

Abstract
At mitosis, focal adhesions disassemble and the signal transduction from focal adhesions is inactivated. We have found that components of focal adhesions including focal adhesion kinase (FAK), paxillin, and p130CAS (CAS) are serine/threonine phosphorylated during mitosis when all three proteins are tyrosine dephosphorylated. Mitosis-specific phosphorylation continues past cytokinesis and is reversed during post-mitotic cell spreading.
We have found two significant alterations in FAK-mediated signal transduction during mitosis. First, the association of FAK with CAS or c-Src is greatly inhibited, with levels decreasing to 16 and 13% of the interphase levels, respectively. Second, mitotic FAK shows decreased binding to a peptide mimicking the cytoplasmic domain of beta-integrin when compared with FAK of interphase cells. Mitosis-specific phosphorylation is responsible for the disruption of FAK/CAS binding because dephosphorylation of mitotic FAK in vitro by protein serine/threonine phosphatase 1 restores the ability of FAK to associate with CAS, though not with c-Src. These results suggest that mitosis-specific modification of FAK uncouples signal transduction pathways involving integrin, CAS, and c-Src, and may maintain FAK in an inactive state until post-mitotic spreading.
Keywords: FAK, CAS, paxillin, mitosis, phosphorylation, c-Src
During mitosis, normal adherent cells show massive changes in their adhesive interactions with the extracellular matrix (ECM).1 During prophase, cells round up and lose attachments to a substrate. After cytokinesis, cells start to reattach and spread. The attachment to the substrate during early G1 phase is essential for cells to pass through the restriction point at each cell cycle (O'Neill et al., 1986), indicating that cell adhesion transmits a signal for cell cycle progression. The cycle of attachment and detachment during the cell cycle is thus critical to proliferation of normal adherent cells.
Focal adhesions are responsible for the cell-substrate adhesion of fibroblasts and other adhesive animal cells (reviewed in Schaller and Parsons, 1994; Jockusch et al., 1995; Burridge and Chrzanowska-Wodnicka, 1996; Craig and Johnson, 1996). They are specialized sites that link the extracellular matrix and the actin cytoskeleton. Since bundles of actin filaments (stress fibers) are anchored to focal adhesions, focal adhesions regulate the organization of stress fibers, thereby controlling cell morphology. In addition to this structural role, focal adhesions are involved in integrin-mediated signal transduction. This is evident from the fact that focal adhesions contain a variety of enzymes involved in signal transduction including protein kinase C, tyrosine kinases, and tyrosine phosphatases (Rohrschneider, 1980; Hanks et al., 1992; Schaller et al., 1992; Liao and Jaken, 1993; Serra-Pages et al., 1995; Shen et al., 1998). Thus, the loss of adhesion during mitosis should not only lead to the disassembly of focal adhesions, but also cause inactivation of integrin-mediated signal transduction. While the importance of the modulation of focal adhesion activities in synchrony with the cell cycle is recognized, little is known regarding the mechanisms responsible for the alterations in these activities during mitosis and post-mitotic spreading. Although mitotic cells and trypsinized cells are both rounded with disassembled focal adhesions and stress fibers, they are clearly different in terms of their responsiveness to ECM. Trypsinized cells immediately begin to reattach and spread when replated on ECM-coated substrates, focal adhesions are reassembled, and certain signals are transduced to allow cell spreading and proliferation. In contrast, mitotic cells stay detached until the completion of cytokinesis, even if they are placed on ECM. There must be distinctive mechanisms in mitotic cells that suppress the formation of focal adhesions and inhibit signal transduction.
Focal adhesion kinase (FAK) appears to be a primary mediator of integrin-mediated signal transduction (reviewed in Parsons et al., 1994; Schaller and Parsons, 1994; Richardson and Parsons, 1995; Hanks and Polte, 1997). FAK is a tyrosine kinase prominently localized at focal adhesions (Hanks et al., 1992; Schaller et al., 1992), and tyrosine phosphorylated FAK binds to the SH2 domains of Src (Schaller et al., 1994; Xing et al., 1994; Eide et al., 1995; Schlaepfer et al., 1997) and Grb2 (Schlaepfer et al., 1994, 1997). In addition, FAK is known to bind to a variety of other signaling proteins, including paxillin (Turner and Miller, 1994; Tachibana et al., 1995), PI-3 kinase (Chen and Guan, 1994; Bachelot et al., 1996; Chen et al., 1996; Saito et al., 1996), p130CAS (CAS) (Polte and Hanks, 1995, 1997; Harte et al., 1996), talin (Chen et al., 1995), and the cytoplasmic domain of integrin beta subunit (Schaller et al., 1995). Among these FAK-associated proteins, both paxillin and CAS are considered as nonenzymatic “docking” proteins (Sakai et al., 1994; Turner, 1994; Turner and Miller, 1994; Burnham et al., 1996; Nojima et al., 1996; Vuori et al., 1996; Klemke et al., 1998). Paxillin is phosphorylated at tyrosine residues during integrin-mediated cell attachment and binds to vinculin, FAK, and Crk (Burridge et al., 1992; Birge et al., 1993; Turner, 1994; Turner and Miller, 1994; Wood et al., 1994; Hildebrand et al., 1995; Schaller and Parsons, 1995; Tachibana et al., 1995). CAS binds to FAK via its SH3 domain (Polte and Hanks, 1995, 1997; Harte et al., 1996), and is also known to associate with other signal transduction molecules, including Src, Crk (Sakai et al., 1994; Petch et al., 1995; Burnham et al., 1996; Nakamoto et al., 1996), and PTP1B (Liu et al., 1996). It is further suggested that the association of Src with either CAS or FAK leads to the activation of Src (Hanks and Polte, 1997). Thus, FAK, CAS, and paxillin are likely to play a central role in signal transduction mediated via focal adhesions.
In this report we investigated possible mechanisms underlying mitosis-induced focal adhesion disassembly and inactivation of integrin-mediated signaling. We found that FAK, paxillin, and CAS each become prominently phosphorylated on serine and/or threonine residues in a mitosis-specific fashion, coincident with their becoming dephosphorylated on tyrosine. We also found that the FAK/ CAS/c-Src signaling complex is dissociated during mitosis. While FAK tyrosine dephosphorylation appears to disrupt the FAK/c-Src interaction, we found that the FAK/CAS dissociation is due to mitosis-specific serine phosphorylation of FAK. We also observed that mitotic FAK shows decreased binding to the cytoplasmic domain of integrin beta subunit. Our findings suggest that mitosis-specific phosphorylation of FAK and other focal adhesion components contributes to the arrest of integrin-associated signaling activities during mitosis.
Materials and Methods
Cell Culture
SV-40 transformed rat embryo cells (REF-2A) were maintained in DME containing 10% newborn calf serum (NCS) in an atmosphere of 5% CO2 and 95% air at 37°C. REF-2A cells showed anchorage dependence for cell growth, and exhibited stress fibers and focal adhesions. Balb/C/3T3 cells and HeLa cells were maintained in DME containing 10% calf serum.
Preparation of Cells and Synchronization for Cell Division
Mitotic cells were prepared as described in Yamashiro et al. (1990). Briefly, 6–10 large dishes (245 × 245 mm; Nalge Nunc International) of REF-2A cells (subconfluent) were treated for 3 h with 0.25 μg/ml nocodazole, and collected by the shake-off method. In some experiments, mitotic cells were collected by the shake-off method without treatment of nocodazole. Trypsinized cells were prepared from one large dish (245 × 245 mm) by incubation with trypsin (0.05% trypsin and 2 mM EDTA in PBS), followed by inactivation of trypsin with the addition of 3% BSA and 0.5 mg/ ml of soybean trypsin inhibitor. Trypsinized cells were collected and incubated for 30 min in DME containing 1% NCS before harvest. Interphase cells were also collected from one large dish (245 × 245 mm).
Cells at later stages of cell division (such as cytokinesis or post-mitotic cell spreading) were prepared as described in Hosoya et al. (1993). Briefly, cells were first treated for 3 h with 0.25 μg/ml nocodazole, and mitotic cells were collected (prometaphase). After being washed with cold DME to remove nocodazole, cells were plated into fresh culture dishes, and incubated at 37°C in DME containing 10% NCS to allow cell cycle progression. Cell morphologies at different mitotic stages were checked by phase-contrast light microscopy. As reported previously (Hosoya et al., 1993), these cells show synchronized cell division in a reasonably short time window. Mitotic cells recovered spindles after 10–20 min of incubation following release of nocodazole arrest, and underwent cytokinesis at 40–60 min. Post-mitotic cell spreading occurred in 80–180 min, at which time both stress fibers and focal adhesions were reassembled. Mitotic 3T3 and HeLa cells were also prepared as above.
In some experiments, REF-2A cells at mitosis were labeled with 32P- orthophosphoric acid as described previously (Yamashiro et al., 1990; Hosoya et al., 1993). Briefly, cells were incubated for 3 h in phosphate-free DME containing 10% dialyzed NCS, 0.25 μg/ml nocodazole, and 0.125 mCi/ml 32P-orthophosphoric acid and mitotic cells were collected. Interphase cells were labeled in the same way except that nocodazole was omitted.
Immunoprecipitation
Immunoprecipitation was performed under the following three conditions. We found that FAK immunoprecipitates prepared under condition II gave consistent results in examination of FAK-associated proteins including CAS, c-Src, paxillin, and talin.
Condition I.
Cells at different stages of mitosis were solubilized in buffer I (10 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% NP-40, 1 mM EDTA, 1 mM EGTA, 0.2 mM sodium vanadate, 20 mM NaF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 0.2 mM PMSF), and stored in a −80°C freezer. After being thawed quickly, the lysates were homogenized by several passes through a 25-gauge needle, and centrifuged at 100,000 g for 15 min. 2–3 μg of mouse mAb against FAK, paxillin (Transduction Laboratories), or rabbit polyclonal antibody (pAb) against FAK or CAS (Santa Cruz Biotechnology, Inc.) was added to the supernatants. After incubation at 4°C for 1.5 h, protein A–Sepharose-conjugated rabbit antibody against mouse IgG (for mAb) or protein A–Sepharose (for pAb) was added, and incubation continued for 1 h. After extensive washing with buffer I, the immunocomplexes were analyzed by SDS-PAGE, followed by immunoblotting.
Condition II.
Cells were lysed with buffer II (50 mM Hepes, 150 mM NaCl, 10% glycerol, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM PMSF, 25 mM NaF, 1 mM sodium vanadate, and 50 mM beta-glycerophosphate), and homogenized by a Dounce homogenizer. After centrifugation at 16,000 g for 20 min, the cell extracts (made to an equal protein concentration of 5–10 mg/ml) were incubated with rabbit pAb against the COOH-terminal peptides of FAK or CAS (Santa Cruz Biotechnology, Inc.) or mouse mAb against paxillin (Transduction Laboratories), followed by incubation with protein A–Sepharose beads (Pharmacia). The immunocomplexes were washed three times with buffer II, analyzed by SDS-PAGE, followed by immunoblotting. In some experiments, protein A–conjugated primary antibodies were used.
Condition III.
Cells were lysed in buffer III (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 0.05% SDS, 0.5% sodium deoxycholate, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM PMSF, 25 mM NaF, 1 mM sodium vanadate, and 50 mM beta-glycerophosphate), and processed as described for condition II.
Immunoblotting
The following antibodies were used for immunoblotting: antibodies against FAK (mAb, Transduction Laboratories; pAb, Santa Cruz Biotechnology, Inc.), paxillin (Transduction Laboratories), phosphotyrosine (PY20; Transduction Laboratories), CAS (Transduction Laboratories), PI-3 kinase (Transduction Laboratories), c-Src (mAb; Upstate Biotechnology; pAb, Santa Cruz Biotechnology, Inc.), Crk (Santa Cruz Biotechnology, Inc.), cyclin B1 (Oncogene Science, Inc.), and talin (Sigma Chemical Co.). For preparation of total cell lysates, cells at different stages of mitosis were lysed in 2× SDS-PAGE sample buffer. The equivalent amounts of total proteins were separated by SDS-PAGE, transferred to PVDF membranes, and analyzed by immunoblotting with the indicated antibodies. FAK immunoprecipitates were separated by SDS-PAGE, and analyzed by immunoblotting with the antibodies against FAK, CAS, c-Src, and phosphotyrosine. The blots were quantitated by densitometry to determine levels of FAK, CAS, c-Src, and phosphotyrosine. The levels of CAS, c-Src, and phosphotyrosine were normalized by the levels of FAK immunoprecipitate. The association of CAS and c-Src, as well as FAK phosphotyrosine levels, from mitotic and trypsinized cells was expressed by ratios to those of interphase FAK.
To determine the level of phosphotyrosine, immunoprecipitates of FAK, paxillin, or CAS were separated on SDS gels, and each was transferred to PVDF membranes. The membranes were first immunoblotted with antiphosphotyrosine antibody (PY20; Transduction Laboratories). After removal of the PY20 antibody, the same membrane blots of paxillin, FAK, and CAS immunoprecipitates were stripped and reprobed with anti-FAK, antipaxillin, and anti-CAS antibodies, respectively. The blots were quantitated by densitometry to determine ratios of phosphotyrosine levels divided by the levels of FAK, paxillin, and CAS.
Protein Phosphatase Treatment
FAK, paxillin, and CAS were immunoprecipitated under condition I from mitotic and interphase cells using specific antibodies, and half of the immunoprecipitates were treated for 30 min at 30°C with one unit of recombinant serine/threonine phosphatase (PP1, α-isoform from rabbit muscle; Calbiochem-Novabiochem Corp.) in 50 mM imidazole buffer, pH 7.0, containing 0.25 mM MnCl2, 5 mM DTT, 100 mM KCl, 2 mM MgCl2, and 0.2 mg/ml BSA. Under this condition, no tyrosine dephosphorylation was observed. Both treated and untreated samples were separated by SDS-PAGE, followed by immunoblotting with anti-FAK (Transduction Laboratories), antipaxillin (Transduction Laboratories), and anti-CAS (Transduction Laboratories) antibodies to examine changes in mobility shifts.
Reconstitution of a FAK/CAS Complex
The reconstitution of a FAK/CAS complex was performed by incubating FAK immunoprecipitates with interphase extracts. First, FAK was immunoprecipitated from mitotic cells under condition I, and the immunocomplex was divided into three equal aliquots. The first was treated with PP1 as described above, while the second two were untreated. All three samples were washed extensively with the immunoprecipitation buffer II and 1 μg of the antigenic peptide (CNLLDVIDQARLKMLG) was added in order to block the residual sites of FAK antibody. The PP1-treated sample and one untreated sample were incubated with an equivalent amount of interphase extracts, which had been prepared by homogenization of interphase cells in the immunoprecipitation buffer II, followed by centrifugation at 16,000 g for 20 min. The incubation with the interphase extracts continued for 30 min at 4°C. The other untreated sample was incubated with the immunoprecipitation buffer II alone. All three were again extensively washed with the same buffer, washed once with PBS, and analyzed by SDS-PAGE followed by Western blotting with antibodies against FAK, CAS, and c-Src.
Binding of FAK to a Cytoplasmic Peptide of Integrin Beta Subunit
Binding of FAK or paxillin to a peptide (called SP1; CKLLMIIHDRREFA) was performed as described by Schaller et al. (1995) as SP1 represents a FAK binding site within the cytoplasmic tail of the integrin beta subunit (Schaller et al., 1995). Briefly, varying concentrations (0.3–3 mg/ml) of the lysates of mitotic, interphase, or trypsinized cells were incubated for 60 min at 4°C with beads which had been conjugated with the SP1 peptide (Schaller et al., 1995). The beads were then washed five times with the lysis buffer, and the bound proteins were solubilized with SDS sample buffer, separated by SDS-PAGE, and analyzed by Western blotting using anti-FAK or antipaxillin antibody.
FAK Kinase Assay
The kinase activity of FAK immunoprecipitates was measured using a synthetic random co-polymer (GluTyr = 4:1, Sigma Chemical Co.) as a substrate. Immunoprecipitated FAK (under condition I or II) and the synthetic peptide were incubated at 30°C in the kinase assay buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM MnCl2, 0.1 mM ATP, and 2 μCi of gamma-[32P]ATP) for 20 min. The reaction mixtures were analyzed by SDS-PAGE followed by autoradiography. The amounts of FAK in immunoprecipitates were normalized by immunoblotting with anti-FAK antibody. The activities were determined by either densitometry of autoradiography or by measuring radioactivity of the phosphorylated co-polymer using a PhosphorImager (Molecular Dynamics Inc.).
Far Western Blot Analysis
The SH2 domain of chicken c-Src (amino acid residues 148–251) was expressed as a glutathione-S-transferase (GST) fusion protein in Escherichia coli and affinity purified by glutathione–agarose adsorption essentially as described by Guan and Dixon (1991). The GST-SrcSH2 fusion protein was used at 10 μg/ml to probe immunoprecipitated FAK immobilized on nitrocellulose membrane as described by Hildebrand et al. (1995). GST-SrcSH2 bound to FAK was detected by antibody against GST (1 μg/ml; Sigma Chemical Co.) followed by ECL with HRP-conjugated protein A (1:1,000; Amersham Life Science). The membrane was then stripped and reprobed with anti-FAK antibody (0.2 μg/ml; Santa Cruz Biotechnology) for normalization.
Other Procedures
SDS-PAGE was performed as described by Blatter et al. (1972) using 12.5% polyacrylamide, except the buffer system of Laemmli (1970) was used. Phosphoamino-acid analysis was performed as described (Hunter and Sefton, 1980). Protein concentrations were determined by the method of Bradford (1976) using BSA as a standard.
Results
Serine Phosphorylation Is Responsible for Mitosis-specific Mobility Shifts of FAK, CAS, and Paxillin
We observed that FAK, CAS, and paxillin showed SDS-PAGE mobility shifts during mitosis. FAK, CAS, and paxillin were immunoprecipitated under condition I from mitotic, interphase, or trypsinized cells, and immunoblotted with specific antibodies. Fig. 1 shows all three proteins of mitotic cells (lane M) showed slower mobilities than their counterparts from interphase and trypsinized cells (lanes I and T, respectively), indicating that these proteins are modified specifically during mitosis. Essentially the same mobility shifts were observed when these proteins were immunoprecipitated under other conditions (II or III, data not shown).
Figure 1.
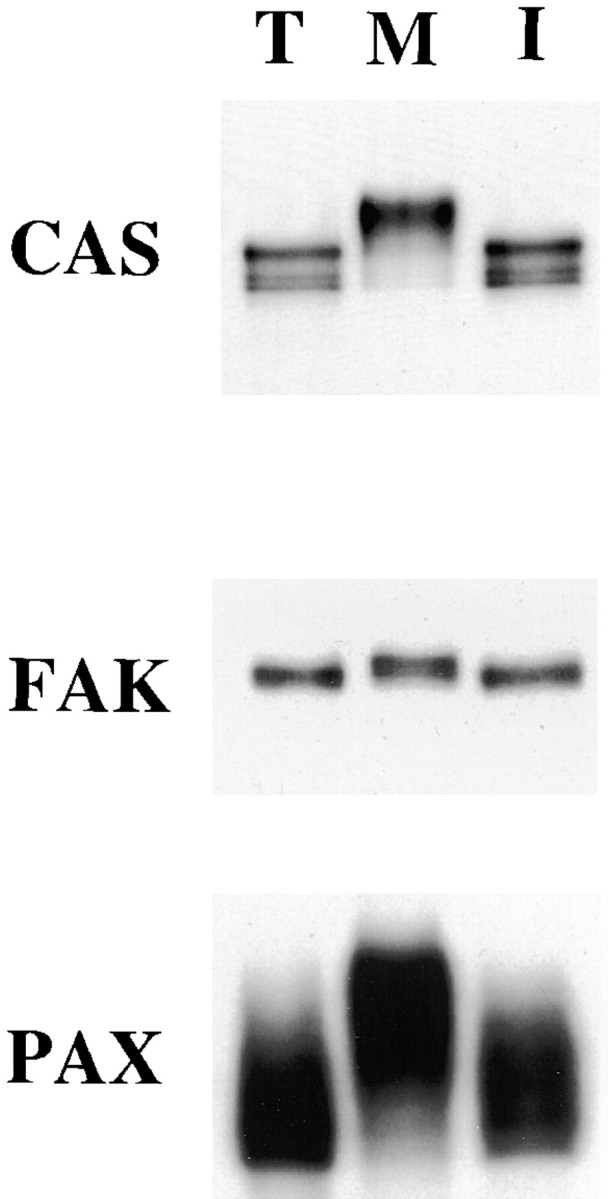
Mitosis-specific modification of CAS, FAK, and paxillin. CAS, FAK, and paxillin (PAX) were immunoprecipitated under condition I from trypsinized (lane T), mitotic (lane M), and interphase (lane I) cells and immunoblotted with the specific antibodies against each protein. All three proteins show slower electrophoretic mobility during mitosis, indicating that FAK, CAS, and paxillin undergo mitosis-specific modification.
To test whether the mitotic modification is serine/threonine phosphorylation, we examined the effect of phosphatase treatment on the mobility shifts. FAK, CAS, and paxillin were again immunoprecipitated from mitotic and interphase cells. One-half of each immunoprecipitate was treated with serine/threonine protein phosphatase 1 (PP1), and immunoblotted with specific antibodies. As Fig. 2 a shows, PP1 treatment eliminated or greatly decreased the mobility shifts shown by mitotic FAK, CAS, and paxillin, indicating that serine/threonine phosphorylation is largely responsible for the mobility shifts.
Figure 2.

Serine phosphorylation is responsible for the mobility shifts. (a) Loss of mobility shifts by the treatment with PP1. FAK, CAS, and paxillin (PAX) were immunoprecipitated under condition I from mitotic (odd numbered lanes) or from interphase (even numbered lanes) cells, and half of the immunoprecipitates (lanes 3, 4, 7, 8, 11, and 12) were treated with PP1 to dephosphorylate phosphoserine and phosphothreonine. Note that PP1 treatment eliminates or greatly decreases the mobility shifts shown by mitotic FAK (compare lanes 1 and 3), CAS (compare lanes 5 and 7), and paxillin (compare lanes 9 and 11). (b) Tyrosine dephosphorylation of FAK, CAS, and paxillin during mitosis. FAK, CAS, and paxillin (PAX) were again immunoprecipitated under condition I from mitotic (lanes M) and interphase (lanes I) cells. The immunoprecipitates were first immunoblotted with PY20 (top; lanes 1, 2, 5, 6, 9, and 10), and then reprobed with FAK, CAS, and paxillin antibodies (bottom; lanes 3, 4, 7, 8, 11, and 12). Note that PY20 reacts strongly with the immunoprecipitates prepared from interphase but not from mitotic cells, indicating that mitotic FAK, CAS, and paxillin are dephosphorylated at tyrosine residues. (c) Phosphate incorporation of FAK, CAS, and paxillin. FAK, CAS, and paxillin were immunoprecipitated under condition I from mitotic (lanes M) and interphase (lanes I) cells that had been labeled in vivo with 32P inorganic phosphate. The 32P-labeled immunoprecipitates were analyzed by SDS-PAGE followed by autoradiography. The levels of 32P incorporation in mitotic FAK, CAS, and paxillin are not greatly increased because serine phosphorylation is negated by tyrosine dephosphorylation.
We then analyzed the tyrosine phosphorylation levels of the three proteins during mitosis. FAK, CAS, and paxillin were immunoprecipitated from mitotic and interphase cells, and blotted with PY20. The same membranes were reprobed with individual antibodies to confirm that approximately equal amounts of the proteins were immunoprecipitated. As Fig. 2 b shows, interphase FAK, CAS, and paxillin strongly reacted with PY20. In contrast, PY20 antibody did not detect mitotic FAK, CAS, and paxillin, indicating that all three proteins were tyrosine dephosphorylated during mitosis. We further examined 32P incorporation of these proteins by immunoprecipitating mitotic and interphase proteins from in vivo 32P-labeled cells, followed by SDS-PAGE and autoradiography. As Fig. 2 c shows, the levels of 32P incorporation were not markedly increased during mitosis. This is because 32P incorporation by mitotic serine/threonine phosphorylation was negated by simultaneous tyrosine dephosphorylation.
Dissociation of FAK/CAS/c-Src Complex during Mitosis
To explore the functional significance of mitosis-specific phosphorylation of FAK, we examined whether the association between FAK and FAK-associated proteins (including CAS, paxillin, c-Src, and talin) was altered. We found that the levels of CAS and c-Src associated with FAK immunoprecipitates were greatly reduced in mitotic cells (Fig. 3 a). In contrast, similar levels of paxillin or talin associated with FAK in mitotic or interphase cells were found (data not shown). Quantitative analyses (Fig. 3 b) reveal the levels of CAS or c-Src associated with mitotic FAK are 16 ± 9% and 13 ± 10% of those associated with interphase FAK. The difference is not due to the rounded morphology of mitotic cells because FAK immunoprecipitated from rounded trypsinized cells was associated with considerable amounts of CAS and c-Src. The levels of CAS and c-Src were 83 ± 23% and 70 ± 34% of the interphase levels, respectively. The dissociation of FAK/CAS/ c-Src complex in mitotic cells is not due to nocodazole treatment because similar dissociation was observed with mitotic cells prepared without nocodazole treatment.
Figure 3.

Dissociation of a FAK/ CAS/c-Src complex during mitosis. (a) FAK was first immunoprecipitated under condition II from trypsinized (lane T), mitotic (lane M), and interphase (lane I) cells, and analyzed by immunoblotting with anti-FAK, anti-CAS, and anti-c-Src. Phosphotyrosine levels of immunoprecipitated FAK were examined by PY20. Note that FAK from mitotic cells showed much reduced association with CAS or c-Src. (b) Quantitative analyses of the association of CAS and c-Src with FAK, and phosphotyrosine levels of FAK immunoprecipitated from trypsinized, mitotic, and interphase cells. The CAS and c-Src association as well as FAK phosphotyrosine levels were expressed as ratios to those of interphase FAK. Data were obtained from five independent experiments. (c) Phosphoamino-acid analyses of FAK immunoprecipitated from 32P-labeled, interphase, mitotic, and trypsinized cells. PS, phosphoserine; PT, phosphothreonine; PY, phosphotyrosine.
Differences in FAK phosphorylation among interphase, mitotic and trypsinized cells were further compared using phosphoamino-acid analysis and immunoblotting with the PY20 antibody. Phosphoamino-acid analysis revealed that FAK from both mitotic and trypsinized cells was tyrosine dephosphorylated relative to interphase FAK (Fig. 3 c). In addition, it showed that mitotic phosphorylation occurred at serine residues but not at threonine residues. Immunoblots with PY20 also revealed that FAK from both trypsinized and mitotic cells had greatly reduced levels of phosphotyrosine when compared with interphase FAK (Fig. 3 a, bottom panel). These results suggest that mitotic serine phosphorylation may be involved in the dissociation of a FAK/CAS/c-Src complex during mitosis. However, quantitative analysis revealed that FAK from trypsinized cells reacted to PY20 to a greater extent than did mitotic FAK; the levels of PY20 reactivity of FAK from trypsinized and mitotic cells are 23 ± 11% and 8 ± 7% of the level of interphase FAK, respectively (Fig. 3 b). Because FAK is tyrosine phosphorylated at multiple sites (Schaller and Parsons, 1994; Richardson and Parsons, 1995; Hanks and Polte, 1997), a possibility remained that residual tyrosine phosphorylation of FAK in trypsinized cells would occur at critical sites, which may be sufficient to retain the association of FAK with CAS or c-Src.
Mitotic Serine Phosphorylation Is Responsible for the Dissociation of FAK and CAS
To test whether mitosis-specific serine phosphorylation or tyrosine dephosphorylation causes the disruption of the FAK/CAS/c-Src complex, we dephosphorylated mitotic FAK by PP1, and examined whether serine dephosphorylated FAK was able to reassociate with CAS or c-Src. FAK was first immunoprecipitated from mitotic cells under condition I. One-third of the immunoprecipitate was used to confirm that neither CAS nor c-Src is associated with mitotic FAK (lane 1 of Fig. 4 a). The rest of the immunoprecipitate was divided in half; one (lane 2) was dephosphorylated by PP1, and the other (lane 3) was untreated. Fig. 4 a shows PP1 treatment resulted in the loss of mobility shift of FAK, confirming dephosphorylation of mitotic FAK. Both PP1 treated and untreated FAK were incubated with interphase extracts. After extensive washing, the binding of CAS or c-Src was examined by immunoblotting. Dephosphorylated FAK (Fig. 4 a, lane 2) was able to reassociate with CAS of interphase extracts (second panel). The level of CAS reassociation was comparable to interphase FAK immunoprecipitates (lane 4). In contrast, the phosphatase untreated mitotic FAK (lane 3) showed reduced binding to CAS. Quantitative analyses revealed that the binding of CAS to dephosphorylated FAK is more than three times higher than for untreated FAK (Fig. 4 b).
Figure 4.

Reconstitution of a FAK/CAS complex following dephosphorylation of mitotic FAK. (a) FAK was immunoprecipitated under condition I from mitotic cells and divided into three equal parts for further treatment: lane 1, neither incubated with interphase extracts nor treated with PP1; lane 2, incubated with interphase extracts following treatment with PP1; lane 3, incubated with interphase extracts. After extensive washing, the association of FAK with CAS was examined by blotting with the anti-FAK antibody or anti-CAS antibody. Phosphotyrosine levels of FAK were examined by PY20. For comparison, FAK immunoprecipitates from interphase cells were blotted with the same antibodies (lane 4). (b) Quantitative analyses. The CAS reassociation and phosphotyrosine levels of FAK are expressed as ratios to those found in FAK incubated with interphase extracts but without prior PP1 treatment (−PP1). Data were obtained from five independent experiments.
The reassociation of FAK with CAS is not due to selective tyrosine rephosphorylation of FAK during incubation with interphase extracts. Immunoblot analyses revealed that the FAK samples, either with or without incubation with interphase extracts, or with or without prior PP1 treatment, showed almost no reactivity to PY20 (third panel, Fig. 4; compare with the reactivity of interphase FAK, lane 4). Quantitative analyses confirmed that the phosphotyrosine levels of all FAK samples were as low as mitotic FAK (Fig. 4 b). We also examined whether the FAK samples showed any difference in the binding to the SH2 domain of chick Src. It seemed possible that PY20 might not detect a specific phosphotyrosine site, such as phosphorylation at Tyr-397 of FAK (the Src SH2 domain is known to bind to FAK phosphorylated at Tyr-397). It was found that the SH2 domain of chick Src showed an equally low binding to all FAK samples, suggesting that differential phosphorylation at Tyr-397 did not occur during incubation with interphase extracts (data not shown). We conclude that mitosis-specific serine phosphorylation of FAK, but not tyrosine dephosphorylation, is responsible for the disruption of the FAK/CAS association.
Unlike the FAK/CAS binding, the association between FAK and c-Src was not reconstituted by serine dephosphorylation of FAK. The anti–c-Src antibody could not detect c-Src in any of the FAK preparations (data not shown). These results suggest that near complete tyrosine dephosphorylation observed during mitosis is likely to be involved in the FAK/c-Src dissociation, and that the low level tyrosine phosphorylation seen in trypsinized cells is responsible for the retained FAK/c-Src association.
Mitotic FAK Shows Reduced Binding to the Cytoplasmic Domain of Integrin Beta Subunit
Both FAK and paxillin have been reported to bind directly to a peptide (SP1) representing a membrane-proximal region of the cytoplasmic domain of the beta subunit of integrin (Schaller et al., 1995). We examined whether mitotic FAK or paxillin exhibited altered binding to the SP1 peptide. SP1 peptide–conjugated beads were incubated in varying concentrations of extracts prepared from mitotic, interphase, and trypsinized cells. After washing, levels of bound FAK and paxillin were detected by immunoblotting. As Fig. 5 a shows, mitotic FAK bound to SP1 peptide–conjugated beads less (lane M) than FAK from interphase (lane I), or trypsinized (lane T) cells. Quantitative analysis (Fig. 5 b) revealed the binding of mitotic FAK is 24–33% of the level shown by interphase FAK, while FAK from trypsinized cells showed 65–85% binding of the interphase level (Fig. 5 b). The binding of paxillin to SP1 beads, in contrast, revealed no difference between mitotic and interphase cells (data not shown).
Figure 5.

Reduced binding of mitotic FAK to a peptide mimicking the cytoplasmic domain of integrin beta subunit. (a) Total cell lysates were prepared from trypsinized (lane T), mitotic (lane M), and interphase (lane I) cells, and were immunoblotted with the anti-FAK antibody to confirm that they contain approximately equal amounts of FAK. SP-1 peptide–conjugated Sepharose beads were incubated with diluted (9×) extracts of trypsinized (lane T), mitotic (lane M), and interphase (lane I) cells. After extensive washing, bound fractions were analyzed by SDS-PAGE, followed by immunoblotting with the anti-FAK antibody. (b) Quantitative analyses of SP1 binding. The extracts from trypsinized (T), mitotic (M), and interphase (I) were serially diluted as indicated and bound FAK was determined by densitometry of immunoblots using the FAK antibody. The levels of bound FAK were expressed as ratios to the level of bound FAK after incubation with 9× diluted interphase extracts.
Reversal of Mitosis-specific Phosphorylation during Post-mitotic Cell Spreading
We examined at which stages of cell division mitosis-specific phosphorylation reversed, because such information would be useful to conceive the physiological roles of mitosis-specific phosphorylation. Nocodazole-arrested mitotic cells were collected and released to allow entry into G1, and total cell lysates were prepared at different stages of metaphase-G1 transition. The mobilities of FAK, CAS, and paxillin were analyzed by immunoblotting. As Fig. 6 shows, the mobility shifts of FAK, CAS, and paxillin continue for 80–120 min after the release of metaphase arrest, with FAK exhibiting the fastest recovery. At 120 min FAK mobility appeared normal, while significant amounts of paxillin and CAS still retained slower mobilities. It should be noted that this time point (120 min) corresponded to post-mitotic cell spreading, long after cytokinesis (which occurred during 40–60 min). Metaphase–anaphase transition was confirmed by a cyclin B immunoblot. The cyclin B band started to disappear at 40 min (Fig. 6).
Figure 6.

Reversal of mobility shifts of FAK, CAS, and paxillin (PAX) during post-mitotic cell spreading. Mobility shifts were examined by immunoblotting of total cell lysates prepared from interphase (I) or mitotic (M) cells, or cells released from mitotic arrest (numbered lanes, n = min removed from nocodazole). Total cell lysates were blotted on PVDF membranes, and the membranes were probed with the antibodies against CAS, FAK, paxillin, or cyclin B, as indicated. Cyclin B1 immunoblot is shown as an indicator of metaphase–anaphase transition. Note that CAS, FAK, and paxillin show reversal of mobility shifts during 80–180 min after the release of mitotic arrest, a time span corresponding to post-mitotic cell spreading.
Because tyrosine phosphorylation plays an important role in signal transduction and organization of focal adhesions, the time course of tyrosine rephosphorylation was also examined. Immunoprecipitates of FAK, paxillin, and CAS prepared at different stages of cell cycle were blotted with PY20, and reprobed with the antibodies against each protein for normalization. As Fig. 7 shows, the proteins were tyrosine rephosphorylated between 80 and 180 min after release of mitotic arrest (corresponding to post- mitotic cell spreading). FAK exhibited the fastest kinetics of tyrosine rephosphorylation and the largest increase in tyrosine phosphorylation at 180 min. The PY20 reactivity of FAK was increased threefold between 80 and 180 min, while paxillin gradually increased only 134%. CAS showed the slowest tyrosine rephosphorylation, the PY20 reactivity of CAS was <15% of the interphase level, even at 120 min. In the next 1 h, however, the PY20 reactivity of CAS increased to 200%.
Figure 7.
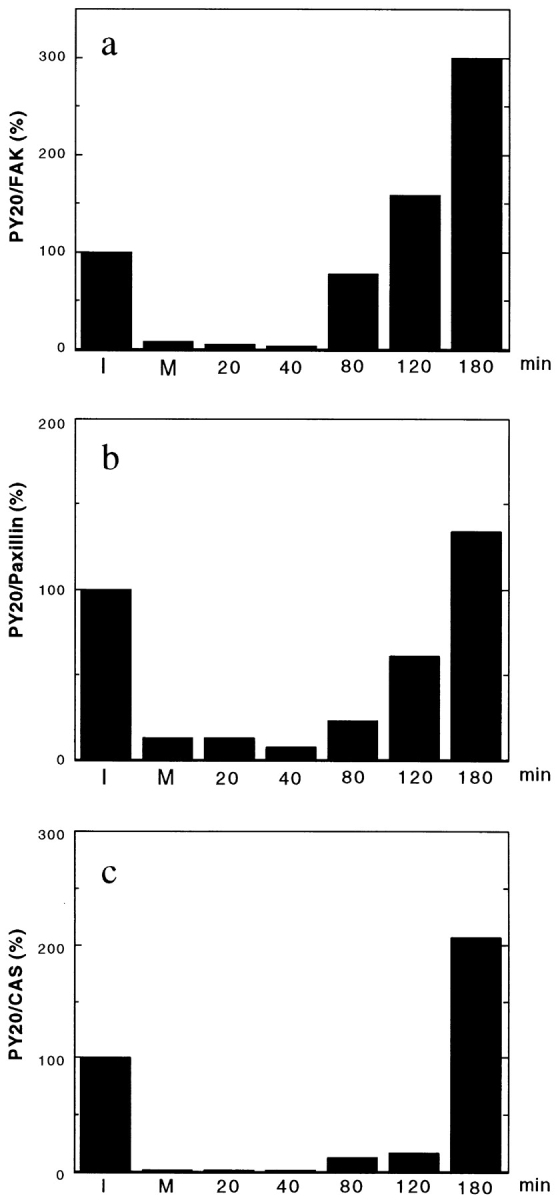
Tyrosine rephosphorylation of FAK (a), paxillin (b), and CAS (c) during post-mitotic cell spreading. FAK, CAS, and paxillin were immunoprecipitated from interphase cells (I), mitotic cells (M), and cells released from mitotic arrest (numbered lanes, n = min removed from nocodazole). The immunoprecipitates were transferred to PVDF membranes, first immunoblotted with PY20, then reprobed with the antibodies against FAK, paxillin, and CAS. The levels of phosphotyrosine are shown by ratios (100% for interphase level) of the levels of PY20 reactivities divided by the levels of FAK, paxillin, or CAS. Note that FAK exhibits the fastest recovery of tyrosine rephosphorylation as well as the greatest increase in tyrosine phosphorylation during 80– 180 min after the release of mitotic arrest.
The increase in tyrosine phosphorylation of FAK during post-mitotic cell spreading resulted in marked activation of FAK-associated tyrosine kinase activity. We measured tyrosine kinase activity associated with FAK immunoprecipitates in different stages of the cell cycle (condition I was used to prepare FAK free from associated c-Src or CAS). The activity of FAK toward poly (Glu/Tyr) at 180 min was twice as high as interphase FAK, while the activity of mitotic FAK was 10 times less than interphase FAK (data not shown). The results are consistent with the report that the enzymatic activity of FAK depends on its level of tyrosine phosphorylation (Calalb et al., 1995).
Discussion
We have demonstrated that FAK undergoes mitosis-specific, serine phosphorylation simultaneously with tyrosine dephosphorylation; during mitosis a FAK/CAS/c-Src complex is dissociated; and mitosis-specific serine phosphorylation of FAK causes the dissociation of FAK/CAS complex while tyrosine dephosphorylation appears to disrupt FAK/c-Src association.
Mechanisms for Dissociation of a FAK/CAS/c-Src Complex during Mitosis
FAK binds directly to the SH3 domain of CAS via proline-rich domains of FAK (Polte and Hanks, 1995, 1997; Harte et al., 1996). It is likely the mitosis-specific serine phosphorylation alters such interactions. It is interesting to note that one of the proline-rich domains contains serine, and it may be possible that phosphorylation would occur at this serine, reducing the interaction between FAK and CAS.
The association of c-Src with FAK or CAS, on the other hand, has been reported to depend on the binding between phosphotyrosine of FAK or CAS and the SH2 domain of c-Src (Schaller et al., 1994; Xing et al., 1994; Eide et al., 1995; Nakamoto et al., 1996; Schlaepfer et al., 1997). However, FAK from trypsinized cells retains a high level of association of c-Src. It seems likely that the residual tyrosine phosphorylation of FAK in trypsinized cells may be able to preserve the association between FAK and c-Src. Furthermore, a recent report has shown, in addition to the SH2-binding site, FAK has a Src SH3-binding site (Thomas et al., 1998). This SH3-mediated interaction could reinforce the retained association between FAK and c-Src in trypsinized cells.
Reduced Binding of Mitotic FAK to the Cytoplasmic Domain of Integrin Beta Subunit
The NH2-terminal noncatalytic domain of FAK was reported to be responsible for its binding to SP1 beads (Schaller et al., 1995). It is worthy to note that this domain does not contain tyrosine phosphorylation sites. Because FAK from both trypsinized and mitotic cells showed low tyrosine phosphorylation, it seems likely that mitosis-specific phosphorylation of FAK is involved in the reduced binding to the cytoplasmic domain of integrin beta subunit. At present, we cannot directly examine the effects of serine dephosphorylation of mitotic FAK on the binding to SP1 beads because purification of mitotic FAK has been unsuccessful. Therefore, we cannot exclude a possibility that tyrosine dephosphorylation is involved in the loss of FAK to SP1 beads.
Mitosis-specific Phosphorylation of CAS and Paxillin
We have demonstrated that CAS and paxillin, like FAK, show mitosis-specific serine phosphorylation. Currently, we do not know the functional consequence of the serine/ threonine phosphorylation of CAS or paxillin. CAS, either from interphase or mitotic cells, does not bind to mitotic FAK (Figs. 3 and 4). This suggests that mitosis-specific phosphorylation of CAS does not seem to be essential for the dissociation between CAS and FAK, although it is possible that phosphorylation of CAS further decreases the binding between FAK and CAS. In addition, the association of paxillin with FAK is not disturbed during mitosis. Both paxillin and CAS function as docking proteins. It is possible that mitosis-specific phosphorylation would disrupt the association with other proteins, which may contribute to the phenotypic alterations seen during mitosis. One interesting possibility would be mitosis-specific phosphorylation of CAS inhibiting cell motility of mitotic cells.
Physiological Significance of the Disruption of FAK/CAS/c-Src Complexes
First, the disruption of FAK/CAS association during mitosis is likely to result in uncoupling of the FAK- and CAS-mediated signal transduction during mitosis. The reduced binding of mitotic FAK to the cytoplasmic domain of integrin beta subunit (Fig. 5) would amplify the uncoupling effects by altering the interactions between ECM and focal adhesions. Further, the dissociation of the FAK/CAS/c-Src complex could abolish FAK-mediated c-Src activation during mitosis because the association of CAS or FAK with c-Src has been suggested to activate Src by displacing the inhibitory tail domain of Src from its catalytic domain (Hanks and Polte, 1997). Src has been reported to be activated during mitosis by dephosphorylation at Tyr-527 of the tail domain (Bagrodia et al., 1991; Shalloway et al., 1992), which appeared to be inconsistent with the above notion. However, the mitotic activation of Src occurs independently of FAK since FAK is neither tyrosine phosphorylated nor associated with Src during mitosis. A substrate of mitotic Src, sam68, does not seem to be involved in FAK-mediated signal transduction (Fumagalli et al., 1994; Taylor and Shalloway, 1994; Taylor et al., 1995).
Second, the uncoupling probably represents an “off” state of the FAK-mediated signaling pathway of mitotic cells, and this off state would be required for mitotic cells to switch “on” the signal transduction pathway at later stages of cell cycle. For proliferation, normal adherent cells must attach to a substrate during early G1 (O'Neill et al., 1986; Lo and Chen, 1994), at which time FAK-mediated signaling is believed to occur. The mitotic inactivation of FAK would thus act to reestablish the requirement of adhesion for the next round of cell cycle progression.
Third, the inactivation of FAK may contribute to the inhibition of cell spreading and/or cell migration in mitosis. FAK has been implicated to play roles in cell spreading (Richardson and Parsons, 1996; Richardson et al., 1997) and in cell migration (Cary et al., 1996; Gilmore and Romer, 1996). The disruption of FAK/CAS/c-Src complex during mitosis could be a mechanism for the inhibition of cell migration during mitosis because the FAK/CAS or CAS/Crk association has been shown to be important for cell migration by FAK (Cary et al., 1998; Klemke et al., 1998).
Mitosis-specific phosphorylation of FAK could block RhoA-mediated activation of FAK during cytokinesis. It has been reported that the activation of RhoA is required for the completion of cytokinesis because botulinum C3 exoenzyme (which inhibits Rho specifically) blocks cytokinesis (Kishi et al., 1993; Mabuchi et al., 1993). Activation of RhoA, on the other hand, is also known to occur upon serum stimulation of 3T3 fibroblasts, which induces the formation of both stress fibers and focal adhesions (Ridley and Hall, 1992, 1994; Ridley, 1995) as well as tyrosine phosphorylation of FAK (Seufferlein and Rozengurt, 1994a,b, 1995). FAK phosphorylation is blocked by botulinum C3 exoenzyme (Rankin et al., 1994), placing FAK downstream of RhoA. Mitotic cells should have a mechanism that suppresses Rho's ability to activate FAK, and mitosis-specific phosphorylation of FAK may be involved in such a mechanism. FAK phosphorylation continues through cytokinesis, and the reversal of the modification occurs during post-mitotic cell spreading (Fig. 6). FAK phosphorylation may act as an internal clock by preventing premature activation of FAK during cytokinesis. The lack of such phosphorylation in trypsinized cells would explain the rapid adhesion shown when they are plated on ECM-coated substrates.
The mitosis-specific modification of FAK, CAS, and paxillin is apparently a general phenomenon. We have observed similar mobility shifts with other types of adherent cultured cells including 3T3 and HeLa cells. A similar mobility shift has been reported for paxillin (Yamaguchi et al., 1997). We are in the process of identifying kinase(s) and phosphatase(s) responsible for the alterations in the phosphorylation states of these proteins. Cdc2 kinase is not responsible for the mitosis-specific phosphorylation (our unpublished data), which is consistent with the time course of phosphorylation, i.e., phosphorylation continues past cytokinesis. The identification and characterization of kinase(s) and phosphatase(s) would help us understand the mechanisms that define the timing of mitotic events including cell rounding during prophase, cytokinesis, and post-mitotic cell spreading.
Acknowledgments
We thank Dr. Frank Deis for critical reading of this manuscript.
This work is supported by grants R37-CA42742 (to F. Matsumura) and 2R01-GM49882 (to S.K. Hanks), from the National Institutes of Health.
Abbreviations used in this paper
- CAS
p130CAS
- ECM
extracellular matrix
- FAK
focal adhesion kinase
- PP1
protein phosphatase 1
Footnotes
Address correspondence to Fumio Matsumura, Dept. of Molecular Biology and Biochemistry, Nelson Labs/Busch Campus, Rutgers University, Piscataway, NJ 08855. Tel.: (732) 445-2838. Fax.: (732) 445-4213. E-mail: matsumura@mbcl.rutgers.edu
References
- Bachelot C, Rameh L, Parsons T, Cantley LC. Association of phosphatidylinositol 3-kinase, via the SH2 domains of p85, with focal adhesion kinase in polyoma middle t-transformed fibroblasts. Biochim Biophys Acta. 1996;1311:45–52. doi: 10.1016/0167-4889(95)00176-x. [DOI] [PubMed] [Google Scholar]
- Bagrodia S, Chackalaparampil I, Kmiecik TE, Shalloway D. Altered tyrosine 527 phosphorylation and mitotic activation of p60c-src. Nature. 1991;349:172–175. doi: 10.1038/349172a0. [DOI] [PubMed] [Google Scholar]
- Birge RB, Fajardo JE, Reichman C, Shoelson SE, Songyang Z, Cantley LC, Hanafusa H. Identification and characterization of a high- affinity interaction between v-Crk and tyrosine-phosphorylated paxillin in CT10-transformed fibroblasts. Mol Cell Biol. 1993;13:4648–4656. doi: 10.1128/mcb.13.8.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatter DP, Garner F, Van Slyke K, Bradley A. Quantitative electrophoresis in polyacrylamide gels of 2-40% J Chromatogr. 1972;64:147–155. [Google Scholar]
- Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Burnham MR, Harte MT, Richardson A, Parsons JT, Bouton AH. The identification of p130cas-binding proteins and their role in cellular transformation. Oncogene. 1996;12:2467–2472. [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Ann Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Burridge K, Turner CE, Romer LH. Tyrosine phosphorylation of paxillin and pp125FAK accompanies cell adhesion to extracellular matrix: a role in cytoskeletal assembly. J Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary LA, Chang JF, Guan JL. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J Cell Sci. 1996;109:1787–1794. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- Cary LA, Han DC, Polte TR, Hanks SK, Guan JL. Identification of p130(Cas) as a mediator of focal adhesion kinase-promoted cell migration. J Cell Biol. 1998;140:211–221. doi: 10.1083/jcb.140.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HC, Guan JL. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1994;91:10148–10152. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HC, Appeddu PA, Parsons JT, Hildebrand JD, Schaller MD, Guan JL. Interaction of focal adhesion kinase with cytoskeletal protein talin. J Biol Chem. 1995;270:16995–16999. doi: 10.1074/jbc.270.28.16995. [DOI] [PubMed] [Google Scholar]
- Chen HC, Appeddu PA, Isoda H, Guan JL. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:26329–26334. doi: 10.1074/jbc.271.42.26329. [DOI] [PubMed] [Google Scholar]
- Craig S, Johnson RP. Assembly of focal adhesions: progress, paradigms, and portents. Curr Opin Cell Biol. 1996;8:74–85. doi: 10.1016/s0955-0674(96)80051-2. [DOI] [PubMed] [Google Scholar]
- Eide BL, Turck CW, Escobedo JA. Identification of Tyr-397 as the primary site of tyrosine phosphorylation and pp60src association in the focal adhesion kinase, pp125FAK. Mol Cell Biol. 1995;15:2819–2827. doi: 10.1128/mcb.15.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli S, Totty NF, Hsuan JJ, Courtneidge SA. A target for Src in mitosis. Nature. 1994;368:871–874. doi: 10.1038/368871a0. [DOI] [PubMed] [Google Scholar]
- Gilmore AP, Romer LH. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol Biol Cell. 1996;7:1209–1224. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Polte TR. Signaling through focal adhesion kinase. Bioessays. 1997;19:137–145. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Calalb MB, Harper MC, Patel SK. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc Natl Acad Sci USA. 1992;89:8487–8491. doi: 10.1073/pnas.89.18.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harte MT, Hildebrand JD, Burnham MR, Bouton AH, Parsons JT. p130Cas, a substrate associated with v-Src and v-Crk, localizes to focal adhesions and binds to focal adhesion kinase. J Biol Chem. 1996;271:13649–13655. doi: 10.1074/jbc.271.23.13649. [DOI] [PubMed] [Google Scholar]
- Hildebrand JD, Schaller MD, Parsons JT. Paxillin, a tyrosine phosphorylated focal adhesion-associated protein binds to the carboxyl terminal domain of focal adhesion kinase. Mol Biol Cell. 1995;6:637–647. doi: 10.1091/mbc.6.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya N, Hosoya H, Yamashiro S, Mohri H, Matsumura F. Localization of caldesmon and its dephosphorylation during cell division. J Cell Biol. 1993;121:1075–1082. doi: 10.1083/jcb.121.5.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci USA. 1980;77:1311–1315. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jockusch BM, Bubeck P, Giehl K, Kroemker M, Moschner J, Rothkegel M, Rudiger M, Schluter K, Stanke G, Winkler J. The molecular architecture of focal adhesions. Annu Rev Cell Dev Biol. 1995;11:397–416. doi: 10.1146/annurev.cb.11.110195.002115. [DOI] [PubMed] [Google Scholar]
- Kishi K, Sasaki T, Kuroda S, Ithoh T, Takai Y. Regulation of cytoplasmic division of Xenopusembryo by rho p21 and its inhibitory GDP/ GTP exchange protein (rho GDP) J Cell Biol. 1993;120:1187–1195. doi: 10.1083/jcb.120.5.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke RL, Leng J, Molander R, Brooks PC, Vuori K, Cheresh DA. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J Cell Biol. 1998;140:961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4 . Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Liao L, Jaken S. Effect of alpha-protein kinase C neutralizing antibodies and the pseudosubstrate peptide on phosphorylation, migration, and growth of REF52 cells. Cell Growth Differ. 1993;4:309–316. [PubMed] [Google Scholar]
- Liu F, Hill DE, Chernoff J. Direct binding of the proline-rich region of protein tyrosine phosphatase 1B to the Src homology 3 domain of p130(Cas) J Biol Chem. 1996;271:31290–31295. doi: 10.1074/jbc.271.49.31290. [DOI] [PubMed] [Google Scholar]
- Lo SH, Chen LB. Focal adhesion as a signal transduction organelle. Cancer Metastasis Rev. 1994;13:9–24. doi: 10.1007/BF00690415. [DOI] [PubMed] [Google Scholar]
- Mabuchi I, Hamaguchi Y, Fujimoto H, Morii N, Mishima M, Narumiya S. A rho-like protein is involved in the organization of the contractile ring in dividing sand dollar eggs. Zygote. 1993;1:325–331. doi: 10.1017/s0967199400001659. [DOI] [PubMed] [Google Scholar]
- Nakamoto T, Sakai R, Ozawa K, Yazaki Y, Hirai H. Direct binding of C-terminal region of p130Cas to SH2 and SH3 domains of Src kinase. J Biol Chem. 1996;271:8959–8965. doi: 10.1074/jbc.271.15.8959. [DOI] [PubMed] [Google Scholar]
- Nojima Y, Mimura T, Morino N, Hamasaki K, Furuya H, Sakai R, Nakamoto T, Yazaki Y, Hirai H. Tyrosine phosphorylation of p130Cas in cell adhesion and transformation. Hum Cell. 1996;9:169–174. [PubMed] [Google Scholar]
- O'Neill C, Jordan P, Ireland G. Evidence for two distinct mechanisms of anchorage stimulation in freshly explanted and 3T3 Swiss mouse fibroblasts. Cell. 1986;44:489–496. doi: 10.1016/0092-8674(86)90470-8. [DOI] [PubMed] [Google Scholar]
- Parsons JT, Schaller MD, Hildebrand J, Leu TH, Richardson A, Otey C. Focal adhesion kinase: structure and signaling. J Cell Sci Suppl. 1994;18:109–113. doi: 10.1242/jcs.1994.supplement_18.16. [DOI] [PubMed] [Google Scholar]
- Petch LA, Bockholt SM, Bouton A, Parsons JT, Burridge K. Adhesion-induced tyrosine phosphorylation of the p130 src substrate. J Cell Sci. 1995;108:1371–1379. doi: 10.1242/jcs.108.4.1371. [DOI] [PubMed] [Google Scholar]
- Polte TR, Hanks SK. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130Cas. Proc Natl Acad Sci USA. 1995;92:10678–10682. doi: 10.1073/pnas.92.23.10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polte TR, Hanks SK. Complexes of focal adhesion kinase (FAK) and Crk-associated substrate (p130(Cas)) are elevated in cytoskeleton-associated fractions following adhesion and Src transformation. Requirements for Src kinase activity and FAK proline-rich motifs. J Biol Chem. 1997;272:5501–5509. doi: 10.1074/jbc.272.9.5501. [DOI] [PubMed] [Google Scholar]
- Rankin S, Morii N, Narumiya S, Rozengurt E. Botulinum C3 exoenzyme blocks the tyrosine phosphorylation of p125FAK and paxillin induced by bombesin and endothelin. FEBS Lett. 1994;354:315–319. doi: 10.1016/0014-5793(94)01148-6. [DOI] [PubMed] [Google Scholar]
- Richardson A, Parsons JT. Signal transduction through integrins: a central role for focal adhesion kinase? . Bioessays. 1995;17:229–236. doi: 10.1002/bies.950170309. [DOI] [PubMed] [Google Scholar]
- Richardson A, Parsons T. A mechanism for regulation of the adhesion-associated protein tyrosine kinase pp125FAK [published erratum appears in Nature.1996. 381:810] Nature. 1996;380:538–540. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- Richardson A, Malik RK, Hildebrand JD, Parsons JT. Inhibition of cell spreading by expression of the C-terminal domain of focal adhesion kinase (FAK) is rescued by coexpression of Src or catalytically inactive FAK: a role for paxillin tyrosine phosphorylation. Mol Cell Biol. 1997;17:6906–6914. doi: 10.1128/mcb.17.12.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. Rho-related proteins: actin cytoskeleton and cell cycle. Curr Opin Genet Dev. 1995;5:24–30. doi: 10.1016/s0959-437x(95)90049-7. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. Signal transduction pathways regulating Rho-mediated stress fibre formation: requirement for a tyrosine kinase. EMBO (Eur Mol Biol Organ) J. 1994;13:2600–2610. doi: 10.1002/j.1460-2075.1994.tb06550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrschneider LR. Adhesion plaques of Rous sarcoma virus-transformed cells contain the src gene product. Proc Natl Acad Sci USA. 1980;77:3514–3518. doi: 10.1073/pnas.77.6.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Mori S, Yokote K, Kanzaki T, Saito Y, Morisaki N. Phosphatidylinositol 3-kinase activity is required for the activation process of focal adhesion kinase by platelet-derived growth factor. Biochem Biophys Res Commun. 1996;224:23–26. doi: 10.1006/bbrc.1996.0978. [DOI] [PubMed] [Google Scholar]
- Sakai R, Iwamatsu A, Hirano N, Ogawa S, Tanaka T, Mano H, Yazaki Y, Hirai H. A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO (Eur Mol Biol Organ) J. 1994;13:3748–3756. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Parsons JT. Focal adhesion kinase and associated proteins. Curr Opin Cell Biol. 1994;6:705–710. doi: 10.1016/0955-0674(94)90097-3. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Parsons JT. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15:2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125FAK: a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci USA. 1992;89:5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Otey CA, Hildebrand JD, Parsons JT. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J Cell Biol. 1995;130:1181–1187. doi: 10.1083/jcb.130.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Broome MA, Hunter T. Fibronectin-stimulated signaling from a focal adhesion kinase-c-Src complex: involvement of the Grb2, p130cas, and Nck adaptor proteins. Mol Cell Biol. 1997;17:1702–1713. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Pages C, Kedersha NL, Fazikas L, Medley Q, Debant A, Streuli M. The LAR transmembrane protein tyrosine phosphatase and a coiled-coil LAR-interacting protein co-localize at focal adhesions. EMBO (Eur Mol Biol Organ) J. 1995;14:2827–2838. doi: 10.1002/j.1460-2075.1995.tb07282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seufferlein T, Rozengurt E. Lysophosphatidic acid stimulates tyrosine phosphorylation of focal adhesion kinase, paxillin, and p130. Signaling pathways and cross-talk with platelet-derived growth factor. J Biol Chem. 1994a;269:9345–9351. [PubMed] [Google Scholar]
- Seufferlein T, Rozengurt E. Sphingosine induces p125FAK and paxillin tyrosine phosphorylation, actin stress fiber formation, and focal contact assembly in Swiss 3T3 cells. J Biol Chem. 1994b;269:27610–27617. [PubMed] [Google Scholar]
- Seufferlein T, Rozengurt E. Sphingosylphosphorylcholine rapidly induces tyrosine phosphorylation of p125FAK and paxillin, rearrangement of the actin cytoskeleton and focal contact assembly. Requirement of p21rho in the signaling pathway. J Biol Chem. 1995;270:24343–24351. doi: 10.1074/jbc.270.41.24343. [DOI] [PubMed] [Google Scholar]
- Shalloway D, Bagrodia S, Chackalaparampil I, Shenoy S, Lin PH, Taylor SJ. c-Src and mitosis. Ciba Found Symp. 1992;170:248–265. doi: 10.1002/9780470514320.ch15. [DOI] [PubMed] [Google Scholar]
- Shen Y, Schneider G, Cloutier JF, Veillette A, Schaller MD. Direct association of protein-tyrosine phosphatase PTP-PEST with paxillin. J Biol Chem. 1998;273:6474–6481. doi: 10.1074/jbc.273.11.6474. [DOI] [PubMed] [Google Scholar]
- Tachibana K, Sato T, D'Avirro N, Morimoto C. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J Exp Med. 1995;182:1089–1099. doi: 10.1084/jem.182.4.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SJ, Shalloway D. An RNA-binding protein associated with Src through its SH2 and SH3 domains in mitosis. Nature. 1994;368:867–871. doi: 10.1038/368867a0. [DOI] [PubMed] [Google Scholar]
- Taylor SJ, Anafi M, Pawson T, Shalloway D. Functional interaction between c-Src and its mitotic target, Sam 68. J Biol Chem. 1995;270:10120–10124. doi: 10.1074/jbc.270.17.10120. [DOI] [PubMed] [Google Scholar]
- Thomas JW, Ellis B, Boerner RJ, Knight WB, White GCN, Schaller MD. SH2- and SH3-mediated interactions between focal adhesion kinase and Src. J Biol Chem. 1998;273:577–583. doi: 10.1074/jbc.273.1.577. [DOI] [PubMed] [Google Scholar]
- Turner CE. Paxillin: a cytoskeletal target for tyrosine kinases. Bioessays. 1994;16:47–52. doi: 10.1002/bies.950160107. [DOI] [PubMed] [Google Scholar]
- Turner CE, Miller JT. Primary sequence of paxillin contains putative SH2 and SH3 domain binding motifs and multiple LIM domains: identification of a vinculin and pp125Fak-binding region. J Cell Sci. 1994;107:1583–1591. doi: 10.1242/jcs.107.6.1583. [DOI] [PubMed] [Google Scholar]
- Vuori K, Hirai H, Aizawa S, Ruoslahti E. Introduction of p130cas signaling complex formation upon integrin- mediated cell adhesion: a role for Src family kinases. Mol Cell Biol. 1996;16:2606–2613. doi: 10.1128/mcb.16.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood CK, Turner CE, Jackson P, Critchley DR. Characterization of the paxillin-binding site and the C-terminal focal adhesion targeting sequence in vinculin. J Cell Sci. 1994;107:709–717. [PubMed] [Google Scholar]
- Xing Z, Chen HC, Nowlen JK, Taylor SJ, Shalloway D, Guan JL. Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol Biol Cell. 1994;5:413–421. doi: 10.1091/mbc.5.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi R, Mazaki Y, Hirota K, Hashimoto S, Sabe H. Mitosis specific serine phosphorylation and downregulation of one of the focal adhesion protein, paxillin. Oncogene. 1997;15:1753–1761. doi: 10.1038/sj.onc.1201345. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Matsumura F. Mitosis-specific phosphorylation of caldesmon: possible molecular mechanism of cell rounding during mitosis. Bioessays. 1991;13:563–568. doi: 10.1002/bies.950131103. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Yamakita Y, Ishikawa R, Matsumura F. Mitosis-specific phosphorylation causes 83K non-muscle caldesmon to dissociate from microfilaments. Nature. 1990;344:675–678. doi: 10.1038/344675a0. [DOI] [PubMed] [Google Scholar]