

Abstract
Previous studies showed that conotruncal heart malformations can arise with the increase or decrease in α1 connexin function in neural crest cells. To elucidate the possible basis for the quantitative requirement for α1 connexin gap junctions in cardiac development, a neural crest outgrowth culture system was used to examine migration of neural crest cells derived from CMV43 transgenic embryos overexpressing α1 connexins, and from α1 connexin knockout (KO) mice and FC transgenic mice expressing a dominant-negative α1 connexin fusion protein. These studies showed that the migration rate of cardiac neural crest was increased in the CMV43 embryos, but decreased in the FC transgenic and α1 connexin KO embryos. Migration changes occurred in step with connexin gene or transgene dosage in the homozygous vs. hemizygous α1 connexin KO and CMV43 embryos, respectively. Dye coupling analysis in neural crest cells in the outgrowth cultures and also in the living embryos showed an elevation of gap junction communication in the CMV43 transgenic mice, while a reduction was observed in the FC transgenic and α1 connexin KO mice. Further analysis using oleamide to downregulate gap junction communication in nontransgenic outgrowth cultures showed that this independent method of reducing gap junction communication in cardiac crest cells also resulted in a reduction in the rate of crest migration. To determine the possible relevance of these findings to neural crest migration in vivo, a lacZ transgene was used to visualize the distribution of cardiac neural crest cells in the outflow tract. These studies showed more lacZ-positive cells in the outflow septum in the CMV43 transgenic mice, while a reduction was observed in the α1 connexin KO mice. Surprisingly, this was accompanied by cell proliferation changes, not in the cardiac neural crest cells, but in the myocardium— an elevation in the CMV43 mice vs. a reduction in the α1 connexin KO mice. The latter observation suggests that cardiac neural crest cells may have a role in modulating growth and development of non–neural crest– derived tissues. Overall, these findings suggest that gap junction communication mediated by α1 connexins plays an important role in cardiac neural crest migration. Furthermore, they indicate that cardiac neural crest perturbation is the likely underlying cause for heart defects in mice with the gain or loss of α1 connexin function.
Keywords: neural crest, gap junction, connexin 43, oleamide, cardiac
Gap junctions provide membrane channels, which allow the passage of ions and small molecules between cells. These channels are made up of a hexameric array of proteins encoded by a multigene family known as the connexins (Willecke et al., 1991; Kumar and Gilula, 1992). Most cells or tissues express multiple connexin genes and show functional coupling via gap junctions (Bennett et al., 1991; Bruzzone et al., 1996). Given the prevalence of gap junctions, it is likely that cell–cell communication via gap junctions mediates cell signaling involved in the maintenance of tissue homeostasis and in regulating growth and proliferation (Loewenstein, 1979). Because gap junctions are found from very early stages of embryogenesis, it has been suggested that they mediate cell–cell signaling events involved in patterning, differentiation, and development (Guthrie and Gilula, 1989; Warner, 1992; Lo, 1996).
The α1 connexin gap junction gene (also referred to as Cx43) is expressed in a developmentally regulated manner in many different regions of the mouse embryo (Ruangvoravat and Lo, 1992; Yancey et al., 1992). Studies of the α1 connexin knockout (KO)1 mice have shown a role for this connexin protein in heart morphogenesis, since mice deficient in α1 connexin gap junctions die soon after birth because of conotruncal heart malformations and pulmonary outflow obstruction (Reaume et al., 1995). However, since very little α1 connexin is normally found in this region of the developing heart (Gourdie et al., 1992), the phenotype of the KO mice has not been very informative as to the role of α1 connexin gap junctions in heart morphogenesis.
Some insights into this question have since come from the analysis of transgenic mice (referred to as CMV43) in which α1 connexin is overexpressed in subpopulations of neural crest cells and their progenitors in the dorsal neural tube. These transgenic animals also exhibited conotruncal heart malformations and outflow tract obstructions (Ewart et al., 1997; Huang et al., 1998). Together, the findings from the CMV43 and α1 connexin KO mice would suggest that the precise level of α1 connexin function is of critical importance in conotruncal heart development, and that this may involve the modulation of cardiac neural crest activity. Further evidence consistent with this possibility has come from recent studies of transgenic mice (FC) expressing a dominant-negative α1 connexin fusion protein (Sullivan and Lo, 1995; Sullivan et al., 1998). Such mice also exhibited conotruncal heart malformations and right ventricular outflow tract obstructions. Of importance to note is the fact that expression of the fusion protein was localized to similar regions of the embryo, that is populations of neural crest cells and their progenitors in the dorsal neural tube (Sullivan et al., 1998).
Neural crest cells are migratory cells that originate from the dorsolateral margins of the neural tube. Along with participating in heart outflow tract morphogenesis, neural crest cells contribute to the development of a diverse array of tissues in the embryo, such as the peripheral nervous system, the thymus, and the craniofacial mesenchyme. Neural crest cells express an abundance of α1 connexins and are functionally well coupled via gap junctions (Lo et al., 1997). Although the involvement of gap junctions in the development of this migratory cell population is unexpected, neural crest cells have been observed to migrate in groups or sheets (Bancroft and Bellairs, 1976; Davis and Trinkaus, 1981). Moreover, gap junctions are dynamic structures that turn over rapidly (Bruzzone et al., 1996). The importance of direct cell–cell interactions in neural crest cell populations has also been intimated by a number of previous studies (Kapur et al., 1995; Kimura et al., 1995; Nakagawa and Takeichi, 1995; Raible and Eisen, 1996). However, the role of gap junctions in cardiac neural crest cells, the subpopulation of crest cells that migrates to the heart and participates in outflow tract remodeling, remains unknown. Gap junctions are not likely involved in the maintenance of neural crest cell viability, since the cardiac phenotype of the α1 connexin KO mice, and the CMV43 and FC transgenic mice do not resemble the classic cardiac crest ablation phenotype (Kirby, 1993; Kirby and Waldo, 1995).
In this study, we have undertaken an analysis of the migration and proliferation of neural crest cells with the gain or loss of α1 connexin function. This has included an examination of cardiac crest cells from the CMV43 transgenic mice exhibiting overexpression of α1 connexins, and crest cells from the α1 connexin KO and the FC transgenic mice with the deletion or inhibition of α1 connexin function, respectively. Using a neural tube explant culture system, we showed that changes in the level of gap junctional communication were associated in parallel with changes in the rate of neural crest migration. Thus, an increase in α1 connexin function increased the rate of crest migration, while the dominant or recessive loss of α1 connexin function slowed neural crest migration. In contrast, neural crest cell proliferation was not affected. These results indicate a role for gap junction communication in the modulation of neural crest cell migration. The in vivo analysis of neural crest cells using a lacZ reporter transgene suggested that there were changes in neural crest abundance in the outflow tract. In the CMV43 transgenic mice, lacZ-positive cells were increased, while in the KO mice they were decreased. This was accompanied by changes in cell proliferation, not in neural crest cells, but in the ventricular myocardium. Myocyte cell proliferation was increased in the CMV43 mice, while it was decreased in the KO mice. These observations in conjunction with previous studies confirm the involvement of cardiac neural crest perturbation in the conotruncal heart defects seen with the perturbation of α1 connexin function. They suggest that gap junction communication plays an important role in modulating cardiac neural crest migration, and through this process, affects events in cardiac development.
Materials and Method
Mouse Breeding and Genotyping
To examine the role of α1 connexin gap junctions in crest migration and proliferation, we used three different mouse lines and the appropriate breeding schemes to obtain embryos with different α1 connexin gene or transgene dosages. For CMV43, a transgenic mouse line in which Cx43 is overexpressed in neural crest cells, both hemizygous and homozygous embryos could be obtained since these transgenic mice are homozygous viable (Ewart et al., 1997). Homozygous embryos were obtained by intercrossing the homozygous CMV43 mice, while hemizygous CMV43 embryos were obtained by breeding hemizygous CMV43 males with CD1 females. From the latter cross, both transgenic and control nontransgenic embryos were obtained. It should be noted that the CMV43 line has been outcrossed into CD1 mice and are maintained in a CD1 background (see Ewart et al., 1997). For the FC transgenic mice expressing a dominant-negative α1 connexin/lacZ fusion protein (Sullivan and Lo, 1995; Sullivan et al., 1998), breeding of hemizygous FC males with nontransgenic CD1 females provided hemizygous transgenic and wild-type nontransgenic control embryos. Because homozygous FC mice are inviable (Sullivan et al., 1998), it was not possible to obtain homozygous embryos for analysis. It should be noted that hemizygous FC intercrosses were not used to obtain homozygous FC embryos, given that PCR analysis cannot distinguish between the homozygous vs. hemizygous genotypes. For the α1 connexin KO mice, interbreeding of the heterozygous KO mice provided wild-type, heterozygous, and homozygous α1 connexin KO mouse embryos.
The genotypes of the mice and embryos were determined by PCR analysis of tail or yolk sac DNA, respectively. For the FC transgenic mice, PCR analysis was carried out using primers that detected the lacZ portion of the α1 connexin/lacZ fusion protein construct (Echelard et al., 1994). For the CMV43 transgenic mice, PCR analysis was carried out using primers to the 3′ untranslated sequence tag included in the construct (Ewart et al., 1997). For the α1 connexin KO mice, PCR analysis using primers to the wild-type α1 connexin gene and neo insert in the α1 connexin knockout allele (Reaume et al., 1995) allowed the identification of wild-type, heterozygous, or homozygous α1 connexin KO embryos. In addition to these three mouse lines exhibiting the perturbation of α1 connexin function, we also used a transgenic mouse line expressing a lacZ reporter gene driven by the α1 connexin promoter (see Lo et al., 1997). This transgene was bred into the CMV43 and α1 connexin KO mice to facilitate the identification of presumptive crest cells in the CMV43 and α1 connexin KO mouse heart.
Neural Crest Outgrowth Cultures
To obtain embryos for neural crest outgrowth studies, mice were mated, and the day the vaginal plug was found was considered E0.5. Embryos from the transgenic or KO mouse lines were collected on E8.5, and the yolk sac from each embryo was harvested for genotyping by PCR analysis. The method used to establish the neural crest outgrowth cultures were adapted from those described by Murphy et al. (1991) and Moiseiwitsch and Lauder (1995). Each embryo was treated with 0.5 mg/ml of collagenase/dispase (Sigma Chemical Co., St. Louis, MO) in PBS for 10–15 min and then bisected longitudinally to expose the hindbrain neural folds. Each half was further transected to provide two fragments spanning the pre- and postotic regions of the hindbrain neural folds. The dorsal ridge of the neuroepithelium was surgically removed from the surrounding tissues and cultured in a 35-mm, fibronectin-coated Petri dish containing DME with high glucose and 10% fetal bovine serum. The cultures were maintained for 24–48 h at 37°C with 5% CO2. Explant cultures were generated from both the preotic and postotic hindbrain neural folds. Because these two populations of neural crest cells exhibited the same migration and proliferation rates in all three mouse models, the data were subsequently pooled. Lineage studies in mouse embryos have confirmed that neural crest cells from the postotic hindbrain neural fold give rise to the cardiac crest cells as has been found in chick embryos (Fukiishi and Morriss-Kay, 1992; for review see Kirby, 1993).
Quantitation of Neural Crest Migration and Proliferation
Digital images of the outgrowth cultures at 24–48 h were acquired by video microscopy. Then NIH Image software was used to measure the area of the outgrowth. This was obtained by taking the area of the entire explant and subtracting from it the area of the dense central neural tube mass. Since the neural tube fragment is attached to the substratum predominantly via its edges where neural crest cells are emerging, we controlled for differences in migration area resulting from random variations in the shape of the explant by dividing the outgrowth area (mm2) by the perimeter (mm) of the explant. This normalized number (in mm) is referred to as the migration index and provides an estimate of the migration rate of the neural crest cells. For monitoring cell proliferation, 48-h explant cultures were incubated with bromodeoxyuridine (BrdU) (10 μM/ml) for 2 h, fixed, and processed for immunodetection using an anti-BrdU antibody and a streptavidin peroxidase detection system (Calbiochem, San Diego, CA) used in conjunction with the True Blue™ peroxidase substrate (Kierkegaard and Perry Laboratories, Gaithersburg, MD). Labeled cells were counted with the aid of NIH Image. To determine the total cell number in the outgrowth after BrdU immunostaining, the cultures were further stained with hematoxylin. This permitted a total cell count to be obtained using NIH Image. The proliferation rate was then calculated as the percent of BrdU-labeled cells in the outgrowth.
Dye Coupling In Vitro and In Vivo
To quantitate dye coupling in the neural crest outgrowths, the DME medium was replaced with phosphate-buffered L-15 medium containing 10% FBS, and the culture dish was placed on a heated microscope stage maintained at 37°C. Individual cells in the outgrowth cultures (24-h cultures) were impaled with microelectrodes filled with 5% carboxyfluorescein. This is followed by iontophoretic injection for 2 min using hyperpolarizing current pulses of 0.5 s duration at a frequency of once per second. The number of dye-filled cells found at the termination of dye injection was recorded. This count included the impaled cell. To examine dye coupling in vivo, E8.5 embryos were harvested and incubated in DME with 10% fetal bovine serum for up to 4 h. Just before microelectrode impalement for the analysis of dye coupling, the embryo was opened along the hindbrain neural fold and immobilized in a thin bed of 0.8% agarose, to which L15 medium was added. For these studies, microelectrode impalement and dye injection into the dorsolateral region of the postotic hindbrain were carried out, and the number of dye-filled cells was recorded as described above.
LacZ Expression and Immunohistochemical Analysis of Mouse Embryos
For detection of β-galactosidase activity, embryos were fixed in 2% paraformaldehyde for 30–45 min and then washed for 30 min twice in PBS. X-gal staining was carried out overnight at room temperature. Stained embryos were postfixed in 3.7% formaldehyde and then examined and photographed as whole-mount specimens. Some samples were further processed for paraffin embedding and sectioning for histological analysis. The tissue sections were either stained with hematoxylin/eosin or further processed for immunohistochemistry with cardiac myosin heavy chain, neurofilament, or proliferating cell nuclear antigen (PCNA) antibodies. For immunostaining, slides with 7-μm sections were treated for 30 min in 3% hydrogen peroxide in methanol and washed for 10 min in tap water. Cardiac myosin antibody was used at a dilution of 1:1,000 (Accurate Antibodies, San Diego, CA), and PCNA antibody was used undiluted (Zymed Laboratories, San Francisco, CA). For staining with cardiac myosin antibody, sections were treated for 15 min in 10 μg/ml of proteinase K and washed briefly in water, then 5 min in 0.1 M Tris, pH 7.4, followed by 5 min in Tris with 2% fetal bovine serum. Immunodetection was carried out using mouse ABC Elite Kit (Vector Laboratories, Burlingame, CA), with the signal visualized with the VIP substrate (Vector Laboratories). Slides were then dehydrated, cleared in xylene, and mounted with Cytoseal (Stephens Scientific, Riverdale, NJ).
Results
Modulation of Neural Crest Migration
Neural tube explant cultures were established from the hindbrain neural folds of E8.5 embryos, spanning the region from which cardiac crest cells are derived. These explants were cultured for a period of 48 h to monitor neural crest outgrowths. After 24 h, a ring of neural crest cells was observed around the neural tube explant, and by 48 h, this ring of cells was enlarged (Fig. 1). To obtain an estimate of the rate of neural crest migration during this culture period, a migration index was determined by measuring the area of the neural crest outgrowth (see Materials and Methods). In the CMV43 transgenic embryos, the migration index was elevated in a transgene dosage–dependent manner. The increase was greatest in the homozygous embryos, while neural crest cells in the hemizygous embryos exhibited a migration index intermediate to that of the homozygous and nontransgenic embryos (Table I). These migration changes can be detected at 24 and 48 h of culture. In contrast to these results, with either the recessive or dominant loss of α1 connexin function, crest migration was inhibited. In the α1 connexin KO mice, the inhibition of migration occurred in step with α1 connexin gene dosage. Thus, the migration index of the heterozygous KO embryos was intermediate to that of the nontransgenic and homozygous KO embryos. Similarly, crest cells from the hemizygous FC transgenic mice exhibited a reduced migration index. Note that the migration differences seen in the nontransgenic outgrowths obtained from the different transgenic and KO crosses are highly reproducible and reflect strain background effects on crest migration (Table I). Overall, these results suggest that changes in the apparent rate of neural crest migration are correlated with changes in α1 connexin gene or transgene dosage. They suggest that α1 connexin gap junctions play a role in modulating neural crest migration.
Figure 1.
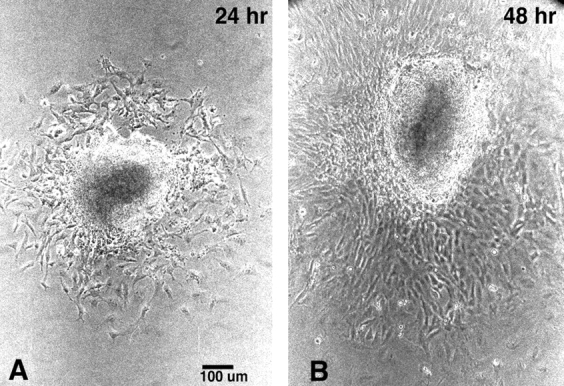
Neural crest migration from neural tube explant cultures. Neural crest cells rapidly emerge from the explanted neural tube fragment and migrate out as a monolayer of cells surrounding the central dense mass of neuroepithelial tissue. Note the increase in the area of the neural crest outgrowth from 24 h (A) to 48 h (B) of culture.
Table I.
Neural Crest Migration and Proliferation in Neural Tube Explant Cultures*
| Strains | Migration index | Percentage of BrdU incorporation‖ | ||||
|---|---|---|---|---|---|---|
| 24 h | 48 h | |||||
| CMV43 | ||||||
| Homozygous | 0.731 ± 0.016 (39) | 2.341 ± 0.061 (38) | 11.0 ± 0.25 (35) | |||
| Hemizygous | 0.547 ± 0.012 (81) | 2.021 ± 0.034 (74) | 10.8 ± 0.26 (37) | |||
| Nontransgenic | 0.345 ± 0.008 (87) | 1.534 ± 0.031 (78) | 11.4 ± 0.26 (44) | |||
| P < 0.0001§ | P < 0.0001§ | |||||
| FC | ||||||
| Hemizygous | 0.443 ± 0.009 (56) | 1.816 ± 0.029 (56) | 12.1 ± 0.33 (40) | |||
| Nontransgenic | 0.673 ± 0.009 (46) | 2.210 ± 0.031 (46) | 11.4 ± 0.35 (29) | |||
| P < 0.0001‡ | P < 0.0001‡ | |||||
| α1 Connexin KO | ||||||
| −/− | 0.276 ± 0.014 (24) | 1.306 ± 0.070 (23) | 11.4 ± 0.78 (11) | |||
| +/− | 0.377 ± 0.010 (65) | 1.632 ± 0.033 (65) | 10.4 ± 0.47 (22) | |||
| +/+ | 0.562 ± 0.010 (35) | 1.936 ± 0.058 (35) | 11.1 ± 0.76 (10) | |||
| P < 0.0001§ | P < 0.0001§ | |||||
Number indicated is the mean ± SD. Number in parentheses = number of outgrowth cultures analyzed.
P value derived from two tailed Student's t test.
P value derived from ANOVA.
No statistically significant differences detected in BrdU incorporation.
Neural Crest Cell Proliferation
In parallel to the analysis of neural crest migration, cell proliferation in the outgrowth was also monitored. For this analysis, BrdU incorporation was examined by immunostaining with a BrdU antibody (Fig. 2 A), after which the cultures were stained with hematoxylin for the determination of total cell numbers in the crest outgrowth (Fig. 2 B). From this analysis, the percentage of BrdU-labeled cells was determined. In contrast to the results of the migration study, we found that all of the transgenic/KO lines exhibited the same level of BrdU incorporation, including the homozygous KO and homozygous CMV43 transgenic embryos. These results indicate that changes in the level of α1 connexin function do not affect neural crest cell proliferation.
Figure 2.

Analysis of cell proliferation in neural crest outgrowth cultures. To quantitate cell proliferation in the neural crest outgrowth, BrdU incorporation was examined. Proliferating cells can be readily distinguished as those that are darkly stained after immunodetection with an anti-BrdU antibody (A). Such cultures were subsequently stained with hematoxylin for counting total cell number in the outgrowth (B). Note that A and B are pictures of the same outgrowth culture.
Dye Coupling Level Changes in Step with α1 Connexin Transgene/Gene Dosage
The correlation of α1 connexin gene or transgene dosage with the rate of neural crest cell migration suggests that gap junction communication may have a direct role in the modulation of crest migration. To examine this possibility, microelectrode impalements and dye injections were carried out to monitor the level of dye coupling in the neural crest outgrowths. These studies showed that dye coupling was elevated in the CMV43 neural crest cells as compared with neural crest cells derived from the nontransgenic embryos (Table II). In the homozygous CMV43 embryos, coupling was increased to a higher level than in the hemizygous CMV43 embryos, thus indicating that coupling levels changed in step with CMV43 transgene dosage (Table II). Analysis of the CMV43 neural crest outgrowths by immunostaining with an α1 connexin antibody showed a marked increase in the abundance of α1 connexin gap junction plaques in regions of cell–cell contact (Fig. 3). In contrast to these results, analysis of outgrowth cultures from the FC and α1 connexin KO mice showed reductions in dye coupling (Table II). In neural crest cells derived from the KO mice, those that were homozygous null mutants showed a lower level of dye coupling than neural crest cells derived from the heterozygous KO embryos (Table II).
Table II.
Dye Coupling in Neural Crest Outgrowths*
| Strains | No. dye-filled cells | |||
|---|---|---|---|---|
| In vitro | In vivo | |||
| CMV43 | ||||
| Homozygous | 8.38 ± 0.207 (58) | 9.63 ± 0.207 (40) | ||
| Hemizygous | 6.55 ± 0.318 (62) | 5.79 ± 0.181 (28) | ||
| Nontransgenic | 3.72 ± 0.335 (36) | 4.20 ± 0.344 (20) | ||
| P ≤ 0.0001§ | P ≤ 0.0001§ | |||
| FC | ||||
| Hemizygous | 1.81 ± 0.092 (78) | 3.25 ± 0.140 (65) | ||
| Nontransgenic | 2.80 ± 0.108 (60) | 4.25 ± 0.204 (28) | ||
| P ≤ 0.0001‡ | P ≤ 0.0001‡ | |||
| α1 Connexin KO | ||||
| −/− | 1.63 ± 0.162 (30) | 2.44 ± 0.182 (16) | ||
| +/− | 1.97 ± 0.102 (68) | 3.63 ± 0.144 (48) | ||
| +/+ | 2.76 ± 0.136 (42) | 4.75 ± 0.192 (24) | ||
| P ≤ 0.0001§ | P ≤ 0.0001§ | |||
Numbers indicated correspond to the mean ± SD. Number in parentheses = number of impalements analyzed.
P value derived from two tailed Student's t test.
P values based on ANOVA.
Figure 3.

Increased α1 connexin gap junction plaques in the CMV43 crest cells. Immunostaining with a α1 connexin antibody revealed increased α1 connexin expression in the homozygous CMV43 neural crest outgrowth (B) as compared with neural crest cells from nontransgenic embryos (A). The punctate pattern of immunostaining is consistent with the localization α1 connexins in gap junction plaques. In the control nontransgenic neural crest outgrowths, very small punctate immunostaining can be observed, although this is hard to record photographically.
To determine whether the dye coupling changes observed in vitro are indicative of the level of dye coupling in vivo, we carried out microelectrode impalements and dye injections directly into presumptive neural crest cells situated dorsolateral to the dorsal neural fold. We had previously shown with such in vivo dye coupling analysis, an elevation of gap junction communication in presumptive crest cells of hemizygous CMV43 embryos (Ewart et al., 1997). Here, we further examined and compared dye coupling levels in presumptive neural crest cells of both hemizygous and homozygous CMV43 transgenic embryos. These studies showed that dye coupling was elevated in step with transgene dosage, with the highest level of dye coupling exhibited in the homozygous embryos (Table II). Similar analysis of the FC and α1 connexin KO mice showed decreases in coupling, and this occurred in step with α1 connexin gene dosage in the KO embryos.
Oleamide Inhibits Gap Junctional Communication and also Crest Migration
The above results show that gap junction communication modulates neural crest migration, with enhanced migration elicited by increased gap junction communication, while decreased migration resulted from reductions in gap junction communication. To further examine whether gap junctions have a direct role in modulating neural crest migration, we examined the effects of oleamide, a compound previously shown to be a potent inhibitor of gap junctional communication (Guan et al., 1997; Boger et al., 1998). For these studies, neural crest outgrowth cultures from nontransgenic embryos were plated in the presence of 50 μM oleamide. At 24 h, the migration index was recorded, and dye coupling was examined. As expected, gap junction communication was decreased by oleamide treatment. More importantly, this was correlated with an inhibition in neural crest migration (Table III). To control for the specificity of the effects seen with oleamide, we further analyzed the effects of treatment with trans-oleamide (trans-9-octadecenamide), a chemical analogue of oleamide previously shown not to have any effects on gap junctional communication. Treatment with trans-oleamide showed no effects on coupling and no effects on migration. We also examined the effects of treatment with trifluoromethylketone, a compound that is known to block the fatty acid amide hydrolase, which inactivates oleamide (Patterson et al., 1996). In contrast to the results obtained with trans-oleamide, treatment with trifluoromethylketone inhibited coupling and crest migration (Table III). A parallel analysis of BrdU incorporation indicated that none of these compounds had any effects on the rate of neural crest cell proliferation. These results are in agreement with those obtained above with the analysis of the transgenic and KO mice and further indicate that gap junction communication has a direct role in the modulation of neural crest migration.
Table III.
Effects of Oleamide Treatment on Dye Coupling and Migration of Neural Crest Cells*
| Treatment | Dye coupling | Migration index | Percentage of BrdU incorporation | |||
|---|---|---|---|---|---|---|
| Control‡ | 3.00 ± 0.134 (34) | 0.68 ± 0.040 (28) | 11.1 ± 1.42 (11) | |||
| Oleamide | 1.56 ± 0.078 (54)§ | 0.49 ± 0.018 (45)§ | 10.4 ± 0.45 (18) | |||
| Trans-oleamide | 2.78 ± 0.141 (34) | 0.71 ± 0.033 (37) | 11.2 ± 0.70 (16) | |||
| Trifluoromethyl ketone | 1.38 ± 0.094 (34)§ | 0.36 ± 0.029 (17)§ |
Numbers indicated are mean ± SD. Number in parentheses in “Dye coupling” column = number of impalements analyzed. Number in parentheses in “Migration index” column = number of outgrowth cultures examined.
The control cultures were treated with the solvent (ethanol) alone.
Compared to control group, P = 0.0001.
Analysis of Neural Crest Cells in the Outflow Tract
Although the above studies showed that gap junction communication levels in presumptive neural crest cells in the embryo are similar to that of neural crest cells in the outgrowths, an important question is whether the migration differences detected in vitro are indicative of changes in the migratory behavior of neural crest cells in vivo. To examine this question, we bred into the CMV43 and α1 connexin KO mice, a lacZ reporter transgene previously shown to label neural crest cells (Lo et al., 1997). Using this reporter gene, it was possible to examine the distribution of cardiac neural crest cells in the heart outflow tract by simply X-gal staining the embryos.
Such an analysis of E14.5 hearts from the CMV43 and α1 connexin KO mice revealed opposite changes in the abundance of lacZ-positive cells in the outflow tract. In comparison to the distribution seen in wild-type hearts (Fig. 4, A and E), the homozygous CMV43 transgenic embryo exhibited an increase in the abundance of lacZ-expressing cells in the outflow tract (Fig. 4, B and F). In contrast, in the α1 connexin KO hearts, the abundance of lacZ-expressing cells was decreased (Fig. 4, C and G). This decrease varied with the apparent severity of the heart phenotype, such that only a very low level of X-gal staining was observed in the outflow tract of hearts showing the most severe conotruncal malformation (compare Fig. 4, C and G vs. D and H; compare region denoted by black arrows showing thinning of the myocardium). Histological analysis showed that these lacZ-expressing cells were in regions previously shown to be populated by cardiac neural crest cells in the outflow septum (Fig. 5; Waldo et al., 1998). It is interesting to note that the aorta in the homozygous KO hearts exhibited an unusual acute bend (Fig. 4, G and H, white arrows), a phenotype consistent with the looping defect recently described for the α1 connexin KO mouse heart (Ya et al., 1998).
Figure 4.

Neural crest cells in fetal hearts of CMV43 and α1 connexin KO mice. The distribution of cardiac neural crest cells in E14.5 fetal hearts were examined using a lacZ reporter transgene driven by the α1 connexin promoter (Lo et al., 1997). Shown are the front and side views of X-gal–stained hearts from wild-type, CMV43, and α1 connexin KO mice. (A and E) Wild-type mouse heart. The white asterisk in A denotes presumptive neural crest cells in the conus, perhaps comprising cells in the closing seam of the aorticopulmonary septum. (B and F) Homozygous CMV43 heart. The black arrow in F denotes mild bulging of the conotruncus. Note a greater abundance of lacZ staining in the conus (B, white asterisk), suggesting an increased abundance of neural crest cells in the outflow septum. (C and G) Homozygous α1 connexin KO heart with severe conotruncal malformation. Note the very low level of lacZ staining in the conus (white asterisk), suggesting that fewer neural crest cells are present in the outflow septum. This heart exhibits a more severe phenotype consisting of very prominent thinning of the conotruncal myocardium (region denoted by black arrows). The white arrow in G denotes acute bend in aorta. (D and H) Homozygous α1 connexin KO heart with mild conotruncal malformation. This heart shows less prominent thinning of the conotruncal myocardium (region denoted by black arrows) as compared with the heart in (C and H). Interestingly, the level of lacZ staining in the conus is close to normal (D, white asterisk). The white arrow in H denotes an acute bend in the aorta. p, pulmonary trunk; a, aorta; rv, right ventricle.
Figure 5.

Presumptive neural crest cells in the outflow septum of E14.5 fetal hearts. Histological analysis of the X-gal–stained E14.5 fetal hearts expressing the lacZ reporter gene used to track the distribution of cardiac neural crest cells shows the presence of presumptive neural crest cells in the wall of the aorta (Ao) and also in the outflow septum (O). Note that compared with the nontransgenic heart (A), there is an increased abundance of lacZ-expressing cells in the outflow septum and the wall of the aorta of the CMV43 heart (C), while lacZ-expressing cells in these same regions appeared to be decreased in the KO heart (B). Section in A was stained with hematoxylin-eosin, while sections in B and C were immunostained with PCNA antibody (B) and myosin heavy chain antibody (C). PI, pulmonary infundibulum.
The examination of sections from these same fetal hearts by immunohistochemistry using an antibody to localize cells expressing the cell proliferation nuclear antigen PCNA (Cox, 1997) showed no changes in the apparent rate of proliferation in the lacZ-expressing presumptive neural crest cells (data not shown). This is consistent with the BrdU analyses of cell proliferation in the neural crest outgrowths. However, we unexpectedly observed marked changes in cell proliferation in the ventricular myocardium. In the CMV43 transgenic mice, there was an increase in PCNA labeling in the myocardium, while a decrease was observed in the homozygous α1 connexin KO mouse heart (Fig. 6). Quantitative analysis of cross sections from the mid-ventricular regions of these hearts showed a doubling of PCNA labeling in the CMV43 hearts (three CMV43 hearts examined showed mean of 52% vs. two wild-type hearts with a mean of 24% PCNA-labeled cells; student's t test show P < 0.0001), but no significant change in PCNA labeling was detected in the homozygous α1 connexin KO hearts (two KO hearts examined with mean of 23%; P = 0.78). However, it should be noted that in the KO hearts, we observed much more regional variability in the level of PCNA labeling. This is likely to account for the inability to detect significant differences in the KO hearts.
Figure 6.

Cell proliferation changes in the ventricular myocardium. Sections of the E14.5 fetal heart were examined for cell proliferation by immunostaining with an antibody to the PCNA. Compared with the wild-type nontransgenic heart (WT), there was a marked increase in PCNA immunostaining in the ventricular myocardium of the homozygous transgenic heart (CMV43). This is indicative of increased myocyte cell proliferation with the overexpression of α1 connexin gap junctions in neural crest cells. In contrast, in the homozygous α1 connexin KO mouse heart (Cx43 −/−), there was a reduction in cells exhibiting PCNA expression, thus indicating a paucity of myocyte cell proliferation with the deletion of α1 connexin function.
Discussion
Gap Junctions and Neural Crest Migration
Using a neural tube explant culture system, we showed that the manipulation of α1 connexin function, whether through reverse genetics or oleamide treatment, elicited changes in neural crest migration in step with changes in the level of gap junction communication. Thus, increased gap junction communication mediated by overexpression of wild-type α1 connexin gap junctions increased the apparent rate of neural crest migration. In contrast, neural crest migration was decreased with either the dominant-negative inhibition of gap junction communication, or the reduction or loss of α1 connexin gap junctions. Treatment of the explant cultures with oleamide, a potent inhibitor of gap junction communication, provided similar results. Thus, oleamide-mediated gap junction inhibition was associated with a reduction in the rate of neural crest migration. As the migration index used in this study represents measurements of the crest outgrowth area, the migration changes detected could reflect alterations in the rate of cell locomotion and/or the directionality of cell movement. To distinguish between these possibilities, the migratory behavior of individual neural crest cells will need to be examined in real time.
The relevance of these in vitro findings to neural crest migratory behavior in vivo is suggested by the analysis of mouse embryos using a lacZ reporter gene previously shown to be expressed in neural crest cells (Lo et al., 1997). LacZ-expressing cells in the outflow septum were increased in the CMV43 transgenic embryos, while they were decreased in the same region in the α1 connexin KO mouse embryos. These changes in lacZ expression are not likely due to alterations in the amount of cell proliferation, as we found no change in PCNA expression in presumptive lacZ-expressing neural crest cells. This result is in agreement with the in vitro analyses of the neural crest outgrowths, which showed no change in BrdU incorporation with either the up or down regulation of gap junction communication. In addition, we also observed no obvious change in cell death in the neural crest outgrowths or in the lacZ-expressing neural crest cells in vivo (Huang, G.Y., C.W. Lo, K. Waldo, and M.L. Kirby, unpublished observations). Together these findings suggest that the modulation of α1 connexin function may perturb the migratory behavior of cardiac neural crest cells in vivo as well as in vitro (also see discussion below).
Gap Junctions and Cardiac Development
Our previous study showed that differentiation of the conotruncal myocardium is altered in the CMV43 and α1 connexin KO mice (Ewart et al., 1997; Huang et al., 1998; Lo and Wessels, 1998). This indicated that gap junction communication in cardiac neural crest cells may have a role to play in myocardialization of the conus, a process by which the muscular outflow septum is formed. Here, we further show that proliferation of the ventricular myocardium was altered in these same animals. Thus, in the CMV43 mice where gap junction communication in neural crest cells is elevated, myocyte proliferation is increased. In contrast, the converse situation was found for the KO mice. It is important to note that the ventricular myocardium is a tissue in which the CMV43 transgene is not expressed (Ewart et al., 1997). Hence the increased myocyte cell proliferation in the CMV43 mice cannot be attributed to myocyte overexpression of α1 connexin gap junctions. Rather these results suggest that cardiac neural crest cells may have a role in the modulation of myocyte cell proliferation and contribute in some manner to the myocyte defects in these mice.
In light of these findings, an important question to consider is how myocyte differentiation and proliferation may be perturbed by alterations in the migratory behavior of neural crest cells. Previous ablation studies have indicated a quantitative requirement for neural crest cells in cardiac development (Nishibatake et al., 1987). It was found that ablating different amounts of cardiac neural crest cells resulted in different cardiac phenotypes. Hence, if α1 connexin perturbation were to alter the efficiency with which neural crest cells home into the heart, or disturb the temporal regulation of cardiac neural crest migration, then cardiac crest abundance could be altered. This could account for the observed changes in lacZ expression in the outflow septum of the CMV43 and α1 connexin KO mice. However, we note that changes in lacZ expression also could result from altered promoter activity associated with the lacZ transgene. In this case, the cardiac defects may not be causally related to the migration perturbations, but rather may arise from changes in neural crest cell activity. At present, we have no evidence to support this interpretation. Yet another possibility to consider is that changes in coronary vessel deployment may contribute to the cardiac defects. Cardiac neural crest derivatives are known to play an important role in the patterning of the coronary vasculature (Hood and Rosenquist, 1992; Waldo et al., 1994), and in fact coronary vessel abnormalities have been observed in both the CMV43 (Ewart et al., 1997) and α1 connexin KO mouse hearts (Waldo, K., M.L. Kirby, and C.W. Lo, unpublished observations).
Resolving these various issues will require a better understanding of the role of neural crest cells in cardiac morphogenesis. Given that cardiac neural crest cells generate very little of the tissue in the heart and outflow tract, their role(s) in cardiac morphogenesis may be indirect. Perhaps changes in neural crest abundance, migration timing, or functional activity may perturb the balance of cytokines required for normal myocyte differentiation and proliferation—cytokines that are produced by crest and/or noncrest cells. For example, changes in neural crest abundance or migration could affect the local concentration of platelet-derived growth factor or endothelins in the developing heart. Both are expressed by noncrest cells found along the cardiac crest migratory pathway, while their respective receptors are expressed by cardiac neural crest cells and also the cardiomyoctes (Soriano, 1997; Clouthier et al., 1998).
Gap Junction Communication and Cell–Cell Signaling in Crest Cells
Although traditionally gap junctions are not considered important in the physiology of migratory cells, the results from our studies indicate that gap junction communication plays an important role in neural crest migration. We propose that during neural crest migration, signals received by cells at the migration front may be relayed via gap junctions to cells throughout the migratory stream, and in this manner, facilitate a coordinated response to migratory cues. This intracellular pathway of cell–cell communication may help increase the efficiency of an otherwise seemingly chaotic process by which neural crest cells find their way to distant target sites.
Given that cardiac neural crest cells also give rise to the enteric ganglia and the thymus capsule (LeDourain, 1973; Bockman and Kirby, 1984; Kuratani and Kirby, 1991; Epstein et al., 1994; LeDourain and Teillet, 1994), one might predict that there should be defects related to these structures with the perturbation of α1 connexin function. In fact, recent studies have revealed enteric ganglionic deficiencies and thymus defects in the α1 connexin KO mice (Lo, C.W., K. Waldo, M.L. Kirby, and L. Spain, unpublished observations). It is interesting to note that it was previously reported that the enteric ganglionic deficiencies in the piebald lethal mice arose from crest migration defects that were non–cell autonomous, that is defects that are non–cell intrinsic (Kapur et al., 1995). Given that this mutation is a deletion of the endothelin receptor B gene, a receptor which is normally expressed in cardiac neural crest cells, the finding of non–cell autonomy was unexpected but entirely consistent with a role for gap junction communication in mediating cell signaling events important in neural crest migration and development (Kapur et al., 1995).
As the α1 connexin gap junctions are also expressed in noncardiac neural crest cells (Lo et al., 1997), it is interesting to note that we have found abnormalities associated with the trigeminal ganglia in the CMV43 (Ewart et al., 1997), and FC and α1 connexin KO mice (Sullivan et al., 1998). Formation of a portion of the trigeminal ganglia and nerves is dependent on preotic crest cells (D'Amico-Martel and Noden, 1983). Hence, the latter findings would suggest a role for α1 connexin gap junctions in development of the preotic hindbrain neural crest cells. This is consistent with the outgrowth studies, which in fact showed migration changes in the preotic crest cells that were indistinguishable from those seen in the postotic crest cells.
A Role for α1 Connexins in Other Migratory Cells?
In light of these observations on neural crest migration, it is interesting to note that another migratory cell population is also affected by the loss of α1 connexin gap junctions, the primordial germ cells. Like neural crest cells, germ cells must traverse long distances to reach their target organ. They also have been described as migrating in a continuous stream, with all of the germ cells in direct cell– cell contact with one another (Gomperts et al., 1994). This has led to the suggestion that cell–cell interactions may play an important role in germ cell migration. In the α1 connexin KO mouse, both sexes show a severe reduction (90% or more) in germ cell numbers (Kidder, G., personal communication). As this deficiency appears to arise very early in development, it is possible that germ cell migration may be perturbed by the loss of α1 connexin function. Consistent with this possibility is our observation of sterility associated with heterozygous α1 connexin KO mice maintained in certain genetic backgrounds (Lo, C.W., unpublished observations). This would indicate that the precise level of α1 connexin function also may be critically important for modulating germ cell migration. Of further significance is that polymorphonuclear leukocytes and lymphoid tissue have been reported to express α1 connexin gap junctions (Krenacs and Rosendaal, 1995; Branes, M.C., J. Conteras, M.B. Bono, and J.C. Saez. 1997. Mol. Cell. Biol. 8:417a). Thus, perhaps gap junctional communication may play a role in the migration and/or homing of cells in the immune system. Overall, these observations suggest the possibility that α1 connexin gap junctions may have a general role in modulating the behavior of motile cells. The future challenge is to identify the signal transduction pathways, and the gap junction–permeable cell signaling molecules involved in the modulation of cell migration. In addition, given that our studies showed a low level of gap junction communication persisting in neural crest cells of homozygous α1 connexin KO mice, in future studies it will be important to examine the possible role of other connexins in the modulation of neural crest migration and cell– cell signaling. Perhaps such studies may reveal a role for gap junctions in other neural crest populations.
Acknowledgments
This work was supported by grants from the Nation Science Foundation (IBN31544) and the National Institutes of Health (HD29573 to C.W. Lo; HL36059, HL51533, and HD17063 to M.L. Kirby, and GM37904 to N.B. Gilula).
Abbreviations used in this paper
- BrdU
bromodeoxyuridine
- KO
knockout
- PCNA
proliferating cell nuclear antigen
References
- Bancroft M, Bellairs R. The neural crest cells of the trunk region of the chick embryo studied by SEM and TEM. ZOON (Upps) 1976;4:73–85. [Google Scholar]
- Bennett MVL, Barrio LC, Bargiello TA, Spray DC, Hertzberg E, Saez JC. Gap junctions: new tools, new answer, new questions. Neuron. 1991;6:305–320. doi: 10.1016/0896-6273(91)90241-q. [DOI] [PubMed] [Google Scholar]
- Bockman DE, Kirby ML. Dependence of thymus development on derivatives of the neural crest. Science. 1984;223:498–500. doi: 10.1126/science.6606851. [DOI] [PubMed] [Google Scholar]
- Boger DL, Patterson JE, Guan X, Cravatt BF, Lerner RA, Gilula NB. Chemical requirements for inhibition of gap junction communication by the biologically active lipid oleamide. Proc Natl Acad Sci USA. 1998;95:4810–4815. doi: 10.1073/pnas.95.9.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- Clouthier DE, Hosoda K, Richardson JA, Williams SC, Yanagisawa H, Kuwaki T, Kumada M, Hammer RE, Yanagisawa M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development (Camb) 1998;125:813–824. doi: 10.1242/dev.125.5.813. [DOI] [PubMed] [Google Scholar]
- Cox LS. Who binds wins: competition for PCNA rings out cell-cycle changes. Trends Cell Biol. 1997;7:493–498. doi: 10.1016/S0962-8924(97)01170-7. [DOI] [PubMed] [Google Scholar]
- D'Amico-Martel A, Noden DM. Contribution of placodal and neural crest cells to avian cranial peripheral ganglia. Am J Anat. 1983;166:445–468. doi: 10.1002/aja.1001660406. [DOI] [PubMed] [Google Scholar]
- Davis EM, Trinkaus JP. Significance of cell-to-cell contacts for the directional movement of neural crest cells within a hydrated collagen lattice. J Embryol Exp Morph. 1981;63:29–51. [PubMed] [Google Scholar]
- Echelard Y, Vassileva G, McMahon AP. Cis-acting regulatory sequences governing wnt-1 expression in the developing mouse CNS. Development (Camb) 1994;120:2213–2224. doi: 10.1242/dev.120.8.2213. [DOI] [PubMed] [Google Scholar]
- Epstein ML, Mikawa T, Brown AMC, McFarlin DR. Mapping the origin of the avian enteric nervous system with a retroviral marker. Dev Dyn. 1994;201:236–244. doi: 10.1002/aja.1002010307. [DOI] [PubMed] [Google Scholar]
- Ewart JL, Cohen MF, Meyer RA, Huang GY, Wessels A, Gourdie RG, Chin AJ, Park SM, Lazatin BO, Villabon S, Lo CW. Heart and neural tube defects in transgenic mice overexpressing the Cx43 gap junction gene. Development (Camb) 1997;124:1281–1292. doi: 10.1242/dev.124.7.1281. [DOI] [PubMed] [Google Scholar]
- Fukiishi Y, Morriss-Kay GM. Migration of cranial neural crest cells to the pharyngeal arches and heart in rat embryos. Cell Tissue Res. 1992;268:1–8. doi: 10.1007/BF00338048. [DOI] [PubMed] [Google Scholar]
- Gomperts M, Garcia-Castro M, Wylie C, Heasman J. Interactions between primoridal germ cells play a role in their migration in mouse embryos. Development (Camb) 1994;120:135–141. doi: 10.1242/dev.120.1.135. [DOI] [PubMed] [Google Scholar]
- Gourdie RG, Green CR, Severs NJ, Thompson RP. Immunolabelling patterns of gap junction connexins in the developing and mature rat heart. Anat Embryol. 1992;185:363–378. doi: 10.1007/BF00188548. [DOI] [PubMed] [Google Scholar]
- Guan X, Cravatt BF, Ehring GR, Hall JE, Boger DL, Lerner RA, Gilula NB. The sleep-inducing lipid oleamide deconvulutes gap junction communication and calcium wave transmission in glial cells. J Cell Biol. 1997;139:1785–1792. doi: 10.1083/jcb.139.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie SC, Gilula NB. Gap junctional communication and development. Trends Neurosci. 1989;12:12–16. doi: 10.1016/0166-2236(89)90150-1. [DOI] [PubMed] [Google Scholar]
- Hood LC, Rosenquist TH. Coronary artery development in the chick: origin and deployment of smooth muscle cells, and the effects of neural crest ablation. Anat Rec. 1992;234:291–230. doi: 10.1002/ar.1092340215. [DOI] [PubMed] [Google Scholar]
- Huang GY, Wessels A, Smith BR, Linask KK, Ewart JL, Lo CW. Alteration in connexin 43 gap junction gene dosage impairs conotruncal heart development. Dev Biol. 1998;198:32–44. doi: 10.1006/dbio.1998.8891. [DOI] [PubMed] [Google Scholar]
- Kapur RP, Sweetser DA, Doggett B, Sieert JR, Palmiter RD. Intercellular signals downstream of endothelin receptor-B mediate colonization of the large intestine by enteric neuroblasts. Development (Camb) 1995;121:3787–3795. doi: 10.1242/dev.121.11.3787. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Matsunami H, Inoue T, Shimamura K, Ueno K, Miyazaki T, Takeichi M. Cadherin-11 expressed in association with mesenchymal morphogenesis in the head, somite, and limb bud of early mouse embryos. Dev Biol. 1995;169:347–358. doi: 10.1006/dbio.1995.1149. [DOI] [PubMed] [Google Scholar]
- Kirby ML. Cellular and molecular contributions of the cardiac neural crest to cardiovascular development. Trends Cardiovasc Med. 1993;3:18–23. doi: 10.1016/1050-1738(93)90023-Y. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Waldo KL. Neural crest and cardiovascular patterning. Circ Res. 1995;77:211–215. doi: 10.1161/01.res.77.2.211. [DOI] [PubMed] [Google Scholar]
- Krenacs T, Rosendaal M. Immunohistological detection of gap junctions in human lymphoid tissue: connexin 43 in follicular dendritic and lymphoendothelial cells. J Histochem Cytochem. 1995;43:1125–1137. doi: 10.1177/43.11.7560895. [DOI] [PubMed] [Google Scholar]
- Kumar NM, Gilula NB. Molecular biology and genetics of gap junction channels. Semin Cell Biol. 1992;3:3–16. doi: 10.1016/s1043-4682(10)80003-0. [DOI] [PubMed] [Google Scholar]
- Kuratani SC, Kirby ML. Initial migration and distribution of the cardiac neural crest in the avian embryo: an introduction to the concept of the circumpharyngeal crest. Am J Anat. 1991;191:215–227. doi: 10.1002/aja.1001910302. [DOI] [PubMed] [Google Scholar]
- LeDourain NM. Cell line segregation during peripheral nervous system ontogeny. Science. 1986;231:1515–1522. doi: 10.1126/science.3952494. [DOI] [PubMed] [Google Scholar]
- LeDourain NM, Teillet MA. The migration of neural crest cells to the wall of the digestive tract in avian embryo. J Embryol Exp Morphol. 1973;30:31–48. [PubMed] [Google Scholar]
- Lo CW. The role of gap junction membrane channels in development. J Bioenerg Biomembr. 1996;28:337–383. doi: 10.1007/BF02110114. [DOI] [PubMed] [Google Scholar]
- Lo CW, Wessels A. Cx43 gap junctions and cardiac development. Trends Cardiovasc Med. 1998;8:266–271. doi: 10.1016/s1050-1738(98)00018-8. [DOI] [PubMed] [Google Scholar]
- Lo CW, Cohen MF, Ewart JL, Lazatin BO, Patel N, Sullivan R, Pauken C, Park SMJ. Cx43gap junction gene expression and gap junctional communication in mouse neural crest cells. Dev Genet. 1997;20:119–132. doi: 10.1002/(SICI)1520-6408(1997)20:2<119::AID-DVG5>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Loewenstein WR. Junctional intercellular communication and the control of growth. Biochem Biophys Acta. 1979;560:1–65. doi: 10.1016/0304-419x(79)90002-7. [DOI] [PubMed] [Google Scholar]
- Moiseiwitsch JRD, Luader JM. Serotonin regulates mouse cranial neural crest migration. Proc Natl Acad Sci USA. 1995;92:7182–7186. doi: 10.1073/pnas.92.16.7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, Reid K, Hilton DJ, Barlett PF. Generation of sensory neurons is stimulated by leukemia inhibitory factor. Proc Natl Acad Sci USA. 1991;88:3498–3501. doi: 10.1073/pnas.88.8.3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Takeichi M. Neural crest cell-cell adhesion controlled by sequential and subpopulation specific expression of novel cadherins. Development (Camb) 1995;121:1321–1332. doi: 10.1242/dev.121.5.1321. [DOI] [PubMed] [Google Scholar]
- Nishibatake M, Kirby ML, Van Mierop LHS. Pathogenesis of persistent truncus arteriosus and dextroposed aorta in the chick embryo after neural crest ablation. Circulation. 1987;75:255–264. doi: 10.1161/01.cir.75.1.255. [DOI] [PubMed] [Google Scholar]
- Patterson JE, Ollmann IR, Cravatt BF, Boger DL, Wong CH, Lerner RA. Inhibition of oleamide hydrolase catalyzed hydrolysis of the endogenous sleep-inducing lipid cis-9-octadecenadmie. J Am Chem Soc. 1996;118:5938–5945. [Google Scholar]
- Raible DW, Eisen JS. Regulative interactions in zebrafish neural crest. Development (Camb) 1996;122:501–507. doi: 10.1242/dev.122.2.501. [DOI] [PubMed] [Google Scholar]
- Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardia malformation in neonatal mice lacking connexin 43. Science. 1995;267:1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- Ruangvoravat CP, Lo CW. Restrictions in gap junctional communication in the Drosophilalarval epidermis. Dev Dyn. 1991;193:70–82. doi: 10.1002/aja.1001930110. [DOI] [PubMed] [Google Scholar]
- Soriano P. The PDGFα receptor is required for neural crest cell development and for normal patterning of somites. Development (Camb) 1997;124:2691–2700. doi: 10.1242/dev.124.14.2691. [DOI] [PubMed] [Google Scholar]
- Sullivan R, Lo CW. Expression of a Cx43/β-gal fusion protein inhibits gap junctional communication in NIH3T3 cells. J Cell Biol. 1995;130:419–429. doi: 10.1083/jcb.130.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan R, Meyer R, Huang GY, Cohen MF, Wessels A, Linask KK, Lo CW. Heart malformations in transgenic mice exhibiting dominant negative inhibition of gap junctional communication. Dev Biol. 1998;204:224–234. doi: 10.1006/dbio.1998.9089. [DOI] [PubMed] [Google Scholar]
- Waldo KL, Kumiski DH, Kirby M. Association of the cardiac neural crest with development of the coronary arteries in the chick embryo. Anat Rec. 1994;239:315–331. doi: 10.1002/ar.1092390310. [DOI] [PubMed] [Google Scholar]
- Waldo K, Miyagawa-Tomita S, Kumiski D, Kirby ML. Cardiac neural crest cells provide new insight into septation of the cardiac outflow tract: aortic sac to ventricular septal closure. Dev Biol. 1998;196:129–144. doi: 10.1006/dbio.1998.8860. [DOI] [PubMed] [Google Scholar]
- Warner A. Gap junctions in development: a perspective. Semin Cell Biol. 1992;108:1039–1051. doi: 10.1016/s1043-4682(10)80009-1. [DOI] [PubMed] [Google Scholar]
- Willecke, K., Henneman, H., E. Dahl, S. Jungbluth, and R. Heynkes. 1991. The diversity of connexin genes encoding gap junctional proteins. Eur. J. Cell Biol. 56:1–7. [PubMed]
- Ya J, Erdtsieck-Ernste EBHW, De Boer PAJ, Van Kempen MJA, Jongsma H, Gros D, Moorman AF, Lamers WH. Heart defects in connexin-43 deficient mice. Circ Res. 1998;82:360–369. doi: 10.1161/01.res.82.3.360. [DOI] [PubMed] [Google Scholar]
- Yancey SB, Biswal S, Revel JP. Spatial and temporal patterns of distribution of the gap junction protein connexin43 during mouse gastrulation and organogenesis. Development (Camb) 1992;114:203–212. doi: 10.1242/dev.114.1.203. [DOI] [PubMed] [Google Scholar]