

Abstract
In the neuroendocrine cell line, PC12, synaptic vesicles can be generated from endosomes by a sorting and vesiculation process that requires the heterotetrameric adaptor protein AP3 and a small molecular weight GTPase of the ADP ribosylation factor (ARF) family. We have now discovered a second pathway that sorts the synaptic vesicle-associated membrane protein (VAMP) into similarly sized vesicles. For this pathway the plasma membrane is the precursor rather than endosomes. Both pathways require cytosol and ATP and are inhibited by GTPγS. The second pathway, however, uses AP2 instead of AP3 and is brefeldin A insensitive. The AP2-dependent pathway is inhibited by depletion of clathrin or by inhibitors of clathrin binding, whereas the AP3 pathway is not. The VAMP-containing, plasma membrane–derived vesicles can be readily separated on sucrose gradients from transferrin (Tf)-containing vesicles generated by incubating Tf-labeled plasma membrane preparations at 37°C. Dynamin- interacting proteins are required for the AP2-mediated vesiculation from the plasma membrane, but not from endosomes. Thus, VAMP is sorted into small vesicles by AP3 and ARF1 at endosomes and by AP2 and clathrin at the plasma membrane.
Keywords: synaptic vesicles, clathrin, plasma membrane, PC12 cells, VAMP
Nerve terminals contain synaptic vesicles of uniform diameter that release their neurotransmitter content by exocytosis. After exocytosis, the vesicle membrane is recycled from the plasma membrane to form a vesicle, which quickly fills up with neurotransmitter. The recycling and refilling processes are very efficient, allowing the nerve terminal to maintain remarkably high rates of neurotransmitter release. Some vesicle recycling might be by a neuron-specific form of exocytosis referred to as “kiss-and-run” (Klingauf et al., 1998). Evidence that a considerable fraction of the recycling process uses conventional, clathrin-mediated endocytotic machinery comes from genetics, morphology, and subcellular fractionation. Mutations in the heterotetrameric adaptor complex, adaptor protein AP21 (Gonzalez-Gaitan and Jackle, 1997), or in the large GTPase, dynamin (van der Bliek et al., 1993; Herskovits et al., 1993), block the reformation of vesicles. Clathrin-coated vesicles are more abundant in rapidly stimulated nerve terminals (Heuser and Reese, 1973). Clathrin-coated vesicles isolated from nerve terminals are enriched in synaptic vesicle membrane proteins (Maycox et al., 1992). Although there is some ambiguity about whether synaptic vesicles are formed from the plasma membrane, the endosome, or both (for review see De Camilli and Takei, 1996), there is little doubt that clathrin and dynamin are involved.
Synaptic vesicle formation involves both vesiculation and sorting of synaptic vesicle membrane proteins. It is a favorable experimental system for the study of vesiculation and sorting processes since the synaptic vesicle membrane is simple and almost completely characterized. This laboratory has developed an in vitro system that reconstitutes the formation of neuroendocrine synaptic vesicle-like microvesicles, here referred to more simply as synaptic vesicles (SVs), by incubating labeled membranes from neuroendocrine PC12 cells in the presence of ATP and brain cytosol (Desnos et al., 1995). The donor organelle for this pathway is an endosome-like organelle (Lichtenstein et al., 1998). To our surprise, depletion of clathrin, AP2, and dynamin from brain cytosol had no detectable effect on SV biogenesis (Faúndez et al., 1997; Horng, J., and R.B. Kelly, unpublished observations). Further analysis revealed that the coat that is recruited to the donor compartment in the presence of ATP and the small GTPase ADP ribosylation factor ARF1, was the heterotetrameric adaptor, AP3 (Faundez et al., 1998). Mice that lack one of the subunits of AP3 have neuronal abnormalities but are viable (Kantheti et al., 1998). This suggests that there is a pathway of SV biogenesis in addition to the one that involves endosomes and AP3.
In vivo evidence has been presented by Huttner and colleagues (Schmidt et al., 1997) that plasma membrane can be a direct precursor of SVs in PC12 cells. Using a vesicle-associated membrane protein (VAMP) mutant that exhibited increased targeting to SVs, N49A/VAMP (Faúndez et al., 1997), we set out to reconstitute vesicle budding from plasma membranes in vitro. The results reported here show that vesicles with the sedimentation properties of SVs, which contain multiple SV marker proteins such as synaptophysin and SV2, and which exclude the Tf receptor (TfR), can also be generated from the plasma membrane in vitro. This alternative pathway of SV biogenesis has few similarities with the endosome-derived pathway since it uses AP2, clathrin, and dynamin-binding proteins. It thus appears as if VAMP-containing SVs that exclude TfR can be formed in two different ways in PC12 cells. Comparing two pathways in the same cell allows us to state with some confidence that recruitment of clathrin or dynamin-related proteins is not essential for the AP3-mediated pathway in vitro, nor is ARF1 essential for the AP2-mediated pathway.
Materials and Methods
125I and ECL reagents were obtained from Amersham Corp. (Arlington Heights, IL). Iodogen came from Pierce Chemical Co. (Rockford, IL). ATP, GTPγS, creatine phosphate, creatine kinase, and Sephadex G25 spin columns were purchased from Boehringer Mannheim Corp. (Indianapolis, IN). Brefeldin A was purchased from Epicentre Technologies (Madison, WI). Percoll was obtained from Sigma Chemical Co. (St. Louis, MO) and rat Tf was purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). Cell culture media and reagents were obtained from the University of California Cell Culture Facility (San Francisco, CA), with the exception of Geniticin (G418), which was obtained from GIBCO BRL (Gaithersburg, MD). All the other reagent grade chemicals were purchased either from Sigma, Fisher Scientific Co. (Fairlawn, NJ), or Calbiochem-Novabiochem Corp. (La Jolla, CA). Female Sprague-Dawley rats were from Bantin and Kingman (Fremont, CA).
Cell Culture
PC12 cell lines were stably transfected to express rat VAMP, to which a T antigen (TAg) epitope was attached at the COOH lumenal end (VAMP-TAg). Unless stated otherwise, the VAMP was the mutant N49A form (Grote et al., 1995). Cells were grown in DME H-21 media supplemented with 10% horse serum, 5% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, and with 250 μg/ml G418. The cells were treated for 24 h before the experiments with 6 mM sodium butyrate to induce the expression of the N49A/VAMP-TAg construct.
Recombinant Proteins
GST control fusion protein was generated by expression of the plasmid pGEX-3X (Pharmacia, Piscataway, NJ). GST-Dyn-PRD was constructed as described in Roos and Kelly (1998). The preparation of constructs encoding the COOH-terminal appendage domains of the βAP3, GST-β3A (810–1094), GST-β3B (799–1081) and GST-β3Amt (β3A with amino acids 820–822 mutated to alanine) has been described elsewhere (Dell'Angelica et al., 1997, 1998). Polyhistidine-tagged clathrin heavy chain fragment (hub) was a gift of F. Brodsky (University of California, San Francisco, CA). Expression of GST fusion proteins followed the recommendations of the manufacturer. Fusion proteins were bound to glutathione–agarose (Sigma), eluted in glutathione elution buffer (20 mM glutathione/50 mM Tris-HCl, pH 8.0/120 mM NaCl), concentrated in a CentriPrep 10 concentrator (Amicon, Beverly, MA) and dialyzed against intracellular buffer (38 mM potassium asparate, 20 mM potassium MOPS, pH 7.2, 5 mM reduced glutathione, 5 mM sodium carbonate, 2.5 mM magnesium sulfate). Fusion protein concentration was determined using the BCA reagent system (Pierce Chemical Co.) and stored at −20°C. Hub was produced as described in Liu et al. (1995). In brief, hub-containing BL21(DE3) bacteria (Novagen, Madison, WI) were induced with 0.8 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h at 30°C. Protein was purified from bacterial lysates in binding buffer (50 mM Tris-HCl, pH 7.9, 0.5 M NaCl, 5 mM imidazole) by binding to a Ni2+ affinity resin, eluted with binding buffer plus 245 mM imidazole, and then dialyzed against intracellular buffer.
Cell Labeling and Subcellular Fractionation
PC12 cells expressing the VAMP-TAg construct (N49A) were labeled with 125I-KT3 mAb to the TAg epitope, following the methods of Desnos et al. (1995). In brief, confluent 15-cm dishes of cells were washed at 4°C with labeling buffer (PBS supplemented with 3% BSA, 0.3 mM CaCl2, 0.3 mM MgCl2, and 1 mg/ml glucose), labeled with 125I-KT3 (5–10 μg/ml) in the same buffer at 4°C for 1 h, and then transferred to 15°C or retained at 4°C for an additional 1 h.
The same conditions were used to label cells with 125I-Tf, except that before the labeling procedure, cells were incubated for 1 h at 37°C in serum-free medium (DME-H21) to deplete their stores of Tf. The concentration of iron-loaded rat Tf in labeling buffer was 20 nM.
After labeling, the cells were extensively washed in the same buffer, scraped, and then sedimented at 800 g for 5 min. Cell pellets were gently resuspended in intracellular buffer containing protease inhibitors. Homogenizations were performed by eight passes through a ball bearing homogenizer (cell cracker; European Molecular Biology Laboratory, Heidelberg, Germany) with 12 μm clearance. A Percoll step gradient was used to obtain membranes free of cytosol. The step gradients were formed by 9 ml of 10% and 3 ml of 50% Percoll (100% Percoll is 1 vol of 10× intracellular buffer and 9 vol of Percoll with protease inhibitors). After collecting the membranes at the 10–50% Percoll interface, a protein assay of the membranes was performed using the Bio-Rad Protein Assay Dye Reagent (Bio-Rad Laboratories, Hercules, CA) using BSA as standard.
In Vitro Budding Assay
VAMP-TAg/N49A PC12 cells were labeled as described above. Aliquots of 1 mg homogenate (or Percoll-washed membranes) were incubated for 30 min at 37°C or 4°C in the presence of an ATP-regenerating system (1 mM ATP, 8 mM creatine phosphate, 5 mg/ml creatine kinase), and 1–4 mg/ml of rat brain cytosol prepared as described (Desnos et al., 1995). In reactions containing peptides, mixtures were preincubated for 15 min at 4°C before warming up. The reactions were stopped by chilling to 4°C for 10 min before fractionation.
After the in vitro incubation a postnuclear supernatant was prepared by sedimenting first at 1,000 g for 5 min (S1) and then at 27,000 g for 35 min (S2). Vesicles in the S2 (150–250 μl, 2–5 mg/ml) were quantified by velocity sedimentation on 5–25% glycerol gradients prepared in intracellular buffer (218,000 g for 75 min at 4°C in a SW55 rotor; Beckman Instruments, Inc., Palo Alto, CA). Fractions (16 or 17) were collected from the bottom and counted on a gamma counter. About 1–2% of the plasma membrane label was recovered in SV-containing fractions of the glycerol gradient.
Immunoadsorption of SVs
SVs isolated by glycerol velocity gradient (fractions 8–10) were pooled and aliquoted. Dynabeads M-450 (Dynal Inc., Great Neck, NY) preincubated with mAbs against syntaxin (Sigma); synaptophysin (Boehringer Mannheim Corp.); SV2 (Buckley and Kelly, 1985); synaptobrevin (69.1); synaptogyrin (80.1, provided by R. Jahn, Yale University, New Haven, CT); or, as a control, mouse γ-globulin (ICN Pharmaceuticals, Inc., Irvine, CA) were added to each aliquot. After 16-h incubation at 4°C, the beads were isolated, and then washed with PBS plus 1% BSA twice for 5 min. Both isolated beads and the supernatant of each reaction were counted on a gamma counter.
Immunodepletion of Clathrin and α-Subunit of AP2
Clathrin heavy chains and AP2 complexes were quantitatively removed from rat brain cytosol using the X22 mAb to clathrin heavy chain and AP6 mAb that binds α-adaptin, respectively (Brodsky, 1985). In brief, 60 μg of antibody were bound overnight to 40 μl of packed protein G–Sepharose in 0.5 ml of intracellular buffer. Unbound Ig was removed by extensive washing in intracellular buffer, and then the antibody-bound affinity matrix was incubated with 0.8 mg of cytosol for 2 h at 4°C with gentle rocking. The Sepharose beads were removed by centrifugation and the cytosol recovered for in vitro reactions. The beads were washed to remove cytosolic proteins. Bead-bound and free cytosolic clathrin or α-adaptin were determined by immunoblotting using an anti–clathrin heavy chain antibody or an anti–α-adaptin antibody (Transduction Laboratories, Lexington, KY). Blots were performed using an enhanced chemiluminescence (Amersham Corp.) system. The cytosol protein concentration before and after the depletion remained constant.
Confocal Immunofluorescence Microscopy
Immunofluorescence procedures and confocal microscopy have been detailed elsewhere (Bonzelius et al., 1994). In brief, N49A/PC12 cells were plated in poly-d-lysine–coated Permanox™ slides (Nunc Inc., Naperville, IL) 2 d before staining. Cells were loaded in vivo with KT3 antibodies (10 μg/ml) in labeling buffer and washed as with labeling buffer with or without 30 mM glycine, pH 2.4, as previously described (Grote and Kelly, 1996). Then cells were fixed in 4% PFA in PBS, permeabilized in 0.02% saponin in PBS, 2% BSA, 1% fish skin gelatin, and then incubated with affinity-purified fluorescent-labeled goat anti–mouse IgG (Cappel Laboratories, Malvern, PA). Observation and image acquisition were performed in a Bio-Rad MRC600 confocal laser scanning microscope (Bio-Rad Laboratories).
Results
Labeling the Plasma Membrane of N49A/PC12 Cells
To study vesicle formation from the plasma membrane, we generated homogenates of PC12 cells in which only SV proteins on the plasma membrane were labeled. The PC12 cell line used in the current study (N49A/PC12) was stably transfected with a luminally tagged VAMP construct that had a point mutation (N49A) in the cytoplasmic tail. This VAMP derivative shows increased targeting to SVs compared with wild type (Grote et al., 1995), and even more specific targeting to SVs than the del61-70 mutation used in a previous study (Desnos et al., 1995). To label the plasma membrane, N49A/PC12 cells were incubated at 4°C with antibodies (KT3) against the lumenal epitope. Immunofluorescent staining was detected around the cell periphery with no detectable staining of any intracellular structures (Fig. 1 A). The peripheral labeling could be stripped away with an acid wash (Fig. 1 B), indicating that, at 4°C, the antibodies only labeled epitopes exposed on the cell surface. In a control experiment, when N49A-transfected cells were labeled at 37°C for 40 min, the antibodies were internalized and an intense, acid strip–resistant intracellular staining could be observed (Fig. 1, C and D). Restriction of label to the cell surface was confirmed by biochemical subcellular fractionation experiments. Intact N49A cells were labeled at 4°C with iodinated KT3 followed by homogenization and fractionation on sucrose density gradients. A peak of radioactivity was recovered at 38% sucrose, which represents labeled plasma membranes (Clift-O'Grady et al., 1998). Thus, incubating cells with antibody at 4°C only labels the cell surface.
Figure 1.

The plasma membrane of N49A/PC12 cells is labeled with KT3 mAb at 4°C. N49A/ PC12 cells were incubated in the presence of KT3 mAb (10 μg/ ml) for 15 min at 4°C. Cells were then warmed up to 37°C for 40 min (C and D) or kept at 4°C (A and B). In B and D, cells were treated with an acid wash (see Materials and Methods) at 4°C before fixation. The immunofluorescence was processed as described. After labeling at 4°C, the label can be removed by an acid wash, but not after warming to 37°C. Bars, 10 μM.
Vesicle Budding from the Plasma Membrane
To reconstitute in vitro vesicle budding from the plasma membrane, we labeled intact N49A cells with iodinated KT3 at 4°C for 2 h. After washing away the unbound antibody, the cells were homogenized and the homogenate incubated at 37°C in the presence of rat brain cytosol and an ATP-regenerating system. After the incubation, the reaction mix was centrifuged to remove large membranes and the supernatant was analyzed by glycerol velocity sedimentation. As shown in Fig. 2 (circles), we observed radioactivity in the SV-containing fractions (8–12) of the glycerol gradient, as well as in free antibody at the top of the gradient (14 and above). SV generated by labeling N49A cells at 15°C followed by an in vitro reaction at 37°C (Desnos et al., 1995) also migrate to the same glycerol fractions (Fig. 2, diamonds). The amount of radioactivity recovered in the SV peak was always about five times less when labeled plasma membrane was used, compared with labeled endosomes. The relatively weak signal was probably due to the limited amount of VAMP on the cell surface compared with the amount internalized at 15°C. The small size of the signal, the shorter incubation time, and the use of a less advantageous mutant VAMP (del61-70) may explain why no budding from plasma membranes was observed in earlier experiments (Desnos et al., 1995). Nonetheless, when expressed as recovery of membrane-bound radioactivity in SVs, the plasma membrane and the endosomal assays have comparable efficiency.
Figure 2.

In vitro budding from cell homogenate labeled at 4°C or 15°C. Cell homogenate (1 mg) was prepared from N49A/PC12 cells labeled with 125I-KT3 at 4°C for 2 h. The homogenate was then incubated with rat brain cytosol (1.5 mg/ml) and ATP at 37°C (closed circles) or 4°C (open circles) for 30 min followed by centrifugation of a high speed supernatant (S2) on a 5–25% glycerol velocity gradient. A significant amount of 125I-KT3 was recovered in fractions 8–12 (closed circles), cosedimenting with the SVs (closed diamonds) generated by an in vitro reaction using homogenate from 15°C-labeled N49A PC12 cells (Desnos et al., 1995). No labeled KT3 was recovered in the SV peak when the in vitro reactions were performed at 4°C (15°C cell labeling, open diamonds; 4°C cell labeling, open circles). The label at the top (right-hand side) of the gradients is free antibody.
Characterization of the Budding Reaction
In vitro vesicle budding is increased in efficiency by the use of cytosol. As shown in Fig. 3 A, the amount of vesicles generated in the budding reaction depended on the concentration of rat brain cytosol, suggesting that cytosolic factors are required for vesicle formation from the plasma membrane. A small vesicle peak was often observed in control reactions with no added rat brain cytosol. Since this background reaction is seen with washed membranes it may be due to coating proteins that remain bound to the plasma membrane after homogenization. As indicated by Western blot, clathrin and α-adaptin can be detected on the washed membranes (not shown). Immunofluorescence also supports the association of dynamin (Estes et al., 1996; Roos and Kelly, 1998) and AP2 (Gonzales-Gaitan and Jackle, 1997) with the plasma membrane at endocytotic “hot spots.”
Figure 3.
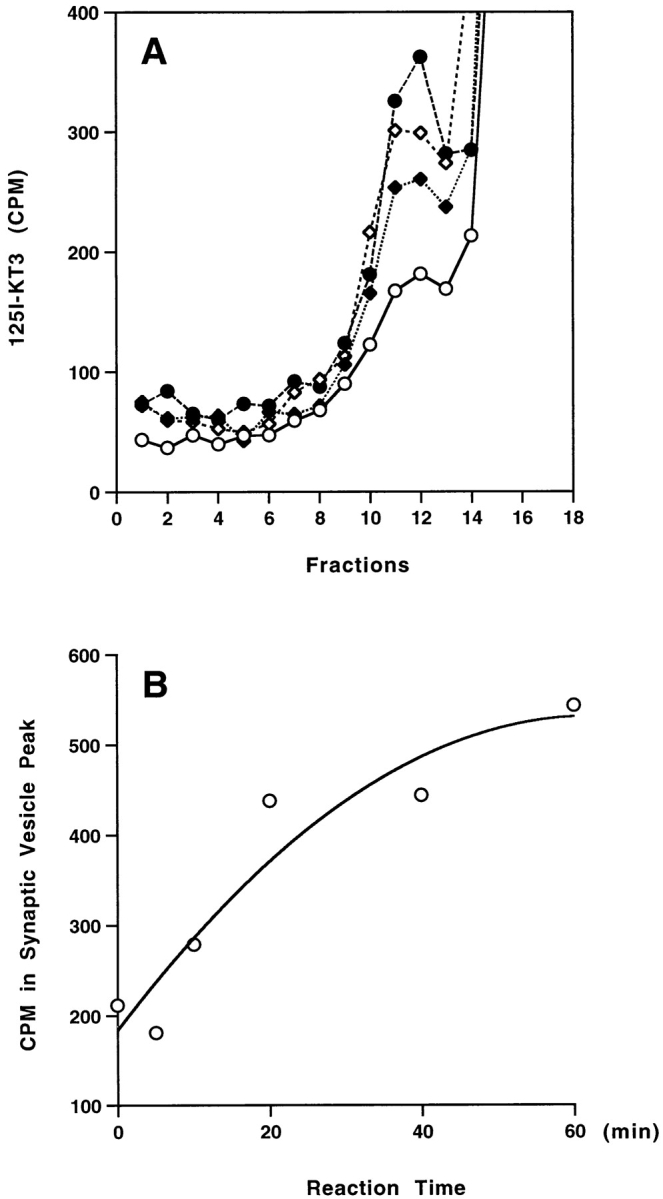
Characterization of the in vitro budding reaction from 4°C-labeled membranes. (A) Effect of rat brain cytosol concentration. Standard reactions were performed using Percoll-washed membranes with different concentrations of rat brain cytosol: 0 mg/ml (open circles), 0.5 mg/ml (closed diamonds), 1 mg/ml (open diamonds), and 2 mg/ml (closed circles). The high speed supernatants (S2) of each reaction were centrifuged on a 5–25% glycerol velocity gradient. The radioactivity of each fraction was plotted against fraction numbers. (B) Kinetics of the in vitro budding reaction. The amount of 125I-KT3 recovered in the SV peak (pool of fractions 8–12) after a complete budding reaction was measured as a function of time. (C) Nucleotide dependence of the budding reactions. Standard in vitro budding reactions were carried out under normal budding conditions (Percoll-washed membranes, rat brain cytosol, and ATP at 37°C), or with 20 μM GTPγS, or in the absence of the ATP regeneration system. (Note that nucleotides in rat brain cytosol were removed by dialysis against intracellular buffer.) Vesicle production (radioactivity in fractions 8–12) was normalized as a percentage of the yield obtained under normal conditions (Control). Reactions at 4°C were subtracted as backgrounds. Error bars represent the range of the results for two independent experiments. The cytosol concentration in all experiments was 1.5 mg/ml.
A time course of vesicle budding from the in vitro reconstitution systems indicated that detectable vesicle production starts within 10 min of incubation and reaches a plateau in ∼40 min (Fig. 3 B), not significantly different from kinetics of the in vitro SV budding from endosomes (Desnos et al., 1995). Vesicle formation was reduced when the ATP-regenerating system was omitted in the reaction mix (Fig. 3 C). The presence of GTPγS in the reconstituted system greatly inhibited the generation of SVs, in line with the possible involvement of dynamin or other GTPases in endocytosis from the plasma membrane (Nuoffer and Balch, 1994; Takei et al., 1995, 1996; Carter et al., 1993).
Newly Formed SVs Contain Multiple SV Proteins and Exclude Tf
Although neuroendocrine cells such as PC12 are not synapse-forming neurons, they have organelles that resemble SVs. Three criteria that are conventionally used to describe the SVs in PC12 cells are their homogeneous size, the presence of a full array of SV proteins, and the absence of other recycling plasma membrane proteins such as the TfR. To determine if SV markers other than VAMP are present on the small vesicles, M-450 Dynabeads (sheep anti–mouse IgG) coated with mAbs against SV proteins synaptophysin, synaptogyrin, or SV2 were used to immunoadsorb the vesicle fractions. As shown in Fig. 4 A, each of these antibodies adsorbed most of the radioactivity associated with the vesicle fractions. As expected, an almost complete adsorption was also achieved by an antibody against VAMP (synaptobrevin; Fig 4 A). However, antibodies against a t-SNARE protein, syntaxin, did not bind vesicles above the level of controls containing mouse γ-globulin confirming earlier observations (Salem et al., 1998). This result shows that the small vesicles formed from the plasma membrane, as expected for SVs, contain a spectrum of SV markers.
Figure 4.
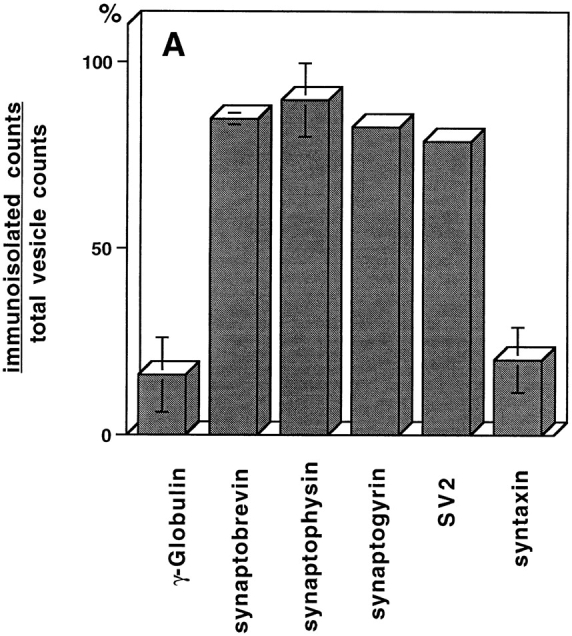
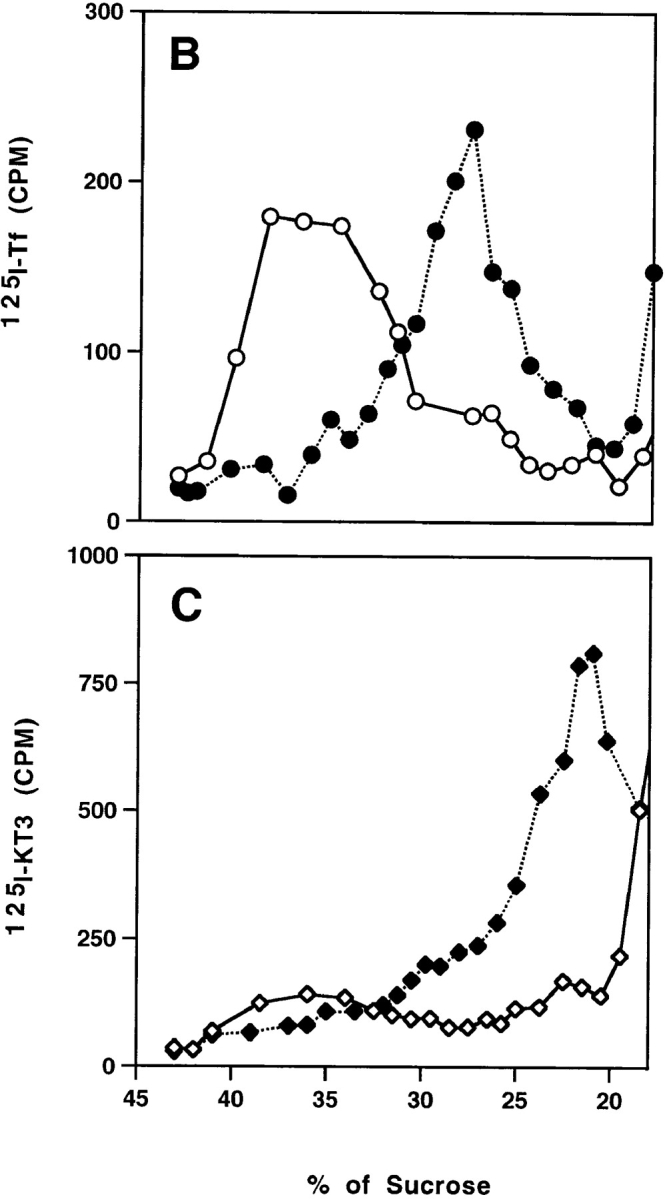
(A) Immunoadsorption of SVs derived from the plasma membrane. Aliquots of SVs were incubated with M-450 Dynabeads (3 mg per reaction) coated with anti-VAMP (synaptobrevin), anti-synaptophysin (SY-38), anti-synaptogyrin (p29), anti-SV2, or anti-syntaxin antibodies. Mouse γ-globulin was used to determine non-specific binding. After extensive wash, the radioactivity associated with the beads was plotted as a percentage of total radioactivity for each reaction. (B and C) TfRs are sorted from SVs from plasma membrane. N49A/PC12 cells were labeled with 125I-Tf (0.2 μg/ml; B) or 125I-KT3 (10 μg/ml; C) at 4°C for 2 h. The cell homogenate was incubated with rat brain cytosol and ATP at 37°C (closed symbols) or 4°C (open symbols). A low speed (5,000 g min) supernatant was made from each reaction and centrifuged on a 10– 45% sucrose velocity gradient. Radioactivity of each gradient fraction was plotted against its sucrose concentration. Radioactivity from 125I-KT3 was recovered at 23% (1.5 sucrose; closed diamonds), similar to the SVs generated from endosomes (Lichtenstein et al., 1998). However, 125I-Tf–labeled membranes were recovered at 28% (0.5 sucrose; closed circles), well separated from the SVs.
To determine if the VAMP-containing vesicles that formed from the plasma membrane are devoid of TfR, PC12 cells were labeled at 4°C with 125I-Tf, homogenized, and then incubated in vitro with brain cytosol and ATP. No radioactivity was recovered in the SV peak region after glycerol velocity sedimentation of the reaction products (data not shown).
The absence of labeled Tf in SV fractions could be because the levels were too low to detect. To determine if other Tf-containing vesicles were made, we analyzed the reaction products differently using a sucrose velocity gradient protocol that resolves SVs from other membrane populations (Lichtenstein et al., 1998). Intact N49A/PC12 cells were labeled with iodinated Tf at 4°C as before, washed, homogenized, and then the homogenate was incubated at 37°C under standard conditions. After the reaction, the plasma membrane was removed from the reaction mix by a lower speed, 5,000 g min centrifugation to allow the analysis of membrane vesicles larger than SVs. The supernatant of the reaction mixture was analyzed on a 10–45% sucrose density gradient. Using this new protocol, Tf-containing vesicles were recovered at 28% ± 0.5 sucrose (Fig. 4 B, closed circles), a density different from that at which the KT3-containing vesicles were recovered (23% ± 1.5 sucrose), as shown in Fig. 4 C (closed diamonds). Our results indicate that VAMP-containing SVs, from which the TfR has been segregated, can bud from the plasma membrane, suggesting that protein sorting may be occurring directly at the plasma membrane.
Small amounts of label, especially in Tf, are recovered between 35% and 40% sucrose in the 5,000 g min supernatants from cells labeled at 4°C (Fig. 4 B). The label is presumably due to small fragments of plasma membrane that are not pelleted. They disappeared during the in vitro incubation, implying that they are potential precursors of the Tf-labeled vesicles at 28% sucrose. The nature of this putative precursor is not yet known.
SV Formation from the Plasma Membrane Requires AP2 and Clathrin But Not ARF1
Although the plasma membrane was the only labeled precursor added to the reaction mix it was possible that the plasma membrane was generating endosomes from which SVs were budding by an AP3 and ARF-dependent mechanism. To examine this possibility we took advantage of the sensitivity of the endosomal budding reaction to a fungal metabolite, brefeldin A (BFA).
ARF1, a small GTP-binding protein, is required for SV biogenesis from endosomes (Faúndez et al., 1997). Its function is to recruit the AP3 adaptor complex as a coat to mediate vesicle budding (Faúndez et al., 1998). A hallmark of ARF1-mediated processes is their sensitivity to BFA, which inhibits GDP–GTP exchange activity of ARF1 (Donaldson et al., 1992). As shown in Fig. 5 (closed circles), SV biogenesis from endosomes was inhibited by BFA in a dose-dependent manner as described previously (Faúndez et al., 1997). However, the vesicle production from the plasma membranes was not inhibited in the presence of BFA ≤500 μg/ml. The difference in BFA sensitivity implies that coat recruitment to allow vesicle budding from the plasma membrane does not require a cytoplasmic ARF1. These observations are consistent with the previous reports showing that BFA inhibits vesicle formation from the TGN, but not from the cell surface (Robinson and Kreis, 1992).
Figure 5.

BFA inhibits in vitro SV formation from endosomes but not from the plasma membrane. Cell homogenates from N49A/PC12 cells labeled with 125I-KT3 at either 4°C for 2 h (open circles) or 15°C for 40 min (closed circles) were incubated with rat brain cytosol (1.5 mg/ml), ATP and the indicated amount of BFA at 4°C for 15 min before warming to 37°C for 30 min. The amount of SV production (radioactivity of fractions 8–12) was expressed as a percentage of the yield obtained from reactions to which no BFA was added.
We were unable to obtain any evidence that clathrin (Faundez et al., 1997) or AP2 (Horng, J.-T., and R.B. Kelly, unpublished) were required for SV formation from endosomes. In contrast, SV formation from the plasma membrane needs clathrin and AP2. When the cell-free reconstitution system was supplied with rat brain cytosol depleted of clathrin heavy chain or AP2 (α-adaptin), the vesicle production was reduced (Fig. 6 A). The extent of depletion was confirmed by Western blotting of rat brain cytosol with clathrin and α-adaptin–specific antibodies (Fig. 6 B). The clathrin involvement was further tested using a bacterially expressed clathrin heavy chain fragment, often called the hub domain. The clathrin hub fragment is the minimal trimerization domain that can self-assemble into clathrin triskelions in vitro (Liu et al., 1995). It interferes with endogenous clathrin function by blocking clathrin polymerization into polyhedral vesicle coats (Liu et al., 1998). An inhibitory effect on vesicle biogenesis was observed upon the addition of the hub fragment (Fig. 6 A). These results suggest that the clathrin/AP2-coating complex is necessary to form KT3-containing vesicles from the plasma membrane.
Figure 6.
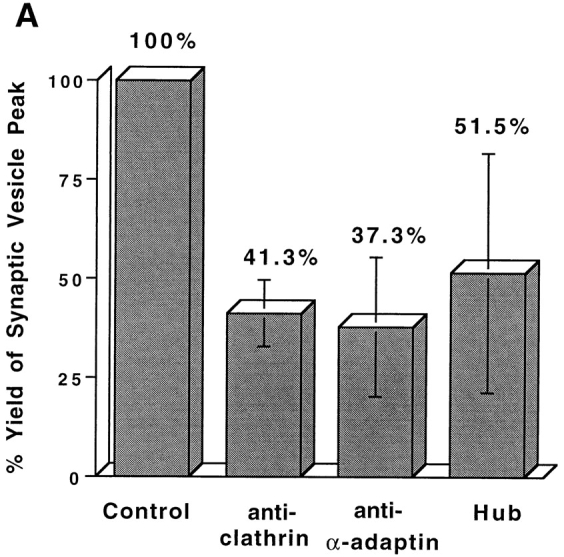
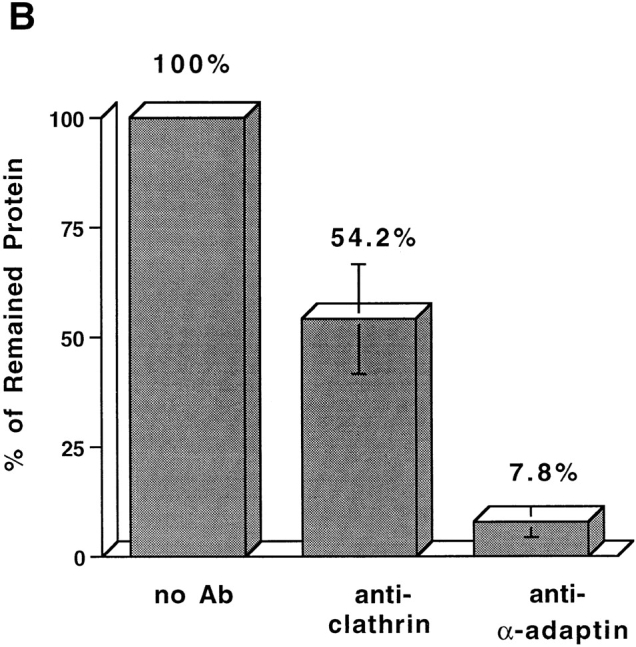
SVs generated from the plasma membrane require clathrin and AP2. (A) Standard in vitro budding reactions from 4°C-labeled, Percoll-washed N49A/PC12 membranes were performed with clathrin-deficient cytosol, with α-adaptin (AP2)-deficient cytosol, or with normal cytosol containing 3 mg/ml clathrin hub fragment. SV production for each reaction was expressed as the percentage of that from control reactions (with only rat brain cytosol). The variability in results with the hub fragment may be due to difficulties in keeping it in solution. (B) Antibodies specific for clathrin heavy chain (X22) or α-adaptin (AP6), respectively, were used to deplete clathrin and AP2 adapter complex from rat brain cytosol. After depletion, 200 μg of rat brain cytosol (supernatant) and immunoprecipitated product (pellet) were fractionated by SDS-PAGE followed by Western blotting with an anti-clathrin heavy chain antibody and an anti–α-adaptin antibody. The remaining clathrin and α-adaptin in the cytosol were plotted as a percentage of total clathrin and α-adaptin before depletion.
In these depletion studies, inhibition of SV formation was never complete. This was expected since SV formation was only stimulated about twofold by the addition of cytosol (Fig. 2). As discussed above, membrane-associated protein machinery may also contribute to the budding activity. To confirm that the inhibition was indeed due to clathrin and AP2 depletion, coat proteins were isolated from brain-coated vesicles by conventional procedures (Manfredi and Bazari, 1987) and added back to the reaction mix. When 80 μg/ml purified clathrin was added back to clathrin-depleted cytosol, the budding activity recovered to 87% of that before clathrin depletion. Similarly, 100 μg/ml AP2 restored the activity to 94%.
Clathrin Is Needed for AP2, But Not AP3-mediated SV Formation In Vitro
Originally it was reported (Simpson et al., 1996) that clathrin did not codistribute with AP3 by immunofluorescence. This was consistent with normal AP3-mediated sorting in yeast cells lacking clathrin (Vowels and Payne, 1998) and our own data from in vitro reconstitution showing clathrin independence (Faundez et al., 1998). However, the COOH- terminal appendage domain of the β3 subunit of AP3 binds clathrin, and AP3 and clathrin can be colocalized on endosomes and TGN by immunoelectron microscopy (Dell'Angelica et al., 1998).
Additional clathrin would not be needed in our in vitro assay if there were already sufficient clathrin on the donor membranes to generate budding. If that clathrin were recruited to the COOH-terminal appendage domains of AP3, then inhibition of budding might be seen in the presence of excess appendage domain. Incubating labeled endosomes with excess appendage domain, however, gave no inhibition of SV formation (Fig. 7 ES).
Figure 7.

The putative appendage domain of β3 subunits of AP3 complex inhibits SV production from plasma membrane but not that from endosome. Standard in vitro budding reactions were performed with 4°C-labeled (PM) or 15°C-labeled (ES), Percoll-washed N49A/PC12 membranes in the presence or absence of 150 μM GST fusion proteins bearing appendage domain of β3A, β3B, or β3Amt. The amount of SV production was normalized to the control reaction incubated with added 150 μM GST.
The absence of inhibition could be attributed to failure to achieve inhibitory concentrations of appendage domain, or the absence of clathrin recruitment from a membrane-bound pool. The first explanation is less likely because the AP3 appendage domains at the same concentration (150 μM) inhibited SV production from labeled plasma membranes by ∼70% (Fig. 7 PM). With 75 μM appendage domain, the inhibition was 40% and with 38 μM, the inhibition was not significant. A mutant, GST-β3Amt, in which residues DLD (820–822 of GST-β3A) are mutated to three alanine residues, has no clathrin binding activity in vitro (Dell'Angelica et al., 1998), and does not show an inhibitory effect on SV formation. The appendage domain of AP2 also inhibited (data not shown). These results are consistent with the evidence that the β3 appendage domain binds clathrin as efficiently as the β2 appendage domain of AP2 and so might inhibit AP2-mediated recruitment (Dell'Angelica et al., 1998).
The data on appendage inhibition are consistent with the other data on clathrin dependence of the AP2 pathway. A concern, however, with the use of appendage domains as inhibitors is the high concentration needed (150 μM) to obtain significant inhibition. Earlier experiments using the AP2 appendage domains reported a very low affinity between appendage and clathrin (Shih et al., 1995) consistent with our findings. The low affinity might not be real but might reflect inefficient folding of the recombinant proteins. It is encouraging that inhibition is not seen with mutant appendages that do not have binding activity (Fig. 7). To resolve definitively whether clathrin is involved in AP3-mediated budding, high affinity inhibitors of clathrin-mediated vesiculation are clearly needed.
The GTPase dynamin has been implicated in endocytosis from the plasma membrane (van der Bliek et al., 1993; Vallee and Okamoto, 1995; De Camilli and Takei, 1996). To explore how dynamin might function in SV formation, a fusion of GST to the proline-rich domain (PRD) of dynamin (GST-Dyn-PRD) was made and used to screen for brain-specific proteins that bound to it (Roos and Kelly, 1998). Four major PRD-binding proteins were found in brain extracts. To determine if dynamin or SH3-containing proteins that bind to the Dyn-PRD were involved in SV biogenesis from PC12 membranes, in vitro budding reactions were incubated with either GST (control) or GST-Dyn-PRD fusion proteins. The GST-Dyn-PRD protein inhibited vesicle formation from plasma membranes by >50%, but had no effect on budding from endosomes (Fig. 8). We conclude that the proteins that bind dynamin PRDs are likely to be involved in the AP2-mediated pathway of vesicle biogenesis, but not the endosomal, AP3-mediated pathway.
Figure 8.
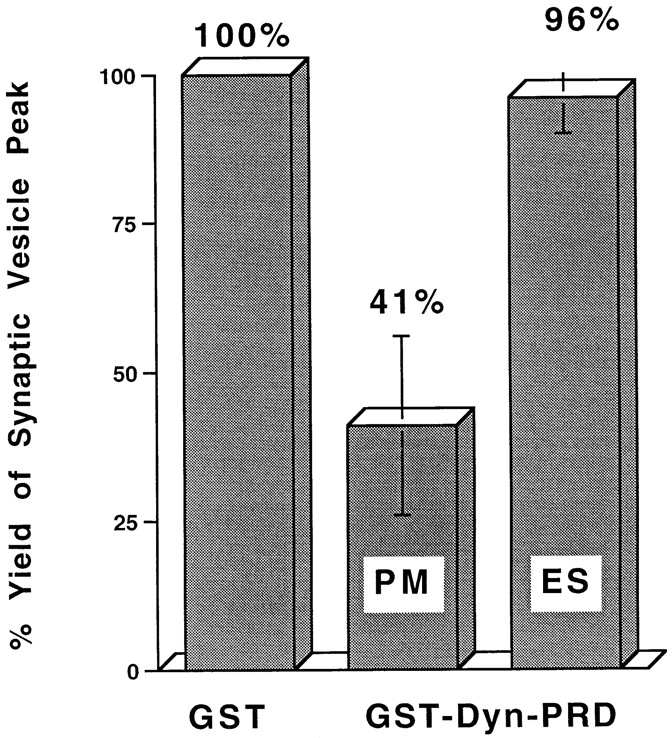
GST-Dyn-PRD inhibits SV formation from the plasma membrane but not that from endosomes. Standard in vitro budding reactions with Percoll-washed N49A/PC12 cell membranes from cells labeled at 4°C (plasma membrane) or 15°C (endosome) were performed with 300 μg/ml of GST-Dyn-PRD or 800 μg/ml GST control protein. SV production (fractions 8–12) with added GST-Dyn-PRD was normalized as a percentage of that with GST control proteins. PM, plasma membrane; ES, endosome.
Discussion
We have reconstituted the biogenesis of SVs from the plasma membrane of PC12 cells in vitro. Unlike the biogenesis of SVs from PC12 endosomes, the formation of plasma membrane–derived SVs requires AP2 and clathrin, but not GTP hydrolysis by ARF1. This mode of small vesicle biogenesis could be a model for the dominant pathway of SV biogenesis in neurons, which is likely to involve AP2 and clathrin on the basis of genetic, morphological, and cell fractionation evidence.
The presence of two distinct biosynthetic pathways in the same cell type throws into sharper relief the differences between the AP2- and AP3-mediated pathways. Most strikingly, AP2-mediated budding requires clathrin recruitment under conditions that AP3 does not. The ability of clathrin triskelions to form pentagons and hexagons has often led to the speculation, occasionally challenged (Kirchhausen et al., 1997), that the function of clathrin is to impart curvature to vesicles budding from a donor membrane. If the AP3 adaptor in the absence of clathrin can form vesicle-sized structures in vitro, then the recruitment of clathrin is not essential for imparting curvature. An alternative explanation is that clathrin recruitment is not necessary because endosomes already have a large pool of clathrin bound to the membranes. For this explanation to be plausible, clathrin recruitment from the membrane pool cannot be sensitive to the AP3 appendage domains. Efforts to make the endosomal budding system dependent on clathrin by stripping the endosome precursors with high salt or Tris washes have not succeeded (Horng, J.-T., and R.B. Kelly, unpublished).
Conversely, the AP3-dependent endosomal pathway, but not the AP2-dependent plasma membrane one, requires the addition of ARF1 in a GTP-bound form. Given the involvement of small GTPases of the ARF family in ER, Golgi, and endosomal budding events it is surprising that recruitment of coat by the plasma membrane should be different. One possible explanation is that the AP2 recruitment mechanism is firmly bound to the plasma membrane. ARF6, for example, is firmly associated with the plasma membrane and is insensitive to BFA (D'Souza-Schorey et al., 1995; Peters et al., 1995).
The discovery of a second potential pathway of SV biogenesis helps us reconcile the findings of Schmidt et al. (1997) with our own (Desnos et al., 1995; Faundez et al., 1997, 1998). By following the internalization of biotinylated synaptophysin, Schmidt et al. (1997) could show that SVs were generated from a plasma membrane precursor, not an endosomal one as we had proposed. It is very likely that the experimental conditions chosen by Schmidt et al. (1997) favored the discovery of the plasma membrane– derived pathway, whereas ours favored the discovery of the endosomal pathway. Using our techniques for monitoring vesicle formation conditions, almost all sSV biogenesis in vivo is sensitive to BFA and so occurs by the AP3 pathway.
Our earlier work had shown that PC12 endosomes containing TfRs were precursors of SVs (Lichtenstein et al., 1998). The original goal of the present paper was to explore the formation of the endosomal precursor from plasma membranes. To our surprise we found that vesicles were formed from plasma membrane in vitro that were remarkably similar in size to SVs. To determine if these small vesicles were endosomal precursors we did the type of double-labeling experiments shown in Fig. 4, B and C. The result was clearcut: the small vesicles do not contain Tf. If the small vesicles were endosomal precursors that subsequently fuse with TfR-containing endosomes, sorting at the plasma membrane would be a futile process. A more likely possibility is that VAMP gets to endosomes by an alternative route. One possibility is the vesicles at 28% sucrose (Fig 4, B and C), which appear to contain both Tf and VAMP.
The absence of detectable Tf in the plasma membrane– derived microvesicles is also consistent with the observations of Schmidt et al. (1997). The simplest explanation of our data and those of Schmidt et al. (1997) is that there are two separate pathways of internalization from the plasma membrane, one that carries both SV proteins and carriers such as the TfR and another that is exclusive for SV proteins. A specialized internalization pathway for SV proteins was first suggested by De Camilli and coworkers to explain the differences between the endosomes in the axons and the cell bodies of neurons (Mundigl et al., 1993), an observation confirmed later in PC12 cells (Bonzelius et al., 1994). Recently evidence that more than one internalization pathway exists has also been obtained by comparing the GLUT4 glucose transporter with the TfR, after internalization at 15°C (Wei et al., 1998). A clathrin-independent pathway of internalization has been known for some time (Sandvig and van Deurs, 1991; Lamaze and Schmid, 1995), but the pathway taken by VAMP-TAg described here is both clathrin and dynamin dependent. If there are two independent pathways of clathrin-mediated internalization from the plasma membrane, some mechanisms must exist to segregate their cargo proteins selectively.
It is not clear why PC12 cells should have two pathways for making SV-like organelles. One possibility is that the two classes of vesicle have different protein compositions. Recent evidence suggests that the AP3-mediated pathway in brain is tightly linked to the formation of a subclass of SVs that contain one of the zinc transporters, ZnT3 (Kantheti et al., 1998). Another possibility is that the two pathways are used at different levels of synaptic activity. A third is that neurons of a certain class, for example with more of a neuroendocrine function, or with large nerve terminals, or with extensive endocytosis of neurotropin receptors, use the AP3-mediated pathway. Fortunately the tools are now available to distinguish these possibilities.
Acknowledgments
We thank Dr. F. Brodsky (University of California, San Francisco, CA [UCSF]) for the generous gift of antibodies to clathrin (X22), AP2 (AP6), and of the polyhistidine-tagged hub fragment. We are also grateful to L. Spector for her help in the preparation of the manuscript.
This work was supported by grants to R.B. Kelly from Wheeler Center for the Neurobiology of Addiction, UCSF, and the National Institutes of Health (NIH; grant Nos. NS19878, NS15927, DA10154). J. Roos is supported by a postdoctoral fellowship from the American Cancer Society. V. Faundez was the recipient of an NIH Fogarty Fellowship.
Abbreviations used in this paper
- AP
adaptor protein
- ARF
ADP ribosylation factor
- BFA
brefeldin A
- PRD
proline-rich domain
- SV
synaptic vesicle
- TAg
T antigen
- Tf
transferrin
- TfR
transferrin receptor
- VAMP
vesicle-associated membrane protein
References
- Bonzelius F, Herman GA, Cardone MH, Mostov KE, Kelly RB. The polymeric immunoglobulin receptor accumulates in specialized endosomes but not synaptic vesicles within the neurites of transfected neuroendocrine PC12 cells. J Cell Biol. 1994;127:1603–1616. doi: 10.1083/jcb.127.6.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky FM. Clathrin structure characterized with monoclonal antibodies. I. Analysis of multiple antigenic sites. J Cell Biol. 1985;101:2047–2054. doi: 10.1083/jcb.101.6.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley K, Kelly RB. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J Cell Biol. 1985;100:1284–1294. doi: 10.1083/jcb.100.4.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter LL, Redelmeier TE, Woollenweber LA, Schmid SL. Multiple GTP-binding protiens participate in clathrin-coated vesicle-mediated endocytosis. J Cell Biol. 1993;120:37–45. doi: 10.1083/jcb.120.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clift-O'Grady, L., C. Desnos, Y. Lichtenstein, V. Faundez, J.-T. Horng, and R.B. Kelly. 1998. A method for the reconstitution of synaptic vesicle biogenesis from PC12 membranes. Methods: A Companion to Methods Enzymol. 16:150–159. [DOI] [PubMed]
- De Camilli P, Takei K. Molecular mechanisms in synaptic vesicle endocytosis and recycling. Neuron. 1996;16:481–486. doi: 10.1016/s0896-6273(00)80068-9. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica EC, Ohno H, Ooi CE, Rabinovich E, Roche KW, Bonifacino JS. AP-3: an adaptor-like protein complex with ubiquitous expression. EMBO (Eur Mol Biol Organ) J. 1997;16:917–928. doi: 10.1093/emboj/16.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Angelica EC, Klumperman J, Stoorvogel W, Bonifacino JS. Association of the AP-3 adaptor complex with clathrin. Science. 1998;280:431–434. doi: 10.1126/science.280.5362.431. [DOI] [PubMed] [Google Scholar]
- Desnos C, Clift-O'Grady L, Kelly RB. Biogenesis of synaptic vesicles in vitro. J Cell Biol. 1995;130:1041–1049. doi: 10.1083/jcb.130.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane-catalyzed exchange of guanine nucleotide onto ARF protein. Nature. 1992;360:350–352. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- D'Souza-Schorey C, Li G, Colombo MI, Stahl PD. A regulatory role for ARF6 in receptor-mediated endocytosis. Science. 1995;267:1175–1178. doi: 10.1126/science.7855600. [DOI] [PubMed] [Google Scholar]
- Estes PS, Roos J, van der Bliek A, Kelly RB, Krishnan KS, Ramaswami M. Traffic of dynamin within individual Drosophila synaptic boutons relative to compartment-specific markers. J Neurosci. 1996;16:5443–5456. doi: 10.1523/JNEUROSCI.16-17-05443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faundez V, Horng JT, Kelly RB. ADP ribosylation factor 1 is required for synaptic vesicle budding in PC12 cells. J Cell Biol. 1997;138:505–515. doi: 10.1083/jcb.138.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faundez V, Horng JT, Kelly RB. A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell. 1998;93:1–20. doi: 10.1016/s0092-8674(00)81170-8. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gaitan M, Jackle H. Role of Drosophila alpha-adaptin in presynaptic vesicle recycling. Cell. 1997;88:767–776. doi: 10.1016/s0092-8674(00)81923-6. [DOI] [PubMed] [Google Scholar]
- Grote E, Kelly RB. Endocytosis of VAMP is facilitated by a synaptic vesicle targeting signal. J Cell Biol. 1996;132:537–547. doi: 10.1083/jcb.132.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote E, Hao JC, Bennett MK, Kelly RB. A targeting signal in VAMP regulating transport to synaptic vesicles. Cell. 1995;81:581–589. doi: 10.1016/0092-8674(95)90079-9. [DOI] [PubMed] [Google Scholar]
- Herskovits JS, Burgess CC, Obar RA, Vallee RB. Effects of mutant rat dynamin on endocytosis. J Cell Biol. 1993;122:565–578. doi: 10.1083/jcb.122.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantheti P, Qiao X, Diaz ME, Peden AA, Meyer GE, Curskado SL, Kapfhamer D, Sufalko D, Robinson MS, Noebels JL, Burmeister M. Mutations in AP-3 delta in the mochu mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron. 1998;21:111–122. doi: 10.1016/s0896-6273(00)80519-x. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T, Bonifacino JS, Riezman H. Linking cargo to vesicle formation: receptor tail interactions with coat proteins. Curr Opin Cell Biol. 1997;9:488–495. doi: 10.1016/s0955-0674(97)80024-5. [DOI] [PubMed] [Google Scholar]
- Klingauf J, Kavalali ET, Tsien RW. Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature. 1998;394:581–585. doi: 10.1038/29079. [DOI] [PubMed] [Google Scholar]
- Lamaze C, Schmid SL. The emergence of clathrin-independent pinocytic pathways. Curr Opin Cell Biol. 1995;7:573–580. doi: 10.1016/0955-0674(95)80015-8. [DOI] [PubMed] [Google Scholar]
- Lichtenstein Y, Desnos C, Faundez V, Kelly RB, Clift-O'Grady L. Vesiculation and sorting from PC12-derived endosomes in vitro. Proc Natl Acad Sci USA. 1998;95:11223–11228. doi: 10.1073/pnas.95.19.11223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SH, Wong ML, Craik CS, Brodsky FM. Regulation of clathrin assembly and trimerization defined using recombinant triskelion hubs. Cell. 1995;83:257–267. doi: 10.1016/0092-8674(95)90167-1. [DOI] [PubMed] [Google Scholar]
- Liu SH, Marks MS, Brodsky FM. A dominant-negative clathrin mutant differentially affects trafficking of molecules with distinct sorting motifs in the class II major histocompatibility complex (MHC) pathway. J Cell Biol. 1998;140:1023–1037. doi: 10.1083/jcb.140.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi JJ, Bazari WL. Purification and characterization of two distinct complexes of assembly polypeptides from calf brain coated vesicles that differ in their polypeptide composition and kinase activities. J Biol Chem. 1987;262:12182–12188. [PubMed] [Google Scholar]
- Maycox PR, Link E, Reetz A, Morris SA, Jahn R. Clathrin-coated vesicles in nervous tissue are involved primarily in synaptic vesicle recycling. J Cell Biol. 1992;118:1379–1388. doi: 10.1083/jcb.118.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundigl O, Matteoli M, Daniell L, Thomas-Reetz A, Metcalf A, Jahn R, DeCamilli P. Synaptic vesicle proteins and early endosomes in cultured hippocampal neurons: Differential effects of Brefeldin A in axon and dendrites. J Cell Biol. 1993;122:1207–1221. doi: 10.1083/jcb.122.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuoffer C, Balch WE. GTPases: multifunctional molecular switches regulating vesicular traffic. Annu Rev Biochem. 1994;63:949–990. doi: 10.1146/annurev.bi.63.070194.004505. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Hsu VW, Ooi CE, Finazzi D, Teal SB, Oorschot V, Donaldson JG, Klausner RD. Overexpression of wild-type and mutant ARF1 and ARF6: Distinct perturbations of nonoverlapping membrane compartments. J Cell Biol. 1995;128:1003–1017. doi: 10.1083/jcb.128.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS, Kreis TE. Recruitment of coat proteins onto Golgi membranes in intact and permeabilized cells: effects of brefeldin A and G protein activators. Cell. 1992;69:129–138. doi: 10.1016/0092-8674(92)90124-u. [DOI] [PubMed] [Google Scholar]
- Roos J, Kelly RB. Dap160, a neural-specific EH- and SH3-domain containing protein that interacts with Drosophila dynamin. J Biol Chem. 1998;273:19108–19119. doi: 10.1074/jbc.273.30.19108. [DOI] [PubMed] [Google Scholar]
- Salem N, Faundez V, Horng J-T, Kelly RB. A v-SNARE participates in synaptic vesicle formation mediated by the AP3 adaptor complex. Nat Neurosci. 1998;1:551–556. doi: 10.1038/2787. [DOI] [PubMed] [Google Scholar]
- Sandvig K, van Deurs B. Endocytosis without clathrin (a minireview) Cell Biol Int Rep. 1991;15:3–8. doi: 10.1016/0309-1651(91)90077-v. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Hannah MJ, Huttner WB. Synaptic-like microvesicles of neuroendocrine cells originate from a novel compartment that is continuous with the plasma membrane and devoid of transferrin receptor. J Cell Biol. 1997;137:445–458. doi: 10.1083/jcb.137.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih W, Gallusser A, Kirchhausen T. A clathrin binding site in the hinge of hte β2 chain of mammlaan AP-2 complexes. J Biol Chem. 1995;270:31083–31090. doi: 10.1074/jbc.270.52.31083. [DOI] [PubMed] [Google Scholar]
- Simpson F, Bright NA, West MA, Newman LS, Darnell RB, Robinson MS. A novel adaptor-related protein complex. J Cell Biol. 1996;133:749–760. doi: 10.1083/jcb.133.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson F, Peden AA, Christopoulou L, Robinson MS. Characterization of the adaptor-related protein complex, AP-3. J Cell Biol. 1997;137:835–845. doi: 10.1083/jcb.137.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei K, McPherson PS, Schmid SL, De Camilli P. Tubular membrane invaginations coated by dynamin rings are induced by GTP-γ-S in nerve terminals. Nature. 1995;374:186–190. doi: 10.1038/374186a0. [DOI] [PubMed] [Google Scholar]
- Takei K, Mundigl O, Daniell L, De Camilli P. The synaptic vesicle cycle: A single vesicle budding step involving clathrin and dynamin. J Cell Biol. 1996;133:1237–1250. doi: 10.1083/jcb.133.6.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee RB, Okamoto PM. The regulation of endocytosis: identifying dynamin's binding partners. Trends Cell Biol. 1995;5:43–47. doi: 10.1016/s0962-8924(00)88937-0. [DOI] [PubMed] [Google Scholar]
- van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol. 1993;122:553–563. doi: 10.1083/jcb.122.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vowels JJ, Payne GS. A dileucine-like sorting signal directs transport into an AP3-dependent, clathrin-independent pathway to the yeast vacuole. EMBO (Eur Mol Biol Organ) J. 1998;17:2482–2493. doi: 10.1093/emboj/17.9.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei ML, Bonzelius F, Scully RM, Kelly RB, Herman GA. GLUT4 and transferrin receptor are differentially sorted along the endocytic pathway in CHO cells. J Cell Biol. 1998;140:565–575. doi: 10.1083/jcb.140.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]