

Abstract
In this paper, we describe the identification and characterization of two novel and essential mitotic spindle proteins, Duo1p and Dam1p. Duo1p was isolated because its overexpression caused defects in mitosis and a mitotic arrest. Duo1p was localized by immunofluorescence, by immunoelectron microscopy, and by tagging with green fluorescent protein (GFP), to intranuclear spindle microtubules and spindle pole bodies. Temperature-sensitive duo1 mutants arrest with short spindles. This arrest is dependent on the mitotic checkpoint. Dam1p was identified by two-hybrid analysis as a protein that binds to Duo1p. By expressing a GFP–Dam1p fusion protein in yeast, Dam1p was also shown to be associated with intranuclear spindle microtubules and spindle pole bodies in vivo. As with Duo1p, overproduction of Dam1p caused mitotic defects. Biochemical experiments demonstrated that Dam1p binds directly to microtubules with micromolar affinity. We suggest that Dam1p might localize Duo1p to intranuclear microtubules and spindle pole bodies to provide a previously unrecognized function (or functions) required for mitosis.
Keywords: mitosis, microtubule, anaphase, cytoskeleton, spindle
The mitotic spindle undergoes a remarkable series of transitions in response to cell cycle control signals. The spindle assembles, it forms bipolar attachments to each chromosome, it orients itself properly within the cell, and then, with extraordinarily high fidelity, it carries out chromosome segregation. Finally, it disassembles.
Proper spindle assembly and function involves coordination of many events and processes including modulation of microtubule dynamics and creation of at least three distinct microtubule populations (kinetochore, polar, and astral microtubules). In addition, connections must be established between different spindle microtubule subpopulations, between spindle microtubules and chromosomes, between spindle microtubules and microtubule-associated proteins and motor proteins, and between spindle microtubules and the cell cortex (Waters and Salmon, 1997). To insure that proper spindle assembly occurs, a cellular surveillance system monitors the spindle and activates a mitotic checkpoint if the spindle is not assembled correctly (Rudner and Murray, 1996; Hardwick, 1998). Once the spindle is assembled properly, a carefully orchestrated set of molecular events results in chromosome to pole movement (anaphase A) and separation of spindle poles (anaphase B).
Genetic approaches to the study of spindle mechanics and regulation in Saccharomyces cerevisiae, Schizosaccharomyces pombe, Aspergillus nidulans, and in a variety of other organisms have complemented biochemical studies using Xenopus extracts and cell biological studies on mammalian and plant cells (Nicklas, 1997; Sobel, 1997). Each different approach has provided an extremely powerful and unique avenue toward identification of mitotic spindle components and elucidation of their functions. Genetic studies in fungal organisms have been particularly valuable both because nontubulin spindle components are typically low in abundance, making their discovery difficult by other means, and because genetic analysis facilitates tests of function in vivo.
Thus, elegant genetic studies have revealed how forces generated by kinesin-related proteins and dynein work both synergistically and antagonistically to assemble and orient spindles and to separate chromosomes (Oakley and Morris, 1980; Gambino et al., 1984; Oakley and Rinehart, 1985; Saunders and Hoyt, 1992; Cottingham and Hoyt, 1997). In addition, γ-tubulin and many other proteins associated with spindle pole bodies have been identified and tested functionally using genetic approaches (Rout and Kilmartin, 1990; Oakley, 1994; Sobel and Snyder, 1995; Spang et al., 1995; Marschall et al., 1996). Finally, a number of spindle accessory proteins have been found and studied functionally by a variety of genetic strategies (Berlin et al., 1990; Pasqualone and Huffaker, 1994; Machin et al., 1995; Pellman et al., 1995; Wang and Huffaker, 1997).
Considering how complex the regulation and mechanics of mitosis appear, it seems likely that a large number of spindle proteins must function in concert with tubulin, the major spindle protein. While many such proteins have been identified, an important question is whether there remain additional proteins that carry out previously unrecognized functions in the spindle. Complete understanding of the mechanisms and regulation of mitosis will require enumeration of all spindle components and determination of their functions.
Here we describe genetic identification of DUO1, a gene that encodes a yeast mitotic spindle protein. We demonstrate that Duo1p is required for proper mitotic spindle function, and we identify an additional spindle protein, Dam1p, which interacts with Duo1p and binds directly to microtubules.
Materials and Methods
Strains and Media
The yeast strains used in this study are listed in Table I. Media were prepared, and standard genetic techniques were carried out according to (Rose et al., 1990). YPD is yeast-rich medium, and SM is synthetic minimal medium supplemented with the appropriate nutrients. The carbon sources used were 2% glucose, 2% galactose and 2% raffinose, or 2% glycerol depending on the experiment.
Table I.
Strains Used
| Name | MAT | Genotype | ||
|---|---|---|---|---|
| DDY757 | a | cry1, ade2-1, his3-11,15, leu2-3,112, ura3-1, trp1-1, can1-100 | ||
| DDY759 | a/α | cry1/cry1, ade2-1/ade2-1, his3-11,15/his3-11,15, leu2-3,112/leu2-3,112, ura3-1/ura3-1 | ||
| trp1-1/trp1-1, can1-100/can1-100 | ||||
| DDY898 | a | his3Δ200, ura3-52 | ||
| DDY1102 | a/α | ade2-1/+, his3Δ200/his3Δ200, leu2-3,112,/leu2-3,112, ura3-52/ura3-52, lys2-801/+ | ||
| DDY1445 | a/α | his3Δ200/his3Δ200, leu2-3,112/leu2-3,112, ura3-52/ura3-52, ade2-1/+ | ||
| DDY1446 | a/α | his3Δ200/his3Δ200, leu2-3,112/leu2-3,112 ura3-52/ura3-52, ade2-1/+ | ||
| Δduo1:HIS3/Δduo1:HIS3, pDD476 | ||||
| DDY1447 | a/α | his3Δ200/his3Δ200, leu2-3,112/leu2-3,112, ura3-52/ura3-52, | ||
| ade2-1/+ Δduo1:HIS3/Δduo1:HIS3, pDD477 | ||||
| DDY1522 | a/α | his3Δ200/his3Δ200, leu2-3,112/leu2-3,112, ura3-52/ura3-52, ade2-1/ADE2, lys2/LYS2, | ||
| duo1Δ::HIS3/DUO1 | ||||
| DDY1523 | α | his3Δ200, leu2-3,112, ura3-52, lys2, DUO1::LEU2 | ||
| DDY1524 | α | his3Δ200, leu2-3,112, ura3-52, lys2, duo1-1::LEU2 | ||
| DDY1525 | α | his3Δ200, leu2-3,112, ura3-52, lys2, duo1-2::LEU2 | ||
| DDY1526 | α | his3Δ200, leu2-3,112, ura3-52, lys2, ade2-101, duo1-2::LEU2, mad2Δ::URA3 | ||
| MAY4425 | a | his3Δ200, leu2-3,112, ura3-52, ade2-101, mad2Δ::URA3 | ||
| (M.A. Hoyt) |
Plasmid Construction and Other DNA Manipulations
All DNA manipulations were carried out by standard methods (Maniatis et al., 1982). Restriction endonucleases and other enzymes were purchased from either New England Biolabs (Beverly, MA) or Boehringer Mannheim Corp. (Indianapolis, IN). Taq DNA polymerase was obtained from Perkin-Elmer/Cetus (Norwalk, CT).
DNA sequencing was performed by the University of California Berkeley Sequencing Facility (Berkeley, CA) through the use of an Applied Biosystems sequencing machine (Foster City, CA). Primers used for both PCR and sequencing were purchased from either the Berkeley Oligo Synthesis Facility (Berkeley, CA) or Operon (Alameda, CA).
Plasmid Isolation and Sequencing
Plasmids were recovered from yeast strains grown on glucose minimal medium plates under conditions that selected for a prototrophic marker carried on the desired plasmid. Strains were grown to saturation in 5 ml of glucose minimal medium, pelleted, and washed once with water. Plasmids were isolated from these cells using the Qiagen Plasmid Miniprep Kit (Chatsworth, CA). Cell lysis was achieved by resuspending yeast in the cell lysis buffer provided with the kit, adding about 100 μl of glass beads, and vortexing for 5–10 min. After lysis, the steps in the Qiagen protocol were followed. The isolated plasmids were then transformed into Escherichia coli, and plasmid DNA was purified using the Qiagen Plasmid Miniprep Kit. This DNA was used for sequencing and subcloning.
General Immunofluorescence and Immunoblot Procedures
Yeast cells were grown to early log phase in minimal or rich medium. For galactose induction, cells were grown to early log phase (∼2 × 106 cells/ml) in medium containing glucose. Using a Millipore 150-ml sterilizing filter flask (Bedford, MA), cells grown on glucose were washed twice with minimal medium without a carbon source and resuspended into medium containing glycerol. After incubating the cells in medium containing glycerol in a shaking water bath for 10–12 h, the cells were washed twice again with minimal medium without a carbon source and then resuspended from the filter surface with minimal medium containing galactose. Galactose induction for the experiment shown in Fig. 1 was instead as described in the Fig. 1 legend. Fixation and immunofluorescence procedures were carried out as described by Drubin et al. (1988). The YOL134 antitubulin antibody was used at 1:200, and the anti-Duo1p antibody (preparation described below) was used at 1:2,000. Fluorescein-conjugated anti-IgG heavy chain secondary antisera were obtained from Cappel/Organon Teknika (Malvern, PA).
Figure 1.

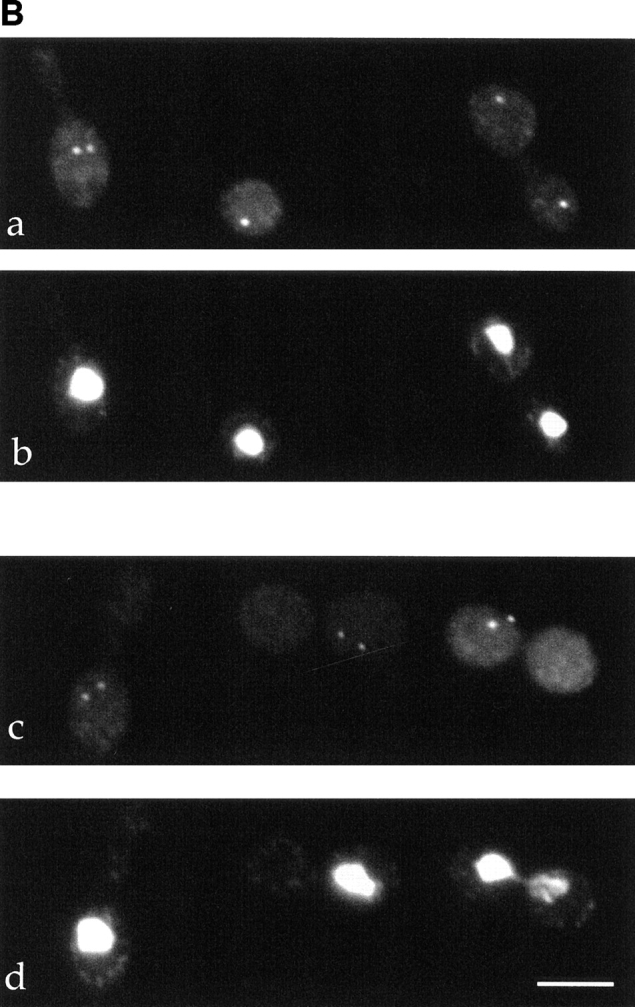

DUO1 overexpression phenotypes. (A) a–c are phase micrographs, d–f are fluorescence micrographs showing microtubule staining, and g–i are fluorescence micrographs showing DNA (DAPI) staining. The first column shows wild-type cells (a, d, and g), the second column shows cells after 8 h of DUO1 overexpression (b, e, and h), and the third column shows cells after 16 h of overexpression (c, f, and i). For overexpression studies, cells were grown in glucose medium until log phase, washed, and grown in raffinose medium for 12 h, and then galactose was added to the raffinose-containing medium. (B) The effect of DUO1 overexpression on spindle pole bodies. a and c show Tub4p staining, and b and d show nuclear (DAPI) staining. a and b show wild-type cells, and c and d show cells overexpressing DUO1 for 16 h. (C) FACS® analysis of nuclear DNA. a shows FACS® data for a wild-type control strain. b shows FACS® data before DUO1 overexpression. c shows FACS® data for the same cell line in b 8 h after overexpression of DUO1 was initiated. Bars, 5 μm.
Immunoblot analysis was performed using standard SDS–polyacrylamide and immunoblot transfer methods (Maniatis et al., 1982). The anti-Duo1p antibody was used at a dilution of 1:2,000 for immunoblot analysis.
Deletion of DUO1
A DUO1 disruption plasmid was constructed in three steps. A 1.2-kb PCR fragment amplified from pDD465 (contains genomic DUO1 fragment) using M13Reverse and oCH18 (CCA TCG ATA TTG AAG ACT TGT TCA) was digested with ClaI and XhoI and ligated into Bluescript KS+. A 0.7-kb NheI-HindIII fragment (HindIII site Klenowed) from pDD465 was then inserted into XbaI and EagI site of the above plasmid (EagI site Klenowed) resulting in vector pDD468. The HIS3 auxotrophic marker of plasmid LV1 was cloned into the BamHI site of pDD468 creating pDD469. A linear PCR fragment was isolated from pDD469 using oIC1 (CTT GGA AAG CCC TGA CAA GGC C) and oIC2 (CTT CTC CAT CCG ATG GAC AAG). This fragment was transformed into the diploid strain DDY1102 by the Li-acetate method to generate DDY1522. Colonies were selected for the integration of the HIS3 marker by plating on minimal medium. Gene disruption was confirmed by PCR using oIC3 (CCC TTT GGC AGA TCC CAG CC), oIC4 (GGT GCA CCA ATG GCA GGT GTC), and a primer internal to HIS3.
Generation of Temperature-sensitive duo1 Mutants
The plasmid pDD467 (containing DUO1 subcloned into pRS315) was mutagenized in vitro using hydroxylamine. Plasmid DNA (20 μg), carrying the selectable marker LEU2 and the DUO1 gene, was incubated at 75°C in a solution containing the mutagen (0.5 M hydroxylamine hydrochloride, 50 mM sodium pyrophosphate, pH 7.0, 100 mM NaCl, and 2 mM EDTA). Reactions were stopped by placing them on ice at the 0-, 20-, 40-, 60-, 80-, and 100-min time points. The mutagen was removed by spinning the reactions through two 1-ml G-25 syringe columns.
To determine the extent of mutagenesis, leuB− bacteria were transformed with the mutagenized plasmids to monitor mutations in the LEU2 gene. The mutation rate was defined as the percentage of bacterial transformants that were unable to grow on M9-leucine medium. Plasmids from the 20- and 40-min time points, which gave 8 and 11% LEU2 mutagenesis, respectively, were used for further studies.
The mutagenized plasmids were transformed into a diploid strain that had both DUO1 loci deleted and carried a genomic copy of DUO1 on a URA3-marked CEN plasmid (pDD466). Transformants were plated on SC-Leu plates (plates containing all amino acids necessary for growth with the exception of leucine) at a density of about 300 colonies per plate. A total of 11,200 colonies were screened for the 20-min time point, and 3,300 colonies were screened for the 40-min time point. After 2–3 d of growth at 25°C, the colonies were replica plated onto 5-fluoroorotic acid (5-FOA)1 plates and incubated at 25 and 37°C. Strains containing a URA3 plasmid are not able to grow on medium containing 5-FOA. This step was carried out to select colonies that had lost the unmutagenized genomic copy of DUO1 marked with URA3 and only carried the mutagenized form marked with LEU2. Cells that did not grow at 37°C but did grow at 25°C on 5-FOA after 3 d were restreaked onto SC-Leu (synthetic complete medium lacking leucine) plates from the SC-Leu plates with the original transformants. These isolates were retested for growth on 5-FOA. The plasmids were recovered from all the 5-FOA selected clones, and DUO1-containing genomic inserts were cloned into an unmutagenized pRS315 vector as a ClaI-HindIII fragment. These constructs were retested. The open reading frames of the temperature-sensitive isolates duo1-1 (pDD476) and duo1-2 (pDD477) were sequenced to identify the mutations.
The duo1-1 and duo1-2 alleles were subsequently integrated into the genome. The Not1 site in the polylinker of pDD466 was made blunt with Klenow and the resulting plasmid was mutagenized using the CLONETECH Transformer™ Site-Directed Mutagenesis Kit (Palo Alto, CA) with mutagenesis oligo oIC7 (AAA AGG ACA CGT TAA GCG GCC GCG TTA AAT TAT TAT TTT T) and selection oligo oIC6 (CGA ATT CCT GCA GTC CGG GGG ATC) to introduce a NotI site 120 bp downstream of the DUO1 open reading frame. The LEU2 gene from pKK582 was cloned into the NotI site to create pIC5. The duo1-1 and duo1-2 mutations were swapped into this construct using the NheI site. Plasmids were cut with NcoI and HindII and transformed into DDY1522. Leu+, His− transformants were selected and confirmed by PCR using oIC10 (GTG TGG ATC ATC CAT AGC CTG) and oIC11 (CAT ATG GTG GGT TCC CAG AAC C). These diploids were then sporulated, and haploid integrants were recovered.
Duo1p Antibody
The DUO1 coding sequence was amplified by PCR using primers oCH35 (GGA CTA GTG AGC AAA GCC AAT TAG ATG) and oCH38 (GCG CGT CTA GAC CCG AAT CTT AAT TAT TTA CC). The product was cloned into pHAT2 in frame with a six histidine tag (pDD471) using the SpeI site in oCH35 and the HindIII site in oCH38. The construct was transformed into BL21 cells, and expression of the fusion protein was induced by addition of 0.4 mM IPTG for 4 h at 37°C. Inclusion bodies were harvested from 6 liters of log phase cells and solubilized by the addition of 8 M urea. The denatured fusion protein was purified on a Qiagen NTA-column as described in the product protocol. The protein was refolded by step-wise dialysis from 8 M urea into PBS (137 mM NaCl, 2.7 mM KCl, 4.5 mM Na2HPO4 7H20, 1.2 mM KH2PO4, pH 8.0) using a 2 M urea reduction per step.
Antibodies were generated by injecting rabbits with the purified protein. An affinity matrix was created by immobilizing the fusion protein on Reacti-gel resin (Pierce Chemical Co., Rockford, IL). The protein was attached to the resin as described in the manual. Sera was circulated over the column, and specific antibodies were purified by MgCl2 elution (Harlow and Lane, 1988). The eluted antibody recognized the injected recombinant protein, a single 32-kD protein in whole yeast cell extracts and a glutathione-S-transferase (GST)–Duo1p fusion protein purified from yeast. Cells overexpressing Duo1p showed a clear increase in immunoreactivity in the 32-kD polypeptide in whole-cell extracts, strongly supporting the conclusion that the 32-kD band was Duo1p.
Duo1p Immunofluorescence and Immunoelectron Microscopy
Cells were processed for immunofluorescence microscopy as described previously (Pringle et al., 1991). For Duo1p staining, the cold methanol/ acetone treatment was replaced by incubating cells with 0.1% SDS in PBS/BSA for 2 min. The monoclonal antitubulin antibody YOL134 was used at a 1:200 dilution, and the anti-DUO1 antibody was used at a 1:2,000 dilution. Detection of the primary antibody was accomplished by applying fluorescein-labeled goat anti–rat or goat anti–rabbit secondary antibodies (Cappel/Organon Teknika, Malvern, PA) at a dilution of 1:1,000. Cy3 secondary anti–rabbit antibodies (Sigma Chemical Co., St. Louis, MO) were used at a 1:500 dilution.
Cells were cryofixed in a Bal-Tec HPM 010 high-pressure freezer (Balzers, Hudson, NH), freeze substituted in 0.2% glutaraldehyde plus 0.1% uranyl acetate in acetone for 2 d at −78°C, and then warmed to −50°C over a 12-h period. Cells were infiltrated with Lowicryl HM20 at −50°C and polymerized in BEEM capsules at −35°C by UV irradiation. Blocks were UV irradiated at room temperature for an additional 48 h to complete the resin curing. Thin (50-nm) sections were cut on a Reichert UltracutE microtome (Vienna, Austria) and picked up on Formvar and carbon-coated nickel grids. The sections were incubated in primary antibody diluted 1:50 for 1 h, rinsed with PBS, incubated in 10 nM goat anti– rabbit secondary diluted 1:20 for 1 h, rinsed with PBS, fixed in 0.5% glutaraldehyde for 5 min, rinsed in dH20, and post-stained with uranyl acetate and lead citrate. Sections were examined in a JEOL 100CX (Peabody, MA) electron microscope.
Purification of GST–Duo1p from Yeast
The entire coding sequence of DUO1 was cloned into a galactose-inducible yeast GST fusion vector and tested for expression. A PCR product using primers oCH35 (GGA CTA GTG AGC AAA GCC AAT TAG ATG) and oCH38 (GCG CGT CTA GAC CCG AAT CTT AAT TAT TTA CC) was cloned into the pEG-KT vector resulting in pDD475. Transformants were grown in medium containing raffinose, and expression was monitored after galactose induction. Expression reached a maximum level after 2 h of induction. A total of 150 liters of cells was grown to log phase in raffinose, induced with galactose for 4 h, and then harvested. Cells were washed once in ice-cold water, frozen in liquid nitrogen, and then stored at −80°C. Cells were lysed in a Waring blender (New Hartford, CT) and resuspended in lysis buffer (50 mM Tris-Cl, pH 7.5, 1% Triton X-100, 150 mM NaCl, 2 mM MgCl2, 1 mM EDTA, 0.5 mM PMSF, and aqueous protease inhibitors [0.5 μg/ml each of antipain, leupeptin, pepstatin A, chymostatin, and aprotinin]). The extract was spun at 10,000 g for 10 min. The supernatant was recovered, preswollen GST beads were added, and the mixture was incubated at 4°C for 4–14 h. Beads were harvested by centrifugation at 1,000 g for 1 min, washed three times in wash buffer (50 mM Tris-Cl, pH 7.5, 1% Triton X-100, 300 mM NaCl, 2 mM MgCl2, 1 mM EDTA, 0.5 mM PMSF, and 0.5 μg/ml aqueous protease inhibitors), and then washed with elution buffer. GST–Duo1p was eluted from beads by the addition of 15 mM glutathione in PME (80 mM Pipes, pH 6.8, 1 mM EGTA, and 1 mM MgCl2) at room temperature for 30–60 min. Beads were collected by centrifugation at 1,000 g for 1 min. The supernatant containing the fusion protein was recovered, flash frozen, and stored at −80°C.
Microtubule Binding Experiments
Purified bovine brain tubulin (80 μM) in PME buffer (80 mM K-Pipes, pH 6.8, 1 mM EGTA, and 1 mM MgCl2) was thawed and prespun in a microfuge for 5 min at 4°C. To assemble microtubules, GTP was added to a final concentration of 1 mM, glycerol was added to a final concentration of 25%, and the reaction was incubated at 35°C for 30 min. After assembly, 20 μM taxol was added to stabilize microtubules. Microtubules were then diluted in PME containing 1 mM GTP and 10 μM taxol.
For cosedimentation assays, Dam1p was translated in vitro and radio-labeled with [S35]methionine using the TNT T7 Quick kit (Promega Corp., Madison, WI). The translation products were precleared by ultracentrifugation for 10 min at 60,000 rpm, 25°C (in a TLA100 rotor; Beckman Instruments, Fullerton, CA). 2 μl of cleared translation product was added to variable concentrations of taxol-stabilized microtubules (0.6–20 μM) in 20-μl reactions. The reactions were incubated for 20 min at 25°C to allow binding to occur and then centrifuged as above to pellet the microtubules. Pellets and supernatants were fractionated on SDS-PAGE gels, and the percentage of Dam1p bound to microtubules in each reaction was determined by analyzing autoradiographs using an IS-1000 densitometer (Alpha Innotech Corporation, San Leandro, CA). As a positive control for microtubule binding, a three-repeat isoform of the microtubule-associated protein tau (Goode et al., 1997) was in vitro translated and tested in parallel to Dam1p.
Two-Hybrid Screen
A two-hybrid screen was carried out as described by Fields and Song (1989). Primers oCH36 (GCG CCC ATG GAG CAA AGC CAA TAA GAT GAT TCG) and oCH37 (GCG GAT CCT AGA TAC ATT CCC G) were used to PCR amplify DUO1 from a plasmid, and the PCR product was cloned into the AS1-CHY2 DNA binding domain vector creating pDD473, which was used as a bait to find binding partners in a cDNA library fused to the GAL4 activation domain. After the initial screen using both HIS3 to select for activation and LacZ expression as reporter of activation, the plasmids of all positive clones were recovered as described above. Over 800,000 transformants were screened using full-length Duo1p as bait. A total of 174 positive clones remained positive after retesting. These clones were sequenced in batches of 40. Presence of plasmid inserts was confirmed by XhoI digestion before sequencing. Every clone that was identified at least twice by sequencing was used to probe all remaining unsequenced isolates by DNA hybridization using an Amersham ECL Southern Analysis kit (Arlington Heights, IL). This scheme allowed us to reduce the number of clones that required sequencing. For hybridization analysis, inserts were PCR-amplified directly from the plasmid recovered from yeast using the primers SY25 (GAG ATC TGG AAT TCG GAT CC) and oCH41 (GGC ATG CCG GTA GAG GTG TGG). 2 ml of each PCR reaction was spotted onto a 12 × 12 grid drawn onto nitrocellulose filters. Filters spotted with the PCR-amplified sequences were probed using the Amersham ECL Southern Analysis kit.
Subcloning of DAM1
The clones that were isolated in the two-hybrid screen were PCR-amplified from genomic DNA isolated from the yeast strain DDY1102. DAM1 was amplified using primers oCH52 (GCG GGA TCC ATG AGC GAA GAT AAA GCT AAA TTA GGG) and oCH56 (CTA GTC TAG AAT CAG TCA GCT CAT C). All 5′ primers contained the sequence for a BamHI restriction site and the original ATG of the individual clone. The 3′ primers were located 100–200 bp downstream of the stop codon and contained an XbaI restriction site. The PCR products were subcloned into the Bluescript SK+ vector (DAM1: pDD478) and into the galactose-inducible green fluorescent protein (GFP) construct pDD113 (DAM1: pDD480).
A DAM1 deletion construct was created by PCR using oCH46 (CTG ATA AGC TCA GCA ATT GCA CCA AAA CAA TAT GAG AAA AGG CTT GTA TTG CCA CTT TCA CCG ATT GTA CTG AGA GTG CAC C) and oCH47 (TTG TGA GGA GGA TAA TTC TTT GGT TGG GTT GGG CGT AGT CAT CTG AAG GGG GGC CTT GTA CTG TGC GGT ATT TCA CAC CGC) and vector pRS313 as a template. The construct was purified by agarose gel electrophoresis and transformed into DDY1102. Correct integration was tested by genomic PCR using primers oCH48 (GCG TTG CCC GGA CAA TAT CG) and oCH49 (CTG CCT TCC TCC CTA TTG C).
Localization and Overexpression of Dam1p
To localize Dam1p using the galactose promoter–driven expression of GFP–Dam1p, the plasmid was transformed into strain DDY757 or DDY759. The strains were grown in minimal medium containing glucose into early log phase, washed in minimal medium lacking a carbon source, and resuspended in minimal medium containing 2% glycerol for 10 h. The cells were then washed with minimal medium lacking a carbon source and resuspended in minimal medium containing 2% galactose. After 4–6 h of fusion protein expression at 30°C, GFP fusion protein localization was observed directly in live cells by fluorescence microscopy. Alternatively, cells were fixed, stained with the YOL134 antitubulin antibody, and examined by fluorescence microscopy, since the GFP–Dam1p fusion protein still fluoresces when fixed. To determine whether Dam1p overexpression was lethal, its open reading frame was subcloned behind the galactose promoter in pDD42 (pDD482) and then transformed into yeast strain DDY759. These yeast strains were streaked onto glucose and galactose plates and incubated at 30°C.
Results
Duo1p Overexpression Causes Spindle Abnormalities
DUO1 (Death Upon Overproduction) (YGL061c) was isolated from a genomic library (Ramer et al., 1992) in a screen for genes that, when overexpressed from a galactose-inducible promoter, cause death and morphological arrest phenotypes suggestive of defects in cytoskeleton function. The DUO1 gene is predicted to encode a protein of 247 amino acids (27.5 kD). Analysis of the protein sequence failed to reveal structural motifs or similarity to other proteins in the public databases. As shown in Fig. 1 A, cells overexpressing DUO1 arrested at the large-budded cell cycle stage. 4′,6-diamidino-2-phenylindole (DAPI) staining revealed that these cells arrested with a single undivided nucleus. This phenotype suggested a defect in mitotic spindle function both because the mitotic checkpoint arrests yeast at the large-budded stage, and because wild-type yeast at the large-budded stage typically contain a divided nucleus (Jacobs et al., 1988; Hoyt et al., 1990; Saunders and Hoyt, 1992; Li et al., 1993; Reijo et al., 1994).
Consistent with the arrest phenotype described above, cells overexpressing DUO1 contain abnormal microtubule arrangements (Fig. 1 A). After 8 h of DUO1 overexpression induced from a galactose-regulated promoter, 91% of yeast cells contained a short mitotic spindle. This block with short mitotic spindles can be seen well when the spindle pole bodies are stained with Tub4p antibodies (Fig. 1 B, c). The spindle pole bodies remain separated by only about the diameter of the nucleus. Since essentially all of the arrested cells contained two distinct spindle pole bodies, we conclude that spindle pole body duplication is unaffected by DUO1 overexpression. FACS® analysis of haploid cells overexpressing Duo1p for 8 h revealed an average DNA content of about 4 N (Fig. 1 C), most likely reflecting continuation of DNA replication in the absence of mitosis, perhaps after cells break through a mitotic checkpoint arrest. Thus, the arrest caused by DUO1 overexpression appears not to have resulted from inhibition of DNA replication. After longer times (16 h) of overexpression, no microtubules were observed by immunofluorescence in 99% of arrested cells (Fig. 1 A). While the loss of microtubules is quite striking, the long time required to develop this phenotype raises questions about its significance.
Duo1p Colocalizes with Spindle Microtubules
A rabbit antibody was raised against bacterially expressed Duo1p containing a six His-tag. After affinity purification, the antibody recognized a single band at the predicted size for Duo1p (32 kD) in yeast whole-cell extracts (Fig. 2 a). Indirect immunofluorescence microscopy experiments using this antibody showed that the protein is located along nuclear microtubules and appears concentrated at spindle pole bodies (Fig. 2). Duo1p was not detected along cytoplasmic microtubules. Consistent with this observation, in G1, a cell cycle stage during which spindle assembly has not yet occurred, Duo1p staining is only seen in the vicinity of the spindle pole body. Localization of a GFP–Duo1p fusion protein yielded the same results (not shown).
Figure 2.





Subcellular localization of Duo1p. To demonstrate antibody specificity, a shows an immunoblot in which a yeast whole cell extract was probed with the Duo1p antiserum. A log phase culture of wild-type strain DDY898 was fixed and stained with antibodies against tubulin (b), Duo1p (c), and with DAPI to visualize DNA (d). The Duo1p localization by immunofluorescence was confirmed by localization of GFP–Duo1p (not shown). (e) A longitudinal EM section through a mitotic spindle, passing through one of the spindle pole bodies (S) and showing numerous spindle microtubules. Duo1p localization is shown by the 10-nm gold particles that appear to associate primarily with microtubules. Bars: (b–d) 5 μm; (e) 0.2 μm.
The spindle localization of Duo1p was confirmed by immunoelectron microscopy. Staining of thin sections of yeast cells with Duo1p antibody and gold-labeled secondary antibodies showed colocalization with spindle microtubules (Fig. 2 e).
To determine whether the localization of Duo1p was dependent on microtubules in vivo, microtubules of wild-type strain DDY898 were depolymerized by the addition of 20 μg/ml benomyl. After incubation for 2 h at 25°C, immunofluorescence revealed that essentially all of the microtubules were depolymerized. With the elimination of microtubules, Duo1p staining also vanished (data not shown).
Temperature-sensitive duo1 Mutants Display a Spindle Elongation Defect
A diploid strain heterozygous for a knockout of DUO1 was constructed as described in Materials and Methods. Upon meiosis, only the Duo1+ spores gave rise to viable progeny, suggesting that DUO1 is an essential gene. Therefore, to examine the loss-of-function phenotype of DUO1 in vivo, we created conditional-lethal alleles. A plasmid carrying the entire DUO1 open reading frame was mutagenized using hydroxylamine. Plasmids conferring temperature sensitivity in a duo1 deletion strain were isolated (see Materials and Methods). We recovered two temperature-sensitive alleles. Each allele carries two mutations. pDD476 (duo1-1) has the mutations E67K and A157V, and pDD477 (duo1-2) has the mutations A117T and M124I. These alleles were subsequently integrated into the genome at the DUO1 locus (see Materials and Methods).
To examine the temperature-sensitive phenotype, strains carrying the mutated gene were grown at 25°C and then shifted to 37°C. The morphologies of wild-type control cells and duo1 mutant (plasmid borne) cells were examined every 3 h for 12 h (see Table II). During this time, cells were maintained in log phase. The cell cycle distribution of wild-type cells as evaluated by cell morphology was not affected dramatically by the temperature shift. The mutant cultures, on the other hand, showed a dramatic reduction in unbudded and small-budded cells and a dramatic increase in large-budded cells within several hours after the shift to the restrictive temperature, suggestive of a mitotic defect. At later times (i.e., 5 h) large-budded cells began to form additional buds.
Table II.
Analysis of duo1 Mutant Arrest Phenotypes
| Strain | Time after temperature shift |

|
No microtubule structures | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| h | ||||||||||||||
| wild type | 0 | 31 | 35 | 34 | ||||||||||
| 3 | 30 | 37 | 33 | |||||||||||
| 6 | 28 | 40 | 32 | |||||||||||
| 9 | 31 | 39 | 30 | |||||||||||
| 12 | 36 | 37 | 27 | |||||||||||
| duo1-1 | 0 | 24 | 31 | 31 | 14 | |||||||||
| 3 | 20 | 18 | 21 | 41 | ||||||||||
| 6 | 21 | 10 | 14 | 55 | ||||||||||
| 9 | 17 | 8 | 8 | 62 | 5 | 2 | ||||||||
| 12 | 11 | 2 | 4 | 65 | 9 | 9 | ||||||||
| duo1-2 | 0 | 25 | 27 | 31 | 13 | 4 | ||||||||
| 3 | 25 | 5 | 10 | 57 | 3 | |||||||||
| 6 | 21 | 5 | 8 | 58 | 5 | 3 | ||||||||
| 9 | 14 | 2 | 4 | 43 | 3 | 35 | ||||||||
| 12 | 13 | 2 | 1 | 30 | 3 | 52 | ||||||||
Wild-type and temperature-sensitive duo1-1 and duo1-2 cells were grown at the permissive temperature and then shifted to the nonpermissive temperature for the indicated times. Spindles were examined by immunofluorescence every 3 h after temperature shift.
When cells carrying integrated duo1-2 were grown at the restrictive temperature for 90 min, essentially all of the large-budded cells contained short spindles (100%, n = 23) (Fig. 3 b). In wild-type cells, most large-budded cells contain elongated spindles (Fig. 3 a). Thus, the earliest defect to develop in the duo1 mutants seems to be the inability to elongate the spindle. When duo1-2 mutant cells were grown for 3 h or 4 h at the restrictive temperature, a large-budded arrest was evident in 81 and 90% of the cells, respectively. After 3 h at the restrictive temperature, 92% (n = 169) of large-budded duo1-2 cells contained a short spindle.
Figure 3.

Immunofluorescence analysis of microtubules in temperature-sensitive duo1 mutants. Wild-type cells, chromosomally integrated duo1-2 mutant cells, and duo-1 mad2Δ double mutant cells were grown to log phase at 25°C, shifted to 37°C for 90 min, and fixed for immunofluorescence microscopy. a–c show microtubule fluorescence for wild-type and duo1-2 and duo1-2 mad2Δ double mutant cells, respectively. d–f show DNA (DAPI) staining for the same cells shown in a–c. Bar, 10 μm.
In response to spindle abnormalities, the spindle assembly checkpoint arrests cells before anaphase. Therefore, the short spindle arrest observed for the duo1 mutants at the restrictive temperature may be the result of a spindle defect that is sensed by the spindle assembly checkpoint. Alternatively, the duo1 mutant might be defective mechanically in spindle elongation. To distinguish between these possibilities, a double mutant of duo1-2 and the mitotic checkpoint mutant mad2Δ was generated. Although the duo1-2 mutation is still lethal when this double mutant was incubated at the restrictive temperature, the large-budded arrest observed with the duo1-2 single mutant was not observed. In addition, when microtubules were visualized in the double mutant by immunofluorescence with antitubulin antibodies (Fig. 3 c), the accumulation of short spindles observed in the duo1-2 single mutant did not occur. After 1.5 and 3 h at the nonpermissive temperature, 92% (n = 13) and 89% (n = 47), respectively, of large-budded duo1-2 mad2Δ double mutant cells contained long spindles. While the duo1-2 mad2Δ double mutant is able to undergo spindle elongation, the resulting elongated spindles are abnormal and have aberrant morphologies (Fig. 3 c). Specifically, many of the spindles in this double mutant, particularly the longer spindles, appear broken or bent. The ability of the duo1-2 mad2Δ spindles to elongate, even though this process is not completely normal, suggests that Duo1p is not specifically required for the process of spindle elongation, but instead contributes to an aspect of spindle function that is sensed by the mitotic checkpoint.
Identification of Dam1p, a Protein That Interacts Physically with Duo1p
To identify proteins that might function with Duo1p, a two-hybrid screen (Fields and Song, 1989) was carried out (see Materials and Methods). Thirteen genes were recovered at least four times (DAM1, LCP5, PUP1, AIP2, CIM5, VMA8, DEP1, YIR004w, YMR012w, YER049w, YDR016c, YGR120c, and YJL036w). Proteins encoded by a subset of these genes (DAM1, LCP5, CIM5, YIR004w, YMR012w, YER049w, YDR016c, and YGR120c) were localized as GFP fusions. One protein showed spindle localization (see below) and was characterized further. The gene for this protein was designated DAM1 for Duo1p and Mps1p interacting factor (YER113w). This gene was identified independently in a screen for mutants that enhanced the defects of mutants in MPS1, a gene encoding a protein kinase implicated in mitotic functions (Bachant, J., M. Jones, and M. Winey, personal communication).
A diploid strain heterozygous for a knockout of DAM1 was constructed (see Materials and Methods). Upon meiosis, only the Dam1+ spores gave rise to viable progeny, suggesting that DAM1 is an essential gene. Dam1p is a protein of 335 amino acids (37.8 kD). Database searches and sequence analysis did not identify any homologues in the public databases or sequence motifs.
To confirm that the two-hybrid interaction between Duo1p and Dam1p was the result of direct physical binding, in vitro–translated Dam1p was incubated with glutathione beads coated with GST or GST–Duo1p. Please note that several translation products, some presumably resulting from internal translation initiation or premature translation arrest, were evident when Dam1p was translated. The beads were pelleted and washed with buffer containing 50 mM NaCl, and the presence of the in vitro– translated Dam1p in the supernatant and pellet fractions was monitored by analysis on protein gels (Fig. 4). Dam1p showed markedly enhanced interaction with GST–Duo1p compared with GST alone. An in vitro–translated fragment of mammalian tau protein (see below) served as a negative control and did not interact with either GST- or GST–Duo1p–coated beads (Fig. 4).
Figure 4.

Binding of Dam1p to Duo1p. GST alone and GST– Duo1p fusion proteins were purified from yeast using glutathione beads (see Materials and Methods). Beads containing these proteins were then incubated with in vitro–translated three-repeat Tau (a negative control, see text) and with Dam1p (full-length Dam1p at 39 kD, with early termination product at ∼29 kD). After incubation, the beads were pelleted, and proteins present in the supernatants (S) and pellets (P) were examined on SDS-PAGE gels. The top panel shows an autoradiograph of the negative control, three-repeat Tau, which did not pellet with the beads. The bottom panel shows that Dam1p appears to bind specifically to GST–Duo1p beads but not to GST beads.
To determine the intracellular localization of Dam1p, a GFP–Dam1p fusion protein was expressed on a plasmid using a galactose-inducible promoter. GFP–Dam1p was found to be associated with intranuclear spindle microtubules and spindle pole bodies throughout the cell cycle, but not with cytoplasmic microtubules (Fig. 5). Similar localization has been observed by expressing an epitope-tagged Dam1p at endogenous levels in yeast (Jones, M., and M. Winey, personal communication). In this case, Dam1p was only detected at spindle pole bodies and along intranuclear microtubules of the shortest spindles observed in the culture.
Figure 5.
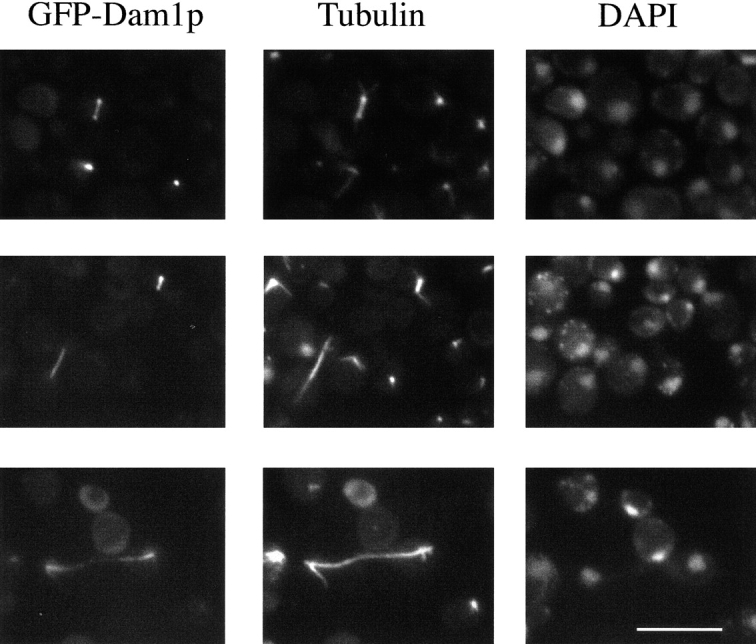
Localization of Dam1p. DDY759 was transformed with a plasmid expressing a galactose-inducible GFP–Dam1p fusion protein. Cells were grown to early log phase in minimal medium containing glucose, and then the cells were washed and shifted to and maintained in minimal medium containing 2% glycerol for 10 h. The cells were then washed and shifted to medium containing 2% galactose. After 5–6 h of induction, cells were fixed and stained with an antitubulin antibody (middle column) and DAPI for DNA staining (right column). The GFP fluorescence from GFP–Dam1p (left column) colocalizes with the intranuclear spindle microtubules and spindle pole bodies. Bar, 10 μm.
Dam1p Overexpression Is Lethal and Causes Spindle Defects
Since overexpression of Duo1p was lethal and caused pronounced mitotic defects, the overexpression phenotype for DAM1 was determined. As with Duo1p, overexpression of Dam1p was lethal. Furthermore, overexpression of Dam1p resulted in a large-budded cell cycle arrest in ∼90% of the cells. The nuclei in the majority of the large-budded cells were undivided or only partially divided, and in either case, the microtubules were organized abnormally. Often, what appeared to be a single long microtubule and one to several short microtubules were organized around a single structure, most likely the spindle pole body (Fig. 6, bottom). In the large-budded cells with partially divided nuclei, the spindle often was bipolar but appeared bent or broken (Fig. 6, top and middle). In some cells, Dam1p localizes to a structure that appears to be a spindle, bent or broken, but which is poorly recognized or not recognized at all by antitubulin antibodies (Fig. 6, top). This may be because GFP–Dam1p physically blocks the antitubulin antibody. After 10 h of DAM1 overexpression, no microtubules were detected by immunofluorescence, a phenotype reminiscent of the effects of prolonged Duo1p overexpression (Fig. 1), and GFP–Dam1p fluorescence was also absent (not shown). Overexpression of wild-type Dam1p and GFP–Dam1p had the same effect on yeast cells, indicating some degree of functionality for the GFP fusion protein.
Figure 6.
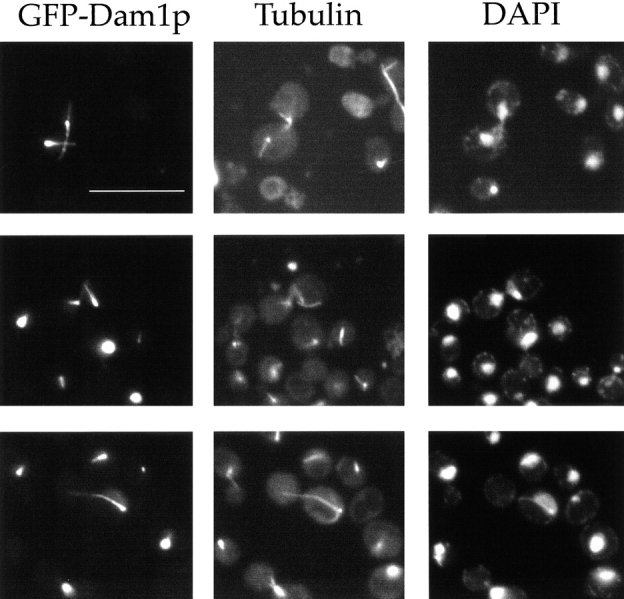
Overexpression of Dam1p results in spindle abnormalities. Dam1p was overexpressed in DDY759 as a GFP–Dam1p construct. Cells are shown after 8 h of overproduction. Fluorescence analysis of microtubule organization in cells overexpressing GFP– Dam1p is shown in the middle panels. Right panels show DNA staining by DAPI. Left panels show GFP–Dam1p fluorescence. Approximately 90% of cells overexpressing Dam1p or GFP–Dam1p arrested at the large-budded stage and displayed abnormal spindles or apparently monopolar spindles. Bar, 10 μm.
Dam1p Binds to Microtubules In Vitro
Attempts to test whether Duo1p binds to microtubules directly were inconclusive. Therefore, the question of what interaction accounts for the colocalization of Duo1p with spindle microtubules remained open. We next tested whether Dam1p binds to microtubules. The experiment in Fig. 7 A shows that in vitro–translated Dam1p binds to microtubules. In vitro translation of Dam1p resulted in three major products, all specific to the Dam1p construct. As discussed earlier in the Results, it is likely that these represent full-length Dam1p and truncated Dam1p products resulting from in frame alternative translation start site usage and/or early translational termination due to RNA secondary structure. While all three Dam1p translation products copelleted with microtubules, the two truncated Dam1p products demonstrated solubility (i.e., lack of pelleting in the absence of microtubules) and concentration-dependent microtubule binding, while the largest product pelleted partially on its own. Therefore, for K d calculations, we focused on the product marked by the arrow in Fig. 7 A. Binding of Dam1p was concentration dependent and saturable. Moreover, when binding reactions were diluted 10-fold after binding reached equilibrium, the percent binding dropped markedly, demonstrating that binding is reversible (not shown). These data show that Dam1p binding to microtubules is specific. The estimated K d of the Dam1p–microtubule interaction is 1 μM, approximately threefold weaker than the interaction between tau and microtubules (Fig. 7 B).
Figure 7.


Dam1p cosediments with microtubules. Different concentrations of taxol-stabilized microtubules were mixed with radio- labeled Dam1p or tau in vitro–translation products. The reactions were then centrifuged at high speeds to pellet the microtubules. The percentage of Dam1p and tau that copelleted with the microtubules in each reaction was determined by fractionating the pellets and supernatants on SDS-PAGE gels and quantitating levels by autoradiography and densitometry. (See text for discussion of three bands resulting when Dam1p is produced by translation in vitro.)
Discussion
Identification of Two Interacting Mitotic Spindle Proteins
In this paper, we described the identification of two novel and essential spindle proteins, Duo1p and Dam1p. Localization of Duo1p by immunofluorescence, immunoelectron microscopy, and localization of a GFP–Duo1p fusion protein in living cells suggested that Duo1p associates exclusively with intranuclear microtubules and spindle pole bodies and not with cytoplasmic microtubules. Two-hybrid and biochemical data demonstrated that Duo1p interacts with another protein, Dam1p. This interaction appeared to be physiologically relevant since Dam1p, like Duo1p, is associated with intranuclear microtubules and spindle pole bodies. Furthermore, overproduction of Dam1p, like overproduction of Duo1p, causes large-budded arrest and spindle morphologies consistent with defects in spindle function.
Cosedimentation assays demonstrated saturable binding of Dam1p to microtubules with micromolar affinity. Since experiments aimed at testing whether Duo1p interacts with microtubules directly were inconclusive, it is possible that Dam1p mediates interaction of Duo1p with spindle microtubules. The isolation of conditional-lethal mutants in the DAM1 gene will allow further elucidation of the mitotic function of Dam1p and tests of the hypothesis that Duo1p interacts with spindle microtubules via an interaction with Dam1p.
Possible Functions and Regulation
In this study, both Duo1p and Dam1p were functionally linked to the mitotic spindle. While two different temperature-sensitive alleles of duo1 are able to duplicate their spindle pole bodies and assemble a mitotic spindle at the nonpermissive temperature, there is a striking accumulation of large-budded cells with short spindles, indicating that spindle elongation does not occur in these mutants. While this observation is consistent with the possibility that Duo1p has a direct role in the mechanics of mitotic spindle elongation, this type of arrest phenotype is also suggestive of a spindle defect that is sensed by the spindle assembly checkpoint. This hypothesis is supported by the observation that unlike the duo1-2 single mutant, a duo1-2 mad2Δ double mutant does not arrest with short spindles.
Below, we consider three possible roles for Duo1p and Dam1p in mitotic spindle function. These roles are not mutually exclusive. First of all, and perhaps most likely, Duo1p and Dam1p might play a physical role in which they contribute to the structure and integrity of the mitotic spindle. This possibility is supported by the observation that mutants in DUO1 can form spindles capable of elongation when the mitotic checkpoint is defective, but that these elongated spindles have abnormal structure. In this regard, it is possible that Duo1p and Dam1p function with, or similarly to, Ase1p and Bik1p, functionally redundant mitotic spindle proteins (Pellman et al., 1995). The phenotypes observed for DUO1 and DAM1 overexpression, and for the arrest of duo1 mutants, is somewhat reminiscent of the arrest phenotype observed for the bik1-S419 ase1-1 double mutant after release from α-factor arrest. This double mutant is able to assemble a bipolar spindle at the restrictive temperature, and although there is not a significant large-budded morphological arrest, 85% of bik1 ase1 cells arrested with a short spindle. At longer times, some of these cells undergo anaphase but form spindles with defective structures. It must be noted that unlike Duo1p and Dam1p, Ase1p is absent from the spindle pole bodies and is restricted to the spindle midzone in cells with elongated spindles.
A second possibility is that Duo1p and Dam1p play a role in either generating or transducing forces required for spindle assembly or elongation. Spindle elongation, also known as anaphase B, requires elongation of intranuclear microtubules via assembly at their plus ends (Masuda and Cande, 1987) and force production from kinesin-related motor proteins located on intranuclear microtubules (Roof et al., 1992; Saunders and Hoyt, 1992). One way in which Duo1p might contribute to these processes is by associating with or regulating motor proteins like Cin8p and Kip1p, or the microtubule-associated protein Stu1p, all of which localize to intranuclear spindle microtubules, such that forces could be transduced between antiparallel arrays. We consider such a role less likely for two reasons. First, Cin8p, Kip1p, and Stu1p are required for spindle assembly (Hoyt et al., 1992; Roof et al., 1992; Pasqualone and Huffaker, 1994), but a bipolar spindle is assembled at the restrictive temperature in duo1 mutants. Furthermore, despite the short spindle arrest observed for a duo1 mutant, a duo1-2 mad2Δ double mutant is able to undergo spindle elongation (Fig. 3).
Third, Duo1p and Dam1p may play a regulatory role, such as promoting the metaphase to anaphase transition. It is intriguing that two of the genes isolated in the two-hybrid screen with DUO1, CIM5 (Ghislain et al., 1993) and PUP1, encode components of the 26S proteosome and its 20S core, respectively. Some of the proteins involved in anaphase, such as Ase1p, are marked for proteolysis by ubiquitination from the anaphase promoting complex (Juang et al., 1997). These observations raise the possibility that one function of Duo1p might be to bring proteosome subunits into proximity with their spindle targets.
Our current view is that Duo1p and possibly Dam1p do not mediate spindle elongation per se, but instead contribute to yet-to-be identified properties of the spindle, at least one of which is monitored by the mitotic checkpoint. Further hints about a role for Duo1p and Dam1p are provided by the independent discovery of DAM1 by J. Bachant, M. Jones, and M. Winey (personal communication). In this case, DAM1 was found in a screen for mutants that enhance the severity of mps1-1. Mps1p is a protein kinase with functions both in spindle pole body duplication (Winey et al., 1991) and the spindle assembly checkpoint (Weiss and Winey, 1996), suggesting possible roles for Dam1p, and maybe Duo1p, in these processes.
While several observations do hint at regulatory roles, the abnormal elongated spindles that are observed in the duo1-2 mad2Δ double mutant suggest that there are additional nonregulatory roles for Duo1p. Further analysis of the duo1 mad2 phenotype, as well as elucidation of interactions, activities, and regulation of Duo1p and Dam1p, promise to provide novel insights into the complex mechanisms that underlie spindle function during mitosis.
Acknowledgments
This work was supported by grants to D. Drubin from the National Institute of General Medical Sciences (GM-50399) and the American Cancer Society (CB-106), to G. Barnes from the National Institute of General Medical Sciences (GM-47842), to B.L. Goode from the National Institute of General Medical Sciences (GM-17715-02), and a National Science Foundation Graduate Research Fellowship to I.M. Cheeseman.
Abbreviations used in this paper
- DAPI
4′, 6-diamidino-2-phenylindole
- 5-FOA
5-fluoroorotic acid
- GFP
green fluorescent protein
- GST
glutathione-S-transferase
Footnotes
The Tub4p antibody was a generous gift from Tim Stearns. We thank Michelle (Shelly) Jones and Mark Winey for sharing results before publication.
Address all correspondence to David G. Drubin, 401 Barker Hall, Department of Molecular and Cell Biology, University of California, Berkeley, CA 94720-3202. Tel.: (510) 642-3692. Fax: (510) 642-6420. E-mail: drubin@uclink4.berkeley.edu
References
- Berlin V, Styles CA, Fink GR. BIK1, a protein required for microtubule function during mating and mitosis in Saccharomyces cerevisiae, colocalizes with tubulin. J Cell Biol. 1990;111:2573–2586. doi: 10.1083/jcb.111.6.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottingham FR, Hoyt MA. Mitotic spindle positioning in Saccharomyces cerevisiaeis accomplished by antagonistically acting microtubule motor proteins. J Cell Biol. 1997;138:1041–1053. doi: 10.1083/jcb.138.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Miller KG, Botstein D. Yeast actin-binding proteins: evidence for a role in morphogenesis. J Cell Biol. 1988;107:2551–2561. doi: 10.1083/jcb.107.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Gambino J, Bergen LG, Morris NR. Effects of mitotic and tubulin mutations on microtubule architecture in actively growing protoplasts of Aspergillus nidulans. . J Cell Biol. 1984;99:830–838. doi: 10.1083/jcb.99.3.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislain M, Udvardy A, Mann C. S. cerevisiae26S protease mutants arrest cell division in G2/metaphase. Nature. 1993;366:358–362. doi: 10.1038/366358a0. [DOI] [PubMed] [Google Scholar]
- Goode BL, Denis PE, Panda D, Radeke MJ, Miller HP, Wilson L, Feinstein SC. Functional interactions between the proline-rich and repeat regions of Tau enhance microtubule binding and assembly. Mol Biol Cell. 1997;8:353–365. doi: 10.1091/mbc.8.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG. The spindle checkpoint. Trends Genet. 1998;14:1–4. doi: 10.1016/S0168-9525(97)01340-1. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Immunoaffinity purification. In Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 511–552.
- Hoyt MA, Stearns T, Botstein D. Chromosome instability mutants of Saccharomyces cerevisiaethat are defective in microtubule-mediated processes. Mol Cell Biol. 1990;10:223–234. doi: 10.1128/mcb.10.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, He L, Loo KK, Saunders WS. Two Saccharomyces cerevisiaekinesin-related gene-products required for mitotic spindle assembly. J Cell Biol. 1992;118:109–120. doi: 10.1083/jcb.118.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs CW, Adams AE, Szaniszlo PJ, Pringle JR. Functions of microtubules in the Saccharomyces cerevisiaecell cycle. J Cell Biol. 1988;107:1409–1426. doi: 10.1083/jcb.107.4.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang YL, Huang J, Peters JM, McLaughlin ME, Tai CY, Pellman D. APC-mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science. 1997;275:1311–1314. doi: 10.1126/science.275.5304.1311. [DOI] [PubMed] [Google Scholar]
- Li R, Havel C, Watson JA, Murray AW. The mitotic feedback control gene MAD2 encodes the α-subunit of a prenyltransferase [published erratum appears in 371:438] Nature. 1993;366:82–84. doi: 10.1038/366082a0. [DOI] [PubMed] [Google Scholar]
- Machin NA, Lee JM, Barnes G. Microtubule stability in budding yeast: characterization and dosage suppression of a benomyl-dependent tubulin mutant. Mol Biol Cell. 1995;6:1241–1259. doi: 10.1091/mbc.6.9.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis, T., E.F. Fritsch, and J. Sambrook. 1982. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 545 pp.
- Marschall LG, Jeng RL, Mulholland J, Stearns T. Analysis of Tub4p, a yeast γ-tubulin-like protein: implications for microtubule-organizing center function. JCell Biol. 1996;134:443–454. doi: 10.1083/jcb.134.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda H, Cande WZ. The role of tubulin polymerization during spindle elongation in vitro. Cell. 1987;49:193–202. doi: 10.1016/0092-8674(87)90560-5. [DOI] [PubMed] [Google Scholar]
- Nicklas RB. How cells get the right chromosomes. Science. 1997;275:632–637. doi: 10.1126/science.275.5300.632. [DOI] [PubMed] [Google Scholar]
- Oakley B, Morris NR. Nuclear movement is β-tubulin dependent in Aspergillus nidulans. . Cell. 1980;19:255–262. doi: 10.1016/0092-8674(80)90407-9. [DOI] [PubMed] [Google Scholar]
- Oakley, B.R. 1994. γ-Tubulin. Wiley-Liss, Inc., New York. 33–45.
- Oakley BR, Rinehart JE. Mitochondria and nuclei move by different mechanisms in Aspergillus nidulans. . J Cell Biol. 1985;101:2392–2397. doi: 10.1083/jcb.101.6.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualone D, Huffaker TC. STU1, a suppressor of a β-tubulin mutation, encodes a novel and essential component of the yeast mitotic spindle. J Cell Biol. 1994;127:1973–1984. doi: 10.1083/jcb.127.6.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellman D, Bagget M, Tu YH, Fink GR, Tu H. Two microtubule-associated proteins required for anaphase spindle movement in Saccharomyces cerevisiae[published erratum appears in 131:561] J Cell Biol. 1995;130:1373–1385. doi: 10.1083/jcb.130.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle JR, Adams AE, Drubin DG, Haarer BK. Immunofluorescence methods for yeast. Methods Enzymol. 1991;194:565–602. doi: 10.1016/0076-6879(91)94043-c. [DOI] [PubMed] [Google Scholar]
- Ramer SW, Elledge SJ, Davis RW. Dominant genetics using a yeast genomic library under the control of a strong inducible promoter. Proc Natl Acad Sci USA. 1992;89:11589–11593. doi: 10.1073/pnas.89.23.11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijo RA, Cooper EM, Beagle GJ, Huffaker TC. Systematic mutational analysis of the yeast β-tubulin gene. Mol Biol Cell. 1994;5:29–43. doi: 10.1091/mbc.5.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roof DM, Meluh PB, Rose MD. Kinesin-related proteins required for assembly of the mitotic spindle. J Cell Biol. 1992;118:95–108. doi: 10.1083/jcb.118.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Hieter. 1990. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 198 pp.
- Rout MP, Kilmartin JV. Components of the yeast spindle and spindle pole body. J Cell Biol. 1990;111:1913–1927. doi: 10.1083/jcb.111.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner AD, Murray AW. The spindle assembly checkpoint. Curr Opin Cell Biol. 1996;8:773–780. doi: 10.1016/s0955-0674(96)80077-9. [DOI] [PubMed] [Google Scholar]
- Saunders WS, Hoyt MA. Kinesin-related proteins required for structural integrity of the mitotic spindle. Cell. 1992;70:451–458. doi: 10.1016/0092-8674(92)90169-d. [DOI] [PubMed] [Google Scholar]
- Sobel SG. Mini review: mitosis and the spindle pole body in Saccharomyces cerevisiae. . J Exp Zool. 1997;277:120–138. doi: 10.1002/(sici)1097-010x(19970201)277:2<120::aid-jez4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Sobel SG, Snyder M. A highly divergent gamma-tubulin gene is essential for cell growth and proper microtubule organization in Saccharomyces cerevisiae. . J Cell Biol. 1995;131:1775–1788. doi: 10.1083/jcb.131.6.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A, Courtney I, Grein K, Matzner M, Schiebel E. The Cdc31p-binding protein Kar1p is a component of the half bridge of the yeast spindle pole body. J Cell Biol. 1995;128:863–877. doi: 10.1083/jcb.128.5.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PJ, Huffaker T. Stu2p: a microtubule-binding protein that is an essential component of the yeast spindle pole body. J Cell Biol. 1997;139:1271–1280. doi: 10.1083/jcb.139.5.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters JC, Salmon E. Pathways of spindle assembly. Curr Opin Cell Biol. 1997;9:37–43. doi: 10.1016/s0955-0674(97)80149-4. [DOI] [PubMed] [Google Scholar]
- Weiss E, Winey M. The Saccharomyces cerevisiaespindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J Cell Biol. 1996;132:111–123. doi: 10.1083/jcb.132.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winey M, Goetsch L, Baum P, Byers B. MPS1 and MPS2: novel yeast genes defining distinct steps of spindle pole body duplication. J Cell Biol. 1991;114:745–754. doi: 10.1083/jcb.114.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]