

Abstract
Heat shock protein 90 (Hsp90), an abundant molecular chaperone in the eukaryotic cytosol, is involved in the folding of a set of cell regulatory proteins and in the re-folding of stress-denatured polypeptides. The basic mechanism of action of Hsp90 is not yet understood. In particular, it has been debated whether Hsp90 function is ATP dependent. A recent crystal structure of the NH2-terminal domain of yeast Hsp90 established the presence of a conserved nucleotide binding site that is identical with the binding site of geldanamycin, a specific inhibitor of Hsp90. The functional significance of nucleotide binding by Hsp90 has remained unclear. Here we present evidence for a slow but clearly detectable ATPase activity in purified Hsp90. Based on a new crystal structure of the NH2-terminal domain of human Hsp90 with bound ADP-Mg and on the structural homology of this domain with the ATPase domain of Escherichia coli DNA gyrase, the residues of Hsp90 critical in ATP binding (D93) and ATP hydrolysis (E47) were identified. The corresponding mutations were made in the yeast Hsp90 homologue, Hsp82, and tested for their ability to functionally replace wild-type Hsp82. Our results show that both ATP binding and hydrolysis are required for Hsp82 function in vivo. The mutant Hsp90 proteins tested are defective in the binding and ATP hydrolysis–dependent cycling of the co-chaperone p23, which is thought to regulate the binding and release of substrate polypeptide from Hsp90. Remarkably, the complete Hsp90 protein is required for ATPase activity and for the interaction with p23, suggesting an intricate allosteric communication between the domains of the Hsp90 dimer. Our results establish Hsp90 as an ATP-dependent chaperone.
Keywords: heat shock protein 90, molecular chaperone, p23, nucleotide binding, geldanamycin
Many newly synthesized polypeptides depend on the assistance of molecular chaperones for their efficient folding. The mechanisms in assisted protein folding of two main classes of molecular chaperones, the Hsp70s and the chaperonins, have been well characterized (Gething and Sambrook, 1992; Craig, 1993; Hartl, 1996; Johnson and Craig, 1997). In contrast, much less is known about the biochemical mechanism of the heat shock protein 90 (Hsp90)1 family, another class of abundant chaperones and constitutively expressed stress proteins whose members have been highly conserved from prokaryotes to eukaryotes (Lindquist and Craig, 1988; Buchner, 1996). The first partial crystal structures of Hsp90 have been published only recently (Prodromou et al., 1997a ,b; Stebbins et al., 1997).
The cellular function of Hsp90 extends beyond the general chaperone properties of this protein. Hsp90 is specifically involved in the folding and conformational regulation of a number of medically relevant signal transduction molecules, including the nuclear receptors for steroid hormones and several protooncogenic kinases (Smith, 1993; Nathan and Lindquist, 1995; Frydman and Höhfeld, 1997; Nathan et al., 1997; Pratt and Toft, 1997). Inhibition of Hsp90 function by the ansamycin antibiotics (geldanamycin [GA], herbimycin A, macbecin) causes the misfolding and degradation of these substrates (Whitesell et al., 1994; Smith et al., 1995; Schneider et al., 1996; Dittmar and Pratt, 1997). In addition, Hsp90 participates in the renaturation of some proteins that have unfolded under conditions of cellular stress (Schneider et al., 1996; Nathan et al., 1997). Although purified Hsp90 can prevent the aggregation of unfolded proteins and in a minimal system in vitro cooperates with Hsp70/Hsp40 in the ATP-dependent refolding of model proteins (Wiech et al., 1992; Jakob et al., 1995; Freeman and Morimoto, 1996; Schumacher et al., 1996; Young et al., 1997; Scheibel et al., 1998), it is likely that Hsp90 fulfills most of its cellular roles in close cooperation with a host of cofactors and cochaperones, including Hop (p60/Sti1p), the immunophilins FKBP52 and FKBP51/54, cyclophilin 40 (Cyp40), cdc37 (p50), and p23, as well as Hsp70 and its cofactors Hsp40, Hip (p48), and Bag1 (Rap46) (Nair et al., 1996; Frydman and Höhfeld, 1997; Pratt and Toft, 1997; Höhfeld, 1998). Models for the ordered, yet dynamic interaction of Hsp90 with these cofactors during the folding of steroid receptors have been proposed on the basis of experiments in crude cytosol extracts and partially purified systems (for reviews see Smith, 1993; Pratt and Toft, 1997). It is generally thought that an unfolded polypeptide substrate associates first with the Hsp70 system (“early complex”) (Smith, 1993) and is then targeted to Hsp90 via Hop, a tetratricopeptide repeat (TPR) protein that interacts with both Hsp70 and Hsp90 (Chen et al., 1996; Dittmar et al., 1996; Dittmar and Pratt, 1997; Johnson et al., 1998). This interaction gives rise to the formation of an “intermediate complex,” containing Hip, Hop, and Hsp70 in addition to Hsp90. Further remodelling results in the “mature complex,” consisting of Hsp90, p23, an immunophilin, or Cyp40. The steroid receptor in this complex is largely folded and competent to bind hormone with high affinity. The acquisition of this high affinity conformation is dependent on the interaction of Hsp90 with the acidic protein p23. Geldanamycin (GA) prevents p23 binding to Hsp90 and inhibits the proper folding of steroid receptor (Johnson et al., 1994; Johnson and Toft, 1995; Smith et al., 1995; Grenert et al., 1997; Sullivan et al., 1997).
The mature Hsp90 multi-chaperone complex dissociates with a t 1/2 of ∼5 min and unliganded steroid receptor re-enters the reaction cycle at the level of the early Hsp70 complex (Smith, 1998). These dynamic interactions are ATP dependent and thought to be driven by the Hsp70 ATPase. Whether Hsp90 itself binds and hydrolyzes ATP has been one of the most controversial issues in the chaperone literature (Csermely and Kahn, 1991; Wiech et al., 1992; Csermely et al., 1993; Johnson and Toft, 1994, 1995; Jakob et al., 1996; Grenert et al., 1997; Scheibel et al., 1997, 1998). The recent crystal structures of the NH2-terminal 25-kD domain of human and yeast Hsp90 revealed that this domain binds not only the ansamycin antibiotic GA but also, in the same site, adenine nucleotide, thus suggesting that GA acts by inhibiting an ATP-dependent function of Hsp90 (Prodromou et al., 1997a ; Stebbins et al., 1997). Interestingly, the NH2-terminal domain functions also in binding unfolded polypeptide (Young et al., 1997; Scheibel et al., 1998), either independently or in conjunction with the COOH-terminal 12-kD domain, which mediates Hsp90 dimerization and the binding of the TPR proteins Hop, FKBP52, FKBP51/54, and Cyp40 (Young et al., 1998). The NH2- and COOH-terminal domains are connected by a ∼35-kD segment of unknown function.
Despite the structural evidence for nucleotide binding to yeast Hsp90, the putative ATPase function of Hsp90 has remained elusive. Here we present the crystal structure of the NH2-terminal domain of human Hsp90 with bound ADP-Mg and demonstrate that Hsp90 indeed has ATP hydrolytic activity. Based on the crystal structure, Hsp90 mutants were made that are defective in ATP binding and ATP hydrolysis. The mutant proteins are unable to cycle the cofactor p23 and fail to replace wild-type Hsp90 in yeast in vivo. Thus, Hsp90 is unequivocally identified as an ATP-dependent molecular chaperone.
Materials and Methods
Crystallization and X-Ray Analysis
The NH2-terminal domain of human Hsp90 (residues 9–236) was expressed and purified as described (Stebbins et al., 1997). Crystals were grown in the presence of 10 mM adenosine-5′-O-(3-thiotriphosphate) (ATPγS) and 0.2 M magnesium chloride, from a buffer of 0.1 M Tris-HCl, pH 8.5, and 30% PEG4000. Data collection, structure determination, and refinement followed the procedures described for the analysis of the same domain with bound geldanamycin (Stebbins et al., 1997).
Cloning Procedures and Site-directed Mutagenesis
Human Hsp90α cDNA in pUC19 (Aligue et al., 1994) and yeast Hsp82 in pTGPD/P82 were used as templates for PCR amplification. The coding region for the yeast homologue of p23 (YKL117W) (Bohen, 1998; Fang et al., 1998) was amplified by PCR from wild-type yeast genomic DNA. In the case of Hsp82, PCR products were subcloned into derivatives of the pET23a Vector (Novagen, Inc., Madison, WI), to generate the recombinant protein fragments containing either a COOH-terminal His6 sequence followed by an EEF immunotag or an NH2-terminal T7 tag. The cDNAs encoding human Hsp90 and Hsp90 mutants, as well as human p23 were cloned into the pAS2-1 vector for the analysis of the Hsp90-p23 interaction in two-hybrid assays (Bartel et al., 1993).
Point mutations in human Hsp90 (E47A, E47D, and D93N) and yeast Hsp82 (E33A, E33D, and D79N) were generated by PCR amplification with mismatch oligonucleotides resulting in site-directed exchanges of codons (Ausubel et al., 1987). The desired exchanges were confirmed by DNA sequencing. Amplified HSP82 cDNA fragments were subcloned into the vector pTGPD/P82 for expression in yeast or into pET23a derivatives for expression in Escherichia coli.
Expression of Hsp82 Mutants in Yeast
The yeast strain ΔPCLDa/α (Nathan and Lindquist, 1995), used throughout these experiments, is homozygous for deletion mutations in both HSP90 genes (HSP82 and HSC82). It is rescued from lethality by the URA3-containing plasmid pKAT6, which expresses wild-type Hsc82. This strain was transformed with wild-type and point-mutated Hsp82 in the expression vector pTGPD/P82 containing the TRP1-marker (Nathan and Lindquist, 1995). Cotransformants were selected on SD/−Trp/−Ura plates at 30°C.
To determine the expression levels of Hsp82 constructs, cells were grown in SD/−Trp/−Ura medium to mid-log phase. Extracts were prepared by agitation of cells with glass beads in ice-cold buffer A (50 mM potassium phosphate, pH 8.0, 500 mM KCl, 15 mM imidazole, 5 mM 2-mercaptoethanol) supplemented with 1 mM PMSF and protease inhibitor cocktail (Boehringer Mannheim GmbH, Mannheim, Germany). After centrifugation, Hsp82 proteins were concentrated by incubation of the supernatants with Ni-NTA. The resin was washed with buffer A and subsequently with buffer B (same as buffer A, but pH 6.0). Finally, the recombinant proteins were eluted with 500 mM imidazole in buffer B and analyzed by SDS-PAGE and immunoblotting with the anti-EEF–specific antibody YL1/2 (Wehland et al., 1984).
To examine the ability of Hsp82 point mutants to support cell viability, wild-type Hsc82 on the URA3-containing pKAT6 plasmid was replaced by a HSP82 mutant gene on the TRP1-containing expression vector pTGPD/P82 (Sikorski and Boeke, 1991). Cotransformants from the SD/ −Trp/−Ura plates were replica plated on 5-FOA (5-fluororotic acid) plates (Sikorski and Boeke, 1991) at 25°C to select for cells that had lost the original pKAT6 plasmid. After a second round of 5-FOA selection, loss of the URA3-contaning plasmid pKAT6 was verified by the inability of these cells to grow on SD/−Ura plates.
Interaction of Hsp90 and p23 In Vivo
The cDNAs encoding human full-length Hsp90 and mutant derivatives were inserted in frame into the pAS2-1 vector (CLONTECH Laboratories, Inc., Palo Alto, CA) containing TRP1 as a marker. The different constructs were used as a “bait” in a two-hybrid assay and were cotransformed into Saccharomyces cerevisiae (strain Y190) together with human p23 in the pACT2 vector (CLONTECH Laboratories, Inc.) carrying a LEU2 marker. Cotransformants were plated on SD/−Trp/−Leu plates. The interaction of the bait construct, or the empty pAS2-1 vector as a control, with the p23 protein was judged by the ability of cotransformants to grow on SD/−Trp/−Leu/−His plus 25 mM 3-AT (3-amino-1,2,4-triazole) selection plates.
Purification of Recombinant Proteins from E. coli
Expression constructs were transformed into E. coli BL21(DE3) LysS cells (Studier et al., 1990). Cells were grown at 18°C in LB medium supplemented with 100 mg/l ampicillin and 34 mg/l chloramphenicol to OD600 = 1 and protein expression induced for 5 h with 0.25 mM IPTG. After harvesting, cell pellets were resuspended and sonicated in buffer A and Ni-NTA chromatography was performed as described. Proteins were purified further by ion-exchange chromatography on MonoQ and by gel filtration on Superose 12 (Pharmacia Biotech, Inc., Piscataway, NJ).
Yeast p23 carrying an NH2-terminal T7 immunotag but lacking a His6 tag was also expressed in E. coli and ammonium sulphate precipitated from the soluble fraction. After dialysis against buffer Y (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 50 mM KCl, 10 mM MgCl2), the protein was purified further by ion-exchange chromatography on Q-Sepharose Fast Flow (Pharmacia Biotech, Inc.) in the same buffer and by gel filtration on Superose 12 in buffer Y.
ATP Binding Assay
Ni-NTA resin was preblocked with buffer Y containing 5 mM ATP and 2 mM MgCl2. After washing with buffer Y, purified NH2-terminal domains of wild-type Hsp82 and the E33A and D79N mutants (35 μM each) were bound to the matrix via their His6 tags by incubation for 15 min at 25°C in buffer Y in the presence of 35 μM [α32P]ATP (4 μCi/mM, Amersham Life Science, Inc., Arlington Heights, IL). The beads were washed twice with 0.5 ml of buffer Y, protein was eluted with 500 mM imidazole in buffer Y, and then bound radioactivity was detected by liquid scintillation counting. Specifity of nucleotide binding was demonstrated by competition with GA.
ATPase Assay
Recombinant full-length Hsp82 wild-type and mutant proteins (10 μM) were incubated at 30°C in 40 mM Hepes-KOH, pH 7.4, 2 mM MgCl2 with 0.01–0.5 mM [α32P]ATP (4 μCi/mM, Amersham Life Science, Inc.) for ≤120 min in a final volume of 20 μl. Reactions were stopped by the addition of 5 mM EDTA and freezing in liquid nitrogen. Separation of ADP from ATP was achieved by thin layer chromatography on PEI–cellulose sheets (Merck, Darmstadt, Germany) in 0.5 M LiCl and 0.5 M formic acid. ATPase activity was monitored by quantitation of [α32P]ADP on a Fuji PhosphorImager (model FLA-2000) using the MacBAS software package. For specific inhibition of the ATPase activity, GA was added to the reaction at a final concentration of 180 μM in 1% DMSO. Conrol reactions received DMSO alone.
Interaction of Hsp82 and p23 In Vitro
Full-length wild-type or mutant Hsp82 proteins (200 μg) were bound to Ni-NTA resin in buffer Y supplemented with 1% (wt/vol) BSA and 25 mM imidazole to minimize unspecific protein interactions. Yeast p23 protein (100 μg) with an NH2-terminal T7 tag was added and the mixture was agitated at 30°C for 45 min in the prescence or absence of nucleotide and GA. After three washes with buffer Y containing 25 mM imidazole, Hsp82 protein was eluted from the resin with 500 mM imidazole in the same buffer, followed by SDS-PAGE, and then immunoblotting with an antibody against the T7 tag of recombinant yeast p23 (Novagen, Inc.).
Miscellaneous Procedures
SDS-PAGE was as described (Laemmli et al., 1970). Immunolotting was performed following the protocol of Towbin et al. (1979). Protein concentrations were determined by the BioRad protein assay (BIO RAD Laboratories GmbH, Munchen, Germany).
Results
Crystal Structure of the NH2-terminal Domain of Human Hsp90 Complexed with ADP-Mg
We have previously reported the structure of the NH2-terminal domain of Hsp90 with bound GA (Stebbins et al., 1997). Here we crystallized the NH2-terminal domain of human Hsp90 (residues 9–236) in the presence of ATPγS. The crystals formed in spacegroup P21 with a = 53.2 Å, b = 44.1 Å, c = 54.0 Å, d = 115.7°. Difference fourier maps showed clear density for ADP and one magnesium ion consistent with full occupancy, but had no electron density for a γ-phosphate, either because the γ-phosphate was disordered or had been lost due to hydrolysis during crystallization. The structure was refined to 1.5 Å resolution with R cryst/R free = 17.8/23.5%.
As was observed with the structure of the NH2-terminal domain of yeast Hsp90 (Prodromou et al., 1997a ), ADP-Mg binds to a site in human Hsp90 where GA also binds (Stebbins et al., 1997). The contacts between the nucleotide and Hsp90 in this complex are very similar to those observed in the yeast complex (Prodromou et al., 1997a ), and involve both direct and water-mediated hydrogen bonds (Fig. 1). The N6 group of the adenine purine forms direct hydrogen bonds with the D93 side chain, and water-mediated hydrogen bonds with the S52 side chain and L48 backbone carbonyl groups. As in the case of the structure with bound GA, the interaction with D93 (D79 in yeast Hsp82) is probably most critical for stable nucleotide binding. The N7 group of the purine interacts via water-mediated hydrogen bonds with the N51 side chain, and the N1 group of the purine with the G97 backbone and D93 and T81 side chain groups. Additional ADP-Mg contacts include a hydrogen bond between the O2′ group and the N106 side chain, a set of hydrogen bonds between the α-phosphate group and the F138, G137 backbone amide and N51 side chain groups. The Mg ion is octahedrally coordinated by the α- and β-phosphate groups of the ADP, the N51 side chain of Hsp90, and three water molecules.
Figure 1.

Structure of the NH2-terminal domain of Hsp90 with bound ADP-Mg. View into the nucleotide-binding pocket. The residues D93 and E47, critical for nucleotide binding and hydrolysis, respectively, are colored in yellow. Image was prepared with the programs MOLSCRIPT (Kraulis, 1991) and RASTER3D (Merrit and Murphy, 1994).
The conformation of the binding site in the ADP-Mg complex is essentially identical to the “open” conformation of this site observed in the GA–Hsp90 complex (Stebbins et al., 1997), except for the region spanning residues 106–124, which differs slightly (0.8–1.2 Å displacements in Cα positions). As was the case with GA binding, the closed conformation, which has backbone displacements of as much as 10 Å, constricting the opening of the binding pocket from 12 Å to 8 Å (Stebbins et al., 1997), would not be compatible with ADP-Mg binding. This raises the possibility that the conformational change is part of a regulatory mechanism that may involve nucleotide binding and turnover.
Interestingly, there is a striking similarity in overall tertiary structure between the NH2-terminal domain of Hsp90 and the NH2-terminal ATP-binding domain of the bacterial type II topoisomerase, DNA gyrase B of E. coli (Wigley et al., 1991). This similarity extends to the conformation of the bound nucleotide and to most of the critical interactions between the nucleotide and the binding pocket. Significantly, a conserved glutamate residue in the gyrase protein, E42, which corresponds to E47 in the NH2-terminal domain of human Hsp90 (E33 in the yeast protein), was identified as the catalytic center for ATP hydrolysis in the gyrase (Jackson and Maxwell, 1993). As shown in Fig. 1, E47 is indeed positioned such that it could play a catalytic role in cleaving the γ-phosphate bond of Hsp90-bound ATP, suggesting that Hsp90 binds and hydrolyzes ATP in a manner similar to DNA gyrase.
Mutations in the ATP Binding Site of Hsp90 Are Incompatible with Cell Growth
To test the possible functional significance of the conserved D93 and E47 residues of human Hsp90 for ATP binding and hydrolysis, respectively, the effects of site-directed mutations in the corresponding residues D79 and E33 of yeast Hsp82 (83% similarity and 70% identity with the human sequence) were analyzed in vivo. Yeast contains two HSP90 genes, HSP82 and HSC82, whose combined deletion is lethal (Borkovich et al., 1989). We therefore used a yeast strain (ΔPCLDa/α) for this analysis that is deleted in both chromosomal HSP90 genes but rescued from lethality by constitutive expression of plasmid-encoded wild-type Hsc82 protein (Nathan and Lindquist, 1995). We first addressed the question whether mutations of D79 and E33 affect the expression and gross stability of Hsp82. The ΔPCLDa/α strain was cotransformed with an additional HSP82 gene expressing either wild-type or mutant proteins (from the same promoter) that could be distinguished from the preexistent Hsc82 by the presence of COOH-terminal His6 and EEF tags (Wehland et al., 1984). Yeast cells expressing either untagged wild-type Hsp82, wild-type Hsp82-His6EEF, or Hsp82-His6EEF proteins containing the mutations E33A, E33D, or D79N were grown to mid-log phase. Cell lysates were prepared, adjusted to equal protein concentrations, and then Hsp82 proteins quantitatively adsorbed to Ni-NTA beads via their His6 tags (see Materials and Methods). All Hsp82-His6EEF proteins were stably expressed as full-length proteins to very similar levels (Fig. 2 A), as judged by Coomassie staining and immunoblotting with anti-EEF antibody. The additional bands at ∼50 and ∼60 kD in the Western blot represent degradation products of the full-length protein that are generated during the isolation procedure, as they are not detected on Western blots of cells that had been solubilized in hot SDS immediately upon harvesting (not shown). Coexpression of mutant proteins at a level equivalent to that of wild-type Hsp82 was without effect on cell growth.
Figure 2.
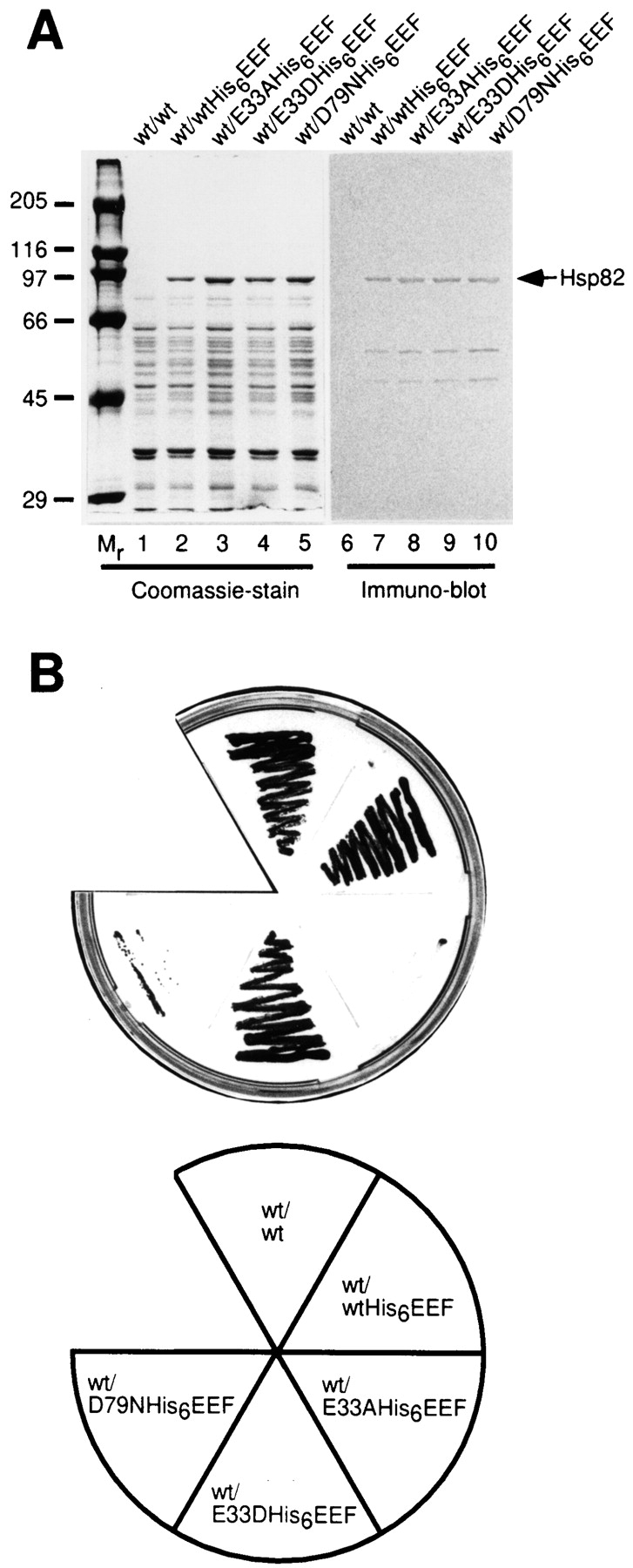
Mutations in the nucleotide-binding site of Hsp90 abolish the function of Hsp90 in vivo. (A) Yeast strain ΔPCLDa/α that expresses wild-type Hsc82 from the plasmid pKAT6 (containing the URA3-marker) was cotransformed with wild-type HSP82 (lane 1), HSP82His6EEF (lane 2), HSP82(E33A)His6EEF (lane 3), HSP82(E33D)His6EEF (lane 4) and HSP82(D79N) His6EEF (lane 5). After growth to mid-log phase in liquid SD/ −Trp/−Ura, cell lysates were prepared, adjusted to equal protein concentrations and His6EEF tagged proteins quantitatively absorbed with Ni-NTA beads followed by SDS-PAGE and Coomassie staining (lanes 1–5). Lanes 6–10 correspond to lanes 1–5 and show an immunoblot with the EEF-specific antibody. Note that only the band at ∼90 kD (and two degradation products at ∼60 and ∼50 kD, generated during isolation) are specifically recognized. (B) The cotransformants described in A were restreaked on 5-FOA plates at 25°C to select for cells that had lost the original wild-type plasmid and were rescued from lethality by a functional hsp82 protein. Only wild-type Hsp82, Hsp82His6EEF, and the E33D mutant were able to support growth.
To establish whether the mutant forms of Hsp82 were able to support cell viability in the absence of wild-type Hsc82, the plasmid shuffling technique (Sikorski and Boeke, 1991) was used to replace the wild-type HSC82 gene in strain ΔPCLDa/α with HSP82-His6EEF, mutant HSP82-His6EEF, or non-tagged wild-type HSP82 as a control. Cotransformed colonies were selected for growth on 5-FOA plates, indicating the functional replacement of wild-type HSC82. Surprisingly, only colonies containing either the wild-type HSP82 gene itself, HSP82-His6EEF, or HSP82(E33D)-His6EEF were viable (Fig. 2 B). In contrast the E33A and the D79N mutants were unable to rescue the ΔPCLDa/α cells from lethality when present as the only source of Hsp82. These results are consistent with the view that ATP binding and hydrolysis are critical functional properties of Hsp90 required in vivo.
The rescued cells expressing wild-type Hsp82 and Hsp82- His6EEF grew with almost identical rates at 30°C (doubling time d = 116 ± 2 min and 112 ± 5 min, respectively), indicating that the His6EEF tag does not compromise the function of Hsp82 in vivo. In contrast, the cells expressing Hsp82(E33D)-His6EEF grew more slowly (d = 132 ± 1 min) but showed no temperature-sensitive phenotype.
D79 Is Required for ATP Binding and E47 for ATP Hydrolysis by Hsp90
To determine the molecular basis for the loss of in vivo function of the mutant Hsp82 proteins analyzed, we expressed the full-length proteins and the respective NH2-terminal domains in soluble form in E. coli and purified them to homogeneity. Based on the available structural information, it seemed unlikely that the residue E33 contributes significantly to ATP or GA binding. Indeed, the NH2-terminal domain of Hsp82(E33A) bound ATP and GA as efficiently as the wild-type protein. In contrast, the NH2-terminal domain of Hsp82(D79N) bound ATP much less efficiently than wild-type protein (Fig. 3 A), confirming the prediction from the structural model.
Figure 3.
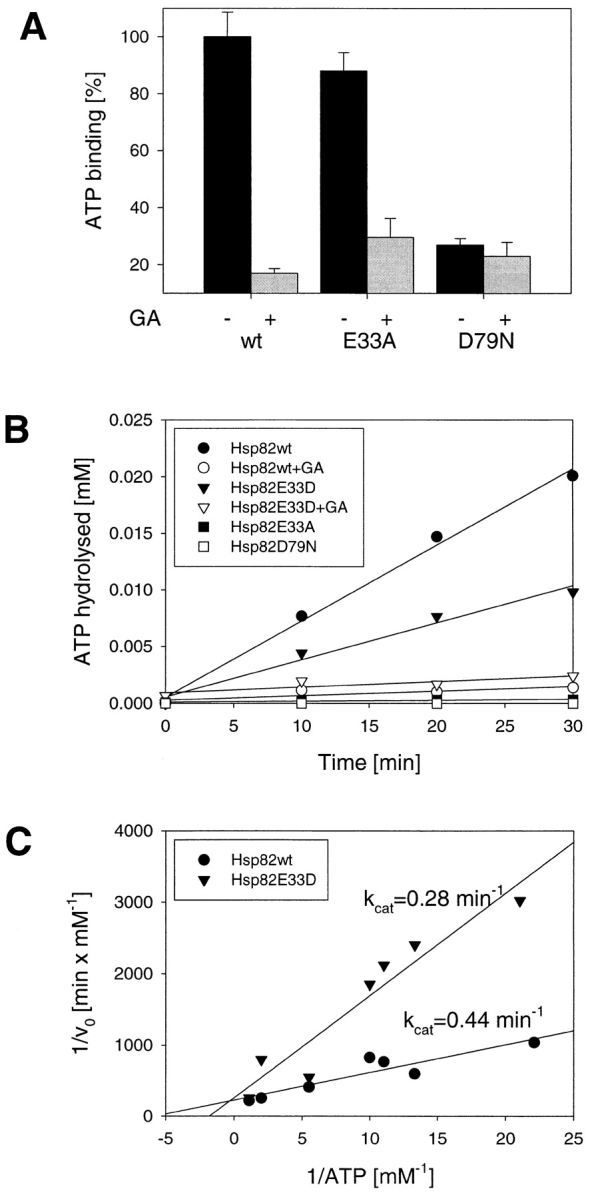
Mutations in the nucleotide binding site of Hsp90 affect ATP binding and hydrolysis differentially. (A) Binding of ATP to the NH2-domains of wild-type and mutant Hsp82 in the presence and absence of GA (see Materials and Methods). Averages of three independent experiments are shown with error bars. (B) A typical plot of ATP hydrolysis versus time at a concentration of 0.05 mM ATP and 10 μM Hsp82. ATPase activity was measured with [α32P]ATP at 30°C as described in Materials and Methods. The ATPase activity of Hsp82 (closed circles) and the E33D mutant (closed triangles) is specifically inhibited by GA (open circles and open triangles, respectively). In contrast, the E33A (closed squares) and D79N mutants (open squares) are devoid of GA-inhibitable ATPase activity. (C) Double reciprocal plot of the rate of ATP hydrolysis versus ATP concentration. K m values for wild-type Hsp82 (closed circles) and for the E33D mutant (closed triangles) were determined as 172 μM and 520 μM, respectively.
Having analyzed the binding of ATP, experiments were performed to establish whether Hsp90 has ATP hydrolytic activity and to test the possible mechanistic significance of residue E33 in this reaction. Any ATPase activity measured with Hsp90 has previously been assigned to background contamination (Scheibel et al., 1997). To avoid such artifacts, we used only highly purified, recombinant Hsp90 proteins. Furthermore, we reasoned that an authentic Hsp90 ATPase activity should be inhibited by GA. ATPase activities were measured with a highly sensitive assay monitoring the production of [α32P]ADP from [α32P]ATP by thin layer chromatography. ATP hydrolysis versus time for the various proteins is plotted in Fig. 3 B, and Fig. 3 C shows the double-reciprocal plot of the data. Wild-type Hsp82 and Hsp82(E33D) hydrolyzed ATP with similar k cat values of 0.44 min−1 and 0.28 min−1, respectively, which is very similar to the ATPase activity of unstimulated mammalian Hsp70 proteins (O'Brien and McKay, 1993; Wei and Hendershot, 1995). However, the K m value of Hsp82(E33D) was about threefold higher than that of wild-type Hsp82 (520 μM vs. 172 μM), indicating a significantly reduced affinity for nucleotide. Addition of GA at 180 μM inhibited the ATPase activity almost completely. A different result was obtained for the E33A and D79N mutants. The D79N mutant was devoid of ATPase activity as expected from its inability to bind ATP specifically. Significantly, the E33A exchange, although preserving the ability of Hsp90 to bind ATP (Fig. 3 A), abolished the capacity of Hsp90 to hydrolyze ATP. Thus, E33 in the yeast protein, corresponding to E47 in human Hsp90, is identified as the catalytic residue of the Hsp90 ATPase, suggesting a mechanism of ATP hydrolysis by Hsp90 similar to that of DNA gyrase (Jackson and Maxwell, 1993).
The Complete Hsp90 Protein Is Required for ATPase Activity
Next, we investigated whether the stably folded NH2-terminal domain of Hsp82 (Stebbins et al., 1997; Prodromou et al., 1997a ) is sufficient for the ATPase activity of Hsp82 or whether additional parts of the molecule are required for ATP hydrolysis. Surprisingly, although the NH2-terminal domain binds ATP (Fig. 3 A), it did not exhibit detectable catalytic activity in the ATPase assay (Fig. 4). Similarly, no ATPase activity was detected with a variant of Hsp82, Hsp82ΔC (Fig. 4), that lacks the COOH-terminal dimerization domain (residues 600–709; Nemoto et al., 1995; Wearsch and Nicchitta, 1996), but can be considered to be native as it was fully soluble after expression in E. coli and active in prevention of rhodanese aggregation (Young et al., 1997). Similarly, the NH2-terminal domain of Hsp82 was properly folded, as it was not only capable of binding ATP and GA (see Fig. 3 A; Prodromou et al., 1997a ; Stebbins et al., 1997) but also preserved the chaperone function in preventing protein aggregation in vitro (Young et al., 1997; Scheibel et al., 1998; data not shown). Thus, the complete Hsp90 protein, presumably as the dimer, is required for ATP hydrolytic activity. Interestingly, the chaperone function of the isolated NH2-terminal domain did not exhibit a detectable dependence on specific nucleotide binding (data not shown), suggesting that ATP hydrolysis may be required for substrate release or that this function depends on the cooperation of Hsp90 with one or several of its cofactors.
Figure 4.

The complete Hsp90 protein is required for ATPase activity. ATP hydrolysis versus time at 0.10 mM ATP and 10 μM Hsp82 proteins at 30°C is shown for full-length Hsp82 (FL82, closed circles), the NH2 domain of Hsp82 (N82, closed triangles) and for ΔC82, lacking the COOH-terminal residues 600–709 (closed squares).
Effect of ATPase Mutations on the Interaction of Hsp90 with p23
Recent studies implicated the small acidic protein p23 as an important cofactor of Hsp90 involved in the maturation of steroid receptor molecules (Dittmar et al., 1997). Interestingly, Toft and colleagues reported that p23 interacts with Hsp90 in an ATP-dependent manner (Sullivan et al., 1997). Initially, it was difficult to reconcile this finding with the view that Hsp90 neither binds nor hydrolyzes ATP (Jakob et al., 1996; Scheibel et al., 1997). The recent structural evidence for ATP binding by Hsp82 (Prodromou et al., 1997a ) together with our demonstration that Hsp90 function is dependent on ATP binding and hydrolysis in vivo provided the basis to resolve this issue. Since point mutations mapping within the first 200 residues of Hsp90 abrogate the interaction between p23 and Hsp90 (Grenert et al., 1997), we performed a two-hybrid assay to test whether the NH2-terminal domain of Hsp90 is sufficient for p23 binding. A set of human Hsp90 constructs (Fig. 5 A) in the pAS2-1 vector served as the bait and was cotransformed with human p23 into S. cerevisiae strain Y190. Surprisingly, neither of the Hsp90 deletion fragments was sufficient to interact with p23 (Fig. 5 B), although the fragments were expressed to similar levels as the full-length Hsp90 protein (Young et al., 1998). Only the full-length Hsp90 polypeptide was able to interact with p23.
Figure 5.


Requirement of full-length Hsp90 with an intact ATP-binding site for the interaction with p23 in vivo. (A) Schematic representation of the domain structure of human Hsp90 established by limited proteolysis (Stebbins et al., 1997) and of the fragments used in the two-hybrid assay. (B) Different fragments of Hsp90 in the pAS2-1 bait vector were cotransformed in S. cerevisiae (strain Y190) with the cDNA encoding human p23 in the pACT2 target vector (see Materials and Methods). To monitor protein–protein interactions, cotransformants were restreaked on SD/−Trp/−Leu/−His plates containing 25 mM 3-AT and incubated for 5 d at 30°C. Cell growth was only observed with full-length Hsp90 protein but not for any of its subfragments or for the vector control without insert. (C) Full-length wild-type Hsp90 and the mutants E47A, E47D, and D93N were analyzed for their ability to interact with p23 by two-hybrid assay as above. Cell growth was observed for wild-type Hsp90, the E47A and E47D mutants, but not for the D93N mutant.
To test whether ATP binding to Hsp90 is required for the interaction with p23, two-hybrid assays were performed with the respective mutant forms of Hsp90 (Fig. 5 C). Cells containing wild-type Hsp90, Hsp90(E47D), and Hsp90(E47A) grew in a manner dependent on the interaction between Hsp90 and p23, suggesting that ATP hydrolysis by Hsp90 is not essential for the interaction with p23 measured in vivo. In contrast, no growth was observed with Hsp90(D93N) in the two-hybrid assay, indicating that Hsp90 that is defective in nucleotide binding is no more able to associate with p23. To establish this conclusion more rigorously, in vitro binding experiments with purified Hsp82 proteins and yeast p23 (Bohen, 1998; Fang et al., 1998) were performed. Full-length wild-type and mutant Hsp82 was purified from E. coli as His6EEF-tagged proteins. Yeast p23 containing an NH2-terminally located T7 immunotag was purified upon expression in E. coli. Wild-type Hsp82 or Hsp82 proteins carrying the mutations E33A, E33D or D79N were bound to Ni-NTA resin and subsequently incubated with p23 (see Materials and Methods). Binding of p23 to the immobilized Hsp82 was analyzed by SDS-PAGE and immunoblotting (Fig. 6). Significant binding to wild-type Hsp82 and Hsp82(E33D) was only observed in the presence of the nonhydrolyzable nucleotide ATPγS and was inhibited by GA. In contrast, Hsp82(E33A) was able to form a complex with p23 in the presence of hydrolyzable ATP, indicating that p23 interacts with the ATP form of Hsp90. No complex formation was observed with the D79N mutation of Hsp82, consistent with the lack of detectable nucleotide binding by this mutant in vitro. These results allow the following conclusions: p23 recognizes the ATP-bound form of Hsp90, but not the ADP-bound form or the nucleotide-free protein. As GA inhibits complex formation in the presence of ATP, the ansamycin antibiotic mimics the ADP state of Hsp90, not the ATP state, at least as far as the interaction with p23 is concerned (see Discussion). In the presence of ATP, mutant Hsp82 carrying the catalytic site mutation E33A is arrested in the p23-bound state. In contrast, wild-type Hsp82 and Hsp82(E33D) are able to cycle between the p23-bound ATP state and the p23-free ADP and non– nucleotide-bound states. Given the ATP dependence of p23 binding it might have been expected that p23 regulates the ATPase of Hsp90. Surprisingly, however, we failed to detect any effect of p23 on the ATPase activity of purified Hsp90 (data not shown).
Figure 6.

p23 binds the ATP-bound state of purified Hsp90 in vitro, not the ADP-bound and nucleotide-free forms. Wild-type and mutant Hsp82 proteins (200 μg each) were bound to Ni-NTA agarose via their His6 tags and incubated for 45 min at 30°C with yeast p23 (100 μg) and different combinations of nucleotides and GA as indicated (lanes 2–22). In lane 1 (control) Ni-NTA beads without bound Hsp82 were used. Protein was eluted from the beads with imidazole and analyzed by 15% SDS-PAGE and immunoblotting for the T7 immunotag of p23 (see Materials and Methods).
Discussion
The question whether Hsp90 function is ATP dependent has been the subject of a long debate. Several groups reported ATP-dependent conformational changes in Hsp90 and ATP-regulated co-factor interactions (Csermely and Kahn, 1991; Csermely et al., 1993; Johnson and Toft, 1994, 1995; Grenert et al., 1997; Sullivan et al., 1997). On the other hand, detailed studies in vitro indicated that the chaperone function of purified Hsp90 is ATP independent (Wiech et al., 1992; Jakob et al., 1996; Wearsch and Nicchitta, 1996; Scheibel et al., 1997), suggesting that the ATP-regulated aspects of Hsp90 function in protein folding are due to the close cooperation between Hsp90 and Hsp70/Hsc70 (Schneider et al., 1996). This view has only changed recently, based on the crystal structure of the nucleotide-bound NH2-terminal domain of yeast Hsp90 (Prodromou et al., 1997a ), and now human Hsp90 (this study). However, whether Hsp90 hydrolyzes ATP had remained elusive (Grenert et al., 1997; Prodromou et al., 1997a ; Scheibel et al., 1998). Using the crystal structure of the NH2-terminal domain as a guide, site-directed mutations were made in Hsp90 that affect ATP binding and hydrolysis differentially. By analyzing these mutants in vivo and in vitro, we now demonstrate not only that Hsp90 has a clearly detectable ATPase activity, but also that both ATP binding and hydrolysis are required for Hsp90 function in vivo. Surprisingly, both the ATPase activity as well as the interaction of Hsp90 with the cofactor p23 require the complete Hsp90 protein. Furthermore, binding and release of p23 by Hsp90 follows an ATP hydrolysis–dependent reaction cycle. These results show that Hsp90 belongs to the group of ATP-regulated molecular chaperones.
ATP binding to Hsp90 is inhibited by the ansamycin antibiotic GA that occupies the ATP binding site with high affinity (Prodromou et al., 1997a ; Stebbins et al., 1997), using essentially the same interactions with the binding pocket as the purine ring of the nucleotide. Thus, the cellular effects of GA must be interpreted as the consequence of the interruption of the Hsp90 ATPase cycle. GA blocks the maturation of specific Hsp90 substrates (for example Whitesell et al., 1994; Smith et al., 1995; Schneider et al., 1996; Whitesell and Cook, 1996; Dittmar and Pratt, 1997; Stancato et al., 1997) and the re-folding of certain stress-denatured polypeptides by the Hsp90 system (Schneider et al., 1996). Whereas in some cases treatment with GA has been reported to result in the dissociation of Hsp90–substrate complexes (for example Whitesell et al., 1994), substrate polypeptides often accumulate on GA-bound Hsp90 in an unfolded conformation that is subject to degradation by the proteasome system (Smith et al., 1995; Schneider et al., 1996; Dittmar and Pratt, 1997). This effect forms the basis for the potential therapeutic application of GA derivatives in depleting the cellular levels of steroid receptors and Hsp90-dependent protooncogenic protein kinases. The accumulation of substrate polypeptide on GA-bound Hsp90 can be explained in light of the recent finding that Hsp90 contains two distinct sites for the interaction with substrate polypeptide (Young et al., 1997; Scheibel et al., 1998), one in the NH2-terminal domain and one in the COOH-terminal domain. GA binding dissociates bound polypeptide from the NH2-terminal domain of Hsp90 but not from the COOH-terminal domain, which seems to have the general capacity to bind unfolded or partially folded polypeptides exposing hydrophobic surfaces (Young et al., 1997). Similarly, ATP but not ADP binding has been shown to lower the affinity of the isolated NH2-terminal domain for peptide substrate (Scheibel et al., 1998). Since the isolated NH2-terminal domain does not hydrolyze ATP, it is indeed likely that ATP binding and not ATP hydrolysis causes the dissociation of substrate. Those substrates that interact with both the NH2- and COOH-terminal domains may maintain their association with Hsp90 in the presence of ATP, whereas other substrates may dissociate completely from Hsp90 upon ATP binding. It is also possible that nucleotide binding has an allosteric effect on the COOH-terminal domain, modulating its polypeptide affinity.
Indeed, the existence of complex, ATP-regulated interactions within the Hsp90 dimer is likely, based on our surprising finding that both the ATPase activity and the interaction of Hsp90 with p23 require the intact Hsp90 molecule. For example, it is conceivable that within the Hsp90 dimer the NH2-terminal domains may contact each other, consistent with recent electron microscopic images of Hsp90 obtained by rotary shadowing (Wearsch and Nicchitta, 1996). It is interesting in this context that the second domain of DNA gyrase contains a lysine residue (K337) that interacts with the γ-phosphate of the bound ATP in the ATPase domain, perhaps providing a mechanism for signaling the hydrolysis of ATP to the molecule as a whole (Wigley et al., 1991). We notice that in our crystal of the ADP-Mg–bound NH2-terminal domain of Hsp90 a lysine residue (K116), coming from a symmetry-related molecule, points into the area where the γ-phosphate would have been. This conserved residue is near the region that differs in the open and closed conformations of the NH2-terminal domain (Stebbins et al., 1997). It is fully solvent exposed in either conformation and has no obvious structural role.
Our findings also help to define the ATP hydrolysis– dependent reaction cycle for the binding and release of p23 by Hsp90. p23 has been described as a component required for the formation of Hsp90 complexes containing high ligand affinity steroid aporeceptor (Johnson et al., 1994; Johnson and Toft, 1995). Before our demonstration of the ATPase acctivity of Hsp90 it has been reported that ATP-Mg is required for p23 binding to Hsp90 and that GA disrupts the Hsp90–p23 complex (Johnson and Toft, 1995; Smith et al., 1995; Grenert et al., 1997; Sullivan et al., 1997). A complete model of the reaction cycle is now emerging from the analysis of the Hsp90 mutants generated in this study. Hsp90 defective in hydrolyzing bound ATP still forms the complex with p23 but is unable to dissociate it. In contrast, the presence of ADP-Mg in the hydrolysis-defective protein does not promote p23 binding. Neither does the nucleotide-free protein associate with p23. Thus, p23 binds to the ATP-bound form of Hsp90 and dissociates upon ATP hydrolysis. GA binding mimics the ADP-bound form of Hsp90 (or the nucleotide-free state) in that it prevents the interaction of p23 with Hsp90 in the presence of ATP. In contrast, GA seems to mimic the ATP-bound form of Hsp90 with regard to substrate binding to the NH2-terminal domain, consistent with the notion that the binding sites on the NH2-terminal domain for p23 and substrate are not identical. Notably, the complete Hsp90 protein, probably as a dimer, is required for p23 binding. Thus, whereas p23 most likely contacts the NH2-terminal domain directly, its interaction with Hsp90 is clearly more complex.
In contrast to Hsp90, p23 (Sba1p) is not essential in yeast but increases the efficiency of expression of heterologous steroid receptor (Bohen, 1998; Fang et al., 1998). Thus, the loss of p23 binding to Hsp82(D79N) cannot be responsible for the lethal phenotype of this mutation. Rather, ATP binding and hydrolysis by Hsp90 is functionally required independently of p23, probably in regulating substrate binding and release. It is possible, however, that with the mutant Hsp82(E33A), that is defective in ATP hydrolysis, trapping of p23 in the Hsp90-bound state may contribute to the functional defect. The mechanism of p23 action is not yet clear. It is possible that p23 generally increases the efficiency of substrate discharge from Hsp90 by stabilizing the ATP state of the Hsp90 NH2-terminal domain that has low affinity for polypeptide. Such a function may be important only for a subset of substrates. Additionally, it has been suggested that p23 itself has chaperone properties and may interact with the substrate directly (Bose et al., 1996). Thus, it is also possible that p23 may function in targeting substrates to Hsp90, similar to the role of Hsp40 (DnaJ) in targeting substrates to Hsp70 (DnaK) (for review see Hartl, 1996). In contrast to Hsp40, p23 does not stimulate the ATPase activity of its partner chaperone and thus does not generate the ADP-bound state of Hsp90 that binds to the substrate more stably. However, such a function may depend on the presence of substrate polypeptide and may so far have escaped detection.
Acknowledgments
The authors thank Dr. S. Lindquist for supplying yeast strain ΔPCLDa/α and the vector pTGPD/P82, and Dr. D. Toft for providing human p23 cDNA. We are grateful to Drs. J. Young and M. Leroux for critically reading the manuscript.
Abbreviations used in this paper
- 3-AT
3-amino-1,2,4-triazole
- 5-FOA
5-fluoroorotic acid
- GA
geldanamycin
- Hsp
heat shock protein
- NTA
nitrilo-triacetic-acid
- SD
synthetic dropout medium
- TPR
tetratricopeptide repeat
References
- Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1987. Current Protocols in Molecular Biology. Wiley and Sons, Inc., New York. 8.5.1–8.5.9.
- Aligue R, Akhavan-Niak H, Russel P. A role for Hsp90 in cell cycle control: Wee 1 tyrosine kinase activity requires interaction with Hsp90. EMBO (Eur Mol Biol Organ) J. 1994;13:6099–6106. doi: 10.1002/j.1460-2075.1994.tb06956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel, P.L., C.-T. Chien, R. Sternglanz, and S. Fields. 1993. Using the two-hybrid system to detect protein–protein interactions. In Cellular Interactions in Development: A Practical Approach. D.A. Hartley, editor. Oxford University Press, Oxford. 153–179.
- Bohen SP. Genetic and biochemical analysis of p23 and ansamycin antibiotics in the function of Hsp90-dependent signaling proteins. Mol Cell Biol. 1998;18:3330–3339. doi: 10.1128/mcb.18.6.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich KA, Farrelly FW, Finkelstein DB, Taulien J, Lindquist S. Hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol Cell Biol. 1989;9:3919–3930. doi: 10.1128/mcb.9.9.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose S, Weikl T, Bugl H, Buchner J. Chaperone function of Hsp90-associated proteins. Science. 1996;274:1715–1717. doi: 10.1126/science.274.5293.1715. [DOI] [PubMed] [Google Scholar]
- Buchner J. Supervising the fold: functional principles of molecular chaperones. FASEB (Fed Am Soc Exp Biol) J. 1996;10:10–19. [PubMed] [Google Scholar]
- Chen SY, Prapapanich V, Rimerman RA, Honore B, Smith DF. Interactions of p60, a mediator of progesterone receptor assembly, with heat shock proteins Hsp90 and Hsp70. Mol Endocrinol. 1996;10:682–693. doi: 10.1210/mend.10.6.8776728. [DOI] [PubMed] [Google Scholar]
- Craig EA. Chaperones: helpers along the pathways to protein folding. Science. 1993;260:1902–1903. doi: 10.1126/science.8100364. [DOI] [PubMed] [Google Scholar]
- Csermely P, Kahn CR. The 90 kD heat-shock protein (Hsp-90) possesses an ATP binding site and autophosphorylation activity. J Biol Chem. 1991;266:4943–4950. [PubMed] [Google Scholar]
- Csermely P, Kajtar J, Hollosi M, Jalsovszky G, Holly S, Kahn CR, Gergely PJ, Soti C, Mihaly K, Somogyi J. ATP induces a conformational change of the 90-kD heat shock protein (hsp90) J Biol Chem. 1993;268:1901–1907. [PubMed] [Google Scholar]
- Dittmar KD, Pratt WB. Folding of the glucocorticoid receptor by the reconstituted hsp90-based chaperone machinery. J Biol Chem. 1997;272:13047–13054. doi: 10.1074/jbc.272.20.13047. [DOI] [PubMed] [Google Scholar]
- Dittmar KD, Hutchison KA, Owens-Grillo JK, Pratt WB. Reconstitution of the steroid receptor-center-dot-hsp90 heterocomplex assembly system of rabbit reticulocyte lysate. J Biol Chem. 1996;271:12833–12839. doi: 10.1074/jbc.271.22.12833. [DOI] [PubMed] [Google Scholar]
- Fang YF, Fliss AE, Rao J, Caplan AJ. Sba1 encodes a yeast Hsp90 cochaperone that is homologous to vertebrate p23 proteins. Mol Cell Biol. 1998;18:3727–3734. doi: 10.1128/mcb.18.7.3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman BC, Morimoto RI. The human cytosolic molecular chaperones Hsp90, Hsp70 (Hsc70) and Hdj-1 have distinct roles in recognition of a non-native protein and protein refolding. EMBO (Eur Mol Biol Organ) J. 1996;15:2969–2979. [PMC free article] [PubMed] [Google Scholar]
- Frydman J, Höhfeld J. Chaperones get in touch: the Hip-Hop connection. Trends Biochem Sci. 1997;22:87–92. doi: 10.1016/s0968-0004(97)01005-0. [DOI] [PubMed] [Google Scholar]
- Gething M-J, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Grenert JP, Sullivan WP, Fadden P, Haystead TA, Clark J, Mimnaugh E, Krutzsch H, Ochel H-J, Schulte TW, Sausville E, et al. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem. 1997;272:23843–23850. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–580. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- Höhfeld J. Regulation of the heat shock cognate hsc70 in the mammalian cell: the characterization of the anti-apoptotic protein BAG-1 provides novel insights. Biol Chem. 1998;379:269–274. [PubMed] [Google Scholar]
- Jackson AP, Maxwell A. Identifying the catalytic residue of the ATPase reaction of DNA gyrase. Proc Natl Acad Sci USA. 1993;90:11232–11236. doi: 10.1073/pnas.90.23.11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob U, Lilie H, Meyer I, Buchner J. Transient interaction of Hsp90 with early unfolding intermediates of citrate synthase—Implications for heat shock in vivo. J Biol Chem. 1995;270:7288–7294. doi: 10.1074/jbc.270.13.7288. [DOI] [PubMed] [Google Scholar]
- Jakob U, Scheibel T, Bose S, Reinstein J, Buchner J. Assessment of the ATP binding properties of Hsp90. J Biol Chem. 1996;271:10035–10041. doi: 10.1074/jbc.271.17.10035. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Schumacher RJ, Ross ED, Toft DO. Hop modulates hsp70/hsp90 interactions in protein folding. J Biol Chem. 1998;273:3679–3686. doi: 10.1074/jbc.273.6.3679. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Craig EA. Protein folding in vivo: unraveling complex pathways. Cell. 1997;90:201–204. doi: 10.1016/s0092-8674(00)80327-x. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Toft DO. A novel chaperone complex for steroid receptors involving heat shock proteins, immunophilins, and p23. J Biol Chem. 1994;269:24989–24993. [PubMed] [Google Scholar]
- Johnson JL, Toft DO. Binding of p23 and Hsp90 during assembly with progesterone-receptor. Mol Endocrinol. 1995;9:670–678. doi: 10.1210/mend.9.6.8592513. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Beito TG, Krco CJ, Toft DO. Characterization of a novel 23-kilodalton protein of unactive progesterone receptor complexes. Mol Cell Biol. 1994;14:1956–1963. doi: 10.1128/mcb.14.3.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis PJ. Molscript: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- Merrit EA, Murphy ME. Raster3D Version 2.0: a program for photorealistic molecular graphics. Acta Crystallogr Sect D Biol Crystallogr. 1994;50:946–950. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- Nair SC, Toran EJ, Rimerman RA, Hjermstad S, Smithgall TE, Smith DF. A pathway of multi-chaperone interactions common to diverse regulatory proteins: estrogen receptor, Fes tyrosine kinase, heat shock transcription factor Hsf1, and the aryl hydrocarbon receptor. Cell Stress Chaperones. 1996;1:237–250. doi: 10.1379/1466-1268(1996)001<0237:apomci>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan DF, Lindquist S. Mutational analysis of hsp90 function: interactions with a steroid receptor and a protein kinase. Mol Cell Biol. 1995;15:3917–3925. doi: 10.1128/mcb.15.7.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan DF, Vos MH, Lindquist S. In vivo functions of the Saccharomyces cerevisiaeHsp90 chaperone. Proc Natl Acad Sci USA. 1997;94:12949–12956. doi: 10.1073/pnas.94.24.12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto T, Oharanemoto Y, Ota M, Takagi T, Yokoyama K. Mechanism of dimer formation of the 90-kD heat-shock protein. Eur J Biochem. 1995;233:1–8. doi: 10.1111/j.1432-1033.1995.001_1.x. [DOI] [PubMed] [Google Scholar]
- O'Brien MC, McKay DB. Threonine 204 of the chaperone protein Hsc70 influences the structure of the active site, but is not essential for ATP hydrolysis. J Biol Chem. 1993;268:24323–24329. [PubMed] [Google Scholar]
- Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997a;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, Piper PW, Pearl LH. A molecular clamp in the crystal structure of the N-terminal domain of the yeast Hsp90 chaperone. Nat Struct Biol. 1997b;4:477–482. doi: 10.1038/nsb0697-477. [DOI] [PubMed] [Google Scholar]
- Scheibel T, Neuhofen S, Weikl T, Mayr C, Reinstein J, Vogel PD, Buchner J. ATP-binding properties of human Hsp90. J Biol Chem. 1997;272:18608–18613. doi: 10.1074/jbc.272.30.18608. [DOI] [PubMed] [Google Scholar]
- Scheibel T, Weikl T, Buchner J. Two chaperone sites in Hsp90 differing in substrate specificity and ATP dependence. Proc Natl Acad Sci USA. 1998;95:1495–1499. doi: 10.1073/pnas.95.4.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Sepp-Lorenzino L, Nimmesgern E, Ouerfelli O, Danishefsky S, Rosen N, Hartl FU. Pharmacological shifting of a balance between protein refolding and degradation mediated by Hsp90. Proc Natl Acad Sci USA. 1996;93:14536–14541. doi: 10.1073/pnas.93.25.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher RJ, Hansen WJ, Freeman BC, Alnemri E, Litwack G, Toft DO. Cooperative action of hsp70, hsp90, and DnaJ proteins in protein renaturation. Biochemistry. 1996;35:14889–14898. doi: 10.1021/bi961825h. [DOI] [PubMed] [Google Scholar]
- Sikorski R, Boeke JD. In vitro mutagenesis and plasmid shuffling: from cloned gene to mutant yeast. Methods Enzymol. 1991;194:302–318. doi: 10.1016/0076-6879(91)94023-6. [DOI] [PubMed] [Google Scholar]
- Smith DF. Dynamics of heat shock protein 90-progesterone receptor binding and the disactivation loop model for steroid receptor complexes. Mol Endocrinol. 1993;7:1418–1429. doi: 10.1210/mend.7.11.7906860. [DOI] [PubMed] [Google Scholar]
- Smith DF. Sequence motifs shared between chaperone components participating in the assembly of progesterone receptor complexes. Biol Chem. 1998;379:283–288. doi: 10.1515/bchm.1998.379.3.283. [DOI] [PubMed] [Google Scholar]
- Smith DF, Whitesell L, Nair SC, Chen SY, Prapapanich V, Rimerman RA. Progesterone receptor structure and function altered by geldanamycin, an hsp90-binding agent. Mol Cell Biol. 1995;15:6804–6812. doi: 10.1128/mcb.15.12.6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancato LF, Silverstein AM, Owens-Grillo JK, Chow YH, Jove R, Pratt WB. The hsp90 binding antibiotic geldanamycin decreases Raf levels and epidermal growth factor signaling without disrupting formation of signaling complexes or reducing the specific enzymatic activity of Raf kinase. J Biol Chem. 1997;272:4013–4020. doi: 10.1074/jbc.272.7.4013. [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89:239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Sullivan W, Stensgard B, Caucutt G, Bartha B, McMahon N, Alnemri ES, Litwack G, Toft D. Nucleotides and two functional states of hsp90. J Biol Chem. 1997;272:8007–8012. doi: 10.1074/jbc.272.12.8007. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehlin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wearsch PA, Nicchitta CV. Endoplasmic reticulum chaperone GRP94 subunit assembly is regulated through a defined oligomerization domain. Biochemistry. 1996;35:16760–16769. doi: 10.1021/bi962068q. [DOI] [PubMed] [Google Scholar]
- Wehland J, Schröder HC, Weber K. Amino sequence requirements in the epitope recognized by the α-tubulin-specific ray monoclonal antibody YL 1/2. EMBO (Eur Mol Biol Organ) J. 1984;3:1295–1300. doi: 10.1002/j.1460-2075.1984.tb01965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Hendershot LM. Characterization of the nucleotide binding properties and ATPase activity of recombinant hamster Bip purified from bacteria. J Biol Chem. 1995;270:26670–26676. doi: 10.1074/jbc.270.44.26670. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Cook P. Stable and specific binding of heat shock protein 90 by geldanamycin disrupts glucocorticoid receptor function in intact cells. Mol Endocrinol. 1996;10:705–712. doi: 10.1210/mend.10.6.8776730. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiech H, Buchner J, Zimmermann R, Jacob U. Hsp90 chaperones protein folding in vitro. Nature. 1992;358:169–170. doi: 10.1038/358169a0. [DOI] [PubMed] [Google Scholar]
- Wigley DB, Davies GJ, Dodson EJ, Maxwell A, Dodson G. Crystal structure of an N-terminal fragment of the DNA gyrase B protein. Nature. 1991;351:624–629. doi: 10.1038/351624a0. [DOI] [PubMed] [Google Scholar]
- Young JC, Schneider C, Hartl FU. In vitro evidence that hsp90 contains two independent chaperone sites. FEBS (Fed Eur Biochem Soc) Lett. 1997;418:139–143. doi: 10.1016/s0014-5793(97)01363-x. [DOI] [PubMed] [Google Scholar]
- Young JC, Obermann WMJ, Hartl FU. Specific binding of tetratricopeptide-repeat proteins to the C-terminal 12 kD domain of Hsp90. J Biol Chem. 1998;273:18007–18010. doi: 10.1074/jbc.273.29.18007. [DOI] [PubMed] [Google Scholar]