

Abstract
Wounding of skin activates epidermal cell migration over exposed dermal collagen and fibronectin and over laminin 5 secreted into the provisional basement membrane. Gap junctional intercellular communication (GJIC) has been proposed to integrate the individual motile cells into a synchronized colony. We found that outgrowths of human keratinocytes in wounds or epibole cultures display parallel changes in the expression of laminin 5, integrin α3β1, E-cadherin, and the gap junctional protein connexin 43. Adhesion of keratinocytes on laminin 5, collagen, and fibronectin was found to differentially regulate GJIC. When keratinocytes were adhered on laminin 5, both structural (assembly of connexin 43 in gap junctions) and functional (dye transfer) assays showed a two- to threefold increase compared with collagen and five- to eightfold over fibronectin. Based on studies with immobilized integrin antibody and integrin-transfected Chinese hamster ovary cells, the interaction of integrin α3β1 with laminin 5 was sufficient to promote GJIC. Mapping of intermediate steps in the pathway linking α3β1–laminin 5 interactions to GJIC indicated that protein trafficking and Rho signaling were both required. We suggest that adhesion of epithelial cells to laminin 5 in the basement membrane via α3β1 promotes GJIC that integrates individual cells into synchronized epiboles.
Keywords: gap junctions, integrins, laminin 5, epidermis, intracellular protein trafficking
Wounding of the epidermis activates cell migration across interstitial connective tissue components exposed in the wound bed, initiates assembly of a provisional basement membrane (BM),1 increases proliferation, and promotes changes in cell–cell adhesion and cell–cell communication (Clark, 1985; Grinell, 1992; Gailit and Clark, 1994; Zambruno et al., 1995; Martin, 1997). It has been suggested that gap junctional intercellular communication (GJIC) may regulate certain aspects of the wound healing process including synchronization of the migration of cells (Larson, 1990; Goliger and Paul, 1995). Laminin 5 is the primary adhesive ligand present in normal adult epidermal BM (Carter et al., 1991; Rousselle et al., 1991) and is secreted by keratinocytes in the provisional wound bed in tissue (Ryan et al., 1994; Kainulainen et al., 1998) and in culture (Carter et al., 1990a ; Carter et al., 1991). Keratinocyte adhesion to laminin 5 is mediated by two different integrin adhesion receptors (Carter et al., 1990a ; Carter et al., 1991; Xia et al., 1996; Fuchs et al., 1997): integrin α6β4 mediates anchorage to laminin 5 in hemidesmosomes (HDs) of homeostatic tissue (Carter et al., 1990a ; Stepp et al., 1990; Jones et al., 1991; Kurpakus et al., 1991; Sonnenberg et al., 1991; Fuchs et al., 1997), and integrin α3β1 mediates cell motility in wounds and culture (Ryan et al., 1994; Kainulainen et al., 1998). Integrin–laminin 5 interactions mediate transmembrane signaling (Xia et al., 1996; Giancotti, 1997; Shaw et al., 1997) and can inhibit expression of differentiation markers (Watt et al., 1993; Symington and Carter, 1995).
The BM performs a barrier function that separates the epithelial cells from interstitial connective tissue in the dermis. Wounding of the epithelium and BM exposes the interstitial tissue and activates repair of the BM. Keratinocytes synthesize and deposit laminin 5 into the provisional BM of the wound bed (Ryan et al., 1994; Kainulainen et al., 1998), redistribute HDs components (Gipson et al., 1993), reorganize integrins, migrate across the dermis exposed in the wound bed (Grinell, 1992; Hertle et al., 1992; Ryan et al., 1994; Zambruno et al., 1995; Kainulainen et al., 1998), and alter intercellular adhesion and communication (Goliger and Paul, 1995; Saitoh et al., 1997). Based on in vitro studies, interaction of epithelial cells with collagen-coated and fibronectin-coated substrates is mediated by integrins α2β1 and α5β1, respectively (Carter et al., 1990a ,b). However, migrating keratinocytes rapidly mask the fibronectin and collagen with deposited laminin 5 and shift their adhesion to α3β1 (Carter et al., 1990a ; Carter et al., 1991; Fuchs et al., 1997). Studies with cultured human keratinocytes (Zhang and Kramer, 1996) as well as in mice in which expression of laminin 5 has been disrupted (Ryan, M.C., K. Lee, Y. Myashito, S. Gil, and W.G. Carter, manuscript in preparation) suggest that deposition of de novo synthesized laminin 5 is required for epithelial cell survival. The interactions of β1 and β4 integrins with ligands result in intracellular signals that are transduced across the plasma membrane of epithelial cells (Xia et al., 1996; Giancotti, 1997; Shaw et al., 1997). Integrin–extracellular matrix (ECM) interactions modulate intracellular pH, activate protein kinase C, MAP kinase, and focal adhesion kinase, and increase cytoplasmic calcium, protein tyrosine phosphorylation, and gene expression (Clark and Brugge, 1995). Conceivably, integrin-mediated cell interactions with laminin 5, collagen, and fibronectin in the wound bed may signal different epithelial cell functions.
Gap junctions are specialized membrane domains that are composed of nonspecific channels that connect neighboring cells. Gap junctions provide cell-to-cell pathways for molecules less than ∼1,000 D (Loewenstein, 1981). In many cell types, the assembly of gap junctions depends on prior cell–cell adhesions mediated by cadherins (Musil et al., 1990; Jongen et al., 1991; Fujimoto et al., 1997). Furthermore, proteoglycans and glycosaminoglycans have been shown to induce gap junction synthesis and function in primary liver cells (Spray et al., 1987). Several recent studies suggest that GJIC is important in cell growth control and differentiation (Beyer, 1993; Goodenough et al., 1996; Kumar and Gilula, 1996). Vertebrate gap junctions are composed of proteins from the “connexin” gene family (Beyer, 1993) and are designated with numerical suffixes referring to the molecular mass of the deduced sequence in kilodaltons (e.g., Cx43) or an α/β nomenclature (Kumar and Gilula, 1996). The predominant connexin in human epidermis and in cultures of human keratinocytes is Cx43 (Fitzgerald et al., 1994). Consistent with the role of GJIC in cell growth control, connexin proteins are differentially expressed in skin with lower expression in the proliferative regions and more expression upon differentiation (Risek et al., 1992; Goliger and Paul, 1994; Salomon et al., 1994). At the wound edge, connexin expression is decreased but expression is enhanced at unwounded adjacent areas and upon wound closure when the cells differentiate (Goliger and Paul, 1995; Saitoh et al., 1997). After healing, gap junctional protein expression returns to normal levels. These changes correlate with the perceived roles of gap junctions and have led to the idea that gap junctions may play a regulatory role in wound repair (Larson, 1990; Goliger and Paul, 1995).
We have hypothesized that interactions of keratinocytes with laminin 5 may promote specific cell functions in wound repair and quiescent epithelium not promoted by dermal ECM ligands (Carter et al., 1990a ,b; Carter et al., 1991; Gil et al., 1994; Xia et al., 1996). Here, we examined the interdependence of integrin-mediated adhesion to laminin 5 on downstream GJIC. We report that adhesion of keratinocytes and other cells to laminin 5 via integrin α3β1 promotes GJIC when compared with adhesion on collagen or fibronectin. We suggest that α3β1–laminin 5 interactions promote GJIC and integrate individual epithelial cells into a synchronous epibole.
Materials and Methods
Cells and Cell Culture
Normal human foreskin keratinocytes (HFKs) were prepared as described by Boyce and Ham (1983). FEPE1L8 cells are HFKs that were transformed with the genome of human papilloma virus 16 (Kaur et al., 1989). Both HFKs and FEPE1L8 cells were maintained in serum-free keratinocyte growth medium (KGM; Clonetics Corp., San Diego, CA) containing insulin, EGF, hydrocortisone, and bovine pituitary extract. Parental and integrin-transfected CHO K1 cells were grown in DME medium with 10% FBS.
Epidermal epibole cultures, used as wound models, were prepared as follows. Punch biopsies (2 mm) were taken from fresh neonatal foreskins and attached onto coverglass coated with collagen type I (100 μg collagen/ ml H2O dried down to a film) dermal-side down and fed with KGM containing 5% FBS. Both epidermal keratinocytes (∼90%) and dermal fibroblasts (∼10%) grew out of the explants onto the culture surface for over 2 wk and segregated into separate populations forming confluent epiboles of 2–3 cm in diameter.
Expression of Dominant Negative (DN)-Rho and Integrins in Cultured Cells
FEPE1L8 cells were infected with DN-Rho as follows. Plasmid expression vectors were prepared by standard methods of subcloning pEXV-RhoN19 (Qiu et al., 1995a ,b; a gift from Dr. Frank Gertler, FHCRC, Seattle, WA) into the EcoRI site of the retroviral vector, pLXSN (a gift from Dr. Dusty Miller, FHCRC, Seattle, WA). The vector contains a SV-40 promoter and a neomycin resistance gene. Infection of pLXSN-RhoN19 into cells was done according to previously described methods (Miller, 1993). In brief, purified pLXSN-RhoN19 plasmid at a concentration of 2.5 mg/ml was transfected using the Lipofectamine standard protocol (GIBCO BRL, Gaithersburg, MD) into NIH3T3 PA317 packaging cell line (a gift from Dr. Dusty Miller, also available through American Type Culture Collection, Rockville, MD; CRL9078). Packaging cells were passed 2 d after transfection into selection media containing 100 mM geneticin (G418; GIBCO BRL). Surviving cells were allowed to grow to 80% confluence and culture media containing the packaged virus was then collected and kept frozen. Culture media containing virus was layered onto an 80% confluent 10-cm plate of FEPE1L8 cells for 6 h at 37°C. Infection media was then washed off and fresh KGM was added to the cells. The next day, cells were passed and allowed to attach and spread. G418 at a concentration of 75 mM was added to FEPE1L8 cells for selection.
CHOs were transfected with cDNAs encoding integrin subunits α2, α3, and α6/β4 (Symington et al., 1993; Kamata et al., 1994) as follows. In brief, electroporation was used to cotransfect CHO K1 cells with integrin subunit cDNAs in PBJ-1 vector (10 μg) and pCDneo plasmid (1 μg). Cells were selected in medium containing geniticin (G418; GIBCO BRL) at 700 μg/ml, and then maintained in medium containing 100 μg/ml geneticin.
Antibodies, ECM, and Antibody/ECM-coated Surfaces
The antibodies against integrin subunits α2 (P4B4, P1H5) and α3 (P1F2, P1B5) have been described previously (Wayner and Carter, 1987; Carter et al., 1990a ,b). Anti-α6 integrin subunit G0H3 was from Amac, Inc. (Westbrook, ME). Noninhibitory mAbs to the α3 chain of laminin 5 (C2-5) and fibronectin (P1H11) have been described previously (Xia et al., 1996). Monoclonal D2-1 binds an epitope in the carboxy-terminal G domain of the α3 chain of laminin 5 and will be described elsewhere (Gil, S., E. Harper, and W.G. Carter, manuscript in preparation). Monoclonal anti–E cadherin antibody (HECD1) was a generous gift from Dr. Masatoshi Tacheichi (Kyoto University, Kyoto, Japan). Monoclonal anti–type VII collagen antibody (L3D) was a generous gift from Dr. Ray Gammon (Virginia Mason, Seattle, WA). mAb 3068 against Cx43 (Chemicon, Temecula, CA) was used for studies with human cells. Rabbit polyclonal antibodies against the amino-terminal 20 residues of connexin Cx43 (AT-2, a generous gift of Barbara Yancey; described in Yancey et al., 1989) and against the last 16 amino acids of Cx43 (PNRF; Hossain et al., 1998) were used for studies with CHO cells.
Poly-l-lysine (Sigma Chemical Co., St. Louis, MO) was bound to tissue culture plates at 0.5 mg/ml in PBS overnight at 4°C. Human plasma fibronectin and human placental collagen type I and IV were prepared as described previously (Wayner and Carter, 1987) and were bound to tissue culture plates at 10 μg/ml in PBS. Collagen I and IV did not differ in their ability to promote dye transfer and both bind to α2β1, so hereafter they are simply referred to as collagen. The dishes were subsequently blocked with 0.25% heat-denatured BSA for 2 h. Laminin 5–coated tissue culture plates were prepared as described previously (Xia et al., 1996). Inhibition studies with anti–laminin 5 antibodies have shown that laminin 5 is the major adhesive ligand present on these plates (Xia et al., 1996).
ECM-coated beads were prepared by one of two methods. Purified collagen and fibronectin were bound directly to 1-μm latex beads per manufacturer's instructions (Molecular Probes, Eugene, OR). Laminin 5– and fibronectin-coated beads were also prepared by incubating goat anti– mouse antibody-coated microspheres (0.9 μm; Bangs Laboratories, Fishers, IN) with a noninhibitory mAb to laminin 5 (C2-5) or fibronectin (P1H11) for 1 h followed by three washes with PBS and incubation with three changes of culture supernatant from HFK cultures or three changes of 10 μg/ml fibronectin, respectively. HFK culture supernatant contains soluble laminin 5 (Xia et al., 1996).
Antibody-coated surfaces were prepared by incubating affinity-purified rabbit anti–mouse IgG at 10 μg/ml PBS on tissue culture dishes for 2 h followed by one PBS wash and incubation of monoclonal antiintegrin antibodies for 2 h. The plates were then blocked for 1 h with 0.5% BSA in PBS. The mAbs used were against integrin subunits α2 (P4B4, P1H5), α3 (P1F2, P1B5), and α6 (G0H3).
Microinjection of Epibole Cell Cultures
Injections were carried out on a Nikon Diaphot TE300 inverted microscope with phase and fluorescence optics using a Narashigi micromanipulator. Micropipettes were drawn to between 50 and 80 mOhm (in 3 M KCl) from World Precision Kwik-Fil borosilicate glass capillaries (1.0 mm OD × 0.75 mm). A filtered 3% solution of Lucifer yellow CH (lithium salt; Sigma Chemical Co.) in 0.15 M LiCl was injected until the injected cell was brightly fluorescent and then the pipette was removed. After 3 min, the epibole cultures were photographed (Tri-X film, Kodak, ASA 400) with phase and fluorescence optics (excitation 425/40, emission 540/40).
Dye Transfer Assay
A 10-cm plate of HFKs or CHO cells was labeled with 0.5 μM calcein-AM (Molecular Probes), the cell-permeant ester of calcein which is cleaved to membrane-impermeant calcein by cellular esterases. Three other culture plates were labeled with 0.25 μM DiI. After washing twice with PBS, the two populations of cells were each trypsin/EDTA suspended, treated with trypsin inhibitor, and pelleted. The cells were suspended in the appropriate media, mixed, plated on appropriate ECM-coated dishes, and placed in a 37°C incubator. Under these conditions, integrins are still capable of rapid ECM binding. Cells were plated in excess to ensure confluency. Usually after 2 h of incubation, unattached cells were removed by gentle washing and dye transfer was assayed with a Zeiss microscope equipped with fluorescence optics. Calcein was viewed with a fluorescein filter set, whereas rhodamine filters were used to view DiI labeling. Phase and fluorescent views for calcein and DiI were recorded using a 35-mm camera with TMax film (Kodak) at 800 ASA and digital images were generated using a Nikon Coolscan film scanner. The assignment of a cell as an acceptor of dye via transfer rather than a poorly loaded or leaking donor is checked by digitally overlaying scanned images of DiI and calcein fluorescence. If a cell adjacent to a calcein-loaded, DiI-negative cell contains both punctate DiI and more diffuse calcein fluorescence, gap junction assembly and dye transfer occurred. If a DiI-labeled cell adjacent to a calcein-loaded cell does not contain calcein, then dye transfer did not occur at that interface. (For a more complete description of this assay see Lampe, 1994.) The fraction of cells that transferred dye was determined by dividing the number of DiI-labeled cells that contained calcein (i.e., transfers) by the number of cell interfaces between calcein-loaded and DiI cells (i.e., total). Error bars represent the standard deviation as determined by ANOVA using the general linear model procedure (Statistical Analysis System, version 6.08; SAS Instruments, Cary, NC).
Northern Analysis of Cx43 mRNA Levels
Total RNA from HFKs adherent on collagen or laminin 5 for 2 h was isolated using TRIzol reagent (GIBCO BRL) according to manufacturer's instructions. Northern blots were obtained by electrophoresis of total RNA, 20 μg/lane, in formaldehyde-agarose gels (1.0% agarose, 6.6% formaldehyde, 20 mM MOPS, pH 7.0, 2.5 mM sodium acetate, 1 mM EDTA). Gels were blotted to Zeta-Probe nylon membranes (Bio-Rad Laboratories, Richmond, CA) by capillary action using 20× SSC. Blots were hybridized in 0.25 M NaCl, 7% SDS, 1 mM EDTA, 0.25 M sodium phosphate, pH 7.0, 150 mg/ml salmon sperm DNA, 50% formamide at 48°C. Filters were washed at 55°C with 0.2× SSC, 0.1% SDS. Cx43 cDNA clone G2 (the entire rat heart Cx43 coding sequence, kindly provided by E. Beyer, University of Chicago, Chicago, IL) was radiolabeled using the Prime-it RmT random primer labeling kit (Stratagene Inc., La Jolla, CA) and used as a hybridization probe for Northern analysis.
Immunoblotting
Control and treated cells were washed once with PBS and solubilized directly with 2% SDS Laemmli sample buffer (whole cell extract) or 1% Triton X-100 in PBS, both of which contained 5 mM EDTA, 50 μM VO4, 10 mM NaF, and protease inhibitors (2 mM PMSF, 1 μg/ml pepstatin, 10 μg/ml aprotinin, and 1 μg/ml leupeptin). The Triton X-100 insoluble fraction was solubilized with 2% SDS sample buffer and the DNA was sheared through a 26-gauge needle. SDS-PAGE was performed on 10% polyacrylamide gels (Laemmli, 1970). Proteins were transferred to nitrocellulose and Cx43 was detected using either anti-Cx43 polyclonal antibody (AT-2; Yancey et al., 1989) for CHO cells or mAb 3068 anti-Cx43 for HFKs. Primary antibody was detected using affinity-purified, peroxidase-conjugated goat anti-primary antibody (Chemicon International, Temecula, CA). Peroxidase detection used ECL (Amersham, Arlington Heights, IL) chemiluminescence and direct exposure to Hyperfilm MP (Amersham). Densitometry was performed on a Macintosh 6100/66 using a Hewlett Packard ScanJet 3c to collect the image and the public domain NIH Image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/).
Immunofluorescence Microscopy
Immunofluorescence was performed on keratinocytes or CHO cells. Cells were washed once in PBS, incubated for 10 min in PBS containing 1% Triton X-100, and then fixed for 20 min with 2% formaldehyde in 0.1 M sucrose, 0.1 M cacodylate, pH 7.2 buffer. The cells were permeabilized with 1% Triton X-100 in PBS for 10 min and then blocked for 1 h with 0.5% BSA, 3% goat serum in PBS. PNRF (Cx43), C2-5 (laminin 5), or P1F2 (integrin α3β1) antibodies were incubated with the cells for 1 h. The cells were washed and bound antibody was reacted with FITC-conjugated, goat anti–rabbit or anti–mouse antibody. After washing, immunofluorescence was visualized using a Zeiss microscope equipped with epifluorescence. Images were photographed with TMax 400 ASA film.
Preparation and Immunostaining of Human Wounds
Wounds of the skin were prepared in normal human donors as described previously (Olerud et al., 1995), and as follows. After informed consent, 1-mm-deep incisional wounds were prepared in the medial forearm or lateral leg with a Simplate II bleeding time device (General Diagnostics, Durham, NC). At times ranging from 1 h to 28 d after the incision, the wounds were removed from the donors with a 4-mm punch biopsy performed using local 1% lidocaine for anesthesia. Biopsies were immediately placed in OCT and snap frozen in isopentane cooled in liquid nitrogen. 6-μm cryostat sections were cut, mounted on glass slides, fixed (2% vol/vol formaldehyde in cacodylate buffer for 10 min), permeabilized (0.5% vol/vol Triton X-100 in PBS for 10 min), reacted with the indicated primary antibodies, and detected with peroxidase-conjugated secondary antibodies as described previously (Carter et al., 1990a ; Carter et al., 1991).
Results
Parallel Changes in Laminin 5, α3β1, E-cadherin, and Connexin Cx43 in Wounds
We investigated the possible interdependence of changes in the expression and localization of the junctional components laminin 5, integrin α3β1, E-cadherin, and Cx43 in response to activation by injury of human epidermis (Fig. 1). Laminin 5, detected with mAb C2-5 against the α3 chain, was expressed in the normal BM distant to the wound (Fig. 1 A, unfilled arrow). Incisional wounding of the skin disrupts the normal BM in the wound bed and exposes dermal fibronectin (Fig. 1 F) and collagen (not shown) present throughout the dermis. By 24 h after wounding, laminin 5 was deposited into the provisional BM of the wound bed by migrating epithelial cells (Fig. 1 A, unfilled arrowhead). The wound edge is marked with filled arrows. Similar results were observed with staining with mAbs that interact with the β3 and γ2 chains of laminin 5 (results not shown). For comparison, type VII collagen, which has been reported to interact with laminin 5 (Chen et al., 1997; Rousselle et al., 1997), was expressed in the mature BM distant to (Fig. 1 B, unfilled arrow) and adjacent to the wound edge (filled arrowhead) but was absent from the provisional BM (unfilled arrowhead). Thus, laminin 5 does not depend on interactions with type VII collagen for localization to the provisional BM. Significantly, we detected a precursor form of the α3 chain of laminin 5 with mAb D2-1 (Fig. 1 C and a subsequent section at higher magnification in I). The precursor α3 chain is expressed in the cytoplasm of epithelial cells in the wound bed (Fig. 1, C and I, unfilled arrowheads) but is reduced or undetectable in the normal BM distant from the wound edge (Fig. 1, C and I, unfilled arrows). Most importantly, precursor α3 detected by mAb D2-1 is expressed in the BM immediately adjacent to the wound edge at 24 h (Fig. 1, C and I, filled arrowheads) and as early as 8 h after wounding (not shown).
Figure 1.
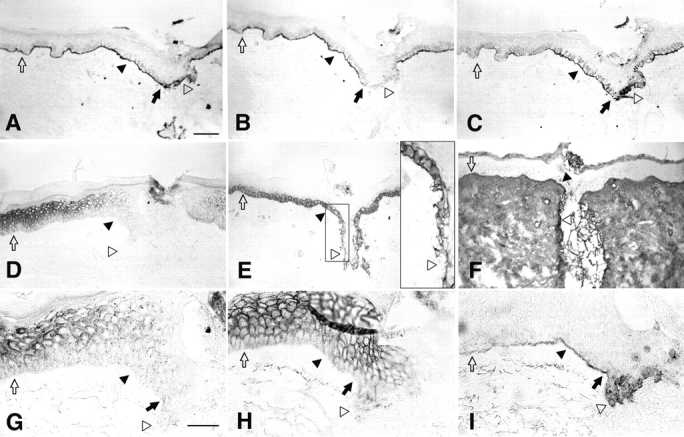
Immunostaining of human wounds identifies parallel changes in laminin 5, integrin α3β1, E-cadherin, and Cx43. Incisional wounds were prepared in normal human skin and then collected by punch biopsy at the indicated times (see Materials and Methods). Fixed cryostat sections (6 μm) were reacted with antibodies specific to components of cell as indicated for each panel. A–F are at the same lower magnification (bar, 100 μm) and G–I are at higher magnification (bar, 50 μm). (A) α3 chain of laminin 5 (mAb C2-5), 24 h wound. (B) Type VII collagen (mAb L3D), 24 h wound. (C) Precursor α3 chain of laminin 5 (mAb D2-1), 24 h wound. (D) Cx43 (PNRF), 24 h wound. (E) Integrin α3 chain (mAb P1B5), 53 h wound (the area within the small rectangle has been enlarged in the large rectangle inset to show the polarization of α3β1 to the basal plasma membrane in the wound bed). (F) Fibronectin (mAb P1H11), 53 h wound. (G) Cx43 (PNRF), 24 h wound. (H) E-cadherin (mAb HECD1), 24 h wound. (I) Precursor α3 chain of laminin 5 (mAb D2-1), 24 h wound. Different regions of the BM and epidermis were identified as follows: wound bed, unfilled arrowhead; normal epidermis distant to the wound, unfilled arrow; wound edge, filled arrow; epidermis adjacent to the wound bed, filled arrowhead.
Connexin Cx43 is expressed at high levels in the more differentiated spinous and granular cell layers and in lower amounts in basal cells of the normal epidermis distant from the wound (Fig. 1 D and at higher magnification in G, unfilled arrow). Within 24 h of injury, expression of Cx43 decreased in the wound bed (unfilled arrowheads) and in all epithelial cell layers adjacent to the wound (filled arrowheads). As with laminin 5, the altered expression of Cx43 was detected as early as 8 h after wounding (not shown). It has been suggested that cadherin-mediated intercellular adhesion is required for GJIC (Musil et al., 1990; Jongen et al., 1991; Fujimoto et al., 1997). Consistently, expression of E-cadherin (Fig. 1 H) in intercellular contacts was highest in differentiated cell layers distal to the wound but was still detectable in the basal cell layer (unfilled arrow) and was downregulated in the wound bed (unfilled arrowhead).
Integrin α3 subunit (Fig. 1 E) and integrin α2 subunit (not shown) localized to the basal, lateral, and apical plasma membrane in the normal basal cells distant from the wound (unfilled arrow). After wounding, activated cells in the wound bed localized the integrin α subunits to the basal plasma membrane presumably in contact with ligands in the wound bed. In summary, these results indicate that laminin 5, Cx43, α3β1, and E-cadherin in wounds display parallel changes in expression and/or localization in the wound bed and in the basal cells immediately adjacent to the wound within 24 h of wounding.
GJIC Is Differentially Regulated in Leading and Following Cells of Epibole Cultures
Parallel changes in cell junctional components were also observed in in vitro epibole cultures of skin (Fig. 2). Punches of human neonatal foreskin explanted onto collagen yielded outgrowths of primary keratinocytes that migrated as an integrated tongue of cells (Fig. 2 A). An assay of GJIC was performed by microinjection of Lucifer yellow into cells at both the leading edge of the tongue and the following cells (Fig. 2 B). In three separate experiments (with >10 injections/experiment), the cells at the leading edge did not transfer a detectable amount of dye to neighbors whereas the following cells transferred dye efficiently. Immunofluorescence of cells at the leading edge of the tongue localized Cx43 to the perinuclear cytoplasm (Fig. 2 C, arrows). In contrast, Cx43 assembled into apparent punctate gap junctions in cells three to four rows back from the leading edge (Fig. 2 C, arrowheads). The dramatic differences in transfer of dye and immunofluorescence established that cells at the leading edge of the migratory tongue did not assemble functional gap junctions whereas the following cells assembled gap junctions and transferred dye to neighboring cells.
Figure 2.
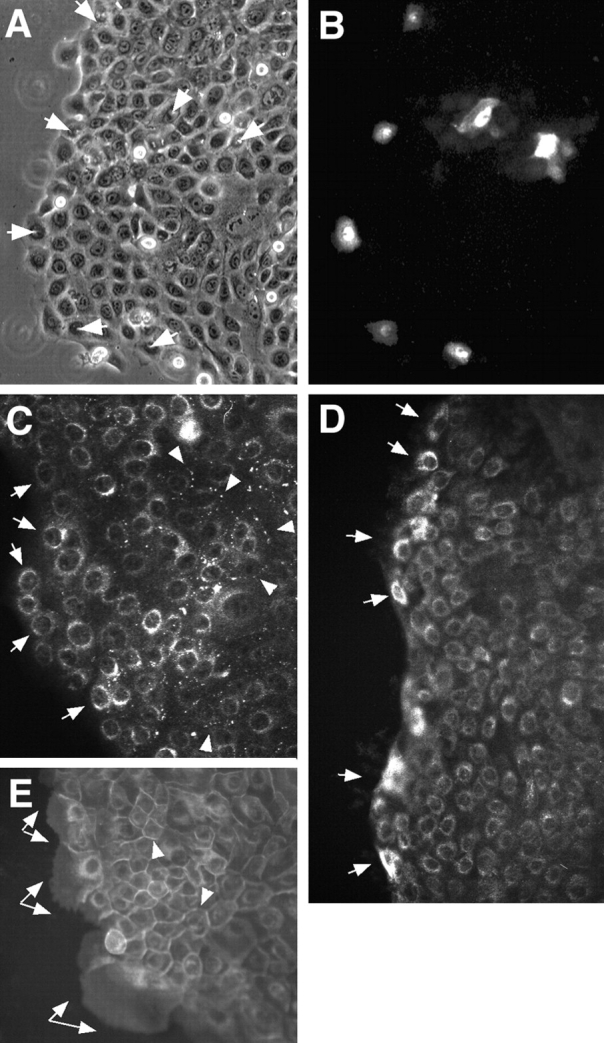
Dye transfer in epibole cultures detects differences in GJIC in leading and following migratory keratinocytes. Explants of neonatal foreskin were grown on collagen for 5 d. Epibole outgrowth of keratinocytes formed a migratory tongue of keratinocytes on the collagen surface with a leading migratory edge and following cells. Lucifer yellow dye was microinjected into keratinocytes in the leading edge and following cells three to four rows back from the leading edge. Dye was allowed to transfer for ∼5 min. (A) Phase view of the epidermal tongue. Injected cells are denoted by arrows. (B) The corresponding Lucifer yellow fluorescence view. (C) Cx43 immunofluorescence of an adjacent area of the epibole culture. Note that cells at the leading edge expressed only perinuclear Cx43 (arrows) whereas following cells also assembled gap junctions (arrowheads). (D) Laminin 5 (C2-5) was increased in expression in keratinocytes at the leading edge of the epibole (arrows). (E) Integrin α3 chain (P1F2) was present in prominent protrusions of the plasma membrane of cells at the leading edge of the outgrowth (arrows) in contrast to staining at cell–cell junctions in the following cells (arrowheads).
Localization of laminin 5 and integrin α3β1 in the epibole cultures was also consistent with the staining in wounded tissue. Laminin 5 expression was increased in keratinocytes at the leading edge of the epibole (Fig. 2 D, arrows). Most of the laminin 5 staining within cells at the leading edge could be extracted with 0.5% Triton X-100 detergent (results not shown) indicating that it was present in the cytoplasm. Integrin α3β1 (Fig. 2 E, arrows) was present in prominent protrusions of the plasma membrane of the cells at the leading edge in contrast to staining at cell–cell junctions in the following cells (Fig. 2 E, arrowheads).
We conclude that keratinocytes at the leading edge of skin wounds or epibole outgrowths display parallel changes in expression or localization of laminin 5, α3β1, and Cx43. Cells at the leading edge of the tongue versus the following cells are exposed to a different environment of cell– substrate, cell–cell interactions, and intercellular communication. In one model, expression and deposition of laminin 5 in the provisional wound bed may partially mask dermal fibronectin and collagen ligands exposed in the wound bed as described previously in culture (Carter et al., 1990a ; Carter et al., 1991). These results support the hypothesis that GJIC and integrin-dependent substrate adhesion may be interdependent functions.
Laminin 5 Promotes GJIC
The possible interdependence of cell–substrate adhesion and GJIC was investigated in an in vitro model. Laminin 5, collagen, and fibronectin were chosen to examine their influence on GJIC. HFKs were assayed for GJIC by loading separate plates of adherent cells with either of two different fluorescent dyes, calcein or DiI, suspending the labeled cells with trypsin, mixing the cells in a 1:4 ratio (calcein/DiI), and then adhering the cells onto laminin 5, collagen, or fibronectin ligands. Calcein, which can be readily loaded into cells via its acetoxymethylester form, is hydrophilic and small enough to pass between cells via gap junctions, and DiI is a lipophilic molecule that marks cells that could function as dye acceptors (Lampe, 1994). Labeled HFKs plated at high density on either collagen- or laminin 5–coated surfaces attached within 1–2 min and spread within 5–20 min. After 30 min, no difference in the amount of attachment or spreading could be observed on either ligand. With fibronectin-coated plates, HFK attachment took 5–10 min. After 2.5 h of incubation, the HFKs had extensive cell–cell contacts on all three ECM ligands (Fig. 3 A, top). Note that in all cases these cells were plated in excess numbers so they would be essentially 100% confluent in order to ensure that extensive cell–cell interfaces formed. By comparison, HFKs plated on tissue culture plastic take >1 h to bind and much longer to spread (not shown). After plating on the different ECM ligands for 2.5 h, phase images (Fig. 3 A, top) and fluorescence images of calcein (Fig. 3 A, bottom) and DiI (not shown) were collected. A representative example of HFKs plated on laminin 5 is presented in color in Fig. 3 B to illustrate the dye transfer assay. Cells with bright green calcein fluorescence in these views represent cells initially loaded with calcein (donors) and cells with red DiI fluorescence are potential recipients of dye via transfer at a cell–cell interface seen in the phase image. The assignment of a cell as a recipient of dye via transfer rather than a poorly loaded or leaking donor was checked by digitally overlaying scanned images of red DiI and green calcein fluorescence. If a cell in contact with a calcein-loaded, DiI-negative cell contained both punctate red DiI and more diffuse green calcein yellow punctate fluorescence was produced in a recipient cell. This indicated that gap junction assembly and dye transfer had occurred (arrows). If a DiI-labeled cell adjacent to a calcein-loaded cell did not contain calcein, then dye transfer did not occur at that interface (arrowheads). The ratio of recipients to total cell interfaces was calculated for HFK attachment to collagen, fibronectin, and laminin 5 in at least three separate experiments and the results are shown in Fig. 3 C. Dye transfer was approximately two- and fivefold better on laminin 5 than on collagen and fibronectin, respectively. These results clearly and reproducibly indicated that adhesion to laminin 5 promoted GJIC when compared with collagen or fibronectin.
Figure 3.
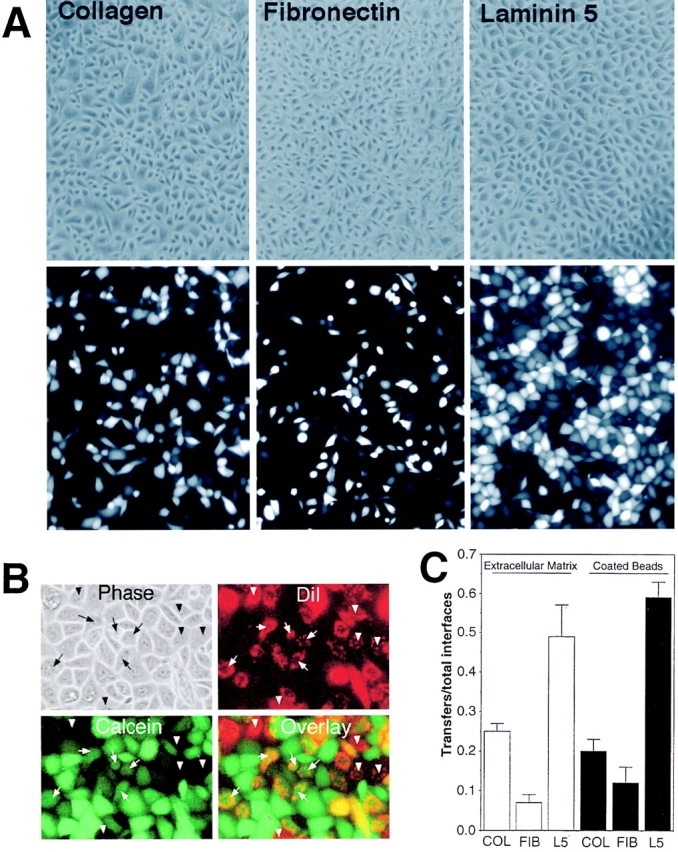
Interaction of laminin 5 with HFKs promotes dye transfer better then collagen or fibronectin. (A) ECM regulation of dye transfer. HFKs containing calcein were mixed with DiI-labeled cells and plated on collagen, fibronectin, and laminin 5 as labeled. Note that cells were plated at a high cell density to ensure confluency. The top row shows the phase view and the bottom row calcein fluorescence of the same field. Cells adherent to laminin 5 transferred calcein dye to recipient cells more readily then cells on collagen or fibronectin. (B) Dye transfer assay. Phase, calcein (green), DiI (red), and an overlay of calcein and DiI (Overlay) fluorescence views are shown. Note that several cells in the overlay show punctate yellow fluorescence indicating dye transfer occurred. Five examples of cells that received calcein via dye transfer are marked by an arrow. Five examples of DiI-labeled cells that did not receive any calcein are marked by arrowheads. (C) Laminin 5 ECM or laminin 5–coated beads promote dye transfer. Calcein-labeled HFKs were plated on collagen, fibronectin, or laminin 5 at confluent cell densities as seen in A and dye transfer levels were compared (unfilled bars). Dye transfer was quantitated as the number of interfaces showing dye transfer over the total number of interfaces between calcein- and DiI-labeled cells (mean ± standard deviation). In an alternative approach, HFKs were attached to poly-l-lysine– coated surfaces and then laminin 5–, collagen-, or fibronectin-coated beads were added to the apical surface of the attached cells and dye transfer was quantitated (filled bars). Using either approach, laminin 5 promoted GJIC better then collagen or fibronectin.
We also determined whether exogenously added ECM components that interact with the apical surface of cells can elicit similar integrin-based responses when compared with substrate-coated ligands. We used latex microspheres coated with purified collagen, fibronectin, or laminin 5. Alternatively, we used fibronectin and laminin 5 bound to microspheres via linker antibodies. When cells were first attached to poly-l-lysine–coated substratum for 0.5 h and then treated for 2 h with ECM-coated beads, the extent of dye transfer was similar to that observed with the ECM-coated culture dishes; cells treated with laminin 5 beads showed three- and fivefold more interfaces showing dye transfer than cells treated with collagen or fibronectin, respectively (Fig. 3 C).
Laminin 5 Promotes Assembly of Cx43 into Triton-insoluble Gap Junctions
We determined if adhesion to collagen and laminin 5 had different effects on expression of mRNA encoding Cx43. Total RNA was purified from cells incubated on each substrate for 2.5 h, quantitated, and electrophoresed. After checking for equal loading with ethidium bromide staining, the gel was blotted and probed with a 32P-labeled cDNA for Cx43. No significant difference between cells plated on collagen or laminin 5 was observed (Fig. 4 A, Cx43 mRNA is ∼3 kb). Reprobing the same blot for changes in mRNA encoding the α3 chain of laminin 5 also showed essentially no difference (not shown).
Figure 4.

Adhesion to laminin 5 promotes assembly of gap junctions but not transcriptional regulation of Cx43. (A) Northern analysis of HFK cells plated on collagen or laminin 5. mRNA was isolated from HFKs attached to collagen or laminin 5 and assayed by Northern blot with a cDNA probe homologous to Cx43 mRNA. The mRNA for Cx43 is 3.0 kb as expected. (B) Laminin 5 promotes assembly of Cx43 into Triton X-100–insoluble gap junctions: effects of BFA. HFKs were attached to surfaces coated with either laminin 5 or collagen for 2.5 h. Adherent HFKs were solubilized with Laemmli sample buffer either before (Whole cell) or after extraction with 1% Triton X-100 pretreatment (Triton insoluble). Triton extraction does not solubilize Cx43 that has assembled into gap junctions. Whole cell and Triton insoluble cell extracts were immunoblotted with anti-Cx43 antibody (mAb 3068). Cell extracts immunoblotted in the fifth through eighth lanes were derived from twice as many cells as extracts in the first through fourth lanes. Treatment with the protein trafficking inhibitor BFA inhibited the laminin 5–mediated assembly of gap junctions.
The quantities of total Cx43 protein found in whole cell extracts did not vary when the HFKs were plated on collagen or laminin 5 for 2.5 h as determined by immunoblot analysis (Fig. 4 B). Brefeldin A (BFA), an inhibitor of intracellular protein trafficking, also did not affect the whole cell level of Cx43 on either ligand. Cx43 can be fractionated into Triton X-100–soluble and –insoluble fractions. The Triton-insoluble fraction has been shown to be enriched for connexin subunits assembled into gap junctional structures (Musil and Goodenough, 1991). The assembled gap junctions contain phosphorylated forms of Cx43 which migrate more slowly in SDS-PAGE as multiple bands (Musil and Goodenough, 1991). In HFKs, the nonphosphorylated form of Cx43 is the predominant detectable species present in the whole cell extracts. However, Triton extraction enriches for slower migrating, presumably phosphorylated species, present in the Triton-insoluble gap junctional structures. The fact that the site of gap junction formation, the cell–cell interface, is Triton resistant indicates that relative comparisons of the Triton insolubility of Cx43 should yield relative levels of gap junction formation. HFKs plated on laminin 5 had fourfold more Triton-insoluble Cx43 than cells plated on collagen (Fig. 4 B, compare the fifth and seventh lanes) and this increase in Triton insolubility appeared to be dependent upon protein trafficking since BFA eliminated more than half of the increase (Fig. 4 B, eighth lane).
Integrin α3β1 Is Sufficient to Mediate Laminin 5 Induction of GJIC
We investigated which integrins mediate signals from laminin 5 that increase assembly and communication of gap junctions. To this end, antibodies to different integrins and control proteins were immobilized on culture dishes. Soluble integrin antibodies (Symington et al., 1993) and beads coated with antibodies to specific integrins have been shown to elicit cellular responses (e.g., focal adhesion kinase phosphorylation) normally initiated by ligand–integrin interaction (Miyamoto et al., 1995). A mixture of calcein- and DiI-loaded HFKs was plated on the dishes and dye transfer was recorded after 2–3 h of incubation. Cells readily adhered to the antibody-coated dishes if the antibody was to an extracellular portion of an integrin receptor and a confluent monolayer was produced. The best dye transfer was observed when cells were plated on immobilized mAbs against the integrin α3 (P1B5, P1F2; Fig. 5 A). Antibodies specific for α2 integrin (P4B4, P1H5), a collagen adhesion receptor, did not enhance dye transfer. An antibody to α6 (G0H3), a receptor for laminin 5, supported dye transfer at about the same level as antibodies to α2. In controls, if nuclear protein-specific antibodies or antibodies that do not react with the cells were used, very little cell attachment and hence no dye transfer was observed (not shown).
Figure 5.

Integrin α3β1 interaction with laminin 5 is sufficient to promote dye transfer. (A) Dye transfer in HFKs plated on immobilized antiintegrin antibodies. The indicated antiintegrin mAbs were immobilized on plastic, blocked with BSA, and used as an adhesive substrate for HFKs that had been labeled with either calcein or DiI. Adhesion and spreading occurred on all antibody ligands. The resulting dye transfer was quantitated as shown. (B) Dye transfer in integrin-transfected CHO cells. CHO cells transfected with human α2 or α3 cDNAs were plated on collagen and laminin 5, respectively. The top row shows the phase view and the bottom row calcein fluorescence. Calcein transfer was elevated in α3-transfected CHO cells plated on laminin 5. (C) Quantitation of dye transfer in integrin-transfected CHO cells. CHOs transfected with integrins α2, α3, and α6 were plated on collagen (filled bars) or laminin 5 (unfilled bars). Dye transfer was much higher on laminin 5 than collagen.
Integrin function in dye transfer was also investigated using CHO cell lines that express either human α2 (collagen receptor), α3 (laminin 5 receptor), or α6β4 (laminin 5 receptor) (Symington et al., 1993; Kamata et al., 1994). GJIC in each of the transfectants or parental cells was investigated using the calcein/DiI dye transfer method with cells plated on either laminin 5 or collagen for 1.5 h. When the α3 transfectant was plated on laminin 5, essentially all cells showed dye transfer (Fig. 5 B). However, when the α2 and α3 transfectants were plated on collagen or the α2 transfectant was plated on laminin 5, they transferred dye at a much lower level (Fig. 5 C, range 25–32%). The parental CHO cells have only low levels of endogenous α2 and α3 (Symington et al., 1993) and are deficient in the ECM signaling required to stimulate gap junctional communication. CHO cells transfected with α6β4 bound to laminin 5 and transferred dye at an intermediate level compared with α2β1 and α3β1, but they did not bind well to collagen so dye transfer was not measured.
Intermediate Processes in the Pathway Linking α3β1–Laminin 5 Interaction to GJIC
We wished to map cellular processes and signals that may be required intermediates in the complex process linking cell–substrate adhesion via laminin 5–α3β1 to GJIC. Therefore, we examined intracellular trafficking of proteins as a requirement for assembly of gap junctions and the Rho family of GTP binding proteins in the signaling pathway.
Intracellular Trafficking of Connexins.
CHO cells were treated with BFA to test whether the increased gap junctional assembly observed when cells were plated on laminin 5 was dependent on protein trafficking. Equal numbers of α2- and α3-transfected CHO cells were plated on collagen or laminin 5 for 1 h in the presence or absence of BFA. The level of total Cx43 protein did not vary when the α2-transfected CHO cells were plated on collagen or α3-transfected CHO cells were plated on laminin 5 for 1 h as determined by immunoblot analysis (Fig. 6 A, first and third lanes). BFA treatment also did not significantly affect the level of total Cx43 protein (Fig. 6 A, second and fourth lanes), but did apparently affect the phosphorylation/migration of Cx43 as has been reported previously (Musil and Goodenough, 1991; Laird et al., 1995). In contrast, densitometry of the Western blots indicated that the α3-transfected CHO cells plated on laminin 5 had 10-fold more Triton-insoluble Cx43 than α2-transfected CHO cells plated on collagen (Fig. 6 A, compare the fifth and seventh lanes). Again, this increase in the Triton insolubility of Cx43 appeared to be at least partially dependent upon protein trafficking since BFA eliminated 46% of the increase. BFA also reduced the fraction of cells transferring dye from the control value of 0.94 to 0.35. To confirm that the increase in Triton insolubility was due to increased gap junction assembly, α3-CHO cells were attached to laminin 5 or collagen, extracted with Triton, and then processed for immunofluorescence with an anti-Cx43 antibody. Cells on both ligands showed some perinuclear labeling. However, typical punctate gap junctional staining was clearly evident when the cells were plated on laminin 5 whereas little was observed on collagen (Fig. 6 B). The results in Figs. 5 and 6 indicate that interaction of integrin α3 with laminin 5 in transfected CHO cells is sufficient to promote GJIC and assembly of Cx43 into Triton-insoluble gap junctions. This implies that increased GJIC on laminin 5 may result from increased assembly of gap junctions. However, increased GJIC could also result from changes in the channel properties of the Cx43 complexes that are already at the cell surface or increased trafficking of a protein other than Cx43 necessary for gap junction formation.
Figure 6.

Protein trafficking mediates assembly of Triton insoluble gap junctions in α3-transfected CHO cells on laminin 5. (A) Western immunoblot for Cx43 of cell lysates from CHO cells plated on collagen or laminin 5 in the presence or absence of BFA for 1.5 h. As indicated, α3-transfected CHO cells plated on laminin 5 and α2-transfected CHO cells plated on collagen were either solubilized directly with Laemmli sample buffer (Whole cell) or after extraction with 1% Triton X-100 (Triton-insoluble) to enrich for gap junctional structures. The cell extracts were immunoblotted with anti-Cx43 rabbit antibody AT2. (B) Cx43 immunofluorescence. α3-transfected cells were plated on collagen or laminin 5, as indicated. The adherent cells were extracted with Triton X-100 before fixation and labeled with anti-Cx43 (PNRF) and FITC-conjugated secondary. Note the extensive punctate gap junctional fluorescence at cell–cell interfaces present in cells attached to laminin 5.
Rho-mediated Signaling in the Pathway.
FEPE1L8 cells are a human keratinocyte cell line that has been immortalized by transfection with human papilloma virus 16. FEPE1L8 cells retain their ability to differentiate in response to calcium, they stratify in organotypic cultures, form focal adhesions, and express the same integrin profile as HFKs, but produce little laminin 5 (Kaur and Carter, 1992). The fact that these cells produce little laminin 5 makes them dependent upon exogenous ECM ligands for rapid adhesion. FEPE1L8 cells show a fivefold increase in GJIC on exogenous laminin 5 versus collagen and a sixfold increase versus fibronectin (Fig. 7 A). In addition, the increase in GJIC observed on laminin 5 could be largely inhibited if the Rho inhibitor Toxin B was included during the incubation (Fig. 7 B).
Figure 7.

RhoA regulates dye transfer in cells adherent on laminin 5. (A) The FEPE1L8 human keratinocyte cell line was attached to surfaces coated with collagen, laminin 5, and fibronectin as indicated and dye transfer was quantitated (filled bars). (B) FEPE1L8 cells (hatched bars) were plated on laminin 5 in the presence or absence of LPA and Toxin B as indicated and dye transfer was quantitated. (C) FEPE1L8 cells transfected with a dominant negative Rho (DN-Rho; see Materials and Methods) were plated on collagen or laminin 5 in the presence or absence of LPA and Toxin B as indicated. Dye transfer was quantitated (unfilled bars). Note that LPA increases the extent of dye transfer whereas Toxin B–treated cells and the DN-Rho–transfected cells do not transfer dye well.
Treatment of FEPE1L8 cells with lysophosphatidic acid (LPA), a known activator of Rho, increased GJIC on both collagen and laminin 5 (Fig. 7 B). Consistent with the LPA-induced increase in GJIC observed in FEPE1L8 cells, treatment of HFKs with LPA increased GJIC (transfers/total interfaces) from 0.49 ± 0.08 to 0.79 ± 0.08 on laminin 5 and from 0.25 ± 0.02 to 0.63 ± 0.09 on collagen. The LPA-dependent increase observed in FEPE1L8 cells could be reduced by Toxin B treatment (Fig. 7 B). In a similar manner, if integrin α3-transfected CHO cells bound to laminin 5 were treated with Toxin B, we observed a 60% reduction in GJIC (results not shown).
To more clearly determine if Rho signaling could be playing a role in integrin-based regulation of GJIC, we transfected FEPE1L8 cells with a T17N mutant RhoA (DN-Rho) that preferentially binds GDP and when overexpressed has been shown to function in a dominant negative manner (Qiu et al., 1995b ). Western blotting of DN-Rho–transfected and vector-transfected cells with anti-RhoA antibody (Santa Cruz Biotechnology, Santa Cruz, CA) demonstrated a 105% increase in Rho expression in the DN-Rho cells. The DN-Rho cells also did not spread well on either collagen or laminin 5, confirming a biological effect of the mutant Rho expression. However, by plating the cells at confluence we were able to ensure that cell–cell contacts occurred, an obvious prerequisite to gap junction assembly. Interaction of these cells with laminin 5 failed to increase GJIC above that observed on collagen (Fig. 7 C, under DN-Rho). Taken together, these results indicate that Rho activity is necessary for the laminin 5–mediated upregulation of GJIC.
Discussion
This study establishes that cell interactions with laminin 5 via integrin α3β1 are sufficient to signal assembly of gap junctions and promote GJIC. The pathway connecting cell–substrate adhesion to intercellular communication requires multiple intermediate or parallel signals from RhoA and protein trafficking. Thus, adhesion of basal epithelium to laminin 5 in the BM instructs downstream cellular signaling and function distinct from interactions with exogenous connective tissue ligands.
Laminin 5 is identified as a promoter of gap junction assembly and intercellular communication based on structural (Triton insolubility) and functional (dye transfer) assays in three different cultured cell populations. We feel this is a strong statement of the generality of the intracellular signaling between cell–substrate and cell–cell communication. Integrin α3β1 is sufficient to transmit signals from laminin 5 to connexins based on studies with integrin-transfected CHO cells and immobilized integrin antibodies. Immobilized integrin α3 antibodies were 2–3.5- fold better at supporting GJIC between HFKs than antibodies to α2. CHO cell transfectants expressing integrin α3 showed approximately fourfold better GJIC on laminin 5 than α2 transfectants on collagen and ninefold better than the parental cell line on collagen or laminin 5. For the CHOs, we conclude that α3β1 interaction with laminin 5 is sufficient to enhance GJIC. However, these studies have not eliminated the possible contribution of α6β4– laminin 5 interactions to the promotion of GJIC in HFKs or FEPE1L8 cells. The three different cell types (HFKs, FEPE1L8s, and CHOs) used to investigate laminin 5–α3β1 promotion of GJIC did not vary in their qualitative responses to exogenous ECM ligands but did vary in the quantitative responses: HFKs had a two- to threefold increase in GJIC on laminin 5 compared with collagen whereas FEPE1L8s and CHOs were fivefold different. Another potentially important difference between these cells is their ability to synthesize laminin 5: HFKs readily express laminin 5, FEPE1L8 cells synthesize only limited levels of laminin 5 (Kaur et al., 1992), and CHOs do not synthesize laminin 5 that can be detected with any of our antibody reagents. HFKs deposit endogenous laminin 5 on exogenous collagen and fibronectin (Carter et al., 1990a ; Carter et al., 1991). The secreted laminin 5 likely generates a mixed signal from both the endogenous and exogenous ligands. Thus, the production of laminin 5 by HFKs may lead to a reduced level of enhancement on laminin 5 relative to collagen or fibronectin. Alternatively, collagen and fibronectin may have a low but detectable ability to promote GJIC when compared with laminin 5.
Multiple intermediate functions are probably required to link laminin 5–α3β1 in cell substrate adhesion to gap junctions in intercellular communication. Gap and adherens junctions appear to be interdependent possibly because adherens junctions are required to establish cell–cell interphases. Antibodies to either Cx43 or N-cadherin inhibited both gap junction and adherens junction assembly (Meyer et al., 1992). Cell lines that were deficient in E-cadherin showed much less gap junctional communication and transfection of these lines with E-cadherin increased cell adhesion and GJIC (Musil et al., 1990; Jongen et al., 1991; Miner et al., 1995). It is also possible that α3β1–laminin 5 may promote assembly of adherens junctions before assembly of gap junctions. Antibodies that inhibit cadherins block calcium-induced differentiation and prevent loss of integrin mRNA, suggesting that the function of cadherins and integrins may be linked (Hodivala and Watt, 1994). Consistently, antibodies that inhibit α3β1–laminin 5 interactions promote differentiation of keratinocytes (Symington and Carter, 1995). Recently, it has been reported that integrin and cadherin synergy regulates motility (Huttenlocher et al., 1998). It is conceivable that α3β1–laminin 5 interactions could promote formation of adherens junctions and thereby upregulate GJIC. In contrast, α2β1– collagen interactions at the leading edge of the wound or in epibole cultures may not promote the formation of adherens junctions or GJIC.
Protein trafficking is also required for GJIC on laminin 5 since BFA inhibited accumulation of Cx43 in Triton insoluble gap junctions on laminin 5. We also examined other possible pharmacological effectors of the signaling pathway between laminin 5 and GJIC including wortmannin, genistein, orthovanadate, forskolin, LPA, and Clostridium difficule Toxin B. Only Toxin B inhibited the laminin 5–dependent increase in GJIC whereas LPA and forskolin increased communication. Toxin B inhibits members of the Rho family of GTPases: Rho, Rac, and Cdc42. The Rho subgroup of the Ras superfamily are small GTPase binding proteins that participate in different cell functions by regulating the actin cytoskeleton (Symons, 1996; Tapon and Hall, 1997). Expression of a T19N (dominant negative) form of RhoA in FEPE1L8 cells inhibited GJIC on laminin 5 indicating that RhoA is the Rho family member that plays the key role in GJIC. Recently, Rho and Rac were shown to be necessary for establishment of cadherin-dependent cell–cell contacts (Braga et al., 1997). Taken together, these results indicate that RhoA activity is necessary for the laminin 5–mediated upregulation of GJIC. However, at this time it is not possible to determine if Toxin B exerts its inhibitory function on Rho and GJIC at the level of cell–substrate adhesion, cell– cell interfaces, protein trafficking, or other functions in the communication pathway between laminin 5–α3β1 and GJIC.
The adhesive ligands, antibodies, and drugs used in these studies have effects on cell shape that may affect GJIC. For example, Toxin B disrupted cell–cell adhesion in HFKs, consistent with the described role of RhoA in E-cadherin–mediated intercellular adhesion (Hordijk et al., 1997; Takaishi et al., 1997). In contrast, Toxin B does not have a major effect on cell–substrate adhesion of HFKs because interactions with laminin 5 are mediated in part by α6β4 and are insensitive to drugs that disrupt actin filaments (Xia et al., 1996). We attempted to reduce the effects of shape changes on GJIC by performing dye transfer assays at confluent cell densities where changes in cell shape will not limit intercellular contact and indirectly affect GJIC.
Differences in the kinetics of initial cell adhesion, spreading, and cell–cell contact on the different ligands could contribute to differences in rates of assembly of gap junctions and GJIC. However, our results argue against a simple kinetic explanation. (a) We detect some differences in the rates of attachment and spreading of HFKs on laminin 5 and collagen in the first 5–10 min of plating, but no differences are apparent after 30 min. However, we detect significant differences in GJIC (Figs. 3 and 7) and gap junction assembly (Figs. 4 and 6) 2–3 h after initial adhesion, long after any differences in adhesion or spreading can be detected. (b) In Fig. 3 C, ECM-coated beads were added to the apical surface of HFKs that had already been attached to poly-l-lysine and allowed to spread. Even with this equivalent starting point, GJIC was threefold higher in response to laminin 5–coated beads than beads coated with collagen or fibronectin. (c) Although we have not detected differences in the kinetics of HFK attachment to immobilized antibodies, attachment of HFKs to immobilized anti-α3 mAbs, but not immobilized anti-α2 or anti-α6, increased GJIC (Fig. 5 A). (d) The differences in gap junction assembly and GJIC reported for the epibole cultures (Fig. 2) in the leading cells contacting collagen verses the following cells on laminin 5 were assayed after 7 d of culture.
Published studies indicate that the function and expression of gap junctions are downregulated adjacent to the wound bed within 2–6 h after injury but before keratinocytes begin to migrate into the wound (Goliger and Paul, 1995). We also observed decreased expression of Cx43 and upregulation of laminin 5 in basal keratinocytes adjacent to the wound only 8 h after injury. The rapid changes in these two junctional components suggest that they may be linked to each other and participate in the subsequent migration into the wound bed. This possible linkage is further supported by subsequent changes: 24 h after wounding, connexin immunostaining was reported to increase and then gradually return to prewound levels after several days (Gabbiani et al., 1978; Goliger and Paul, 1995; Saitoh et al., 1997). Our results indicate that laminin 5 continues to be expressed at an elevated level in the wound bed for 3–5 d and declines to baseline by 7 d (Ryan et al., 1994; Gil, S., E. Harper, and W.G. Carter, manuscript in preparation). This suggests that both the initial activation of laminin 5 expression and the subsequent suppression may be linked to the changes in GJIC.
The results presented here establish a communication path from cell–substrate adhesion via laminin 5–α3β1 to GJIC. We suggest that gap junctions and laminin 5 may participate in bidirectional communication to regulate pre- and postmigratory responses in the wound as follows. (a) Wounding of skin disrupts epidermal GJIC and cell–cell adhesion in quiescent epidermis and may initiate wound repair, including synthesis and deposition of laminin 5 before migration begins. (b) Keratinocytes at the leading edge of the migrating tongue would contact dermal ECM ligands (collagen, fibronectin) in the wound bed which do not promote GJIC. (c) One possible scenario would include deposition of the precursor form of laminin 5 (recognized by mAb D2-1) into the provisional BM of the wound bed by the leading edge cells that could partially mask the exposed dermal ligands and facilitate α3β1–laminin 5 interactions that promote GJIC by the following cells. (d) As the wound heals and gap junctional expression returns to homeostatic levels (Gabbiani et al., 1978; Goliger and Paul, 1995; Saitoh et al., 1997), expression of laminin 5 would also reduce to prewound levels (Ryan et al., 1994). In one model, interaction of laminin 5 with α3β1 is replaced by interaction with α6β4 as the wound heals (Carter et al., 1990a ; Carter et al., 1991; Xia et al., 1996; Fuchs et al., 1997) and α3β1 relocates from a polarized localization in the BM zone of the wound bed to the preactivation location at lateral cell–cell junctions. In an alternative model, α3β1 may function in conjunction with α6β4 to mediate adhesion to laminin 5 in the normal BM. Consistent with the second model, α3β1 does codistribute with laminin 5 at the ultrastructural level in the BM zone (Carter et al., 1991) particularly in the deep rete ridge of epidermis in palms and feet (Symington et al., 1993) suggesting that α3β1 may contribute to adhesion to the BM of both normal epidermis as well as wounds. Also, in support of the later model, targeted disruption of integrin α3 in mice generates blisters in the legs and footpads (DiPersio et al., 1997). It is of related interest that laminin 5 is initially synthesized and deposited into the provisional BM as a precursor protein recognized by mAb D2-1 (Fig. 1). The precursor form of laminin 5 is subsequently proteolytically cleaved and converted to the mature α3 chain of laminin 5 present in the assembling BM (Fig. 1; Gil, S., E. Harper, and W.G. Carter, manuscript in preparation). Recent in vitro studies have suggested that proteolytic cleavage of the carboxy terminus of the α3 chain of laminin 5 with plasmin promotes downstream assembly of HDs (Goldfinger et al., 1998). Thus, modification of the precursor laminin 5 may be related to both changes in keratinocyte adhesion in the wound and to changes in GJIC.
In conclusion, we have shown that assembly of gap junctions and intercellular communication are upregulated when integrin α3β1 interacts with laminin 5 in vitro and that consistent correlative changes are observed in wounds in vivo. Neither collagen nor fibronectin duplicates this communication path. The promotion of GJIC is dependent on intracellular protein trafficking involved in assembly of gap junctions, and Rho-mediated signaling. We suggest that the intracellular signaling resulting from α3β1–laminin 5 interaction instructs assembly of gap junctions and increases GJIC in order to coordinate the functions of individual epithelial cells in the wound and in adjacent normal epidermis.
Abbreviations used in this paper
- BFA
Brefeldin A
- BM
basement membrane
- Cx43
connexin 43
- DN
dominant negative
- ECM
extracellular matrix
- GJIC
gap junctional intercellular communication
- HD
hemidesmosome
- HFK
human foreskin keratinocyte
- KGM
keratinocyte growth medium
- LPA
lysophosphatidic acid
Footnotes
The authors would like to acknowledge financial support from National Institutes of Health grants GM55632 (P.D. Lampe), GM47157 (Y. Takada), CA49259 (W.G. Carter), and AR-21557 (W.G. Carter).
Address correspondence related to ECM–integrin interaction and signaling to William Carter, Fred Hutchinson Cancer Research Center, A3-015, 1100 Fairview Avenue North, Seattle, WA 98109. Tel.: (206) 667-4478. Fax: (206) 667-3331. E-mail: wcarter@fhcrc.org Address correspondence related to gap junctions to Paul Lampe, Fred Hutchinson Cancer Research Center, DE-320, 1100 Fairview Avenue North, Seattle, WA 98109. Tel.: (206) 667-4123. Fax: (206) 667-2537. E-mail: plampe@fhcrc.org
References
- Beyer, E.C. 1993. Gap junctions. Int. Rev. Cytol. 137C:1–37. [PubMed]
- Boyce ST, Ham RG. Calcium-regulated differentiation of normal human epidermal keratinocytes in chemically defined clonal culture and serum-free serial culture. J Invest Dermatol. 1983;81:335–405. doi: 10.1111/1523-1747.ep12540422. [DOI] [PubMed] [Google Scholar]
- Braga VMM, Macheskky LM, Hall A, Hotchin NA. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell–cell contacts. J Cell Biol. 1997;137:1421–1431. doi: 10.1083/jcb.137.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter WG, Kaur P, Gil SG, Gahr PJ, Wayner EA. Distinct functions for integrins α3β1 in focal adhesions and α6β4/bullous pemphigoid antigen in a new stable anchoring contact (SAC) of keratinocytes: relationship to hemidesmosomes. J Cell Biol. 1990a;111:3141–3154. doi: 10.1083/jcb.111.6.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter WG, Wayner EA, Bouchard TS, Kaur P. The role of integrins α2β1 and α3β1 in cell–cell and cell–substrate adhesion of human epidermal cells. J Cell Biol. 1990b;110:1387–1404. doi: 10.1083/jcb.110.4.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter WG, Ryan MC, Gahr PJ. Epiligrin, a new cell adhesion ligand for integrin α3β1 in epithelial basement membranes. Cell. 1991;65:599–610. doi: 10.1016/0092-8674(91)90092-d. [DOI] [PubMed] [Google Scholar]
- Chen M, Marinkovich MP, Veis A, Cai X, Rao CN, Ea OT, Woodley DT. Interactions of the amino-terminal noncollagenous (NC1) domain of type VII collagen with extracellular matrix components. A potential role in epidermal-dermal adherence in human skin. J Biol Chem. 1997;272:14516–14522. doi: 10.1074/jbc.272.23.14516. [DOI] [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Clark RAF. Cutaneous tissue repair: basic biological considerations. J Am Acad Dermatol. 1985;13:701–725. doi: 10.1016/s0190-9622(85)70213-7. [DOI] [PubMed] [Google Scholar]
- DiPersio CM, Hodivala-Dilke KM, Jaenisch R, Kreidberg JA, Hynes RO. α3β1 integrin is required for normal development of the epidermal basement membrane. J Cell Biol. 1997;137:729–742. doi: 10.1083/jcb.137.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald DJ, Fusenig NE, Boukamp P, Piccoli C, Mesnil M, Yamasaki H. Expression and function of connexin in normal and transformed human keratinocytes in culture. Carcinogenesis. 1994;15:1859–1865. doi: 10.1093/carcin/15.9.1859. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Dowling J, Segre J, Lo S-H, Yu Q-C. Integrators of epidermal growth and differentiation: distinct functions for beta 1 and beta 4 integrins. Curr Opin Gen Develop. 1997;7:672–682. doi: 10.1016/s0959-437x(97)80016-0. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Nagafuchi A, Tsukita S, Kuraoka A, Ohokuma A, Shibata Y. Dynamics of connexins, E-cadherin and alpha-catenin on cell membranes during gap junction formation. J Cell Sci. 1997;110:311–322. doi: 10.1242/jcs.110.3.311. [DOI] [PubMed] [Google Scholar]
- Gabbiani G, Chaponnier C, Huttner I. Cytoplasmic filaments and gap junctions in epithelial cells and myofibroblasts during wound healing. J Cell Biol. 1978;76:561–568. doi: 10.1083/jcb.76.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailit J, Clark RA. Wound repair in the context of extracellular matrix. Curr Opin Cell Biol. 1994;6:717–725. doi: 10.1016/0955-0674(94)90099-x. [DOI] [PubMed] [Google Scholar]
- Giancotti FG. Integrin signaling: specificity and control of cell survival and cell cycle progression. Curr Opin Cell Biol. 1997;9:691–700. doi: 10.1016/s0955-0674(97)80123-8. [DOI] [PubMed] [Google Scholar]
- Gil SG, Brown TA, Ryan MC, Carter WG. Junctional epidermolysis bullosis: defects in expression of epiligrin/nicein/kalinin and integrin beta 4 that inhibit hemidesmosome formation. J Invest Dermatol. 1994;103:31S–38S. doi: 10.1111/1523-1747.ep12398953. [DOI] [PubMed] [Google Scholar]
- Gipson IK, Spurr-Michaud S, Tisdale A, Elwell J, Stepp MA. Redistribution of the hemidesmosome components alpha 6 beta 4 integrin and bullous pemphigoid antigens during epithelial wound healing. Exp Cell Res. 1993;207:86–98. doi: 10.1006/excr.1993.1166. [DOI] [PubMed] [Google Scholar]
- Goldfinger LE, Stack MS, Jones JC. Processing of laminin-5 and its functional consequences: role of plasmin and tissue-type plasminogen activator. J Cell Biol. 1998;141:255–265. doi: 10.1083/jcb.141.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goliger JA, Paul DL. Expression of gap junction proteins Cx26, Cx31.1, Cx37, and Cx43 in developing and mature rat epidermis. Dev Dynam. 1994;200:1–13. doi: 10.1002/aja.1002000102. [DOI] [PubMed] [Google Scholar]
- Goliger JA, Paul DL. Wounding alters epidermal connexin expression and gap junction-mediated intercellular communication. Mol Biol Cell. 1995;6:1491–1501. doi: 10.1091/mbc.6.11.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem. 1996;65:475–502. doi: 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- Grinell F. Wound repair, keratinocyte activation and integrin modulation. J Cell Sci. 1992;101:1–5. doi: 10.1242/jcs.101.1.1. [DOI] [PubMed] [Google Scholar]
- Hertle MD, Kubler MD, Leigh IM, Watt FM. Aberrant integrin expression during epidermal wound healing and in psoriatic epidermis. J Clin Invest. 1992;89:1892–1901. doi: 10.1172/JCI115794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodivala KJ, Watt FM. Evidence that cadherins play a role in the downregulation of integrin expression that occurs during keratinocyte terminal differentiation. J Cell Biol. 1994;124:589–600. doi: 10.1083/jcb.124.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hordijk PL, ten Klooster JP, van der Kammen RA, Michiels F, Oomen LC, Collard JG. Inhibition of invasion of epithelial cells by Tiam1-Rac signaling. Science. 1997;278:1464–1466. doi: 10.1126/science.278.5342.1464. [DOI] [PubMed] [Google Scholar]
- Hossain MZ, Ao P, Boynton AL. Rapid disruption of gap junctional communication and phosphorylation of connexin43 by platelet-derived growth factor in T51B rat liver epithelial cells expressing platelet-derived growth factor receptor. J Cell Physiol. 1998;174:66–77. doi: 10.1002/(SICI)1097-4652(199801)174:1<66::AID-JCP8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A, Lakonishok M, Kinder M, Wu S, Truong T, Knudsen KA, Horwitz AF. Integrin and cadherin synergy regulates contact inhibition of migration and motile activity. J Cell Biol. 1998;141:515–526. doi: 10.1083/jcb.141.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JC, Kurpakus MA, Cooper HM, Quaranta V. A function for the integrin alpha 6 beta 4 in the hemidesmosome. Cell Reg. 1991;2:427–438. doi: 10.1091/mbc.2.6.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongen WMF, Fitzgerald DJ, Asamoto M, Piccoli C, Slaga TJ, Gros D, Takeichi M, Yamasaki H. Regulation of connexin43-mediated gap junctional intercellular communication by Ca2+in mouse epidermal cells is controlled by E-cadherin. J Cell Biol. 1991;114:545–555. doi: 10.1083/jcb.114.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kainulainen T, Hakkinen L, Hamidi S, Larjava K, Kallioinen M, Peltonen J, Salo T, Larjava H, Oikarinen A. Laminin-5 expression is independent of the injury and the microenvironment during reepithelialization of wounds. J Histochem Cytochem. 1998;46:353–360. doi: 10.1177/002215549804600309. [DOI] [PubMed] [Google Scholar]
- Kamata T, Puzon W, Takada Y. Identification of putative ligand binding sites within I domain of integrin alpha 2 beta 1 (VLA-2, CD49b/ CD29) [published erratum appears in J. Biol. Chem.271:19008] J Biol Chem. 1994;269:9659–9663. [PubMed] [Google Scholar]
- Kaur P, Carter WG. Integrin expression and differentiation in transformed human epidermal cells is regulated by fibroblasts. J Cell Sci. 1992;103:755–763. doi: 10.1242/jcs.103.3.755. [DOI] [PubMed] [Google Scholar]
- Kaur P, McDougall JK, Cone R. Immortalisation of primary human epithelial cells by cloned cervical carcinoma DNA containing human papillomavirus 16 E6/7 ORFs. J Gen Virol. 1989;70:1261–1266. doi: 10.1099/0022-1317-70-5-1261. [DOI] [PubMed] [Google Scholar]
- Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- Kurpakus MA, Quaranta V, Jones JC. Surface relocation of α6 β4 integrins and assembly of hemidesmosomes in an in vitro model of wound healing. J Cell Biol. 1991;115:1737–1750. doi: 10.1083/jcb.115.6.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4 . Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Laird DL, Castillo M, Kasprzak L. Gap junction turnover, intracellular trafficking, and phosphorylation of connexin43 in Brefeldin A–treated rat mammary tumor cells. J Cell Biol. 1995;131:1193–1203. doi: 10.1083/jcb.131.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe PD. Analyzing phorbol ester effects on gap junction communication: a dramatic inhibition of assembly. J Cell Biol. 1994;127:1895–1905. doi: 10.1083/jcb.127.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, D.M. 1990. Junctional communication and the wound healing response. In Cell Intercommunication. W.C. DeMello, ed. CRC Press, Boca Raton, FL. 93–109.
- Loewenstein WR. Junctional intercellular communication: the cell-to-cell membrane channel. Physiol Rev. 1981;61:829–913. doi: 10.1152/physrev.1981.61.4.829. [DOI] [PubMed] [Google Scholar]
- Martin P. Wound healing: aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- Meyer RA, Laird DW, Revel J-P, Johnson RG. Inhibition of gap junction and adherens junction assembly by connexin and A-CAM antibodies. J Cell Biol. 1992;119:179–189. doi: 10.1083/jcb.119.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AD, Miller DG, Garcia JV, Lynch CM. Use of retroviral vectors for gene transfer and expression. Methods Enzymol. 1993;217:581–599. doi: 10.1016/0076-6879(93)17090-r. [DOI] [PubMed] [Google Scholar]
- Miner P, Lampe PD, Atkinson MA, Johnson RG. Extracellular calcium and cadherins regulate the process of gap junction assembly between cells in culture. Prog Cell Res. 1995;4:331–334. [Google Scholar]
- Miyamoto S, Akiyama SK, Yamada KM. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science. 1995;267:883–885. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil LS, Cunningham BA, Edelman GM, Goodenough DA. Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J Cell Biol. 1990;111:2077–2088. doi: 10.1083/jcb.111.5.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olerud JE, Odland GF, Burgess EM, Wyss CR, Fisher LD, Matsen FA., III A model for the study of wounds in normal elderly adults and patients with peripheral vascular disease or diabetes mellitus. J Surg Res. 1995;59:349–360. doi: 10.1006/jsre.1995.1175. [DOI] [PubMed] [Google Scholar]
- Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature. 1995a;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- Qiu RG, Chen J, McCormick K, Symons M. A role for Rho in Ras transformation. Proc Natl Acad Sci USA. 1995b;92:11781–11785. doi: 10.1073/pnas.92.25.11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risek B, Klier FG, Gilula NB. Multiple gap junction genes are utilized during rat skin and hair development. Development. 1992;116:639–651. doi: 10.1242/dev.116.3.639. [DOI] [PubMed] [Google Scholar]
- Rousselle P, Lunstrum GP, Keene DR, Burgeson RE. Kalinin: an epithelium-specific basement membrane adhesion molecule that is a component of anchoring filaments. J Cell Biol. 1991;114:567–576. doi: 10.1083/jcb.114.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousselle P, Keene DR, Ruggiero F, Champliaud MF, Rest M, Burgeson RE. Laminin 5 binds the NC-1 domain of type VII collagen. J Cell Biol. 1997;138:719–728. doi: 10.1083/jcb.138.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MC, Tizard R, VonDevanter DR, Carter WG. Cloning of the LamA3 gene encoding the α3 chain of the adhesive ligand epiligrin: expression in wound repair. J Biol Chem. 1994;269:22779–22787. [PubMed] [Google Scholar]
- Saitoh M, Oyamada M, Oyamada Y, Kaku T, Mori M. Changes in the expression of gap junction proteins (connexins) in hamster tongue epithelium during would healing and carcinogenesis. Carcinogenesis. 1997;18:1319–1328. doi: 10.1093/carcin/18.7.1319. [DOI] [PubMed] [Google Scholar]
- Salomon D, Masgrau E, Vischer S, Ullrich S, Dupont E, Sappino P, Saurat J-H, Meda P. Topography of mammalian connexins in human skin. J Invest Dermatol. 1994;103:240–247. doi: 10.1111/1523-1747.ep12393218. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Rabinovitz I, Wang HH, Toker A, Mercurio AM. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell. 1997;91:949–960. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- Sonnenberg A, Calafat J, Janssen H, Daams H, van der Raaij-Helmer LM, Falcioni R, Kennel SJ, Aplin JD, Baker J, Loizidou M, et al. Integrin α6/β4 complex is located in hemidesmosomes, suggesting a major role in epidermal cell-basement membrane adhesion. J Cell Biol. 1991;113:907–917. doi: 10.1083/jcb.113.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spray DC, Fujita M, Saez JC, Choi H, Watanabe T, Hertzberg E, Rosenberg LC, Reid LM. Proteoglycans and glycosaminoglycans induce gap junction synthesis and function in primary liver cultures. J Cell Biol. 1987;105:541–551. doi: 10.1083/jcb.105.1.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepp MA, Spurr-Michaud S, Tisdale A, Elwell J, Gipson IK. Alpha 6 beta 4 integrin heterodimer is a component of hemidesmosomes. Proc Natl Acad Sci USA. 1990;87:8970–8974. doi: 10.1073/pnas.87.22.8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington BE, Carter WG. Modulation of epidermal differentiation by epiligrin and integrin alpha 3 beta 1. J Cell Sci. 1995;108:831–838. doi: 10.1242/jcs.108.2.831. [DOI] [PubMed] [Google Scholar]
- Symington BE, Takada Y, Carter WG. Interaction of integrins α3β1 and α2β1: potential role in keratinocyte intercellular adhesion. J Cell Biol. 1993;120:523–535. doi: 10.1083/jcb.120.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons M. Rho family GTPases: the cytoskeleton and beyond. Trends Biochem Sci. 1996;21:178–181. [PubMed] [Google Scholar]
- Takaishi K, Sasaki T, Kotani H, Nishioka H, Takai Y. Regulation of cell–cell adhesion by rac and rho small G proteins in MDCK cells. J Cell Biol. 1997;39:1047–1059. doi: 10.1083/jcb.139.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N, Hall A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- Watt FM, Kubler MD, Hotchin NA, Nicholson LJ, Adams JC. Regulation of keratinocyte terminal differentiation by integrin-extracellular matrix interactions. J Cell Sci. 1993;106:175–182. doi: 10.1242/jcs.106.1.175. [DOI] [PubMed] [Google Scholar]
- Wayner EA, Carter WG. Identification of multiple cell adhesion receptors for collagen and fibronectin in human fibrosarcoma cells possessing unique alpha and common beta subunits. J Cell Biol. 1987;105:1873–1884. doi: 10.1083/jcb.105.4.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Gil SG, Carter WG. Anchorage mediated by integrin α6β4 to laminin 5 (epiligrin) regulates tyrosine phosphorylation of a membrane-associated 80-kD protein. J Cell Biol. 1996;132:727–740. doi: 10.1083/jcb.132.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey SB, John SA, Lal R, Austin BJ, Revel JP. The 43-kD polypeptide of heart gap junctions: immunolocalization (I), topology (II), and functional domains (III) J Cell Biol. 1989;108:2241–2254. doi: 10.1083/jcb.108.6.2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambruno G, Marchisio PC, Marconi A, Vaschieri, Melchiori A, Giannetti A, De Luca M. TGFβ1 modulates β1 and β5 integrin receptors and induces de novo expression of the αvβ6 heterodimer in normal human keratinocytes: implications for wound healing. J Cell Biol. 1995;129:853–865. doi: 10.1083/jcb.129.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kramer RH. Laminin 5 deposition promotes keratinocyte motility. Exp Cell Res. 1996;227:309–322. doi: 10.1006/excr.1996.0280. [DOI] [PubMed] [Google Scholar]