

Abstract
Previous data have shown that reducing agents disrupt the structure of vaccinia virus (vv). Here, we have analyzed the disulfide bonding of vv proteins in detail. In vv-infected cells cytoplasmically synthesized vv core proteins became disulfide bonded in the newly assembled intracellular mature viruses (IMVs). vv membrane proteins also assembled disulfide bonds, but independent of IMV formation and to a large extent on their cytoplasmic domains. If disulfide bonding was prevented, virus assembly was only partially impaired as shown by electron microscopy as well as a biochemical assay of IMV formation. Under these conditions, however, the membranes around the isolated particles appeared less stable and detached from the underlying core. During the viral infection process the membrane proteins remained disulfide bonded, whereas the core proteins were reduced, concomitant with delivery of the cores into the cytoplasm. Our data show that vv has evolved an unique system for the assembly of cytoplasmic disulfide bonds that are localized both on the exterior and interior parts of the IMV.
Keywords: poxviridae, viral assembly, vaccinia virus, disulfide bonding, reducing agents
The formation of disulfide bonds during (re)folding of proteins in vitro has been well documented (reviewed in 15). Most of the recent interest in protein folding, however, has concentrated on the folding and disulfide bond formation in (mammalian) cells. It is now well accepted that disulfide bonding and folding of proteins occur in the lumen of the ER. The unique (GSH/GSSG)1 ratio (of ∼3:1) within the lumen of this compartment is thought to provide it with a redox potential that, in contrast to the cytosol (ratio 100:1), is oxidizing enough to favor disulfide bonding (24). Moreover, the ER lumen contains a variety of molecular chaperones that assist in the folding reactions (for review see 17, 19, 20). Finally, this compartment is equipped with an abundant nonmembrane protein, protein disulfide isomerase (PDI), that from in vitro experiments (3, 5), as well as from genetic evidence (36), plays an important role in the disulfide bonding of proteins in the ER. PDI contains two, thioredoxin-like motifs, a sequence of 11 amino acids that aligns with a corresponding sequence in the Escherichia coli thioredoxin protein (reviewed in 15, 16). This latter protein may be involved in catalyzing reduction (21). Other proteins that contain thioredoxin motifs include glutaredoxin (21), E. coli DsbA (the E. coli equivalent of PDI), and the ER proteins ERp72 and ERp60. These proteins contain the characteristic active site sequence composed of CXXC in which the cysteine residues act as redox-active groups (16).
Viral membrane proteins have been used extensively to study the disulfide bonding and folding processes in the ER lumen (reviewed in 10). They appear to follow the same rules as cellular proteins in that they are inserted in a cotranslational manner into the ER where they undergo folding and oligomerization reactions. Moreover, they provide an advantage over cellular proteins in being abundantly expressed upon viral infection, thus facilitating detailed studies of these processes. Recent results using the well-characterized influenza virus hemagglutinin (HA) protein, have shown that upon addition of DTT to living cells, newly synthesized HA remains reduced in the lumen of the ER while already synthesized and disulfide bonded molecules become reduced as long as they resided inside the ER (2). This observation has subsequently been tested with other proteins, mostly with similar results (for example see 37, 45, 56).
Disulfide bonding can occur within one molecule (intramolecular) where two cysteines are bridged by a disulfide bond allowing the molecule to fold. Other types of disulfide bonds may be intermolecular, whose role can be to link different subunits of a complex or to assemble subunits of one protein into a higher homo-oligomeric form. An example of this latter process is the posttranslational dimerization of the equine arteritis virus Gs membrane protein (9). The general consensus is that this kind of disulfide bonding also occurs in the lumen of the ER.
Vaccinia virus (vv), the best studied member of the poxviridae, is the largest and most complex of animal viruses known, measuring ∼350 nm in its largest dimension. It contains a dsDNA genome of ∼190 kB encoding for >200 proteins, of which ∼100 seem to be associated with the virion (14). vv is unique in that during its life cycle two infectious forms are made, the intracellular mature virus (IMV) and the extracellular enveloped virus (EEV; 40). We have recently shown that the IMV membranes are derived from the intermediate compartment (IC) located between the ER and the Golgi complex (53), a finding that is consistent with the fact that at least three of its membrane proteins insert cotranslationally into the RER and are retained in the IC in infected cells (34, 51). Although the detailed structure of the IMV is unknown, it is generally accepted that the virion is composed of a membrane-enclosed, brick-shaped core that contains four particularly abundant proteins, 4a (gene A10L), 4b (A3L), and the 11-kD (F17R) and 25-kD (L4R) putative DNA-binding proteins (see Fig. 1 and Table I). The surface of the viral core is studded with a spike-like structure, that comprises, at least in part, an abundant 39-kD protein (A4L; 8, 50). The IC-derived cisternal membranes that surround the core contain three highly abundant membrane proteins of 16 (A14L), 21 (A17L), and 8 (A13L) kD (26, 51), as well as a peripheral membrane protein p14 (A27L; 47, 49, 54). Furthermore the IMV membrane contains a set of less abundant membrane proteins such as p32 (D8L; 43), p35 (H3L; 4), and a 27-kD myristoylated protein (L1R; 60; see Fig. 1 and Table I).
Figure 1.

Schematic representation of the IMV structure. The brick-shaped core in the central part is made up predominantly by the proteins 4a, 4b, p25, and p11. The core is studded with a layer of spikes that consists at least in part of the p39 protein. The particle is surrounded by two cisternal membrane layers that we believe do not fused with themselves (which is depicted in this figure as a hypothetical overlapping of the cisternal membranes). The inner of the two membranes contains mainly p21 and p16, the outer p35, p32, p8, and the peripheral membrane protein p14 (see also 50). In this model the cisternal envelope is akin to the two membranes of the nuclear envelope, continuous but distinct in protein composition.
Table I.
Overview of the Main vv Proteins Described in This Study
| Name | Molecular mass* | Gene | Location‡ | Reference | ||||
|---|---|---|---|---|---|---|---|---|
| p16 | 15–16 kD | A14L | M | 49 | ||||
| p21 | 23–25 kD | A17L | M | 47 | ||||
| p32 | 32 kD | D8L | M | 38, 43 | ||||
| p35 | 35 kD | H3L | M | 4 | ||||
| 4a | 65 kD | A10L | C | 59 | ||||
| p25 | 25 kD | L4R | C | 61 | ||||
| p39 | 40–45 kD | A4L | C | 39 |
Predicted molecular mass in SDS-PAGE of the monomeric (and mature) form of the proteins.
Location in the virion; M, membrane; C, core.
The IMV structure is highly dependent on disulfide bonds. It is well known that the removal of the viral membranes from the underlying core structure requires a mixture of a nonionic detergent and a reducing agent such as β-mercaptoethanol (12, 22, 52). More recent evidence for the sensitivity of the viral structure to reducing agents comes from the work of Roos et al. (50) showing that treatment of intact IMV with DTT induces the enveloping cisternal membranes to peel off the underlying core, as assessed by a novel cryo-EM technique. At the biochemical level several studies have indeed shown that vv membrane proteins may be disulfide bonded. Thus, the two abundant membrane proteins p21 and p16 as well as p32 have been shown to form disulfide bonded dimers (38, 47, 49). Since these latter proteins are associated with the IMV membrane, their disulfide bonding could logically be expected to occur in the lumen of the ER, following the accepted rules of disulfide bonding in vivo (see above). However, our recent detailed characterization of the topology of some of those proteins appeared to show that the cysteines potentially involved in disulfide bonding may be located in the cytoplasm (for example see 34, 51). Several studies have also suggested that cytoplasmically synthesized vv core proteins may form disulfide bonds. The work by Oie and Ichihashi (44) and Ichihashi et al. (25) described disulfide bonding of several proteins that were not further identified, but that from their characteristic migration in SDS-PAGE could be expected to be viral core proteins. These combined data seemed at odds with the current dogma on the intracellular formation of disulfide bonds. Until now we had rationalized by assuming that such cytoplasmic disulfide bonds were the result of after lysis oxidation, since in most of the relevant studies no alkylating reagent was used to prevent such (artifactual) disulfide bonding.
In this paper, we have investigated the vv disulfide bonds in more detail. Under conditions in which artifactual disulfide bonding is prevented, we show that the three vv core proteins that were analyzed are disulfide bonded in the IMV (and EEV) but not when IMV assembly is prevented. We also show that three vv membrane proteins form disulfide bonded dimers and, unexpectedly, in some cases on their cytoplasmic domains. We present data to show that these disulfide bonds seem required to assemble the membranes tightly around the core. When vv infects a new cell, these disulfide bonds are reduced on the core proteins whereas those on the membrane proteins remain disulfide bonded.
Materials and Methods
Cells, Viruses, Antibodies, and Plasmids
HeLa and BSC40 cells were grown as previously described (53). The WR strain of vaccinia virus was propagated as described (11). The vv recombinant expressing the MHV-M protein, as well as the COOH and NH2 terminus-specific antibodies to the M protein were described before (51). The following antibodies were used throughout this study; a peptide antibody to p16 (A14L; 51), to p11(F17R; 26), antibodies to p25 (L4R), and 4a (A10L) were a kind gift of Dr. Hruby (Oregon State University, Corvalis, OR; 57, 58); the antibodies to p39 (A4L) were a gift of Dr. Esteban (National Centre of Biotechnology, Madrid, Spain; 39); and the antibodies to p32 (D8L) were a gift of Dr. Niles (SUNY Medical School, Buffalo, NY; 43). To make antibodies to p21 (A17L) and p35 (H3L) peptides matching amino acid 17 through 37 of p21 and amino acid 31 through 43 of the H3L sequence were synthesized. Antibodies were made according to Cudmore et al. (8). One OD260 unit of purified WR was treated with NP-40 and DTT to make viral cores as described by Cudmore et al. (8). The cores were fixed for 30 min at room temperature with 0.5% glutaraldehyde in 10 mM Tris-Cl, pH 9, and an antibody, further referred to as anti-core antibody was generated as described by Cudmore et al. (8).
Preparation of post-nuclear supernatants (PNS) of infected cells and of NEM-treated WR and detection of vv proteins by Western blots.
For the preparation of PNS of infected cells, Hela cells grown in 10-cm dishes were infected with a MOI of 10 for 60 min at 37°C and then incubated overnight in the presence or absence of 100 μg/ml rifampicin (Sigma; from a 100 mg/ml stock in DMSO kept at −20°C). The cells were put on ice, washed twice with ice-cold PBS and once with PBS containing 20 mM N-ethyl maleimide (NEM, prepared fresh; Sigma). The cells were incubated for 20 min on ice in PBS with NEM, scraped from the dish and pelleted at 1,500 rpm in a Hereaus minifuge for 10 min. The pellet was resuspended in 300 μl of 10 mM Tris-Cl, pH 9, containing NEM and broken in a 2-ml dounce homogenizer by 12 strokes. The nuclei were removed by a 2-min spin at 2,000 rpm in an eppendorf centrifuge. To isolate NEM-treated WR, four 175-cm2 flasks of HeLa cells were infected at low MOI for 2 to 3 d. PNS of infected cells was prepared as above, the PNS was loaded on top of 8 ml of a 36% (wt/vol) sucrose cushion in 10 mM Tris-Cl, pH 9, and the virus pelleted by centrifugation in a SW40 rotor at 24 krpm for 30 min. The pellet was resuspended in 10 mM Tris-Cl, pH 9, containing 20 mM NEM. PNS or virus were loaded onto 15% SDS-PAGE after boiling for 3 min in LSB with or without 5% β-ME and 100 mM DTT, the proteins blotted unto nitrocellulose and detected as described (13).
[3H]NEM Labeling
To 10 μl of a purified virus preparation (with a protein concentration of 2 mg/ml or 30 OD260/ml; 26) diluted in 165 μl of 10 mM Tris-Cl, pH 9, 10 μCi of [3H]NEM (Amersham) was added and the mixture incubated on ice for 20 min. To terminate the labeling, 40 μl of freshly prepared 100 mM NEM in 10 mM Tris-Cl, pH 9, was added and the virus spun for 30 min at 65 krpm in a TLA 100.2 tabletop centrifuge. The pelleted virus was resuspended in 40 μl sample buffer containing 5% β-ME. 5 μm of [3H]NEM-labeled virus was loaded on 15% SDS-PAGE and analyzed by autoradiography. [35S]methionine–labeled and purified virus was made as before (8). Labeled proteins were immunoprecipitated from 35S-labeled virus as followed; to 100 μl of labeled virus SDS was added to a final concentration of 1% and incubated for 30 min at 37°C. Samples were diluted in lysis buffer (32) to a final concentration of 0.1% SDS and proteins immunoprecipitated from them using specific antibodies. To demonstrate that NEM had access to intracellular viruses, HeLa cells were infected for two 2 d, washed twice with ice-cold PBS, and washed twice PBS with or with 20 mM NEM. The cells were incubated for 20 min on ice in PBS with or without NEM, scraped, and collected by pelleting. PNS was prepared as above in 10 mM Tris-Cl, pH 9, with or without NEM and the virus concentrated by pelleting through a sucrose cushion as described above. The pelleted virus was resuspended in Tris-Cl, pH 9, without NEM. 3 and 8 μg of total protein (as determined by OD260 measurement; see 28) of virus preparation from NEM or non-NEM-treated cells were labeled with 5 μCi of [3H]NEM as above. Equal amounts of virus as used for the [3H]NEM labeling were analyzed in 15% SDS-PAGE and the proteins detected by silver staining (26).
Pulse–Chase Experiments
HeLa cells grown in 3.5-cm dishes were infected with a multiplicity of infection (MOI) of 10. At 5.5 h after infection the cells were incubated for 30 min in DME without methionine (Sigma), and when indicated with or without 5 mM DTT. The cells were pulse-labeled for indicated times with 50 μCi per dish of [35S]methionine (Amersham) and chased in DME with 5% FCS. After the labeling the cells were washed twice with ice-cold PBS, once with PBS-20 mM N-ethyl maleimide (NEM), incubated for 20 min on ice in PBS with NEM, lysed in detergent solution with NEM and processed for immunoprecipitation as described (32).
Protease Digestion of Virus and PNS
Protease digestion of purified virus preparations was essentially as previously described (51). In brief, purified virus was diluted in 10 mM Tris-Cl, pH 9, and mixed with an equal volume of a two times concentrated trypsin solution. After digestion for 30 min at 30°C, aprotinin was added to a final concentration of 4 μg/ml and the samples spun for 30 min at 4°C at full speed in an Eppendorf centrifuge. Both the supernatant and the pellet were analyzed in 10% SDS-PAGE and the proteins detected by Western blots. Protease digestion of PNS was as described in Salmons et al. (51). After digestion and addition of aprotinin, the digested samples were centrifuged for 30 min at 150,000 g. Supernatant and pellet were analyzed by Western blots and ECL.
DTT Treatment of Infected HeLa Cells
HeLa cells grown to confluency in two parallel dishes were infected with an MOI of 10. At 4 h after infection the medium was replaced with new medium with or without 5 mM freshly prepared DTT. Medium was replaced each hour with or without freshly prepared DTT. For EPON embedding the cells were fixed at 8 h after infection and embedded as described (13). For the isolation of virions grown in the presence or absence of DTT, at 6 h after infection the medium was replaced by one volume of DMEM/5% FCS and 1 vol of MEM without methionine containing 50 μCi of [35S]methionine with or without DTT. This medium was replaced each hour with medium with or without DTT containing 50 μCi [35S]methionine until 9 h after infection. To remove unpenetrated virus, cells were rinsed extensively with cold PBS (or when indicated cold PBS with 20 mM NEM) and incubated for one hour on ice with 0.2 mg/ml of trypsin (Worthington Biochemical Corp.) in PBS. Digestion was terminated by the addition of aprotinin (Sigma) to a final concentration of 0.4 μg/ml. The cells were squirted from the dish and washed three times with PBS/aprotinin. The cells were broken by 10 strokes in a dounce homogenizer in 10 mM Tris-Cl, pH 9, and nuclei removed by centrifugation at 2,000 rpm in a Hereaus minifuge for 5 min. The PNS was layered on top of a 36% sucrose (wt/vol) cushion in 10 mM Tris-Cl, pH 9, and the virus pelleted by centrifugation of 30 min at 24 krpm in a SW40 rotor. The pellet was resuspended in 10 mM Tris-Cl, pH 9.
Negative staining EM after immunolabeling of the particles isolated from nontreated or DTT-treated cells was performed as described (13).
Plaque Assays and Immunofluorescence
Equal amounts of TCA precipitable counts of viruses from untreated or DTT-treated cells were diluted in 2 ml of serum-free DME and serial 10-fold dilutions were made. The dilutions were plaque-titrated on BSC-40 cells, cells fixed at 48 h after infection and the plaques counted. The titer was expressed as plaque forming units (PFU) per 1,000 cpm or as PFU per ml. HeLa cells grown on 11-mm-diam coverslips were infected with an MOI of 5, 10, and 20 for 60 min at 37°C, the inoculum was removed and replaced by DME with 5% FCS. The cells were fixed at 6 h after infection by a 4-min incubation at −20°C in methanol. The synthesis of viral late proteins was detected by indirect immunofluorescence using the anti-core antibody at 1:1,000 and donkey anti–rabbit coupled to FITC (Dianova: Jackson ImmunoResearch Labs) at 1:150. Viral DNA was labeled using Hoechst (Sigma) at 5 μg/ml. The percentage of infected cells was calculated as follows: of each fixed coverslip 50 cells were counted. They were considered positive by immunofluorescence if the anti-core antibody clearly labeled perinuclear structures typical of viral factories. They were scored DNA-positive, if clear cytoplasmic DNA labeling was detected.
Entry Experiments
For the entry experiments, WR was propagated in four 24 × 24-cm dishes of HeLa cells and purified as described by Jensen et al. (26). After banding the isolated virus in a 25–40% (wt/vol) sucrose gradient, the virus band was collected by fractionation and the fractions containing the virus was determined by a Biorad protein assay. The peak fractions were pooled and titered by plaque assay (generally the titer was ∼5 × 107 to 108 PFU/ml). For the entry assay HeLa cells were grown in 3.5-cm dishes, washed once with cold DME containing 20 mM Hepes, pH 7.4, and left on ice in this medium for 10–20 min. Virus (on the average an MOI of 10–20 was used) was diluted in 800 μl of DME with Hepes, sonicated for 1 min and cooled on ice. The virus was applied to the cooled cells and left for 60 min on ice. The dishes were then incubated for 60 min at 37°C, put on ice and washed twice with PBS and once with PBS/NEM. The cells were incubated for 60 min on ice in PBS containing 0.2 mg/ml of trypsin to remove unpenetrated virus from the plasma membrane. The trypsin digestion was terminated by adding 0.4 μg/ml of aprotinin (Sigma). After 10 min of incubation the cells were gently squirted off the dishes and collected by centrifugation for 30 s at 6,000 rpm in an Eppendorf centrifuge. The pellet was washed three times with PBS containing aprotinin. Subsequently, the cells were resuspended in 300 μl 10 mM Tris-Cl, pH 9, containing aprotinin and broken by 10 strokes of a dounce homogenizer. The nuclei were removed and the proteins concentrated by acetone precipitation overnight at −20°C. The samples were run on 15% SDS-PAGE and the proteins detected by Western blotting.
Measurements of the Total Intracellular the GSH/GSSG Concentrations
For the determination of the total intracellular GSH and GSSG concentrations Ellman's reagent was used. 5 × 107 HeLa spinner cells were infected at an MOI of 10 or mock infected. At 20 h after infection the cells were washed with PBS and collected by a centrifugation for 10 min at 1,500 rpm. Pelleted cells were resuspended in 300 μl of water and broken in a dounce-homogenizer and the nuclei removed. Ellman's reagent and NADPH (both from Sigma) were dissolved at 1 mg/ml and 4 mg/ml, respectively, in 1% NaHCO3. Subsequently, they were diluted to a final concentration of 0.13 mg/ml (Ellman's) and 0.1 mg/ml (NADPH), respectively, in 0.1 M phosphate buffer, pH 7, containing 1 mM EDTA. To 900 μl of this latter mixture 100 μl of sample or standard (see below) was added and 10 μl of glutathione-reductase (Boehringer) that was diluted 1:10 just before use in phosphate buffer/EDTA. Subsequently, the increase in OD412 was measured over a period of 3 min. As a standard a known concentration of GSSG was used. To determine the GSSG concentration only, PNS of infected or uninfected HeLa cells was prepared after pretreating the cells with 10 mM NEM.
Results
vv Membrane Proteins form Disulfide-bonded Dimers
Since previous papers describing the disulfide bonding of vv proteins omitted to use an alkylating reagent before cell lysis and virus isolation, it could not be excluded that the observed disulfide bonds were due to an artifactual post-lysis oxidation of the proteins. Throughout this study we therefore used the alkylating reagent NEM before cell lysis. Disulfide bonding was analyzed under three different conditions of infection. First, IMV was isolated and purified using our standard protocols, except that the infected cells were treated with NEM (or iodoacetamide; data not shown) before lysis, in order to assess the disulfide bonding as it occurred in the (isolated) IMV. Second, cells were infected in the presence of rifampicin, a drug that reversibly blocks the IMV formation at an early assembly stage but does not interfere with the synthesis of late proteins (40); this allowed us to assess whether or not the disulfide bonding depended on IMV assembly. Finally, cell lysates were also prepared from infected cells that were not treated with rifampicin to study the vv proteins in the infected cells at all stages of assembly.
The membrane proteins p21, p16, and p32 (see Fig. 1 and Table I) appeared to form disulfide-bonded dimers under all three conditions, since they occurred in monomer and dimer forms under nonreducing conditions (Fig. 2 a). The amount of dimer formation was somewhat variable; in the case of p21 and p16 ∼50% of the protein dimerized (see also below) while of p32 no more than 30% of the protein was in the dimer form. As noted before (48, 49, 55), the dimers of both p16 and p21 migrated at molecular masses that were smaller than the sum of two monomers. We presume that this is a consequence of a folded conformation of the dimer and not due to heterodimerization. By two-dimensional (2-D) gel analysis we have shown before that the dimers of p21 and p16 run at the same pI as their respective monomers (26). Since heterodimer formation would result in a shift of the pI relative to the monomeric protein, these 2-D gel data show that both proteins form homodimers.
Figure 2.
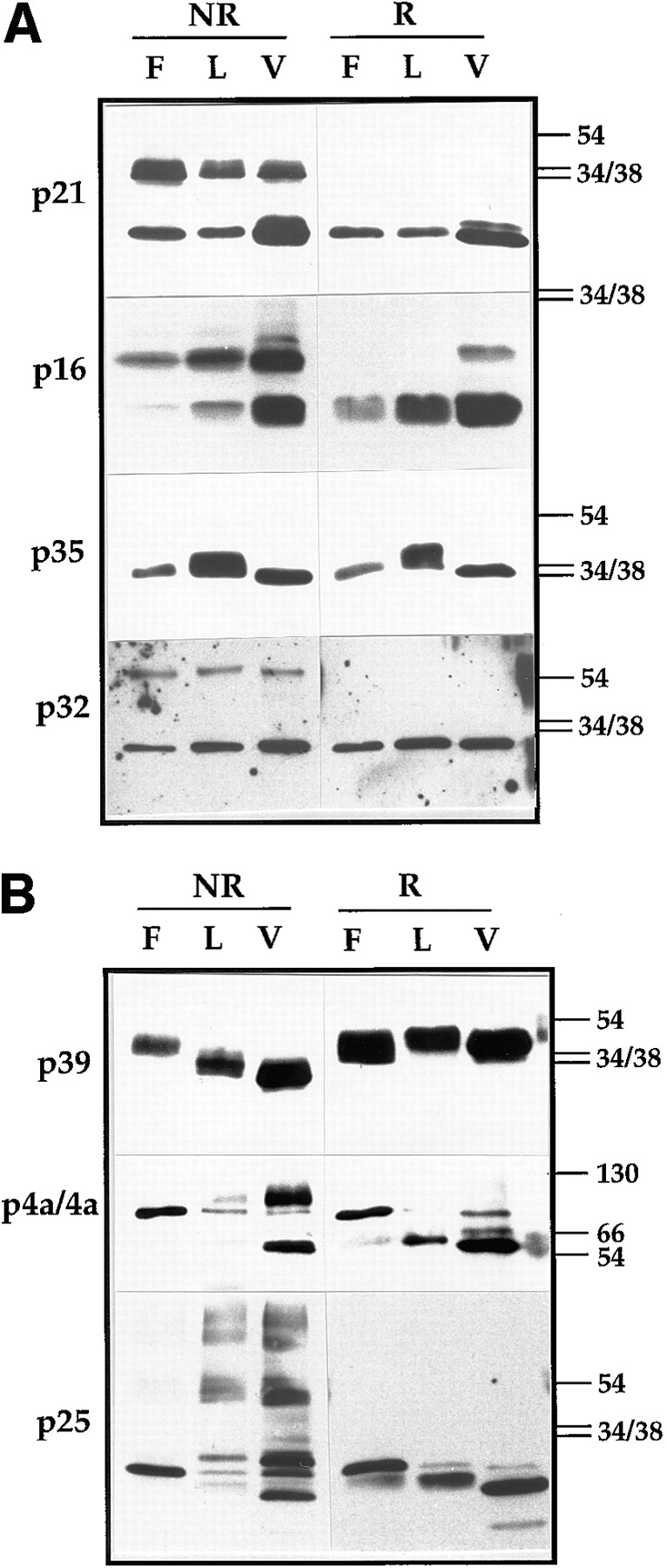
Western blots of vv proteins analyzed under reducing and nonreducing conditions. In A analysis of four IMV membrane proteins, p21 (A17L), p16 (A14L), p35 (H3R), and p32 (D8L). Cell lysates of infected HeLa cells treated (F) or not treated (L) with rifampicin or purified virus (V) were run on 15% SDS-PAGE. Before electrophoresis the samples were boiled for 3 min in LSB with (R) or without (NR) 5% β-ME and 100 mM DTT. The proteins were detected by Western blotting using the respective antibodies. On the right side of the figure the positions of the 54-, 38-, and 34-kD marker proteins is indicated. In B, three vv core proteins 4a (A10L), p39 (A4L), and p25 (L4R) were analyzed as described in A. Note that in rifampicin-blocked cell lysates for both 4a and p25 only their precursor form (of 110 and 28 kD, respectively) can be detected that is subsequently cleaved to the 65- and 25-kD mature forms, respectively, in the IMV. Some of the uncleaved form, can however, still be detected in the isolated virions. The positions of the 130-, 66-, 54-, 38-, and 34-kD marker proteins are indicated on the right.
In contrast to these three proteins, the migration of p35 was not altered when comparing reducing to nonreducing gels, arguing that this membrane protein does not form disulfide bonds (Fig. 2 a). The migration of this latter protein, however, varied slightly when comparing e.g., the rifampicin-blocked cell lysates to the IMV. The reason of these variations in molecular mass are presently not clear.
The dimers of p21 and p16 (but not of p32, or the disulfide bonded forms of the core proteins; see below) were highly resistant to reduction (see also 48, 49); boiling in sample buffer containing 5% β-mercaptoethanol only resulted in little reduction of their dimers. Addition to the standard sample buffer of higher concentrations of SDS or urea (up to 4 M), replacing β-ME by freshly prepared DTT or increasing the boiling time also did not result in a quantitative reduction of either p21 or p16 (not shown). For reasons we do not understand, the most complete reduction of these two proteins was obtained by using a mixture of 5% β-ME and 100 mM freshly prepared DTT (see Discussion).
Taken together, these data show that three vv membrane proteins form disulfide bonded dimers, in early vaccinia membranes as well as in the mature IMV.
vv Core Proteins Are Disulfide Bonded in the IMV Only
Next, we used the same approach to analyze some of the vv core proteins. In contrast to the three membrane proteins, the core proteins 4a, p25, and p39 (Fig. 1 and Table I) occurred only in their reduced form when the IMV formation was blocked (in rifampicin-blocked cell lysates; Fig. 2 b). However, in the purified IMV (and to some extent in cell lysates that had not been blocked with rifampicin) these three core proteins appeared to form disulfide bonds. This was most obvious for p39, whose migration was clearly faster in IMV analyzed under nonreducing conditions. The faster migration of the unreduced versus the reduced protein is typical for intramolecular disulfide bonding, resulting from a more folded conformation of the oxidized form (for example see 1).
In infected cells both p25 and 4a are initially made as precursors of 28 and 110 kD, respectively, which are subsequently cleaved upon IMV formation into their mature forms of 25 and 65 kD, respectively (57, 58). Indeed, in rifampicin treated cell lysates both proteins migrated at the position of their precursor form and the migration of both proteins was the same when analyzed with or without prior reduction. However, in the IMV the mature forms of these two proteins showed additional bands when analyzed without reduction. The protein 4a occurred both as a monomer of 65 kD, as well as a dimer of ∼130 kD when β-ME and DTT were omitted from the sample buffer (Fig. 2 b). That the dimer indeed ran at ∼130 kD can be better appreciated in Fig. 8, where 4a was analyzed in a 10% SDS-PAGE (see below). After reduction 4a migrated predominantly in its (reduced) mature form of 65 kD, while the 130 kD band was clearly absent. Two additional bands were seen in this reduced sample, the upper one represents the uncleaved precursor, p4a (note the comigration of this band with the form made in rifampicin-blocked cell lysates), of which small amounts can be detected in mature virions, while the identity of the lower of the two remained open. When analyzing p25 in isolated virions under nonreducing conditions the protein showed a very unusual pattern and appeared as several (disulfide bonded) species some of which migrated as a smear rather than discrete bands (Fig. 2 b). When analyzed after reduction, the protein migrated predominantly at the position expected for its mature 25-kD form, but also, as for 4a, some of the uncleaved precursor could still be detected.
Figure 8.

The proteins from viruses isolated from DTT-treated cells are not disulfide bonded. Infected HeLa cells were treated (DTT +) or not (DTT −) with 5mM DTT from 4 h after infection and the medium replaced each hour as described in the Results. 9 h after infection, virions were isolated from the cells as described in Materials and Methods. Isolated virions were analyzed on 10% SDS-PAGE and the viral proteins p21, p16, 4a, and p25 were detected by Western blotting. Virions isolated from untreated cells (V−) were analyzed after boiling in sample buffer with (DTT +) or without (DTT −) 5% β-ME and 100 mM DTT, while virions isolated from treated (V +) cells were only analyzed after boiling in sample buffer without (DTT −) these reducing agents. On the right the positions of the 34-, 38-, 54-, 84-, 116-, and 180-kD marker proteins are indicated.
Collectively, these results show that the vv core proteins are also able to make disulfide bonds, but in contrast to the membrane proteins, do so only in the assembled IMV.
[3H]NEM Labeling Shows That NEM Has Access to vv Core Proteins
The above results strongly suggest that vv membrane and core proteins form disulfide bonds. Care was taken in each of these experiments to incubate the infected cells at least 20 min with NEM on ice before cell lysis, thus excluding the possibility that the disulfide bonding resulted from post-lysis oxidation. However, the mature IMV is a highly compact structure and we had to rule out that NEM might not have access to vv proteins, especially to those within the viral core. Therefore, we performed two experiments to address this issue. First, isolated and purified IMVs were labeled with [3H]NEM for 20 min on ice and the labeled protein pattern compared with [35S]methionine-labeled virions. [3H]NEM labeling showed a discrete pattern of bands, the most abundant of which comigrated with the core proteins 4a, p11, and p25 (Fig. 3 a). Some of the other minor bands were identified as p21, p16, and p35 (not shown). The [3H]NEM labeling of both p21 and p16 increased significantly when the IMV was treated with DTT before the labeling (not shown).
Figure 3.

[3H]NEM has access to the viral core proteins. In A, isolated and purified IMVs were labeled for 20 min on ice with [3H]NEM (3H). Infected cells were labeled for 3 d with [35S]methionine and the labeled virions isolated and purified (35S). 35S-labeled virions were disrupted by incubation for 30 min at 37°C with 1% SDS and subsequently diluted in lysis buffer to a final concentration of 0.1% SDS. From these lysed particles the protein 4a (4a), p25 (p25), and p11 (p11) were immunoprecipitated. Analysis was on 15% SDS-PAGE followed by autoradiography. M-14C-labeled marker proteins of 14, 30, 45, 69, 93, and 200 kD. In B, infected cells were treated (NEM +) or not treated (NEM −) with 20 mM NEM for 20 min on ice before the preparation of PNS in buffer with or without NEM. The virions were concentrated by pelleting through a sucrose cushion at 24 krpm in a SW40 rotor for 30 min. Pellets were resuspended in buffer without NEM. Equal amounts of pelleted virions were incubated with 5 μCi of [3H]NEM and the labeled virions analyzed on 15% SDS-PAGE followed by autoradiography. In the left lane (of + or − NEM) 3 μg of total protein, in the right lane 8 μg of protein was used for [3H]NEM labeling. 35S virus control lane consisting of [35S]methionine-labeled, purified virions. M-14C-labeled marker proteins of 69, 45, 30, and 14 kD. In C, the same amounts of virions as used for the [3H]NEM labeling (3 μg of protein [left] and 8 μg [right], respectively) isolated from cells that were pretreated (NEM +) or not pretreated (NEM −) with NEM were run on 15% SDS-PAGE and the proteins were detected by silver staining.
To demonstrate that NEM also had access to the viral proteins in infected cells we carried out the following experiment. Infected cells were incubated with or without 20 mM NEM on ice before lysis. Subsequently, a PNS was prepared either in the presence or absence of NEM and the virions were concentrated from it by pelleting through a sucrose cushion. This pelleting step also served to separate the virions from unbound NEM. The pelleted viruses were then resuspended in NEM-free buffer and labeled on ice with [3H]NEM. As shown in Fig. 3 b, IMVs isolated from untreated cells were readily labeled with [3H]NEM and showed the same characteristic labeling pattern as in Fig. 3 a. In contrast, when infected cells were incubated with NEM before virus isolation, the virion were not labeled at all by [3H]NEM. The corresponding silver stained gel of the same virion preparations showed that the absence of labeling was not due to a lack of (viral) proteins (Fig. 3 c). We interpret these results to mean that upon preteatment of infected cells with (cold) NEM, this component readily binds to viral proteins, efficiently inhibiting the subsequent [3H]NEM labeling.
In conclusion, these data show that NEM has readily access to viral (core) proteins both in purified virions as well as in infected cells. This also rules out that the IMV could form artifactual disulfide bonds by a post-lysis oxidation in our experiments.
Kinetics of Disulfide Bonding
We next asked whether the vv disulfide bonds were built up concomitant with, or after protein synthesis. For this we chose to follow the kinetics of dimer formation of p16. This protein has been shown to be a very abundant membrane protein of the IMV that inserts into RER in a cotranslational manner and is retained in the IC in infected cells (51).
Infected cells were pulse-labeled at 6 h after infection and chased for up to 4 h. Small amounts of the dimer of p16 were first detected after 60 min, but was seen more clearly at 2 h of chase. At 4 h of chase ∼50% of p16 was in the dimer form (Fig. 4). The band migrating intermediate between the monomer and the dimer of p16 is due to the addition to some of N-linked glycosylation to a fraction of the molecules (Krijnse Locker, J., manuscript in preparation). A similar analysis of the core protein p39 showed that its disulfide bonding also occurred posttranslationally. The half time of its formation was ∼2 h, similar to the half time of IMV formation (data not shown).
Figure 4.

Disulfide bonding occurs posttranslationally. Infected HeLa cells were pulse-labeled (0 min chase) for 2 min at 6 h after infection and chased for the indicated time. Cell lysates were prepared and the p16 protein immunoprecipitated from them. The samples were run on 15% SDS-PAGE after boiling for 3 min in LSB with or without β-ME and DTT. The proteins were detected by autoradiography. After the pulse only the monomeric p16 is seen. After 60 min of chase the disulfide bonded dimer is detected as assessed by the absence of the 25-kD band when analyzed after reduction. The band migrating with a molecular mass between the monomer and the dimer is a N-glycosylated form of p16 that will be described elsewhere (Krijnse Locker, J., manuscript in preparation). M-14C-labeled marker proteins, of which the 14- and 30-kD proteins are indicated.
These data show that disulfide bonding occurs posttranslationally, adding further support to the evidence that they do not arise from artifactual post-lysis oxidation.
The Unique Cysteine of p32 Is Exposed on the Surface of the Virus as Well as in the Cytosol
Previous detailed analyses of the topology of the membrane proteins p21 and p16 strongly suggested that their relevant cysteines were not, as expected, exposed towards the lumen but are most likely on the cytosolic side of the membrane. For instance, we have proposed that p21 crosses the membrane four times, exposing its NH2 and COOH terminus towards the cytoplasm (34). If this topology is correct, p21 exposes two of its three cysteines (at positions 101 and 178) into the cytoplasm while its third cysteine (at position 131) is buried in the membrane (34). However, the possibility that p21 may span the membrane twice instead of four times, thereby exposing its cysteine at position 101 towards the lumen (where it could potentially dimerize with itself) cannot be excluded at present.
According to the previously proposed topology of p16, this protein spans the membrane twice and appears to adopt two different orientations in the membrane; one in which the NH2 and its 22–amino acid long COOH terminus (which includes both its cysteine at position 71 and a functional N-glycosylation site) are lumenal and one with the opposite orientation (51; Krijnse Locker et al., manuscript in preparation). That p16 may adopt two different topologies in infected cells was based on two independent pieces of evidence. First, using two different morphological assays we were able to show that a considerable fraction of p16 exposes the COOH terminus towards the cytosol (51). Second, since p16 is also partially N-glycosylated, its COOH terminus (that contains the N-glycosylation site) must also be exposed towards the lumen. A recently developed assay to quantitate the two different topologies of p16 in infected cells (Krijnse Locker and Griffiths, manuscript in preparation), showed that ∼50% of the disulfide bonded dimer of p16 exposes its COOH terminus towards the cytoplasm, implying that disulfide bonding of this protein occurs, at least in part, on its cytoplasmic domain.
Hydrophobicity plots of p32 revealed a single hydrophobic segment of 20 amino acids in its extreme COOH terminus that could serve as membrane-anchor (38). Thus, the protein seems to belong to a recently described class of membrane proteins with COOH-terminal hydrophobic domains that insert posttranslationally, thereby exposing most of their NH2 terminus towards the cytoplasm (35). If p32 inserts into the membrane accordingly, the protein would expose its single cysteine in the NH2 terminus at position 262 towards the cytoplasm as well as on the outer surface of the IMV (see also 54). To test this possibility, we carried out two different set of experiments. First, purified IMV was treated with increasing concentrations of trypsin and the digestion of p32 was assayed by Western blot using an antibody recognizing amino acid 77 to 294 (43). Under conditions in which the internally located core protein 4a was completely protease-protected, the entire population of both the dimer as well as the monomer of p32 were digested (Fig. 5 a). In fact, after pelleting the IMV from the digestion mixture, a new 28-kD fragment that was recognized by the p32 antibody was now detected in the supernatant, while the digested virion was totally devoid of immunoreactive p32 protein (Fig. 5 a). The well characterized peripheral membrane protein p14 was completely digested even at the lowest protease concentration tested and no protected fragment was detected in the supernatant.
Figure 5.

The NH2 terminus of p32 is exposed on the IMV surface and towards the cytoplasm of infected cells. (A) Purified IMV was treated for 30 min at 30°C with the indicated concentrations of trypsin. Digested IMV was pelleted by a 30-min centrifugation at 15,000 g. The pelleted virus (pellet) and the remaining supernatant were analyzed on 10% SDS-PAGE followed by Western blots using antibodies to p32, 4a, and p14. P14 and 4a were analyzed after reduction while p32 was analyzed under nonreducing conditions only. The position of the monomer of p32 is indicated with one and that of the dimer with two asterisks. (B) Post nuclear supernatants of rifampicin-treated cells infected with a vv recombinant expressing the MHV-M protein were prepared at 7 h after infection. PNS was treated with the indicated concentrations of trypsin at 30°C for 30 min. Supernatant was separated from the membranes (pellet) by a centrifugation for 30 min at 150,000 g and both fractions were analyzed on 15% SDS-PAGE. The proteins were detected by Western blots using antibodies to p32, the NH2 (M-N) or COOH terminus (M-C) of the MHV-M protein. The unglycosylated (M0) and glycosylated forms (M3 and M4) of the M protein are indicated (see also 33). Note that the COOH-terminal specific peptide serum recognizes only the M0 and M4 forms.
In a second set of experiments, post-nuclear supernatants of infected cells that were treated with rifampicin (to block virion formation), were digested with trypsin. Subsequently, the membranes were pelleted and the digestion of p32 followed by Western blots. In these experiments we made use of a vv recombinant expressing the mouse hepatitis virus (MHV) M protein as a control. The topology of this latter membrane protein has been well established, exposing its extreme COOH terminus of ∼18 amino acids towards the cytoplasm and its NH2 terminus lumenally (32). Indeed, with increasing concentrations of trypsin, reactivity to a peptide antibody raised against the COOH-terminal 18 amino acids of the MHV-M protein was completely lost (Fig. 5 b). In contrast, at all trypsin concentrations tested, the antibody to the NH2 terminus recognized the characteristic triplet representing the unglycosylated and two of the O-glycosylated forms of M (see also 33), showing that the membranes were intact (Fig. 5 b). When analyzing p32 under the same conditions, both the dimer as well as the monomer were completely digested. However, the same 28-kD fragment that was observed for the virus (see also 43), was again found in the supernatant, apparently released from the membranes (Fig. 5 b).
These experiments show that the entire NH2 terminus is not only exposed on the surface of the IMV, but also into the cytoplasm of infected cells. Since its single cysteine at position 262 is contained in this part of the molecule, it follows that disulfide bonding of p32 must occur on its cytoplasmic domain that is exposed on the surface of the IMV. Together with the available data on p21 and p16, these results suggest that cytosolic disulfide bonding may be a common feature of vv membrane proteins.
In the Presence of 5 mM DTT IMVs Are Formed That Are Less Stable
To address the possible function of the disulfide bonds in the IMV, it was necessary to reversibly inhibit their formation in infected cells. For this, advantage was taken of a previously described observation that the addition of 5 mM DTT to living cells not only keeps newly synthesized membrane proteins reduced but is also capable of reducing already oxidized proteins residing in the ER (2). 4 h after infection 5 mM DTT was added to one dish of cells, while a parallel dish was kept free of DTT. 4 h after infection was chosen since this time point is about one hour before the onset of viral assembly. At 8 h after infection the cells were fixed, embedded into Epon and the IMV-formation was assessed by EM. Since the DTT incubations were done over relatively long periods of time, the medium of the cells was replaced each hour with medium to which freshly prepared DTT was added just before use.
At first glance, the addition of DTT had no obvious effect on IMV assembly, since by thin section EM we readily observed many apparently normal looking immature viruses (IVs), intermediates and IMVs (Fig. 6; see 53). However, upon closer examination a substantial fraction of IMVs could be detected that resembled virions in various stages of apparent disassembly (Fig. 6, b–f). Profiles were seen of particles in which the membranes had become less rigid and detaching from the underlying core. Occasionally we also observed particles indistinguishable from viral cores (not shown). Compared with the control cells, IVs and intermediates (53) appeared totally normal. None of these aberrant particles with detaching membranes or naked cores could be detected in the untreated control cells, implying that they resulted from the prolonged DTT incubation.
Figure 6.

In the presence of DTT apparently normal looking IMVs are formed but in some of them the viral membranes detach from the underlying core. A shows Epon sections of control, untreated HeLa cells infected with vv for 8 h. IMVs and immature viruses (IVs) are evident. In the inset on the left the outer and inner membrane profiles of the IMV are indicated by arrows. The inset on the right shows an intermediate stage between the IV and IMV (arrowhead) next to a IV. B and C show vv-infected cells treated with 5 mM DTT from 4 h after infection onwards and fixed at 8 h after infection (see Results for details). In B, one sees both normal looking IMVs (star) as well as aberrant IMVs (arrowheads), where the viral core is seen as a distinct entity separated from the outer membranes. This is more evident at higher magnifications in C–F; in C a normal IV is apparent. The arrowheads indicate the viral core that in all these examples appears as a distinct entity. Bars, 100 nm.
These results argue that in the presence of DTT mostly normal looking IMVs are formed, but in some the membranes fail to assemble compactly around the core.
Cleavage of the Core Protein 4a Shows that in the Presence of DTT the IMV Formation Occurs with the Same Kinetics
In the final stage of IMV assembly, just before the IMV closure, at least three core proteins are proteolytically cleaved (30, 41). Estimation of the cleavage of the precursor of a protein such as the core protein 4a using pulse– chase experiments can thus be used as an indirect measure of the extent of IMV formation. To test the effect of DTT on IMV assembly using this assay, infected cells were left untreated or treated with DTT from 4 h after infection onwards. Subsequently, they were pulse labeled for 15 min at 6 h after infection, chased for 2 or 3 h and cell lysates were then processed for immunoprecipitation, using antibodies to 4a and its precursor. Again, during the course of this experiment the medium was replaced several times with medium containing freshly prepared DTT (see legend to Fig. 7). As shown in Fig. 7 both in the presence and absence of DTT the precursor p4a of 110 kD was efficiently cleaved into its 65-kD mature form (4a) during the chase. PhosphorImager analyses showed that, consistent with previously published results (41; Fig. 7), after 2 h of chase 35% of 4a was cleaved in the absence and 40% in the presence of DTT, while these percentages amounted to 48 and 57%, respectively, after 3 h of chase (not shown). These same analyses also showed that the overall synthesis of 4a was reduced by ∼40% in the presence of DTT, which is most likely the result of the long incubations with this drug. Importantly, however, 4a and p4a did not seem to undergo substantial degradation, suggesting that the newly formed cores were quite stable, both with and without DTT.
Figure 7.

In the presence of DTT p4a is cleaved to its mature (4a) form. Infected HeLa cells were treated (DTT +) or not treated (DTT +) from 4 h after infection onwards with 5 mM DTT. In each case, when medium with DTT was applied to the cells, freshly prepared DTT was added to the medium just before use. At 5 h after infection the medium was replaced with medium with or without DTT. At 5.5 h after infection the cells were starved in methionine-free DMEM with or without DTT. Pulse-labeling for 15 min (chase 0 h) was at 6 h after infection and consisted of replacing the methionine-free medium by the same medium containing 50 μCi of [35S]methionine with or without DTT. After the pulse, cells were chased for 2 and 3 h (chase 2 and 3 h) in medium with or without DTT that was replaced each hour. At the end of the pulse or chase cell lysates were prepared and 4a and its precursor form (p4a; both indicated) were immunoprecipitated from them. Immunoprecipitates were run on 10% SDS-PAGE and the proteins detected by autoradiography. M-14C-labeled marker protein of 93 kD.
Although this experiment suggests that in the presence of DTT less IMVs are formed, it also shows that the kinetics of IMV assembly is the same, if not slightly faster than in untreated control cells.
Thus, by two independent assays, 5 mM DTT does not appear to significantly affect the assembly of the IMV.
Addition of DTT to Infected Cells Prevents the Formation vv Disulfide Bonds
The above results implied that the vv-disulfide bonds may have no obvious role in IMV formation, although they might be required for the tight sealing of the vv membranes around the particle. A possibility that remained to be ruled out was that the disulfide bonds of vv proteins might resist the DTT treatment. We decided therefore to isolate the virions from infected cells and to analyze the disulfide bonding of their proteins. For this, infected cells were left untreated or were treated with DTT from 4 until 9 h after infection and IMVs were isolated after extensive washing of the cells with PBS containing NEM to quench the DTT. Again during the 5-h DTT treatment the medium with DTT was replaced every hour with medium containing fresh DTT. After virus isolation the disulfide bonding of selected membrane and core proteins was analyzed by Western blots. DTT indeed had its expected effects on the vv proteins; the core proteins 4a, p25 as well as the membrane proteins p21 and p16 migrated in their reduced form only in particles isolated from DTT-treated cells (Fig. 8). As expected, in the virions from control cells the same pattern of disulfide bonding was observed as described above (Fig. 8). Since in this particular experiment we used 10% instead of 15% SDS-PAGE, the dimer of 4a was much better separated from its monomer. Again, small amounts of the precursor of 4a were present in this virus preparation and it is more evident in this analysis that the dimer of 4a is substantially larger than the 110-kD precursor form.
When the particles isolated from DTT-treated cells were examined by negative staining EM, we observed that most of them were not intact (in contrast to untreated preparations where most of them were intact). By combining this approach with immunogold labeling using specific antibodies, we found that almost all of the isolated particles appeared to have lost or opened up their membranes. The brick-shaped particles from DTT-treated cells, labeled heavily for p39, an abundant antigen on the surface of the vv core (Fig. 9, a and d; 8, 50), while IMV particles isolated from nontreated cells (as expected; 8) could not be labeled with this antibody (Fig. 9 b). In the particles isolated from DTT-treated cells, the viral membranes often remained attached to the core and were heavily labeled with antibodies to the membrane protein p16. We have shown before that in intact IMVs this protein is located on the inner of the two cisternal membranes and hidden from antibody accessibility (Fig. 9 c; see 51).
Figure 9.

Negative staining EM of particles isolated from untreated (B) or DTT treated cells (A, C, and D). Particles from infected HeLa cells untreated or treated with DTT were isolated as described in Materials and Methods. The particles were absorbed to 300 mesh formvar and carbon-coated grids and immuno-labeled with antibodies to p39 (A, B, and D) and to p16 (C), followed by protein A–gold. Whereas virions isolated from untreated cells are not labeled with p39 (B), the particles from treated cells label heavily with the antibody (A and D). The anti-p16 antibody labels fragments that are still attached to the particles isolated from DTT-treated cells. These fragments often appear as tubular structures (C). Bars, 100 nm.
Thus, while the disulfide bonds may not be essential for the assembly process in infected cells, they appear to be necessary for maintaining the stability/integrity of the particles when released from cells.
Viruses of DTT-treated Cells Are Less Infectious
We next asked whether the particles isolated from DTT-treated cells were still infectious. To relate the infectivity to the number of viruses made, infected and DTT-treated as well as nontreated cells were metabolically labeled from 6 to 9 h after infection and the titers, measured by plaque assay, were expressed as PFU relative to trichloroacetic acid-precipitable, radioactive counts. In these experiments the cells were not quenched with NEM before virus isolation since treatment of the IMV with DTT in vitro, followed by NEM or iodoacetamide incubation, completely abolished infectivity (data not shown). The average of three independent experiments showed clearly that the IMV isolated from DTT-treated cells had a title that was 10-fold reduced relative to virus isolated from control cells (Table II). We have shown before in several studies that when purified IMV is treated for 30 min at 37°C with 20 mM DTT, the membranes separate in part from the core (see 8, 50, 54). Structurally, this in vitro DTT treatment is very similar to what we observed with the virions isolated from DTT-treated cells. We next asked therefore whether isolated IMV treated with DTT in vitro was still infectious. Remarkably, and in contrast to the virus from DTT-treated cells, IMV that had been treated in vitro with DTT had the same title (and sometimes even slightly higher; not shown) as the untreated virus (Table II).
Table II.
Infectivity of vv Isolated from DTT-treated or -untreated Cells or after DTT Treatment of Isolated IMV
Average titer over three independent experiments, determined by plaque assay on BSC40 cells.
Control is IMV isolated from untreated cells.
The infectivity of the IMV isolated from treated and nontreated infected cells was also assayed by Immunofluorescence microscopy. In vv life cycle DNA replication starts from 2 h after infection onwards, while late protein synthesis commences at 5–6 h of infection. The characteristic pattern of immunofluorescence at 6 h after infection is the complete overlap of the sites where viral DNA and late proteins accumulate. HeLa cells grown on coverslips were therefore infected with different multiplicity of infections (MOIs) ranging from 5 to 20 (see Materials and Methods) and fixed at 6 h after infection. The extent of infection was assayed by double indirect immunofluorescence using an antibody to vv cores (see Materials and Methods) that recognizes newly synthesized core proteins, as well as Hoechst dye to label the viral DNA. As expected, the cytoplasm of all cells were labeled for both DNA as well as the anti-core antibody when using virus from untreated cells, even at the lowest multiplicity of infection (Table III). Similarly, when this same virus preparation was treated with DTT in vitro all cells were infected at 6 h after infection (data not shown). However, when similar amounts of infectious units were used of virus from DTT-treated cells, even at the highest MOI the amount of cells showing DNA replication, as well as protein synthesis, was significantly reduced (Table III). More specifically, we found that, compared with control virus, at an MOI of 5 the percentage of DNA-positive cells was reduced by 73% and late protein-expressing cells by 98%. Apparently, the infection cycle of these viruses was severely delayed.
Table III.
Infectivity of vv Isolated from DTT-treated or Control Cells Determined by Indirect Immunofluorescence*
| MOI‡ | DNA positive§ | IF positive‖ | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 10 | 20 | 5 | 10 | 20 | |||||||
| Control ¶ | 98% | 100% | 100% | 90% | 100% | 100% | ||||||
| DTT** | 27% | 34% | 52% | 2% | 11% | 12% | ||||||
HeLa cells grown on coverslips were infected with different MOIs for 60 min at 37°C and fixed at 6 h after infection. The infectivity is expressed as the percentage of a total of 50 cells that labeled positive for viral DNA or viral late proteins.
To calculate the MOI, the virus-titer was calculated as PFU/ml.
Fixed cells were stained with Hoechst and scored positive if clear cytoplasmic DNA labeling typical of viral DNA was observed.
Fixed cells were labeled with a rabbit antibody to the viral core proteins and with donkey anti-rabbit coupled to FITC and scored positive if typical viral factories in the perinuclear region could be detected. IF, indirect immunofluorescence.
IMV isolated from untreated cells.
IMV isolated from DTT-treated cells.
During the Early Stages of IMV Infection the Core Proteins Become Reduced
Since reduction of the IMV in vitro, as well as in vivo led to a form of uncoating of the virus, we were curious to know whether reduction of vv proteins was also required for disassembly in vivo, that is during the infection process. Highly purified IMV stocks were therefore bound to cells on ice and subsequently shifted to 37°C for 1 h. Bound (but nonpenetrated) particles were removed by trypsin treatment on ice (see Materials and Methods). Subsequently, the infected cells were lysed and the vv proteins analyzed under reducing and nonreducing condition using Western blots. Under these conditions the two core proteins 4a and p25 migrated exclusively at the position of the reduced form (Fig. 10). To our surprise, the membrane proteins p16 and p21 did not appear to become reduced during early times of infection. Instead both proteins migrated with a slightly higher molecular mass than their respective dimers seen under nonreducing conditions (Fig. 10). When analyzed after reduction these forms of both p16 and p21 migrated slightly slower than their respective monomers (Fig. 10). Since by 2-D gel analysis, these newly generated forms of p21 and p16 appeared to have the same pI as the proteins in isolated virions (not shown), we presume that this slower migration is due to a (unconventional) conformational change and not the consequence of their binding to some other (unknown) protein.
Figure 10.

Upon viral entry the core proteins become reduced while the membrane proteins do not. HeLa cells were infected with purified virus preparations for 60 min at 37°C and cell lysates were prepared as described in Materials and Methods. The cell lysates (L) or purified virions (V) that were not exposed to cells, were run on 15% SDS-PAGE after boiling in LSB with (DTT +) or without (DTT −) 5% β-ME and 100 mM DTT. The membrane proteins p21 and p16 and the core proteins 4a and p25 were detected by Western blot.
It thus seems likely that either the membrane proteins of vv are inherently resistant to the reducing environment of the cytoplasm or they are never exposed to the cytoplasm during the entry stage.
Discussion
It is now a well established cell biological dogma that the redox state within the lumen of the endoplasmic reticulum facilitates the formation of disulfide bonds while the cytoplasm is generally considered to be too reducing for this process (17, 20, 24). Unexpectedly, we show here that vaccinia virus seems to differ from this generalization since a number of its proteins that do not reside in the lumen of the ER become disulfide bonded during the assembly process.These unusual disulfide bonds were detected not only on viral core proteins but also on the cytoplasmic surface of viral integral membrane proteins.
Three vv core proteins were engaged in (posttranslational) intramolecular (p39), intermolecular (4a) and complex type (p25) disulfide bonding. These disulfide bonds occur in the newly assembled (intracellular) IMV particle since they are not formed in the presence of rifampicin which arrests the assembly of the IMV. This is further supported by the fact that the mature form of the core protein 4a, that is made concomitant with the assembly of the IMV (30, 31, 41), forms dimers while its cytosolic precursor, p4a does not. The model that emerges from these results is that after the vaccinia virus has sealed, it is able to provide an environment within the particle that promotes the assembly of disulfide bonds of the core proteins, an environment that is inaccessible to GSH/GSSG, the cytoplasmic redox buffer. Indeed, treatment in vitro of purified IMV with concentrations up to 10 mM GSH failed to reduce the core proteins as analyzed by Western blots (data not shown).
In addition to the core proteins we also show that three of the four vv membrane proteins tested, p16, p21, and p32 become disulfide bonded. Our detailed topology studies of these membrane proteins strongly suggest that these disulfide bonds do not (exclusively) occur in the lumen of the endoplasmic reticulum. In agreement with the data of Niles and Seto (43) we show that the disulfide bonded dimer, as well as the monomer of p32 is exposed on the surface of the IMV. This protein adopts a similar topology in infected cells in which the entire NH2 terminus, including its single cysteine, of both the dimer and the monomer forms are located in the cytoplasm, since both were completely digested after protease treatment. These results indirectly but strongly argue that the disulfide bonding of this protein must occur on the cytosolic side of the membrane that will become the outer surface of the virus, where p32 is accessible to antibody labeling (54).
The available data on p16 strongly suggest that this protein acquires two topologies in infected cells and at least part of its molecules dimerizes by means of cytoplasmic disulfide bonds. As mentioned, the available data for p21 are less clear. If the topology we have previously proposed for this protein is correct, it would span the membrane four times and also be disulfide bonded in the cytoplasm (see Results). The disulfide bonding of these membrane proteins may be an inherent property of the proteins since it can occur in infected cells under conditions where assembly is blocked (rifampicin) and upon independent expression in HeLa cells (not shown).
A computer search analysis has revealed that the vv genome contains 19 proteins with a CXXC sequence, that, as outlined in the Introduction, makes up the active site sequence of proteins with a thioredoxin motif. Two of those proteins, the gene products of O2L and G4L, have been shown to share homology to human glutaredoxin (18, 46). Moreover, we have noticed that the gene product of G4L also shares ∼50% homology over a stretch of ∼50 amino acids (which includes the CXXC sequence) with yeast PDI. Preliminary data, however, have shown that neither of the two vv proteins is required for the disulfide bonding of vv proteins (data not shown).We also considered the possibility that the vv infection alters the cytoplasmic redox providing an environment that is more favorable for the assembly of cytoplasmic disulfide bonds. However, we were able to show that the ratio between reduced and oxidized glutathione is not altered in infected cells (see Materials and Methods).
Since the redox state of the cytoplasm of vv infected cells remains normal, it suggests that those membrane proteins that acquire cytoplasmic disulfide bonds possess an inherent property to acquire these bonds. In vitro studies have shown that the formation of intramolecular disulfide bonds depends on the conformation, as well as on the inherent disulfide oxidation-reduction potential of the protein (for example see 7). Intracellular disulfide bonding is influenced by the cellular redox potential, that is the consequence mainly of the ratio of reduced and oxidized glutathione. The redox state of the cytoplasm does not generally favor disulfide bond formation. However, based on the in vitro studies the putative disulfide bonding of proteins under cytosolic (reducing) redox conditions can be rationalized by assuming that the redox potential of the membrane proteins themselves must be higher than the cytosolic redox (for example see 6) and that, in addition, the conformation/organization of the proteins in the virally modified IC membranes favors the intermolecular disulfide bonding and folding. Consistent with the first idea could be the fact that, as analyzed by Western blots, the viral membrane proteins are highly resistant to reduction and that quantitative reduction requires the addition of 5% β-ME as well as high amounts of DTT (see also 49). This observation may also explain why cytosolic disulfide bonds of vv membrane proteins would naturally resist the (physiological) reducing environment of the cytosol.
Collectively, our data argue the vv disulfide bonds play a key role in the vaccinia viral life cycle. Apparently normal looking IMV particles that lack detectable disulfide bonds were seen in cells infected in the presence of DTT. These data imply that assembly per se is not dependent on disulfide bonding and must therefore rely on other types of protein interactions. However, the virions made in the presence of DTT were not completely assembled since they tended to loose their membranes in vivo and (more clearly) upon their isolation. Together with our recent data showing that treatment of purified virions with DTT opens up the viral membranes (which we argue are not fused into a continuous bilayer around the virus; see Fig. 1), these results clearly establish an important role of disulfide bonds in vv membrane proteins. Both the in vitro as well as the in vivo data show that in their absence the viral membranes are unable to remain tight around the particle and instead open up and detach from the underlying core. At present the reason why the three membrane proteins form homodimers as well as monomers is not clear. This phenomenon is obviously needed for the stability of the particle.
The role of the disulfide bonds of the vv core proteins is less clear at present. An obvious idea that comes to mind is that disulfide bonding during assembly will generate a stable core, while reduction during entry makes it unstable and primes it for disassembly. However, some results are difficult to reconcile with this idea. For instance, in DTT treated cells the cores are reduced but do not seem to fall apart. Also, cores resist prolonged treatment of IMVs with DTT in vitro (unpublished observations) and DTT-treated IMVs retain full infectivity. And finally, it is known that when uncoating of the cores after entry in prevented, the cores may persist for hours in the cytoplasm (for example see 27). When the IMV infects cells a viral core particle indistinguishable from those produced by treating IMVs with NP-40 and DTT (23) appears in the cytoplasm. Within minutes this core starts to make a defined set of so-called early mRNAs, a process that has been extensively studied in vitro using NP-40/DTT-treated virions. From such studies it is clear that, in vitro, this process of early transcription requires a reducing environment (42). Although it is very likely that the process of transcription itself requires reducing conditions, we would like to propose that the reduction of the core may play a role as well. Such a role can for example consist of a conformational change of the core structure that is required to activate the transcription machinery (that is contained in the core; 40). Alternatively, such a structural change could (also) be needed to allow the newly synthesized mRNA to exit the core, a process that has been shown to occur in vitro (29). Since the questions of changes that accompany transcription in vitro or the overall process in vivo have never been seriously addressed, a testing of our proposed scenario would require further advances in our understanding if these aspects of vaccinia virus life cycle.
In summary, disulfide bonds are an established mechanism for stabilizing proteins in extra-cytoplasmic locations. Our data show that vv has evolved the novel ability to use two different kinds of unusual disulfide bonds in its life cycle. The first are represented in a number of viral core proteins: these appear to be assembled only after the intracellular virions have sealed themselves from the cytoplasm, in effect forming an extra-cytoplasmic compartment that favors disulfide bonding. The second bonds are seen in three vv membrane proteins that seem to have evolved the capacity to fold their (cytoplasmic domains) into a conformation that allows intermolecular disulfide bonds to assemble that can resist the reducing environment of the cytoplasm.
Acknowledgments
We are grateful to Heinz Faulstich and Suze Zobeley for their help with the GSH determination for this study. Tom Creighton was a constant source of help and inspiration during the early parts of this work. Sibylle Schleich is acknowledged for her excellent help with the EM, Petra Riedinger for Fig. 1, and Reiner Saffrich for his tremendous patience in microinjecting vv-infected cells. We thank Hélène Defacque and Ari Helenius for their critical reading of the manuscript. The two reviewers, who, with their critiques substantially improved the quality of the manuscript, are also acknowledged.
Abbreviations used in this paper
- 2-D
two-dimensional
- EEV
extracellular enveloped virus
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- HA
hemagglutinin
- IC
intermediate compartment
- IMV
intracellular mature virus
- MOI
multiplicity of infection
- PDI
protein disulfide isomerase
- PFU
plaque-forming units
- PNS
post-nuclear supernatant
- vv
vaccinia virus
Footnotes
Address correspondence to Jacomine Krijnse Locker, EMBL, Cell Biology Programme, Meyerhofstrasse 1, 69117 Heidelberg, Germany. Tel.: 49 6221 387508. Fax: 49 6221 387306. E-mail: krijnse@embl-heidelberg.de
Part of this work was supported by a Human Frontier Science Program fellowship to J. Krijnse Locker.
References
- 2.Braakman I, Hoover-Litty H, Wagner KR, Helenius A. Folding of influenza hemagglutinin in the endoplasmic reticulum. J Cell Biol. 1991;114:401–411. doi: 10.1083/jcb.114.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braakman I, Helenius I, Helenius A. Manipulating disulfide bond formation and protein folding in the endoplasmic reticulum. EMBO (Eur Mol Biol Organ) J. 1992;11:1717–1722. doi: 10.1002/j.1460-2075.1992.tb05223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bulleid NJ, Freedman RB. Defective co-translational formation of disulfide bonds in protein disulphide-isomerase-deficient microsomes. Nature. 1988;335:649–651. doi: 10.1038/335649a0. [DOI] [PubMed] [Google Scholar]
- 5.Chertov OY, Telezhinskaya IN, Zaitseva EV, Golubeva TB, Zinov'ev VV, Ovechkina LG, Mazkova LB, Malygin EG. Amino acid sequence determination of vaccinia virus immunodominant protein p35 and identification of the gene. Biomed Sci. 1991;2:151–154. [PubMed] [Google Scholar]
- 6.Creighton TE, Hillson DA, Freedman RB. Catalysis by protein disulfide isomerase of the unfolding and refolding of proteins with disulphide bonds. J Mol Biol. 1980;142:43–62. doi: 10.1016/0022-2836(80)90205-3. [DOI] [PubMed] [Google Scholar]
- 7.Creighton, T.E. 1983. Pathways and energetics of protein disulfide formation. In Functions of Glutathione: Biochemical, Physiological, Toxilogical, and Clinical Aspects. A. Larsson et al., editors. Raven Press, New York. 205–213.
- 8.Creighton, T.E. 1995. Proteins, Structures and Molecular Properties. W.H. Freeman and Company, New York. 98–100.
- 9.Cudmore S, Blasco R, Vincentelli R, Esteban M, Sodeik B, Griffiths G, Krijnse J, Locker A vaccinia virus core protein, p39, is membrane associated. J Virol. 1996;70:6909–6921. doi: 10.1128/jvi.70.10.6909-6921.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Vries AAF, Raamsman MJB, van Dijk HA, Horzinek MC, Rottier PJM. The small envelope glycoprotein (Gs) of equine arteritis virus folds into three distinct monomers and a disulfide-linked dimer. J Virol. 1995;69:3441–3448. doi: 10.1128/jvi.69.6.3441-3448.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doms RW, Lamb RA, Rose JK, Helenius A. Folding and assembly of viral membrane proteins. Virology. 1993;193:545–562. doi: 10.1006/viro.1993.1164. [DOI] [PubMed] [Google Scholar]
- 12.Earl PL, Moss B. Preparation of cell cultures and vaccinia virus stocks. Curr Prot Mol Biol. 1991;16:1–7. doi: 10.1002/cpmb.33. [DOI] [PubMed] [Google Scholar]
- 13.Easterbrook KB. Controlled degradation of vaccinia virions in vitro: an electron microscope study. J Ultrastruct Res. 1966;14:484–496. doi: 10.1016/s0022-5320(66)80077-1. [DOI] [PubMed] [Google Scholar]
- 14.Ericsson M, Cudmore S, Shuman S, Condit RC, Griffiths G, Krijnse J, Locker Characterization of ts 16, a temperature sensitive mutant of vaccinia virus. J Virol. 1995;69:7072–7086. doi: 10.1128/jvi.69.11.7072-7086.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Essani K, Dales S. Biogenesis of vaccinia: evidence for more than 100 polypeptides in the virion. Virology. 1979;95:385–394. doi: 10.1016/0042-6822(79)90493-8. [DOI] [PubMed] [Google Scholar]
- 16.Freedman RB. The formation of protein disulfide bonds. Curr Opin Struct Biol. 1995;5:85–91. doi: 10.1016/0959-440x(95)80013-q. [DOI] [PubMed] [Google Scholar]
- 17.Freedman RB, Hirst TR, Tuite MF. Protein disulfide isomerase: building bridges in protein folding. Trends Biochem Sci. 1994;19:331–336. doi: 10.1016/0968-0004(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 18.Gaut JR, Hendershot LM. The modification and assembly of proteins in the endoplasmic reticulum. Curr Opin Cell Biol. 1993;5:589–595. doi: 10.1016/0955-0674(93)90127-c. [DOI] [PubMed] [Google Scholar]
- 19.Gvakharia BO, Koonin EK, Mathews CK. Vaccinia virus G4L gene encodes a second glutaredoxin. Virology. 1996;226:408–411. doi: 10.1006/viro.1996.0669. [DOI] [PubMed] [Google Scholar]
- 20.Hammond C, Helenius A. Quality control in the secretory pathway. Curr Opin Cell Biol. 1995;7:523–529. doi: 10.1016/0955-0674(95)80009-3. [DOI] [PubMed] [Google Scholar]
- 21.Helenius A, Marquardt T, Braakman I. The endoplasmic reticulum as a protein-folding compartment. Trends Cell Biol. 1992;2:227–231. doi: 10.1016/0962-8924(92)90309-b. [DOI] [PubMed] [Google Scholar]
- 22.Holmgren A. Thioredoxin and glutaredoxin systems. J Biol Chem. 1989;264:13963–13966. [PubMed] [Google Scholar]
- 23.Holowczak JA, Joklik WK. Studies of the structural proteins of virions and cores. Virology. 1967;33:717–725. doi: 10.1016/0042-6822(67)90072-4. [DOI] [PubMed] [Google Scholar]
- 24.Holowczak JA. Uncoating of poxviruses. I. Detection and characterization of subviral particles in the uncoating process. Virology. 1972;50:216–232. doi: 10.1016/0042-6822(72)90362-5. [DOI] [PubMed] [Google Scholar]
- 25.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 26.Ichihashi Y, Oie M, Tsuruhara T. Location of DNA-binding proteins and disulphide-linked proteins in vaccinia virus structural elements. J Virol. 1984;50:929–938. doi: 10.1128/jvi.50.3.929-938.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen ON, Houthaeve T, Shevchenko A, Cudmore S, Mann M, Griffiths G, Krijnse J, Locker Identification of the major membrane and core proteins of vaccinia virus by two-dimensional electrophoresis. J Virol. 1996;70:7485–7497. doi: 10.1128/jvi.70.11.7485-7497.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joklik WK. The intracellular uncoating of poxvirus DNA. II The molecular basis of the uncoating process. J Mol Biol. 1964;8:277–288. doi: 10.1016/s0022-2836(64)80137-6. [DOI] [PubMed] [Google Scholar]
- 29.Joklik WK. The purification of four strains of poxvirus. Virology. 1962;18:9–18. doi: 10.1016/0042-6822(62)90172-1. [DOI] [PubMed] [Google Scholar]
- 30.Kates J, Beeson J. Ribonucleic acid synthesis in vaccinia virus. I. The mechanism of synthesis and release of RNA in vaccinia cores. J Mol Biol. 1970;50:1–18. doi: 10.1016/0022-2836(70)90100-2. [DOI] [PubMed] [Google Scholar]
- 31.Katz E, Moss B. Formation of a vaccinia virus structural polypeptide from a high molecular weight precursor: inhibition of rifampicin. Proc Natl Acad Sci USA. 1970;66:677–684. doi: 10.1073/pnas.66.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katz E, Moss B. Vaccinia virus structural polypeptide derived from a high molecular weight precursor: formation and integration into virus particles. J Virol. 1970;6:717–726. doi: 10.1128/jvi.6.6.717-726.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krijnse Locker, J., J.K. Rose, M.C. Horzinek, and P.J.M. Rottier. Membrane assembly of the triple-spanning coronavirus M protein. Individual transmembrane domains show preferred orientation. J Biol Chem. 1992;267:21911–21918. doi: 10.1016/S0021-9258(19)36699-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krijnse Locker, J., G. Griffiths, M.C. Horzinek, and P.J.M. Rottier. O-glycosylation of the coronavirus M protein. Differential localization of sialyltransferases in N- and O-glycosylation. J Biol Chem. 1992;267:14094–14101. doi: 10.1016/S0021-9258(19)49683-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krijnse Locker, J., S. Schleich, D. Rodriguez, B. Goud, E.J. Snijder, and G. Griffiths. The role of a 21-kDa viral membrane protein in the assembly of vaccinia virus from the intermediate compartment. J Biol Chem. 1996;271:14950–14958. doi: 10.1074/jbc.271.25.14950. [DOI] [PubMed] [Google Scholar]
- 36.Kutay U, Hartmann E, Rapoport TA. A class of membrane proteins with a C-terminal anchor. Trends Biochem Sci. 1993;3:72–75. doi: 10.1016/0962-8924(93)90066-a. [DOI] [PubMed] [Google Scholar]
- 37.LaMantia ML, Lennarz WJ. The essential function of yeast protein disulfide isomerase does not reside in its isomerase activity. Cell. 1993;74:899–908. doi: 10.1016/0092-8674(93)90469-7. [DOI] [PubMed] [Google Scholar]
- 38.Lodish HF, Kong N. The secretory pathway is normal in dithiothreitol-treated cells, but disulfide-bonded proteins are reduced and reversibly retained in the endoplasmic reticulum. J Biol Chem. 1993;268:20598–20605. [PubMed] [Google Scholar]
- 39.Maa J-S, Rodriguez JF, Esteban M. Structural and functional characterization of a cell surface binding protein of vaccinia virus. J Biol Chem. 1990;265:1569–1577. [PubMed] [Google Scholar]
- 40.Maa J-S, Esteban M. Structural and functional studies of a 39,000-Mr immunodominant protein of vaccinia virus. J Virol. 1987;61:3910–3919. doi: 10.1128/jvi.61.12.3910-3919.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moss, B. 1990. Poxviridae and their replication. In Fields Virology. B.N. Fields, D.M. Knipe, R.M. Chanock, M.S. Hirsch, J.L. Melnick, T.P. Monath, and B. Roizman, editors. Raven Press, New York. 2079–2111.
- 42.Moss B, Rosenblum EN. Protein cleavage and poxvirus morphogenesis: tryptic peptide analysis of core precursors accumulated by blocking assembly with rifampicin. J Mol Biol. 1973;81:267–269. doi: 10.1016/0022-2836(73)90195-2. [DOI] [PubMed] [Google Scholar]
- 43.Munyon W, Paoletti E, Grace JT. RNA polymerase activity in purified infectious vaccinia virus. Proc Natl Acad Sci USA. 1967;58:2280–2287. doi: 10.1073/pnas.58.6.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niles EG, Seto J. Vaccinia virus gene D8 codes for a virion transmembrane protein. J Virol. 1988;62:3772–3778. doi: 10.1128/jvi.62.10.3772-3778.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oie M, Ichihashi Y. Characterization of vaccinia polypeptides. Virology. 1981;113:263–276. doi: 10.1016/0042-6822(81)90153-7. [DOI] [PubMed] [Google Scholar]
- 46.Opstelten D-J, de Groote P, Horzinek MC, Vennema H, Rottier PJM. Disulfide bonds in folding and transport of mouse hepatitis coronavirus glycoproteins. J Virol. 1993;67:7394–7401. doi: 10.1128/jvi.67.12.7394-7401.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajagopal I, Ahn B-Y, Moss B, Mathews CK. Roles of vaccinia virus ribonucleotide reductase and glutaredoxin in DNA precursor biosynthesis. J Biol Chem. 1995;270:27415–27418. doi: 10.1074/jbc.270.46.27415. [DOI] [PubMed] [Google Scholar]
- 48.Rodriguez D, Rodriguez JR, Esteban M. The vaccinia virus 14-kilodalton fusion protein forms a stable complex with the processed protein encoded by the vaccinia virus A17L. J Virol. 1993;67:3435–3440. doi: 10.1128/jvi.67.6.3435-3440.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rodriguez D, Esteban M, Rodriguez JR. Vaccinia virus A17L gene product is essential for an early step in virion morphogenesis. J Virol. 1995;69:4640–4648. doi: 10.1128/jvi.69.8.4640-4648.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodriguez JR, Risco C, Carrascosa JL, Esteban M, Rodriguez D. Characterization of early stages in vaccinia virus membrane biogenesis: implication of the 21kDa and a newly identified 15kDa envelope protein. J Virol. 1997;71:1821–1833. doi: 10.1128/jvi.71.3.1821-1833.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roos N, Cyrklaff M, Cudmore S, Blasco R, Krijnse J, Locker, Griffiths G. A novel immunogold cryoelectron microscopic approach to investigate the structure of the intracellular and extracellular forms of vaccinia virus. EMBO (Eur Mol Biol Organ) J. 1996;15:2343–2355. [PMC free article] [PubMed] [Google Scholar]
- 52.Salmons T, Kuhn A, Wylie F, Schleich S, Rodriguez JR, Rodriguez D, Esteban M, Griffiths G, Krijnse J, Locker Vaccinia virus membrane proteins p8 and p16 are co-translationally inserted into the rough ER and retained in the intermediate compartment. J Virol. 1997;71:7404–7420. doi: 10.1128/jvi.71.10.7404-7420.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarov I, Joklik WK. Studies on the nature and location of capsid polypeptides of vaccinia virions. Virology. 1972;50:579–592. doi: 10.1016/0042-6822(72)90409-6. [DOI] [PubMed] [Google Scholar]
- 54.Sodeik B, Doms RW, Ericsson M, Hiller G, Machamer CE, van't Hof W, van Meer G, Moss B, Griffiths G. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J Cell Biol. 1993;121:521–541. doi: 10.1083/jcb.121.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sodeik B, Cudmore S, Ericsson M, Esteban M, Niles EG, Griffiths G. Incorporation of p14 and p32 into the membrane of the intracellular mature virus. J Virol. 1995;69:3560–3574. doi: 10.1128/jvi.69.6.3560-3574.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takahashi T, Oie M, Ichihashi Y. N-terminal amino acid sequences of vaccinia virus structural proteins. Virology. 1994;202:844–852. doi: 10.1006/viro.1994.1406. [DOI] [PubMed] [Google Scholar]
- 57.Tatu U, Braakman I, Helenius A. Membrane glycoprotein folding, oligomerization and intracellular transport: effects of dithiothreitol in living cells. EMBO (Eur Mol Biol Organ) J. 1993;12:2151–2157. doi: 10.1002/j.1460-2075.1993.tb05863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van Slyke JK, Whitehead SS, Wilson EM, Hruby DE. The multistep proteolytic maturation pathway utilized by vaccinia virus P4a protein: a degenerate conserved cleavage motif within core proteins. Virology. 1991;183:467–478. doi: 10.1016/0042-6822(91)90976-i. [DOI] [PubMed] [Google Scholar]
- 59.Van Slyke JK, Franke CA, Hruby DE. Proteolytic maturation of vaccinia virus core proteins: identification of a consensus motif at the N termini of the 4b and 25K virion proteins. J Gen Virol. 1991;72:411–416. doi: 10.1099/0022-1317-72-2-411. [DOI] [PubMed] [Google Scholar]
- 60.Wittek R, Haenggi M, Hiller G. Mapping of a gene coding for a major late structural polypeptide on the vaccinia virus genome. J Virol. 1984;49:371–378. doi: 10.1128/jvi.49.2.371-378.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wollfe EJ, Vijaya S, Moss B. A myristoylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology. 1995;211:53–63. doi: 10.1006/viro.1995.1378. [DOI] [PubMed] [Google Scholar]
- 62.Yang WP, Kao SY, Bauer WR. Biosynthesis and post-translational cleavage of vaccinia virus structural protein VP8. Virology. 1988;167:585–590. [PubMed] [Google Scholar]