

Abstract
In this work, we have used novel mAbs against two proteins of the endoplasmic reticulum and outer nuclear membrane, termed NEP-B78 and p65, in addition to a polyclonal antibody against the inner nuclear membrane protein LBR (lamin B receptor), to study the order and dynamics of NE reassembly in the Xenopus cell-free system. Using these reagents, we demonstrate differences in the timing of recruitment of their cognate membrane proteins to the surface of decondensing chromatin in both the cell-free system and XLK-2 cells. We show unequivocally that, in the cell-free system, two functionally and biochemically distinct vesicle types are necessary for NE assembly. We find that the process of distinct vesicle recruitment to chromatin is an ordered one and that NEP-B78 defines a vesicle population involved in the earliest events of reassembly in this system. Finally, we present evidence that NEP-B78 may be required for the targeting of these vesicles to the surface of decondensing chromatin in this system. The results have important implications for the understanding of the mechanisms of nuclear envelope disassembly and reassembly during mitosis and for the development of systems to identify novel molecules that control these processes.
Keywords: Xenopus, nuclear envelope, NEP-B78, LBR, cell cycle
The nuclear envelope (NE)1 consists of an inner nuclear membrane (INM) and outer nuclear membrane (ONM), separated by a perinuclear space and perforated periodically by nuclear pore complexes. The ONM is continuous with the ER and these membranes are believed to be structurally and functionally similar (Franke, 1974), while the INM is distinct, containing proteins that are not normally present in the ONM and bulk ER (Worman et al., 1988; Foisner and Gerace, 1993; Ashery et al., 1997b). In higher eukaryotes, the process of mitosis requires that the nuclear envelope be disassembled, its constituent components dispersed and then accurately reassembled in each daughter cell. A fundamental question of membrane biology concerns the mechanisms by which the structural and functional asymmetry of the nuclear envelope is maintained during interphase and restored after the disassembly and reassembly of the nucleus during mitosis.
Mitotic disassembly of the nucleus is characterized by fragmentation of the NE in pro-metaphase, giving rise initially to large smooth flattened cisternae and accompanied by a dismantling of nuclear pores. Fragmentation occurs to variable extents to give rise to vesicles and cisternae, which are morphologically indistinguishable from rough ER fragments (Roos, 1973; Zeligs and Wollman, 1979; Hepler and Wolniak, 1984). In Xenopus oocytes, M-phase fragmentation is extensive, and vesicles ranging from 70 to ∼500 nm have been observed (Wilson and Newport, 1988; Vigers and Lohka, 1991; Newport and Dunphy, 1992). Fragmentation of the NE is also accompanied by depolymerization of the lamina into its constituent subunits, some of which are freely soluble in the cytoplasm (Ottaviano and Gerace, 1985; Gerace and Blobel, 1980; FirmbachKraft and Stick, 1993), while others remain associated with fragmented membrane (Gerace and Blobel, 1980; Lourim and Krohne, 1993).
NE reassembly involves the retargeting of membrane to the surface of decondensing chromosomes, membrane fusion and assembly of nuclear pore complexes, followed by repolymerization of the lamina (reviewed in Gerace and Foisner, 1994; Marshall and Wilson, 1997). The mechanism by which membranes are targeted to the chromosome surface is not clear. Proteins proposed to be involved in this process include both soluble and membrane-associated lamins (Burke and Gerace, 1986; Glass and Gerace, 1990; Dabauvalle et al., 1991; Ulitzur et al., 1992; Lourim and Krohne, 1993), integral inner nuclear membrane proteins with affinities for lamins and chromatin, termed lamin-associated proteins (LAPS; Foisner and Gerace, 1993), p58 LBR (lamin B receptor; Collas et al., 1996; Pyrpasopoulou et al., 1996) and otefin (Ashery et al., 1997a). However, the incorporation of the bulk of lamins into the reforming nucleus is a relatively late event occurring by pore-mediated uptake from the cytoplasm (Collas et al., 1996). In addition, Xenopus extracts immunodepleted of >95% of the total lamin complement support normal nuclear envelope reassembly (Newport et al., 1990; Jenkins et al., 1993a). However, low levels of lamins B2 and B3 have been reported to be associated with membranes and suggested to be important for targeting to chromatin (Lourim and Krohne, 1993; Lourim and Krohne, 1994).
Two models have been proposed to explain the disassembly and reassembly of the nuclear membranes during mitosis. In one, the barrier that ensures that inner nuclear membrane proteins do not diffuse throughout the bulk ER during interphase, would be lost in mitosis and nu-clear membrane proteins would thence become dispersed throughout the ER. After mitosis, resorting would occur by a process that is initiated by the binding of targeting proteins at the surface of chromatin, allowing the sequential assembly of specific protein domains in the plane of the membrane and ultimately regenerating the asymmetry that is characteristic of the NE (Gerace and Foisner, 1994). In support of this model, B-type lamins have been found on all ER fragments during mitosis in chicken cells (Stick et al., 1988). In addition, several integral membrane proteins derived from the inner nuclear membrane have been shown to colocalize with bulk ER markers in mitosis (Ellenberg et al., 1997; Yang et al., 1997).
In the second model, nuclear membranes would remain distinct from the bulk ER during fragmentation, and reassembly would involve the selective retargeting of nuclear-specific vesicles to chromosome surfaces after inactivation of the p34cdc2 kinase (Pfaller et al., 1991; Vigers and Lohka, 1991). In this model, the biochemical composition and perhaps intracellular location of presumptive NE membranes would remain different from bulk ER during mitosis. Such vesicle heterogeneity has been reported in a number of systems (Vigers and Lohka, 1991; Chaudhary and Courvalin, 1993; Lourim and Krohne, 1993; Buendia and Courvalin, 1997) and these observations have led to the suggestion that NE disassembly may be domain specific, with mitotic vesicles retaining proteins exclusively from either the inner, outer or pore membrane regions (Gerace and Foisner, 1994; Marshall and Wilson, 1997).
In the Xenopus cell-free system, the nature of NE precursors remains controversial. A size-homogeneous population of vesicles (∼70 nm) has been recovered from extracts, at least when first treated with the membrane fusion inhibitor, GTPγS (Newport and Dunphy, 1992). Isolation of chromatin-associated vesicles and subsequent reconstitution of a replication competent nucleus with membrane-free cytosol has led to one model in which the functions required for envelope assembly resided solely in chromatin-associated vesicles (Newport and Dunphy, 1992). In contrast, others (Vigers and Lohka, 1991) have described the separation of two membrane containing fractions, neither of which was sufficient for NE assembly in the presence of cytosol and chromatin, but which when recombined gave rise to normal nuclei. However, both the membranous nature of the essential elements of these fractions (Yang et al., 1997), together with the notion that discrete functions can be assigned to each fraction (Wiese et al., 1997), have been questioned. The functional and biochemical characterization of NE precursors has thus been limited by the lack of integral membrane markers that would allow a precise molecular and cell biological analysis of the temporal order and dependency requirements for NE reassembly.
We have generated new molecular reagents with the long-term aim of identifying molecules that are responsible for the formation and disassembly of the NE in a Xenopus cell-free system. In this work, we have used novel mAbs against two proteins of the endoplasmic reticulum and outer nuclear membrane, termed NEP-B78 and p65, in addition to a polyclonal antibody (Collas et al., 1996) against the inner nuclear membrane protein LBR (lamin B receptor), to study the order and dynamics of NE re-assembly in the Xenopus cell-free system. Using these reagents we demonstrate differences in the timing of re-cruitment of membrane proteins to the surface of decondensing chromatin and find that the differences reflect the existence of two functionally and biochemically distinct vesicle types that are necessary for NE assembly. We show that the process of vesicle recruitment to chromatin is an ordered process and that NEP-B78 defines a vesicle population involved in the earliest events of reassembly in this system. Finally, we present evidence that NEP-B78 function is required for the targeting of these vesicles to the surface of decondensing chromatin in this system.
Materials and Methods
Antibodies
mAbs were generated as follows. A nuclear membrane precursor fraction equivalent to nuclear envelope precursor binding fraction (NEP-B) was derived from cell-free extracts of Xenopus eggs according to the method of Vigers and Lohka (1991). Sperm pronuclei, assembled in cell-free extracts of Xenopus eggs were isolated as described previously (Jenkins et al., 1993a). 6-wk-old female BALBc mice were inoculated intraperitoneally with samples of NEP-B containing 20 μg of protein suspended in Freund's incomplete adjuvant, at 30-d intervals (group 1). Alternatively, 6-wk-old female BALBc mice were inoculated intraperitoneally with the equivalent of 106 sperm pronuclei suspended in Freund's incomplete adjuvant, again at 30-d intervals (group 2). Before the initial inoculation and then 10 d after each subsequent injection, serum samples were collected from each mouse by tail bleed. Serum samples were tested for reactivity against NEP-B and NE proteins by indirect immunofluorescence microscopy and immunoblotting. After the third inoculation, the two mice from each group showing the best responses were inoculated with isolated sperm pronuclei (group 1) or NEP-B that had been affinity purified on chromatin (group 2). In brief, NEP-B was bound to chromatin. The chromatin was recovered by centrifugation through a 30% sucrose cushion and the bound vesicles were then released from the chromatin by exposure to highly purified maturation promoting factor (MPF). Released vesicles were separated from chromatin and MPF by density gradient centrifugation through sucrose (Ferrigno, 1996). 9 d after the final inoculation, the mice were tail bled and serum samples were tested for reactivity against nuclear envelopes. The best responder from each group was killed, and its spleen removed. Splenocytes were recovered and fused to SP2/0/Ag 14 myeloma cells using standard protocols (Dyer et al., 1997). After fusion, cells were cultured in the presence of DME containing 10% FCS and supplemented with HAT (hypoxanthine, aminopterin, and thymidine) in 96-well microtiter plates. Colonies were screened by indirect immunofluorescence microscopy and those showing strong reactivity against the NEs of Xenopus sperm pronuclei were grown up and subcloned. In total, five hybridoma cell lines were selected from group 1, established and coded 19C5, 7D6, 3E9, 4G12, and 16H12. All five hybridomas secreted antibodies showing strong reactivity with NEs in indirect immunofluorescence assays. In addition, all five hybridomas secreted antibodies that detected a doublet migrating at 78 kD in immunoblotting experiments using isolated sperm pronuclei and purified MP2 vesicles (see below). 50 hybridomas were selected from group 2 that displayed reactivities against a variety of nuclear proteins (Lyon, 1995). A single mAb termed CEL 5C is described below.
Rabbit polyclonal antisera to LBR were provided by Howard Worman (Department of Medicine, College of Physicians and Surgeons, Columbia University, New York), and antibodies were affinity purified using a recombinant NH2-terminal fragment comprising residues 1–214 of LBR as described in Harlow and Lane (1988).
Preparation of Fab Fragments
Approximately 150 ml of mAb 3E9 tissue culture supernatant was applied to a 2-ml protein G–Sepharose column equilibrated in PBS, and eluted with 0.2 M glycine, pH 3.0. The pH of the resulting antibody solution was made 7.0 by the addition of 0.1 vol of 1 M Tris, pH 8.0. To 250 μl of this solution (protein concentration 40 μg/ml) was added 62.5 μl of 0.1 M sodium acetate pH 5.5, 15 μl of 20 mM EDTA, pH 5.5, and 30 μl of 0.5 M cysteine, pH 5.5. 50-μl aliquots of this solution were incubated with 1.2 μl of papain-agarose (Sigma Chemical Co.) at 37°C for 2 h without shaking and the reaction terminated with 0.1 vol of 825 mM iodoacetamide. The aliquots were recombined, papain-agarose removed by centrifugation at 13,000 g and discarded and the antibody fragments applied to a protein A–Sepharose column (100 μl). The flow through fraction containing the Fab fragments was collected and dialyzed against PBS before use.
Preparation of Membrane and Cytosolic Fractions
Cell-free extracts from Xenopus eggs were prepared and fractionated either as described (Smythe and Newport, 1991) with the modification that high speed centrifugation (200,000 g) was performed for 4 h or as described by Vigers and Lohka (1992). In brief, eggs were dejellied in 2% cysteine, pH 8.0, then washed in modified extraction buffer (25 mM potassium gluconate, 10 mM hemi-magnesium gluconate and 20 mM Hepes, pH 7.5; 300 μm PMSF, 15 μg/ml leupeptin, and 1 mM DTT). Abnormal eggs were discarded and the remaining eggs were packed into tubes by centrifugation at 170 g for 40 s in a clinical centrifuge. After removal of excess buffer, the eggs were supplemented with cytochalasin B (to a final concentration of 10 μg/ml), then crushed by centrifugation at 10,000 g for 10 min. The low speed supernatant (LSS) was retained and centrifuged again in the same manner to remove residual lipid and yolk platelets. This extract was further fractionated by centrifugation at 200,000 g for 75 min to produce a supernatant (S200) and a vesicle containing layer overlying a gelatinous pellet. The membranous layer, termed membrane pellet 1 (MP1), was carefully removed and mixed with an equal volume of modified extraction buffer containing 60% sucrose, snap frozen in liquid nitrogen and stored in 10-μl aliquots at −80°C. The S200 supernatant was then divided into two equal volumes. One volume was diluted 1:20 with modified extraction buffer and centrifuged at 15,800 g in a Beckman JS13.1 rotor for 10 min. The resulting supernatant was further centrifuged at 21,000 g in a Beckman SW 50 rotor for 1 h, and the pellet was termed membrane pellet 2 (MP2). MP2 was mixed with an equal volume of modified extraction buffer containing 60% sucrose, snap frozen in liquid nitrogen and stored at −80°C. The other volume of S200 supernatant was centrifuged at 200,000 g for 4 h to yield a membrane-free cytosolic supernatant (S2004h) that was snap frozen in liquid nitrogen and stored in aliquots at −80°C. As necessary, membrane fractions were recovered from their storage conditions by dilution and centrifugation as described by Vigers and Lohka (1992).
Preparation of Demembranated Sperm Chromatin
Acquisition and preparation of demembranated Xenopus sperm was performed as described (Hutchison, 1994).
Assay for Chromatin-binding and NE Assembly
Unfractionated LSS or isolated MP1, MP2, and S2004h fractions were assayed for chromatin-binding and/or NE assembly by the addition of demembranated sperm (1,000 sperm/μl) to LSS or to a reconstituted mixture consisting of 1 μl of membranes in 20 μl of cytosol with an ATP-regeneration system (2 mM ATP, 10 mM creatine phosphate, and 50 μg/ml creatine kinase). Samples were incubated for indicated times at room temperature and processed either for direct fluorescence or indirect immunofluorescence microscopy described below.
Chaotropic Disruption of Oocyte Nuclear Membranes
Stage VI oocytes were isolated from Xenopus ovary into Modified Barth X Medium at 4°C. Nuclei were manually dissected from these oocytes and stored in NIB (Hutchison, 1994). Aliquots of 15 oocyte nuclei were incubated with egg lysis buffer (25 mM potassium gluconate, 10 mM hemi-magnesium gluconate and 20 mM Hepes, pH 7.5, 300 μm PMSF, 15 μg/ml leupeptin, and 1 mM DTT) alone or supplemented with 1 M KCl, 1 M Na2CO3, pH 11.0, 6 M Urea, or 1% (vol/vol) Triton X-100 for 10 min. Proteins remaining associated with the oocyte nuclei were pelleted by centrifugation at 25,000 rpm in a Beckman SW50 rotor for 25 min at 4°C, and those proteins solubilized were collected in the resultant supernatant. Each fraction was used for immunoblotting.
Fluorescence Microscopy of In Vitro Assembly Reactions
For direct fluorescence microscopy, 1–2 μl of each sample was examined after addition of an equal volume of fix-stain buffer (15 mM Pipes, pH 7.2, 80 mM KCl, 5 mM EDTA, 15 mM NaCl, and 3.3% formaldehyde) containing 1 μg/ml Hoechst 33258 and 10 μg/ml 3,3′-dihexyloxocarbocyanine (DHCC) to stain DNA and membranes, respectively. For indirect immunofluorescence microscopy, 5-μl samples were taken from each incubation mix and subjected to one of the following procedures. Samples were fixed by suspension in 100 μl of 0.33× extraction buffer containing 1 mM ethylene glycol bis-(succinic acid N-hydroxysuccinimide ester) (EGS) and incubated at 37°C for 30 min. Samples were centrifuged through a 25% glycerol cushion onto glass coverslips that were air dried. Alternatively samples were diluted 20-fold in extraction buffer and centrifuged through a 25% glycerol cushion onto glass coverslips before fixing in 100 μl of 0.33× extraction buffer containing 1 mM EGS with incubation at 37°C for 30 min. All samples were then incubated with the relevant antibody for 1 h at room temperature. After washing in PBS the coverslips were incubated with fluorescein-labeled goat anti–mouse Ig (diluted 1/40 in PBS containing 1% newborn calf serum [NCS]) at 4°C for 4 h. Samples probed with anti-LBR Ab were incubated with fluorescein-labeled swine anti–rabbit Ig (diluted 1/40 in PBS containing 1% NCS). In costaining experiments, samples were incubated with a combination of the relevant primary antibodies, followed by a mixture of FITC-labeled goat anti–mouse Ig (diluted 1/40 in PBS containing 1% NCS) and TRITC-labeled swine anti– rabbit Ig (diluted 1/40 in PBS containing 1% NCS) at 4°C for 4 h. After a final wash in PBS, the coverslips were mounted on glass slides (in 50% glycerol/PBS containing 1 μg/ml DAPI) and sealed. These slides were then examined with a Zeiss microscope fitted with a ultraviolet source.
XLK-2 Cell Culture and Indirect Immunofluorescence Protocols
A Xenopus tissue culture cell line XLK-2, derived from embryonic kidney cells, was grown in 50% Leibovitz medium supplemented with 10% (vol/ vol) FCS, penicillin, and streptomycin. Cultures were established at an initial density of 3 × 105 cells per 90-mm petri dish, were maintained at room temperature and were sub-cultured at 7-d intervals.
For indirect immunofluorescence, XLK-2 cells were grown on 11-mm-diam glass coverslips. Cells were fixed in freshly prepared 4% paraformaldehyde (wt/vol) in PBS for 20 min at room temperature and permeabilized as follows: each coverslip was placed in 200 ml of ice-cold permeabilization buffer (5 mM magnesium acetate, 2 mM EGTA, 50 mM potassium acetate, 2 mM DTT, 50 mM Hepes-KOH, pH 7.5, 10 μg/ml leupeptin, and 10 μg/ml aprotonin) in a 24-well Costar plate. Permeabilization was achieved by incubation with 0.1% Triton X-100 at 4°C for 2 min and was terminated by the addition of BSA to 1 mg/ml.
For indirect immunofluorescence, fixed and permeabilized cells were incubated with undiluted mAb supernatants or anti-LBR (diluted 1:50 in PBS containing 1% NCS; PBS/NCS) for 1 h at room temperature. The cells were then washed 5× in PBS, drained and incubated with secondary antibodies (FITC goat anti–mouse Ig diluted 1/40 in PBS/NCS or TRITC goat anti–sheep Ig diluted 1/40 in PBS/NCS) for 1 h at room temperature. The cells were washed 5× in PBS, drained and mounted in MOWIOL containing 1 μg/ml DAPI. Slides were viewed using a Zeiss Axiovert 10 equipped for epifluorescence and fitted with ×63 1.4 N/A and ×40 1.3 N/A PlanNeofluor lenses. Images were collected using a 12 bit CCD camera using IPLab software. Montages were assembled in Adobe Photoshop 4.0.
Electron Microscopy and Immunogold Labeling
TEM.
Vesicle binding assays were established as described above. After 30, 60, and 120 min incubation, samples were fixed as follows. 15 μl of the reaction mixture was dropped into 1 ml of PBS containing 4% paraformaldehyde (prepared on the same day) and 1% glutaraldehyde. Fixation in this solution was for 1 h at room temperature. The fixed sample was washed twice in PBS and then post-fixed with 1 ml of 1% OsO4 in PBS for 1 h at room temperature. After osmification, the sample was washed twice in distilled water and the dehydrated through a 50–100% ethanol series. The alcohol was removed and the samples embedded in Spurr's resin. Sections were cut on a Reichert OMY-3 ultramicrotome and mounted on pioloform coated copper grids. Sections were viewed with a Joel-1200 EX transmission electron microscope at 80 kV accelerating voltage.
Field Emission In Lens Scanning Electron Microscopy.
Germinal vesicles were isolated manually from stage six oocytes and spread on silicon chips as described previously (Goldberg and Allen, 1992). MP1 or MP2 vesicles were collected on silicon chips by sedimentation through 250 mM sucrose at 800 g for 10 min at 4°C. Samples were fixed in 4% paraformaldehyde and 0.1% glutaraldehyde in Viki buffer (80 mM Pipes, pH 6.8, 1 mM MgCl2, 150 mM sucrose) for 10 min at room temperature. Fixation was terminated by quenching with 100 mM glycine and after rinsing in PBS, the samples were blocked by incubation in PBS containing 1% (vol/vol) fish scale gelatin (Sigma Chemical Co.) for 1 h at room temperature. For primary antibody incubations, mAbs CEL5C and 3E9 were used as undiluted antibody supernatants supplemented with fish scale gelatin to 1% (vol/vol). Rabbit anti-LBR was diluted 1:10 in PBS containing 1% (vol/ vol) fish scale gelatin. Samples were incubated with primary antibodies for 2 h at room temperature. The specimens were washed six times for 5 min each with PBS containing 1% (vol/vol) fish scale gelatin before incubation with secondary antibody for 1 h at room temperature. Secondary antibodies (10-nm gold-conjugated sheep anti–mouse or sheep anti–rabbit Ig; Amersham) were diluted 1:10 in PBS containing 1% (vol/vol) fish scale gelatin. Specimens were washed six times as previously and post-fixed in 2% glutaraldehyde in Viki buffer for 18 h at 4°C. The specimens were washed for 1 min in 0.2 M sodium cacodylate and incubated with 1% (wt/ vol) osmium tetroxide in 0.2 M sodium cacodylate for 10 min at room temperature. The samples were washed in double distilled water before incubation in 1% (wt/vol) uranyl acetate for 10 min at room temperature. The specimens were washed and dehydrated through 30, 50, 70, 95, and 100% (vol/vol) ethanol. After alcohol dehydration, the specimens were rinsed in arklone and subjected to critical point drying in arklone purged with liquid CO2. After drying, the samples were coated with chromium and viewed in a Topcon Field Emission In Lens Scanning Electron Microscope (FEISEM) at 30 kV accelerating voltage.
Gel Electrophoresis and Immunoblotting
Protein samples were resolved on 8% or 10% SDS-PAGE at 200 V (25 mA), then transferred onto nitrocellulose filters in transfer buffer (25 mM Tris, 192 mM glycine, and 20% methanol) for 1 h at 100 V (250 mA). The filter was blocked in BLOTTO (4% dried milk in blot rinse buffer [BRB]: 10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, and 0.1% Tween-20) for 1 h at room temperature and probed with undiluted NEP-B78 mAb, p65-specific ascites (diluted 1/1,000 in BRB with 1% NCS) or LBR Ab (diluted 1/200 in BRB with 1% NCS) for 1 h at room temperature. Blots probed with NEP-B78- or p65-specific antibodies were incubated with rabbit anti–mouse peroxidase (diluted 1/400 in BRB containing 1% NCS) for 1 h at room temperature. Blots probed for the presence of LBR were incubated under the same conditions with swine anti–rabbit peroxidase (diluted 1/400 in BRB containing 1% NCS). After final washes in BRB and TBS (10 mM Tris-HCl and 500 mM NaCl, pH 8.0) the filters were developed for visualization by enhanced chemiluminescence.
Disruption of Vesicle/Chromatin Interactions and NE Reassembly with mAbs
10-μl aliquots of MP2 were thawed and suspended in 300 μl of tissue culture supernatant containing the indicated mAbs. Each suspension was mixed end-over-end at 6 rpm for 90 min at room temperature. MP2 was recovered from each antibody supernatant by centrifugation at 13,000 g for 1 min. 10 min before use, 100 μl of membrane-free cytosol (S2004h) was thawed and supplemented with an energy regenerating system and 200,000 demembranated sperm heads. 40 μl was withdrawn and divided into four 10-μl aliquots for chromatin binding assays, referred to as incubation 1. A further 40 μl was withdrawn and supplemented with the equivalent of 4 μl of MP1. This mixture was divided in four 10-μl aliquots for nuclear envelope reassembly assays, referred to as incubation 2. The equivalent of 1 μl of antibody-treated MP2 was added to each 10-μl incubation (1 and 2). Incubation 1 was for 30 min at room temperature. Incubation 2 was for 90 min at room temperature. At the end of each incubation, 3 μl was withdrawn, mixed with 6 μl of DAPI wet mount containing 1 μg/ml DHCC and mounted under a glass coverslip. The remaining 7 μl was suspended in 200 μl of 1 mM EGS and incubated for 30 min at 37°C. EGS fixed samples were processed for indirect immunofluorescence as described previously (Hutchison, 1994).
Analysis of MP1 and MP2 Membranes by Sucrose Density Gradient Centrifugation
100-μl aliquots of MP1 or MP2 in storage sucrose buffer were suspended in 325 μl of modified egg extraction buffer containing 2 M sucrose in a TLS-55 rotor centrifuge tube. In each case, this mix was overlaid with 255 μl of modified egg extraction buffer containing 1.1 M sucrose, 510 μl of modified egg extraction buffer containing 0.9 M sucrose, and finally, 340 μl of modified egg extraction buffer containing 0.7 M sucrose. The sucrose step gradients were centrifuged at 200,000 g for 2 h at 4°C in a Beckman TLS-55 rotor. The gradients were fractionated to give fourteen 110-μl aliquots that were analyzed by SDS-PAGE and immunoblotting for the presence of NEP-B78 and LBRx.
Results
We set out to study the dynamics of NE assembly in the Xenopus cell-free system (Smythe and Newport, 1991). The formation of replication-competent nuclei in this cell-free system is dependent on assembly factors present in egg cytosol in addition to an ER membrane fraction obtained by high speed centrifugation of crushed egg extract (Smythe and Newport, 1991). We obtained a polyclonal antibody raised to the highly conserved integral, inner nuclear membrane protein p58/LBR (Collas et al., 1996; Pyrpasopoulou et al., 1996) and in addition generated a set of novel mAbs as follows. First, a nuclear membrane precursor fraction was purified from cell-free extracts of Xenopus eggs and used to immunize BALBc mice. Splenocytes from immunized mice were fused to SP2/0 myeloma cells and selected upon the basis of indirect immunofluorescence performed on in vitro assembled nuclei. Five cell lines were established, termed 3E9, 4G12, 7D6, 16H12, and 19C5, each of which secreted antibodies that stained nuclear membranes in the indirect immunofluorescence screen. A sixth cell line, CEL5C, which also secreted antibodies that stained nuclear membranes, was established from mice that were immunized with whole nuclei assembled in vitro (Lyon, 1995).
The antigen recognized by four of the mAbs was characterized by immunoblotting. Egg extract (LSS) and whole, isolated, in vitro assembled nuclei were resolved on SDS-PAGE and blotted against one of mAbs 3E9, 4G12, 16H12, or CEL5C (Fig. 1 A). mAbs 3E9, 4G12 and 16H12 each detected a protein of M r ∼78 kD in LSS and isolated nuclei, which in some gels could be resolved into a closely migrating doublet (e.g., Fig. 1 A, 16H12). In contrast, mAb CEL5C detected a single band in LSS and isolated nuclei, migrating with a M r of 65 kD. LSS was then fractionated into a nuclear envelope precursor membrane fraction and a membrane-free supernatant (Smythe and Newport, 1991). Each fraction was resolved on SDS-PAGE and blotted with the mAbs as well as affinity-purified antibodies to LBR (Fig. 1 B). Both the 78- and the 65-kD protein were only detected in the membrane fraction and were absent from the membrane-free supernatant, indicating that both antigens were membrane proteins (Fig. 1 B). As expected, affinity-purified antibodies to LBR (Fig. 1 B) recognized a single, major ∼56-kD protein exclusively in the membrane fraction (Fig. 1 B) and we have designated this protein, LBRx, for LBR-cross reacting protein. Since mAbs 3E9, 16H12, and 4G12 were all raised against a nuclear envelope precursor vesicle fraction (termed NEP-B; Vigers and Lohka, 1991) that binds to chromatin in vitro, and each antibody detects the same 78-kD membrane protein, we adopted the term NEP-B78 to describe this protein. The 65-kD protein is present in two membrane fractions (see below) and is hereafter referred to as p65.
Figure 1.
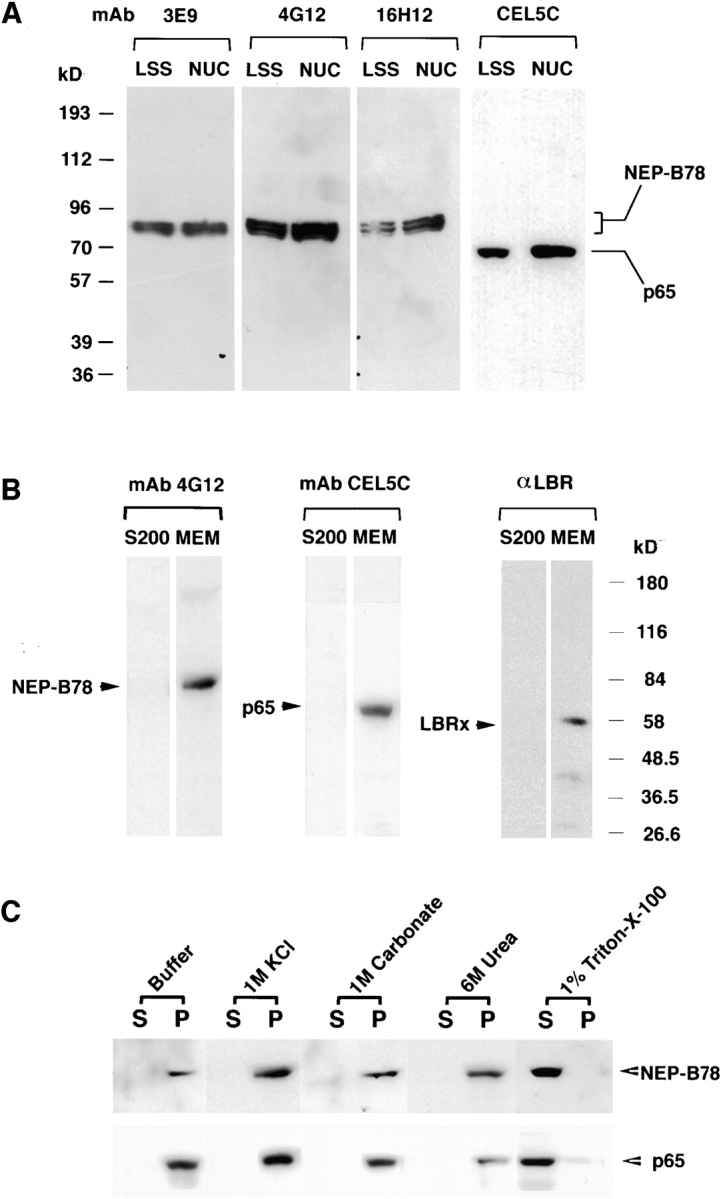
Immunoblotting analysis of NEP-B78, p65, and LBRx. (A) Egg extracts (LSS) or isolated sperm pronuclei (NUC) were resolved by 8% SDS-PAGE, transferred to nitrocellulose and blotted with one of mAbs 3E9, 4G12, 16H12 or CEL5C. 3E9, 4G12 and 16H12 all detected a protein migrating with a M r of 78 kD (termed NEP-B78) in each fraction. CEL5C detected a single band migrating at 65 kD (termed p65) in each fraction. The positions of prestained molecular mass markers are shown at the left-hand side of the panel. (B) Xenopus LSS was fractionated by centrifugation at 200,000 g for 4 h to yield membrane-free supernatant (S200) and a nuclear membrane precursor fraction (MEM) that is sufficient for complete NE assembly (Smythe and Newport, 1991). Equivalent volumes of each fraction were resolved by SDS-PAGE and subjected to immunoblotting with the indicated antibodies for NEP-B78, p65, or LBRx. (C) Oocyte germinal vesicles were manually dissected from stage six oocytes and incubated with egg lysis buffer (Buffer), 1 M KCl, 1 M Na2CO3 (1 M carbonate), 6 M Urea or 1% Triton X-100 (all in egg lysis buffer). Soluble proteins (S) were separated from insoluble proteins (P) by centrifugation, resolved on 8% SDS-PAGE and blotted with either 4G12 or CEL5C. Only Triton X-100 was capable of solubilizing NEP-B78 and p65.
To determine the nature of the association of p65 and NEP-B78 proteins with membranes, membranes isolated from LSS (not shown) or isolated stage VI oocyte germinal vesicles were extracted with egg lysis buffer or egg lysis buffer containing either chaotropic agents (1 M KCl; 1.0 M carbonate, pH 11.0; 6 M Urea) or 1% (vol/vol) Triton X-100. All samples were subjected to centrifugation and the resultant supernatants and pellets analyzed by SDS- PAGE and Western blotting. The results, shown in Fig. 1 C, clearly demonstrate that only Triton X-100 is capable of solubilizing NEP-B78 and p65, indicating that both are integral membrane proteins.
To further characterize these antibodies and their cognate antigens in the Xenopus system, we carried out immunogold labeling of isolated Xenopus oocyte germinal vesicles in conjunction with FEISEM. Germinal vesicles (GVs) were isolated manually from stage VI oocytes, spread on silicon chips and labeled with mAbs 3E9 or CEL5C, or rabbit anti-LBR followed by 10-nm gold–conjugated secondary antibodies (Fig. 2). Using this approach, the nucleoplasmic face of the inner membrane and cytoplasmic face of the outer nuclear membrane are both revealed (Goldberg and Allen, 1992). The distribution of gold-labeled antibodies can be detected either in backscatter, revealing white spots corresponding to the position of 10-nm gold or using the secondary electron detector to reveal antibody-gold particles directly on the membrane surface. Importantly, nuclear membrane topology can be readily distinguished by reference to nuclear pore morphology (Goldberg and Allen, 1992).Thus the area shown in Fig. 2, A and C (left-hand panels) can be distinguished as the cytoplasmic face of the outer nuclear membrane because all of the NPCs in the surrounding area display cytoplasmic ring structures while the area displayed in Fig. 2 B is distinguished as the nucleoplasmic face of inner nuclear membrane by the presence of nuclear pore baskets. In this experiment, NEP-B78 was observed in clusters between NPCs at the outer nuclear membrane (Fig. 2 A) but not at the inner nuclear membrane (data not shown). In contrast, specific LBRx labeling was observed in clusters between nuclear pores on the inner nuclear envelope (Fig. 2 B) but not at the outer nuclear membrane (data not shown). Labeling of the inner nuclear membrane with rabbit anti-LBR was prevented when incubations were performed in the presence of excess recombinant human LBR1-214 (data not shown). Finally, p65 was densely distributed between NPCs on the outer nuclear membrane (Fig. 2 C) but not the inner nuclear membrane (data not shown). The data suggest that NEP-B78 and p65 are both located on the outer nuclear membrane while LBRx is located on the inner nuclear membrane of oocyte germinal vesicles.
Figure 2.
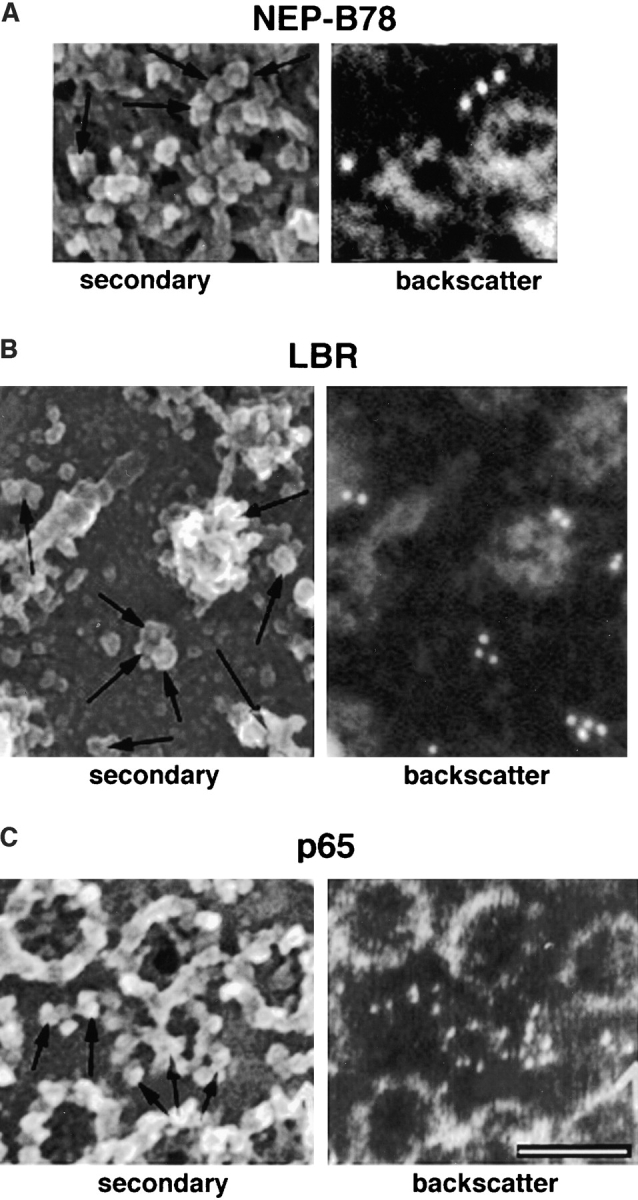
Immunogold electron microscopy reveals the distribution of NEP-B78, LBRx, and p65 between the inner and outer nuclear envelope. Germinal vesicles (GVs) were isolated manually from stage VI oocytes and spread on silicon chips. Using this approach, areas of inner and outer nuclear membrane are both revealed and can be readily distinguished by reference to nuclear pore morphology (Goldberg and Allen, 1992). Spread GVs were labeled with mAb 3E9 (A), rabbit anti-LBR (B), or mAb CEL5C (C) followed by 10-nm gold-conjugated secondary antibodies. Using this approach the distribution of gold labeled antibodies can be detected either in backscatter (revealing white spots corresponding to the position of 10-nm gold; right-hand panels in A–C) or using the secondary electron detector (revealing antibody gold particles of ∼20-nm diam, arrows in left-hand panels in A–C). Bars, 200 nm.
NEP-B78 Is Incorporated into the Nuclear Envelope Concomitantly with p65 but before LBRx in Xenopus Cell-free Extracts and XLK-2 Cells
In cell-free extracts derived from Xenopus eggs, replication competent nuclei are assembled over a 60-min period (Hutchison et al., 1987). Assembly involves the cytosol- dependent decondensation of chromatin followed by membrane recruitment to chromatin surfaces. Subsequent membrane fusion and surface area expansion is accompanied by the appearance of nuclear pores and the lamina, before the onset of DNA replication. Typically in these extracts, membrane recruitment to decondensing chromatin can be detected after 10–15 min. To compare the timing of recruitment of NEP-B78, p65, and LBRx, nuclear reassembly assays in unfractionated extract were analyzed by indirect immunofluorescence microscopy using antibodies against each protein (Fig. 3). Nuclei displaying rim staining with mAbs 4G12 (detecting NEP-B78; Fig. 3, left-hand panels) and CEL5C (detecting p65; Fig. 3, right-hand panels) were readily observed 15 min after the addition of sperm chromatin to Xenopus egg extracts. As the NEs expanded over the following 45-min period, the intensity of staining with both 4G12 and CEL5C increased. In contrast, LBRx was only detected in the NE of assembling nuclei some 30 min after the addition of demembranated sperm chromatin to LSS (Fig. 3, middle panels). These data suggest that the association of NEP-B78- and p65-containing vesicles with decondensing chromatin is one of the earliest events of NE assembly and is distinct from the association of LBRx-containing vesicles.
Figure 3.

Recruitment of NEP-B78, LBRx, and p65 to chromatin periphery during NE reassembly in vitro. Demembranated sperm chromatin was incubated in Xenopus LSS and samples were taken at the indicated time points and processed for immunofluorescence (see Materials and Methods). In each case DNA was stained with DAPI and proteins detected using fluorescein-labeled secondary antibody. NEP-B78 (left-hand micrographs), LBRx (center micrographs), or p65 (right-hand micrographs). Bar, 20 μm.
We extended our observations concerning the timing of recruitment of NEP-B78- and LBRx-containing membranes to the Xenopus cultured cell line, XLK-2. Cells were fixed using 4% paraformaldehyde and 0.1% Triton X-100 and analyzed by indirect immunofluorescence microscopy using antibodies against each protein. We found that in metaphase cells, both LBR and NEP-B78 antibodies reacted with reticular structures in the cytoplasm in regions peripheral to condensed chromosomes, although NEP-B78 staining was very weak (Fig. 4, a, e, i, and m). NEP-B78 association with chromatin was first observed in anaphase (Fig. 4, j and n) at which stage LBRx-containing membranes appeared to remain in the cytoplasm (Fig. 4, b and f). Labeling of the chromatin periphery by LBR antibodies could be observed at telophase with a substantial amount of label still decorating the cytoplasm (Fig. 4, c and g). In contrast, NEP-B78 showed predominantly perichromatin staining at this time (Fig. 4, k and o). Both antibodies gave rise to nuclear staining in early G1 cells (Fig. 4, d, h, l, and p). These data support and extend the observation that the association of NEP-B78-containing membranes with decondensing chromatin is one of the earliest events of NE assembly and is temporally distinct from the association of LBRx-containing membranes.
Figure 4.

The association of NEP-B78 and LBRx with reforming nuclear envelopes in XLK-2 cells. XLK-2 cells were fixed and stained with either rabbit anti-LBR followed by FITC goat anti–rabbit Ig (a–h) or mAb 3E9 followed by FITC-goat anti– mouse Ig (i–m). Fluorescence images of cells in metaphase (a, e, i, and m), anaphase (b, f, j, and n), telophase (c, g, k, and o) or early G1 (d, h, i, and p) were collected with a 12 bit CCD camera attached to a Zeiss Axioskop microscope. In each pair of micrographs, the distribution of DNA is revealed by DAPI staining (a–d and i–l). Bars, 10 μm.
Xenopus NE Precursor Membranes Consist of Functionally and Biochemically Distinct Vesicle Types
To investigate the possibility that two distinct vesicle populations mediate nuclear membrane assembly and that one population binds to chromatin before the other, we fractionated cell-free extracts of Xenopus eggs. Vigers and Lohka (1991) described the separation of two particulate fractions from LSS, which were required to reconstitute NE assembly, by differential and density dependent centrifugation. However, in the absence of biochemical markers, they were unable to unequivocally identify the nature of the components in each fraction that were necessary for NE reassembly. To test the hypothesis that NEP-B78 and LBRx defined the activities of two distinct NE-precursor vesicles, we repeated the fractionation procedure developed by Vigers and Lohka (1991). Two membrane containing fractions (termed MP1 and MP2) were separated and assayed individually or in combination for their ability to form a structurally and functionally competent NE when incubated with membrane-free cytosol (here termed S2004h) and demembranated sperm chromatin. These assays were analyzed by fluorescence microscopy using Hoechst 33258 to stain chromatin and DHCC for total membranes (Fig. 5, left- and right-hand columns, respectively). This revealed that MP1 did not contain membranes with the ability to bind to chromatin in the absence of MP2 (Fig. 5 A). In contrast, membranes did bind to chromatin upon incubation with MP2 alone (Fig. 5 B). The incubation of MP2 with cytosol and chromatin did not undergo any further morphological changes normally associated with complete NE reassembly (Wiese et al., 1997) even after prolonged incubation, and failed to initiate DNA replication (data not shown). When both vesicle-containing fractions were introduced into the assay system, full nuclear envelope assembly was supported as judged by continuous rim staining with DHCC and morphology characteristic of fully formed nuclei (Fig. 5 C) that underwent DNA replication (data not shown). No vesicles were observed upon incubation of demembranated sperm in S2004h (Fig. 5 D). These results indicate that the MP2 fraction was enriched in a vesicle type capable of binding to chromatin although, even after prolonged incubation, nuclear envelope formation did not occur and DNA replication was not supported. At the level of resolution of the light microscope, it was not possible to determine at which point NE assembly was arrested.
Figure 5.

Reconstitution of NE assembly with purified particulate fractions. Demembranated sperm was incubated in cytosol with the addition of MP1 (A), MP2 (B), MP1+ MP2 l (C), or no additions (D) for 90 min at room temperature. Samples were processed for fluorescence microscopy (see Materials and Methods). In each case the left-hand panel shows DNA visualized with Hoechst 33258 and the right-hand panel shows the same sperm stained with 3,3′-dihexyloxocarbocyanine (DHCC) to reveal membranes. Bar, 10 μm.
To do this, samples from each of the assays described above were therefore fixed and sectioned for visualization by electron microscopy (Fig. 6) as described in Materials and Methods. Fig. 6 A shows typical structures resulting from incubations of sperm with MP2 and cytosol. The decondensed chromatin was extensively covered with a single layer of vesicles. At higher resolution, vesicles appeared distorted in two distinct ways. At points of contact with chromatin, vesicles appeared crenulated (white arrows, Fig. 6 B), while vesicles with adjacent surfaces appeared to adopt complementary shapes (black arrows, Fig. 6 C). However, even vesicles tightly juxtaposed did not fuse with one another (shown in detail in Fig. 6, B and C).
Figure 6.

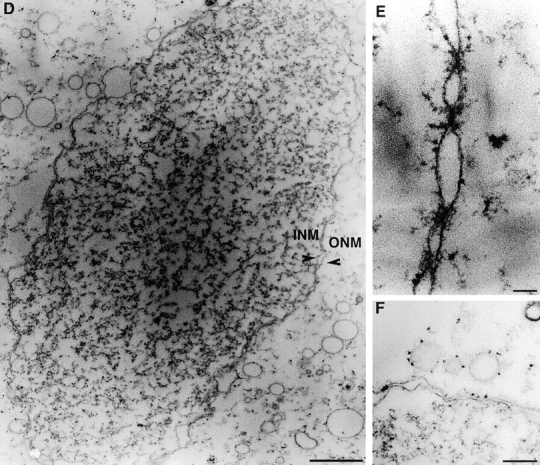
Electron microscope analysis of vesicle binding and fusion. MP2 was incubated with sperm chromatin and cytosol alone (A–C) or with sperm chromatin, cytosol, and MP1 (D–E) as described in Materials and Methods. Incubations for the samples displayed were for 2 h at room temperature after which time the extracts were fixed and sectioned for TEM. A–C show MP2 vesicles bound to the surface of partially decondensed sperm chromatin. White arrows show details of membrane morphology at sites of interaction with chromatin (B). Black arrows show typical membrane morphology at sites adjacent to other vesicles (C). D–F show nuclear envelope structures typically observed in fully reconstituted extracts. Double unit membranes formed a continuous boundary around decondensed chromatin (D) and contained structures resembling nuclear pores (E) and were studded with ribosome-like particles (F, arrowheads). INM, inner nuclear membrane; ONM, outer nuclear membrane. Bars: (A and D) 1 μm; (B and E) 100 nm; (C and F) 200 nm.
As expected, when sperm chromatin was incubated with MP1 and cytosol alone, no vesicles were bound to chromatin (data not shown). However, when both MP1 and MP2 were incubated with chromatin and cytosol, complete formation of a nuclear envelope around the decondensed chromatin was clearly observed (Fig. 6 D), with continuous and distinct inner and outer nuclear membranes (labeled INM and ONM, respectively) and the formation of nuclear pores (Fig. 6 E, nuclear pore indicated by arrow). In addition, ribosome-like structures were seen encrusting the cytoplasmic face of the outer nuclear membrane as well as on vesicles within the surrounding cytosol (Fig. 6 F, arrows). These results clearly show that while vesicles in the MP2 fraction can bind to chromatin, they lack the ability to fuse to neighboring chromatin-bound vesicles or other vesicles within this fraction that remain unbound.
To determine whether we could detect any biochemical differences between membrane fractions required for NE assembly, all fractions obtained by differential centrifugation were subjected to SDS-PAGE and either stained with Coomassie blue for protein or immunoblotted with antibodies to NEP-B78, LBR, p65, and B-type lamins. Coomassie blue staining of total and fractionated membranes indicated that MP1 and MP2 comprised different but partially overlapping sets of proteins (Fig. 7 A). Fractionation of a defined volume of eggs yields differing volumes of the three fractions of interest. Therefore to investigate the distribution of specific proteins between these fractions, volumes of each fraction, proportional to their ratios in a single egg, were examined (Fig. 7, B and C). Western blotting analysis (Fig. 7 B) revealed that antibodies to NEP-B78, LBR, and p65 detected their cognate proteins in an unfractionated extract (lane 1), and as expected for integral membrane proteins, were undetectable in the cytosolic fraction obtained by prolonged high speed centrifugation (lane 4). We found that p65 (Fig. 7 B, middle panels), was distributed approximately equally between the MP1 and MP2 fractions. Importantly, NEP-B78 was found exclusively in the MP2 fraction (Fig. 7 B, upper panels), while LBRx was observed only in the MP1 fraction (Fig. 7 B, lower panels). Isolated MP1 and MP2 fractions were also blotted for the presence of B-type lamins using the mAb L6 8A7 that detects lamins B1, B2, and B3. In Xenopus egg extracts, the major lamin isoform present is B3, which is largely soluble and present in the cytosolic fraction (Jenkins et al., 1993b; Meier et al., 1991). However, both lamin B3 and low levels of lamin B2 have been detected in isolated Xenopus membrane fractions (Lourim and Krohne, 1993). Using mAb L6 8A7 to detect both B3 and B2, we found that a band of the expected mobility for lamin B3 was indeed present in MP1 membranes but not in MP2. In addition, we observed an additional band with reduced electrophoretic mobility in both MP1 and MP2 (Fig. 7 C) presumably corresponding to lamin B2 (Lourim and Krohne, 1993). The results indicate that the fractionation procedure gives rise to membrane preparations that are biochemically distinct.
Figure 7.

Biochemical characterization of MP1 and MP2 fractions; localization of NEP-B78, p65, LBRx, and B-type lamins in egg extract fractions. (A) Total egg extract was subjected to fractionation by centrifugation as described (Smythe and Newport, 1991) to produce a total NE precursor membrane fraction (tot) or as described in (Vigers and Lohka, 1991) to yield MP1 and MP2 fractions. Equal protein loadings of each fraction were subjected to SDS-PAGE and stained with Coomassie blue. (B) Total egg extract (LSS) was subjected to fractionation by centrifugation as described (Vigers and Lohka, 1991) to yield MP1 and MP2 fractions and a membrane-free cytosol (S2004h). Volumes of each fraction, proportional to that found in a single egg, were resolved on an 8% polyacrylamide gel and subjected to immunoblotting for NEP-B78 (top) using mAb 4G12, p65 (middle), or LBRx (bottom). In each case, lane 1 contains low speed supernatant (LSS) and lanes 2–4 contain MP1, MP2 and cytosol, respectively. (C) MP1 and MP2 fractions were resolved on a 10% gel and subjected to immunoblotting for B-type lamins using the mAb L6 8A7.
To further characterize the discrete vesicles involved in NE assembly in the cell-free system, we performed a morphological analysis of NEP-B78- and LBRx-containing vesicles by immunogold labeling of membranes in MP1 and MP2 fractions in conjunction with FEISEM. Membranes were treated with either mAb 3E9 (to detect NEP-B78) or anti-LBR antibodies, followed by gold-tagged secondary antibodies, and examined using FEISEM (Fig. 8 A). We detected LBRx in larger ∼600-nm-diam “rough” vesicles that were enriched in the MP1 fraction (Fig. 8 A, upper right-hand panel). Importantly, no gold particles were observed in MP1 treated with mAb 3E9 (Fig. 8 A, upper left-hand panel). Conversely, we found NEP-B78 in “smooth” vesicles and tubular structures with variable diameter, which were enriched in MP2. No labeling of MP2 vesicles was observed with anti-LBR antibodies (Fig. 8 A, lower panels).
Figure 8.

Characterization of NEP-B78- and LBRx-containing vesicles in MP2 and MP1 fractions by (A) immunogold labeling and FEISEM and (B) sucrose density gradient centrifugation. (A) MP1 (top panels) or MP2 vesicles (bottom panels) were isolated on silicon chips, fixed with 4% paraformaldehyde and 0.1% glutaraldehyde and labeled with either mAb 3E9 followed by 10-nm gold–conjugated sheep anti–mouse Ig (left-hand panels) or rabbit anti–LBR followed by 10-nm gold–conjugated sheep anti–rabbit Ig (right-hand panels). Samples were imaged on a Topcon DS130F scanning electron microscope using both secondary electron and backscatter detectors. The micrographs displayed in A show backscatter images superimposed over secondary images. Before superimposition, the backscatter images were chromatically inverted so that 10-nm gold particles appear as black dots. (B) Aliquots of MP1 and MP2 were overlaid with a sucrose density gradient and subjected to centrifugation as described in Materials and Methods. 14 fractions were retrieved, from the top of the centrifuge tube (lane 1) to the bottom (lane 14), and analyzed by SDS-PAGE and Western blotting for the presence of NEP-B78 (top) using mAb 3E9 or LBR (bottom). Bars, 200 nm.
NEP-B78 and LBRx containing membranes were also subjected to flotation analysis by sucrose density gradient centrifugation. Isolated MP1 and MP2 fractions were overlaid with solutions of decreasing sucrose concentration and centrifuged for 2 h at 200,000 g. After gradient fractionation, samples were subjected to SDS-PAGE and immunoblotting to detect either NEP-B78 or LBRx (Fig. 8 B). We found that flotation of MP2 resulted in the fractionation of NEP-B78-containing membranes across a wide range of sucrose concentrations, presumably reflecting the variation in density and size of membranes in MP2. As expected from the data in Fig. 7, no LBRx was detected in fractions derived from MP2 (data not shown). Similarly, no NEP-B78 was detected in fractions derived from MP1 (not shown). However, in contrast to the wide distribution of NEP-B78 in MP2, LBRx from MP1 was found in a sharp peak at about 1.0 M sucrose, reflecting the homogeneous size and density of LBRx-containing membranes.
Discrete Vesicle Populations Are Required for NE Assembly
We then used the isolated membrane fractions separately and in combination in nuclear assembly assays that we analyzed by indirect immunofluorescence microscopy using all three antibodies as probes. Fig. 9, a shows the results obtained when incubation mixtures containing cytosol plus MP1 alone, cytosol plus MP2 alone, cytosol plus MP1 and MP2 or cytosol alone were incubated in the presence of demembranated sperm chromatin, and an energy regeneration system. The results show that NEP-B78 was recruited to the surface of chromatin when MP2 vesicles alone were used (Fig. 9 A, MP2), but not when MP1 vesicles alone were present (Fig. 9 A, MP1), and consequently was found in the complete nuclear envelope that is formed when MP1 and MP2 were present together (Fig. 9 A, MP1 + MP2). Interestingly, recruitment of p65 was also only observed if MP2 vesicles were included in the assembly incubation. Importantly, recruitment of LBRx was not observed when incubations contained either MP2 alone or MP1 alone, but as expected, recruitment of LBRx was observed in the reconstituted mixture giving a morphologically normal nucleus. These results were obtained using a standard protocol for the detection by indirect immunofluorescence of nuclear antigens in the Xenopus cell-free system (Hutchison, 1994) and involves cross-linking with EGS before dilution and centrifugation of assembled nuclei onto coverslips. The absence of p65 and LBRx staining after incubation of chromatin and MP1 alone indicated that nonspecific cross-linking of membranes to chromatin with EGS was unlikely. However, we repeated this experiment with an altered protocol in which chromatin, after incubation with cytosol and the relevant membrane fraction, was diluted and pelleted onto coverslips before cross-linking with EGS to eliminate this possibility. In this experiment, NEP-B78 and LBRx were visualized after double immunofluorescence labeling with mAb 3E9 and anti-LBR antibodies. Colocalization between these antibodies was observed only in fully reconstituted nuclei (MP1 + MP2; Fig. 9 B). We have also performed colocalization between lamin B3 and LBRx again with the result that colocalization was only observed in fully reconstituted nuclei (data not shown). The results in Fig. 9 B, confirm that recruitment of LBRx from the MP1 fraction is only observed in incubations also containing MP2.
Figure 9.

Recruitment of NEP-B78, p65, and LBRx to chromatin using isolated membrane fractions. Nuclear assembly assays were performed in which demembranated sperm was incubated in cytosol (S2004h) with MP1, MP2, MP1 + MP2 or with no additions for 90 min and prepared for immunofluorescence microscopy (see Materials and Methods). (A) Samples were diluted in 0.33× extract buffer containing EGS, incubated at 37°C for 30 min and centrifuged onto coverslips before incubation with antibodies as described in Materials and Methods. Assays were probed for the presence of NEP-B78 (left-hand micrographs) using mAb 4G12, p65 (center micrographs) or LBRx (right-hand micrographs), and in each case the left-hand column shows DNA stained with DAPI and the right-hand column shows the corresponding fluorescein-labeled images. (B) Samples were diluted in 0.33× extract buffer, and centrifuged onto coverslips before incubation with EGS followed by antibodies for double immunofluorescence microscopy as described in Materials and Methods. DNA was stained with DAPI (left-hand panels), NEP-B78 was detected using FITC-labeled secondary antibodies (middle panels) and LBRx was detected using TRITC-labeled secondary antibodies (right-hand panels). Bars, 20 μm.
Three conclusions can be drawn from the data presented in Figs. 7 and 9. First, NEP-B78 is present exclusively in the membrane preparation containing vesicles that bind initially to chromatin, and is a constituent protein of such vesicles. Since NEP-B78 is an integral membrane protein that is incorporated into the NE and this protein is found exclusively in the MP2 fraction, then this fraction must contain a physiologically relevant and discrete population of membrane vesicles involved in NE assembly. Second, LBRx was shown to be present only in MP1 (Fig. 7 B) and was not present on the vesicles that initially target chromatin. Therefore, the inclusion of this protein into the NE indicates that there are also vesicles within the MP1 fraction that are essential for complete NE assembly. In addition, LBRx-containing membranes must require some component, present in the MP2 fraction, to mediate their incorporation into an NE undergoing reassembly. Finally, although p65 is present in both MP1 and MP2 fractions, this does not indicate the existence of a unique population of p65-containing vesicles distributed between both fractions. Instead, p65 must be present in two functionally distinct types of vesicle as p65 positive vesicles in MP1 are incapable of chromatin binding whereas those in MP2 are capable of this function.
Antibodies to NEP-B78 but Not p65 Interfere with Vesicle Targeting to Chromatin
The data presented in Figs. 3, 7, and 9 indicated that both p65 and NEP-B78 are present in vesicles that are targeted to chromatin in the first step of NE reassembly. To determine whether either of these proteins played any role in the process of NE reassembly, we incubated aliquots of MP2 fraction either with mAb 3E9 or mAb CEL5C. After reisolation of membranes, vesicle recruitment was assayed by the addition of cytosol and chromatin (Fig. 10 A), while NE assembly was assayed by the readdition of cytosol, chromatin, and the essential MP1 fraction (Fig. 10 B). In all cases, membranes were visualized using the membrane dye, DHCC. Preincubation of MP2 membranes with mAb 3E9, but not CEL5C or an irrelevant control antibody (CK18), abolished recruitment of membranes present in the MP2 fraction to chromatin periphery. Similarly, treatment of MP2 membranes with the NEP-B78 specific mAb also blocked recruitment of MP1-derived membrane and nuclear envelope reassembly failed to occur (Fig. 10 B). Antibodies to p65 had no effect on NE reassembly, nor did Fab fragments derived from mAb 3E9 (not shown). These results were confirmed by indirect immunofluorescence assays. Nuclei assembled in the presence of MP1, MP2, and cytosol, with and without prior treatment with 3E9 were assayed for recruitment of LBRx-containing vesicles. 3E9 treatment of MP2 vesicles completely inhibited subsequent incorporation of LBRx into the NE in this reconstituted extract.
Figure 10.

Effect of NEP-B78 and p65 mAbs on vesicle recruitment and nuclear assembly. Aliquots of MP2 membranes were incubated with either anti-cytokeratin 18 mAb (CK-18) as control or anti-NEP-B78 mAB 3E9 or the anti-p65 mAb CEL5C and reisolated as described in Materials and Methods. (A) Vesicle recruitment assays were performed using demembranated sperm chromatin and cytosol supplemented with indicated antibody- treated MP2 alone. (B and C) NE assembly assays were performed using sperm chromatin, cytosol plus MP1 and indicated antibody-treated MP2. In A and B, DNA was stained with Hoechst 33258 (left-hand panels) and membranes are stained with DHCC (right-hand panels). In (C) MP2 previously treated with control CK-18 mAb or 3E9 was incubated with cytosol, MP1 and sperm chromatin, fixed for immunofluorescence and probed for the presence of LBRx using an FITC-labeled secondary antibody. Left-hand panels show DNA stained with DAPI with corresponding fluorescein-labeled images to the right.
Taken together, the results obtained above indicate that there is a specific vesicle population, for which NEP-B78 is a unique molecular marker, which is targeted to surfaces of chromatin as the first step in the reassembly of the NE, and that NEP-B78 function may be required for the targeting process. In addition, membrane fusion and consequent envelope growth is dependent on the subsequent recruitment of LBRx-containing membrane, which is itself dependent on the prior recruitment of vesicles from MP2.
Discussion
Distinct Membrane Vesicles Are Required for Nuclear Envelope Reassembly around Sperm Chromatin In Vitro
In this paper, we have used antibodies that we have generated against integral membrane proteins termed NEP-B78 and p65, together with an affinity-purified anti-LBR antibody to investigate the dynamics of NE reassembly in the Xenopus cell-free system as well as Xenopus tissue culture cells. We have established that NEP-B78 becomes incorporated into the NE during the process of nuclear envelope assembly in cell-free extracts of Xenopus eggs. We found that this protein is present exclusively in an isolated membrane fraction (MP2) that contains vesicles with chromatin binding activity. This fraction was necessary and sufficient for recruitment of NEP-B78 to chromatin surfaces. Since NEP-B78 is a constituent of a mature NE, and while the MP2 fraction is not sufficient to generate an intact NE or replication competent nucleus, nonetheless MP2 contains a population of membrane vesicles that must be incorporated into a mature NE.
We found that preincubation of MP2 with antibodies against NEP-B78 blocked all vesicle recruitment to chromatin. Importantly, preincubation with antibodies to p65 had no effect, suggesting that the inhibition by mAb 3E9 is not simply due to steric hindrance, but that some aspect of NEP-B78 function may be required for the process of targeting MP2 vesicles to chromatin. Interestingly, monovalent Fab fragments derived from mAb 3E9 did not block targeting of vesicles, indicating that mAb 3E9 is not a direct inhibitor of NEP-B78 function. This was not due to loss of epitope recognition as a consequence of mAb proteolysis to generate the Fab fragments, as these Fabs were capable of immunoprecipitating NEP-B78 from detergent-solubilized membranes (data not shown). It seems unlikely that mAb 3E9 cross-links vesicles to yield large membrane aggregates incapable of interacting with chromatin, given the relative size of vesicles and antibody complexes (see Fig. 8). One possibility is that incubation of vesicles in the presence of divalent mAbs result in aggregation of NEP-B78 in the plane of the membrane, resulting in loss of function.
As pretreatment of MP2 with mAb 3E9 essentially blocked all MP2 binding, this indicates that only vesicles containing NEP-B78 can be involved in the initial chromatin-binding step. In addition, the absence of any membrane associated with chromatin, when MP2 is treated with mAb 3E9, together with electron micrographs showing that MP2 vesicles bind to chromatin as a monolayer (Fig. 6, A–C), indicate that NEP-B78-containing vesicles must be the first to be targeted to chromatin surfaces during the reassembly process.
We have also characterized an additional membrane fraction (MP1) that is essential for NE assembly. This fraction could be distinguished from MP2, in that membranes contained within it are unable to bind to chromatin in isolation. However, a component in this fraction supports vesicle fusion, flattening of the NE double membrane, maturation of pore complex assembly and DNA replication. We found that this fraction is the sole source of a Xenopus protein recognized by antibodies that detect human and sea urchin LBR (Collas et al., 1996). On the basis of its membrane location in egg extracts and NE location in XLK-2 cells, molecular mass, incorporation into the NE, recognition by affinity-purified anti-LBR antibodies, and specific localization in the inner nuclear membrane in oocyte germinal vesicles, we suggest that this protein is the Xenopus homologue of the lamin B receptor, or a very closely related protein, which we have termed LBRx. Importantly, since we found that this protein is incorporated into the NE during reassembly (Fig. 3), we conclude that MP1 membranes containing LBRx are also essential for the reconstitution of a replication-competent nucleus.
In this cell-free system, we consistently observed that the recruitment of NEP-B78 to the periphery of chromatin preceded the appearance of LBRx (Fig. 3). We wished to extend these observations to a Xenopus cell line. In XLK-2 cells, we found that NEP-B78 was present on the outer NE, consistent with our SEM observations of germinal vesicles, as well as in the peripheral ER (Hutchison, C.J., S. Drummond, C.E. Lyon, S. McLean, and C. Smythe, manuscript in preparation). However, we have found that the protein residing in the NE differs from that in the ER and we have developed conditions that permit the observation of the NE form of the protein only (Fig. 4 p; Hutchison et al., in preparation). In these cells, we also observed that NEP-B78 recruitment appeared to precede that of LBRx (Fig. 4). NEP-B78 was clearly associated with chromosomes in anaphase cells while LBRx recruitment appeared to begin in telophase with significant amounts of LBRx remaining in cytoplasmic structures (Fig. 4 g) at this point. However, we cannot exclude the possibility that in cells, the levels of LBRx expression vastly exceed that of NEP-B78 and thus the later appearance of LBRx at the chromatin periphery is the consequence of this. In this circumstance, it might be difficult to specifically detect a small fraction of the total LBRx recruited to chromosomes at earlier stages in mitosis. The recruitment of LBR to the NE has also recently been examined (Ellenberg et al., 1997), by expressing a GFP-LBR chimera in living cells. Those authors reported that LBR recruitment could be observed in late anaphase. The significance of the difference between the two observations is unclear but may reflect issues raised above, differences in experimental approaches, species differences or effects associated with the antibody used in this study.
Importantly, in the cell-free system, we also found that recruitment of MP1 to chromatin did not occur when the binding of MP2 was blocked by inhibition with mAb 3E9. Furthermore, the incorporation of LBRx at the surface of chromatin was also blocked by treatment of MP2 vesicles with mAb 3E9. Together, these results demonstrate that the recruitment of MP1 membrane is dependent on the prior recruitment of NEP-B78-containing vesicles present in MP2 and that at least in this system, the arrival of LBRx-containing vesicles at the periphery of chromatin is a consequence of the prior association of a different vesicle type.
Previous work has suggested that at least two membrane fractions termed NEP-A and NEP-B, are required to reconstitute NE formation in Xenopus egg extracts. NEP-B contained vesicles with the ability to bind chromatin. NEP-A contained fusogenic activity and was required for further expansion of the NE surface area (Vigers and Lohka, 1991). However, in two other investigations, a single vesicle fraction provided both chromatin binding and fusogenic activity (Newport and Dunphy, 1992; Wiese et al., 1997). This discrepancy, together with the absence of any molecular markers for the NEP-A and NEP-B fractions, has led to the suggestion that the fractionation procedure developed by Vigers and Lohka generated a functionally homogeneous vesicle population, separated from a detergent-sensitive particulate fraction necessary for NE reassembly (Wiese et al., 1997; Yang et al., 1997). The results presented in this work clearly demonstrate that two functionally and biochemically distinct membrane populations are indeed required for NE assembly. We propose that NEP-B vesicles support chromatin binding but not vesicle fusion and contain NEP-B78. In addition, they consist of smooth vesicles that distribute widely on sucrose density gradients, and are highly enriched in the MP2 fraction described here. NEP-A describes a NE precursor membrane population unable to bind chromatin in isolation, but capable of supporting vesicle-vesicle fusion at the surface of chromatin. NEP-A vesicles are rough and on flotation gradients are recovered in a narrow peak at ∼1.0 M sucrose. In addition, they are highly enriched in MP1 and contain LBRx.
Proteins That Target the Association of NE Precursor Vesicles with Chromatin
A number of proteins have been proposed to be responsible for targeting vesicles to chromatin at the end of mitosis. These include peripheral membrane proteins such as lamins (Burke and Gerace, 1986) or integral membrane proteins such as otefin (Ashery et al., 1997a), LBR (Collas et al., 1996; Pyrpasopoulou et al., 1996), or LAPS (Foisner and Gerace, 1993). Both LAP2 and LBR are capable of binding to B-type lamins and to chromatin in vitro (Foisner and Gerace, 1993; Worman et al., 1988; Ye and Worman, 1996). These associations are reversible and are prevented after specific phosphorylation by the mitotic kinase cdc2. Both LAP2- and LBR-positive vesicles associate rapidly with chromosomes and the reassembling NE during anaphase (Chaudhary and Courvalin, 1993; Foisner and Gerace, 1993). In sea urchin eggs, antibodies against LBR were found to block the recruitment of membrane containing an LBR-like protein to chromatin, arguing for an important role for that protein in the targeting process (Collas et al., 1996). However, it is unclear whether or not LBR vesicles were the first to arrive at the surface of chromatin in that system. Due to the limited amounts of LBR antibodies available to us, we were unable to determine whether these antibodies had any effect on NE reassembly in our system. We predict that interfering with LBRx function will have no effect on the initial recruitment of vesicles to chromatin but may interfere with subsequent steps. In the experiments reported here, it is clear that LBRx is contained in membranes that are recruited after the initial recruitment of NEP-B78-containing vesicles. However, it is important to stress that our results do not exclude the possibility that LBRx, or indeed LAPs, may subsequently participate in binding and remodelling of chromatin after their arrival at the surface. More likely, the initial interactions between NEP-B78 containing vesicles and chromatin cause surface remodelling of the chromatin that in turn permits LBRx-chromatin binding activity. Indeed, evidence for surface remodelling of chromatin at sites of interaction with NEP-B78 vesicles is presented in Fig. 6 C.
Lamin B3 is the major form of lamin protein found in Xenopus egg extracts (Benavente et al., 1985; Stick and Hausen, 1985). Blocking lamin function either by antibody inhibition or by specific immunodepletion fails to inhibit NE assembly arguing that lamin proteins do not play a role in targeting vesicles to chromatin (Newport et al., 1990; Meier et al., 1991). Recent reports suggest that low residual levels of lamin B3, in addition to lamin B2 remain associated with membranes (Lourim and Krohne, 1993). Using mAb L6 8A7 to detect both B3 and B2, we found that a band of the expected mobility for lamin B3 was indeed present in MP1 membranes but not in MP2. In addition, we observed an additional band with reduced electrophoretic mobility in both MP1 and MP2, presumably corresponding to lamin B2 (Fig. 7 C). The inability of MP1 vesicles to bind to chromatin directly suggests that these proteins are not sufficient to bring about chromatin targeting. Lamin B2 might be necessary for MP2 binding since this protein binds to chromatin in vitro (Hoger et al., 1991) and mAbs against lamin B2 have been reported to inhibit NE assembly in vitro (Dabauvalle et al., 1991). However, the levels of this protein in MP2 are very low compared with the levels of membrane-associated lamin B3 and in other experimental systems, B-type lamins appear unnecessary for membrane targeting (Collas et al., 1996; Pyrpasopoulou et al., 1996).
Previous reports suggesting the ability of membrane vesicles to carry out both binding and fusion (Newport and Dunphy, 1992; Wiese et al., 1997) may be explained by the possibility that other fractionation procedures failed to separate NEP-A and -B type vesicles. The molecular mechanism underlying the mutual recognition of NEP-A and -B type vesicles is unknown but might be expected to reside in elements of fusogenic machinery that must be present on both vesicle types. Therefore, in different buffers, interactions between the fusogenic machinery may lead to cofractionation of NEP-A and NEP-B. In addition, previous experiments reporting the coordinated recruitment of rough and smooth vesicles were performed using artificially remodelled chromatin template (Wiese et al., 1997). The use of artificially remodelled sperm chromatin may eliminate the ordered recruitment of NEP-B and NEP-A.
Post-mitotic Regeneration of NE Asymmetry
We found that both NEP-B78 and p65 could be recruited to chromatin surfaces when incubated with MP2 alone, indicating that vesicles with chromatin binding activity must also contain proteins that ultimately reside almost exclusively in the outer nuclear membrane (Hutchison et al., in preparation). It is likely, therefore, that NEP-B vesicles contain proteins that will ultimately reside on the outer envelope together with proteins that play an essential role at the inner NE. As it is unlikely that inner and outer NE proteins reside in distinct domains on each vesicle, it is clear that during the assembly process, a sorting mechanism must exist to generate the asymmetric protein composition of a mature NE. We suggest that the establishment of chromatin-membrane macromolecular complexes is the first step in the generation of NE asymmetry. Sites of membrane-chromatin association act as anchor points for the recruitment of other inner NE proteins from the same vesicle, as well as from incoming fusogenic vesicles and may act to exclude proteins of the outer NE. However, it is important not to exclude the possibility that proteins involved in modelling the early stages of chromatin-membrane architecture may not be required in later stages of assembly and may be free to diffuse/migrate elsewhere.
Fusion of the nuclear envelope vesicles requires ATP and GTP and is inhibited by the alkylating agent N-ethylmaleimide (NEM; Newport and Dunphy, 1992). It remains to be established whether this reflects an inhibition of the heterotypic fusion machinery (NEM-sensitive factor, NSF), soluble NSF attachment proteins (SNAPS), and SNAP receptors (reviewed in Rothman and Warren, 1994) or another fusion machine, such as that responsible for homotypic fusion and yeast karyogamy (Latterich and Schekman, 1994; Acharya et al., 1995; Rabouille et al., 1995). However, our ability to separate functionally discrete precursors in this cell-free system should allow us to develop a system to test these possibilities.
How Are Discrete Vesicle Populations Generated in M-phase?
In higher eukaryotes, the process of mitosis requires that the nuclear envelope be disassembled and reassembled in each daughter cell. In Xenopus oocytes, M-phase fragmentation is extensive, and vesicles ranging from 70 to ∼500 nm have been observed (Wilson and Newport, 1988; Vigers and Lohka, 1991; Newport and Dunphy, 1992). Vesicle heterogeneity has been reported in a number of systems (Vigers and Lohka, 1991; Chaudhary and Courvalin, 1993; Lourim and Krohne, 1993; Buendia and Courvalin, 1997) and these observations have led to the suggestion that NE disassembly may be domain specific, with mitotic vesicles retaining proteins exclusively from either the inner, outer or pore membrane regions (Gerace and Foisner, 1994; Marshall and Wilson, 1997). More recently, confocal microscopy in NRK cells (Yang et al., 1997), and photobleaching of GFP-LBR chimeras in COS-7 cells (Ellenberg et al., 1997) have indicated that marker proteins of the inner NE can be detected throughout the bulk ER in mitosis, with little observable membrane fragmentation, and have led to the alternative hypothesis that NE disassembly is simply the consequence of the loss of the barrier (the nuclear pore complex) that ensures that inner nuclear membrane proteins do not diffuse throughout the bulk ER during interphase. A clear prediction of this hypothesis is that the protein composition of bulk ER in mitosis will be identical and NE proteins should be detectable throughout this organelle. It is clear from the results presented here that the products of NE breakdown, which act as precursors for reassembly after inactivation of the mitotic cdc2 kinase, are not functionally nor biochemically homogeneous (Figs. 6–9). Thus our data are not consistent with a simple model of diffusion into the bulk ER.
Wiese et al. (1997) have developed a more complex model of the above suggesting that the processes of membrane fragmentation and vesiculation occur concomitantly with diffusion between the inner, outer and pore domains of the NE. This might be expected to give rise to vesicles with differing protein compositions, depending on relative kinetics of diffusional events and fragmentation rates. If diffusion predominates over fragmentation, then vesicle protein compositions will be very similar. Where fragmentation predominates over diffusion, vesicles may be expected to have distinct protein compositions and functions. The absence of significant amounts of NEP-B78 in NEP-A, and the absence of significant amounts of LBRx in NEP-B, together with the clear functional distinctions observed between these two populations suggest that, during M-phase in this system, the degree of diffusion, if any, is very low compared with the rate of fragmentation.
This work has important implications for the understanding of the mechanisms of nuclear envelope disassembly and reassembly during mitosis and for the development of systems to identify novel molecules that control these processes. Future work must be directed towards identifying molecular components that permit the generation of NEPs as well as those that allow their reconstitution into an intact NE. The identification of specific proteins in discrete vesicle types presented here is a first step in the elucidation of these mechanisms.
Acknowledgments
We wish to thank Shona McLean, Richard Paley, Martin Kierans and Fred Brewster for technical assistance; Howard Worman for anti-LBR antibodies and LBR expression constructs; and Elizabeth Smythe and Darren Cross for critical reading of the manuscript.
Abbreviations used in this paper
- BRB
blot rinse buffer
- DHCC
3,3′- dihexyloxocarbocyanine
- EGS
ethylene glycol bis-(succinic acid N-hydroxysuccinimide ester)
- INM
inner nuclear membrane
- LAPS
lamin-associated proteins
- LBR
lamin B receptor
- LSS
low speed supernatant
- MP1
membrane pellet 1
- MPF
maturation promoting factor
- NCS
newborn calf serum
- NE
nuclear envelope
- NEM
N-ethylmaleimide
- NEP-B
nuclear envelope precursor binding factor
- NSF
NEM-sensitive factor
- ONM
outer nuclear membrane
- SNAPS
soluble NSF attachment proteins
Footnotes
Address correspondence to Carl Smythe, MRC Protein Phosphorylation Unit, MSI/WTB Complex, Dow Street, Dundee DD1 5EH, Scotland, UK. Tel.: 44 1382 345095. Fax: 44 1382 223778. E-mail: cgwsmythe@bad.dundee.ac.uk or Christopher Hutchison, Department of Biological Sciences, University of Dundee, Dundee, DD1 4HN, Scotland, UK. Tel.: 44 1382 344728. E-mail: c.j.hutchison@dundee.ac.uk
C.J. Hutchison and C. Smythe are members of the Dundee Eukaryotic Membrane Dynamics MRC Cooperative Group at the University of Dundee. This work was supported by the Medical Research Council, Biotechnology and Biological Sciences Research Council, and the Wellcome Trust. C.J. Hutchison is the recipient of a Wellcome Trust Research Leave Fellowship. S. Drummond is the recipient of a BBSRC-CASE quota studentship.
Dr. Ferrigno's current address is SM922, Dana-Farber Cancer Institute, 44 Binney St., Boston, MA 02115.
References
- Acharya U, Jacobs R, Peters JM, Watson N, Farquhar MG, Malhotra V. The formation of Golgi stacks from vesiculated Golgi membranes requires two distinct fusion events. Cell. 1995;82:895–904. doi: 10.1016/0092-8674(95)90269-4. [DOI] [PubMed] [Google Scholar]
- Ashery PR, Ulitzur N, Arbel A, Goldberg M, Weiss AM, Maus N, Fisher PA, Gruenbaum Y. Localization and posttranslational modifications of otefin, a protein required for vesicle attachment to chromatin, during Drosophila melanogasterdevelopment. Mol Cell Biol. 1997;17:4114–4123. doi: 10.1128/mcb.17.7.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashery PR, Weiss AM, Feinstein N, Gruenbaum Y. Distinct regions specify the targeting of otefin to the nucleoplasmic side of the nuclear envelope. J Biol Chem. 1997;272:2493–2499. doi: 10.1074/jbc.272.4.2493. [DOI] [PubMed] [Google Scholar]
- Benavente R, Krohne G, Franke WW. Cell type-specific expression of nuclear lamina proteins during development of Xenopus laevis. . Cell. 1985;41:177–190. doi: 10.1016/0092-8674(85)90072-8. [DOI] [PubMed] [Google Scholar]
- Buendia B, Courvalin JC. Domain-specific disassembly and reassembly of nuclear membranes during mitosis. Exp Cell Res. 1997;230:133–144. doi: 10.1006/excr.1996.3395. [DOI] [PubMed] [Google Scholar]
- Burke B, Gerace L. A cell free system to study reassembly of the nuclear envelope at the end of mitosis. Cell. 1986;44:639–652. doi: 10.1016/0092-8674(86)90273-4. [DOI] [PubMed] [Google Scholar]
- Chaudhary N, Courvalin JC. Stepwise reassembly of the nuclear envelope at the end of mitosis. J Cell Biol. 1993;122:295–306. doi: 10.1083/jcb.122.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collas P, Courvalin JC, Poccia D. Targeting of membranes to sea urchin sperm chromatin is mediated by a lamin B receptor-like integral membrane protein. J Cell Biol. 1996;135:1715–1725. doi: 10.1083/jcb.135.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabauvalle MC, Loos K, Merkert H, Cheer U. Spontaneous assembly of pore complex-containing membranes (“annulate lamellae”) in Xenopusegg extract in the absence of chromatin. J Cell Biol. 1991;112:1073–1082. doi: 10.1083/jcb.112.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberg J, Siggia ED, Moreira JE, Smith CL, Presley JF, Worman HJ, Lippincott SJ. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrigno, P. 1996. Regulation of mitotic progression by protein phosphorylation: events downstream of p34cdc2. Ph.D. Thesis. University of Dundee.
- FirmbachKraft I, Stick R. The role of CaaX-dependent modifications in membrane association of Xenopusnuclear lamin B3 during meiosis and the fate of B3 in transfected mitotic cells. J Cell Biol. 1993;123:1661–1670. doi: 10.1083/jcb.123.6.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foisner R, Gerace L. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell. 1993;73:1267–1279. doi: 10.1016/0092-8674(93)90355-t. [DOI] [PubMed] [Google Scholar]
- Franke WW. Structure, biochemistry, and functions of the nuclear envelope. Int Rev Cytol. 1974;4:71–236. [PubMed] [Google Scholar]
- Gerace L, Blobel G. The nuclear envelope lamina is reversibly depolymerized during mitosis. Cell. 1980;19:277–287. doi: 10.1016/0092-8674(80)90409-2. [DOI] [PubMed] [Google Scholar]
- Gerace L, Foisner R. Integral membrane proteins and dynamic organization of the nuclear envelope. Trends Cell Biol. 1994;4:127–131. doi: 10.1016/0962-8924(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Glass JR, Gerace L. Lamins A and C bind and assemble at the surface of mitotic chromosomes. J Cell Biol. 1990;111:1047–1057. doi: 10.1083/jcb.111.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Hepler PK, Wolniak SM. Membranes in the mitotic apparatus: their structure and function. Int Rev Cytol. 1984;90:169–238. doi: 10.1016/s0074-7696(08)61490-4. [DOI] [PubMed] [Google Scholar]
- Hutchison, C.J. 1994. The use of cell-free extracts of Xenopus eggs for studying DNA replication in vitro. In The Cell Cycle: A Practical Approach. P. Fantes and R. Brookes, editors. Oxford IRL Press, UK. 177–195.
- Hutchison CJ, Cox R, Drepaul RS, Gomperts M, Ford CC. Periodic DNA synthesis in cell-free extracts of Xenopuseggs. EMBO (Eur Mol Biol Organ) J. 1987;6:2003–2010. doi: 10.1002/j.1460-2075.1987.tb02464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins H, Holman T, Lyon C, Lane B, Stick R, Hutchison C. Nuclei that lack a lamina accumulate karyophilic proteins and assemble a nuclear matrix. J Cell Sci. 1993;106:275–285. doi: 10.1242/jcs.106.1.275. [DOI] [PubMed] [Google Scholar]
- Latterich M, Schekman R. The karyogamy gene KAR2 and novel proteins are required for ER-membrane fusion. Cell. 1994;78:87–98. doi: 10.1016/0092-8674(94)90575-4. [DOI] [PubMed] [Google Scholar]
- Lourim D, Krohne G. Lamin-dependent nuclear envelope reassembly following mitosis: An argument. Trends Cell Biol. 1994;4:314–318. doi: 10.1016/0962-8924(94)90228-3. [DOI] [PubMed] [Google Scholar]
- Lourim D, Krohne G. Membrane-associated lamins in Xenopusegg extracts: Identification of two vesicle populations. J Cell Biol. 1993;123:501–512. doi: 10.1083/jcb.123.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon, C. 1995. Immunological studies of the molecular basis of nuclear envelope structure. Ph.D. Thesis. University of Dundee.
- Marshall ICB, Wilson KL. Nuclear envelope assembly after mitosis. Trends Cell Biol. 1997;7:69–74. doi: 10.1016/S0962-8924(96)10047-7. [DOI] [PubMed] [Google Scholar]
- Newport J, Dunphy W. Characterization of the membrane binding and fusion events during nuclear envelope assembly using purified components. J Cell Biol. 1992;116:295–306. doi: 10.1083/jcb.116.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport JW, Wilson KL, Dunphy WG. A lamin-independent pathway for nuclear envelope assembly. J Cell Biol. 1990;111:2247–2259. doi: 10.1083/jcb.111.6.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaviano Y, Gerace L. Phosphorylation of the nuclear lamins during interphase and mitosis. J Biol Chem. 1985;260:624–632. [PubMed] [Google Scholar]
- Pfaller R, Smythe C, Newport JW. Assembly/disassembly of the nuclear envelope membrane: cell cycle-dependent binding of nuclear membrane vesicles to chromatin in vitro. Cell. 1991;65:209–217. doi: 10.1016/0092-8674(91)90155-r. [DOI] [PubMed] [Google Scholar]
- Pyrpasopoulou A, Meier J, Maison C, Simos G, Georgatos SD. The lamin B receptor (LBR) provides essential chromatin docking sites at the nuclear envelope. EMBO (Eur Mol Biol Organ) J. 1996;15:7108–7119. [PMC free article] [PubMed] [Google Scholar]
- Rabouille C, Levine TP, Peters JM, Warren G. An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell. 1995;82:905–914. doi: 10.1016/0092-8674(95)90270-8. [DOI] [PubMed] [Google Scholar]
- Roos UP. Light and electron microscopy of rat kangaroo cells in mitosis. I. Formation and breakdown of the mitotic apparatus. Chromosoma. 1973;40:43–82. doi: 10.1007/BF00319836. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Warren G. Implications of the snare hypothesis for intracellular membrane topology and dynamics. Curr Biol. 1994;4:220–233. doi: 10.1016/s0960-9822(00)00051-8. [DOI] [PubMed] [Google Scholar]
- Smythe C, Newport J. Systems for the study of nuclear assembly, DNA replication, and nuclear breakdown in Xenopus laevisegg extracts. Methods Cell Biol. 1991;35:449–468. doi: 10.1016/s0091-679x(08)60583-x. [DOI] [PubMed] [Google Scholar]
- Stick R, Angres B, Lehner CF, Nigg EA. The fates of chicken nuclear lamin proteins during mitosis: evidence for a reversible redistribution of lamin B2 between inner nuclear membrane and elements of the endoplasmic reticulum. J Cell Biol. 1988;107:397–406. doi: 10.1083/jcb.107.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitzur N, Harel A, Feinstein N, Gruenbaum Y. Lamin activity is essential for nuclear envelope assembly in a Drosophilaembryo cell-free extract. J Cell Biol. 1992;119:17–25. doi: 10.1083/jcb.119.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigers GP, Lohka MJ. A distinct vesicle population targets membranes and pore complexes to the nuclear envelope in Xenopuseggs. J Cell Biol. 1991;112:545–556. doi: 10.1083/jcb.112.4.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigers GPA, Lohka MJ. Regulation of nuclear envelope precursor functions during cell division. J Cell Sci. 1992;102:273–284. doi: 10.1242/jcs.102.2.273. [DOI] [PubMed] [Google Scholar]
- Wiese C, Goldberg MW, Allen TD, Wilson KL. Nuclear envelope assembly in Xenopusextracts visualized by scanning EM reveals a transport-dependent “envelope smoothing” event. J Cell Sci. 1997;110:1489–1502. doi: 10.1242/jcs.110.13.1489. [DOI] [PubMed] [Google Scholar]
- Wilson KL, Newport J. A trypsin-sensitive receptor on membrane vesicles is required for nuclear envelope formation in vitro. J Cell Biol. 1988;107:57–68. doi: 10.1083/jcb.107.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worman HJ, Yuan J, Blobel G, Georgatos SD. A lamin B receptor in the nuclear envelope. Proc Natl Acad Sci USA. 1988;85:8531–8534. doi: 10.1073/pnas.85.22.8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Guan T, Gerace L. Integral membrane proteins of the nuclear envelope are dispersed throughout the endoplasmic reticulum during mitosis. J Cell Biol. 1997;137:1199–1210. doi: 10.1083/jcb.137.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Worman HJ. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J Biol Chem. 1996;271:14653–14656. doi: 10.1074/jbc.271.25.14653. [DOI] [PubMed] [Google Scholar]
- Zeligs JD, Wollman SH. Mitosis in thyroid epithelial cells in vivo. V. Characteristic apical protrusions are associated with mitosis in unstimulated glands. J Ultrastruct Res. 1979;68:308–316. doi: 10.1016/s0022-5320(79)90162-x. [DOI] [PubMed] [Google Scholar]