

Abstract
Laminins are the major noncollagenous glycoproteins of all basal laminae (BLs). They are α/β/γ heterotrimers assembled from 10 known chains, and they subserve both structural and signaling roles. Previously described mutations in laminin chain genes result in diverse disorders that are manifested postnatally and therefore provide little insight into laminin's roles in embryonic development. Here, we show that the laminin α5 chain is required during embryogenesis. The α5 chain is present in virtually all BLs of early somite stage embryos and then becomes restricted to specific BLs as development proceeds, including those of the surface ectoderm and placental vasculature. BLs that lose α5 retain or acquire other α chains. Embryos lacking laminin α5 die late in embryogenesis. They exhibit multiple developmental defects, including failure of anterior neural tube closure (exencephaly), failure of digit septation (syndactyly), and dysmorphogenesis of the placental labyrinth. These defects are all attributable to defects in BLs that are α5 positive in controls and that appear ultrastructurally abnormal in its absence. Other laminin α chains accumulate in these BLs, but this compensation is apparently functionally inadequate. Our results identify new roles for laminins and BLs in diverse developmental processes.
Keywords: basement membrane, development, placenta, knockout mice, limb deformities
Cells in most tissues of vertebrates and invertebrates bear coats of basal laminae (BLs),1 thin layers of extracellular matrix whose main components are laminin, collagen IV, entactin/nidogen, and sulfated proteoglycans (Timpl, 1996; Timpl and Brown, 1996). Laminins are glycoproteins that self-assemble to form the major noncollagenous network of all BLs (Chung et al., 1979; Timpl et al., 1979; Yurchenco and O'Rear, 1994). All laminins studied to date are heterotrimers composed of one α, one β, and one γ chain (Timpl, 1996). Five α, three β, and two γ chains have now been identified, which can associate to form at least 11 heterotrimers. Different BLs contain distinct complements of laminin trimers, allowing BLs to subserve distinct functions while maintaining a uniform structure.
In adults, BLs provide structural support for tissues, serve as scaffolds to organize regeneration after tissue damage, and underlie physiological functions such as renal glomerular filtration (Timpl, 1989; Yurchenco and O'Rear, 1994). The importance of laminin to the proper function of BLs is underscored by the phenotypes of known mutations in laminin chain genes: the laminin α2 gene is mutated in some congenital muscular dystrophies (Sunada et al., 1994; Xu et al., 1994; Helbling-Leclerc et al., 1995); mutations in any one of the α3, β3, or γ2 chain genes causes junctional epidermolysis bullosa, a skin blistering disease (Aberdam et al., 1994; Pulkkinen et al., 1994; McGrath et al., 1995; Kuster et al., 1997); and targeted mutagenesis of the laminin β2 chain gene impairs both neuromuscular and renal function (Noakes et al., 1995a ,b). BLs and the laminins they contain are also likely to play critical roles in embryos by providing signals for cell proliferation, migration, and differentiation and by serving as cell attachment sites (Timpl, 1989; Yurchenco and O'Rear, 1994). However, no hitherto described mutations in laminin genes lead to major embryonic defects. Thus, either other laminin chains play predominant roles during development, or laminins might be dispensable for embryogenesis.
One good candidate for a laminin chain that could be involved in developmental processes is the laminin α5 chain (Miner et al., 1995). This chain associates with the γ1 chain and either β1 or β2 to form laminins 10 and 11, respectively (Miner et al., 1997). The laminin α5 gene is expressed in many fetal and adult tissues, including kidney, lung, skeletal muscle, skin, and intestine, and laminin α5 protein is present in specific BLs within these tissues (Durbeej et al., 1996; Lentz et al., 1997; Miner et al., 1997; Patton et al., 1997; Sorokin et al., 1997a ,b). Recently, purified laminin 11 has been shown to induce responses from cultured neurons and Schwann cells that are qualitatively different from those induced by laminins 1, 2, or 4 (α1/β1/γ1, α2/β1/γ1, and α2/β2/γ1, respectively) (Patton et al., 1997, 1998). These results suggested that laminin α5 might have unique developmental roles.
To explore roles of laminin α5 in embryos, we have documented its localization and generated mice lacking this chain. We show that laminin α5 is present in most embryonic and extraembryonic BLs at early stages, that it becomes restricted to a distinct subset of BLs as development proceeds, and that its loss leads to fetal lethality. Numerous defects were apparent in mutant homozygotes, including failure of neural tube closure (exencephaly), failure of digit septation (syndactyly), and dysmorphogenesis of the placenta. In each case, we consider mechanisms that may explain how loss of laminin α5 disrupts normal development.
Materials and Methods
Histology
Immunofluorescence microscopy was performed on cryostat sections as described previously (Miner et al., 1997). Antisera to mouse laminins α4 and α5 were described previously (Miner et al., 1997). The recombinant fragment used to generate anti–laminin α5 antiserum comprised amino acids 1415–1642 (Miner et al., 1995), which are COOH-terminal to the region deleted in Lama5 −/− embryos. Several other antibodies were provided by generous colleagues: anti–laminin α2 from Peter Yurchenco (Robert Wood Johnson Medical School, Piscataway, NJ; Cheng et al., 1997); anti–laminin-5 (α3β3γ2) from Robert Burgeson (CBRC, Harvard Medical School, Charlestown, MA; Marinkovich et al., 1992); anti–keratin 14 from Elaine Fuchs (University of Chicago, Chicago, IL; Stoler et al., 1988); and anti–laminin β1 (5A2) and anti-α1 (8B3) from Dale Abrahamson (University of Alabama at Birmingham; Abrahamson et al., 1989). We showed previously that 5A2 specifically recognizes the β1 chain (Martin et al., 1995) and have now used similar methods to show that 8B3 recognizes mouse laminin α1 (data not shown). A commercial antibody to mouse laminin γ1 (MAB1914) was from Chemicon International (Temecula, CA). Cy3- and FITC-conjugated secondary antibodies were from ICN/Cappel (Costa Mesa, CA).
For semithin and thin sectioning, embryos were fixed in 4% paraformaldehyde, 4% glutaraldehyde in 0.1 M cacodylate buffer and processed as described (Noakes et al., 1995b ). 2-μm sections were cut with glass knives and stained with toluidine blue for light microscopy. Thin sections were cut with a diamond knife and stained with lead citrate plus uranyl acetate for electron microscopy.
To label and detect dividing cells in embryos, we used the 5-Bromo-2′-deoxy-uridine (BrdU) Labeling and Detection Kit II (Boehringer Mannheim Corp., Indianapolis, IN). Pregnant females were injected intraperitoneally with 0.15 ml of 10 mM BrdU per 10 g of body weight. After 1 h, the mice were killed, and the embryos were removed, frozen, and sectioned at 7 μm on a cryostat. The label was detected in nuclei according to the manufacturer's instructions.
To stain cartilage in embryos, we used Alcian blue 8GX (Sigma Chemical Co., St. Louis, MO). Fixation, staining, and clearing were performed as described (Jegalian and De Robertis, 1992).
Generation and Genotyping of Mutant Mice
A λ clone containing exons encoding parts of domains VI, V, and IVb of laminin α5 was obtained by screening a 129sv mouse genomic library (Stratagene, La Jolla, CA) with the 5′ 900 bp of the cDNA previously described by Miner et al. (1995). To construct a targeting vector, two consecutive XbaI fragments totaling 3.5 kb and encoding 113 amino acids (129– 241 in Miner et al., 1995) were replaced with an in frame lacZ cDNA and a PGK neo cassette. The neo cassette was derived from the vector pPNT, which also supplied PGK HSV-tk for negative selection (Tybulewicz et al., 1991; see Fig. 2 A). The mutated chromosomal segment was transferred to R1 ES cells by electroporation, and transfectants were selected with G418 (400 μg/ml) and FIAU (0.2 μM). Approximately 450 clones were screened by PCR, and a single homologous recombinant clone was obtained. Cells from this clone were injected into C57BL/6J blastocysts by standard methods. Three chimeras transmitted the mutation to their offspring. Homozygotes derived from each of these founders exhibited the same phenotypes, and all phenotypes segregated with the mutation, even after five generations of crossing to C57BL/6J mice. We never detected lacZ activity in heterozygotes or homozygotes, presumably because the signal sequence of the laminin α5 protein forces the catalytic domain of the lacZ protein into the endoplasmic reticulum, where it is inactive.
Figure 2.


Targeted mutagenesis of the Lama5 gene. (A) The targeting vector deleted exons encoding 113 amino acids and replaced them with an in frame lacZ cDNA and the PGK neo selectable marker. N, NcoI; X, XbaI. (B) Southern analysis of genomic DNA from E13.5 embryos demonstrates the existence of the three expected genotypes, confirming that targeting was successful and that homozygous mutants were alive at this age. The probe, shown in A, was from outside the short arm of the targeting vector. (C and D) Presence of laminin α5 protein in control (C) but not mutant (D) distal limb ectodermal BL at E13.5, detected immunohistochemically with antisera to epitopes outside of the targeted region. Bar, 50 μm.
Mice were genotyped either by Southern blot or by PCR. For Southern analysis, genomic DNA was prepared from embryos, digested with NcoI, fractionated on an agarose gel, transferred to nitrocellulose, and probed with a fragment just outside the short arm of homology (Fig. 2 A). For PCR, lacZ-specific primers were used to identify the mutated allele, and Lama5 primers from within the deleted region were used to identify the wild-type allele.
Ra/+ mice were purchased from The Jackson Laboratory (Bar Harbor, ME).
Results
Widespread Expression of Laminin α5 in Embryos
We began this study by determining the distribution of the laminin α5 chain in embryonic tissues. Sections were double-labeled with a previously characterized rabbit antiserum to α5 (Miner et al., 1997) and a monoclonal antibody to the γ1 chain. γ1 is present in all BLs described to date and therefore serves as a general marker for them.
Laminin α5 was present in virtually all γ1-positive BLs at embryonic day (E) 8.5 (Fig. 1, A and B). These included the BL underlying the neural folds and the surface ectoderm, as well as BLs associated with gut epithelium. Thus, laminin α5 is not only a major α chain in adult BLs (Miner et al., 1997) but also a prominent component of BLs at early somite stages. At later stages, however, the distribution of α5 became restricted to a subset of BLs. In the spinal cord, for example, α5 was found throughout the pial BL at E9.5 (Fig. 1 E), but levels then decreased in dorsal regions, and by E13.5, this chain was confined to the floorplate at the ventral midline (Fig. 1 H). On the other hand, α5 remained abundant in the surface ectodermal BL throughout embryogenesis (Fig. 1 and data not shown).
Figure 1.
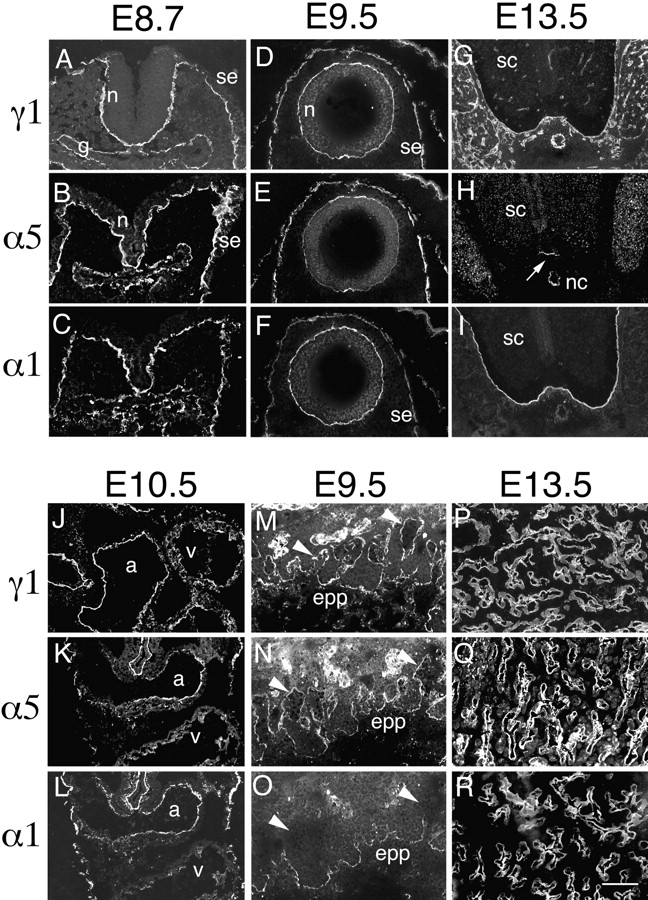
Distribution of laminin α1 and α5 chains in embryonic and extraembryonic BLs. Sections of embryos at the indicated ages were labeled with antibodies specific for the laminin α1, α5, or γ1 chains. The γ1 chain is present in, and thus serves to mark, all BLs. (A–C) BLs underlying the unclosed neuroepithelium (n), the surface ectoderm (se), and the gut epithelium (g) contain both α1 and α5 chains at E8.7. (D–F) After neural tube closure, α5 levels decrease in the neuroepithelial BL, and α1 levels decrease in the surface ectodermal BL. (G–I) By E13.5, α5 is confined to the BL adjacent to the floorplate of the spinal cord (sc) (arrow in H) and to the notochord (nc), whereas α1 is found throughout the pial BL but is absent from the notochord. Neither chain is present in BLs of blood vessels within and outside the spinal cord. These vascular BLs contain laminin α4 (not shown). (J–L) In E10.5 heart, both α1 and α5 are found in the atria (a) and in the ventricles (v), though levels of α1 are low in ventricle. (M–O) In the nascent placental labyrinth, the BLs at the base of embryonic blood vessels in the ectoplacental plate (epp) contain both α1 and α5, whereas the tips of vessels that have migrated towards the maternal blood spaces (arrowheads) contain α5 but lack α1. N and O are from a single, doubly labeled section. (P–R) In E13.5 placental labyrinth, the fetal blood vessel BLs contain both α1 and α5 throughout their lengths. Bar, 10 μm for A–C and P–R, 20 μm for all other panels.
The presence of BLs rich in γ1 but poor in α5 at later stages suggested that other α chains were being expressed. We tested this idea by staining sections with antibodies specific for the α1–α4 chains. Levels of α2–α4 were low at E8.5–9.5, but the laminin α1 chain was codistributed with α5 in most BLs at this stage (Fig. 1, C and F, and data not shown). Later, as α5 became restricted to a subset of BLs, α1 became restricted to a largely complementary subset, so the extent of their colocalization decreased. For example, the ectodermal and neuroectodermal BLs were rich in both α1 and α5 at the earliest stages examined, but each chain became restricted to distinct regions as development proceeded. Whereas α5 remained abundant in ectodermal BL but became restricted to the floor plate of the neural tube, α1 remained abundant in the neural tube (Fig. 1, H and I) but was gradually lost from the ectoderm. Though α3 was not detected at E8.5–10.5, it became a major component of ectodermal BL by E12.5, as shown previously (Aberdam et al., 1994) and discussed below.
In some BLs, levels of both α1 and α5 were low. In heart, for example, α1 and α5 were both detected in atria at E9.5 and later ages, but ventricular expression of α1 and α5 was low and regionally restricted throughout the embryonic period (Fig. 1, J–L). Likewise, small γ1-positive blood vessels that arose within the brain, spinal cord, and mesenchymal regions lacked both α1 and α5 (Fig. 1, G–I; see also Klein et al., 1990). At least one of the other known α chains, α2–α4, was found in these regions. For example, BLs of ventricular muscle were rich in α2, and blood vessels were rich in α4 (data not shown).
We also examined the distribution of the laminin α1 and α5 chains in extraembryonic tissues. At all stages examined, Reichert's membrane was rich in laminin α1 but contained comparatively little α5, whereas the yolk sac and amnion contained both α1 and α5 at E10.5 and later ages (data not shown). The few blood vessel BLs found inside the nascent placental labyrinth at E9.5 contained γ1 and α5 but not α1, whereas all three chains were found in vessel BLs near their origins in the ectoplacental plate (Fig. 1, M–O). Finally, at E13.5, the BLs of fetal vessels in the more mature placental labyrinth were rich in γ1 and both α5 and α1 (Fig. 1, P–R). Thus, α5 is a prominent component of many extraembryonic as well as embryonic BLs.
Production of Laminin α5–deficient Mice
To determine the function of laminin α5, we mutated the laminin α5 gene (Lama5) using a targeting vector that deleted exons encoding portions of the NH2-terminal domains VI and V (Fig. 2 A). With this strategy, only 211 of 3,718 amino acids were upstream of the deletion; transcription originating from the Lama5 promoter should be terminated by SV-40 sequences in the vector; and translation of any resulting mRNAs should be terminated by multiple stop codons. The targeting vector was transferred to R1 ES cells (Nagy et al., 1993) by electroporation, and a single homologous recombinant clone was obtained from 450 screened. Cells from this clone were injected into C57BL/6J blastocysts to produce three germline chimeras. Heterozygotes, which displayed no obvious abnormalities, were back-crossed to wild-type C57BL/6J mice for at least three generations to obtain a more defined genetic background. Initial and back-crossed heterozygotes were bred, but no live homozygotes were detected among ∼40 offspring, indicating that mice require laminin α5 to complete development.
To determine when homozygotes were dying, we killed timed pregnant females at E8.5–17.5. PCR (not shown), and Southern blot analyses (Fig. 2 B) showed that homozygotes were alive at E13.5. Immunostaining of tissues from confirmed homozygotes showed a complete absence of laminin α5 protein (Fig. 2, C and D). Of 267 E8.5–16.5 embryos scored, 61 (23%) were Lama5 −/−, suggesting that most homozygotes survived well past the implantation stage. However, defects became apparent after E9, and some homozygotes were dead in litters taken between E13.5 and 16.5. No homozygotes lived past E17. Consistent with the broad distribution of laminin α5, defects were visible in many tissues, including the limb, neural tube, and placenta. In the following sections, we describe these defects and consider mechanisms that could account for them. Defects in some internal organs, including lung, heart, intestine, and kidney, were also observed; these will be described elsewhere (Miner, J.H., manuscript in preparation).
Syndactyly
In normal mice, distal extremities of limbs are initially paddle-shaped, then septation occurs to form digits. The distal limbs of Lama5 −/− embryos were also initially paddle-shaped, but then became club-like and failed to form separate digits, a phenomenon known as syndactyly. This defect was visible at E12.5, as soon as septation began in controls. It became more obvious at later ages and was apparent in all Lama5 −/− mice examined (Fig. 3, A and B). The forelimbs were more severely affected than the hindlimbs; little if any septation occurred in mutant forelimbs, but digits 1 and 5 exhibited partial separation in hindlimbs by E14.5.
Figure 3.
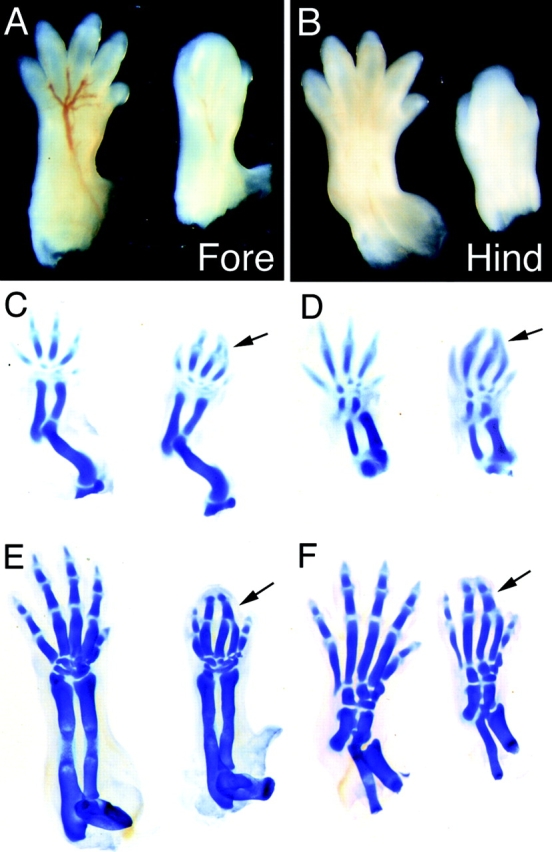
Syndactyly in Lama5 −/− embryos. Controls are on the left and mutants on the right. (A and B) Whole mount dorsal views of E14.5 fore- and hindlimbs. Digits 1 and 5 are partially septated in the mutant hindlimb. (C and D) Alcian blue staining at E13.5 demonstrates proper patterning in the mutant. Digits 2 and 3 are closely apposed but are not fused (arrows). (E and F) Alcian blue staining at E15.5 shows complete fusion of mutant digits 2 and 3 (arrows) and the absence of distal phalanges from digits 2–4 in the mutant forelimb.
Syndactyly could result either from a failure of pattern formation within the distal limb, or from failure to form septa between digits that were otherwise well formed. To distinguish between these alternatives, we stained limbs from mutant and control embryos with Alcian blue, which selectively stains cartilage. Digit pattern was established normally in Lama5 −/− embryos at E13.5 (Fig. 3, C and D), indicating that α5 was not required for the complex developmental processes that initially pattern the distal limb. Later, however, the phalanges of digits 2 and 3 fused (Fig. 3, E and F). This sequence suggests that the defective cartilage pattern is secondary to, rather than a cause of, the syndactyly in Lama5 mutants.
Genesis of the Limb Defect
We used light and electron microscopy to learn how absence of laminin α5 results in syndactyly. The following description refers to forelimbs, but similar results were obtained in hindlimbs. In normal development, the forelimb bud is visible at E9. It is covered by a continuous ectodermal epithelium, which overlies the mesenchymal cells of the dermis. The basal surface of the epithelial cells is coated by a laminin α5–rich BL, which separates ectoderm from dermis (Fig. 2 C). The ectodermal and dermal layers appeared similar in mutants and controls during early embryogenesis (not shown).
As the limb bud elongates, cells are added to the ectoderm so that it completely covers the limb at all times. In mutants, however, ∼200-μm gaps appeared in the distal epidermis slightly ventral to the tip of each digit condensation (Fig. 4, A and B). Mesenchymal cells migrated or were extruded through these gaps. Some of these cells may have been sloughed into the amniotic fluid, but many remained associated with the external (peridermal) surface of the epithelium and migrated from the hole to form a continuous, multilayered coat over each digit tip (Fig. 4, B and C). As a result, the surface ectoderm thickened and became covered on both sides by a dermis at the limb tip (Fig. 4 C), and a second BL formed at the ectopic ectodermal/dermal interface (Fig. 4 D). Apparently, either the surface ectoderm “doubled back” on itself to maintain a basal relationship with the displaced mesenchymal cells, or, alternatively, the presence of displaced mesenchyme on the apical side of the epithelial cells induced a repolarization and/or a reorganization of some of these cells, producing a second epithelial/mesenchymal interface. In either case, the additional cells in the epithelium were clearly of epidermal lineage, as shown by immunostaining with an antibody to the keratinocyte-specific antigen keratin 14 (Stoler et al., 1988). This antibody stained all surface ectodermal cells as well as the full width of the thickened mutant epithelium at this stage (data not shown). Because the interdigital mesenchyme is intimately involved in digit septation (see Jiang et al., 1998 and references therein), its depletion from the interior might be expected to impede this process. Depleted mesenchyme could also lead to the observed digit fusion, and the doubled ectoderm could physically impede proper septation. Together, these abnormalities appear sufficient to account for the syndactyly observed in Lama5 −/− embryos.
Figure 4.
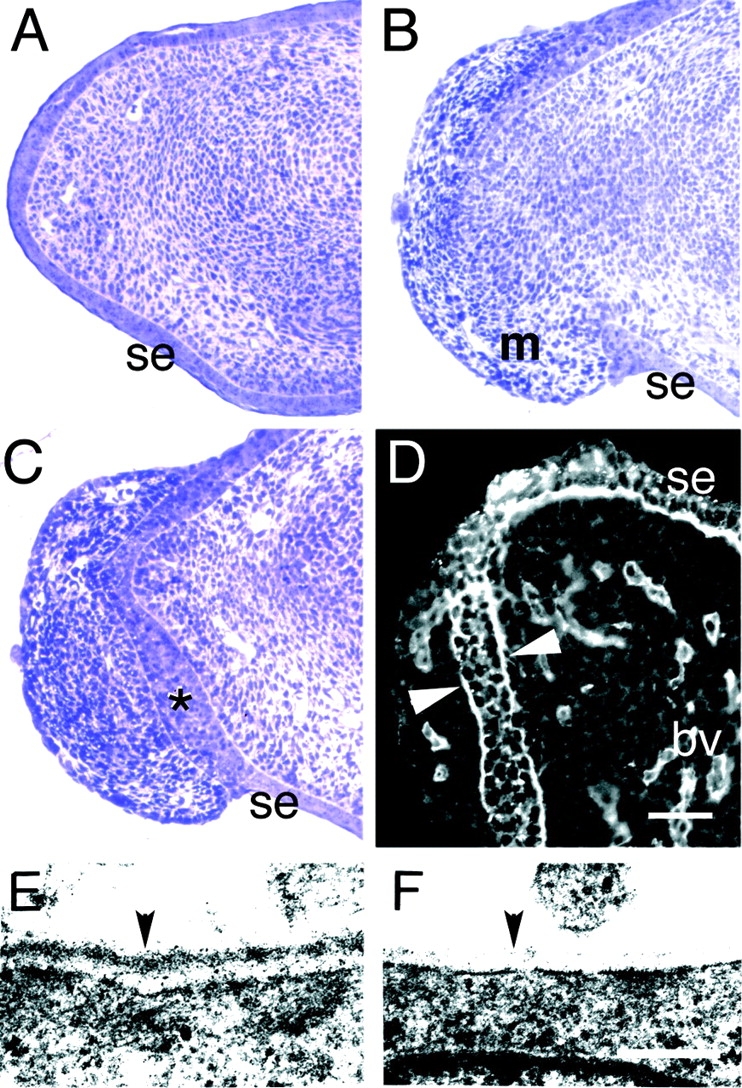
Discontinuity of epidermis and BL in the distal limb. (A–C) Toluidine blue–stained semithin sections of distal limb from control (A) and mutant (B and C) E14.5 embryos. The surface ectoderm (se) is continuous in controls (A). In mutants, in contrast, the ectoderm has been breached, and extruded mesenchymal cells (m) have migrated along the outer surface of the limb (B). A nearby section from the same mutant limb shows a thickened surface ectoderm (*) between the outer and inner mesenchymal populations (C). (D) Immunostaining of Lama5 −/− limb with an antibody to laminin γ1 demonstrates the presence of BL material on both sides of the thickened ectoderm (arrowheads), suggesting that the ectoderm has maintained a proper relationship with the displaced mesenchymal cells. (E and F) Ultrastructural analysis of distal limb BL at E14.5 shows a dense, continuous BL (arrowhead) in the control (E) but a patchy, discontinuous BL in the mutant (F). Bars: (in D) 62.5 μm for A–C, 50 μm for D; (in F) 0.25 μm for E and F.
How does loss of laminin α5 lead to the discontinuities and a thickened epithelium at the limb tip? Ultrastructural analyses showed that the BL associated with the mutant ectoderm in the limb tip was patchy and discontinuous compared with that seen in controls (Fig. 4, E and F). In contrast, the ectodermal BL in the proximal part of the limb was similar in mutants and controls (data not shown). We surmise that the discontinuities observed in the distal BL rendered it unable to maintain either its own integrity or that of the surface ectoderm, leading to rupture of the ectoderm under the stress of distal limb outgrowth. As a result, mesenchymal cells from within the limb passed through the hole and migrated along the outer surface of the limb, thereby both depleting the interior of mesenchyme and capping the limb tip.
We also considered the possibility that syndactyly resulted from defects in the apical ectodermal ridge (AER). The AER is a morphologically distinct epithelium that runs from anterior to posterior at the distal margin of the limb bud. It forms in the ectoderm at E10.5 and is necessary for limb outgrowth (Martin, 1998). That limb outgrowth occurs in the Lama5 mutants indicates that AER function is not severely disrupted by the absence of α5 from the ectodermal BL. On the other hand, we did observe that the most distal phalanges of mutant forelimb digits 2, 3, and 4 were missing or severely shortened (Fig. 3 E). Thus, the discontinuities at the distal tips of the limbs may lead to subtle defects in the AER that contribute to defects in both outgrowth and patterning (see also Jiang et al., 1998). Such subtle defects in the AER could combine with the mesenchymal and ectodermal abnormalities discussed above to account for both syndactyly and the missing distal phalanges.
Exencephaly
In ∼60% of Lama5 −/− fetuses examined, the brain was enlarged and misshapen, and it was not covered by skin or skull (Fig. 5, A and B); the remaining mutants appeared to have grossly normal heads (data not shown). This partially penetrant defect is termed exencephaly, a condition in which the cranial vault fails to develop, and the tissues of the brain are exposed (Wallace and Knights, 1978). We do not know why this defect is partially penetrant, but we do note that partial penetrance has been observed in other mutant strains that exhibit exencephaly (Wallace and Knights, 1978; Macdonald et al., 1989; Vogelweid et al., 1993; Harris and Juriloff, 1997).
Figure 5.
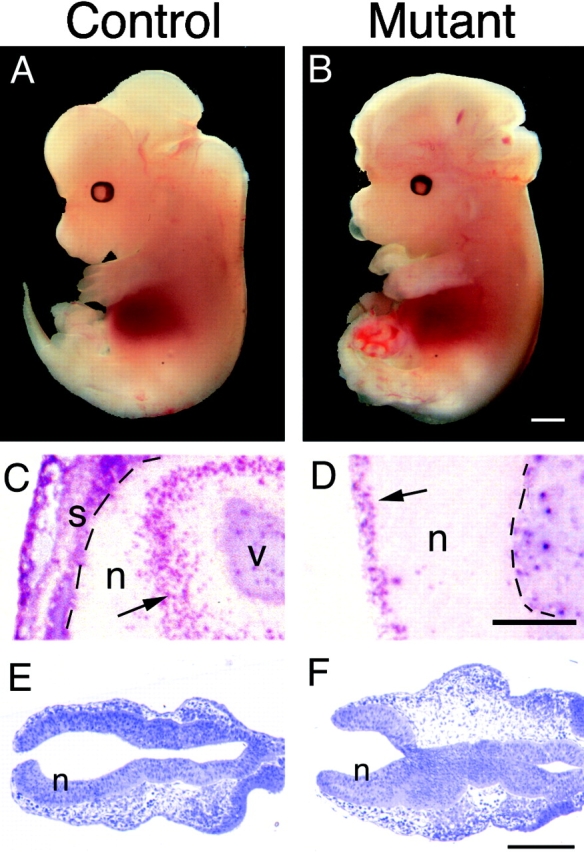
Exencephaly in Lama5 −/− embryos. (A and B) Whole mount views of control and mutant embryos at E13.5. Exencephalics lack the skin and skull that normally enclose the brain and have a topologically “inside out” brain that does not develop properly. The skin and skull of the control was dissected away to allow direct comparison of mutant and normal brains. (C and D) Immunohistochemical localization of proliferating cells with anti-BrdU and alkaline phosphatase chemistry after labeling for 1 h in utero with BrdU. (C) In normal neural tissue (n), BrdU-labeled cells (arrow) are confined to the ventricular zone. v, ventricle. (D) In exencephalics, labeled cells (arrow) are found on the outer surface of the neural tissue. This surface would have been ventricular had the neural tube closed, but now it is in direct contact with amniotic fluid. Skin and skull (s) overlay the brain in the control but are missing from the mutant. Pia is indicated by dashed lines. (E and F) Toluidine blue–stained sections through the anterior of E8.7 control and mutant embryos; the neural tube is unclosed at this stage in both control and mutant. Electron micrographs shown in Fig. 6 were obtained from these regions. Bars: (B) 1 mm; (D) 0.25 mm; (F) 0.2 mm.
Exencephaly is caused by failure of the anterior neural tube to close. As a result, the left and right sheets of nonneural surface ectoderm do not fuse at the dorsal midline or detach from the neural tube, but remain contiguous with the neuroepithelium throughout development. Histological analyses of embryos at multiple gestational ages confirmed that this aspect of exencephaly was evident in the affected embryos (data not shown).
A major consequence of exencephaly is that the neuroepithelium is topologically “inside out.” In normal mice, neurons are born in a ventricular zone of proliferating cells that abuts an interior sealed ventricle, and then they migrate toward the outer pial surface to form the cortical plate. The topology of exencephalic brains predicts that the proliferating cells should lie on the outer surface or abut a “pseudoventricle” that is actually continuous with the extraembryonic space. To assess the polarity of the neuroepithelium in Lama5 −/− embryos, we labeled dividing cells in E13.5 embryos with BrdU for 1 h and detected incorporation immunohistochemically in frozen sections. Control brains and brains of Lama5 −/− mice that were not exencephalic showed characteristic labeling near the ventricles, where proliferating cells normally reside at this stage (Fig. 5 C and data not shown). In exencephalic brains, labeling was observed in cells on the outer surface (Fig. 5 D) as well as near the pseudoventricles (not shown). These patterns confirm that the cranial defect observed in the absence of laminin α5 is exencephaly.
Genesis of the Neural Defect
To determine how a lack of laminin α5 leads to exencephaly, we examined the ultrastructure of the cranial neural tube and its neighboring ectoderm in controls and mutants at E8.7, the age just before the anterior neuropore closes in normal embryos (Kaufman, 1992). Low-magnification views of sections representative of those that were used for electron microscopy demonstrated that the anterior neuropore was nearly but not completely closed in both control and mutant embryos (Fig. 5, E and F). If the absence of laminin α5 led to significant ultrastructural defects, we expected them to be detectable at this stage.
In control embryos, the basal surfaces of the ectoderm and neuroectoderm were coated by a continuous BL, but discontinuities were apparent at the junction of ectoderm and neuroectoderm, the area through which neural crest cells later migrate from the dorsal neural tube to the periphery (Fig. 6, B–E). The mutant BL was similar to that of controls in being continuous beneath the neuroectoderm and ectoderm but discontinuous at the ectodermal–neuroectodermal junction (Fig. 6, F, H, and I). Lama5 −/− ectodermal BL differed from that of controls, however, along a strip bordering the neural folds: the ectodermal BL of controls was continuous, whereas that of mutants was thin and patchy (Fig. 6 G). This difference was seen in several mutant/control littermate pairs. Interestingly, Schoenwolf and colleagues have suggested that this strip of ectoderm is involved in generating forces that are necessary to close the neural tube (Schoenwolf and Smith, 1990; Hackett et al., 1997). We speculate that weakness of the BL in this region decreases the amount of lateral force the ectoderm can generate on the neural folds, thereby leading to sporadic failure of cranial neural tube closure.
Figure 6.
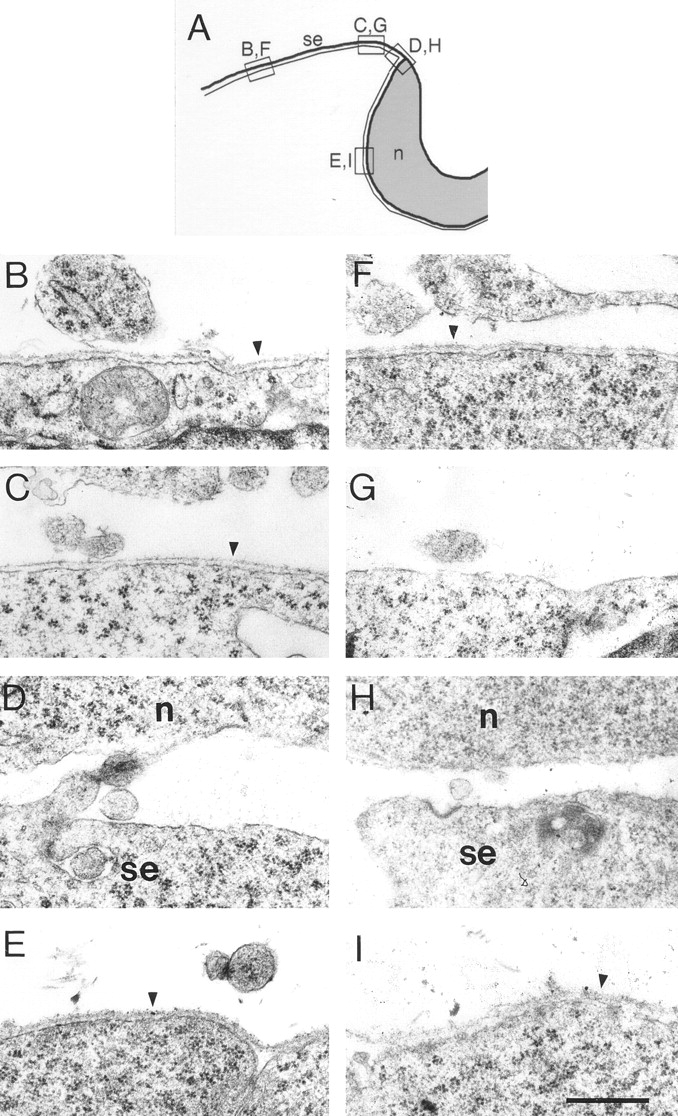
Electron micrographs of epithelial BLs in the heads of control (B–E) and mutant (F–I) embryos at E8.7. (A) Drawing indicating the approximate origins of the sections shown in B–I. The narrow line represents the BL. (B and F) Lateral cranial surface ectoderm. (C and G) Surface ectoderm near the dorsal midline. (D and H) Junction of surface ectoderm and neuroepithelium. (E and I) Neuroepithelium. BLs are intact in both mutants and controls beneath lateral ectoderm and neuroepithelium. BLs are discontinuous in both mutants and controls near the junction of ectoderm and neuroepithelium. Beneath the surface ectoderm near the neural folds, however, control BL is intact, whereas mutant BL is disrupted. This region of the ectoderm has been shown to be important in neural tube closure (see text). n, neuroepithelium; se, surface ectoderm; arrowheads, BL. Bar, 0.5 μm.
Molecular Compensation
In some laminin mutants, loss of one chain results in compensatory upregulation of other chains. For example, laminin β1 is found ectopically in the mature glomerular basement membrane in kidneys of mice with a targeted mutation in the laminin β2 chain gene. This compensatory response produces a structurally intact BL that nevertheless does not function properly as a filter (Noakes et al., 1995b ). Likewise, loss of laminin α2 in dy/dy mice results in upregulation of α4, which is, however, insufficient to prevent muscular dystrophy (Patton et al., 1997). In view of these precedents, we assessed the consequences of laminin α5 deficiency on the composition of ectodermal BL. In control embryos, α3, α5, and γ1 were present in ectodermal BL at E13.5, but α1, α2, and α4 were undetectable (Fig. 7, A–E). In mutants, the entire surface ectoderm was coated by a laminin γ1–positive BL, consistent with electron microscopic observations that the discontinuities in this structure were small and sparse (data not shown), except at the particular sites discussed above. The α5 chain was absent, as expected, but the α3 chain was retained at apparently normal levels. In addition, the α1, α2, and α4 chains were present in many but not all segments of mutant ectodermal BL (Fig. 7, F–J, and data not shown). Thus, absence of laminin α5 led to retention and/or ectopic accumulation of the α1, α2, and α4 chains. The preservation of the mutant BL may result from the retention of α3-containing laminins or the ectopic accumulation of α1-, α2-, and α4-containing laminins. Conversely, the localized defects in mutant BL that lead to exencephaly and syndactyly may reflect either regional differences in the extent of compensation or uniform weakness that renders the BL vulnerable in regions subjected to greatest stress.
Figure 7.
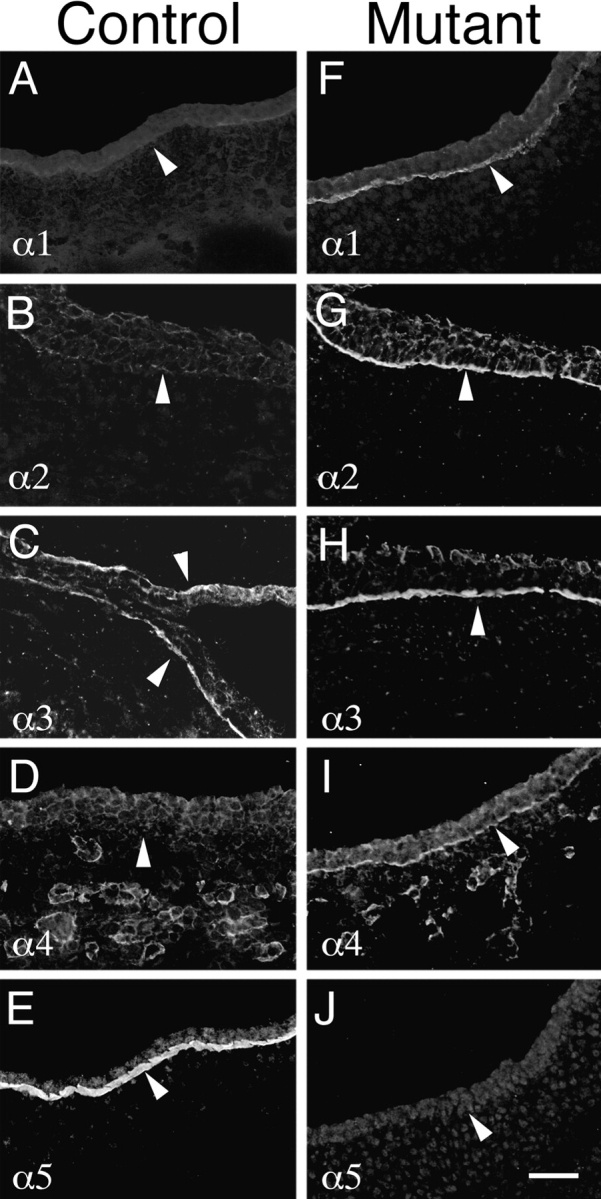
Molecular compensation for loss of laminin α5 in Lama5 −/− ectoderm. E13.5 controls (A–E) and mutants (F–J) were stained with antisera specific for the five known laminin α chains. Micrographs show surface ectoderm from the flank. Normal ectodermal BL (arrowheads) contained only α3 and α5 at this age, but mutant BL contained the α1–α4 chains. Thus, α1, α2, and α4 were upregulated in response to loss of α5. Bar, 50 μm.
Placental Dysmorphogenesis
Syndactyly and exencephaly seemed unlikely to account for the death of homozygous mutants at E14–17. Moreover, even mutants without exencephaly died by E17. Cardiac abnormalities were also observed, but these were relatively minor and only seen in some of the Lama5 −/− embryos (Miner, J.H., manuscript in preparation). We therefore examined extraembryonic tissues from mutant and control embryos. Reichert's membrane and the yolk sac of Lama5 −/− embryos were similar to those in controls, but the placental labyrinth was clearly malformed. The labyrinth is the part of the placenta in which the fetal and maternal circulations come into close proximity to exchange gases, nutrients, and wastes. In mice, the placental labyrinth is composed primarily of trophoblasts and endothelial cells, both derived from the embryo. The endothelial cells form the vessels through which the embryonic blood circulates. Three layers of trophoblasts surround these vessels and line the adjacent spaces through which the maternal blood flows (Cross et al., 1994; Hogan et al., 1994). The endothelium and the innermost trophoblast layer are separated from each other by a BL that, as shown in Fig. 1, is normally rich in laminin α5.
The placental labyrinth was significantly smaller in mutants than controls by E13.5 and remained smaller at later ages (data not shown). Immunostaining with antibodies to laminin γ1 (to localize BLs) and to PECAM (to localize endothelial cells) revealed that the network of fetal blood vessels was present and associated with BLs in the mutants at E13.5. However, the complexity of vessel branching was markedly reduced in mutant homozygotes, and the diameter of the vessels was significantly increased (Fig. 8, A and B). This defect was still apparent at E16.5, indicating that branching of the blood vessels was not merely developmentally delayed. The resulting simplification of the labyrinth reduces the surface area available for exchange of molecules between the fetal and maternal bloodstreams, and could thereby lead to placental insufficiency in mutants. Consistent with this possibility, mutant embryos were almost always smaller than their normal littermates after E14.
Figure 8.
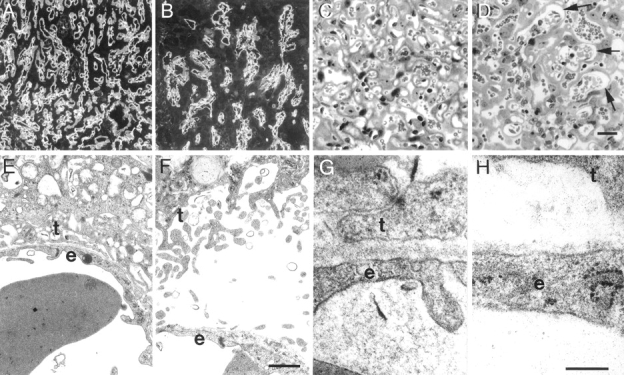
Placental dysmorphogenesis in the absence of laminin α5. (A and B) Immunostaining for laminin γ1 at E13.5 reveals the network of fetal blood vessel BLs in the placental labyrinth. The mutant vessels (B) are fewer, less branched, and wider bore than those in the control (A). (C and D) Toluidine blue staining of E16.5 control (C) and mutant (D) labyrinth shows that large bore mutant vessels (arrows in D) are still present at this later stage. The endothelium appears to have detached from the trophoblasts in these vessels. (E and F) Transmission electron micrographs show definitively that some mutant fetal vessels (F) have become detached from the trophoblasts, leaving a cell-free space between the endothelium (e) and the trophoblasts (t), while in the control (E) the two cell layers remain closely apposed. (G and H) Higher power micrographs of vascular BL. In control (G), both endothelium and trophoblast are tightly adherent to this BL, whereas in the mutant (H), the BL is patchy and has lost attachment to the trophoblasts. Bars: (in D) 100 μm for A and B, 25 μm for C and D; (in F) 1 μm for E and F; (in H) 0.25 μm for G and H.
High-resolution light microscopic and ultrastructural studies of plastic embedded tissue revealed an additional defect in mutant labyrinths. In controls, the fetal endothelium was separated from the trophoblasts only by a BL, to which both cell layers were tightly adherent (Fig. 8, C and E). In mutants, in contrast, the two cell layers were frequently separated from each other by a cell-free space. In these regions of separation, the BL was associated exclusively with the endothelial cell, leaving the basal surface of the trophoblast uncoated (Fig. 8, D and F). Ultrastructurally, the mutant BL was heterogeneous in width and exhibited splits and discontinuities (Fig. 8, G and H, and data not shown). Thus, the trophoblasts failed to form stable adhesions with the laminin α5-deficient BL, suggesting that α5-containing laminins are necessary to maintain trophoblast adhesion to the BL. The abnormal separation of the fetal blood vessels from the trophoblasts would have serious consequences for placental function and, together with the branching defects discussed above, provide a plausible explanation for death of the mutant embryos.
Phenotype of Lama5/ragged Trans-heterozygotes
The naturally occurring dominant mutation ragged (Ra) leads to sparse fur in heterozygotes (Ra/+). Ragged homozygotes (Ra/Ra) are almost completely hairless and usually die before weaning (Carter and Phillips, 1954). We (Miner et al., 1997) and Durkin et al. (1997) previously noted that Lama5 maps to a region of mouse chromosome 2 very close to the ragged gene and raised the possibility that Ra is, in fact, a neomorphic allele of the Lama5 gene. If this were the case, Ra/Lama5 mice might resemble Ra/ Ra mice because only mutant protein would be produced in both cases. To test this possibility, we mated Ra/+ mice with Lama5 +/− mice. We identified 11 mice out of 36 offspring from 4 litters that were genotypically Lama5 +/− (as determined by PCR) and phenotypically Ra/+ (by coat abnormality). This frequency was not significantly different from that expected (9/36). None of these 11 mice had the severe abnormalities observed in Ra/Ra homozygotes, and none died before weaning. Moreover, these mice had apparently normal amounts of laminin α5 protein in their kidney BLs, as assessed immunohistochemically (data not shown). It remains formally possible that Ra is a dominant-negative mutation in Lama5 that is only lethal or severe when present in two copies. However, we suspect that the ragged defect does not result from a mutation in the Lama5 gene.
Discussion
Mutations have been generated or discovered for 5 of the 10 laminin genes identified to date (see introduction). In all of these cases, embryogenesis proceeds essentially normally, and defects only become severe at or after birth. In contrast, the absence of laminin α5 leads to embryonic lethality. Lama5 −/− homozygotes appear normal until about E9, but development is then progressively aberrant, and no embryos live beyond E16.5. Multiple defects were observed, consistent with the wide expression pattern of Lama5. Here, we focused on the most severe defects: those observed in limb, head, and placenta. In all three cases, ultrastructural analysis suggests that the phenotypes result fairly directly from defects in BLs that normally contain laminin α5.
Despite the multiple defects observed in Lama5 −/− embryos, we are unsure of why they die by E17. Exencephaly is an unlikely cause, since mice routinely live to birth in other models of exencephaly (Wallace and Knights, 1978; Macdonald et al., 1989; Vogelweid et al., 1993), and the Lama5 −/− fetuses with normal neural tube closure also die by E17. Similarly, syndactyly is unlikely to be lethal. The observed placental defects may be at least partly responsible for mortality. First, the percentage of the placenta that contained the labyrinth of fetal blood vessels and maternal blood spaces was reduced in mutants. Second, the complexity of the fetal capillary network was significantly reduced compared with controls; the presence of α5 but not α1 in the BLs at the distal tips of blood vessels in the nascent placental labyrinth (Fig. 1) is consistent with it having a role in elaboration of the fetal vessel network. Third, portions of many fetal vessels were no longer juxtaposed to the trophoblasts by E16.5. Fourth, the vessel BLs themselves were aberrant, varying in width and exhibiting splits and discontinuities. Thus, laminin α5 may be important in placental endothelial cell migration and blood vessel branching, in trophoblast adhesion to BL, and in formation of a proper BL. Together, these defects would be expected to impair exchange of gases, nutrients, and wastes between the fetal and maternal circulation, and thus contribute to death of the fetuses by E17. We plan to test whether these placental defects are wholly responsible for fetal death by aggregation of Lama5 −/− ES cells with tetraploid wild-type embryos, which can contribute to formation of the placenta but not to the embryo itself (Nagy et al., 1993).
In addition to exencephaly, syndactyly, and placentopathy, several defects were seen in internal organs of Lama5 −/− embryos, including small or absent kidneys, small or absent eyes, defects in lung lobe septation and bronchiolar branching, and reduced size of the left ventricle of the heart (Miner, J.H., manuscript in preparation). Some of these defects were variable in severity and penetrance, but all can be associated with BLs that normally contain laminin α5. Though none of these defects alone appears sufficient to be responsible for fetal death, together with exencephaly and syndactyly, they may compromise the health of the fetus enough that an otherwise tolerable level of placental malfunction becomes lethal.
The limb and cranial defects in Lama5 mutants may have a common pathogenesis. Both are associated with localized structural deficiencies in regions of BLs that are likely to be subjected to greater mechanical stress than are neighboring regions. In the limb, robust outgrowth occurs in the distal portion of the limb bud under the direction of the AER. In Lama5 mutants, this outgrowth may stress the laminin α5–deficient BL in this region, leading in turn to rupture of the overlying ectoderm and extrusion of mesenchymal cells. More proximal segments of the ectoderm and its underlying BL did not exhibit obvious defects, and the proximal limb was normal. Likewise, neural tube closure was defective in cranial regions, but not in more caudal sections of the neural tube, consistent with the notion that elevation and closure of the large cranial neural folds that give rise to the brain generates more stress than elevation and closure of the much smaller caudal neural folds that give rise to the spinal cord. This stress may be maximal in the strip of ectoderm just lateral to the neural folds, which Schoenwolf and colleagues have shown to play a critical role in neural tube closure (Hackett et al., 1997). In their experiments, removal of this portion of the ectoderm in chick embryos impaired elevation of the neural plate and prevented its bending towards the dorsal midline. It is in this region that the BL is discontinuous in Lama5 −/− mice but not in controls. We favor the idea that impaired interactions with the defective BL decrease the amount of lateral force the overlying ectoderm can generate. Thus, in both limb and head, mechanical stresses on weakened, α5-deficient BLs could result in rupture of the BL, which in turn would impair function of the overlying ectoderm.
This explanation is essentially mechanical. Another explanation, not mutually exclusive, is that laminin α5 has unique signaling functions necessary for development. Indeed, laminins are known to be signaling molecules that interact with numerous signaling receptors required for cellular integrity and motility, the best studied of which are the integrins (Mercurio, 1995). Thus, some defects observed in the mutants could reflect lack of activation of laminin α5 receptors, or lack of some other ligand that is absent because the BL is disrupted. For example, the neuroepithelial BL is rich in laminin α5 at early stages, and this laminin might be required for the neuroepithelium to generate intrinsic forces necessary, along with ectoderm-derived forces, for neural tube closure (for review see Schoenwolf and Smith, 1990). Likewise, matrix-bound factors such as FGFs and Wnts have been implicated in patterning of the vertebrate limb (Martin, 1998); their accumulation or localization might be perturbed in laminin α5–deficient BLs. In any event, our results show that BLs and the laminins they contain are required for neurulation and for proper digit septation.
The finding that laminin α5 deficiency leads to localized structural defects in the BL is noteworthy, in that BLs lacking some other major components do not exhibit obvious structural defects. For example, laminin β2 is the only β chain so far found in renal glomerular or neuromuscular synaptic BLs, but these BLs appear structurally normal in mice lacking laminin β2, apparently because laminin β1 substitutes for the absent β2 chain (Noakes et al., 1995a ,b; Patton et al., 1997). These BLs are functionally defective, however, so synaptic and glomerular functions are impaired. Thus, molecular compensation permits formation of a structurally intact but functionally inadequate BL. Likewise, in humans and mice lacking the collagen α3– α5(IV) chains, which are normally the major collagen chains of the glomerular BL, the α1 and α2(IV) chains substitute to form an ultrastructurally normal BL. Eventually, however, this BL becomes severely damaged, and the kidney ceases to function properly (Kashtan and Kim, 1992; Cosgrove et al., 1996; Miner and Sanes, 1996; Kalluri et al., 1997). In Lama5 −/− mice, in contrast, at least some BLs are ultrastructurally defective despite ectopic deposition of other α chains. It remains to be determined whether the severity of these defects reflects quantitatively inadequate compensation by other α chains, or whether α5 is uniquely qualified to maintain the structural integrity of BLs.
Acknowledgments
We thank D. Abrahamson, P. Yurchenco, R. Burgeson, and E. Fuchs for antibodies; A. Nagy for ES cells; M. Nichol for ES cell culture and blastocyst injections; C. Kenoyer, J. Mudd, E. Ryan, and R. Lewis for technical assistance; C. Borgmeyer, J. Gross, and S. Weng for care of mice; T. Tolley (supported by National Institutes of Health PO1HL29594) for help with histology; and R.M. Grady, Y. Sadovsky, D.M. Nelson, Z. Werb, and R. Kopan for helpful discussions.
This work was supported by grants from the National Institutes of Health to J. Miner and to J.R. Sanes.
Abbreviations used in this paper
- AER
apical ectodermal ridge
- BL
basal lamina
- BrdU
5-bromo-2′-deoxy-uridine
- E
embryonic day
References
- Aberdam D, Aguzzi A, Baudoin C, Galliano MF, Ortonne JP, Meneguzzi G. Developmental expression of nicein adhesion protein (laminin-5) subunits suggests multiple morphogenic roles. Cell Adhes Commun. 1994;2:115–129. doi: 10.3109/15419069409004431. [DOI] [PubMed] [Google Scholar]
- Abrahamson DR, Irwin MH, St. John PL, Perry EW, Accavitti MA, Heck LW, Couchman JR. Selective immunoreactivities of kidney basement membranes to monoclonal antibodies against laminin: localization of the end of the long arm and the short arms to discrete microdomains. J Cell Biol. 1989;109:3477–3491. doi: 10.1083/jcb.109.6.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter TC, Phillips RJS. Ragged, a semidominant coat texture mutant. J Hered. 1954;45:151–154. [Google Scholar]
- Cheng Y-S, Champliaud M-F, Burgeson RE, Marinkovich MP, Yurchenco PD. Self-assembly of laminin isoforms. J Biol Chem. 1997;272:31525–31532. doi: 10.1074/jbc.272.50.31525. [DOI] [PubMed] [Google Scholar]
- Chung AE, Jaffe R, Freeman IL, Vergnes JP, Braginsk JE, Carlin B. Properties of a basement membrane related glycoprotein synthesized by a mouse embryonal carcinoma-derived cell line. Cell. 1979;16:277–287. doi: 10.1016/0092-8674(79)90005-9. [DOI] [PubMed] [Google Scholar]
- Cosgrove D, Meehan DT, Grunkemeyer JA, Kornak JM, Sayers R, Hunter WJ, Samuelson GC. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 1996;10:2981–2992. doi: 10.1101/gad.10.23.2981. [DOI] [PubMed] [Google Scholar]
- Cross JC, Werb Z, Fisher SJ. Implantation and the placenta: key pieces of the development puzzle. Science. 1994;266:1508–1518. doi: 10.1126/science.7985020. [DOI] [PubMed] [Google Scholar]
- Durbeej M, Fecker L, Hjalt T, Zhang H-Y, Salmivirta K, Klein G, Timpl R, Sorokin L, Ebendal T, Ekblom P, Ekblom M. Expression of laminin α1, α5 and β2 chains during embryogenesis of the kidney and vasculature. Matrix Biol. 1996;15:397–413. doi: 10.1016/s0945-053x(96)90159-6. [DOI] [PubMed] [Google Scholar]
- Durkin ME, Loechel F, Mattei M-G, Gilpin BJ, Albrechtsen R, Wewer UM. Tissue-specific expression of the human laminin α5-chain, and mapping of the gene to human chromosome 20q13.2-13.3 and to distal mouse chromosome 2 near the locus for the ragged (Ra)mutation. FEBS Lett. 1997;411:296–300. doi: 10.1016/s0014-5793(97)00686-8. [DOI] [PubMed] [Google Scholar]
- Hackett DA, Smith JL, Schoenwolf GC. Epidermal ectoderm is required for full elevation and for convergence during bending of the avian neural plate. Dev Dyn. 1997;210:397–406. doi: 10.1002/(SICI)1097-0177(199712)210:4<397::AID-AJA4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Harris MJ, Juriloff DM. Genetic landmarks for defects in mouse neural tube closure. Teratology. 1997;56:177–187. doi: 10.1002/(SICI)1096-9926(199709)56:3<177::AID-TERA1>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tome FM, Schwartz K, Fardeau M, Tryggvason K, et al. Mutations in the laminin α2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. 1995;11:216–218. doi: 10.1038/ng1095-216. [DOI] [PubMed] [Google Scholar]
- Hogan, B., R. Beddington, F. Costantini, and E. Lacy. 1994. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 497 pp.
- Jegalian BG, De Robertis EM. Homeotic transformations in the mouse induced by overexpression of a human Hox3.3 transgene. Cell. 1992;71:901–910. doi: 10.1016/0092-8674(92)90387-r. [DOI] [PubMed] [Google Scholar]
- Jiang R, Lan Y, Chapman HD, Shawber C, Norton CR, Serreze DV, Weinmaster G, Gridley T. Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev. 1998;12:1046–1057. doi: 10.1101/gad.12.7.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Shield CF, III, Todd P, Hudson BG, Neilson EG. Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest. 1997;99:2470–2478. doi: 10.1172/JCI119431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashtan CE, Kim Y. Distribution of the α1 and α2 chains of collagen IV and of collagens V and VI in Alport syndrome. Kidney Int. 1992;42:115–126. doi: 10.1038/ki.1992.269. [DOI] [PubMed] [Google Scholar]
- Kaufman, M.H. 1992. The Atlas of Mouse Development. Academic Press Limited, London. 525 pp.
- Klein G, Ekblom M, Fecker L, Timpl R, Ekblom P. Differential expression of laminin A and B chains during development of embryonic mouse organs. Development (Camb) 1990;110:823–837. doi: 10.1242/dev.110.3.823. [DOI] [PubMed] [Google Scholar]
- Kuster JE, Guarnieri MH, Ault JG, Flaherty L, Swiatek PJ. IAP insertion in the murine LamB3 gene results in junctional epidermolysis bullosa. Mamm Genome. 1997;8:673–681. doi: 10.1007/s003359900535. [DOI] [PubMed] [Google Scholar]
- Lentz SI, Miner JH, Sanes JR, Snider WD. Distribution of the ten known laminin chains in the pathways and targets of developing sensory axons. J Comp Neurol. 1997;378:547–561. doi: 10.1002/(sici)1096-9861(19970224)378:4<547::aid-cne9>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Macdonald KB, Juriloff DM, Harris MJ. Developmental study of neural tube closure in a mouse stock with a high incidence of exencephaly. Teratology. 1989;39:195–213. doi: 10.1002/tera.1420390211. [DOI] [PubMed] [Google Scholar]
- Marinkovich MP, Lunstrum GP, Burgeson RE. The anchoring filament protein kalinin is synthesized and secreted as a high molecular weight precursor. J Biol Chem. 1992;267:17900–17906. [PubMed] [Google Scholar]
- Martin GR. The roles of FGFs in the early development of vertebrate limbs. Genes Dev. 1998;12:1571–1586. doi: 10.1101/gad.12.11.1571. [DOI] [PubMed] [Google Scholar]
- Martin PT, Ettinger AJ, Sanes JR. A synaptic localization domain in the synaptic cleft protein, s-laminin/laminin β2. Science. 1995;269:413–416. doi: 10.1126/science.7618109. [DOI] [PubMed] [Google Scholar]
- McGrath JA, Kivirikko S, Ciatti S, Moss C, Dunnill GS, Eady RA, Rodeck CH, Christiano AM, Uitto J. A homozygous nonsense mutation in the α3 chain gene of laminin 5 (LAMA3) in Herlitz junctional epidermolysis bullosa: prenatal exclusion in a fetus at risk. Genomics. 1995;29:282–284. doi: 10.1006/geno.1995.1246. [DOI] [PubMed] [Google Scholar]
- Mercurio AM. Laminin receptors: achieving specificity through cooperation. Trends Cell Biol. 1995;5:419–423. doi: 10.1016/s0962-8924(00)89100-x. [DOI] [PubMed] [Google Scholar]
- Miner JH, Sanes JR. Molecular and functional defects in kidneys of mice lacking collagen α3(IV): implications for Alport syndrome. J Cell Biol. 1996;135:1403–1413. doi: 10.1083/jcb.135.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH, Lewis RM, Sanes JR. Molecular cloning of a novel laminin chain, α5, and widespread expression in adult mouse tissues. J Biol Chem. 1995;270:28523–28526. doi: 10.1074/jbc.270.48.28523. [DOI] [PubMed] [Google Scholar]
- Miner JH, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jenkins NA, Copeland NG, Sanes JR. The laminin α chains: expression, developmental transitions, and chromosomal locations of α1-5, identification of heterotrimeric laminins 8-11, and cloning of a novel α3 isoform. J Cell Biol. 1997;137:685–701. doi: 10.1083/jcb.137.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/ laminin β2. Nature. 1995a;374:258–262. doi: 10.1038/374258a0. [DOI] [PubMed] [Google Scholar]
- Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP. The renal glomerulus of mice lacking s-laminin/laminin β2: nephrosis despite molecular compensation by laminin β1. Nat Genet. 1995b;10:400–406. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. 1997;139:1507–1521. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton BL, Chiu AY, Sanes JR. Synaptic laminin prevents glial entry into the synaptic cleft. Nature. 1998;393:698–701. doi: 10.1038/31502. [DOI] [PubMed] [Google Scholar]
- Pulkkinen L, Christiano AM, Gerecke D, Wagman DW, Burgeson RE, Pittelkow MR, Uitto J. A homozygous nonsense mutation in the β3 gene of laminin 5 (LAMB3) in Herlitz junctional epidermolysis bullosa. Genomics. 1994;24:357–360. doi: 10.1006/geno.1994.1627. [DOI] [PubMed] [Google Scholar]
- Schoenwolf GC, Smith JL. Mechanisms of neurulation: traditional viewpoint and recent advances. Development (Camb) 1990;109:243–270. doi: 10.1242/dev.109.2.243. [DOI] [PubMed] [Google Scholar]
- Sorokin LM, Pausch F, Durbeej M, Ekblom P. Differential expression of five laminin α (1-5) chains in developing and adult mouse kidney. Dev Dyn. 1997a;210:446–462. doi: 10.1002/(SICI)1097-0177(199712)210:4<446::AID-AJA8>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Sorokin LM, Pausch F, Frieser M, Kroger S, Ohage E, Deutzmann R. Developmental regulation of the laminin α5 chain suggests a role in epithelial and endothelial cell maturation. Dev Biol. 1997b;189:285–300. doi: 10.1006/dbio.1997.8668. [DOI] [PubMed] [Google Scholar]
- Stoler A, Kopan R, Duvic M, Fuchs E. Use of monospecific antisera and cRNA probes to localize the major changes in keratin expression during normal and abnormal epidermal differentiation. J Cell Biol. 1988;107:427–446. doi: 10.1083/jcb.107.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunada Y, Bernier SM, Kozak CA, Yamada Y, Campbell KP. Deficiency of merosin in dystrophic dy mice and genetic linkage of laminin M chain gene to dylocus. J Biol Chem. 1994;269:13729–13732. [PubMed] [Google Scholar]
- Timpl R. Structure and biological activity of basement membrane proteins. Eur J Biochem. 1989;180:487–502. doi: 10.1111/j.1432-1033.1989.tb14673.x. [DOI] [PubMed] [Google Scholar]
- Timpl R. Macromolecular organization of basement membranes. Curr Opin Cell Biol. 1996;8:618–624. doi: 10.1016/s0955-0674(96)80102-5. [DOI] [PubMed] [Google Scholar]
- Timpl R, Brown JC. Supramolecular assembly of basement membranes. BioEssays. 1996;18:123–132. doi: 10.1002/bies.950180208. [DOI] [PubMed] [Google Scholar]
- Timpl R, Rohde H, Robey PG, Rennard SI, Foidart JM, Martin GR. Laminin—a glycoprotein from basement membranes. J Biol Chem. 1979;254:9933–9937. [PubMed] [Google Scholar]
- Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Vogelweid CM, Vogt DW, Besch-Williford CL, Walker SE. New Zealand white mice: an experimental model of exencephaly. Lab Anim Sci. 1993;43:58–60. [PubMed] [Google Scholar]
- Wallace ME, Knights PJ. Inheritance and morphology of exencephaly, a neonatal lethal recessive with partial penetrance, in the house mouse. Genet Res (Camb) 1978;32:135–149. doi: 10.1017/s0016672300018632. [DOI] [PubMed] [Google Scholar]
- Xu H, Wu X-R, Wewer UM, Engvall E. Murine muscular dystrophy caused by a mutation in the laminin α2 (Lama2)gene. Nat Genet. 1994;8:297–302. doi: 10.1038/ng1194-297. [DOI] [PubMed] [Google Scholar]
- Yurchenco PD, O'Rear JJ. Basal lamina assembly. Curr Opin Cell Biol. 1994;6:674–681. doi: 10.1016/0955-0674(94)90093-0. [DOI] [PubMed] [Google Scholar]