

Abstract
To study the effect of continued telomere shortening on chromosome stability, we have analyzed the telomere length of two individual chromosomes (chromosomes 2 and 11) in fibroblasts derived from wild-type mice and from mice lacking the mouse telomerase RNA (mTER) gene using quantitative fluorescence in situ hybridization. Telomere length at both chromosomes decreased with increasing generations of mTER−/− mice. At the 6th mouse generation, this telomere shortening resulted in significantly shorter chromosome 2 telomeres than the average telomere length of all chromosomes. Interestingly, the most frequent fusions found in mTER−/− cells were homologous fusions involving chromosome 2. Immortal cultures derived from the primary mTER−/− cells showed a dramatic accumulation of fusions and translocations, revealing that continued growth in the absence of telomerase is a potent inducer of chromosomal instability. Chromosomes 2 and 11 were frequently involved in these abnormalities suggesting that, in the absence of telomerase, chromosomal instability is determined in part by chromosome-specific telomere length. At various points during the growth of the immortal mTER−/− cells, telomere length was stabilized in a chromosome-specific man-ner. This telomere-maintenance in the absence of telomerase could provide the basis for the ability of mTER−/− cells to grow indefinitely and form tumors.
Keywords: telomerase-deficient mice, telomeres, chromosome fusions, Q-FISH, cancer
Eukaryotic linear chromosomes are capped by a special structure known as the telomere. In vertebrates, telomeric DNA consists of tandem repeats of the sequence TTAGGG (reviewed in Blackburn, 1991). These specialized structures constitute the final 10 kb of all human chromosomes, and the final 12–80 kb of all mouse chromosomes (Lansdorp et al., 1996; Zijlmans et al., 1997). More than 50 years ago, Barbara McClintock observed that chromosome ends lacking telomeres have a tendency to fuse (McClintock, 1941). Recent studies in yeast and mice have proven that telomeres are essential to maintain chromosomal stability (Sandell and Zakian, 1993; Blasco et al., 1997; Lee et al., 1998; Naito et al., 1998; Nakamura et al., 1998).
In most human cells, telomeres shorten with each cell division due to the incomplete replication of linear DNA molecules and the absence of telomere-elongating mechanisms (reviewed in Greider, 1996). Indeed, telomere shortening to a critical length has been proposed to limit the life span of somatic cells in humans (Harley et al., 1990; Counter et al., 1992). Cell types that proliferate indefinitely, such as unicellular eukaryotes, germline cells, and immortal cells, maintain their telomeres at a constant length. In such cells the enzyme telomerase seems to be the main mechanism for the maintenance of telomere length (Greider and Blackburn, 1985; Morin, 1989; Yu et al., 1990; Singer and Gottschling, 1994; McEachern and Blackburn, 1996; Nakamura et al., 1997).
Telomerase is a reverse transcriptase that elongates telomeres by synthesizing TTAGGG repeats onto the 3′ ends of chromosomes. Thus, telomerase can compensate for the incomplete replication of telomeres (reviewed in Greider, 1996). Telomerase is formed by a protein catalytic subunit with similarity to other reverse transcriptases (Lingner et al., 1997; Harrington et al., 1997a; Kilian et al., 1997; Meyerson et al., 1997; Nakamura et al., 1997; Martín-Rivera et al., 1998), an RNA molecule that contains the template for the synthesis of telomeric repeats (Greider and Blackburn, 1989; Singer and Gottschling, 1994; Blasco et al., 1995; Feng et al., 1995; McEachern and Blackburn, 1995), and other associated proteins (Collins et al., 1995; Harrington et al., 1997b; Nakayama et al., 1997; Gandhi and Collins, 1998).
It has been recently shown that the introduction of the telomerase catalytic subunit in primary human cells is sufficient to restore enzymatic activity, to elongate and maintain telomeres, and, in some cell types, to sustain immortal growth (Bodnar et al., 1998; Wang et al., 1998; Kiyono et al., 1998). Yeast and mouse strains that lack any of the essential telomerase components do not have detectable telomerase activity and undergo telomere shortening and loss of viability after a variable number of generations, indicating that active telomerase is essential to maintain telomere length in vivo (Singer and Gottschling, 1994; McEachern and Blackburn, 1996; Blasco et al., 1997; Nakamura et al., 1997, 1998). Interestingly, it is possible to isolate survivor yeast strains that are able to stabilize their chromosomes by mechanisms that involve telomere elongation by recombination or chromosome circularization (Lundblad and Blackburn, 1993; McEachern and Blackburn, 1996; Nakamura et al., 1997, 1998; Naito et al., 1998). Telomerase-independent telomere elongation has been also described in some immortal human cell lines that do not have detectable telomerase activity (Bryan et al., 1995, 1997).
The analysis of telomere length in primary cells from mouse telomerase RNA (mTER)1 knock out mice that lack telomerase activity has revealed that telomeres shorten at a rate of 4.8 ± 2.4 kb per generation, and that this is accompanied by an increase in chromosomal instability (Blasco et al., 1997). The loss of telomere repeats was not obvious from conventional telomere length measurements by Southern analysis but was readily apparent using quantitative fluorescence in situ hybridization (Q-FISH; Lansdorp et al., 1996; Zijlmans et al., 1997; Martens et al., 1998). With Q-FISH, the fluorescence intensity at individual telomeres is calculated from digital images using image analysis techniques after quantitative hybridization of denatured telomere target sequences with directly labeled and highly efficient (CCCTAA)3 peptide nucleic acid probes. Q-FISH has become the method of choice for the analysis of telomere length in the mouse (Lansdorp, 1997) and for further details the reader is referred to the references above.
Despite lacking telomerase activity, mTER−/− cells have divided >500 times in culture (this paper), suggesting that telomerase activity per se is not essential for immortalization and/or that there might be telomerase-independent mechanisms maintaining telomeres in these telomerase-deficient cells. Here, we have analyzed telomere length dynamics at the level of individual chromosomes in primary and immortal cells that lack the mTER gene. Furthermore, we have characterized the type and frequency of fusions and translocations that accumulate in these cells as a consequence of proliferation in the absence of telomerase activity. The results of this analysis underscore the importance of telomeres in the maintenance of genomic stability.
Materials and Methods
Cell Culture
Mouse embryonic fibroblasts (MEFs), were prepared from day 13.5 embryos derived from wild-type (wt) and mTER−/− mice from different generations. The details are explained elsewhere (Blasco et al., 1997). The first culture directly obtained from embryos was considered as population doubling 2 (PD 2). Serial cultures were done according to the 3T3 protocol (Todaro and Green, 1963) in dishes of 10 cm diam, seeding 106 cells every 3 d. The doubling number of each passage was calculated using the formula PD = log (nf/n0)/log2; where n0 is the initial number of cells (106) and nf is the final number of cells. The details on the establishment of the immortal cell lines are explained elsewhere (Blasco et al., 1997).
Fluorescence In Situ Hybridization with PNA-telomere Probe
Plates containing primary MEFs or established cell lines were treated with colcemid (0.1 μg/ml) for 4–5 h and subsequently trypsinized and spun for 8 min at 120 g. After hypotonic swelling in sodium citrate (0.03 M) for 25 min at 37°C, cells were fixed in methanol/acetic acid (3:1). After 2–3 additional changes of fixative, cell suspensions were dropped on wet, clean slides and dried overnight. FISH with Cy-3 labeled (CCCTAA)3 peptide-nucleic acid, and subsequent quantitative analysis of digital images were performed as described (Zijlmans et al., 1997). The slide coordinates of the metaphase images captured were noted to relocate the same metaphase after FISH with minor satellite DNA (see below) and mouse chromosome probes (Oncor).
Quantitative Image Analysis
Digital images were recorded with a MicroImager MI1400-12 camera (Xillix) on an Axioplan fluorescence microscope (Zeiss). Microscope control and image acquisition was performed with a dedicated software (SSM; Xillix). Separate DAPI and Cy-3 images were subjected to telomere fluorescence analysis by using ad dedicated computer program, TFLTELO (Martens et al., 1998). Chromosomes and telomeres were identified through segmentation of the DAPI image and the Cy-3 image, respectively. Both images were combined and corrected for pixel shifts. The integrated fluorescence intensity for each telomere was calculated after correction for image acquisition exposure time. Finally, the integrated fluorescence intensity of individual telomeres is expressed in a table for each chromosome, which can be subjected to editing. Each metaphase of 40 chromosomes (in the mouse) yields 160 telomere spots and a typical analysis of 15–20 metaphases produces several thousand telomere fluorescence values. Because of the large number of data points in Q-FISH analysis, the standard error of mean telomere fluorescence estimates is typically small (for example, less than a few percent of the average) despite considerable variation in individual telomere fluorescence values.
Details of the calibration procedures used to reliably measure telomere fluorescence intensity are explained elsewhere (Martens et al., 1998). In brief, two levels of calibration values were used. First, to correct for daily variations in lamp intensity and alignment, images of fluorescent beads (orange beads, size 0.2 μm; Molecular Probes) were acquired and similarly analyzed with the analysis software. Second, relative telomere fluorescence units (TFU) were extrapolated from the plasmid calibration. For this, we hybridized and analyzed plasmids with a defined (TTAGGG)n length of 0.15, 0.4, 0.8, and 1.6 kb. There was a linear correlation (r2 = 0.99) for plasmid fluorescence intensity and (TTAGGG)n length with a slope of 48.7 (Martens et al., 1998). Therefore, the calibration corrected telomere fluorescence intensity (ccTFI) of each telomere was calculated according to the formula: ccTFI = (Bea1/Bea2) × (TFI/48.7), where Bea1 equals the fluorescence intensity of beads when plasmids were analyzed, Bea2 equals the fluorescence intensity of beads when sample x was analyzed, and TFI equals unmodified fluorescence intensity of a telomere in sample x.
A restriction of this calibration method is that the actual telomeres are outside the range of (TTAGGG)n length of the plasmids. The assumption is made that the linear correlation obtained between fluorescence intensity and telomere insert size in plasmid is maintained in the higher range in chromosomal DNA.
FISH with Minor Satellite DNA
The murine minor satellite DNA probe was a gift from Drs. J.B. Rattner and Shu-Lin Liu (Calgary University, Calgary, Canada). The minor satellite probe labels all centromeres near the kinetochore except mouse chromosome Y (Wong and Rattner, 1988). The probe was labeled with Spectrum-green dUTP by nick translation and then precipitated with ethanol, using salmon sperm DNA as carrier. The probe was dissolved in a hybridization buffer containing 30% deionized formamide, 2× SSC, 10% dextran sulphate and 50 mM phosphate buffer (pH 7.0) to a concentration of 20 ng/μl. The slides used for telomere FISH were washed and then hybridized with the minor satellite DNA probe. The hybridization was performed as described (Hande et al., 1996). Slides were incubated with pepsin (0.005%) in 10 mM HCl for 10 min at 37°C, washed with PBS containing 50 mM MgCl2, and then treated with 1% formaldehyde in PBS/MgCl2 for 10 min at room temperature. After one more wash in PBS, slides were dehydrated in a 70, 90, and 100% ethanol series. The labeled probe was diluted with hybridization buffer to a final concentration of 4 ng/μl, and 20 μl were added on each slide. The probe and the target DNA were denatured simultaneously at 80°C, for 3.5–4.0 min. Hybridization was carried out overnight at 37°C in a moist chamber. After hybridization, the slides were washed three times with 30% formamide, 2× SSC buffer (pH 7.0) for 5 min at 37°C, followed by two washes in 2× SSC at room temperature. The slides were dehydrated and embedded with Vectashield mounting medium (Vector Labs) containing 1 μg/ml propidium iodide. About 15–25 metaphases per group that had already been used for telomere measurements were relocated and observed under the microscope for characterizing the end-to-end fusions.
FISH with Mouse Chromosome Paint Probes
Chromosome painting probes for chromosomes 2 and 11 were used to follow telomere length in these chromosomes. The same slides that were analyzed after FISH with telomere probe and minor satellite DNA were processed for the third FISH with chromosome painting probes. The chromosome painting probes were purchased from Oncor. FISH was performed according to the manufacturer's instructions. About 15–25 metaphases were relocated using the known coordinates in the microscope and analyzed.
Results
Telomere Dynamics of Individual Chromosomes in mTER− /− Telomerase-deficient Embryos from Different Generations
Primary cells (passage 1 mouse embryonic fibroblasts, MEFs) derived from wt embryos and from mTER−/− embryos from the 1st (G1) to the 6th (G6) generation were obtained following the scheme previously described (Blasco et al., 1997). The telomere length of individual chromosomes (chromosomes 2 and 11) was measured using Q-FISH and chromosome painting. Chromosome 2 was chosen because it consistently has relatively short telomeres in several mouse strains (Hande, M.P., and P. Lansdorp, unpublished results; this paper). Chromosome 11 is the mouse homologue of human chromosome 17, which was found to have relatively short telomeres in all individuals analyzed to date (Martens et al., 1998).
Fig. 1 shows the mean and standard error of telomere fluorescence intensity of all telomeres together (average of q- and p-arms), and also of q- and p-arms separately from primary MEFs of both wt and mTER−/− embryos of the 2nd (KO2-G2), 4th (KO7-G4), and 6th generation (littermate embryos KO9-G6 and KO11-G6; littermate embryos KO1-G6 to KO4-G6 and embryo KO5-G6). Despite considerable variation between individual telomere fluorescence values (see for example Fig. 2 D), the large number of data points (>1,000) resulted in insignificant standard error values in the telomere values of all chromosomes. The standard error was also small for individual telomeres on chromosomes 2 and 11, with smaller number of data points (50–100; see Figs. 1 A and 2, B and C). The standard error rather than the standard deviation is shown for clarity and presentation purposes only. The average telomere fluorescence of all chromosomes decreased linearly during successive generations of mTER−/− mice. The average telomere shortening was 3.9 kb per generation (calculated as described in Fig. 1 B). This shortening affected both q-telomeres (telomeres of the q-arms) that showed a shortening of 4.17 kb per generation, and p-telomeres (telomeres of the p-arms) that showed a shortening of 3.7 kb per generation. As a result, the difference in telomere length between p- and q-arm telomeres was maintained throughout the six mouse generations (Fig. 1 B). The loss of telomere repeats in mTER−/− mice resulted in an average length of 14.5 and 22.4 kb for p- and q-telomeres, respectively, in cells derived from the 6th generation. When we measured the mean telomere fluorescence of chromosome 2, the estimated rate of telomere shortening per generation was 3.4 kb for both 2q- and 2p-telomeres (Fig. 1, A and B). This telomere shortening resulted in 6th generation 2p- and 2q-telomeres of an average length of 7.6 kb and 16.2 kb, respectively (embryo KO9-G6 had an estimated 2p-telomere length of only 0.15 ± 0.1 kb), shorter than the average of all chromosomes. In the case of chromosome 11, the average telomere fluorescence of 11q and 11p-telomeres decreased at an average rate of 5.2 and 5.6 kb per generation, respectively, up to the 4th generation (Fig. 1, A and B). Interestingly, from the 4th (embryo KO-G4) to the 6th generation (the average of seven different embryos) we did not detect the expected telomere shortening in any of chromosome 11 telomeres (Fig. 1 B). In contrast, there was a 6-kb increase in the telomere length at the 6th generation (Fig. 1, A and B). Altogether, these results suggest that in the absence of telomerase activity, telomere shortening occurs at a similar rate in all chromosome ends. However, it appears that mechanisms that prevent telomere shortening in the absence of telomerase act differentially on different telomeres. In our study, chromosome 11 telomeres did not show the predicted shortening with increasing generations in seven different embryos, while chromosome 2 telomeres continued to shorten throughout the six generations of mTER−/− mice (see Discussion). In this study, we cannot rule out that telomerase independent mechanisms of telomere maintenance are also operating in early generation mTER−/− or wt mice.
Figure 1.

Telomere dynamics in wild type and mTER−/− primary cells from different mouse generations. (A) Telomere fluorescence of all chromosomes (top), chromosome 2 (center), and chromosome 11 (bottom) from primary MEFs derived from embryos of the indicated genotype and generation. Fluorescence is expressed in TFU, where 1 TFU corresponds to 1 kb of TTAGGG repeats in plasmid DNA (Martens et al., 1998). Each value represents the mean of 15 or more metaphases. Primary cells from wt and mTER−/− embryos from 2nd (KO2-G2), 4th (KO7-G4), and 6th (KO9-G6, KO11-G6, KO1-G6, KO2-G6, KO3-G6, KO4-G6, KO5-G6) generation were used in the study. Black squares, average of q- and p-telomeres; white diamonds, q-telomeres; and open circles, p-telomeres. all chr., All chromosomes; chr.2, chromosome 2; chr.11, chromosome 11. The standard error is indicated with a bar. Despite the wide heterogeneity in individual telomere fluorescence intensity values (see for example Fig. 2 D), the standard errors of the mean were usually very small due to the large number of data points (See Materials and Methods for details). As a result, the error bars are not always visible in the graphs (i.e., A). (B) The average telomere length of q- and p-telomeres together, and of q-telomeres and p-telomeres, separately, in the different embryos studied from each generation was plotted and the data was analysed by least square methods to calculate the average telomere shortening per generation. In the case of chromosome 11 telomeres, we obtained a low r2 value (0.67) when we included all the data up to the G6 generation. The low r2 value indicates that it is not correct to include G6 chromosome 11 telomere values to calculation of rate of shortening, further suggesting that chromosome 11 did not suffer the predicted telomere shortening from G4 to G6. To calculate the telomere shortening per generation in chromosome 11, we excluded G6 telomeres (B) obtaining a linear equation with r2 close to 1. Primary cells from wt and mTER−/− embryos from 2nd (G2), 4th (G4), and 6th (G6) generation were used in the study. Black squares, average of q- and p-telomeres; white diamonds, q-telomeres; open circles, p-telomeres.
Figure 2.

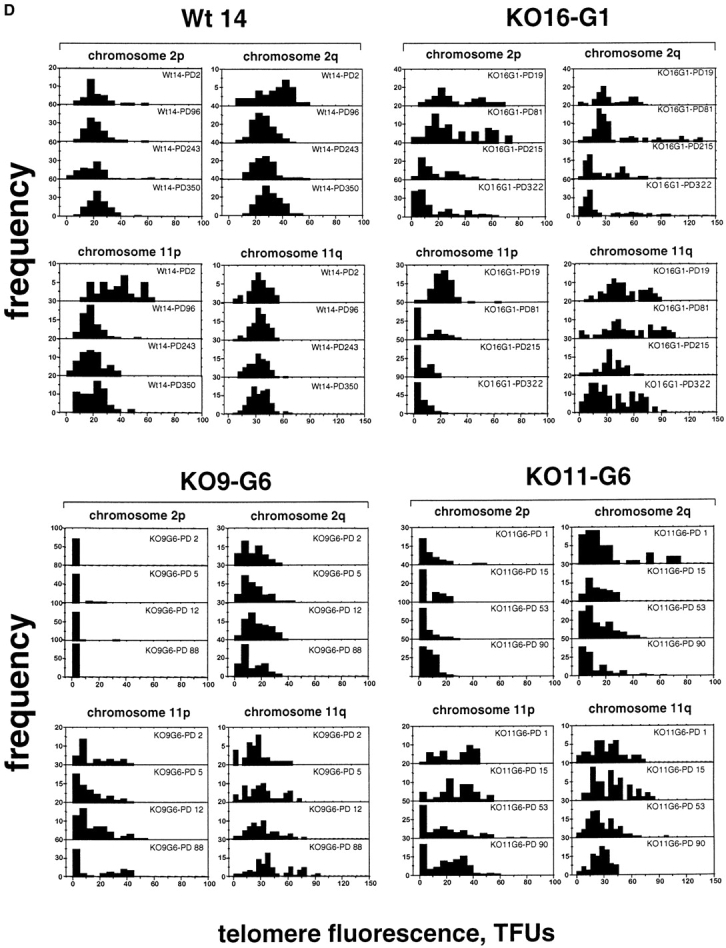
Telomere dynamics in wt and mTER−/− cell lines. (A–C) The telomere fluorescence, measured as TFU, of all telomeres (A), chromosome 2 telomeres (B), and chromosome 11 telomeres (C) in wt (Wt14) and mTER−/− cell lines at different PDs is shown. Black bars, fluorescence of q-telomeres; gray bars, fluorescence of p-telomeres. Despite the wide heterogeneity in individual telomere fluorescence intensity values, and due to the large number of data points used in the analysis, the error bars are not always visible in the graphs (i.e., A). (D) The distribution of telomere fluorescence intensity values of 2p-, 2q-, 11p-, and 11q-telomeres (black bars) in wt cell line (Wt14) and the indicated mTER−/− cell lines at increasing PDs.
Interestingly, in the different mTER−/− cells derived from 6th generation embryos, we observed a marked heterogeneity in the mean telomere fluorescence. This heterogeneity affected both chromosome 2 and 11 telomeres and was also observed in cells derived from littermate embryos. This variation in telomere length could be the basis for the variable penetrance of the phenotypes described in 6th generation mTER−/− mice (Lee et al., 1998; Herrera et al., 1999).
Telomere Dynamics of Individual Chromosomes in Spontaneously Immortalized mTER− /− Cell Lines
Serial passage of mouse embryonic fibroblasts allows the selection of oligoclonal populations with the capacity to stably proliferate in culture. We had previously described that serial passage of mTER−/− MEFs according to a 3T3 protocol resulted in the selection of immortal cell lines in a manner similar to that of mTER+/+ MEFs, indicating that telomerase activity is not essential for the immortalization of mouse cells (Blasco et al., 1997). Although some differences were observed in the growth rate between wt and mTER−/− cell lines, all cultures have shown a continued growth that exceeded 500 PDs for G1 MEFs and 250 PDs for G6 MEFs (not shown). To understand the basis for the continuous growth of these telomerase negative cells we have analyzed their telomere dynamics. Fig. 2 A shows the mean telomere fluorescence of q- (black bars) and p-telomeres (gray bars), separately, during increasing PDs of wt and mTER−/− cells. We calculated that telomeres of wt cells, Wt14, underwent a modest shortening at an estimated rate of 24.8 bp per PD (Fig. 2 A). The occurrence of telomere shortening in wt MEFs could indicate that the level of telomerase activity present in these cells is not sufficient to prevent telomere erosion as it has been proposed previously for other cell types (Counter et al., 1994; Chiu et al., 1996) or, alternatively, that telomere length in these cultured cells is not tightly regulated around a fixed length.
In contrast to wt cells, mTER−/− cell lines derived from 1st (KO16-G1 and KO19-G1), 2nd (KO2-G2), and 4th generation (KO7-G4) embryos, showed a marked decrease in the telomere fluorescence of both p- and q-telomeres (Fig. 2 A). The estimated average telomere loss in the different cell lines ranged between 65 and 108 bp per PD, similar to the shortening rate described for human cells that do not express telomerase. This indicates that in these mTER−/− cells that had escaped senescence and are immortal, the mean telomere length continues to shorten with increasing passage number. Interestingly, the rate of telomere shortening at both p- and q-telomeres decreased at later passages (PDs 215 and 322) of the KO16-G1 cell line, suggesting the activation of telomere maintenance mechanisms when telomeres shorten to a critical length (Fig. 2 A). In the case of two different mTER−/− cell lines, KO9-G6 and KO11-G6, derived from 6th generation embryos, the telomere fluorescence at both p- and q-telomeres was maintained or increased during the different PDs analyzed (Fig. 2 A). In KO9-G6 cells, p- and q-telomeres were maintained at an average length of 10.8 and 24.8 kb, respectively, and in KO11-G6 cells at an average length of 16.3 and 25.7 kb. These observations point to telomerase-independent mechanisms for the maintenance of telomeres in the immortal cells derived from the 6th generation mTER−/− MEFs.
Representative FISH images of metaphase spreads from wt and mTER−/− cell lines at early and late passages are shown in Fig. 3. As previously described for immortal MEF cultures (Zindy et al., 1997), most of the cell lines studied here were aneuploid at late passages (Fig. 3, A, C, and D). Fig. 3 A shows two metaphases of Wt14 cells before, PD 2, and after immortalization, PD 243. At PD 243, all chromosome ends had TTAGGG repeats and the cells did not show an increase of end-to-end fusions except for a very long chromosome that was clonal (indicated by an arrowhead in Fig. 3 A). Metaphases of KO16-G1 and KO7-G4 mTER−/− cell lines (Fig. 3, B and C, respectively) show a decrease in telomere fluorescence when early and late PDs are compared. In contrast, KO9-G6 cells showed a similar telomere fluorescence signal at both early, PD 2, and late, PD 88, PDs, in agreement with the observation that the mean telomere length is maintained in these cells (Fig. 3 D, see above). Finally, all mTER−/− cell lines contain many chromosomes lacking detectable telomere signal at late PDs, as well as a significant increase of end-to-end fusions (Fig. 3 arrows; see below).
Figure 3.


Metaphase spreads from wt and mTER−/− cell lines at selected PDs. (A) Representative metaphase spreads from wt cells (Wt14) at the indicated PD. The arrowhead points to a long chromosome present in all the metaphases analyzed at PD 243. (B) Metaphase spreads from 1st generation mTER−/− cells, KO16-G1, at the indicated PDs. Note the weaker telomere fluorescence at PD 215 compared with PD 19. A chromosome with intrachromosomal TTAGGG signal is indicated with a white arrowhead. (C) Metaphase spreads from 4th generation mTER−/− cell line, KO7-G4, at the indicated PDs. Note that telomere fluorescence decreased at PD 159 compared with PD 2. (D) Metaphase spreads from 6th generation mTER−/− cell line, KO9-G6. Note the strong heterogeneity in telomere fluorescence. Red arrows, chromosomal ends lacking detectable telomere fluorescence. White arrows, end-to-end fusions. These are representative images from individual metaphase spreads after FISH showing fluorescent spots on telomeres for illustration purpose only.
We have also studied the telomere fluorescence of chromosomes 2 and 11 as a function of the accumulated number of cell doublings (Fig. 2, B and C). When telomere fluorescence of both chromosomes 2 and 11 was measured in the wt cell line Wt14, we observed a slight decrease with increasing PDs, (Fig. 2, B and C). The calculated average rate of telomere shortening for chromosomes 2 and 11 in the Wt14 cells was 10 and 11 bp per PD, respectively. Interestingly, when we studied telomere dynamics of chromosome 2 and 11 in the different generation of mTER−/− cell lines, we could not detect the predicted pattern of telomere shortening with increasing PDs (Fig. 2, B and C). The length of p- and q-telomeres at chromosomes 2 and 11 with increasing PDs suggests the activation of telomere maintenance mechanisms at different points during the growth of the cell lines. In this regard, it is interesting to note that in KO16-G1 cells, 11q-telomeres did not shorten from PD 19 to PD 81 or from PD 215 to PD 322. However, 11p-telomeres continued to shorten to an average length of only 5.6 kb at PD 215 and then were stabilized. The involvement of 11p telomere in the chromosomal instability of this cell line will be discussed later in this paper. The maintenance of telomere length was particularly clear in cumulative PDs of the two cell lines derived from the 6th generation mTER−/− embryos (Fig. 2, B and C). Interestingly, in these cells telomeres from different chromosomes were maintained at different length and, in general, chromosome 2 telomeres were stabilized at shorter lengths than chromosome 11 telomeres (Fig. 2, B and C).
Fig. 2 D shows the distribution of fluorescence intensity values for 2q, 2p, 11q, and 11p telomeres with increasing PDs in wt (Wt14) and in mTER−/− cell lines from the first (KO16-G1) and from the 6th (KO9-G6 and KO11-G6) generation. The telomere fluorescence in wt cells at 2p, 2q, 11p, and 11q telomeres with increasing PDs remained similar, in agreement with the fact that, overall, telomeres were maintained in this cell line. In contrast, the number of telomeres with low fluorescence values (0–10 TFU) increased with passage number in the mTER−/− cell lines. Interestingly, in the mTER−/− cell lines the heterogeneity in fluorescence intensity values increased with increasing PDs for some telomeres (i.e., 11q telomeres in KO9-G6 cell line), again suggesting the existence of alternative telomere maintenance mechanisms in these cells.
Analysis of End-to-End Fusions
To analyze the nature of the chromosomal fusions promoted by the absence of telomerase, we performed FISH on wt and mTER−/− metaphases using telomeric and centromeric probes, as well as chromosomes 2 and 11 painting probes (Materials and Methods). Fig. 4 shows the diagrams of the different fusions characterized in this study together with representative images. End-to-end fusions were classified into different types according to their structure as shown in Fig. 4. Types I, II, and III involve p-to-p arms fusions. Type I fusions contain telomeric repeats at the fusion point (Fig. 4 a) and two copies of minor satellite centromere repeat sequences (b). Type II fusions do not contain detectable telomeric sequences at the fusion point (Fig. 4 a) and yield two centromere signals (b). Type III fusions lack telomeric signals at the fusion point (Fig. 4 a) and only have one centromere signal (b). Type IV and V fusions involve q-to-q arm fusion, and have or lack detectable telomeric signals at the fusion point, respectively. Finally, type VI involves p-to-q arm fusion. In some cases, we performed chromosome painting to determine whether the fusions were homologous (for example, chromosome 2-to-chromosome 2 in panel c of type II fusions) or nonhomologous (for example, chromosome 11 to an undetermined chromosome in panel c of type VI fusions).
Figure 4.

Examples of the different end-to-end fusions detected in mTER−/− cells. The chromosomal arms involved in the fusion are inferred by the morphology of the fused chromosome. Chromosomes probed with telomeric PNA to characterize the fusion according to the presence or absence of detectable telomeric sequences at the fusion point are depicted in panels a. Chromosomes probed with minor satellite DNA to characterize the fusion according to the number and location of centromeres are depicted in panels b (in p-arm and p-arm/q-arm fusions). Panels b (in q-arm examples) depict DAPI staining of the fusion. Panels c depict chromosome painting with whole- chromosome DNA from chromosome 2 (example of type II fusion) and chromosome 11 (examples of type III fusion and p-arm/q-arm fusion). Chromosomes are counterstained with DAPI for telomere probe and propidium iodide for minor satellite and chromosome probes.
Chromosomal Instability in Primary mTER− /− Cells
No fusions were detected in metaphases analyzed from early passage primary wt cells (Table I). In the case of mTER−/− primary cells, the frequency of fusions increased significantly from 0.07 fusions per metaphase in mTER−/− cells from the 1st generation (KO19-G1) to an average of 1.04 fusions (range from 0.5 to 1.72) per metaphase in seven independently derived mTER−/− cells from 6th generation embryos (KO9-G6, KO11-G6, and KO1-G6 to KO5-G6; Table I). Interestingly, cytogenetic analysis of the cells derived from seven independent 6th generation embryos revealed that an average of 41% of the fusions was type II fusions. Chromosome painting showed that 70% of these type II fusions were homologous fusions involving 2p (2p-to-2p fusions; see Fig. 4 for example) and only 2% involved chromosome 11. The dramatic increase in chromosome 2 but not chromosome 11p-arm fusions in 6th generation mTER−/− cells, is probably the consequence of 2p-telomeres shortening from 26.0 ± 2.8 kb in the wt cells, to an average of 7 kb, shorter than the average of all telomeres in cells from the 6th generation. In this regard, in 6th generation cells derived from embryo KO9-G6, 2p-telomeres were only an estimated 0.15 kb long and 100% of type II fusions were 2p-to-2p fusions. The homologous nature of these fusions indicates that they are likely to be the result of a failure to separate sister chromatids during mitosis (see model in Fig. 5). Interestingly, fusions involving chromosome 2 were stably maintained in two different G6 cell lines studied, KO9-G6 and KO11-G6, at least for >80 PDs (Table II, see Discussion). Other fusions found in primary KO9-G6 cells included type I fusions (35%), and less frequently, type III (8%) and type V (18.4%) fusions (see Fig. 4 for examples and Table I for data). Taken together, chromosome 2 seems to be more frequently involved in chromosome fusions than other chromosomes in the mTER−/− MEFs, although we can not rule out that other chromosomes might occasionally be involved in fusions in mTER−/− cells (Lee et al., 1998).
Table I.
Different Types of End-to-End Fusions Detected in mTER−/− Primary MEFs
| Metaphases analyzed | Type of fusion | Fusions per metaphase | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p-arm | q-arm | |||||||||||||
| Generation | Type I | Type II | Type III | Type IV | Type V | |||||||||
| wt | 25 | 0 | 0 | 0 | 0 | 0 | <0.04 | |||||||
| KO19-G1 | 15 | 0 | 0 | 1 | 0 | 0 | 0.07 | |||||||
| KO2-G2 | 31 | 4 | 0 | 0 | 1 | 4 | 0.29 | |||||||
| KO-G4 | 25 | 7 | 0 | 0 | 0 | 7 | 0.56 | |||||||
| KO9-G6 | 25 | 17 | 24 (24) | 1 | 0 | 1 | 1.72 | |||||||
| KO11-G6 | 11 | 4 | 2 (2) | 0 | 1 | 2 | 0.82 | |||||||
| KO1-G6 | 15 | 0 | 7 (1) | 1 | 1 | 6 | 1.0 | |||||||
| KO2-G6 | 13 | 1 | 2 (1) | 0 | 0 | 3 | 0.5 | |||||||
| KO3-G6 | 13 | 4 | 4 (2) | 1 | 0 | 3 | 0.9 | |||||||
| KO4-G6 | 13 | 2 | 5 (2) | 4 | 0 | 3 | 1.1 | |||||||
| KO5-G6 | 12 | 7 | 3 (1) | 2 | 0 | 3 | 1.25 | |||||||
Figure 5.

Models for the generation of chromosomal fusions by telomere loss. (Top) Replication and segregation of a mouse chromatid with normal telomeres. (Middle and bottom) Shortening of p- or q-arm telomeres to a critical length leads to fusion of sister chromatids after replication. The subsequent failure in the separation of these fusions might result in a daughter cell (1) harboring a Robertsonian-like configuration (middle) or a dicentric chromosome (bottom) and a second daughter cell (2) that has lost a chromosome. Fused chromosomes may undergo successive cycles of breakage-fusion-bridge and are inherently unstable (reviewed in de Lange, 1995).
Table II.
Chromosome End-to-End Fusions in Wild-type and mTER−/− Cell Lines as a Function of the Population Doubling
| Cell line and population doubling | Metaphases analyzed | Type of fusion | Fusions per metaphase | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p-arm | q-arm | |||||||||||||
| Type I | Type II | Type III | Type IV | Type V | ||||||||||
| Wt14 | ||||||||||||||
| PD 20 | 25 | 0 | 0 | 0 | 0 | 0 | 0 | |||||||
| PD 96 | 25 | 4 | 0 | 0 | 0 | 0.16* | ||||||||
| PD 243 | 26 | 5 | 0 | 0 | 0 | 0 | 0.19 | |||||||
| PD 350 | 25 | 4 | 0 | 1 | 0 | 0 | 0.2 | |||||||
| KO16-G1 | ||||||||||||||
| PD 19 | 27 | 0 | 0 | 0 | 0 | 0 | 0 | |||||||
| PD 81 | 26 | 5 | 0 | 33 (0;25) | 1 | 1 | 1.54 | |||||||
| PD 215 | 30 | 6 | 0 | 105 (0;46) | 0 | 1 | 3.73 | |||||||
| PD 322 | 20 | 5 | 18 | 121 (0;38) | 7 | 23 | 8.70 | |||||||
| KO19-G1 | ||||||||||||||
| PD 2 | 15 | 0 | 0 | 1 | 0 | 0 | 0.82 | |||||||
| PD 280 | 15 | 4 | 13 | 56 (2;5) | 2 | 16 | 6.07 | |||||||
| KO2-G2 | ||||||||||||||
| PD 2 | 31 | 4 | 0 | 0 | 1 | 4 | 0.29 | |||||||
| PD 221 | 25 | 4 | 17 (4;1) | 95 (3;2) | 0 | 27 | 5.72 | |||||||
| KO7-G4 | ||||||||||||||
| PD 2 | 25 | 7 | 0 | 0 | 0 | 7 | 0.56 | |||||||
| PD 159 | 20 | 6 | 13 (3;1) | 105 (4;7) | 3 | 8 | 6.75 | |||||||
| KO9-G6 | ||||||||||||||
| PD 2 | 25 | 17 | 24 (24;0) | 1 | 0 | 1 | 1.72 | |||||||
| PD 5 | 31 | 4 | 21 (21;0) | 5 (0;1) | 1 | 0 | 1.00 | |||||||
| PD 12 | 29 | 3 | 27 (27;0) | 19 (0;9) | 0 | 0 | 1.69 | |||||||
| PD 88 | 20 | 0 | 24 (20;0) | 34 (0;3) | 0 | 5 | 3.15 | |||||||
| KO11-G6 | ||||||||||||||
| PD 1 | 11 | 4 | 2 (2;0) | 0 | 1 | 2 | 0.82 | |||||||
| PD 15 | 13 | 4 | 5 (5;0) | 4 | 1 | 3 | 1.31 | |||||||
| PD 53 | 14 | 5 | 9 (9;0) | 70 (10;6) | 4 | 8 | 6.86 | |||||||
| PD 90 | 20 | 2 | 10 (7;1) | 104 (11;10) | 3 | 7 | 6.7 | |||||||
One terminal translocation detected after PD 96 and found to be clonal.
Chromosomal Instability in mTER− /− Cell Lines
To study the consequences of continuous proliferation in the absence of telomerase activity on chromosomal stability, we also analyzed chromosomal aberrations in wt and mTER−/− cell lines as a function of the accumulated number of PDs (Table II). The wt cell line, Wt14, did not show any end-to-end chromosome fusions in the first 20 PDs, except for a very long chromosome that was clonal. We determined that this long chromosome was the result of a terminal translocation between chromosome 11 and another chromosome (see arrowhead in Fig. 3 A), and was stably transmitted throughout all the PDs analyzed. In later passages of the Wt14 cell line (PD 350), a low percentage of p-arm fusions, 0.2 per metaphase, was also detected. These fusions could be a consequence of the moderate telomere shortening detected in these cells (see above).
A dramatic increase in end-to-end fusions was observed with increasing PDs in all the mTER−/− cell lines studied (see also Fig. 3, B–D for examples of metaphases). This chromosomal instability furthermore increased with the generation number (Table II). The high frequency of p-arm fusions (89%) versus q-arm fusions (11%) in the cell lines could be due to the fact that mouse p-telomeres are shorter than q-telomeres. Whereas type I and type II fusions were the most common fusions in primary cells (Table I), type III fusions (with only one pair of centromere signals) were the most abundant in the cell lines (65%; see also Table II). Interestingly, 80% of the type II fusions detected in the two different G6 cell lines, KO9-G6 and KO11-G6 involved chromosome 2, in agreement with the observation that chromosome 2 telomeres were shorter than the average of all telomeres in the 6th generation MEFs and/or that there is an increased stability of fusions involving chromosome 2 relative to fusions involving other chromosomes. By chromosome painting, we determined that type III fusions present in mTER−/− cell lines, usually involve nonhomologous chromosomes. Interestingly, 20% of these fusions involved chromosome 11p fused to other chromosomes. In the KO16-G1 PD 81 cell line, >75% of all type III fusions involved chromosome 11, in agreement with the fact that 11p-telomeres were specially short in this particular cell line (for example, Figs. 2 A and 4). Type VI fusions, visualized as chromosome rings, were also present in mTER−/− cell lines (for examples see Fig. 4, B1 and B2). Other chromosomal rearrangements appear at late passage in mTER−/− cells. For example, in the case of KO16-G1 (PD 215) cells, these rearrangements included reciprocal and terminal translocations involving chromosomes 2 or 11. The frequency of such chromosome exchanges was 0.06 and 0.1 per metaphase, respectively (not shown).
Fusion between nonhomologous chromosomes could originate by the simultaneous existence of two different chromosomes with critically short telomeres. To estimate the minimal telomere length that triggers chromosome fusions, we have calculated the mean telomere length of intrachromosomal telomere repeats in all fusions involving the p-arm of one chromosome and q-arm of a different chromosome (Type VI fusions; see Fig. 4) and in terminal translocations detected at PD 215 and at PD 322 of the KO16-G1 cell line (Type I fusions were excluded from the analysis). The average length of intrachromosomal telomere repeats (not considering the type I fusions) was 2.3 kb (ranging between 0.1 and 5.7 kb), indicating that this length is not sufficient to prevent chromosome fusions in mouse cells. Altogether, these results suggest that end-to-end fusions and other chromosomal aberrations detected in the mTER−/− cell lines are the result of telomere shortening to a critical length, and that chromosomes 2 and 11 are commonly involved in these fusions.
Discussion
To study the role of telomeres and telomerase, we have analyzed telomere length and chromosomal stability in mouse cells deficient for telomerase activity. Previously, we have shown that the average telomere length of mice genetically deficient in the RNA component of telomerase (mTER) decreases with each generation number (Blasco et al., 1997). However, somatic cells derived from these mice have been able to grow in culture beyond 500 PDs and were shown to be transformed and form tumors in nude mice (Blasco et al., 1997; this paper). These findings have raised the question of how telomeres are maintained in late generation mTER−/− cells in the absence of telomerase. Indeed, deletion of the mTER gene in embryonic stem (ES) cells was recently reported to result in a severe growth defect after >300 divisions, suggesting that the growth of ES cells is telomerase dependent (Niida et al., 1998). In our study we found no growth inhibition in primary somatic cells from mTER−/− mice, suggesting that the telomerase requirement for continued growth may vary between cell types. To address questions about the mechanism of telomere maintenance in primary somatic cells from mTER−/− mice, the length of individual telomeres was measured using quantitative FISH and chromosome painting. Chromosome painting with whole chromosome probes unequivocally identifies mouse chromosomes and has allowed us to follow telomere length of particular chromosomes and their involvement in chromosome fusions.
Telomere Dynamics in Primary Cells
We concentrated our attention on chromosomes 2 and 11. Initial banding analysis of mTER−/− 6th generation MEFs used in an earlier study (Blasco et al., 1997) suggested that chromosome 2 was frequently involved in fusions (unpublished results). Therefore, we sought to analyze the telomere length dynamics of this chromosome in mTER−/− cells. Chromosome 11 is the mouse homologue of human chromosome 17. Recently, it was found that chromosome 17p has relatively short telomeres in normal human cells (Martens et al., 1998). The average telomere length of all chromosomes decreased uniformly at a rate of 3.9 kb per mouse generation in a number of independent groups of telomeres such as p-telomeres and q-telomeres, as well as individual 2p- and 2q-telomeres. This uniform and constant rate of shortening indicates that probably all telomeres are subjected to the same erosive processes in vivo. In the case of chromosome 2p, telomeres shortened to an average length of 7 kb in the 6th generation; however, some 6th generation cells (KO9-G6) had an average 2p telomere length of only 150 bp. Heterogeneity in telomere length was also observed for 2q, 11p, and 11q telomeres in different KO-G6 mice. Such variation between littermates could be the basis for the fact that only a percentage of KO-G6 mice show defects associated with telomere loss (Lee et al., 1998; Herrera el al., 1999). Interestingly, the average telomere length of chromosome 11 increased ∼6 kb from the 4th to the 6th generation. This telomere lengthening in the absence of telomerase activity is suggestive of alternative telomere maintenance mechanisms. Such mechanisms may act in a telomere chromosome-specific manner because telomere 11p and 11q but not chromosome 2 telomeres showed this type of elongation upon analysis of the average telomere length in seven different 6th generation mTER−/− embryos.
Telomere Dynamics in Immortal Cells
Additional evidence for the operation of telomerase-independent telomere maintenance mechanisms was obtained from the study of serially passaged cells. Serial passage of mouse embryonic fibroblasts allows the selection of oligoclonal populations with the capacity to stably proliferate in culture. This experimental system allows the analysis of telomere dynamics in a context less restrictive than the entire organism. The telomere length in wt embryonic fibroblasts was stabilized after 100 PDs after an initial modest decline. In contrast, the average telomere length of mTER−/− cultures derived from embryos of the 1st or 4th generation continued to decrease. As the cells spontaneously escaped senescence (around PD 10), this shortening occurred at a similar rate to the one estimated in primary cells during successive generations of mTER−/− mice (Blasco et al., 1997). When individual telomeres were followed during successive passages, a variable telomere shortening was observed, and not all telomeres were affected to the same extent. These observations support the existence of telomerase-independent and chromosome-specific telomere maintenance mechanisms in these cells.
Previous studies in yeasts have suggested the existence of telomerase-independent mechanisms that are sufficient to ensure yeast viability (McEachern and Blackburn, 1996; Nakamura et al., 1998). Moreover, there are a variety of human established cell lines that do not show detectable telomerase activity (Bryan et al., 1995, 1997). Telomere maintenance in these cells has been proposed to occur through alternative mechanisms (ALT; Bryan et al., 1995, 1997). The fact that human ALT cells were not engineered to be genetically deficient for telomerase genes left open the possibility that discrete bursts of telomerase activity could be compensating for telomere shortening in these cells. However, our studies in mTER−/− mouse cells unequivocally prove the existence of mechanisms in mammalian cells that are capable of maintaining telomeres in the absence of telomerase. The genes responsible for telomerase-independent telomere maintenance are currently not known. Several genes involved in DNA recombination, i.e., Rad 52 (McEachern and Blackburn, 1996) or in DNA repair, such as DNA-PK, ATM, Rad 3, tel1+, and TEL 1, have been proposed as candidates because their mutation results in accelerated loss of telomeres (Greenwell et al., 1995; Boulton and Jackson, 1996; Metcalfe et al., 1996; Nugent et al., 1998; Naito et al., 1998). Telomerase-deficient mTER−/− cells are a good system to identify genes involved in telomere maintenance that could be essential to sustain tumor growth in the absence of telomerase. The targeting of both telomerase and gene products involved in alternative telomere-maintenance mechanisms could be used to prevent cell proliferation beyond a threshold telomere length. Such telomere-directed strategies may offer a promising approach to cancer therapy.
Telomere Shortening and Chromosomal Instability
In 1941, Barbara McClintock made the observation that chromosomes that lack telomeres have a tendency to fuse and concluded that one of the functions of the telomeric complex is to prevent end-to-end fusions of chromosomes. It has been shown recently that telomere binding proteins are of crucial importance to prevent end-to-end fusions in human chromosomes (Van Steensel et al., 1998). In addition, the study of Tetrahymena mutants with an altered telomerase RNA template suggests an important role of telomeres in chromatid separation during anaphase (Kirk et al., 1997). Similarly, other studies have suggested a role of telomeres and telomere binding proteins in chromosome segregation and meiosis (Bass et al., 1997; Chikashige et al., 1997; Chua et al., 1997; Conrad et al., 1997, Cooper et al., 1998; Naito et al., 1998).
We have performed a detailed cytogenetic analysis to examine the relationship between chromosomal fusions and telomere shortening in mammalian cells. We could not observe chromosomal fusions in primary cells derived from wt embryos, thus estimating that the frequency of fusions is lower than 0.04 per metaphase. In contrast, primary cells derived from mTER−/− embryos showed a dramatic increase in chromosomal fusions. The frequency of chromosomal fusions increased with the generation number from <0.04 fusions per metaphase in wt mice to 1.72 fusion per metaphase in some mTER−/− cells from the 6th generation (∼40 times the estimated maximum frequency in wt mice). We performed a similar study in serially cultured cells. The loss of control of the chromosomal number or aneuploidy is an alteration commonly observed in cultured cells (Zindy et al., 1997). Indeed, most of the established cell populations that we have generated in this study were aneuploid, independently of the presence or absence of telomerase activity. Despite being aneuploid, established wt cells presented a very low level of chromosomal fusions (0.20 fusions per metaphase in cultures that have undergone 350 PDs). In comparison, mTER−/− cells showed a very high proportion of chromosomal fusions (for example, 8–9 fusions per metaphase in mTER−/− cultures of the 1st generation that have undergone 325 PDs), >40-fold the number of fusions found in wt cell lines. These observations indicate that telomere shortening promotes the accumulation of chromosomal fusions both in vivo and in vitro. Recently, it was shown that the junctions of human dicentric chromosomes are typically characterized by the absence or a low number of telomere repeats (Wan et al., 1999), providing further support for a critical role of telomere shortening in chromosomal instability.
To investigate the mechanisms by which telomeres prevent end-to-end fusions, we performed a detailed FISH analysis of the chromosomal fusions observed in mTER−/− cells using whole chromosome and centromeric minor satellite probes. The results obtained from this study are different from the initial characterization of end-to-end fusions carried out in mTER−/− mice (Lee et al., 1998). In these studies, chromosome 3 was found to be frequently involved in fusions of chromosomes derived from splenocytes of one mTER−/− G6 mouse. Possible explanations for this discrepancy range from cell type specificity to technical artefacts. In our study with primary mouse embryonic fibroblasts, most of the end-to-end chromosome fusions detected in mTER−/− primary cells from the 6th generation, i.e., KO9-G6, unequivocally involved 2p-telomeres as determined by chromosome painting and in agreement with the fact that 2p-telomeres were shorter than average in these cells. Moreover, the emergence of 2p-to-2p fusions with increasing passage number in the KO11-G6 cell line also supports the involvement of chromosome 2 in the initial chromosomal instability detected in mTER−/− cells. It is tempting to speculate that the frequent occurrence of this particular abnormality is related to the relatively short length of telomeres on this particular chromosome arm in several mouse strains (Hande, M.P., M.A. Blasco, and P. Lansdorp, unpublished observations) as it seems unlikely that fusions involving 2p may have occurred as isolated clonal events in seven independently derived embryos. While the majority of the primary fusions seem to involve chromosome 2, we cannot rule out the involvement of other chromosomes, such as chromosome 3 or 14 (Lee et al., 1998).
As illustrated in the model shown in Fig. 5, telomere loss or telomere shortening to a critical length could trigger either end-to-end fusions between sister chromatids or a failure to separate them during mitosis. As a consequence of either situation, the sister chromatids could be drawn to the same spindle pole at anaphase resulting in chromosome nondisjunction or missegregation (see Fig. 5 for model). Alternatively, the fusion product could break at fragile sites and trigger breakage-fusion-bridge cycles. In the first case, one of the daughter cells (daughter cell 1 in the model) will end up with a Robertsonian fusion-like chromosome (fusion types I or II) or a dicentric chromosome (fusion types IV or V), and the other cell (daughter cell 2) will lose the chromosome. Further rounds of cell division might result in the maintenance of the fusion provided that the sister chromatids segregate normally with two centromeres, or that one of the centromeres is inactivated (type III fusions). In this regard, in 6th generation mTER−/− cells 16% of the p-fusions are type II fusions and were stably transmitted for >88 PDs. Interestingly, q-arm fusions were very infrequent in mouse cells. Although the fact that q-arm telomeres are generally longer than p-arm telomeres may in part responsible for this observation, q-arm fusions may also be more likely to form an anaphase bridge and may thus be less stable than p-arm fusion products. Altogether, these results indicate that telomeres protect chromosomes from end-to-end fusion events in vivo by maintaining telomere length above a critical threshold. This observation supports the important role of telomeres in chromosome segregation as previously proposed (Kirk et al., 1997; Bass et al., 1997; Chikashige et al., 1997; Chua et al., 1997; Conrad et al., 1997).
When chromosomal instability was studied in the spontaneously immortalized mTER−/− cell lines, we found a dramatic increase in nonhomologous type III end-to-end fusions, in p-to-q arms fusions and in various types of translocations. Again, 2p and 11p telomeres were involved in many of these fusions in the different cell lines studied (derived from different embryos), suggesting that the involvement of these chromosomes in fusions is a general phenomena for mouse fibroblasts. The existence of these chromosomal abnormalities in mTER−/− cell lines can be explained by breakage-fusion-bridge cycles triggered by different chromosomes with critically short telomeres (reviewed in de Lange, 1995).
In general, the types of chromosomal aberrations detected in mTER−/− cell lines are similar to the ones described in tumor cells, supporting the notion that telomere loss is one of the main inducers of chromosomal instability in tumors (reviewed in de Lange, 1995). In agreement with this, tumors generally have short telomeres (reviewed in de Lange, 1995) probably because telomere loss is not compensated at the earlier steps of tumorigenesis. Telomere shortening would lead to chromosomal instability associated with end-to-end fusions and translocations of the types described in this work, favoring the allelic loss of tumor suppressor genes, such as p53 in the case of mouse chromosome 11 or human chromosome 17 (Martens et al., 1998). Our data agrees with the idea that when telomeres are critically short and there is a high degree of chromosomal instability telomere-maintenance mechanisms are activated and selected (see Fig. 6 for model). These mechanisms, which can be either telomerase dependent (reviewed in Shay and Bacchetti, 1997) or independent (this paper and Bryan et al., 1995, 1997), can prevent telomeres from further shortening, allowing the immortal growth of cells that have critically short telomeres and a high degree of genetic instability. In this regard, mTER−/− mice from late generations show an increased cancer incidence than the wt counterparts (Rudolph et al., 1999).
Figure 6.

Model of telomere dynamics and chromosomal instability during tumor progression. Telomere shortening could occur during the initial stages of tumor progression if cells divide in the absence of compensating telomere lengthening mechanisms. Telomere shortening to a critical length eventually triggers chromosomal instability as described in this paper. At this point, telomere maintenance mechanisms can be activated and selected to allow immortal growth. The preferred mechanism to maintain telomeres in tumor cells is the activation of the enzyme telomerase (reviewed in Shay and Bacchetti, 1996). However, the results shown in this paper indicate that in the absence of telomerase activity alternative telomere-maintaining mechanisms are activated as a consequence telomere shortening.
Acknowledgments
We are indebted to Juan Martín-Caballero for deriving the different mTER−/− mouse generations and to Jessica Freire for the genotyping work. We specially thank Manuel Serrano, Fermín Goytisolo, Carlos Martinez, and Carol Greider for their support.
We are grateful to Dr. Carol Greider in whose laboratory M.A. Blasco derived the mTER−/− cell lines with the support of National Institutes of Health (NIH) grant POI-CA 13106 to Carol Greider. E. Samper is recipient of a predoctoral fellowship from The Regional Government of Madrid. Research in the laboratory of P.M. Lansdorp is supported by NIH grants ROIAI29524 and GM56162, and by a grant from the National Cancer Institute of Canada with funds from The Terry Fox Run. Research at the laboratory of M.A. Blasco is funded by grants PM95-0014 and PM97-0133 from the Ministry of Education and Culture, Spain, by grant 08.1/0030/98 from The Regional Government of Madrid and by the Department of Immunology and Oncology. The Department of Immunology and Oncology was founded and is supported by the Spanish Research Council (CSIC) and by Pharmacia and Upjohn.
Abbreviations used in this paper
- MEF
mouse embryonic fibroblast
- mTER
mouse telomerase RNA gene
- PD
population doubling
- Q-FISH
quantitative fluorescence in situ hybridization
- TFU
telomere fluorescence units
- wt
wild-type
Footnotes
Address correspondence to María A. Blasco, Department of Immunology and Oncology, Centro Nacional de Biotecnología/CSIC, Campus Cantoblanco, Madrid E-28049, Spain. Tel.: 34 91 585 4846. Fax: 34 91 372 0493. E-mail: mblasco@cnb.uam.es
References
- Bass HW, Marshall WF, Sedat JW, Agard DA, Cande WZ. Telomeres cluster de novo before the initiation of synapsis: a three-dimensional spatial analysis of telomere positions before and during meiotic prophase. J Cell Biol. 1997;137:5–18. doi: 10.1083/jcb.137.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH. Structure and function of telomeres. Nature. 1991;350:569–573. doi: 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- Blasco MA, Funk WD, Villeponteau B, Greider CW. Functional characterization and developmental regulation of mouse telomerase RNA. Science. 1995;269:1267–1270. doi: 10.1126/science.7544492. [DOI] [PubMed] [Google Scholar]
- Blasco MA, Lee H-W, Hande P, Samper E, Lansdorp P, DePinho R, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu C-P, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Boulton SJ, Jackson SP. Identification of a Saccharomyces cerevisiaeKu80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res. 1996;24:4639–4648. doi: 10.1093/nar/24.23.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO (Eur Mol Biol Organ) J. 1995;14:4240–4248. doi: 10.1002/j.1460-2075.1995.tb00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan TM, Marusic L, Bacchetti S, Namba M, Reddel RR. The telomere lengthening mechanism in telomerase-negative immortal human cells does not involve the telomerase RNA subunit. Hum Mol Genet. 1997;6:921–926. doi: 10.1093/hmg/6.6.921. [DOI] [PubMed] [Google Scholar]
- Chikashige Y, Ding DQ, Imai Y, Yamamoto M, Haraguchi T, Hiraoka Y. Meiotic nuclear reorganization: switching the position of centromeres and telomeres in the fission yeast Schizosaccharomyces pombe. . EMBO (Eur Mol Biol Organ) J. 1997;16:193–202. doi: 10.1093/emboj/16.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C-P, Dragowska W, Kim NW, Vaziri H, Yui J, Thomas TE, Harley CB, Lansdorp PM. Differential expression of telomerase activity in hematopoietic progenitors from adult human bone marrow. Stem Cells. 1996;14:239–248. doi: 10.1002/stem.140239. [DOI] [PubMed] [Google Scholar]
- Chua PR, Roeder GS. Tam1, a telomere-associated meiotic protein, functions in chromosome synapsis and crossover interference. Genes Dev. 1997;11:1786–1800. doi: 10.1101/gad.11.14.1786. [DOI] [PubMed] [Google Scholar]
- Collins K, Kobayashi R, Greider CW. Purification of Tetrahymenatelomerase and cloning of genes encoding the two protein components of the enzyme. Cell. 1995;81:677–686. doi: 10.1016/0092-8674(95)90529-4. [DOI] [PubMed] [Google Scholar]
- Conrad MN, Dominguez AM, Dresser MW. Ndj1p, a meiotic telomere protein required for normal chromosome synapsis and segregation in yeast. Science. 1997;276:1252–1255. doi: 10.1126/science.276.5316.1252. [DOI] [PubMed] [Google Scholar]
- Cooper JP, Watanabe Y, Nurse P. Fission yeast taz1 protein is required for meiotic telomere clustering and recombination. Nature. 1998;392:828–831. doi: 10.1038/33947. [DOI] [PubMed] [Google Scholar]
- Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO (Eur Mol Biol Organ) J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counter CM, Botelho FM, Wang P, Harley CB, Bacchetti S. Stabilization of short telomeres and telomerase activity accompany immortalization of Epstein-Barr virus-transformed human B lymphocytes. J Virol. 1994;68:3410–3414. doi: 10.1128/jvi.68.5.3410-3414.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange, T. 1995. Telomere dynamics and genome instability in human cancer. In Telomeres. Cold Spring Harbor Press, Cold Spring Harbor, NY. 265–293.
- Feng J, Funk WD, Wang S, Weinrich SL, Avilion AA, Chiu CP, Adams RR, Chang E, Allsopp RC, Yu J, Le S, West MD, Harley CB, Andrews WH, Greider CW, Villeponteau B. The RNA component of human telomerase. Science. 1995;269:1236–1241. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- Gandhi L, Collins K. Interaction of recombinant Tetrahymenatelomerase proteins p80 and p95 with telomerase RNA and telomeric DNA substrates. Genes Dev. 1998;12:721–733. doi: 10.1101/gad.12.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD. TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human Ataxia Telangiectasia gene. Cell. 1995;82:823–829. doi: 10.1016/0092-8674(95)90479-4. [DOI] [PubMed] [Google Scholar]
- Greider CW. Telomere length regulation. Annu Rev Biochem. 1996;65:337–365. doi: 10.1146/annurev.bi.65.070196.002005. [DOI] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989;337:331–337. doi: 10.1038/337331a0. [DOI] [PubMed] [Google Scholar]
- Hande MP, Boei JJWA, Natarajan AT. Induction and persistence of cytogenetic damage in mouse splenocytes following whole-body X-irradiation analysed by fluorescence in situ hybridisation. Int J Radiat Biol. 1996;69:437–446. doi: 10.1080/095530096145733. [DOI] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during aging of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Harrington L, Zhou W, McPhail T, Oulton R, Yeung D, Mar V, Bass MB, Robinson MO. Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes Dev. 1997a;11:3109–3115. doi: 10.1101/gad.11.23.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington L, McPhail T, Mar V, Zhou W, Oulton R, Bass MB, Arruda I, Robinson MO. A mammalian telomerase-associated protein. Science. 1997b;275:973–977. doi: 10.1126/science.275.5302.973. [DOI] [PubMed] [Google Scholar]
- Herrera E, Samper E, Blasco MA. Telomerase deficiency and telomere loss in mTR−/−embryos is associated with failure to close the neural tube. EMBO (Eur Mol Biol Organ) J. 1999;18:5. doi: 10.1093/emboj/18.5.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian A, Bowtell DDL, Abud HE, Hime GR, Venter DJ, Keese PK, Duncan EL, Reddel RR, Jefferson RA. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet. 1997;6:2011–2019. doi: 10.1093/hmg/6.12.2011. [DOI] [PubMed] [Google Scholar]
- Kirk KE, Harmon BP, Reichardt IK, Sedat JW, Blackburn EH. Block in anaphase chromosome separation caused by a telomerase template mutation. Science. 1997;275:1478–1481. doi: 10.1126/science.275.5305.1478. [DOI] [PubMed] [Google Scholar]
- Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4ainactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- Lansdorp PM. Lessons from mice without telomerase. J Cell Biol. 1997;139:309–312. doi: 10.1083/jcb.139.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdorp PM, Verwoerd NP, van de Rijke FM, Dragowska V, Little MT, Dirks RW, Raap AK, Tanke HJ. Heterogeneity in telomere length of human chromosomes. Hum Mol Genet. 1996;5:685–691. doi: 10.1093/hmg/5.5.685. [DOI] [PubMed] [Google Scholar]
- Lee H-W, Blasco MA, Gottlieb GJ, Horner JW, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- Lingner J, Hughes TR, Schevchenko A, Mann M, Lundblad V, Cech T. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276:561–567. doi: 10.1126/science.276.5312.561. [DOI] [PubMed] [Google Scholar]
- Lundblad V, Blackburn EH. An alternative pathway for yeast telomere maintenance rescues est1-senescence. Cell. 1993;73:347–360. doi: 10.1016/0092-8674(93)90234-h. [DOI] [PubMed] [Google Scholar]
- Martens UM, Zijlmans JM, Poon S, Dragowska W, Yui J, Chavez E, Ward R, Lansdorp PM. Short telomeres on human chromosome 17p. Nat Genet. 1998;18:76–80. doi: 10.1038/ng0198-018. [DOI] [PubMed] [Google Scholar]
- Martín-Rivera L, Herrera E, Albar JP, Blasco MA. Expression of mouse telomerase catalytic subunit in embryos and adult tissues. Proc Natl Acad Sci USA. 1998;95:10471–10476. doi: 10.1073/pnas.95.18.10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. The stability of broken ends of chromosomes in Zea mays. Genetics. 1941;26:234–282. doi: 10.1093/genetics/26.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEachern MJ, Blackburn EH. Runaway telomere elongation caused by telomerase RNA gene mutations. Nature. 1995;376:403–409. doi: 10.1038/376403a0. [DOI] [PubMed] [Google Scholar]
- McEachern MJ, Blackburn EH. Cap-prevented recombination between terminal telomeric repeats arrays (telomere CPR) maintains telomeres in Kluyveromyces lactislacking telomerase. Genes Dev. 1996;10:1822–1834. doi: 10.1101/gad.10.14.1822. [DOI] [PubMed] [Google Scholar]
- Metcalfe JA, Parkhill J, Campbell Y, Stacey M, Biggs P, Byrd PJ, Taylor AM. Accelerated telomere shortening in Ataxia Telangiectasia. Nat Genet. 1996;13:350–353. doi: 10.1038/ng0796-350. [DOI] [PubMed] [Google Scholar]
- Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Dickinson S, Caddle, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, et al. hEST2, the putative human telomerase catalytic subunit gene, is upregulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- Morin GB. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989;59:521–529. doi: 10.1016/0092-8674(89)90035-4. [DOI] [PubMed] [Google Scholar]
- Naito T, Matsuura A, Ishikawa F. Circular chromosome formation in a fission yeast mutant defective in two ATM homologues. Nat Genet. 1998;20:203–206. doi: 10.1038/2517. [DOI] [PubMed] [Google Scholar]
- Nakamura TM, Morin GB, Chapman KB, Weinrich SAL, Andrews WH, Lingner J, Harley CB, Cech T. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- Nakamura TM, Cooper JP, Cech T. Two modes of survival of fission yeast without telomerase. Science. 1998;282:493–496. doi: 10.1126/science.282.5388.493. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Saito M, Nakamura H, Matsura A, Ishikawa F. TLP1: a gene encoding a protein component of mammalian telomerase is a novel member of WD repeat family. Cell. 1997;88:875–884. doi: 10.1016/s0092-8674(00)81933-9. [DOI] [PubMed] [Google Scholar]
- Niida H, Matsumoto T, Satoh H, Shiwa M, Tokutake Y, Furuici Y, Shinkai Y. Severe growth defect in mouse cells lacking the telomerase RNA component. Nat Genet. 1998;19:203–206. doi: 10.1038/580. [DOI] [PubMed] [Google Scholar]
- Nugent CI, Bosco G, Ross L, Evans SK, Salinger AP, Moore JK, Harber J, Lundblad V. Telomere maintenance is dependent on activities required for end-repair of double strand breaks. Curr Biol. 1998;8:657–660. doi: 10.1016/s0960-9822(98)70253-2. [DOI] [PubMed] [Google Scholar]
- Rudolph, K.L., S. Chang, H.W. Lee, M.A. Blasco, G. Gottlieb, C.W. Greider, and R.A. dePinho. 1999. Progressive organ dysfunction and increased cancer incidence with age in telomerase deficient mice. Cell. In press. [DOI] [PubMed]
- Sandell LL, Zakian VA. Loss of a yeast telomere: arrest, recovery and chromosome loss. Cell. 1993;75:729–739. doi: 10.1016/0092-8674(93)90493-a. [DOI] [PubMed] [Google Scholar]
- Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Singer MS, Gottschling DE. TLC1: template RNA component of the Saccharomyces cerevisiaetelomerase. Science. 1994;266:387–388. doi: 10.1126/science.7545955. [DOI] [PubMed] [Google Scholar]
- Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- Wan TSK, Martens UM, Poon SSS, Tsao S-W, Chan LC, Lansdorp PM. Absence or low number of telomere repeats at junctions of dicentric chromosomes. Genes Chromosomes Cancer. 1999;24:83–86. doi: 10.1002/(sici)1098-2264(199901)24:1<83::aid-gcc12>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Wang J, Xie LY, Allan S, Beach D, Hannon GJ. Myc activates telomerase. Genes Dev. 1998;12:1769–1774. doi: 10.1101/gad.12.12.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AKC, Rattner JB. Sequence organisation and cytological localization of the minor satellite of mouse. Nucleic Acid Res. 1988;16:11645–11661. doi: 10.1093/nar/16.24.11645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G-L, Bradley JD, Attardi LD, Blackburn EH. In vivo alteration of telomere sequences and senescence caused by mutated Tetrahymena telomerase RNAs. Nature. 1990;344:126–132. doi: 10.1038/344126a0. [DOI] [PubMed] [Google Scholar]
- Zijlmans JM, Martens UM, Poon S, Raap AK, Tanke HJ, Ward RK, Lansdorp PM. Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc Natl Acad Sci USA. 1997;94:7423–7428. doi: 10.1073/pnas.94.14.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]