

Abstract
Two alleles at a new locus, central pair–associated complex 1 (CPC1), were selected in a screen for Chlamydomonas flagellar motility mutations. These mutations disrupt structures associated with central pair microtubules and reduce flagellar beat frequency, but do not prevent changes in flagellar activity associated with either photophobic responses or phototactic accumulation of live cells. Comparison of cpc1 and pf6 axonemes shows that cpc1 affects a row of projections along C1 microtubules distinct from those missing in pf6, and a row of thin fibers that form an arc between the two central pair microtubules. Electron microscopic images of the central pair in axonemes from radial spoke–defective strains reveal previously undescribed central pair structures, including projections extending laterally toward radial spoke heads, and a diagonal link between the C2 microtubule and the cpc1 projection. By SDS-PAGE, cpc1 axonemes show reductions of 350-, 265-, and 79-kD proteins. When extracted from wild-type axonemes, these three proteins cosediment on sucrose gradients with three other central pair proteins (135, 125, and 56 kD) in a 16S complex. Characterization of cpc1 provides new insights into the structure and biochemistry of the central pair apparatus, and into its function as a regulator of dynein-based motility.
Keywords: cilia, flagella, motility, microtubule, Chlamydomonas
Most motile eukaryotic cilia and flagella are built on a scaffold of nine outer doublet microtubules and two central singlet microtubules. Disruption of the central pair and its associated structures has been linked with flagellar paralysis in humans (Chapelin et al., 1997), sea urchins (Morris and Scholey, 1997), and the protist Chlamydomonas (Witman et al., 1978), but the exact role of the central pair apparatus in generating or regulating flagellar bending is not known (reviewed in Smith and Lefebvre, 1997). Indirect evidence suggests that central pair structures interact with radial spokes, which in turn transmit regulatory signals to the multiple isoforms of dynein that are attached to outer doublet microtubules. In Chlamydomonas, flagellar paralysis caused by either radial spoke or central pair assembly mutations can be partially overcome by bypass suppressor mutations in genes that encode outer row (Huang et al., 1982; Porter et al., 1994; Rupp et al., 1996) or inner row (Porter et al., 1992; Myster et al., 1997) dynein heavy chains, or that disrupt a regulatory complex associated with inner row dyneins (Piperno et al., 1992). The flagellar waveform in these suppressed central pair or radial spoke mutant strains is less highly asymmetric than in wild-type Chlamydomonas, suggesting that central pair–radial spoke interactions regulate waveform (Brokaw and Luck, 1985). However, more recent studies (Frey et al., 1997; Wakabayashi et al., 1997) have shown that both asymmetric and symmetric planar waveforms can be generated in central pair or radial spoke defective axonemes if they are reactivated in vitro at very low ATP concentrations. This switch between highly asymmetric and symmetric waveforms, which is triggered by increases in free calcium concentration to above 10−5 M, clearly does not require a functional central pair apparatus, but normal flagellar beat regulation does.
In addition to the four loci required for assembly of both central pair microtubules in Chlamydomonas (PF15, PF18, PF19, and PF20), mutations at two other loci only partially disrupt central pair structure and provide additional clues to central pair function. Mutations at the PF16 locus prevent assembly of three known central pair proteins, the resulting flagella occasionally twitch or slowly propagate bends (Dutcher et al., 1984). The pf16 gene product is a 57-kD protein with sequence similarity to armadillo repeat proteins (Smith and Lefebvre, 1996) whose precise role in central pair structure and function has not been determined, but the C1 microtubule is unstable in demembranated pf16 axonemes. This property was used to identify 10 of the 23 proteins missing in axonemes of mutants such as pf18, in which the entire central pair complex is unstable, as C1-associated proteins. pf6 mutations result in the absence of three other central pair proteins from either flagella or demembranated flagellar axonemes, with concomitant loss of an 18-nm projection from the C1 microtubule (Dutcher et al., 1984). Chlamydomonas cells harboring the pf6 mutation have flagella that occasionally beat, but too slowly to propel the cells. In both mutations, seemingly minor disruptions of central pair structure result in nearly complete inhibition of flagellar bend propagation, highlighting a critical function of the central pair apparatus in normal motility.
Using a beat-independent measure of flagellar dynein activity, we have shown (Smith and Sale, 1992; Howard et al., 1994; Habermacher and Sale, 1997) that dynein-driven microtubule sliding rates are reduced in radial spoke and central pair assembly mutants, and that sliding can be restored to wild-type levels with protein kinase inhibitors. The reduction of sliding rate in these mutants correlates with the level of phosphorylation of an inner arm dynein intermediate chain (IC138). Mutations that alter IC138 phosphorylation, or that prevent assembly of the IC138-associated inner arm I1 dynein, block phototactic changes in flagellar motility in vivo (King and Dutcher, 1997). One role for central pair–radial spoke interactions, therefore, is the regulation of an inner row dynein-directed kinase/ phosphatase cascade that is important for tactic regulation, and preliminary evidence suggests that some of the signaling enzymes themselves are located in the central pair apparatus (Yang, P., L. Fox, and W.S. Sale. 1998. Mol. Biol. Cell. 9:130a; Roush, A.M., and W.S. Sale. 1998. Mol. Biol. Cell. 9:397a).
In a search for new insertionally generated motility- defective mutants, two strains were selected that swim progressively but at a reduced speed, and lack a central pair–associated structure. Here we show that these strains harbor allelic mutations at a new locus, central pair–associated complex 1 (CPC1), and lack a projection from the C1 microtubule. Characterization of the cpc1 structural defect by EM has revealed previously uncharacterized central pair–associated structures, including two connections between this C1 projection and the C2 microtubule, and rows of densities that project from the lateral surface of each central pair microtubule toward nearby radial spoke heads. Biochemically, cpc1 axonemes lack three proteins which can be extracted from wild-type axonemes as components of a larger complex. These results reveal the central pair apparatus to be more complex than previously known, and provide new tools for further studies of central pair structure and function.
Materials and Methods
Strains
Chlamydomonas reinhardtii wild-type strain 137c (CC 124) and mutant strains pf6 (CC 1029), pf14 (CC 1032), pf16 (CC 1034), pf18 (CC 1036), pf27 (CC 1387), arg2 (CC 1930), and arg7 (CC 1931) were obtained from Elizabeth Harris at the Chlamydomonas genetics center (Duke University, Durham, NC); oda1 was provided by Ritsu Kamiya (University of Tokyo, Tokyo, Japan). The nit1-305cw15 strain used for mutagenesis was provided by Paul Lefebvre (University of Minnesota, St. Paul, MN). Stocks were maintained on minimal medium supplemented with arginine. All genetic manipulations including diploid generation followed standard procedures (Harris, 1989).
Mutagenesis
Motility mutants were generated by transformation of nit1-305cw15 cells with plasmid pMN24 linearized at EcoRI, as previously described (Tam and Lefebvre, 1993). Transformation mixes were spread on plates of minimal medium lacking a reduced nitrogen source (NH4NO3 replaced with NaNO3) and transformant colonies were picked into minimal liquid medium in 96-well microtiter plates. Wells were examined on an Olympus stereo microscope under dark-field illumination, and slow-swimming strains were clonally selected on agar plates before further examination. Plasmid rescue followed the procedure of Tam and Lefebvre (1995) using Escherichia coli DH5α MCR cells (Life Technologies).
Motility
Flagellar beat frequency was measured as previously described (Mitchell and Kang, 1991). A 625-nm cutoff red filter, used to reduce phototactic responses during beat frequency measurements, was removed to induce photophobic reversal episodes. The ability of cells to respond to phototactic stimuli was tested with a simple photoaccumulation assay (King and Dutcher, 1997). Reactivation of axonemes was performed by a modification of the cell model reactivation procedure of Horst and Witman (1995) as follows. Demembranated cell models (1 μl) were diluted into 10−4 M Ca2+ reactivation buffer containing 1 mM ATP (20 μl) and introduced into a slide chamber made with double-stick tape and a coverslip. These conditions result in deflagellation of >90% of the cells, and simultaneous reactivation of the resulting axonemes. After 15 s, cell bodies and most nonadherent axonemes were removed by perfusion of 60 μl of reactivation buffer (containing either 10−4 M or 10−8 M Ca2+ and 1 mM ATP) through the chamber. All reactivation solutions were formulated as in Bessen et al. (1980) with substitution of potassium acetate for potassium chloride. Reactivated axonemes were observed on a Zeiss Axioskop under stroboscopic dark-field illumination and recorded at flash rates between 50 and 150 Hz on Tmax 3200 film transported at 20 cm/s. Negatives at an original magnification of 90 were scanned into a personal computer at 1,200 dpi, images were enlarged electronically, and space between sequential images was reduced for figure presentation.
Electron Microscopy
Whole cells were prepared for electron microscopy by fixation in glutaraldehyde as previously described (Hoops and Witman, 1983), while demembranated flagellar axonemes were fixed in the added presence of tannic acid (Porter et al., 1992). All samples were embedded in Araldite, and gold-silver (for cross-section images) or silver-gray (for longitudinal images) thin sections stained with uranyl acetate and lead citrate were examined in a JEOL 100CXII microscope operated at 80 kV. Negatives taken at an original magnification of 40,000 were used for image averaging. The methods for digitizing and averaging central pairs were previously described (Mastronarde et al., 1992) with the following modifications. Axoneme profiles that contained central pair microtubules in good cross-section were digitized at a scale of 0.9 nm/pixel. The central pair microtubules and projections were extracted into a 120 × 120 pixel box, the images averaged, and automatically aligned to this average. Base-tip orientation in longitudinal images was determined from the appearance of outer row dyneins, which tilt toward the base of the axoneme (Sale and Satir, 1977; Warner et al., 1977).
Fractionation of Flagella
Cells grown in acetate-enriched medium were deflagellated with 5 mM dibucaine (Witman et al., 1978). Flagella were demembranated by resuspension in HMDEK (30 mM Hepes, 5 mM MgSO4, 1 mM DTT, 0.5 mM EGTA, 25 mM potassium acetate, 1 mM PMSF, pH 7.4) and mixing with an equal volume of HMDEK containing 0.4% NP-40. To prepare pf16 axonemes that displayed instability of the C1 microtubule, HMDEK was replaced by HMDEN (10 mM Hepes, 5 mM MgSO4, 1 mM DTT, 0.5 mM EDTA, 30 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, pH 7.4). To remove C2, axonemes were extracted with HMDEK containing 0.6 M NaCl for 20 min on ice, centrifuged for 20 min at 4°C, and resuspended in HMDEK before fixation for EM or additional extraction steps to solubilize C1. The C1 microtubule and its associated structures were solubilized by treating NaCl-extracted axonemes with 0.2 M KI in HMDEK. Proteins in the KI extract were further separated by dialysis into HMDEK and centrifugation on a 5–20% sucrose gradient (prepared with HMDEK) at 35,000 rpm for 16 h in an SW41 rotor. Gradients were collected from the bottom of the tube in 0.5-ml fractions. Sedimentation values were determined using thyroglobulin (19.2S) and catalase (11.3S) as standards. Protein composition was determined by SDS-PAGE on 6% acrylamide gels run with resolving gels that contained 3 M urea and were stained with Coomassie blue or silver (Wray et al., 1981), as indicated. Dynein heavy chains (average M r = 500,000) and Benchmark Protein Ladder (Life Technologies) were used as protein size standards. The 90- and 80-kD markers did not migrate true to size in the presence of 3 M urea and are unlabeled in figures. For immunoblotting, proteins were transferred to PVDF membranes and probed with affinity-purified (H2) polyclonal anti-HIPYR antibody (Sawin et al., 1992) at a 1:30 dilution, or with affinity-purified polyclonal anti-Klp1 antibody (Bernstein et al., 1994) at a 1:5,000 dilution. Antibodies were detected with peroxidase-labeled goat anti–rabbit (BioRad) developed with ECL reagents (Amersham).
Results
Mutant Selection and Genetic Characterization
To identify genes involved in regulation of dynein activity, insertional mutagenesis was used to create motility-defective strains. In a screen of 7,000 transformants, 68 motility-defective strains were selected. Two of these strains, 6E9 and 60A8, were found to have a flagellar beat frequency during normal forward swimming of ∼40 Hz (compared to 60 Hz for wild-type cells under the same conditions). Both retained an ability to respond to photophobic stimulation with a rapid, transient change from an asymmetric bending pattern to a symmetric bending pattern, and to accumulate when presented with a phototactic gradient. By electron microscopy, both strains were seen to be defective in assembly of the same central pair–associated structure, as detailed below. No recombinants (fast swimmers) were observed among 400 zygotes from a cross between these two strains, indicating linkage within an interval of <1 map unit. To directly test allelism, each mutant was crossed into an Arg− background and diploids were selected from a cross between 6E9arg7 and 60A8arg2 strains. The 6E9arg7/ 60A8arg2 diploids had an average beat frequency of 47.1 ± 3.1 Hz (n = 30), compared with 62.4 ± 2.5 Hz (n = 30) for wild-type (+arg7/+arg2) diploids. These two mutations are therefore allelic, and have been designated cpc1-1 and cpc1-2 (for central pair–associated complex).
The CPC1 locus can be placed at 8.3 map units from its centromere based on tetratype frequencies between cpc1 and three separate centromere markers (Table I). These and additional crosses failed to show significant linkage between cpc1 and at least one marker on each of the 17 recognized Chlamydomonas linkage groups. By Southern blot analysis with a vector-specific probe, all randomly selected slow-swimming products from a third generation backcross of cpc1-2 carried a single copy of the vector (pUC119) used for mutagenesis (n = 4), indicating linkage between the mutation and the vector insertion site. The vector and an adjacent 1.4-kb genomic fragment were therefore released from genomic DNA by PstI digestion and cloned into E. coli by plasmid rescue (Tam and Lefebvre, 1995). This insert should be useful for mapping the CPC1 locus by RFLP analysis and for future characterization of the gene product.
Table I.
Distance of CPC1 from Its Centromere
| Centromere marker | Linkage group | Tetratype ratio | Distance | |||
|---|---|---|---|---|---|---|
| cM | ||||||
| oda2 (pf28) | XI | 16:17:7 | 8.7 | |||
| pf27 | XII/XIII | 3:3:1 | 7.1 | |||
| oda1 | XVIII | 2:4:1 | 7.1 | |||
| Total | 21:24:9 | 8.3 |
Motility
Previous analysis of motility defects associated with inner and outer row dynein assembly mutations showed that only inner row dynein defects have a marked effect on asymmetric waveforms, but that both types of defect greatly reduce bend amplitude during symmetric beating (Brokaw and Kamiya, 1987). Images of wild-type and cpc1 axonemes reactivated at either 10−8 M or 10−4 M Ca2+ (Fig. 1) show that cpc1 axonemes beat asymmetrically at low Ca2+ concentrations and symmetrically at high Ca2+ concentrations, and display no radical differences from wild-type waveforms under either condition. The combination of reduced beat frequency with apparently normal waveform in cpc1 flagella is phenotypically similar to motility parameters of some outer row dynein defects such as sup1 and sup2 (Brokaw and Luck, 1985). To see whether cpc1 acts exclusively through a modulation of outer row dynein activity, we compared beat frequencies of strains carrying cpc1 alone, an outer row dynein assembly mutation alone (pf28), or both mutations (pf28cpc1). As illustrated in Fig. 2, cpc1 reduced beat frequency almost identically in a pf28 background (30% reduction) and a wild-type background (38% reduction), and thus does not act exclusively through effects on outer row dyneins.
Figure 1.

Dark-field stroboscopic images of reactivated axonemes used for comparison of waveforms. Wild-type (A) and cpc1 (B) axonemes reactivated at 10−8 M Ca2+ beat with highly asymmetric waveforms. Both axonemes are freely swimming near the coverslip. Wild-type (C) and cpc1 (D) axonemes reactivated at 10−4 M Ca2+ beat with symmetric waveforms. Both axonemes have adhered to the coverslip at their proximal ends. All panels were photographed at a flash rate of 50 Hz. Bar in A, 10 μm.
Figure 2.

Flagellar beat frequencies were measured on freely swimming cells. The cpc1 defect reduces beat frequency by 38% from wild-type (WT) levels. Absence of outer row dyneins (pf28) reduces beat frequency by 60%, and the combination of cpc1 and absence of outer row dyneins (pf28cpc1) results in a further decrease to 17 Hz, or 30% below pf28 alone. Each bar shows mean and standard deviation (n = 31).
Central Pair Structure
As seen in electron micrographs of wild-type axonemal cross-sections (Fig. 3 A), the two central pair microtubules can be differentiated by the presence of two long projections (1a and 1b) on the C1 tubule and two short projections (2a and 2b) on the C2 tubule. These and all other micrographs have been printed as if viewed from inside the cell (base to tip of the axoneme) with A tubules of each outer doublet pointing clockwise, and have been rotated so that the C1 central pair tubule is on the left. The two C1-associated projections make perpendicular connections to the C1 microtubule surface. Thinner arcs (previously described as a sheath around the central pair; Warner, 1976) apparently connect the tip of each projection to the lateral side of C1. The two long C1-associated projections can be distinguished from each other by a thickening at the tip of 1a that is not seen at the tip of 1b. Axonemes of cpc1 (Fig. 3 B) consistently lack the 1b projection and its associated arc, and frequently lack the 2b projection as well. To determine whether a similar structural defect is present in vivo, whole cells were fixed for electron microscopy and images of intact flagella were examined (Fig. 3 C). These images lack the contrast seen in axonemal images because tannic acid does not penetrate through cell membranes, but they nonetheless show that the 1b projection and its associated arc are missing from flagella as well as axonemes of this mutant. The presence or absence of the 2b projection in cpc1 flagella could not be determined from these preparations.
Figure 3.
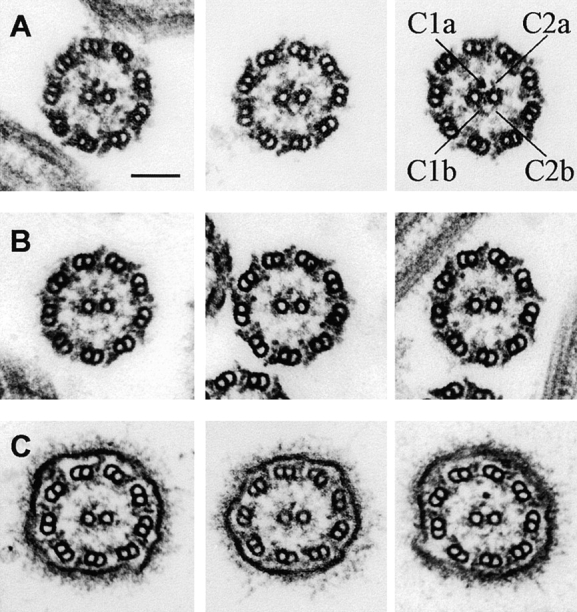
Electron micrographs of thin sections through demembranated wild-type axonemes (A) show several electron-dense projections from the two central pair microtubules. All micrographs are oriented as if viewed from inside the cell and rotated so that the C1 central microtubule is to the left. The longest central pair projections (1a, 1b, 2a, and 2b) have been labeled in the third panel. In similar micrographs of cpc1 axonemes (B) the central pair microtubules lack the 1b and 2b densities. Images of intact cpc1 flagella (C) show that the C1b projection is not lost during axoneme preparation, but rather fails to assemble in this mutant. Bar, 100 nm.
Further attempts to clarify central pair–associated structures in both wild-type and mutant axonemes were hampered by the close proximity of radial spoke heads to the central pair. To avoid this problem, axonemes were isolated from radial spoke assembly mutant pf14 and from double mutants between pf14, and three mutations that alter central pair structure, cpc1, pf6, and pf16. Two individual pf14 axonemes, a composite average of 16 similar images, and a cartoon summarizing densities associated with pf14 central pair microtubules are shown in Fig. 4. In addition to the four projections labeled in Fig. 3 A, the averaged image reveals densely stained material at ∼11 o'clock (1c) and 8 o'clock (1d) on C1, at 2 o'clock (2c) on C2, and in a broad zone between the two microtubules. Based on the image average and on additional information from double mutants (see following sections), this mid-zone has been further divided into a bipartite intermicrotubule bridge and a diagonal linker between projection 1b and the C2 microtubule. Although there is an electron lucent gap between the tip of the 1a and 2a projections, the tips of 1b and 2b always appear to be joined by a continuous thin arc extending from the 1d density on C1 through 180° to the lateral margin of C2. This arc of sheath material and the 2b projection together form a triangle of nearly uniform density at the bottom of C2, whereas the sheath generally appears separated by a less dense region from the 1a and 1b projections. Careful examination of favorable images of wild-type axonemes confirm that all of these structural details are also present in wild-type axonemes such as those in Fig. 3 A and are not artifacts created by the loss of radial spokes.
Figure 4.

Cross-sections through pf14 axonemes (A and B) and an average of 16 similar images (C) reveal additional details of central pair structure that are summarized diagramatically (D). These include four densities (1a, 1b, 1c, and 1d) attached to the C1 microtubule, three densities (2a, 2b, and 2c) attached to the C2 microtubule, a bipartite bridge and a diagonal link spanning the gap between the microtubules, and sheath material that forms an arc from 1c to the tip of 1a, and from 1d to the tip of 1b. Individual cross-sections through cpc1pf14 axonemes (E and F) show that the 1b projection is invariably missing in cpc1 axonemes, whereas the 2b projection is sometimes retained (filled arrowhead in E). An average of 12 images (G) reveals the presence of a densely staining region at the site where 1b attaches to the C1 microtubule in wild-type axonemes. A difference image (H) made by subtracting G from C produces a dark image of the missing structures and a bright region where rotation of C1 relative to C2 has produced an apparent overlap of the 1a and 2a projections (asterisk). Cross-sections through pf6pf14 axonemes (I and J), an average of 25 images (K), and a difference image (L) show that only projection 1a and its associated sheath material are affected by the pf6 mutation. Bars, 100 nm in A and 20 nm in C.
When axonemes of cpc1pf14 double mutants were examined, two types of images were observed. In all of the images, projection 1b and its associated arc were missing, but a small residual density remained at the site of attachment of 1b onto the C1 microtubule. In approximately half of the images a triangular region of electron density remained in the 2b position (Fig. 4 E, filled arrowhead), while in the remaining images the 2b density was missing (Fig. 4 F, empty arrowhead). We interpret these images as evidence that in cpc1 the 2b projection is either unstable and frequently lost during preparation of axonemes, or its assembly is partially prevented. An average of 12 cross-sections (Fig. 4 G) and a difference image created by subtracting mutant from wild-type averages (Fig. 4 H) reveal several details of the mutant phenotype. The long 1b projection and arc are clearly missing in the average, as is a portion of the diagonal link, but the base of the diagonal link remains associated with C2 in these images, and a density is also retained on the surface of C1 at the base of the 1b projection. In addition to the loss of specific densities the relative orientations of C1 and C2 have changed in cpc1, with C1 rotating clockwise such that the 1a and 2a projections now appear to overlap at their tips, creating a lighter region in the difference image (Fig. 4 H, asterisk).
Similar images of pf6pf14 axonemes clearly show that pf6 and cpc1 affect different central pair structures. In pf6pf14 axonemes the 1a projection and its associated sheath are missing but there is no apparent instability of 1b or of any C2-associated structures (Fig. 4, I–L). The image average (Fig. 4 K) and difference (Fig. 4 L) show no change in the interdoublet bridge, other C1-associated densities, or the relative orientation of C1 and C2 microtubules in this mutant.
Although projections 1b and 2b appear physically connected at their tips in wild-type axonemes, it is possible that the images of these structures overlap, but the structures themselves could be noninteracting. The disrupted assembly or reduced stability of 2b in cpc1 axonemes could result either because a normal interaction of 2b with the tip of 1b is absent, if these structures do interact, or because the cpc1 gene product is part of (or needed for the stability of) each structure alone. To test for the presence of direct interactions between these projections, we created axonemes in which only one or the other central pair microtubule had disassembled, and then looked for retention of 1b-2b connections, or for loss of one when the other was removed. Extraction of Chlamydomonas axonemes with 0.6 M NaCl has been shown to selectively destabilize C2, as well as to extract most of the inner and outer dynein arms, without removing C1 (Piperno and Luck, 1979). When this procedure was applied to pf14 axonemes, most axonemal cross-sections displayed either 0 (33%) or 1 (44%) central microtubule, and the one remaining tubule was invariably C1 (n = 208). In half of the remaining images (12%) a part of the C2 tubule and its associated projections was visible and 11% retained both central pair microtubules (Fig. 5 A). In the absence of C2, C1 appears to retain all of its projections along with material in the interdoublet bridge-diagonal link region, but does not retain material with the appearance of 2b (Fig. 5, B–E). Although we did not attempt to make image averages of these extracted axonemes due to variability in the extent of extraction, none of the structures that directly interact with C1 appear to have been removed by 0.6 M NaCl. C1 microtubules lacking the 1b projection were never seen, whereas the 2b projection was often missing even in cross-sections where other C2 structures remained.
Figure 5.
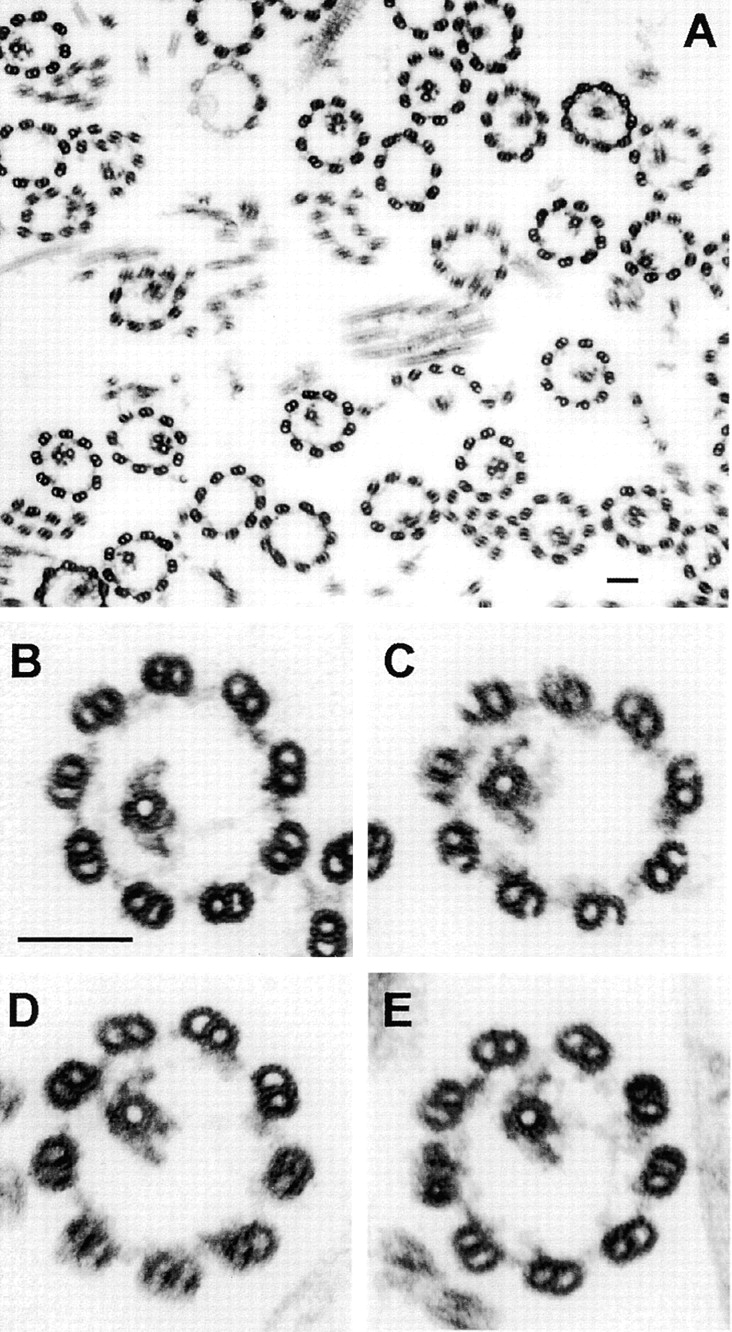
Cross-sections through pf14 axonemes that were extracted with 0.6 M NaCl. (A) Survey view of extracted axonemes. (B–E) Cross-sections in which C2 has been solubilized. These C1 microtubules retain all of the electron densities identified as C1-associated in Fig. 4. Bars, 100 nm in both A and B.
To create axonemes lacking C1 but retaining C2, samples were prepared from a pf16pf14 double mutant strain. Although intact pf16 flagella retain both central pair microtubules, the C1 tubule is reportedly unstable under the detergent extraction conditions used to prepare axonemes (Dutcher et al., 1984). To our surprise, >90% of pf16 axonemes prepared by our standard isolation procedure retained both central pair tubules. However, when pf16 or pf16pf14 flagella were demembranated in a buffer containing sodium chloride (Smith and Lefebvre, 1996) rather than potassium acetate, central pair complexes lost variable amounts of C1-associated structures. Both central pair tubules were intact in only 10% of pf16pf14 axonemal cross-sections compared with 17% in which part of C1 was missing, 38% that contain only C2, and 35% that lacked both central pair tubules (n = 230). The most complete structures observed (under conditions used here for axoneme preparation) lacked only the 1a projection and its associated sheath, and showed a reduced interdoublet bridge density (Fig. 6 A). Fig. 6, B–D, illustrates increasingly disrupted C1 structures and shows that the 1b projection and its associated sheath remain attached to C2, even when no C1 microtubule remains in the section plane, through connections at both the base and tip of 1b (Fig. 6 C). In some images (e.g., the last panel of Fig. 6 C), 2b has been lost and the only remaining connection between 1b and the C2 tubule is the diagonal link at the base of the 1b projection. When no C1-associated structures were evident (Fig. 6 D), most C2-associated structures were also absent and proper orientation of C2 could not be accurately determined. Densities similar to projection 2a were sometimes present, but densities with the characteristic triangular shape of 2b were never observed. Because we never observed an identifiable 2b density unless 1b was also visible, the pf16pf14 data favor the hypothesis that the tips of 1b and 2b do not just overlap, but physically interact.
Figure 6.
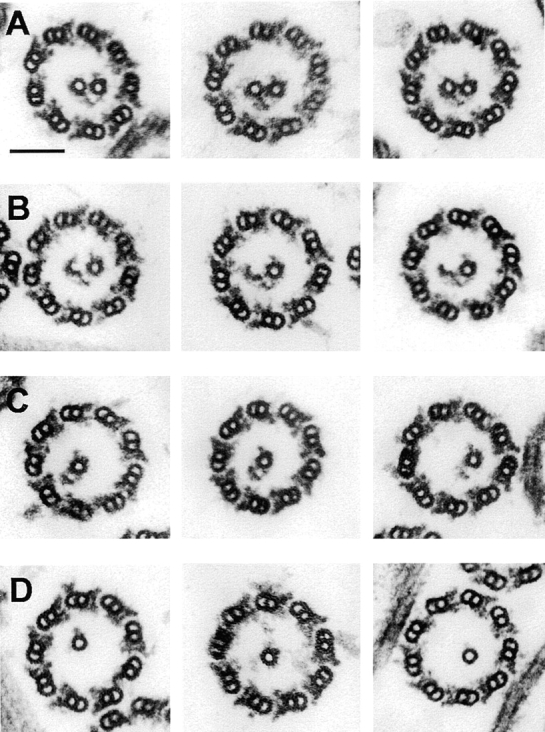
Selected images of cross-sections through pf16pf14 axonemes which illustrate apparent progressive steps in the loss of C1 and its associated structures. In row A, the 1a projection and its associated sheath are missing, and little remains at the 1c position, but the 1b and 1d densities are retained. In row B, several protofilaments are missing from the wall of the C1 microtubules. In rows C and D, C1 is completely missing. The 1b projection can remain attached to the C2 microtubule through apparent connections at its base to the diagonal link and at its tip to 2b (seen most clearly in row C). Bar, 100 nm.
Longitudinal images were used to determine the periodicity and angular orientation of the 1b projection and the other densities defined by cross-section analysis, and to see whether the 1b and 2b densities are longitudinally aligned. Longitudinal sections that graze the lateral surface of C1 show that projection 1a and its associated sheath fibers (extending from the right-hand edge of C1 in the longitudinal image of Fig. 7 A) have a 16-nm repeat period and tilt toward the tip of the axoneme at ∼24°. Also visible in Fig. 7 A are two rows of rectangular or rhomboidal densities, corresponding to densities 1c and 1d in cross-sectional images, which overlie the walls of C1, and project out from its left-hand margin, respectively. They are indicated in the longitudinal image by arrows and by parallel lines marking their 32-nm repeat period. When the section plane included both 1a and 1b projections (Fig. 7 B), their identical periodicity was revealed. Sections passing perpendicular to the central pair microtubule axes and including the intermicrotubule bridge (Fig. 7, C and D) show the presence of closely spaced rope-like material with a 16-nm repeat overlying the central pair microtubule image in the bridge region (diagonal lines in Fig. 7 C). This material, which may include both the diagonal link and the intermicrotubule bridge, appears to follow a left-handed helix with a pitch matching that of the underlying dimer lattice of the C2 microtubule (Linck et al., 1981). Both the 2a and 2b projections repeat at a 16-nm period, as seen in the lower half of the longitudinal image in Fig. 7 D.
Figure 7.
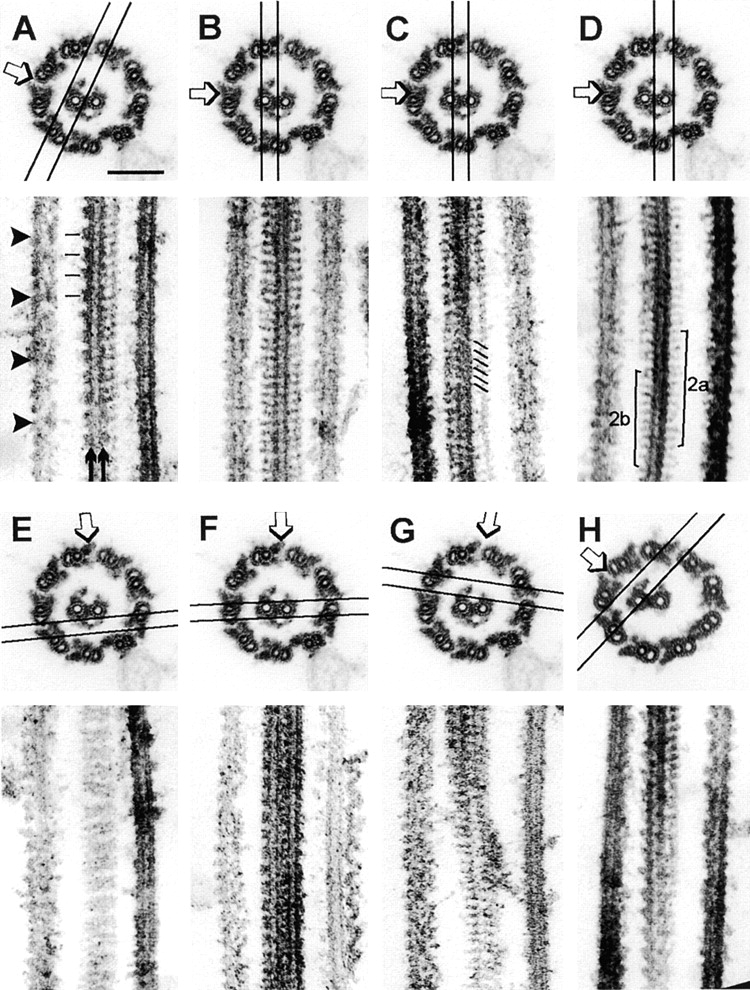
Analysis of central pair structure in longitudinal thin sections. The top of each panel indicates the plane of section and direction of view for the longitudinal image in the bottom. Cross-sections are printed looking from base to tip of the axoneme, longitudinal sections are printed with the base of the axoneme toward the bottom of the page. (A–G) pf14 axonemes; (H) cpc1pf14 axonemes. In A, arrows point to two rows of rectangular densities (corresponding to 1d and 1c in Fig. 4 D) that repeat every 32 nm (indicated by parallel lines); arrowheads emphasize the 96-nm periodicity of inner row dyneins. In B, the section plane contains microtubule C1, with projections that correspond to densities 1a and 1b along the right-hand and left-hand edges. In C, diagonal lines indicate material with a 16-nm repeat periodicity in the intermicrotubule bridge region, superimposed on an image of the C2 microtubule. D includes the C2 microtubule and projections 2a and 2b (brackets), as well as material in the bridge region. The longitudinal section in E passes tangentially through the sheath and reveals pairing of sheath fibers. In F, the 1c or 1d densities project with a 32-nm period from the left edge of C1, but material projecting from C2 does not show a distinct periodicity. The oblique section in G passes through the lumen of C1 and C2 (top of longitudinal image) out to the tips of 1a and 2a (bottom of image). In H, absence of the 1b/sheath complex changes the outline of 1-d densities to a saw-tooth (compare with the similar section through a wild-type central pair in A). Bar in A, 100 nm.
Longitudinal sections that graze the 1b/2b/sheath complex (Fig. 7 E) reveal pairs of thin fibers that run perpendicular to the axonemal axis. In oblique sections (not shown) pairs of these fibers give the appearance of inserting into each 1d projection (left edge of central pair in Fig. 7, A and F). Such pairing of sheath fibers is not seen in sections passing through the tips of the 1a and 2a projections (bottom of Fig. 7 G), but can be observed where 1a-associated sheath fibers insert into 1c densities (near the top of the longitudinal section in Fig. 7 G). Sheath fibers appear paired at their attachment sites near the 1c and 1d densities. Those associated with the tip of 1a do not appear to connect to the tip of 2a (Fig. 7 G), whereas those associated with 1b and 2b do form continuous filaments (Fig. 7 E). Since sheath fibers terminating near projection 1d are absent in cpc1 axonemes, we looked at the effects of this mutation on the appearance of the 1d projection in longitudinal views (Fig. 7 H). In this grazing oblique section, the rectangular outline of 1d seen in wild-type central pairs (Fig. 7 A) is shifted to a saw-toothed outline, suggesting that 1d (and perhaps 1c as well) are saw-toothed projections that repeat at 32 nm, but superposition of these structures with the base of each sheath fiber generates the rectangular images seen in Fig. 7, A and F. In summary, most of the electron-dense structures defined in our cross-section analysis, including those disrupted by cpc1 and pf6 mutations, are discrete projections with 16-nm repeat periods. Two exceptions, densities 1c and 1d, both have 32-nm repeat periods. The sheath fibers (16-nm repeat) insert in pairs at or near the 1c and 1d densities, and form continuous arcs connecting the tips of projections 1b and 2b.
Biochemical Defects in cpc1
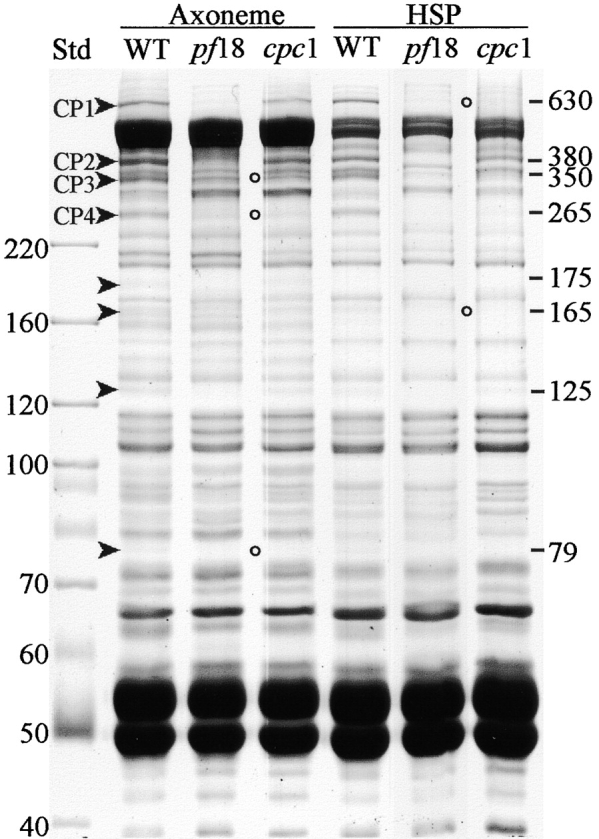
To find out which central pair proteins were missing from cpc1, SDS-PAGE was used to compare axonemes isolated from wild-type, central pair assembly mutant pf18, and cpc1 cells. As shown in Fig. 8, eight bands with estimated sizes ranging from 630 to 79 kD could be recognized as depleted in pf18 axonemes using this 1-d gel system (marked with arrowheads), including four bands >200 kD that apparently correspond to the four high molecular weight proteins previously identified as central pair components on 1-d gels (Witman et al., 1978; Adams et al., 1981) and designated CP1–CP4 (Dutcher et al., 1984). Three of the eight proteins with apparent sizes of 350 (CP3), 265 (CP4), and 79 kD were greatly reduced or missing from cpc1 axonemes (open circles). Two others, 630 kD (CP1), and 380 kD (CP2), were slightly reduced in amount. CP1, CP2, and CP4 have been characterized as C1-associated proteins based on their loss from pf16 axonemes and resistance to high salt extraction from wild-type axonemes, whereas CP3 has been similarly identified as a C2-associated protein (Dutcher et al., 1984). Under our conditions, extraction of wild-type axonemes with 0.6 M NaCl to create a high salt pellet (HSP) fraction (Fig. 8, HSP WT lane) depleted only one of the eight identifiable central pair– associated bands (175 kD), but did not remove the 350-kD band. Either this band is not the same as CP3, or our extraction conditions differ in an unknown way from those of Dutcher et al. (1984). We favor the second explanation and have kept the designation CP3 for this band. Seven central pair proteins were retained in our experiments and thus reside in structures associated with C1 following salt extraction (Fig. 5). Similar extraction of cpc1 axonemes removed CP1 (630 kD) and a 165-kD band (open circles in Fig. 8, HSP cpc1 lane) in addition to the 175-kD band that was extracted from wild-type axonemes.
Figure 8.
SDS-PAGE analysis of the cpc1 defect. Demembranated flagellar axonemes (Axoneme lanes) from wild-type cells (WT) or a central pair assembly mutant (pf18) show that eight bands are missing when the central pair is absent (arrowheads; estimated M r shown along the right margin). Three of these proteins are depleted or missing from cpc1 axonemes (open circles). The HSP lanes show insoluble high salt pellet fractions produced by extracting axonemes with 0.6 M NaCl. The WT HSP sample retains the three proteins missing from cpc1 axonemes. Extraction of cpc1 axonemes removes two additional bands of 630 and 165 kD that are not removed from wild-type axonemes by high salt treatment (cpc1 HSP, open circles). Gel was stained with Coomassie blue. Molecular mass standards (kD) are indicated along the left margin.
Three kinesin-like proteins have been localized to the Chlamydomonas central pair apparatus by blot analysis of central pair mutants (Bernstein et al., 1994; Fox et al., 1994) and by immunoelectron microscopy (Bernstein et al., 1994; Johnson et al., 1994). Klp1 (83 kD) is missing from central pair assembly mutants such as pf15 and has been localized to the C2 microtubule both by virtue of its presence in pf16 axonemes and by immunoelectron microscopy (Bernstein et al., 1994). Blot analysis with affinity-purified anti-Klp1 showed that normal amounts of this antigen were present in cpc1 axonemes (not shown) and thus Klp1 is not associated with projection 2b. A 110-kD antigen recognized by a polyclonal antibody to Drosophila kinesin, αHD, has also been immunolocalized to a single central pair microtubule (Johnson et al., 1994), but remains associated with axonemes in all central pair assembly mutants tested. A second 110-kD antigen recognized by polyclonal sera raised against peptides spanning either of two highly conserved kinesin head domain sequences, HIPYR and LAGSE, is missing from axonemes of central pair mutants such as pf18 (Fox et al., 1994; Johnson et al., 1994) but has not been localized within the central pair structure. We tested for the presence of this kinesin-like protein in cpc1 axonemes by blot analysis using affinity-purified anti-HIPYR antibody. As seen in Fig. 9, this antibody detects multiple bands in wild-type axonemes, and two of these (105 and 85 kD) are depleted in pf18 axonemes. The 105-kD HIPYR antigen, which most likely corresponds to the 110-kD central pair antigen previously detected with this antibody, is missing from the pf16 sample and thus should be designated a C1-associated protein, but is present in cpc1 axonemes. The 85-kD band, which does not correspond to any previously reported kinesin-like proteins in Chlamydomonas flagella, was retained in both pf16 and cpc1 axonemes at wild-type levels and is, like Klp1, a C2-associated protein that is not part of projection 2b.
Figure 9.
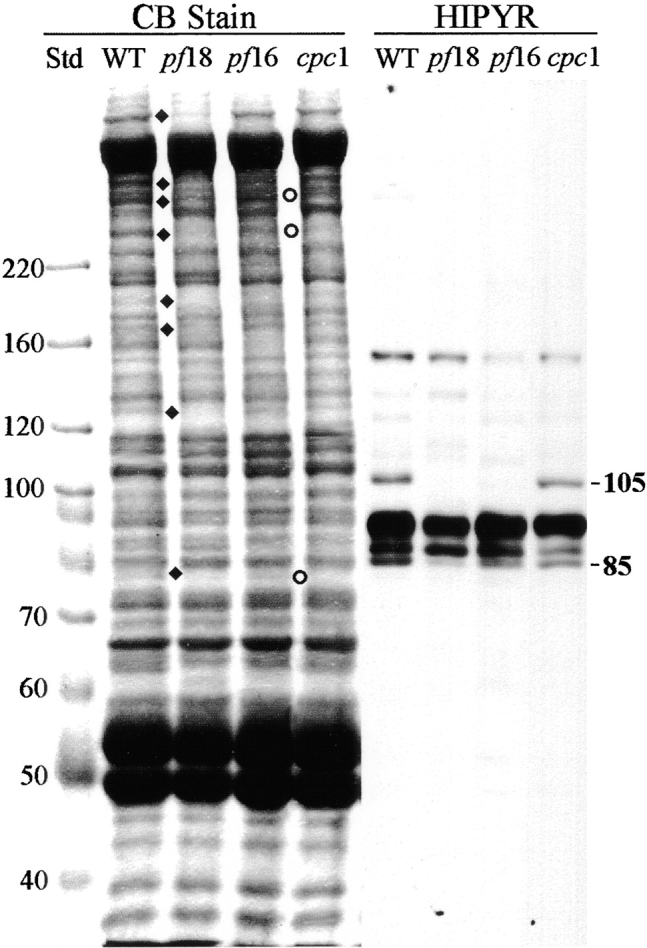
Kinesin-like central pair proteins are not disrupted by the cpc1 defect. Axoneme samples from wild-type (WT), pf18, pf16, and cpc1 strains were separated by SDS-PAGE as in Fig. 8, and either stained with Coomassie blue (CB lanes) or transferred and probed with affinity-purified polyclonal anti-HIPYR antibody (HIPYR lanes). Bands missing from pf18 axonemes are indicated by diamonds, bands missing from cpc1 by circles. The apparent M r of two bands recognized by anti-HIPYR and missing from pf18 axonemes are shown along the right margin. The 105-kD band is missing from pf16 axonemes, whereas neither band is missing from cpc1 axonemes. Molecular mass standards (kD) are indicated along the left margin.
To determine whether the three proteins missing from Coomassie blue–stained gels of cpc1 axonemes represent subunits of a single structure, C1-associated proteins were solubilized and fractionated by centrifugation on a sucrose gradient. Wild-type axonemes were extracted with 0.6 M NaCl to remove dyneins and most C2-associated proteins, and the resulting high salt pellet (HSP) was further extracted with 0.2 M KI and centrifuged to generate pellet (KIP) and supernatant (KIS) fractions (Fig. 10). All seven C1 proteins identifiable with this gel system are quantitatively solubilized by 0.2 M KI treatment (Fig. 10, KIS lane). Electron microscopic examination of the KIP fraction confirmed that C1 microtubules and associated structures were removed by this treatment (i.e., no central pair material remained). After dialysis to remove salt, the KI soluble material was centrifuged through a sucrose gradient and fractions were examined by SDS-PAGE (Fig. 11). The three proteins missing in cpc1 (CP3, CP4, and 79 kD, marked with circles) consistently cosedimented along with three additional proteins of 135, 125, and 56 kD (marked with diamonds), and reached a peak at approximately 16S (fractions 7 and 8). When 16S peak fractions from two separate experiments were analyzed on 12% acrylamide gels, no additional cosedimenting proteins of lower M r were observed. A second and much smaller peak of 350- and 265-kD bands, without any smaller subunits, cosedimented at 9S (fraction 16). These bands may represent another smaller complex containing only CP3 and CP4, or a complex of unrelated proteins of similar apparent size to CP3 and CP4. The remaining C1-associated bands identified on our 1-d gels sedimented elsewhere in the gradient.
Figure 10.
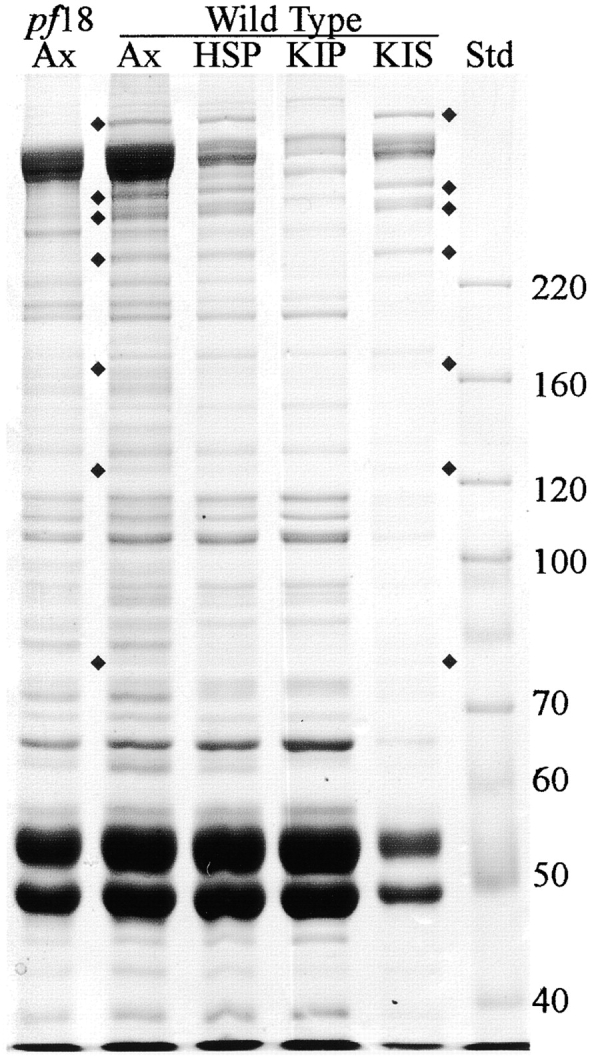
Coomassie blue–stained gel of pf18 axonemes and various fractions of wild-type axonemes demonstrating selective extraction of C1-associated proteins. Ax, unextracted axonemes; HSP, pellet following extraction with high salt (0.6 M NaCl); KIP, pellet following extraction of the HSP fraction with 0.2 M KI; KIS, supernatant from extraction with 0.2 M KI; Std, M r standards. All seven central pair proteins identified as C1-associated based on their retention in the HSP fraction, indicated by diamonds, are also present in the KIS fraction. Molecular mass standards (kD) are indicated along the right margin.
Figure 11.
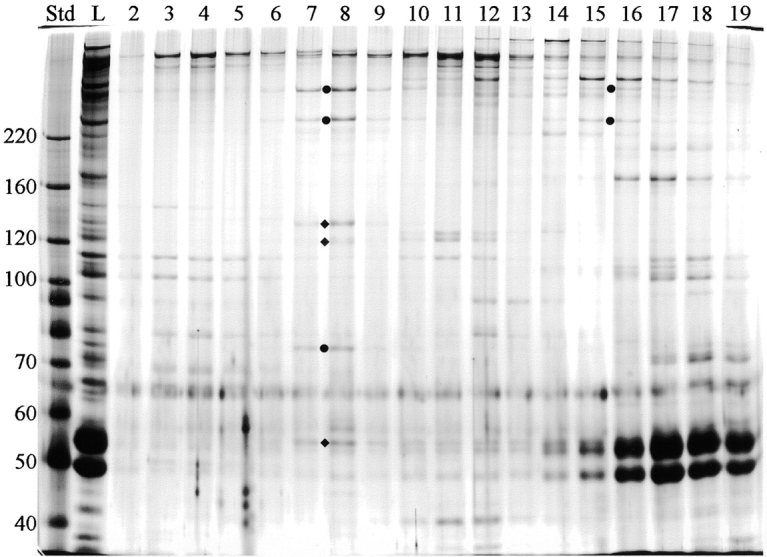
Sucrose gradient fractionation of C1-associated proteins. The KIS fraction shown in Fig. 10 was centrifuged on a 5–20% sucrose gradient and collected from the bottom in 24 fractions. Equal volumes of fractions 2–19 were analyzed by SDS-PAGE and silver stained. All three proteins missing from cpc1 axonemes (filled circles) cosediment in a complex at 16S (fractions 7 and 8) along with three additional proteins (diamonds). Std, mass standards; L, material loaded onto the gradient. The mass of markers (kD) is indicated along the left margin.
Discussion
Flagellar Motility
Previous genetic studies of Chlamydomonas central pair structure and biochemistry (Warr et al., 1966; Witman et al., 1978; Adams et al., 1981; Dutcher et al., 1984) focused on mutations that have severe effects on flagellar motility. Extensive screening identified four loci required for central pair assembly (PF15, PF18, PF19, and PF20) and two loci needed for C1 stability (PF16) or assembly of a C1-associated projection (PF6). By selecting for more moderate motility defects and using electron microscopy as a secondary screen, we have identified two alleles at a new locus, CPC1, that also prevent assembly of a C1-associated structure. Unlike other characterized central pair mutants, cpc1 mutants are able to swim progressively and hence can be tested for retention of specific beat parameters and forms of regulation. Here we report that cpc1 mutant flagella beat at about two-thirds of the wild-type frequency, and respond at a gross level to physiological stimuli such as phototactic and photophobic changes in light intensity in vivo and to the corresponding changes in Ca2+ concentration in vitro (Fig. 1). Preliminary measures of waveform and beat frequency parameters in reactivated axonemes suggest that motility at high Ca2+ concentrations is completely normal in this mutant (Mitchell, D.R., unpublished observations), which would indicate that the structures missing in cpc1 are specifically involved in asymmetric forward swimming motility. Beat frequency measurements on forward swimming cells show that cpc1 reduces frequency through a mechanism that does not depend on outer row dyneins (Fig. 2). Additional tests will be needed to characterize the more subtle effects of this mutation on waveform and other bend parameters, which should more directly define the function of this central pair component.
Central Pair Structure
Although the central pair apparatus is often represented diagramatically as a symmetric structure with just two rows of projections from each microtubule, data presented here and in earlier work (reviewed in Smith and Lefebvre, 1997) are more consistent with a high degree of asymmetry. The C1 microtubule has at least five unique complexes attached to its surface: the long 1a and 1b projections with 16-nm repeat periodicities, the much shorter 1c and 1d projections with 32-nm repeat periodicities, and the bridge between C1 and C2. The C2 microtubule likewise has five unique attachments: projections 2a and 2b and the bridge with 16-nm repeat periodicities, and projection 2c and the diagonal link, whose periodicities have not yet been determined. Although we designate 1c and 1d as single densities, both can be further subdivided in the averaged transverse images (Fig. 4), and both are altered by loss of sheath fibers in either pf6 (Fig. 4 K) or cpc1 (Figs. 4 G and 7 H).
The 1a projection appears to be attached directly to the microtubule surface at one end and to the sheath at the other end (Fig. 4), and its loss in pf6 axonemes leaves little or no residual density on the microtubule surface. Attachment of 1b is more complex, since the density lost in cpc1 does not extend in a straight line to the microtubule surface, but instead includes material transposed clockwise by one protofilament on the C1 microtubule surface and leaves a residual density at the base of the projection. The base of 1b also interacts with the diagonal link and thus is connected to C2 as well as C1 (Fig. 4 C and Fig. 6). At the tip of 1b there are apparent connections to both the sheath and the 2b projection, although it is equally possible that the sheath is a continuous structure and is alone responsible for connecting the tips of the 1b and 2b projections. Loss of 1b (cpc1) results in both loss of the associated sheath material and instability or disrupted assembly of 2b.
Cross-sections of the central pair apparatus from other distantly related organisms such as Tetrahymena (Sugrue et al., 1991) show a remarkably similar pattern to that seen in Chlamydomonas. Structures associated specifically with the C1 microtubule at a 32-nm periodicity have been noted previously in negatively stained Tetrahymena axonemes (Chasey, 1972), squid sperm axonemes (Linck et al., 1981), and in quick-freeze deep-etched Chlamydomonas axonemes (Goodenough and Heuser, 1985). In each case, 16-nm projections were seen along both central pair microtubules, suggesting a highly conserved structural arrangement for most central pair components.
Central Pair Biochemistry
Dutcher et al. (1984) identified 23 central pair–associated proteins by comparing wild-type and pf18 axonemal proteins on 1- and 2-d gels, and presented evidence that 10 of these were likely to be C1-associated because they were reduced or missing in pf16 axonemes and were not extracted when C2 was solubilized by 0.6 M NaCl. Of these 10 proteins, three were identified as missing in pf6 axonemes and thus presumably reside in the 1a projection (97, 66, and 20 kD) and three were missing from pf16 flagella (128, 57, and 32 kD), leaving only four known C1 proteins (CP1, CP2, CP4, and 110 kD) that might be a part of the remaining C1-associated structures. Only one of these four proteins, CP4, is missing from cpc1 axonemes. We characterize CP3 and bands of 165, 125, and 79 kD as additional C1 proteins, and show that two of these proteins (CP3 and 79 kD) are also missing in cpc1. The three proteins missing in cpc1 axonemes, CP3, CP4, and 79 kD, likely reside in the projection 1b/sheath complex along with proteins of 135, 125, and 56 kD, which were not identified as missing in gels of cpc1 axonemes, but which cosediment with the directly identified proteins in a 16S complex (Fig. 11).
No definitive location can be given for C1-associated CP1, CP2, or 165-kD bands, but indirect evidence suggests that CP1 and 165 kD are in the diagonal link between projection 1b and C2. This structure should be biochemically definable as a protein or proteins that are present in both pf16 axonemes (Fig. 6) and cpc1 axonemes (Fig. 4), and also retained after high salt extraction of wild-type axonemes (Fig. 5), but should be lost from cpc1 axonemes when the C2 microtubule is removed by high salt extraction. Any connection between this link and C1 is already disrupted by the cpc1 defect before exposure to high salt. CP1 and 165 kD fit all of these criteria: both are present in axonemes from pf16 (Fig. 9) and cpc1 (Figs. 8 and 9) but are lost from cpc1 axonemes on salt extraction (Fig. 8).
Our data extend previous antibody studies that identified several kinesin-like proteins in the central pair apparatus (Bernstein et al., 1994; Fox et al., 1994; Johnson et al., 1994). Although Klp1 is the only central pair protein proven to be related to kinesins at the sequence level, preliminary molecular data support the existence of at least three other flagellar kinesins (Bernstein, 1995) increasing the likelihood that central pair kinesin-like antigens are truly kinesin family members. Based on our Western blots (Fig. 9), the 105-kD kinesin-like protein recognized by anti-HIPYR (110 kD in Fox et al., 1994) is missing from pf16 as well as pf18 axonemes and is thus a C1 protein. Since it is not depleted in either cpc1 (Fig. 9) or pf6 axonemes (Mitchell, D.R., unpublished observations), this 105-kD kinesin-like protein is not part of 1a, 1b, or the sheath. The 110-kD protein recognized by αHD antiserum may represent a second C1-associated kinesin, since it was localized to a single central pair microtubule that probably corresponds to C1 (Bernstein, 1995). This 110-kD antigen was not depleted in axonemes of pf18 or other central pair assembly mutants (Johnson et al., 1994) and is therefore a different protein from the 105-kD HIPYR antigen. A third kinesin-like protein, Klp1, has been immunolocalized to the lateral side of C2 in a region that most closely corresponds to densities 2b or 2c (see Fig. 10 of Bernstein et al., 1994). However, retention of Klp1 in cpc1 axonemes (Mitchell, D.R., unpublished observations) argues against a 2b location. The 85-kD HIPYR antigen (Fig. 9) also has the properties of a C2-associated kinesin-like protein, since it is missing from pf18 and retained by both pf16 and cpc1. In summary, there may be two kinesin-like proteins attached to each central pair microtubule, but additional data will be needed for precise localizations and to determine their roles in flagellar motility.
Central Pair Function
While not addressed in this paper, interactions between spoke heads and central pair projections have been observed previously in mussel gill cilia (Warner and Satir, 1974), Tetrahymena cilia (Goodenough and Heuser, 1985), and Chlamydomonas flagella (Mastronarde et al., 1992). These interactions result in tilting of spokes in bent regions of gill cilia, where spoke heads interact with central pair projections in positions equivalent to the tips of projections 1a and 1b in Chlamydomonas (Fig. 15 in Warner and Satir, 1974). In this image, as well as in Fig. 7 of Goodenough and Heuser (1985) and Fig. 9 of Mastronarde et al. (1992), each T-shaped spoke head appears to interact with a pair of projections or sheath elements. In Chlamydomonas, the two spokes in each 96-nm repeat interval are separated by 32 nm, so that each spoke head can align precisely with pairs of adjacent structures that have a 16-nm repeat. If the central pair apparatus functions strictly through interactions with radial spokes, then it will be important to identify structures involved in those interactions and characterize their biochemical components. How signals are transmitted from these paired structures through radial spokes is not known, nor is it clear why so many apparently different central pair projections should be required. At present, we hypothesize that the inherent asymmetry of the central pair apparatus allows simultaneous unique interactions of different central pair projections with spokes on different doublets, and that these interactions in turn are involved in doublet-specific, kinase-mediated regulation of dynein activity.
Central pair microtubules represent a unique system for studying both microtubule-associated proteins and dynein regulation. Central pair structures with both 16- and 32-nm periodicities overlie the 8-nm tubulin dimer lattice and form complex interactions that, because of their regular periodicity, lend themselves to modern structural approaches such as digital image averaging. When combined with the ability to select mutations that disrupt parts of the central pair apparatus, and to clone and characterize the loci involved, these approaches should lead to rapid advances in our understanding of this flagellar structure and its role in motility. The recent identification of the pf15 gene product as a subunit of the microtubule severing protein katanin (Smith, E., and P. Lefebvre. 1998. Mol. Biol. Cell. 9:278a), and discovery of both a type 1 protein phosphatase, and an A kinase anchoring protein associated with the Chlamydomonas central pair (Sale, W., unpublished observations), suggest that the regulatory roles of this complicated structure are just beginning to be recognized. Further biochemical and molecular characterization of the CPC1 gene product should expand our understanding of the mechanisms through which radial spoke–central pair interactions regulate flagellar dynein activity, and may provide useful parallels to dynein regulation in other systems as well.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM44228 to D. Mitchell and GM51173 to W. Sale).
Footnotes
Address correspondence to David R. Mitchell, Department of Anatomy, SUNY Health Science Center, 750 E. Adams Street, Syracuse, NY 13210. Tel.: (315) 464-8575. Fax: (315) 464-8535. E-mail: mitcheld@vax.cs.hscsyr.edu
D. Mitchell would like to express his appreciation to all members of the Sale lab, where most of this work was done, and acknowledge contributions from Kim Brown (plasmid rescue and Southern blotting). We would also like to thank M. Bernstein for provided anti-Klp1 antibodies, and Eileen O'Toole for the image averaging.
References
- Adams GMW, Huang B, Piperno G, Luck DJL. Central-pair microtubular complex of Chlamydomonasflagella: polypeptide composition as revealed by analysis of mutants. J Cell Biol. 1981;91:69–76. doi: 10.1083/jcb.91.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein M. Flagellar kinesins: new moves with an old beat. Cell Motil Cytoskelet. 1995;32:125–128. doi: 10.1002/cm.970320211. [DOI] [PubMed] [Google Scholar]
- Bernstein M, Beech PL, Katz SG, Rosenbaum JL. A new kinesin-like protein (Klp1) localized to a single microtubule of the Chlamydomonasflagellum. J Cell Biol. 1994;125:1313–1326. doi: 10.1083/jcb.125.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen M, Fay RB, Witman GB. Calcium control of waveform in isolated flagellar axomenes of Chlamydomonas. . J Cell Biol. 1980;86:446–455. doi: 10.1083/jcb.86.2.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brokaw CJ, Kamiya R. Bending patterns of Chlamydomonasflagella. IV. Mutants with defects in inner and outer dynein arms indicate differences in dynein arm function. Cell Motil Cytoskelet. 1987;8:68–75. doi: 10.1002/cm.970080110. [DOI] [PubMed] [Google Scholar]
- Brokaw CJ, Luck DJL. Bending patterns of Chlamydomonasflagella. III. A radial spoke head deficient mutant and a central pair deficient mutant. Cell Motil. 1985;5:195–208. doi: 10.1002/cm.970050303. [DOI] [PubMed] [Google Scholar]
- Chapelin C, Coste A, Reinert P, Boucherat M, Millepied MC, Poron F, Escudier E. Incidence of primary ciliary dyskinesia in children with recurrent respiratory diseases. Ann Otol Rhinol Laryngol. 1997;106:854–858. doi: 10.1177/000348949710601008. [DOI] [PubMed] [Google Scholar]
- Chasey D. Further observations on the ultrastructure of cilia from Tetrahymena pyriformis. . Exp Cell Res. 1972;74:471–479. doi: 10.1016/0014-4827(72)90403-x. [DOI] [PubMed] [Google Scholar]
- Dutcher SK, Huang B, Luck DJL. Genetic dissection of the central pair microtubules of the flagella of Chlamydomonas reinhardtii. . J Cell Biol. 1984;98:229–236. doi: 10.1083/jcb.98.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox LA, Sawin KE, Sale WS. Kinesin-related proteins in eukaryotic flagella. J Cell Sci. 1994;107:1545–1550. doi: 10.1242/jcs.107.6.1545. [DOI] [PubMed] [Google Scholar]
- Frey E, Brokaw CJ, Omoto CK. Reactivation at low ATP distinguishes among classes of paralyzed flagellamutants. Cell Motil Cytoskelet. 1997;38:91–99. doi: 10.1002/(SICI)1097-0169(1997)38:1<91::AID-CM8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Goodenough UW, Heuser JE. Substructure of inner dynein arms, radial spokes, and the central pair/projection complex. J Cell Biol. 1985;100:2008–2018. doi: 10.1083/jcb.100.6.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habermacher G, Sale WS. Regulation of flagellar dynein by phosphorylation of a 138-kD inner arm dynein intermediate chain. J Cell Biol. 1997;136:167–176. doi: 10.1083/jcb.136.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, E.H. 1989. The Chlamydomonas sourcebook. Academic Press Inc., San Diego. 399–446.
- Hoops HJ, Witman GB. Outer doublet heterogeneity reveals structural polarity related to beat direction in Chlamydomonasflagella. J Cell Biol. 1983;97:902–908. doi: 10.1083/jcb.97.3.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horst, C.J., and G.B. Witman. 1995. Reactivation of Chlamydomonas cell models. In Cilia and Flagella. W. Dentler and G. Witman, editors. Academic Press Inc., San Diego. 207–210. [DOI] [PubMed]
- Howard DR, Habermacher G, Glass DB, Smith EF, Sale WS. Regulation of Chlamydomonasflagellar dynein by an axonemal protein kinase. J Cell Biol. 1994;127:1683–1692. doi: 10.1083/jcb.127.6.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Ramanis Z, Luck DJL. Suppressor mutations in Chlamydomonasreveal a regulatory mechanism for flagellar function. Cell. 1982;28:115–124. doi: 10.1016/0092-8674(82)90381-6. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Haas MA, Rosenbaum JL. Localization of a kinesin-related protein to the central pair apparatus of the Chlamydomonas reinhardtiiflagellum. J Cell Sci. 1994;107:1551–1556. doi: 10.1242/jcs.107.6.1551. [DOI] [PubMed] [Google Scholar]
- King SJ, Dutcher SK. Phosphoregulation of an inner dynein arm complex in Chlamydomonas reinhardtiiis altered in phototactic mutant strains. J Cell Biol. 1997;136:177–191. doi: 10.1083/jcb.136.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linck RW, Olson GE, Langevin GL. Arrangement of tubulin subunits and microtubule-associated proteins in the central pair microtubule apparatus of squid (Loligo pealei)sperm flagella. J Cell Biol. 1981;89:309–322. doi: 10.1083/jcb.89.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN, O'Toole ET, McDonald KL, McIntosh JR, Porter ME. Arrangement of inner dynein arms in wild-type and mutant flagella of Chlamydomonas. . J Cell Biol. 1992;118:1145–1162. doi: 10.1083/jcb.118.5.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DR, Kang Y. Identification of oda6 as a Chlamydomonasdynein mutant by rescue with the wild-type gene. J Cell Biol. 1991;113:835–842. doi: 10.1083/jcb.113.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RL, Scholey JM. Heterotrimeric kinesin-II is required for the assembly of motile 9+2 ciliary axonemes on sea urchin embryos. J Cell Biol. 1997;138:1009–1022. doi: 10.1083/jcb.138.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myster SH, Knott JA, O'Toole E, Porter ME. The Chlamydomonas Dhc1gene encodes a dynein heavy chain subunit required for assembly of the I1 inner arm complex. Mol Biol Cell. 1997;8:607–620. doi: 10.1091/mbc.8.4.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piperno G, Luck DJL. Axonemal adenosine triphosphatases from flagella of Chlamydomonas reinhardtii. . J Biol Chem. 1979;254:3084–3090. [PubMed] [Google Scholar]
- Piperno G, Mead K, Shestak W. The inner dynein arms I2 interact with a “dynein regulatory complex” in Chlamydomonasflagella. J Cell Biol. 1992;118:1455–1463. doi: 10.1083/jcb.118.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter ME, Power J, Dutcher SK. Extragenic suppressors of paralyzed flagellar mutations in Chlamydomonas reinhardtiiidentify loci that alter the inner dynein arms. J Cell Biol. 1992;118:1163–1176. doi: 10.1083/jcb.118.5.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter ME, Knott JA, Gardner LC, Mitchell DR, Dutcher SK. Mutations in the SUP-PF-1 locus of Chlamydomonas reinhardtiiidentify a regulatory domain in the beta dynein heavy chain. J Cell Biol. 1994;126:1495–1507. doi: 10.1083/jcb.126.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp G, O'Toole E, Gardner LC, Mitchell BF, Porter ME. The sup-pf-2 mutations of Chlamydomonasalter the activity of the outer dynein arms by modification of the gamma-dynein heavy chain. J Cell Biol. 1996;135:1853–1865. doi: 10.1083/jcb.135.6.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale WS, Satir P. Direction of active sliding of microtubules in Tetrahymenacilia. Proc Natl Acad Sci USA. 1977;74:2045–2049. doi: 10.1073/pnas.74.5.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin KE, Mitchison TJ, Wordeman LG. Evidence for kinesin-related proteins in the mitotic apparatus using peptide antibodies. J Cell Sci. 1992;101:303–313. doi: 10.1242/jcs.101.2.303. [DOI] [PubMed] [Google Scholar]
- Smith EF, Lefebvre PA. PF16 encodes a protein with armadillo repeats and localizes to a single microtubule of the central apparatus in Chlamydomonasflagella. J Cell Biol. 1996;132:359–370. doi: 10.1083/jcb.132.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EF, Lefebvre PA. The role of central apparatus components in flagellar motility and microtubule assembly. Cell Motil Cytoskelet. 1997;38:1–8. doi: 10.1002/(SICI)1097-0169(1997)38:1<1::AID-CM1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Smith EF, Sale WS. Regulation of dynein-driven microtubule sliding by the radial spokes in flagella. Science. 1992;257:1557–1559. doi: 10.1126/science.1387971. [DOI] [PubMed] [Google Scholar]
- Sugrue P, Avolio J, Satir P, Holwill MEJ. Computer modeling of Tetrahymenaaxonemes at macromolecular resolution. J Cell Sci. 1991;98:5–16. doi: 10.1242/jcs.98.1.5. [DOI] [PubMed] [Google Scholar]
- Tam L-W, Lefebvre PA. Cloning of flagellar genes in Chlamydomonas reinhardtiiby DNA insertional mutagenesis. Genetics. 1993;135:375–384. doi: 10.1093/genetics/135.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam, L.-W., and P.A. Lefebvre. 1995. Insertional mutagenesis and isolation of tagged genes in Chlamydomonas. In Cilia and Flagella. W. Dentler and G. Witman, editors. Academic Press Inc., San Diego. 519–523. [DOI] [PubMed]
- Wakabayashi K, Yagi T, Kamiya R. Ca2+-dependent waveform conversion in the flagellar axoneme of Chlamydomonasmutants lacking the central-pair radial spoke system. Cell Motil Cytoskelet. 1997;38:22–28. doi: 10.1002/(SICI)1097-0169(1997)38:1<22::AID-CM3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Warner FD. Ciliary inter-microtubule bridges. J Cell Sci. 1976;20:101–114. doi: 10.1242/jcs.20.1.101. [DOI] [PubMed] [Google Scholar]
- Warner FD, Satir P. The structural basis of ciliary bend formation. Radial spoke positional changes accompanying microtubule sliding. J Cell Biol. 1974;63:35–63. doi: 10.1083/jcb.63.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner FD, Mitchell DR, Perkins CR. Structural conformation of the ciliary ATPase dynein. J Mol Biol. 1977;114:367–384. doi: 10.1016/0022-2836(77)90255-8. [DOI] [PubMed] [Google Scholar]
- Warr JR, McVittae A, Randall J, Hopkins JM. Genetic control of flagellar structure in Chlamydomonas reinhardtii. . Genet Res. 1966;7:335–351. [Google Scholar]
- Witman GB, Plummer J, Sander G. Chlamydomonasflagellar mutants lacking radial spokes and central tubules. J Cell Biol. 1978;76:729–747. doi: 10.1083/jcb.76.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray W, Boulikas T, Wray VP, Hancock R. Silver staining of proteins in polyacrylamide gels. Anal Biochem. 1981;118:197–203. doi: 10.1016/0003-2697(81)90179-2. [DOI] [PubMed] [Google Scholar]