

Abstract
Naturally occurring sympathetic neuron death is the result of two apoptotic signaling events: one normally suppressed by NGF/TrkA survival signals, and a second activated by the p75 neurotrophin receptor. Here we demonstrate that the p53 tumor suppressor protein, likely as induced by the MEKK-JNK pathway, is an essential component of both of these apoptotic signaling cascades. In cultured neonatal sympathetic neurons, p53 protein levels are elevated in response to both NGF withdrawal and p75NTR activation. NGF withdrawal also results in elevation of a known p53 target, the apoptotic protein Bax. Functional ablation of p53 using the adenovirus E1B55K protein inhibits neuronal apoptosis as induced by either NGF withdrawal or p75 activation. Direct stimulation of the MEKK-JNK pathway using activated MEKK1 has similar effects; p53 and Bax are increased and the subsequent neuronal apoptosis can be rescued by E1B55K. Expression of p53 in sympathetic neurons indicates that p53 functions downstream of JNK and upstream of Bax. Finally, when p53 levels are reduced or absent in p53+/− or p53−/− mice, naturally occurring sympathetic neuron death is inhibited. Thus, p53 is an essential common component of two receptor-mediated signal transduction cascades that converge on the MEKK-JNK pathway to regulate the developmental death of sympathetic neurons.
Keywords: neuronal apoptosis, MEKK, nerve growth factor, brain-derived neurotrophic factor, sympathetic neurons
Naturally occurring neuronal death is an essential component of neural development in which <50% of any given neuronal population is lost as a mechanism for establishing appropriate neuron:target cell connections (reviewed in Oppenheim, 1991). Sympathetic neurons of the peripheral nervous system, which require NGF for their survival (reviewed in Levi-Montalcini, 1987), have been a prototype for analysis of the mechanisms regulating this event. During the first two postnatal weeks, sympathetic neurons compete for limiting amounts of target-derived NGF, which binds to and activates TrkA tyrosine kinase receptors (Kaplan et al., 1991a ,b; Klein et al., 1991) on neuronal teminals, thereby mediating a retrograde neuronal survival signal (Campenot et al., 1982; Riccio et al., 1997; Senger et al., 1997). Traditionally, it has been thought that the absence of such a TrkA retrograde signal was sufficient to cause rapid neuronal apoptosis during developmental death. However, we have recently shown that the lack of such a NGF/TrkA-mediated survival signal is, of itself, insufficient for appropriate sympathetic neuron death. Instead, a second neurotrophin receptor, p75NTR (Johnson et al., 1986; Radeke et al., 1987), is required for this process; in the absence of p75NTR, sympathetic neuron apoptosis is delayed both in culture and in vivo (Bamji et al., 1998). Moreover, ligand-mediated activation of p75NTR is sufficient to cause sympathetic neuron apoptosis (Bamji et al., 1998). Thus, appropriate developmental sympathetic neuron death occurs when p75NTR is activated coincident with suboptimal NGF/TrkA survival signals (reviewed in Miller and Kaplan, 1998). This convergent regulation of neuronal apoptosis provides a mechanism whereby sympathetic neurons are able to recognize not only whether they are receiving adequate amounts of NGF, but also whether or not they are exposed to “inappropriate” neurotrophins, potentially as a function of late or inappropriate target innervation.
The intracellular mechanisms responsible for transducing these receptor-mediated apoptotic cascades in sympathetic neurons remain ill-defined, although activation of the JNK pathway occurs after both p75NTR activation (Casaccia-Bonnefil et al., 1996; Bamji et al., 1998) and NGF withdrawal (Estus et al., 1994; Ham et al., 1995) and, in the case of NGF withdrawal, is a necessary early event in the apoptotic pathway. Moreover, Bax is essential for sympathetic neuron apoptosis both in culture and during naturally occurring death in vivo (Deckwerth et al., 1996; Easton et al., 1997). One protein that is known to regulate transcription of Bax (Miyashita and Reed, 1995) and that has been implicated in cellular apoptosis is the p53 tumor suppressor protein (reviewed in Hainut, 1995; Jacks and Weinberg, 1996; Levine, 1997). However, although p53 has been implicated in neuronal death in response to DNA damage or cellular stress (Li et al., 1994; Sakhi et al., 1994; Wood and Youle, 1995; Morrison et al., 1996; Xiang et al., 1998), this protein has not previously been thought to play a role in naturally occurring cell death nor has it been shown to act downstream of death receptor activation or of the MEKK-JNK pathway.
In this regard, we have previously demonstrated that increased expression of p53 was sufficient to cause sympathetic neuron apoptosis in the presence of NGF (Slack et al., 1996). On the basis of this observation, together with the fact that Bax is essential for naturally occurring sympathetic neuron death (Deckwerth et al., 1996; Easton et al., 1997), we hypothesized that p53 was an essential component of the signaling pathways causing sympathetic neuron apoptosis either in response to NGF withdrawal and/or to p75NTR activation. In this paper, we test this hypothesis, and demonstrate that both p75NTR activation and NGF withdrawal cause increased expression of p53, likely as a consequence of activation of the MEKK-JNK (Derijard et al., 1994; Yan et al., 1994) pathway, and that this convergent regulation of p53 is essential for normal naturally occurring sympathetic neuron death.
Materials and Methods
Primary Neuronal Cultures
Mass cultures of pure sympathetic neurons from the superior cervical ganglion (SCG)1 of postnatal day 1 rats were prepared as previously described (Belliveau et al., 1997; Bamji et al., 1998). Neurons were plated on rat tail collagen coated tissue culture dishes; 6-well plates for biochemistry, and 48-well plates for survival assays. Low density SCG cultures were used for all of the survival assays; for the survival assays in NGF withdrawal and p75 activation experiments, neurons were used at densities of 4,000–6,000 and 2,000–4,000 neurons per well of a 48-well plate, respectively. For biochemistry, ∼40,000–50,000 neurons were plated per well of a 6-well dish. For all experiments not involving viral infection, neurons were initially cultured for 5 d in the presence of 50 ng/ml NGF. At the end of this 5 day selection, neurons were washed several times in neurotrophin-free media before addition of the new neurotrophin- or KCL-containing media.
NGF for these experiments was purified from mouse salivary glands (CedarLane, Hornby, ON). Two sources of recombinant human BDNF (Amgen, Thousand Oaks, CA; Preprotech, Rocky Hill, NJ) were used for these experiments; similar results were obtained with both, as discussed previously (Bamji et al., 1998).
Virus Infections and Survival Assays
Six different recombinant adenoviruses were used for these experiments. Two, those expressing E1B55K, the E1B55K mutant A262 (Yew et al., 1990), and p53 (Slack et al., 1996) have been previously constructed and described. We also constructed adenoviruses (Massie et al., 1995) expressing activated MEKK1 (Eilers et al., 1998), and Bcl-xl (Boise et al., 1993) in the Ad5 backbone (Bett et al., 1994), which drives expression from the CMV promoter. As a control for these viruses, we used a recombinant adenovirus in the same Ad5 backbone expressing E. coliβ-galactosidase, as we have previously described (Slack et al., 1996; gift of Dr. Frank Graham, McMaster University, Hamilton, Ontario).
All recombinant adenoviruses were purified on CsCl gradients, as we have previously described (Slack et al., 1996). Infectious titer was determined by plaque assay on 293 cells (Graham and Prevec, 1991).
For the experiments involving E1B55K rescue from NGF withdrawal, neurons were cultured for 3–4 d in 50 ng/ml NGF, and then were infected overnight with various mois of recombinant adenovirus in serum-free media containing 50 ng/ml NGF. The next day, the virus was removed, and the cells were fed with fresh media containing 10 ng/ml NGF. The following day, cells were washed free of NGF with 3-h-long washes in serum-free, NGF-free media, and then were switched to media with or without 10 ng/ml NGF. 2 d later, neuronal survival was assessed. For the p75 activation experiments, the protocol was similar with the exception that, after the washes to remove NGF, neurons were switched to media containing 50 mM KCl with or without 100 ng/ml BDNF for 2 d.
For the experiments with the activated MEKK1 or p53 adenoviruses, neurons were plated at a density of 10,000–12,000 neurons/well in 48-well plates (Falcon Plastics, Cockeysville, MD) with 20 ng/ml NGF. On the fifth day of culture, medium was removed, the virus was added in a volume of 200 ul DMEM media (GIBCO-BRL, Gaithersburg, MD) containing 2 mM glutamine, 100 U/ml penicillin, 100 ug/ml streptomycin (all from BioWhittaker, Walkersville, MD), and 10% FBS. After 16–18 h of infection, the virus-containing media was removed and was replaced with Ultraculture media containing 2 mM glutamine, 100 U/ml penicillin, 100 μg/mlstreptomycin, and 20 ng/ml NGF. Cultures were maintained for 48 h, washed four times for one hour each with growth factor–free media, and then switched into the same media containing various concentrations of NGF.
Survival assays were performed as previously described (Slack et al., 1996; Bamji et al., 1998) using nonradioactive cell proliferation (MTT) assays (CellTitre 96; Promega Corp., Madison, WI). 50 μl of the MTT reagent was added to each well and left for 90 min followed by the addition of 100 μl of solubilization solution to lyse the cells. Each condition was repeated in triplicate. In each assay, baseline (0% survival) was considered to be 0 ng/ml NGF, and 10 ng/ml NGF was considered to be 100% survival, unless stated otherwise. All other conditions were related to these values.
Western Blot Analysis
For biochemistry, sympathetic neurons were lysed in TBS lysis buffer (Knusel et al., 1994) containing 137 mM NaCl, 20 mM Tris (pH 8.0), 1% (vol/vol) NP-40, 10% (vol/vol) glycerol, 1 mM PMSF, 10 μg/ml aprotinin, 0.2 μg/ml leupeptin, 1.5 mM sodium vanadate, and 0.1% SDS. Cells were collected in cold PBS by gentle scraping to detach them from the collagen substratum, were washed three times with the same buffer, and then were resuspended in 50 to 100 μl of lysis buffer, followed by rocking for 10 min at 4°C. After a 10 minute centrifugation, the lysates were normalized for protein concentration using a BCA Protein Assay Reagent (Pierce Chemical Co., Rockford, IL). Equal amounts of protein (10–50 μg) were then boiled in sample buffer for 5 minutes and separated by SDS-PAGE. After electrophoresis, proteins were transferred to 0.2 μm nitrocellulose for 1.5 h at 0.6 Amps, and the membrane was washed three times with TBS. For all antibodies, the membranes were blocked in 3% nonfat milk in TBST (blotto) for 1.5 h at room temperature. The membranes were then incubated overnight at 4°C with the primary antibodies in blotto: anti-p53 (Oncogene Neurosciences, Manhasset, NY), anti-p53 (Novacastra, Burlingame, CA), anti-p21 (Transduction Laboratories, Lexington, KY), anti-p27 (Transduction Laboratories), anti-Bad (Transduction Laboratories), anti-Bcl2 (Transduction Laboratories), anti-Bclxl (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Bax (Santa Cruz Biotechnology), anti-E1B55K antibody 2A6, anti-human c-myc (PharMingen, San Diego, CA), anti- phosphothr183/tyr184JNK (New England Biolabs, Boston, MA; Derijard et al., 1994), anti-phosphoser473Akt (New England Biolabs), anti-phosphothr183/tyr185Erk (Promega Corp.), anti-TrkA (RTA; gift of Dr. L. Reichardt, University of California, San Francisco, CA; Weskamp and Reichardt, 1991), anti-tyrosine hydroxylase (Chemicon Laboratories, Temecula, CA), or anti-tubulin (Oncogene Neurosciences). After incubation with the primary antibodies, membranes were washed four times with TBST over 40 min, and incubated with the secondary antibody for 1.5 h at room temperature. The secondary antibodies (goat anti–mouse or goat anti–rabbit HRP from Boehringer Mannheim GmhB, Mannheim, Germany) were used at 1/10,000 dilution. After three washes with TBST, detection was carried out using the ECL chemiluminescence reagent from Amersham and XAR x-ray film from Kodak.
Analysis of p53−/− Mice
Mice heterozygous for a targeted mutation in the p53 gene (Donehower et al., 1992) were obtained from GenPharm International (Mountainview, CA). p53−/− mice were maintained in a C57Bl/6 background, as homozygotes or heterozygotes. Progeny from p53−/−× p53+/− or from p53 heterozygote crosses were screened for the mutant allele(s) using PCR. To amplify the mutant allele, PCR was conducted for 35 cycles (94°C for 30 s, 55°C for 60 s, 72°C for 90 s) using the following oligonucleotides: GTGGGAGGGACAAAAGTTCGAGGCC for the 5′ end, and TTTACGGAGCCCTGGCGCTCGATGT for the 3′ end. This results in a 200 nucleotide fragment in mutant mice and no product in wildtype mice. To amplify the wild-type allele, the same oligonucleotide was used as the 5′ primer and ATGGGAGGCTGCCAGTCCTAACCC was used as the 3′ primer. This results in amplification of a 600 nucleotide fragment in heterozygote and wild-type mice, and no product in the p53−/− mice.
For morphometric analysis, the SCG were removed and immersion-fixed in 4% paraformaldehyde in phosphate buffer (PB) for 1 h to overnight at 4°C. Ganglia were cryoprotected in graded sucrose solutions, 7-μm-thick sections were serially cut on a cryostat, and every section was collected and thaw-mounted onto chrom-alum subbed slides. Slides were stained with cresyl violet and morphometric analyses were performed using a computer-based image analysis system (Biocom, Paris, France). Neuronal numbers were determined by counting all neuronal profiles with nucleoli on every third section, as per Coggeshall (1984). This sampling frequency (every 21 μm) ensures that neurons are not double counted, since the average neuronal diameter does not exceed 21 μm in any of the groups examined (Bamji et al., 1998). This method does not correct for split nucleoli. Statistical results were expressed as the mean ± SE of the mean and were tested for significance by a one-tailed Student's t test.
For TUNEL analysis, SCGs were dissected from p53+/− and p53+/+ littermates at postnatal day 7. Ganglia were fixed for 30 min in 4% paraformaldehyde, cryoprotected in graded sucrose solutions, and 7-μm-thick sections cut on a cryostat. Every third section was collected and thaw-mounted onto chrom-alum subbed slides and TUNEL staining immediately performed using the Boehringer-Mannheim in situ cell death detection kit. The number of TUNEL-positive cells on every third section was determined by fluorescence microscopy, and these numbers were used to determine the total number of TUNEL-positive cells per ganglia. Results were tested for significance by a one-tailed Student's t test.
Results
p53 and Bax Are Elevated after NGF Withdrawal from Sympathetic Neurons
Previously, we have demonstrated that an increase in p53 levels is sufficient to cause sympathetic neuron apoptosis in the presence of NGF (Slack et al., 1996). To determine whether endogenous p53 protein levels were ever similarly elevated during sympathetic neuron apoptosis, we cultured sympathetic neurons from neonatal animals, a time when developmental death of these neurons is ongoing. Initially, we examined p53 during sympathetic neuron apoptosis induced by NGF withdrawal; this apoptosis is relatively slow, taking ∼48 h, and is transcription-dependent (Deckwerth and Johnson, 1993; reviewed in Johnson and Deckwerth, 1993). Neurons were cultured for 5 d in the presence of 50 ng/ml NGF, NGF was withdrawn, and then the cellular levels of p53 were quantitated using Western blots with anti-p53 at various timepoints post-withdrawal (Fig. 1 A). This analysis revealed that p53 levels were elevated approximately threefold by 16 h after NGF withdrawal (Fig. 1 A). Elevation of p53 protein was first observed at 12 h (data not shown), and was maintained until at least 36 h post NGF-withdrawal (Fig. 1 A). Interestingly, this timecourse corresponds to the commitment point, after which NGF-withdrawn sympathetic neurons cannot be rescued from apoptotic death (Johnson and Deckwerth, 1993). Thus, NGF withdrawal leads to an increase in p53 levels that correlates with the timecourse of commitment to an apoptotic death.
Figure 1.

p53 and its transcriptional targets, p21 and Bax, are increased during NGF-withdrawal induced apoptosis of sympathetic neurons. (A–G) Western blot analysis of equal amounts of protein derived from sympathetic neurons either maintained in 10 ng/ml NGF (NGF), or at various timepoints from 4 to 48 h after withdrawal of NGF (−NGF).Western blots were probed with antibodies specific to p53 (A), p21 (B), p27 (C), Bad (D), Bcl-2 (E), Bax (F), or Bcl-xl (G). Note that p53, p21, p27, Bad, and Bax all increase after NGF withdrawal, whereas Bcl-2 and Bcl-xl decrease. (H) Western blot analysis of equal amounts of protein derived from sympathetic neurons either maintained in 20 ng/ml NGF, or withdrawn from NGF for 12 h (0 NGF). Western blots were probed with antibodies specific to phosphorylated forms of Akt (P AKT) or ERKS (P Erks). Note that phosphorylation of these TrkA targets was significantly decreased after NGF withdrawal.
To determine whether this increase in p53 had functional consequences, we examined the expression of a well-characterized p53 transcriptional target, the cyclin-dependent kinase p21 (El-Diery et al., 1993). Western blot analysis of equal amounts of protein from sympathetic neurons at various timepoints after NGF withdrawal revealed that p21 levels were increased twofold by 16 h after NGF withdrawal, and that this increase was maintained for at least 36 h (Fig. 1 B). This increase was not specific to p21, since levels of p27, a second cyclin-dependent kinase (Toyoshima and Hunter, 1994), were also increased approximately twofold at 24 and 36 h after NGF withdrawal (Fig. 1 C).
We next investigated the levels of Bax, another transcriptional target of p53 (Miyashita and Reed, 1995) that is known to be essential for naturally occurring sympathetic neuron death (Deckwerth et al., 1996; Easton et al., 1997). Western blot analysis revealed that, like p53, Bax levels increased approximately twofold after NGF withdrawal (Fig. 1 F). This increase was first observed at 16 h, and levels were maximal by 24 h (Fig. 1 F). Interestingly, Western blot analysis revealed that a second proapoptotic member of this family, Bad (Yang et al., 1995), was also increased threefold after NGF withdrawal (Fig. 1 D). Like Bax, this increase in Bad commenced at ∼16 h after withdrawal, the committment point for apoptosis. In contrast to Bad and Bax, Western blot analysis for the anti-apoptotic protein Bcl-2 (Hockenberry et al., 1990) revealed that levels of this protein were relatively constant over this timecourse, decreasing slightly at 24 h and later. A similar decrease was observed for another prosurvival member of this family, Bcl-xl (Boise et al., 1993), which was decreased by 36 h after NGF withdrawal (Fig. 1 G). Thus, these data indicate (a) that two transcriptional targets of p53, p21 and Bax, are increased after NGF withdrawal, and (b) that the balance of proapoptotic to prosurvival members of the Bcl2 family is significantly shifted after removal of NGF.
We also examined signaling proteins that are activated in response to TrkA receptor activation, including the ERKs (Virdee and Tolkovsky, 1995; Creedon et al., 1996) and Akt, a serine/threonine kinase thought to play a key role in promoting NGF-dependent sympathetic neuron survival (Crowder and Freeman, 1998). Western blot analysis with an antibody specific to the phosphorylated form of the serine/threonine kinase Akt revealed that, 12 h after NGF withdrawal, phosphorylation of Akt was greatly reduced (Fig. 1 H). Western blot analysis with an antibody specific to the phosphorylated form of the ERKs revealed a similar decrease in the phosphorylation of these proteins after NGF withdrawal (Fig. 1 H). Thus, NGF withdrawal leads to a decrease in signaling via these TrkA-regulated prosurvival pathways coincident with induction of potentially proaptotic pathways.
Elevated p53 is Necessary for Sympathetic Neuron Apoptosis in Response to NGF Withdrawal
To determine whether this increase in p53 protein levels was an essential component of the apoptotic cascade that follows NGF withdrawal, we took advantage of the adenoviral E1B55K protein, which functionally ablates p53 (Yew et al., 1992). Specifically, neonatal sympathetic neurons were cultured for 3–4 d in 50 ng/ml NGF, and then were infected with one of two recombinant, replication-defective adenoviruses; one of these adenoviruses expressed E1B55K while the second expressed a mutant E1B55K protein (A262) that is defective in its ability to bind and ablate p53 (Yew et al., 1990). As a further control, neurons were infected with a similar moi of recombinant adenovirus expressing β-galactosidase (Slack et al., 1996). 2 d after viral infection, neurons were washed free of NGF and 2 d later, neuronal survival was measured using MTT assays (Fig. 2 A). These experiments demonstrated that E1B55K, but not the E1B55K mutant A262 or β-galactosidase, was able to rescue sympathetic neurons from apoptosis. In 5 independent experiments, E1B55K rescued 49% (100 moi) and 63% (500 moi) of the neurons relative to those kept alive in 10 ng/ml NGF, increases which were highly significant (P < 0.005 in both cases). In none of these experiments did either the A262 or β-galactosidase virus have a significant effect on neuronal survival (Fig. 2 A).
Figure 2.
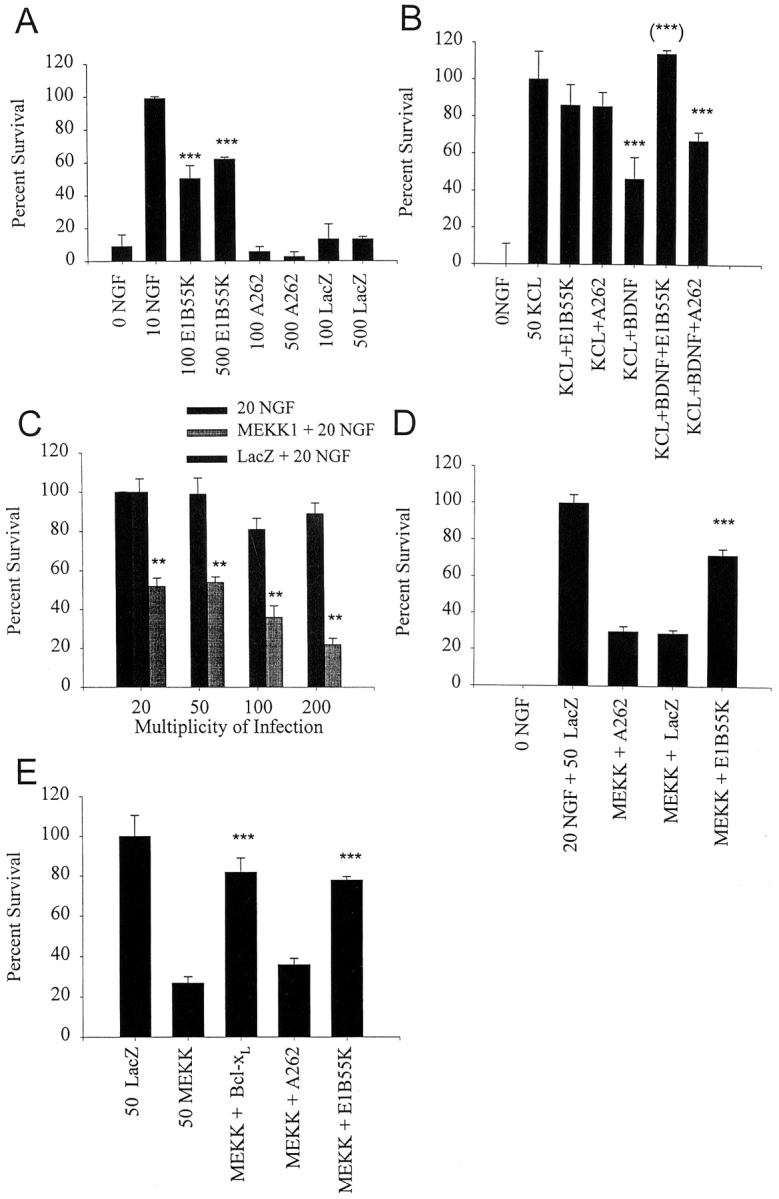
E1B55K-mediated inhibition of p53 rescues sympathetic neuron apoptosis as induced by NGF withdrawal (A), p75NTR activation (B), and activated MEKK (C–E). (A) p53 and NGF withdrawal-induced apoptosis. Sympathetic neurons were infected in parallel with recombinant adenovirus expressing E1B55K, the mutant E1B55K A262, or β-galactosidase (LacZ) at titers of 100 or 500 moi and NGF was withdrawn from the media 2 d later. After 48 h, cell survival was measured by MTT assays. 0 NGF represents uninfected sympathetic neurons that were also withdrawn from NGF, whereas 10 NGF represents uninfected sympathetic neurons maintained in the presence of 10 ng/ml NGF for the course of the experiment. Results are those obtained in a representative experiment performed in triplicate, and represent the mean ± the standard error. Similar results were obtained in five separate experiments. ***Indicates those values that are significantly different from the 0 NGF control (P < 0.005). Note that only those neurons transduced with E1B55K were rescued from NGF withdrawal-induced apoptosis. (B) p53 and p75-induced apoptosis. Sympathetic neurons were infected with recombinant adenovirus expressing E1B55K or the mutant E1B55K A262 at a titer of 500 moi and 2 d later were switched into 50 mM KCl (KCL+E1B55K and KCL+A262) or 50 mM KCl plus 100 ng/ml BDNF (KCL+BDNF+E1B55K and KCL+BDNF+ A262). As controls, uninfected neurons were switched into 50 mM KCl (50 KCL), into 50 mM KCl plus 100 ng/ml BDNF (KCL+BDNF), or were withdrawn from NGF (0 NGF). After 48 h, cell survival was measured using MTT assays. Results are those obtained in a representative experiment performed in triplicate, and represent the mean ± standard error. Similar results were obtained in four separate experiments. ***Indicates those values that are significantly different from 50 mM KCl alone, and (***) indicates those values amongst the points that received BDNF that are significantly different from KCL+BDNF (P < 0.005). Note that, as previously reported (Bamji et al., 1998), BDNF reduces neuronal survival in the presence of KCl, and that E1B55K but not A262 is able to completely rescue this BDNF- mediated apoptosis. (C) MEKK1-induced apoptosis. Sympathetic neurons were infected with recombinant adenovirus expressing activated MEKK1 or β-galactosidase (LacZ) at concentrations of 20 to 200 moi, and were maintained in 20 ng/ml NGF for 4 d after infection. As controls, uninfected neurons were maintained for the entire experiment in 20 ng/ml NGF. Cell survival was measured using MTT assays, and results are those obtained in a representative experiment performed in triplicate, and represent the mean ± standard error. Similar results were obtained in 4 separate experiments. **Indicates those values that are significantly different between MEKK1 and the β-galactosidase control at a given moi (P < 0.005). Note that activated MEKK1 decreases sympathetic neuron survival in a concentration-dependent fashion. (D) p53 and MEKK1-induced apoptosis. Sympathetic neurons were infected with 50 moi of MEKK1 plus 500 moi of E1B55K (MEKK +E1B55K), A262 (MEKK + A262) or β-galactosidase (MEKK + LacZ), were maintained in 20 ng/ml NGF for 4 d after infection, and survival was then measured using MTT assays. As further controls, neurons were infected with 50 moi β-galactosidase virus (20 NGF+ 50 LacZ) for the same time period, or were withdrawn from NGF for the final two days. Results are those obtained in a representative experiment performed in triplicate, and represent the mean ± standard error. Results are normalized so that 0 NGF is 0% survival, and 20 ng/ml NGF plus 50 moi β-galactosidase (20 NGF+ 50 LacZ) is 100% survival. Similar results were obtained in three separate experiments. ***Indicates that E1B55K significantly rescues MEKK1-induced killing of sympathetic neurons (P < 0.005). (E) p53 in JNK-Bax cell death pathway. Sympathetic neurons were infected with 50 moi of the activated MEKK1 adenovirus (50 MEKK) plus or minus 50 moi Bcl-xl (MEKK+ Bcl-xl), 500 moi E1B55K (MEKK+ E1B55K) or 500 moi A262 (MEKK + A262) adenoviruses. As a control, neurons were infected with 50 moi β-galactosidase adenovirus (50 LacZ). 4 d after the initial infection, during which time neurons were maintained in 10 ng/ml NGF, survival was measured using MTT assays. Results are those obtained in a representative experiment performed in triplicate, and represent the mean ± standard error. Results are normalized so that 0 NGF is 0%, and 10 ng/ml NGF plus 50 moi β-galactosidase adenovirus is 100% survival. ***Indicates those values that are significantly different from MEKK alone (P < 0.005).
To ensure that the effects of E1B55K were being mediated via p53, we examined levels of p53 in NGF-withdrawn sympathetic neurons after viral infection; the vector used not only ablates p53 through the actions of E1B55K, but also targets p53 for degradation through the adenoviral E4orf6 product (Yew and Berk, 1992; Querido et al., 1997; Teodoro and Branton, 1997). For these experiments, sympathetic neurons were initially selected in 50 ng/ml NGF for 3 d, were infected with the adenoviruses expressing E1B55K or the mutant E1B55K, and 2 d later were withdrawn from NGF. As a control, neurons were not infected or were infected with the β-galactosidase virus. Analysis of p53 protein levels 36 h after this treatment revealed that levels of p53 protein were similarly elevated in NGF-withdrawn neurons that were uninfected, or that were expressing either β-galactosidase or the mutant E1B55K (Fig. 3 A). In contrast, p53 protein levels were significantly reduced in the neurons expressing E1B55K, as predicted. Reprobing of the same blot with an antibody specific for the cytoskeletal protein tubulin confirmed that equal amounts of protein were present in all of the samples (Fig. 3 B). Moreover, the differential effect of E1B55K versus the A262 mutant was not due to a difference in levels of expression of these two proteins, since the A262 protein was expressed at higher levels than E1B55K (Fig. 3 C). These experiments therefore indicate that elevated p53 protein levels are essential for NGF-withdrawal induced apoptosis of sympathetic neurons.
Figure 3.

E1B55K reduces p53 levels in sympathetic neurons withdrawn from NGF. (A) Western blot analysis for p53 in sympathetic neurons that were withdrawn from NGF for 36 h (0 NGF), that were maintained in 10 ng/ml NGF (10 NGF), or that were infected with 500 moi of recombinant adenovirus expressing A262 or E1B55K, and 2 d later were withdrawn from NGF for 24 h. Equivalent amounts of protein from lysed neurons were electrophoresed, transferred to nitrocellulose filters, and p53 protein levels were assessed using anti-p53. Note that p53 levels increase after NGF withdrawal, and that expression of E1B55K reverses this increase, while expression of A262 has no effect. (B) The same blot as in panel (A) reprobed for the cytoskeletal protein tubulin. (C) Western blot analysis for E1B55K using anti-E1B55K in equal amounts of protein derived from sympathetic neurons infected for 36 h with adenoviruses expressing E1B55K or the E1B55K mutant A262. The antibody used for these studies recognizes both the wild-type and mutant proteins. (D) Western blot analysis for p53 in sympathetic neurons that were maintained in 20 ng/ml NGF, and that were infected with 50 moi of recombinant adenovirus expressing activated MEKK1 (MEKK) with or without 200 moi of E1B55K for 36 h. Equivalent amounts of protein from lysed neurons were electrophoresed, transferred to nitrocellulose filters, and p53 protein levels were assessed using anti-p53. (E) The same blot as in D reprobed for TrkA.
p53 is Downstream of p75NTR and is Essential for p75NTR-mediated Neuronal Apoptosis
We have previously demonstrated that the immediate early protein, c-jun, is hyperphosphorylated in sympathetic neurons after p75NTR activation (Bamji et al., 1998) as it is after NGF withdrawal (Estus et al., 1994; Ham et al., 1995). To determine whether p53 elevation was also a common downstream component of these two apoptotic signaling cascades, we examined p53 levels in sympathetic neurons in conditions where p75NTR activation leads to apoptosis. Specifically, for these experiments, sympathetic neurons were cultured for 5 d in 50 ng/ml NGF and then were switched to KCl, which maintains sympathetic neuron survival in the absence of TrkA activation (Franklin et al., 1995). We then added the neurotrophin BDNF, which selectively binds p75NTR but not Trk receptors on sympathetic neurons (Bamji et al., 1998), and determined cellular p53 levels (Fig. 4 A). As seen with NGF withdrawal, p53 levels were increased at 24 and 36 h in neurons maintained in KCl plus BDNF relative to those in KCl alone. Reprobing of the same blot with an antibody to tyrosine hydroxylase revealed that equal amounts of protein were present in all of the samples (Fig. 4 B). This increase was already apparent at 12 h, a timepoint when p53 levels were only first starting to increase after NGF withdrawal (data not shown). Thus, p53 elevation is downstream of both p75NTR activation and NGF withdrawal.
Figure 4.
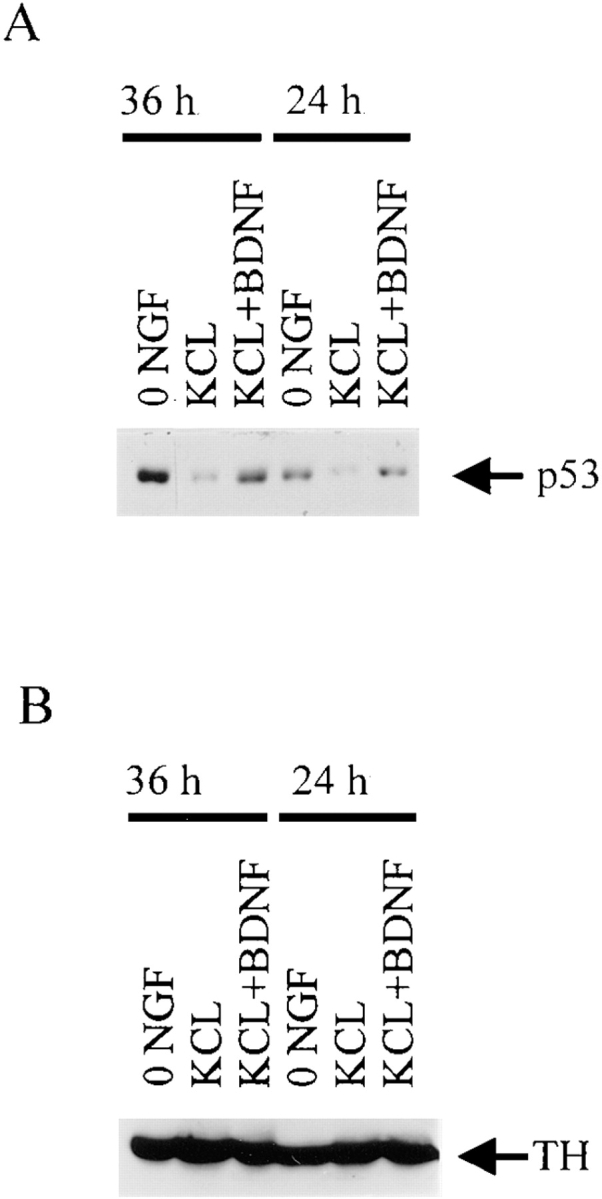
p53 levels increase during apoptosis of sympathetic neurons as induced by BDNF-mediated activation of p75NTR. (A) Western blot analysis for p53 in equal amounts of protein derived from sympathetic neurons that were cultured in 50 ng/ ml NGF for 4 d, and then were washed free of NGF and switched into 50 mM KCl (KCL), 50 mM KCl plus 100 ng/ml BDNF (KCL + BDNF), or media containing no NGF or KCl (0 NGF) for 24 or 36 h. Note that p53 levels in the neurons treated with KCl and BDNF are similar to those in 0 NGF, and are greater than those maintained in KCl alone. (B) The same blot as in A reprobed for the neurotransmitter enzyme, tyrosine hydroxylase (TH) to demonstrate that equal amounts of protein were present in each of the lanes.
To determine whether this increased p53 was essential for p75NTR-mediated apoptosis, we performed rescue experiments using the E1B55K adenovirus. Specifically, neurons were cultured in 50 ng/ml NGF for 3–4 d, were infected overnight with recombinant adenovirus, and 2 d later were switched to media containing 50 mM KCl plus or minus 100 ng/ml BDNF. Neuronal survival was then measured after 48 h using MTT assays (Fig. 2 B). As we have previously reported (Bamji et al., 1998), the addition of BDNF to 50 mM KCl caused a decrease in sympathetic neuron survival of ∼54% (Fig. 2 B). Expression of the mutant E1B55K had no significant effect on this decrease (P =0.11). In contrast, expression of E1B55K rescued 100% of the p75NTR-driven apoptosis (P = 0.012; Fig. 2 B), a rescue similar to that observed for NGF-withdrawal induced apoptosis. Thus, p53 elevation was an essential component of the apoptotic cascades induced in cultured sympathetic neurons by both NGF withdrawal and p75NTR activation.
p53 is Downstream of the MEKK-JNK Pathway, and Is Essential for MEKK-mediated Neuronal Apoptosis
These results indicated that the apoptotic cascades originating from NGF withdrawal and p75NTR activation share two common components; hyperphosphorylation of c-jun and p53 elevation. On the basis of these findings, we hypothesized that p53 was downstream of the MEKK-JNK pathway, which leads to c-jun hyperphosphorylation (Derijard et al., 1994; Yan et al., 1994). To test this hypothesis, we generated a recombinant adenovirus expressing an activated form of MEKK1 which has previously been shown to cause sympathetic neuron apoptosis and c-jun hyperphosphorylation (Eilers et al., 1998). We first confirmed that this virus expressed the recombinant myc epitope-tagged MEKK1 protein, and that it was capable of activating the MEKK1 downstream target, JNK (Yan et al., 1994), in sympathetic neurons. Specifically, sympathetic neurons were grown in 20 ng/ml NGF for 4 d, were infected with 20 moi of recombinant virus expressing activated MEKK1 or β-galactosidase, and then were analyzed 2 d later for expression of the myc-tagged MEKK1 on Western blots with anti-myc (Fig. 5 A). Analysis of equal amounts of protein from β-galactosidase versus MEKK1-infected sympathetic neurons revealed the presence of a myc-immunoreactive protein of the appropriate size, 35 kD, in the MEKK1-infected neurons (Fig. 5 A). To confirm that virally expressed MEKK1 was capable of activating JNK, we performed similar experiments and then analyzed the lysates for phosphorylation of JNK using a phospho-JNK antibody (Fig. 5 B). In this experiment, we compared MEKK1-infected sympathetic neurons to sympathetic neurons withdrawn from NGF, where JNK is known to be activated (Eilers et al., 1998). Western blot analysis revealed that the activated MEKK1 adenovirus caused phosphorylation of JNK to the same level as did NGF withdrawal, relative to neurons maintained in 10 ng/ml NGF alone (Fig. 5 B).
Figure 5.

(A–D) p53 and Bax protein levels increase after activation of the MEKK-JNK pathway in sympathetic neurons. (A) Western blot analysis for c-myc in equal amounts of protein derived from sympathetic neurons infected with 20 moi myc-tagged MEKK1 (20 moi MEKK) or β-galactosidase (control) adenovirus for 48 h. In both cases, neurons were maintained in 20 ng/ml NGF for the entirety of the experiment. (B) Western blot analysis for phospho-JNK in equal amounts of protein derived from sympathetic neurons that were infected with 50 moi MEKK1 adenovirus and maintained for 48 h (50 moi MEKK), or from uninfected sister cultures that were either maintained in 10 ng/ml NGF (10 NGF), or that were withdrawn from NGF for 48 h (0 NGF). Note that the level of phospho-JNK immunoreactivity is similar in neurons withdrawn from NGF or transduced with activated MEKK1. (C) Western blot analysis for p53 in equal amounts of protein derived from sympathetic neurons that were infected with 20 moi MEKK1 adenovirus and maintained in 20 ng/ml NGF for 48 h (20 moi MEKK), or from uninfected sister cultures that were maintained in 10 ng/ml NGF (10 NGF), or that were withdrawn from NGF for 48 h (0 NGF). Note that p53 protein levels are increased by activated MEKK1 as they are by NGF withdrawal. (D) Western blot analysis for Bax in equal amounts of protein derived from sympathetic neurons that were infected with 50 moi MEKK1 adenovirus and maintained in 20 ng/ml NGF for 48 h (50 moi MEKK), or from uninfected sister cultures that were maintained in 10 ng/ml NGF for the same time period. (E and F) Increased expression of p53 in sympathetic neurons causes increased Bax protein, but does not affect phosphorylation of JNK. (E) Western blot analysis for phospho-JNK in equal amounts of protein derived from sympathetic neurons infected with 20 moi p53 adenovirus and maintained in 20 ng/ml NGF for 48 h, or from uninfected sister cultures that were maintained in 20 ng/ml for the same time period. (F) Western blot analysis for Bax in equal amounts of protein derived from sympathetic neurons infected with 20 moi p53 adenovirus and maintained in 20 ng/ml NGF for 48 h, or from uninfected sister cultures maintained in 20 ng/ml NGF for the same timeperiod. (G) Western blot analysis for p53 in equal amounts of protein derived from sympathetic neurons maintained in 20 ng/ml NGF and infected with 20 moi p53 adenovirus or 50 moi activated MEKK adenovirus for 30 h. As a control, neurons were withdrawn from NGF (0 NGF) for 30 h.
We next determined whether adenovirus-mediated expression of activated MEKK1 was sufficient to cause sympathetic neuron apoptosis, as was previously reported with microinjection (Eilers et al., 1998). Specifically, neurons were selected in 20 ng/ml NGF for 5 d, infected with various concentrations of recombinant virus expressing activated MEKK1 or β-galactosidase, maintained in 20 ng/ml NGF for a further 4 d, and then assayed for neuronal survival. Results from four separate experiments revealed that activated MEKK1 led to neuronal apoptosis; at 20, 50, or 100 moi, sympathetic neuron survival was decreased to 52, 54, and 44%, respectively, relative to neurons infected with the same concentration of β-galactosidase virus (Fig. 2 C). Similar results were obtained with neurons maintained in 10 ng/ml and 50 ng/ml NGF (data not shown).
We next determined whether activation of the MEKK-JNK pathway caused elevation of p53 levels, as we had hypothesized. Specifically, sympathetic neurons were maintained in 20 ng/ml NGF for 4 d, were infected with 20 or 50 moi of the activated MEKK1 adenovirus, and were then maintained in 10 ng/ml NGF for a further 2 d before analysis. To ensure that viral infection itself did not induce p53, we infected sister cultures with the β-galactosidase adenovirus. Western blot analysis of equal amounts of protein revealed that p53 levels were increased when sympathetic neurons were infected with 20 or 50 moi of MEKK1 adenovirus, and that the magnitude of this increase was similar to that observed after NGF withdrawal for 48 h (Fig. 5 C). In contrast, no increase in p53 levels was seen upon infection with similar mois of the β-galactosidase adenovirus (data not shown), consistent with the observation that this latter virus had little or no effect on neuronal survival (Fig. 2 C). Thus, activation of the MEKK-JNK pathway led to increased levels of p53 in the presence of NGF.
To determine whether p53 was an essential component of the MEKK1-induced apoptotic signaling cascade, we asked whether the E1B55K adenovirus could rescue the apoptotic effects of activated MEKK1. To perform these experiments, sympathetic neurons were cultured for 5 d in 20 ng/ml NGF, and were infected with 50 moi of the MEKK1 virus plus or minus 500 moi of the E1B55K virus. As a baseline for this study, we first determined whether coinfection with E1B55K reduced the p53 levels induced by activated MEKK1 alone (Fig. 3 D). Western blot analysis of sympathetic neurons infected for 2 d revealed that p53 protein levels were reduced in neurons expressing E1B55K plus activated MEKK1 relative to those expressing MEKK1 alone (Fig. 3 D). Reprobing of the same blot with an antibody specific for TrkA confirmed that equal amounts of protein were present in all of the samples (Fig. 3 E).
To determine whether this reduction in p53 levels mediated by E1B55K rescued sympathetic neurons from apoptosis induced by activated MEKK1, we performed similar experiments and, 4 d after infection, MTT assays were performed. As negative controls in this experiment, we used adenoviruses expressing the mutant A262 protein or β-galactosidase. Results of three separate experiments indicated that the E1B55K adenovirus was able to significantly rescue MEKK1-induced neuronal apoptosis (Fig. 2 D) relative to MEKK1 alone, or relative to MEKK1 plus the β-galactosidase or A262 adenovirus (P < 0.005 for E1B55K in all cases; Fig. 2 D). Thus, elevation of p53 protein levels is necessary for sympathetic neuron apoptosis after activation of the MEKK-JNK pathway.
One potential downstream candidate for the apoptotic effects of p53 in sympathetic neurons after MEKK-JNK pathway activation is the proapoptotic protein Bax. To determine whether Bax levels were increased in response to the MEKK1 adenovirus as they were after NGF withdrawal (Fig. 1 F), we selected sympathetic neurons for 4 d in 20 ng/ml NGF, infected them with 50 moi of the MEKK1 adenovirus, switched them into 10 ng/ml NGF and 2 d later performed Western blots with anti-Bax. This analysis revealed that Bax levels were increased approximately twofold in sympathetic neurons expressing activated MEKK1 (Fig. 5 D), an increase similar in magnitude to that seen after NGF withdrawal (Fig. 1 F).
Previous studies have demonstrated that Bax is necessary for sympathetic neuron apoptosis after NGF withdrawal (Deckwerth et al., 1996). To determine whether proapoptotic proteins like Bax are also necessary for sympathetic neuron apoptosis after activation of the MEKK-JNK pathway, we generated a recombinant adenovirus expressing a prosurvival member of this pathway, Bcl-xl (Boise et al., 1993). Using this adenovirus, we then asked whether altering the balance between prosurvival versus proapoptotic members of this family could rescue MEKK1- induced apoptosis. Specifically, sympathetic neurons were cultured for 5 d, and were infected with 50 moi of the MEKK1 adenovirus plus or minus various concentrations of Bcl-xl virus. For comparison, neurons were infected with 50 moi of MEKK1 virus plus 500 moi E1B55K or A262. Neurons were then maintained in 20 ng/ml NGF for four additional days before measuring survival using MTT assays. This analysis revealed that the Bcl-xl adenovirus was able to rescue the apoptotic effects of activated MEKK1 at 20 (data not shown) or 50 moi (Fig. 2 E). A similar rescue was observed with the E1B55K virus (Fig. 2 E), while no rescue was observed with the A262 virus (Fig. 2 E). Similar results were obtained when the MEKK1 adenovirus was used at 100 moi, although the magnitude of the rescue effect with lower concentrations of Bcl-xl was decreased (data not shown).
p53 Is Upstream of Bax and Downstream of JNK
These experiments demonstrated that JNK, p53 and Bax are all downstream of activated MEKK, and that elevated p53 is essential for MEKK-induced apoptosis. To determine whether p53 is downstream of JNK and upstream of Bax, as we had hypothesized, we took advantage of a recombinant adenovirus expressing human p53 (previously described in Slack et al., 1996). Specifically, sympathetic neurons were cultured for 4 d in 20 ng/ml NGF, were infected with 20 moi of p53-expressing adenovirus and 48 h later, cell lysates were analyzed by Western blots with anti-p53, anti-phosphoJNK or anti-Bax. This analysis revealed that the p53 adenovirus increased expression of p53 to levels similar to those observed after NGF withdrawal (Fig. 5 G), but that this elevated expression of p53 had no effect on JNK phosphorylation in sympathetic neurons maintained in 20 ng/ml NGF (Fig. 5 E). In contrast, infection with the p53 virus caused an increase in the levels of Bax protein (Fig. 5 F) that was similar in magnitude to that observed after NGF withdrawal (Fig. 1 F) or after infection with the activated MEKK1 adenovirus (Fig. 5 D). Together with the previous experiments with activated MEKK1, these experiments indicate that MEKK and JNK are upstream of p53, while Bax is downstream (Fig. 7).
Figure 7.

Model of sympathetic neuron apoptosis induced by NGF withdrawal or p75NTR activation. Both of these apoptotic stimuli increase p53 and Bax protein levels, and require elevated p53 levels to efficiently promote cell death. Neuronal death induced by activated MEKK1 also increases p53 and Bax protein levels, and requires elevated levels of p53 protein. NGF withdrawal-mediated sympathetic neuron death increases JNK and c-jun phosphorylation and activity, and requires c-jun (Estus et al., 1994; Ham et al., 1995). p75NTR activation in sympathetic neurons also leads to hyperphosphorylation of c-jun, presumably via JNK (Bamji et al., 1998). Bax is also essential for sympathetic neuron apoptosis in vivo and in vitro (Deckwerth et al., 1996; Easton et al., 1997). The precise MEKK or JNK family member in these pathways is not known, and other intermediate proteins, such as SEK1 (Sanchez et al., 1994) are likely involved as intermediate steps in this hypothetical cascade. Moreover, although this model shows JNK signaling to p53 via c-jun, recent work indicates that JNK can directly interact with and phosphorylate p53 (Hu et al., 1997). p53 may induce death by increasing Bax protein or activity, or via other p53 target proteins (Polyak et al., 1997).
p53 is Essential for Naturally Occurring Sympathetic Neuron Death In Vivo
Our previous work indicates that naturally occurring sympathetic neuron death is a result both of suboptimal activation of the TrkA receptor and of coincident activation of p75NTR (Bamji et al., 1998). Since, in culture, both of these types of neuronal apoptosis require p53, we hypothesized that p53 would also be essential for sympathetic neuron death in vivo. To test this hypothesis, we examined the SCG of transgenic mice in which the p53 gene was deleted by homologous recombination (Donehower et al., 1992). In control mice, the SCG contains ∼20,000–25,000 neurons at birth, depending on the genetic background. Over the ensuing two weeks approximately half of these neurons are lost so that by P15, control SCGs contain ∼13,000 neurons (Bamji et al., 1998). We therefore chose to analyze the SCG from p53+/+, p53+/− and p53−/− mice at two timepoints; postnatal days 1 and 15, timepoints immediately preceding and after the normal period of naturally occurring cell death (Fig. 6, A and D).
Figure 6.

The number of sympathetic neurons in the superior cervical ganglia is increased when p53 levels are decreased in vivo. (A) Photomicrographs of cresyl-violet stained sections through the SCG of wild-type (p53+/+) versus p53+/− mice of the same genetic background at postnatal day 15, when naturally occurring sympathetic neuron death is complete. (B) Fluorescent photomicrographs of in situ TUNEL labeling in the SCG of p53+/+ versus p53+/− mice of the same genetic background at postnatal day 7, when developmental sympathetic neuron death is ongoing. (C) The number of TUNEL-positive cells in the SCG of P7 mice that are heterozygous (p53+/−) or wild-type (p53+/+) for a p53 mutation created by homologous recombination (Donehower et al., 1992). Results are expressed as the mean ± standard error of the total number of TUNEL-positive cells per SCG (n = 5 for p53+/− andn = 4 for p53+/+). **Indicates that these two values are significantly different (P = 0.016). (D) The number of neurons in the SCG of P1 or P15 mice that are either p53+/+ (control), or heterozygous (p53+/−) or homozygous (p53−/−) for the p53 mutant allele. Results are expressed as the mean ± standard error (n = 3 for P1 p53+/+, 5 for P1 p53+/−, 3 for P1 p53−/−, 5 for P15 p53+/+, 6 for P15 p53+/− and 3 for P15 p53−/−). **Indicates values significantly different from p53+/+ SCG of the same age (P < 0.05). Note that while there is a statistically significant loss of 45% of the neurons in the p53+/+ SCG over this time period (**P < 0.05), there is no significant loss of neurons in either the p53+/− or p53−/− SCG (P > 0.3).
Analysis of the SCG at postnatal day 1 revealed that sympathetic neuron numbers before the period of naturally occurring death were similar regardless of the presence or absence of p53 (Fig. 6 D). The SCG of p53+/+ animals contained 23,976 ± 2764 neurons (n = 3), a number similar to mice of other genetic backgrounds (Bamji et al., 1998), while those from p53+/− and p53−/− animals contained 20,537 ± 2514 (n = 5) and 19016 ± 2675 (n = 3), respectively (Fig. 6 D). All of these numbers were statistically similar (P > 0.1 for all comparisons). However, analysis of the SCG from animals of these same genotypes at P15 (Fig. 6, A and D), after the period of naturally occurring death, revealed significant differences. In control p53+/+ animals the P15 SCG contained 13,163 ± 875 neurons (n = 5; Fig. 6 D). In contrast, the SCG of p53+/− animals contained 20,352 ± 944 neurons (n = 6), and that of p53−/− mice contained 20,600 ± 1709 neurons (n = 3), statistically significant increases of 55 and 56%, respectively (P < 0.005 in both cases). A comparison within the same genotype from P1 to P15 revealed that while the p53+/+ ganglia lost ∼45% of its sympathetic neurons over this timeframe (P < 0.05; Fig. 6 D), there was no significant loss of sympathetic neurons in either the p53+/− or p53−/− SCG over the same period (Fig. 6 D; P > 0.3).
To confirm that this difference in neuron number in the SCG during the period of naturally occurring cell death reflected a deficit in apoptosis in the p53+/− and p53−/− mice, and was not due to an increase in the proliferation of neuronal progenitors that occurs for a short period postnatally in the SCG (Hendry, 1977), we analyzed the total number of apoptotic cells in the SCG at postnatal day 7, when sympathetic neuron apoptosis is ongoing. To perform this analysis, SCG from p53+/− and p53+/+ littermates were sectioned, and every third section was analyzed by in situ TUNEL-labeling. This analysis revealed that TUNEL-positive cells were detected in the ganglia of both p53+/− and p53+/+ animals (Fig. 6 B), but that the total number of apoptotic nuclei was significantly decreased in the p53+/− ganglia (p53+/− SCG, mean = 661 ± 19,n = 5; p53+/+ SCG, mean = 1025 ± 155,n = 4;P = 0.016; Fig. 6 C). Thus, the magnitude of sympathetic neuron apoptosis is decreased when p53 levels are decreased in vivo.
Discussion
In this study, we have examined the role of the p53 tumor suppressor in naturally occurring sympathetic neuron death. Our findings support an essential role for p53 in this process and, more specifically, support the following conclusions. First, after NGF withdrawal of neonatal sympathetic neurons, p53 levels are increased with a timecourse that is consistent with it playing a role in the “commitment” to apoptosis. The magnitude of this increase is similar to that required to induce sympathetic neuron apoptosis using adenovirus-mediated transduction of p53 (Slack et al., 1996). Second, levels of two p53 transcriptional targets, p21 and Bax, are also increased; the latter of these two, Bax, has already been shown to be essential for sympathetic neuron apoptosis after NGF withdrawal (Deckwerth et al., 1996). Third, p53 levels are also increased when sympathetic neuron apoptosis is induced by p75NTR activation, with a similar timecourse to that observed after NGF withdrawal. Fourth, a decrease in levels of p53 mediated by the E1B55K protein is sufficient to inhibit sympathetic neuron apoptosis induced either by NGF withdrawal or by p75NTR activation, indicating that increased p53 levels are both necessary and sufficient (Slack et al., 1996) for sympathetic neuron apoptosis. Fifth, NGF withdrawal and p75NTR activation may induce apoptosis via a pathway involving MEKK-JNK-p53-Bax (Fig. 7) since (a) expression of activated MEKK1, which is sufficient to cause sympathetic neuron apoptosis, leads to elevated levels of p53 and Bax, (b) expression of p53 leads to elevated Bax levels, but does not affect JNK activation, and (c) elevated p53 is essential for MEKK-induced apoptosis. Finally, the physiological relevance of these observations is indicated by our findings that, when p53 is reduced or absent, naturally occurring sympathetic neuron death is inhibited in vivo. Thus, our data indicate that p53 is a common, essential target during developmental sympathetic neuron death that is downstream of the MEKK-JNK pathway and that may well integrate apoptotic signals deriving both from p75NTR activation and from a lack of NGF/ TrkA receptor activation.
The p53 tumor suppressor protein encodes a transcriptional regulator that functions to control cell proliferation and apoptosis in a cell context-dependent fashion (reviewed in Levine, 1997; Jacks and Weinberg, 1998). The precise mechanism by which p53 mediates apoptosis is not well understood, but it is believed to proceed by a number of mechanisms including direct transactivation, transcriptional repression, and direct involvement in DNA cleavage (Lane, 1993; Caelles et al., 1994; Elledge and Lee, 1995; Miyashita and Reed, 1995). Within the nervous system, p53 is upregulated in neurons after a number of traumatic insults, including kainic acid and ischemia (Li et al., 1994; Sakhi et al., 1994; Wood and Youle, 1995), and is necessary for excitotoxicity-induced apoptosis of hippocampal and cortical neurons (Morrison et al., 1996; Xiang et al., 1998). However, p53 has not been thought to be involved in naturally occurring neuronal cell death, nor has it been thought to be an essential downstream effector of death receptor activation. In particular, a number of previous studies have failed to implicate p53 in sympathetic neuron death. In one study, Martinou et al. (1995) demonstrated that microinjection of the adenoviral protein E1B19K, but not E1B55K, rescued sympathetic neurons from apoptosis due to NGF withdrawal. The inability to rescue sympathetic neuron apoptosis with microinjected E1B55K as opposed to adenovirally expressed E1B55K is likely due to the lower levels of expression obtained using microinjection. In this regard, we have ourselves compared the rescue of sympathetic neurons using adenovirally expressed E1B19K versus E1B55K, and E1B19K was able to rescue sympathetic neurons at titers ∼10-fold lower than those needed for E1B55K (Pacquet, L., F. Miller, and D. Kaplan, unpublished data). In a second set of studies, cultured p53−/− sympathetic neurons died after NGF withdrawal, leading to the conclusion that p53 played no role in developmental sympathetic neuron death (Davies and Rosenthal, 1994; Sadoul et al., 1996). The studies presented here demonstrate that neurons with lowered p53 levels still die in vivo, although the magnitude of this death is significantly lower than in controls; these results do not necessarily contradict the finding that p53−/− neurons die in culture after NGF withdrawal. Moreover, results might also differ in response to acute inhibition of p53 (as mediated via E1B55K) versus a chronic loss of p53 (as in p53−/− mice). For example, the developmental absence of p53 may well cause compensatory upregulation of parallel apoptotic pathways. Precedent for such compensation derives from our previous studies with another tumor suppressor protein, pRb. In Rb−/− mice, cortical neurons are unable to differentiate appropriately and, as a consequence, undergo neuronal apoptosis in vivo (Slack et al., 1998). However, when the same Rb−/− cortical progenitors are cultured and undergo the transition to postmitotic neurons in vitro, they differentiate normally and do not undergo apoptosis (Slack et al., 1998); this difference is explained by a selective upregulation of p107 and p130, two other Rb family members, in culture but not in vivo (Ruth Slack, personal communication). A similar explanation could be invoked for cultured p53−/− neurons; sympathetic neurons express p73 (Pozniak, C., and F. Miller, unpublished data), an apoptotic p53 family member (Jost et al., 1997; Kaghad et al., 1997) that might well compensate for the lack of p53 in a mutant p53 background.
What is the signal transduction pathway that derives either from p75NTR activation or from a lack of activation of TrkA and that leads to increased p53? Our data implicate the MEKK-JNK pathway (Derijard et al., 1994; Yan et al., 1994; Fig. 7). This pathway is activated in sympathetic neurons in response to either NGF withdrawal (Ham et al., 1995; Eilers et al., 1998) or p75NTR activation (Bamji et al., 1998), and the resultant hyperphosphorylation of c-jun is necessary for NGF-withdrawal induced sympathetic neuron death (Estus et al., 1994; Ham et al., 1995). The data presented here indicate that activation of the MEKK-JNK pathway causes sympathetic neuron death via a p53-dependent mechanism, thereby providing a potential direct link from p75 to p53. Moreover, our finding that the stress-induced (Herdegen et al., 1997) MEKK-JNK pathway acts via p53 in neurons may explain why excitotoxicity-induced death of CNS neurons can be inhibited by deletion of either the JNK3 (Yang et al., 1997) or p53 genes (Morrison et al., 1996; Xiang et al., 1998).
Are there other pathways that cause elevated p53 after NGF withdrawal or p75NTR activation? A second candidate upstream pathway involves deregulation of the cell cycle; in cycling cells, p53 is viewed as a “fail-safe” mechanism that causes cellular apoptosis when proliferative mechanisms are deregulated (reviewed in Levine, 1997; Jacks and Weinberg, 1996). Such cell cycle deregulation has been proposed to occur in sympathetic neurons after NGF withdrawal (Rubin et al., 1993; Freeman et al., 1994; Park et al., 1997). Therefore, it is possible that NGF withdrawal causes two different signals, MEKK-JNK activation and cell cycle deregulation, that together converge on p53. Thus, p53 may function as a sensor to induce neuronal apoptosis when the sum of these upstream signals reaches a critical level.
What are the downstream events that are responsible for p53-mediated neuronal apoptosis? One potential downstream mechanism involves the protein Bax, which is known to be essential for sympathetic neuron apoptosis (Deckwerth et al., 1996), and for p53-dependent death of cortical neurons (Xiang et al., 1998). Data presented here indicate that, after the commitment of sympathetic neurons to apoptosis, the balance of proapoptotic versus prosurvival members of the bcl-2 family is considerably shifted to the proapoptotic, and that one of the changes contributing to this shift is an increase in Bax protein levels. Moreover, we demonstrate that increased expression of p53 alone is sufficient to cause increased Bax protein levels in sympathetic neurons. Since Bax is a direct transcriptional target of p53 (Miyashita and Reed, 1995), we propose that elevated p53 protein levels cause increased p53-dependent transcription of Bax, and that this is sufficient to tip the balance towards neuronal apoptosis. In support of this hypothesis, exogenous p53 is unable to mediate apoptosis of Bax−/− cortical neurons (Xiang et al., 1998), Bax−/− sympathetic neurons do not undergo apoptosis in response to NGF withdrawal (Deckwerth et al., 1996), and increased expression of Bcl2 or Bcl-xl (Garcia et al., 1992; Gonzalez-Garcia et al., 1995) in sympathetic neurons rescues NGF withdrawal induced cell death.
The in vivo data presented here support a number of additional conclusions. First, our studies demonstrate that the postnatal period of naturally occurring sympathetic neuron death is inhibited, but not completely eliminated when p53 levels are lowered, indicating that p53 is essential for normal apoptosis in the SCG, but also suggesting that other, potentially parallel death pathways are important. Such pathways may involve other p53 family members such as p73 (Jost et al., 1997; Kaghad et al., 1997) or p51 (Osada et al., 1998; Trink et al., 1998). Second, our data showing decreased apoptosis in the p53+/− SCG support the idea that it is not the presence or absence of p53 that determines apoptosis, but it is instead the levels of p53 that are important. Precedent for such a gene dosage effect has been documented with regard to p53 and tumorigenesis (Donehower et al., 1992; Harvey et al., 1993). Third, our in vivo data demonstrate that, although p53 may be essential embryonically for regulating the proliferation and survival of sympathetic neuroblasts (Vogel and Parada, 1998), that by birth any resultant deficits are not obvious, since neuron numbers in the newborn p53+/+ versus p53−/− SCG are not significantly different.
What is the biological rationale for p53 acting as a “threshold” to neuronal apoptosis? During the period of naturally occurring death, sympathetic neurons compete for limiting amounts of trophic support to ultimately ensure an appropriate target innervation density. The net result of this competition is a loss of ∼40–50% of sympathetic neurons over the first 2 wk of postnatal life. Our previous data indicate that both TrkA and p75NTR are required for this process (Bamji et al., 1998). Moreover, these same studies demonstrate that robust activation of TrkA antagonizes p75NTR-derived apoptotic signals and that, conversely, p75NTR can override TrkA-derived survival signals. On the basis of the current study, we propose that p53 is a convergent downstream target that “sums” opposing signals deriving from TrkA versus p75NTR activation, and that apoptosis is triggered when the balance of “death” signals deriving from p75NTR is greater than the “survival” signals deriving from TrkA. In cellular terms, this ensures that all neurons that are incapable of sequestering sufficient target-derived NGF are rapidly and efficiently eliminated from competition.
One final issue derives from the emerging similarities between sympathetic neuron apoptosis as induced by NGF withdrawal and p75NTR activation. In both cases, the MEKK-JNK pathway is induced, and in both cases, p53 is required for apoptosis. In the case of p75, while this receptor has been demonstrated to signal via a number of pathways, including ceramide production (Dobrowsky et al.,1994), JNK activation (Casaccia-Bonnefil et al., 1996), c-jun hyperphosphorylation (Bamji et al., 1998) and NFkB activation (Carter et al., 1996), the results presented here are the first to identify a required component, p53, of the p75NTR apoptotic pathway. Given the similarities between NGF withdrawal and p75NTR activation, we propose that NGF-withdrawal-induced apoptosis may be, to a large extent, a p75NTR-mediated process. In particular, our previous work (Bamji et al., 1998) indicates that NGF-withdrawal induced apoptosis of sympathetic neurons is greatly delayed in the absence of p75NTR. We propose that, during naturally occurring sympathetic neuron death, p75NTR may well provide the basis for a constitutive apoptotic signal in sympathetic neurons via one of two different mechanisms. First, there may be an autocrine p75NTR-dependent apoptotic loop; sympathetic neurons synthesize BDNF (Causing et al., 1997) that they process and secrete (Causing, C.G., R. Aloyz, and F.D. Miller, unpublished observations). Since BDNF can bind to p75NTR to cause sympathetic neuron apoptosis (Bamji et al., 1998), and since BDNF is required in vivo for a portion of appropriate naturally occurring sympathetic neuron death (Bamji et al., 1998), then this may indicate the presence of an ongoing autocrine BDNF:p75NTR death signal. A second possibility is that p75NTR functions to mediate a constitutive death signal in the absence of ligand, as previously suggested (Rabizadeh et al., 1993). However, it is clear that addition of exogenous BDNF is essential for a maximal p75NTR-dependent apoptotic signal (Bamji et al., 1998), indicating that even if a constitutive death mechanism(s) is in place, activation of p75NTR by exogenous, potentially target-derived, neurotrophins is likely to play a key role. In either case, exposure to an appropriate neurotrophin, in this case NGF, would lead to TrkA activation, thereby providing a mechanism for overriding p75 and p53-mediated apoptosis.
Acknowledgments
We would like to thank Phil Branton for his generous gift of the E1B55K and A262 adenoviruses, Pierre Laneuville for help in establishing the p53−/− colony, Jonathan Ham for providing us with the activated MEKK1 construct, Luc Paquet for performing the recombination of the MEKK1 adenovirus, Farid Said for his advice and assistance with generation of recombinant adenovirus, and Christine Laliberte for excellent technical assistance.
This study was supported by grants from the Canadian Medical Research Council (MRC) and the Canadian NeuroSciences Network to F.D. Miller and D.R. Kaplan. F.D. Miller is a Killam Scholar and D.R. Kaplan is a recipient of the Harold Johns and Canadian Cancer Society Research Scientist Award. During the course of this work, S.X. Bamji and C.D. Pozniak were funded by MRC studentships, and J. Atwal by a McGill Major studentship.
Abbreviations used in this paper
- PB
phosphate buffer
- moi
multiplicity of infection
- SCG
superior cervical ganglion
Footnotes
The first three authors contributed equally to this work.
References
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belliveau DJ, Krivko I, Kohn J, Lachance C, Pozniak C, Rusakov D, Kaplan D, Miller FD. NGF and NT-3 both activate TrkA on sympathetic neurons but differentially regulate survival and neuritogenesis. J Cell Biol. 1997;136:374–388. doi: 10.1083/jcb.136.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett AJ, Haddara W, Prevec L, Graham FL. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc Natl Acad Sci USA. 1994;91:8802–8806. doi: 10.1073/pnas.91.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindstem T, Turka LA, Mao X, Nunez G, Thompson CB. Bcl-xl, a bcl-2 related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Caelles C, Helmberg A, Karin M. p53-dependent apoptosis in the absence of transcriptional activation of p53 target genes. Nature. 1994;370:220–223. doi: 10.1038/370220a0. [DOI] [PubMed] [Google Scholar]
- Campenot RB. Development of sympathetic neurons in compartmentalized cultures. 1. Local control of neurite growth by nerve growth factor. Dev Biol. 1982;93:1–12. doi: 10.1016/0012-1606(82)90232-9. [DOI] [PubMed] [Google Scholar]
- Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R, Baeuerle PA, Barde Y-A. Selective activation of NF-kappaB by nerve growth factor through the neurotrophin receptor p75. Science. 1996;272:542–545. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factors with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron. 1997;18:257–267. doi: 10.1016/s0896-6273(00)80266-4. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE. An empirical method for converting nucleolar counts to neuronal numbers. J Neurosci Meth. 1984;12:125–132. doi: 10.1016/0165-0270(84)90011-6. [DOI] [PubMed] [Google Scholar]
- Creedon DJ, Johnson DM, Jr, Lawrence JC., Jr Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J Biol Chem. 1996;271:20713–20718. doi: 10.1074/jbc.271.34.20713. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;15:2833–2843. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AM, Rosenthal A. Neurons from mouse embryos with a null mutation in the tumour suppressor gene p53 undergo normal cell death in the absence of neurotrophins. Neurosci Lett. 1994;182:112–114. doi: 10.1016/0304-3940(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu I-H, Barrett T, Su B, Deng T, Karin M, David RJ. JNK1: a protein kinase stimulated by UV light and ha-ras that binds and phosphorylates the c-jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Jenkins GM, Hannun YA. Neurotrophins induce sphingomyelin hydrolysis: modulation by co-expression of p75 with Trk receptors. J Biol Chem. 1995;270:22135–22142. doi: 10.1074/jbc.270.38.22135. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Easton RM, Deckwerth TL, Parsadanian AS, Johnson EM., Jr Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from Bax deletion. J Neurosci. 1997;17:9656–9666. doi: 10.1523/JNEUROSCI.17-24-09656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers A, Whitfield J, Babij C, Rubin LL, Ham J. Role of the jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J Neurosci. 1998;18:1713–1724. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer E, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;72:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Elledge RM, Lee W-H. Life and death by p53. Bioessays. 1995;17:923–930. doi: 10.1002/bies.950171105. [DOI] [PubMed] [Google Scholar]
- Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman RS, Estus S, Johnson EM., Jr Analysis of cell-related gene expression in postmitotic neurons: selective induction of Cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- Franklin JL, Sans-Rodriguez C, Juhasz A, Deckwerth TL, Johnson EM., Jr Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growth: requirement for Ca2+ influx but not trk activation. J Neurosci. 1995;15:643–664. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia I, Martinou I, Tsujimoto Y, Martinou J-C. Prevention of programmed cell death of sympathetic neurons by the Bcl2 protooncogene. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia M, Garcia I, Ding L, O'Shea S, Boise LH, Thompson CB, Nunez G. Bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA. 1995;92:4304–4308. doi: 10.1073/pnas.92.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, F.L., and L. Prevec. 1991. Manipulation of adenovirus vectors in Methods in Molecular Biology, EJ Murray ed. The Humana Press, Totone, NJ. 109–128. [DOI] [PubMed]
- Hainut P. The tumor suppressor protein p53: a receptor to genotoxic stress that controls cell growth and survival. Curr Opin Oncol. 1995;7:76–82. [PubMed] [Google Scholar]
- Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Harvey M, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A, Donehower LA. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat Genet. 1993;5:225–229. doi: 10.1038/ng1193-225. [DOI] [PubMed] [Google Scholar]
- Hendry IA. Cell division in the developing sympathetic nervous system. J Neurocytol. 1977;6:299–309. doi: 10.1007/BF01175193. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Skene P, Bahr M. The c-Jun protein-transcriptional mediator of neuronal survival, regeneration and death. Trends Neurosci. 1997;20:227–231. doi: 10.1016/s0166-2236(96)01000-4. [DOI] [PubMed] [Google Scholar]
- Hockenberry D, Nunez G, Milliman CL, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;377:695–701. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- Hu MC, Qiu WR, Wang YP. JNK1, JNK2, and JNK3 are p53 N-terminal serine 34 kinases. Oncogene. 1997;15:2277–2287. doi: 10.1038/sj.onc.1201401. [DOI] [PubMed] [Google Scholar]
- Jacks T, Weinberg RA. Cell-cycle control and its watchman. Nature. 1996;381:643–644. doi: 10.1038/381643a0. [DOI] [PubMed] [Google Scholar]
- Johnson D, Lanahan A, Buck CR, Sehgal A, Morgan C, Mercer E, Bothwell M, Chao M. Expression and structure of the human NGF receptor. Cell. 1986;47:545–554. doi: 10.1016/0092-8674(86)90619-7. [DOI] [PubMed] [Google Scholar]
- Johnson EM, Jr, Deckwerth TL. Molecular mechanisms of developmental neuronal death. Ann Rev Neurosci. 1993;16:31–46. doi: 10.1146/annurev.ne.16.030193.000335. [DOI] [PubMed] [Google Scholar]
- Jost CA, Marin MC, Kaelin WG., Jr p73 is a human p53-related protein that can induce apoptosis. Nature. 1997;389:191–194. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991a;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Martin-Zanca D, Parada LF. Tryosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991b;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- Klein R, Jing S, Nanduri V, O'Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- Knusel B, Rabin SJ, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DP. A death in the life of p53. Nature. 1993;362:786–787. doi: 10.1038/362786a0. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Li Y, Chopp M, Zhang ZG, Zaloga C, Niewenhuis L, Gautam S. p53-immunoreactive protein and p53 mRNA expression after transient middle cerebral artery occlusion in rats. Stroke. 1994;25:849–855. doi: 10.1161/01.str.25.4.849. [DOI] [PubMed] [Google Scholar]
- Martinou I, Fernandez PA, Missotten M, White E, Allet B, Sadoul R, Martinou JC. Viral proteins E1B19K and p35 protect sympathetic neurons from cell death induced by NGF deprivation. J Cell Biol. 1995;128:201–208. doi: 10.1083/jcb.128.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massie B, Dionne J, Lamarche N, Fleurent J, Langelier Y. Improved adenovirus vector provides herpes simplex virus ribonucleotide reductase R1 and R2 subunits very efficiently. Biotechnology. 1995;13:602–608. doi: 10.1038/nbt0695-602. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR. Life and death decisions: a biological role for the p75 neurotrophin receptor. Cell Death Differ. 1998;5:343–345. doi: 10.1038/sj.cdd.4400385. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC. p53 is a direct transcriptional activator of the human Bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Osada M, Ohba M, Kawahara C, Ishioka C, Kanamaru R, Katoh I, Ikawa Y, Nimura Y, Nakagawara A, Obinata M, Ikawa S. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat Med. 1998;4:839–843. doi: 10.1038/nm0798-839. [DOI] [PubMed] [Google Scholar]
- Park DS, Farinelli SE, Greene LA. Inhibitors of cycline-dependent kinases promote survival of postmitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem. 1996;271:8161–8169. doi: 10.1074/jbc.271.14.8161. [DOI] [PubMed] [Google Scholar]
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- Querido E, Marcellus RC, Lai A, Charbonneau R, Teodoro JG, Ketner G, Branton PL. Regulation of p53 levels by the E1B 55-Kilodalton protein and E4orf6 in adenovirus-infected cells. J Virol. 1997;71:3788–3798. doi: 10.1128/jvi.71.5.3788-3798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM, Butcher LL, Bredesen DE. Induction of apoptosis by the low-affinity NGF receptor. Science. 1993;261:345–348. doi: 10.1126/science.8332899. [DOI] [PubMed] [Google Scholar]
- Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM. Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature. 1987;325:593–596. doi: 10.1038/325593a0. [DOI] [PubMed] [Google Scholar]
- Riccio A, Pierchala BA, Ciarallo CL, Ginty DD. An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science. 1997;277:1097–1100. doi: 10.1126/science.277.5329.1097. [DOI] [PubMed] [Google Scholar]
- Rubin LL, Philpott KL, Brooks SF. The molecular mechanisms of neuronal apoptosis. Curr Opin Neurobiol. 1993;3:391–394. doi: 10.1016/0959-4388(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Sadoul R, Quiquerez AL, Martinou I, Fernandez PA, Martinou JC. p53 protein in sympathetic neurons: cytoplasmic localization and no apparent function in apoptosis. J Neurosci Res. 1996;43:594–601. doi: 10.1002/(SICI)1097-4547(19960301)43:5<594::AID-JNR9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS. p53 induction is associated with neuronal damage in the central nervous system. Proc Natl Acad Sci USA. 1994;91:7525–7529. doi: 10.1073/pnas.91.16.7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez I, Hughes RT, Mayer BJ, Yee K, Woodgett JR, Avruch J, Kyriakis JM, Zon LI. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- Senger DL, Campenot RB. Rapid retrograde tyrosine phosphorylation of trkA in response to NGF in rat sympathetic neurons. J Cell Biol. 1997;138:411–421. doi: 10.1083/jcb.138.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmuller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1–12. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack RS, El-Bizri H, Wong J, Belliveau DJ, Miller FD. A critical temporal requirement for the retinoblastoma protein family during neuronal determination. J Cell Biol. 1998;140:1497–1509. doi: 10.1083/jcb.140.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodoro JG, Branton PE. Regulation of p53-dependent apoptosis, transcriptional repression, and cell transformation by phosphorylation of the 55-kilodalton E1B protein of human adenovirus type 5. J Virol. 1997;71:3620–3627. doi: 10.1128/jvi.71.5.3620-3627.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- Trink B, Okami K, Wu L, Sriuranpong V, Jen J, Sidransky D. A new human p53 homologue. Nat Med. 1998;4:747–748. doi: 10.1038/nm0798-747. [DOI] [PubMed] [Google Scholar]
- Virdee K, Tolkovsky AM. Activation of p44 and p42 Map kinases is not essential for the survival of rat sympathetic neurons. Eur J Neurosci. 1995;7:2159–2169. doi: 10.1111/j.1460-9568.1995.tb00637.x. [DOI] [PubMed] [Google Scholar]
- Vogel KS, Parada LF. Sympathetic neuron survival and proliferation are prolonged by loss of p53 and neurofibromin. Mol Cell Neurosci. 1998;11:19–28. doi: 10.1006/mcne.1998.0670. [DOI] [PubMed] [Google Scholar]
- Weskamp G, Reichardt LF. Evidence that biological activity of NGF is mediated through a novel subclass of high affinity receptors. Neuron. 1991;6:649–663. doi: 10.1016/0896-6273(91)90067-a. [DOI] [PubMed] [Google Scholar]
- White E. Life, death, and the pursuit of apoptosis. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- Wood KA, Youle RJ. The role of free radicals and p53 in neuron apoptosis in vivo. J Neurosci. 1995;15:5851–5857. doi: 10.1523/JNEUROSCI.15-08-05851.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Dai T, Deak JC, Kyriakis JM, Zon LI, Woodgett JR, Templeton DJ. Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature. 1994;372:798–800. doi: 10.1038/372798a0. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel EJ, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-xl and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Yang D, Kuan C-Y, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the JNK3 gene. Nature. 1997;289:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Yew PR, Kao CC, Berk AJ. Dissection of functional domains in the adenovirus 2 early 1B 55K polypeptide by suppressor-linker insertional mutagenesis. Virology. 1990;179:795–805. doi: 10.1016/0042-6822(90)90147-j. [DOI] [PubMed] [Google Scholar]
- Yew PR, Berk AJ. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature. 1992;337:82–85. doi: 10.1038/357082a0. [DOI] [PubMed] [Google Scholar]