iii, an example has been selected in which several events occurred repeatedly at a single point within the acquisition period, and in iv, FCCP was applied at the end of the acquisition time to demonstrate the full range of TMRE signal with complete dissipation of the mitochondrial potential in comparison with the small transient event that preceded it.
We have adopted several strategies in an attempt to define the properties of these events more precisely. First, to achieve improved spatial resolution, some experiments were performed using a Zeiss confocal system. In this imaging system, identical events were observed, and the improved spatial resolution made it possible to see that some events clearly arose from changes in signal over single mitochondria (Fig. 3 a). Again, because the change in signal is small, it is more readily revealed by the differential, which is shown in Fig. 3 a, iii. The lower series (Fig. 3 b) shows an enlargement of the images of the area that included the mitochondrion concerned, and it would seem that, at least in this focal plane, an event arose from depolarization of a single mitochondrion in the whole cell. In these experiments, the temporal resolution was still limited, and so some experiments were performed using a fast readout cooled CCD imaging system, with which rates of 7–15 Hz were possible while retaining reasonably good spatial resolution. The spatial resolution was further improved in this system by using neonatal cardiomyocytes grown in culture. As these cells lose their rod-shaped morphology and flatten out with time in culture, resolution of individual mitochondria is possible, even with this nonconfocal optical system. In such experiments, transient depolarizations could be resolved at the level of a single mitochondrion (Fig. 3 c).
A more detailed analysis of these events was then possible because the combination of improved spatial and temporal resolution permitted unambiguous identification of transients as arising from single mitochondria. These could be identified, particularly in the thin peripheries of the cells, as discrete, rod-shaped objects clearly distinct from the background. Measurements of the time constant of the recovery phase of events that clearly arose from single mitochondria were routinely fit by a single exponential function, giving a mean value of 422 ms (± 157, SD) for 50 clearly distinct events in 12 cells. Examples from four discrete events measured over single identifiable mitochondria are plotted in Fig. 4 a (such as that illustrated in Fig. 3 c).
An important consideration in the interpretation of all these data is the true reversibility of these events, i.e., does the transient reversibility of the signal truly reflect mitochondrial repolarization, or is the mitochondrial depolarization in fact irreversible, while the decline of the signal simply reflects diffusion of dye away from the site of release? This is perhaps most simply addressed by examination of single mitochondria with time either in the neonatal cells or in the confocal images. First, it should be clear from Figs. 3 and 4 that recovery of the signal is associated with clearly identified retention of dye within the mitochondrion studied. If the event signaled complete dissipation of potential and diffusion of dye away from the site of release, the mitochondrion should effectively disappear from view because the localization of TMRE is determined by its distribution in response to the maintained potentials between compartments. Even if the signal included a potential independent component from dye bound within the mitochondria, it seems unlikely that the signal would almost invariably return to the same baseline level after a depolarizing event.
Perhaps a more convincing functional indication of reversibility is the observation of events seen repeatedly over the same single mitochondrion; these must of necessity involve a true repolarization after each event if a further event is to occur. Such repeated events were routinely seen and are illustrated specifically in Fig. 4, a (iv) and c (see below), but they are also apparent in a number of other image sequences (Figs. 2, 5, 6, and 7).
Figure 5.
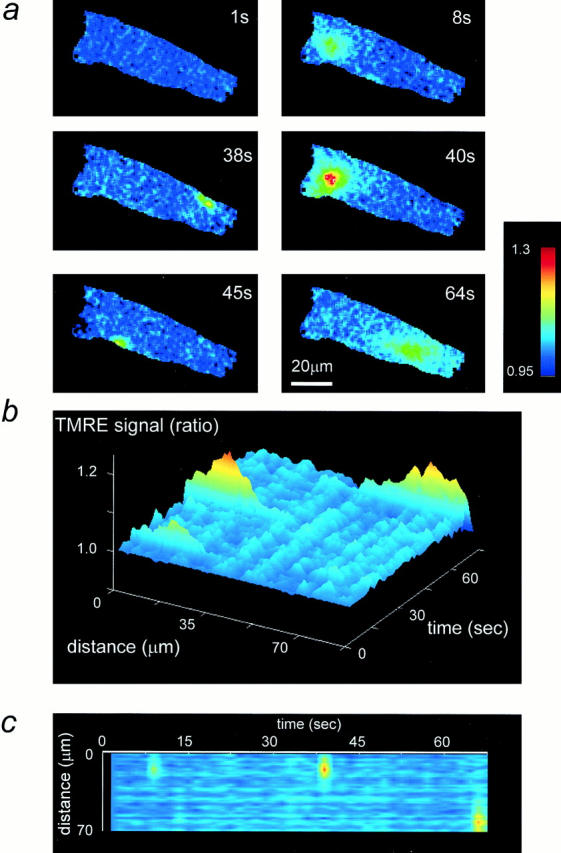
Localized transient depolarization of Δψm in processed image sequences. (a) A series of six images taken from a time series obtained over ∼65 s is shown with the time of acquisition of each within the series as indicated. These images were processed as described and ratioed against a virtual image constructed from the darkest value for each pixel throughout the time series. b and c show the surface and line images, respectively, for this cell, showing changes in intensity along the axis of the cell with time. Note that such processing of course excludes the small events seen close to the edge of these cells at 38 and 45 s.
Figure 6.
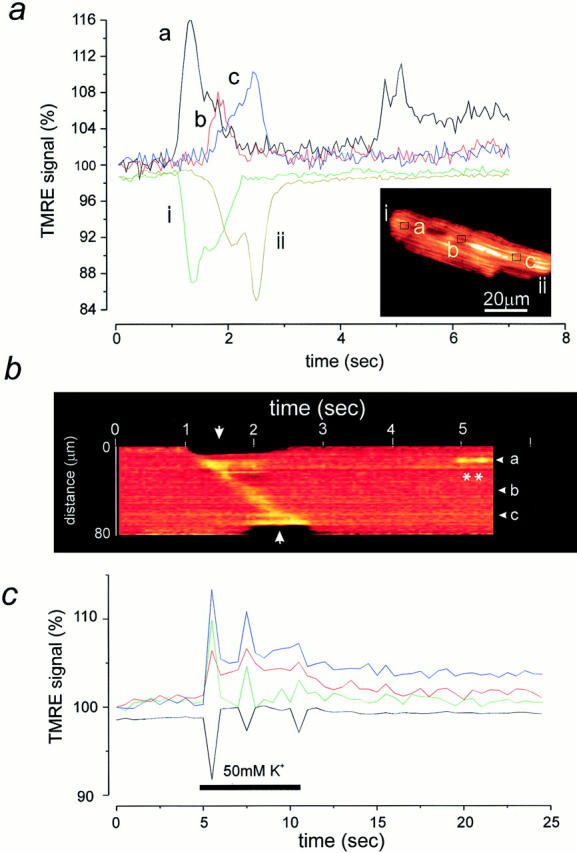
Contractile waves are associated with waves of mitochondrial depolarization. (a) A spontaneous contractile wave propagating across the cell (inset) was associated with a propagating wave of mitochondrial depolarization that coincided with the contraction. The intensity of signal at the areas indicated on the image are shown as a function of time. Note that the decrease in signal at the cell edges (i and ii) due to the contraction coincided well with the increased signal seen at the corresponding part of the cell. In addition, at position a, a doublet of brief transient events were seen later in the image sequence. It is noteworthy that these had a similar amplitude as the transient associated with the contraction. In b, a line image is shown to further illustrate the spatial characteristics of the propagation of the wave of mitochondrial depolarization, seen here as an increase in signal that moves diagonally across the image, starting at one edge of the cell (the upper boundary of this image) and moving towards the other (at the lower edge of this sequence). The transient events seen later (position a) are indicated here as well (asterisks). In c, a plot of intensity with time is shown to illustrate the time course of the mitochondrial response to depolarization of a cell with 50 mM KCl (applied for the period indicated by the white bar). The depolarization caused a series of diminishing twitches, each of which was associated with mitochondrial depolarizations. Note also that the largest twitch was associated with the largest increase in TMRE signal.
Figure 7.

Transient mitochondrial depolarizations are dependent on SR calcium stores. a and b show some characteristic surface and line images, illustrating the occurrence of flicker in a cell under control conditions. After exposure to 30 μM ryanodine (c and d), the flicker was almost completely abolished.
In Fig. 4, b and c, we have also illustrated single and repetitive transient events using a different device to convey the spatial information that is lacking in the time domain plots shown above. These three-dimensional representations were obtained by following the intensity profile along a line drawn along the length of a single mitochondrion with time. Thus, in Fig. 4 b, i, and in the equivalent image seen in Fig 4 b, ii (viewed as if from above), the intensity of signal representing a mitochondrion is seen as a brighter band of signal 3–4 μm in width (the length of the mitochondrion), which is fairly consistent with time. The transient depolarization appears superimposed upon the constant background. In Fig. 4 c, i and ii, a similar image sequence is shown, but in this case, two events were clearly seen over the same single mitochondrion. Again, note that the mitochondrial dimensions are clearly seen as a bright band, about 5 μm in length, while superimposed upon this are two bright flashes over distributions in space, which are exactly equivalent on the two occasions. These images serve to illustrate two points: (a) The signal is truly reversible; the retention of signal over the clearly defined length of the mitochondrion should be clear after recovery of the signal, and that two events can occur over a single mitochondrion clearly confirms the true functional recovery of potential after the initial transient. (b) The signal is associated with the spread of dye away from the mitochondrion and its reuptake and requenching, as the lateral spread of signal, should be clear during the transient. This accounts for the more broadly distributed “puffs” of signal seen above in the differential images (Fig. 2, a and b) because these revealed those pixels over which the signal has changed.
Fig. 5 shows a further example of a time series (obtained using the slow readout camera) from a single cell in which both highly localized (e.g., the images at 38 and 45 s) and more widespread events (the images at 8, 40, and 64 s) were seen. This figure illustrates the way we have processed and analyzed the data to be presented below (see Materials and Methods). To remove nonuniformities of fluorescence signal through the cell, all images of a time series were ratioed against a virtual image created from the darkest value of the series for each pixel. The rationale is based on the assumption that the darkest signal is equivalent to the most polarized state of the mitochondria, and that the alternative choice as a ratio denominator, the first image of the series, might itself contain transient events. It should also be clear that cells in which any twitch or movement occurred were of necessity excluded from this kind of analysis. In Fig. 5 a, some sample images and their time within the series are represented. Note that, after this kind of processing, the increased signal no longer shows a mitochondrial shape, reflecting the movement of dye away from the mitochondria into the surrounding cytosol. To illustrate the changing signal in a full sequence typically containing 50 images, line images were constructed as illustrated in Fig. 5, b and c. These show again that the transients could be very restricted in space and time but were sometimes more widespread. It is important to note that none of the events described here were associated with cell movement or contraction, and it must be stressed that the phenomenon appeared in quiescent, nontwitching cells that showed no signs of Ca2+ overload.
A simple quantitative measure of the amount of transient activity is given by creating an average image from a three-dimensional data set of normalized images, always obtained under identical conditions of time and intensity of illumination, and measuring the average intensity over the resultant image of the cell. In a randomly selected series of 32 control cells, the mean value obtained (which we have chosen to call the index of variation; see Materials and Methods) was 1.23 ± 0.13 (mean ± SD).
Waves of Mitochondrial Depolarization Accompany Contractile Waves
In most preparations of rat cardiomyocytes, a small proportion of cells show spontaneous contractile waves that are associated with spontaneous propagating calcium waves. In such cells loaded with TMRE, the contractile wave was associated with a propagating wave of increased TMRE signal, implying a wave of mitochondrial depolarization following in the wake of a rise in [Ca2+]i (Fig. 6, a and b). In Fig. 6 a, we have plotted the intensity over small groups of pixels as a function of time, as indicated by the lowercase letters next to the traces and on an image extracted from the series from which the data were obtained (inset). Small areas were selected at various points along the length of the cell, and the progression of the increase in signal from one end of the cell to the other should be clear, taking ∼2 s in all. In addition, the time course of the related contraction is illustrated simply by selecting an area that spans the edge of the cell and the background, so that any contraction of the cell effectively reduces the signal as the cell moves out of the region of interest. Note that the increase in signal at “a” parallels the twitch at the same end of the cell at “i,” and similarly, an increase at the other end of the cell “c” accompanies the arrival of the contraction at “ii.” The propagation of the wave of mitochondrial depolarization with contraction is also illustrated in a different way to demonstrate the spatial and temporal evolution of the signal with the twitch in Fig. 6 b, in which a line image has been constructed. Movement of the cell precludes ratiometric image processing for these data. Again, a line has been selected along the length of the cell, and the intensity profile along that line is then displayed as a color-coded image. The relative positions of the selected points in Fig. 6 a (labels a–c) are indicated at the margin of the line image. Note that the twitch is seen as a “waist” on the image as the edges of the cell contract inwards, and the progression of the mitochondrial depolarization is seen as a diagonal band running from the upper border of the cell (arrow), progressing along the cell with time, and reaching the other end of the cell ∼1.5 s later (arrow at lower edge). Two brief transient events were seen in this cell later in the sequence, as seen also in Fig. 6 a, trace a (and b, asterisks). These were not associated with any movement of the cell. Note that they are of very similar amplitude to the increase in the TMRE signal during the twitch, and also that the event consists effectively of a doublet, two events closely apposed in time. Such waves of mitochondrial depolarization invariably accompanied spontaneous twitches and were documented in more than 30 cells.
In an attempt to reproduce this phenomenon experimentally, cells were depolarized with 50 mM potassium, which raised [Ca2+]i and caused a series of contractions of the cells. These were again associated with transient increases in TMRE signal (Fig. 6 c), which clearly correlate with the twitches again measured at the edge of the cell (black trace). 10 mM caffeine was also used to release SR calcium. This invariably caused a contraction and a substantial increase in TMRE signal, but a significant (albeit significantly smaller) TMRE response was also seen in cells in which the SR store had been depleted (see below) and has since been seen in cell types that fail to show a rise in [Ca2+]i in response to caffeine (unpublished observations), suggesting that a part of the TMRE response to caffeine may involve a direct interaction between caffeine and TMRE. This was confirmed because caffeine showed a direct interaction with TMRE in cell-free preparation (in TMRE in a cuvette in a spectrofluorimeter, c.f., interactions between caffeine and Indo-1; Toescu et al., 1992).
The correlation of a propagating transient increase in TMRE signal with contractile waves strongly suggests that calcium release associated with a propagating calcium wave is associated with mitochondrial calcium uptake (see also Chacon et al., 1996). The influx of calcium into mitochondria will in turn promote local mitochondrial depolarization, as described in some other cell types in response to elevated [Ca2+]i either in response to calcium influx or to calcium release from ER (e.g., see Duchen, 1992; Loew et al., 1994; Peuchen et al., 1996).
Transient Mitochondrial Depolarizations Are Dependent on SR Calcium Release
Spontaneous transient localized changes in [Ca2+]i have been described in cardiomyocytes and in a variety of other cell types (Cheng et al., 1993; Lipp and Niggli, 1994; Yao et al., 1995; Bootman et al., 1997; for review see Berridge, 1997). We therefore asked whether the transient mitochondrial depolarizations reported here might be the response to such [Ca2+]i signals. Removal of extracellular calcium (with the addition of EGTA, 1 mM) had no apparent impact in the short term (<30 min) on the occurrence of focal mitochondrial depolarization (n = 22; in a sample of five cells, the mean index of variation [±SD] was 1.15 ± 0.075, not significantly different from the control series). Similarly, elevating external calcium to 10 mM had no obvious short-term consequences. However, calcium overload of the cells, seen with prolonged (0.5–2 h) incubation in high (10 mM) calcium and associated with spontaneous twitching of the cells, was associated with a marked increase initially in localized events and then in spreading waves of mitochondrial depolarization, which is consistent with the well-established association of calcium overload with the occurrence of spontaneous propagating calcium waves (n = 22). Since most of these cells were twitching, a full quantitative analysis of these data was not feasible according to our scheme.
These observations alone suggest that any association between the mitochondrial depolarization and intracellular calcium are independent of calcium influx but may nevertheless be associated with calcium release from the SR, since calcium overload causes spontaneous release of SR calcium stores (e.g., Orchard et al., 1983). The specific role of SR calcium stores was tested by using the Ca2+-ATPase inhibitor thapsigargin to deplete the stores. The cells were bathed in 500 nM thapsigargin, and in early experiments, cells were also initially exposed to caffeine (10 mM) to empty the stores, which were then unable to refill as demonstrated by the loss of a twitch response to caffeine. After incubation in thapsigargin for >45 min, caffeine exposure failed to elicit a twitch, suggesting that the stores had become depleted simply as a consequence of the action of thapsigargin. Following this regime, the flickering depolarization was effectively abolished (mean signal from ratio series, 1.05 ± 0.003; n = 13; P < 0.01). Similarly, depletion of SR calcium stores after prolonged incubation (>60 min) in calcium-free saline also dramatically attenuated the events (mean ratio of 1.04 ± 0.005; n = 4; P < 0.05), and a role for changing [Ca2+]i was further confirmed by the dramatic reduction in the flickering transients on loading the cells with BAPTA-AM (5 μM for 30 min). The index of variation (±SD) was reduced to 1.06 ± 0.021, n = 9 (highly significantly different [P < 0.005] from the mean of 1.23).
The specificity of the release pathway through the ryanodine-sensitive calcium release channel was tested using ryanodine at concentrations that block the SR calcium release channel (30 μM). On initial incubation with ryanodine, the incidence of spontaneous events appeared first to increase (n = 9). Because this response to ryanodine was transient, quantification is difficult, but some of the largest individual events that we have ever seen were recorded early after exposure to ryanodine, with individual transient events reaching 60% increase in signal. However, after more prolonged incubation (>10 min), the events were effectively completely abolished (index of variation: 1.03 ± 0.035; n = 9; P < 0.0005; Fig. 7, c and d).
Transient Mitochondrial Depolarizations Depend on Mitochondrial Calcium Uptake
Together, these data strongly implicate focal SR calcium release as the primary event underlying the localized mitochondrial depolarizations. To determine the role of calcium influx into the mitochondria through the uniporter, cells were microinjected with a novel inhibitor of the uniporter, DAPPAC (Crompton and Andreeva, 1994). Unlike ruthenium red, which itself also inhibits calcium flux through a variety of calcium channels (including the SR calcium release channel [Miyamoto and Racker, 1982] and the mitochondrial calcium uniporter), DAPPAC is selective for the uniporter. Cells were coinjected cells with fura-2–free acid. This provided both a direct index of successful microinjection and also permitted measurement of changing [Ca2+]i. After the microinjection, cells were challenged with 10 mM caffeine, and it was clear that the caffeine-induced change in [Ca2+]i and associated contraction were maintained despite the microinjected DAPPAC (n = 12; Fig. 8 a). Studies of purified SR vesicles also suggest that DAPPAC does not block SR calcium release (Leyssens, A., and M. Crompton, unpublished observation). Clearly, the effective concentration achieved within the cell after such a maneuver is unpredictable, but in five of eight cells successfully microinjected, the focal mitochondrial depolarizations were dramatically reduced (Fig 8 c; index of variation: 1.00 ± 0.004; n = 5; P < 0.001).
The quantitative results of all the manipulations described above are summarized in Fig. 9.
Figure 9.

Quantitative comparisons between manipulations. The histogram illustrates the mean values obtained for the signal measured over the images averaged from time series under each of the conditions discussed and indicated below the columns. The number of cells from which the values were derived are indicated by each column. Significance at the level of <0.05 is indicated by * and at the level of 0.001 by **.
Discussion
In the present study, we have demonstrated transient depolarizations of mitochondrial potential localized to discrete volumes of single rat cardiomyocytes. We have also shown that these clearly reflect the response of mitochondria or mitochondrial clusters to microdomains of high calcium caused by the spontaneous release of calcium from the SR. Over recent years, interest has grown in the dynamics and detail of mitochondrial calcium uptake in situ. For example, Rizzuto et al. (1993) have presented data that strongly suggests that, at least in some cell types, mitochondria may be localized in close enough apposition to ER or SR to be exposed effectively to micromolar concentrations of calcium in the vicinity of a calcium release site. Thus, even though the mean rise in [Ca2+]i averaged through the cytosol may not reach a concentration expected to have much influence on mitochondrial function, microdomains of calcium close to release sites achieve concentrations high enough to promote mitochondrial calcium uptake sufficient to have some impact on mitochondrial function.
Intramitochondrial calcium is a major regulator of oxidative phosphorylation because the major rate-limiting enzymes of the citric acid cycle are upregulated by calcium (Denton and McCormack, 1990; Duchen, 1992; Hajnoczky et al., 1995). This provides a simple but elegant mechanism that serves to couple energy supply and demand to cell function. Increased work by the myocyte is inextricably linked to an increase in cell calcium, which in turn signals an increased ATP requirement to the mitochondria. The present observations suggest that SR and mitochondria may be intimately associated as a functional unit and also raises the possibility that mitochondrial function in different parts of the cell may be regulated depending on local changes in [Ca2+]i. Of course, these observations have been made in quiescent cells, and in a functional, beating heart, the coordination of calcium signaling will tend to reduce the likelihood of spatial nonuniformities in calcium signaling or their mitochondrial responses. However, we have also demonstrated waves of mitochondrial depolarization accompanying contractile waves, suggesting that propagating calcium signals under more physiological conditions will also be associated with coordinated and systematic mitochondrial calcium uptake through the cell (see also Bassani et al., 1993; Chacon et al., 1996).
Whether or not mitochondria take up calcium from SR release sites will be determined by a combination of quantitative and geometric considerations. Ultrastructural evidence suggests that mitochondria may be located very close to SR release sites in cardiac myocytes, where they will be exposed to the highest concentrations of calcium as it is released from a concentrated store and before diffusion or propagation through the cytosol. The concentration of calcium achieved close to those release sites will then determine the effective mitochondrial response. In the present study, we have measured changes in mitochondrial potential, not in mitochondrial calcium. The absolute changes in intramitochondrial calcium that result from these brief transient events—and by implication, from beat to beat SR calcium release—may be small. If the mitochondrial inner membrane has a high electrical resistance, then a very small calcium flux may suffice to cause a substantial potential change. Changes in Δψm may also be shaped by the mitochondrial response to a rise in intramitochondrial calcium, which activates dehydrogenases, stimulates respiration, and therefore hastens repolarization. Indeed, the mitochondrial depolarization itself will also stimulate the respiratory rate and will thus directly influence the shape of mitochondrial potential change.
We cannot distinguish, from the present data set, whether the transient depolarizations are associated with transient changes in [Ca2+]m or whether the changes in [Ca2+]m are longer lasting and slower to decay, as the depolarization will accompany only the period of calcium flux. Attempts to measure intramitochondrial calcium more directly have provided somewhat equivocal data but have suggested significant mitochondrial uptake of SR calcium release in the beating heart. Thus, Isenberg et al. (1993), using electron probe spin microscopy, suggested beat to beat variation in the mitochondrial calcium content, as did Chacon et al. (1996), using confocal microscopy of mitochondrial fluo-3 fluorescence. However, Di-Lisa et al. (1993), using the quench of cytosolic Indo-1 by manganese to reveal the remaining Indo-1 signal within mitochondria, suggested that a steady state of intramitochondrial calcium concentration is achieved in response to the calcium release during the contractile cycle, increasing with increasing rate. Perhaps a conclusive answer still awaits less equivocal and direct measurements of intramitochondrial calcium within paced cells.
While we cannot be more quantitative about absolute mitochondrial potential using this approach, a rough index of the dynamic range of the TMRE signals comes from the response to complete dissipation of Δψm in response to FCCP. The mean response to FCCP was an increase of signal by 260%. If we were to assume a linear calibration (as suggested in fact for rhodamine 123 dequench; Emaus et al., 1986) and accept that the 260% represents a complete depolarization from a resting mitochondrial membrane potential of (say) −160 mV, then the typical change during the transient events of ∼20% would represent only an ∼10–15-mV depolarization. These values are obviously highly speculative, but they make the point that the responses are small and subtle compared with the full range of mitochondrial potential. Indeed, the signals are larger than would be expected from studies in isolated mitochondria, in which uptake from as much as 17 μM caused only a 2–3-mV mitochondrial depolarization (Goldstone et al., 1987). Rapid mitochondrial calcium uptake is hard to study more directly, and it is possible that measurements of potential change serve to amplify a small net calcium flux. Thus, the observation of focal mitochondrial responses to focal calcium release may provide a valuable if indirect way to study the mechanisms of mitochondrial calcium uptake under physiological conditions (see Gunter and Gunter, 1994) and open the way to examine, for example, the role of the newly identified rapid uptake pathway in cell signaling (Sparagna et al., 1995).
If mitochondria accumulate significant calcium loads in response to local calcium release, then an obvious corollary would be that mitochondrial uptake might serve to limit the spatial spread of SR calcium release. In a cell that shows calcium-induced calcium release, this could have profound functional consequences should mitochondrial uptake be disturbed (e.g., during anoxia) because the increased spread of a spark-like event could initiate waves that would in turn lead to aberrant contractures and dysrhythmias.
Isolated mitochondria will take up an imposed calcium load, and that uptake is associated with mitochondrial depolarization. However, when that load is associated with oxidative stress, the calcium uptake may precipitate opening of a large conductance pathway through the mitochondrial inner membrane known as the permeability transition pore (PTP) or mitochondrial megachannel (Crompton and Costi, 1990; Bernardi et al., 1994; Zoratti and Szabo, 1995). This may be a catastrophic event for the cell, leading to leakage of mitochondrial contents into the cytosol, and possibly culminating in cell death, either through mitochondrial uncoupling and ATP depletion or by release of apoptosis-inducing factors (Kroemer et al., 1997). Thus, mitochondrial calcium uptake in a heart that is in a state of oxidative stress, such as during reperfusion after an ischaemic or anoxic episode, may precipitate further damage (Duchen, et al., 1993; Griffiths and Halestrap, 1993). However, recent studies published while the present paper was in preparation have also suggested that the PTP may be active in cells in a lower conductance mode, acting as a transient and reversible channel (Ichas et al., 1997). Thus, the transient responses described here may represent either the electrogenic movement of calcium or transient openings of the PTP, while full and irreversible PTP opening may follow as a slower pathological consequence if the rise in calcium is accompanied by oxidative stress. These issues will be developed further in a separate study.
Acknowledgments
We thank D. Lilian Patterson for her unflagging help with preparation and maintenance of myocytes and Richard Carroll for help with the preparation of neonatal myocytes. Much of this work progressed through the efforts of undergraduate BSc students undertaking final year projects, and we thank Madeleine Craske, Martin Anderson, Robin Rastoghi, Naureen Latif and Alessandro dos Santos for their hard work. We also thank Steven Bolsover, Eric Boitier, D. “Jake” Jacobson, and John Carroll for their helpful comments on the manuscript.
We thank the Wellcome Trust and the British Heart Foundation for support.
Abbreviations used in this paper
- DAPPAC
diaminopentane pentammine cobalt
- FCCP
carbonyl cyanide p-trifluoromethoxy-phenylhydrazone
- PTP
permeability transition pore
- SR
sarcoplasmic reticulum
- TMRE
tetra-methyl rhodamine-ethyl ester
Footnotes
We would like to dedicate this paper to the late Dr. Louis Bohm, whose generosity helped to make much of this work possible.
Some animated sequences of images that have been used to provide data for this paper can be viewed on the web at http://www.geribolsover.physiol. ucl.ac.uk/Duchen/jcb.html
Address all correspondence to Michael R. Duchen, Department of Physiology, University College London, Gower St., London WC1E 6BT, UK. Tel.: 44 171 419 3207. Fax: 44 171 916 3239. E-mail: m.duchen@ucl.ac.uk
References
- Bassani RA, Bassani JW, Bers DM. Ca2+cycling between sarcoplasmic reticulum and mitochondria in rabbit cardiac myocytes. J Physiol (Lond) 1993;460:603–621. doi: 10.1113/jphysiol.1993.sp019489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Broekemeier KM, Pfeiffer DR. Recent progress on regulation of the mitochondrial permeability transition pore, a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr. 1994;26:509–517. doi: 10.1007/BF00762735. [DOI] [PubMed] [Google Scholar]
- Berridge M. Elementary and global aspects of calcium signalling. J Physiol (Lond) 1997;499:291–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootman M, Niggli E, Berridge M, Lipp P. Imaging the hierarchical calcium signalling system in HeLa cells. J Physiol (Lond) 1997;499:307–314. doi: 10.1113/jphysiol.1997.sp021928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting JR, Phan TV, Kamali E, Dowben RM. Fluorescent cationic probes of mitochondria. Metrics and mechanism of interaction. Biophys J. 1993;56:979–993. doi: 10.1016/S0006-3495(89)82743-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon E, Ohata H, Harper IS, Trollinger DR, Herman B, Lemasters JJ. Mitochondrial free calcium transients during excitation-contraction coupling in rabbit cardiac myocytes. FEBS Lett. 1996;382:31–36. doi: 10.1016/0014-5793(96)00138-x. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Crompton M, Andreeva L. On the interactions of Ca2+ and cyclosporin A with a mitochondrial inner membrane pore: a study using cobaltammine complex inhibitors of the Ca2+uniporter. Biochem J. 1994;302:181–185. doi: 10.1042/bj3020181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Costi AA. Heart mitochondrial Ca2+-dependent pore of possible relevance to re-perfusion-induced injury. Evidence that ADP facilitates pore interconversion between the closed and open states. Biochem J. 1990;266:33–39. doi: 10.1042/bj2660033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming D, Heads R, Brand N, Yellon D, Latchman D. The ability of heat stress and metabolic preconditioning to protect primary rat cardiac myocytes. Basic Res Cardiol. 1996;91:79–85. doi: 10.1007/BF00788868. [DOI] [PubMed] [Google Scholar]
- Denton RM, McCormack JG. Ca2+as a second messenger within mitochondria of the heart and other tissues. Annu Rev Physiol. 1990;52:451–466. doi: 10.1146/annurev.ph.52.030190.002315. [DOI] [PubMed] [Google Scholar]
- Di-Lisa F, Gambassi G, Spurgeon H, Hansford RG. Intramitochondrial free calcium in cardiac myocytes in relation to dehydrogenase activation. Cardiovasc Res. 1993;27:1840–1844. doi: 10.1093/cvr/27.10.1840. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Ca2+-dependent changes in the mitochondrial energetics of single mouse sensory neurons. Biochem J. 1992;283:41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Relative mitochondrial membrane potential and [Ca2+]iin Type I cells isolated from the rabbit carotid body. J Physiol (Lond) 1992;450:31–61. doi: 10.1113/jphysiol.1992.sp019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, McGuinness O, Brown LA, Crompton M. On the involvement of the cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc Res. 1993;27:1790–1794. doi: 10.1093/cvr/27.10.1790. [DOI] [PubMed] [Google Scholar]
- Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as a probe of transmembrane potential in isolated rat liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta. 1986;850:436–448. doi: 10.1016/0005-2728(86)90112-x. [DOI] [PubMed] [Google Scholar]
- Goldstone TP, Roos I, Crompton M. Effects of adrenergic agonists and mitochondrial energy state on the Ca2+transport systems of mitochondria. Biochemistry. 1987;26:246–254. doi: 10.1021/bi00375a034. [DOI] [PubMed] [Google Scholar]
- Griffiths E, Halestrap A. Protection by cyclosporin A of ischemic/ reperfusion damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- Gunter KK, Gunter TE. Transport of calcium by mitochondria. J Bioenerg Biomembr. 1994;26:471–485. doi: 10.1007/BF00762732. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Isenberg G, Han S, Schiefer A, Wendt-Gallitelli MF. Changes in mitochondrial calcium concentration during the cardiac contraction cycle. Cardiovasc Res. 1993;27:1800–1809. doi: 10.1093/cvr/27.10.1800. [DOI] [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevisoocytes. Nature. 1995;377:438–441. doi: 10.1038/377438a0. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- Leyssens A, Nowicky AV, Patterson DL, Crompton M, Duchen MR. The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]istudied in isolated rat cardiomyocytes. J Physiol (Lond) 1996;496:111–128. doi: 10.1113/jphysiol.1996.sp021669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipp P, Niggli E. Modulation of Ca2+release in cultured neonatal rat cardiac myocytes. Insight from subcellular release patterns revealed by confocal microscopy. Circ Res. 1994;74:979–990. doi: 10.1161/01.res.74.5.979. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local stochastic release of Ca2+in voltage clamped heart cells: visualisation with confocal microscopy. J Physiol (Lond) 1994;480:21–30. doi: 10.1113/jphysiol.1994.sp020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loew LM, Carrington W, Tuft RA, Fay FS. Physiological cytosolic Ca2+transients evoke concurrent mitochondrial depolarizations. Proc Natl Acad Sci USA. 1994;91:12579–12583. doi: 10.1073/pnas.91.26.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto H, Racker E. Mechanism of calcium release from skeletal sarcoplasmic reticulum. J Membr Biol. 1982;66:193–201. doi: 10.1007/BF01868494. [DOI] [PubMed] [Google Scholar]
- Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature. 1967;213:137–139. doi: 10.1038/213137a0. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Crompton M. Mitochondrial calcium transport. FEBS Lett. 1980;111:261–268. doi: 10.1016/0014-5793(80)80806-4. [DOI] [PubMed] [Google Scholar]
- Orchard CH, Eisner DA, Allen DG. Oscillations of intracellular Ca2+in mammalian cardiac muscle. Nature. 1983;304:735–738. doi: 10.1038/304735a0. [DOI] [PubMed] [Google Scholar]
- Peuchen S, Duchen MR, Clark CB. Energy metabolism of adult astrocytes in vitro. . Neuroscience. 1996;7:855–870. doi: 10.1016/0306-4522(95)00480-7. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- Simpson PB, Russell JT. Mitochondria support inositol 1,4,5-trisphosphate-mediated Ca2+waves in cultured oligodendrocytes. J Biol Chem. 1996;271:33493–33501. doi: 10.1074/jbc.271.52.33493. [DOI] [PubMed] [Google Scholar]
- Sparagna GC, Gunter KK, Shen SS, Gunter TE. Mitochondrial calcium-uptake from physiological-type pulses of calcium—a description of the rapid uptake mode. J Biol Chem. 1995;270:27510–27515. doi: 10.1074/jbc.270.46.27510. [DOI] [PubMed] [Google Scholar]
- Toescu EC, O'Neill SC, Petersen OH, Eisner DA. Caffeine inhibits the agonist-evoked cytosolic Ca2+signal in mouse pancreatic acinar cells by blocking inositol trisphosphate production. J Biol Chem. 1992;267:23467–23470. [PubMed] [Google Scholar]
- Yao Y, Choi J, Parker I. Quantal puffs of intracellular Ca2+ evoked by inositol trisphosphate in Xenopusoocytes. J Physiol (Lond) 1995;482:533–553. doi: 10.1113/jphysiol.1995.sp020538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]