

Abstract
FGF-2 and VEGF are potent angiogenesis inducers in vivo and in vitro. Here we show that FGF-2 induces VEGF expression in vascular endothelial cells through autocrine and paracrine mechanisms. Addition of recombinant FGF-2 to cultured endothelial cells or upregulation of endogenous FGF-2 results in increased VEGF expression. Neutralizing monoclonal antibody to VEGF inhibits FGF-2–induced endothelial cell proliferation. Endogenous 18-kD FGF-2 production upregulates VEGF expression through extracellular interaction with cell membrane receptors; high-M r FGF-2 (22–24-kD) acts via intracellular mechanism(s). During angiogenesis induced by FGF-2 in the mouse cornea, the endothelial cells of forming capillaries express VEGF mRNA and protein. Systemic administration of neutralizing VEGF antibody dramatically reduces FGF-2-induced angiogenesis. Because occasional fibroblasts or other cell types present in the corneal stroma show no significant expression of VEGF mRNA, these findings demonstrate that endothelial cell-derived VEGF is an important autocrine mediator of FGF-2-induced angiogenesis. Thus, angiogenesis in vivo can be modulated by a novel mechanism that involves the autocrine action of vascular endothelial cell-derived FGF-2 and VEGF.
Angiogenesis, the formation of capillaries from preexisting blood vessels, occurs in a variety of physiological and pathological settings, including embryonic development, wound healing, and tumor growth (Folkman and Klagsbrun, 1987; Folkman, 1991, 1992; Breier et al., 1997; Risau, 1997). Numerous studies have shown that angiogenesis is required for the growth and metastasis of solid tumors (Weidner et al., 1991; Horak et al., 1992; Macchiarini et al., 1992; Wakui et al., 1992; Vartanian et al., 1994). A number of cytokines and growth factors modulate angiogenesis in vitro and in vivo with a paracrine mode of action. Among these factors, basic fibroblast growth factor (FGF-2)1 and vascular endothelial growth factor (VEGF) are the most potent angiogenesis inducers (for reviews see Basilico and Moscatelli, 1992; Ferrara et al., 1992; Bikfalvi et al., 1997; Ferrara and Davis-Smyth, 1997).
FGF-2 is the prototype member of a family of 13 structurally related, heparin-binding growth factors (for recent review see Bikfalvi et al., 1997). It is expressed ubiquitously in cells of mesodermal and neuroectodermal origin, and in a variety of tumor cells. In vitro, FGF-2 is a potent mitogen for different cell types, including vascular endothelial cells and fibroblasts. With cultured endothelial cells, FGF-2 induces an angiogenic phenotype consisting of increased proliferation, migration, proteinase production, and expression of specific integrins (Moscatelli et al., 1986a ; Klein et al., 1993, 1996). In vivo FGF-2 is a potent inducer of angiogenesis and has pleiotropic effects on development and differentiation in various organs (Basilico and Moscatelli, 1992; Bikfalvi et al., 1997). However, mice genetically deficient in FGF-2 (FGF-2 knockout) have no apparent defects related to impaired angiogenesis (Ortega et al., 1998; Zhou et al., 1998).
FGF-2 exists in four different molecular weight isoforms: 18-kD or low molecular weight (LMW) FGF-2, and 22-, 22.5-, and 24-kD or high molecular weight (HMW) FGF-2. The HMW forms derive from alternative translation initiation (CUG) codons and contain the complete LMW sequence in addition to an NH2-terminal extension of varying length (Moscatelli et al., 1987; Sommer et al., 1987; Florkiewicz and Sommer, 1989; Prats et al., 1989). The different forms of FGF-2 have been associated with different cell functions and cellular compartmentalization. LMW FGF-2 is released by the cells and stimulates cell migration, proliferation, and FGF receptor downregulation through binding to surface receptors; HMW FGF-2 forms primarily localize to the nucleus and modulate cell proliferation (Bugler et al., 1991; Mignatti et al., 1991; Florkiewicz et al., 1991; Quarto et al., 1991a ,b; Renko et al., 1991; Bikfalvi et al., 1995).
The biological activity of FGF-2 is mediated through a dual receptor system consisting of four high-affinity, tyrosine kinase receptors and low-affinity, heparan sulfate proteoglycans located at the cell surface (Moscatelli, 1987; Flaumenhaft et al., 1989; Lee et al., 1989; Dionne et al., 1990; Keegan et al., 1991; Partanen et al., 1991). However, FGF-2 lacks a signal peptide that directs secretion through the classical secretory pathway (Abraham et al., 1986). Although the mechanism(s) by which FGF-2 is released from cells is/are still unknown (Mignatti and Rifkin, 1991; Mignatti et al. 1991, 1992), FGF-2 is found extracellularly and modulates several cell functions in an autocrine manner (Sato and Rifkin, 1988; Mignatti et al., 1991; Sato et al., 1991; Peverali et al., 1994; Bikfalvi et al., 1995; Klein et al., 1996). Because of its high affinity for heparin and heparan sulfate glycosaminoglycans, significant amounts of FGF-2 are associated with the extracellular matrix of in vitro cell cultures (Vlodavski et al., 1987; Bashkin et al., 1989; Flaumenhaft et al., 1989; Rojeli et al., 1989). In vivo, FGF-2 has been detected in the basal lamina of blood capillaries, primarily at sites of vessel branching, and in the endothelium of the capillaries of some tumors (Folkman et al., 1988; DiMario et al., 1989; Cordon et al. 1990; Schulze-Osthoff et al., 1990), suggesting that endothelial cell-derived FGF-2 may mediate angiogenesis with an autocrine mode of action. This hypothesis is supported by the observation that FGF-2 has an autocrine effect on several cell functions required for angiogenesis, including proliferation, migration, proteinase production, and integrin expression (Sato and Rifkin, 1988; Mignatti et al., 1991; Sato et al., 1991; Peverali et al., 1994; Bikfalvi et al., 1995; Klein et al., 1996).
VEGF, the prototype member of a family of four structurally related growth factors, is a potent mitogen for micro- and macrovascular endothelial cells but lacks appreciable mitogenic activity for other cell types (Ferrara and Davis-Smyth, 1997; Senger et al., 1993). VEGF exists in five molecular species of 121, 145, 165, 189, and 206 amino acids (VEGF121, VEGF145, VEGF165, VEGF189, VEGF206) that derive from exon splicing of a single gene (Houck et al., 1991; Keck et al., 1989; Leung et al., 1991; Tisher et al., 1991; Poltorak et al., 1997). VEGF165, a 46-kD homodimeric glycoprotein, is the predominant isoform produced by a variety of normal and transformed cells. VEGF121 is a freely soluble protein. The other VEGF isoforms show increased affinity for heparan sulfate proteoglycans: VEGF165 is soluble although a fraction can remain bound to the extracellular matrix; VEGF189 and VEGF206 are almost exclusively sequestered in the extracellular matrix (Ferrara et al., 1992; Houck et al., 1992; Park et al., 1993).
The promoter region of VEGF contains hypoxia-responsive elements in addition to several potential Ap-1, Ap-2 and Sp-1 binding sites, indicating that VEGF transcription can be enhanced in response to multiple stimuli (Tisher et al., 1991; Levy et al., 1995; Liu et al., 1995). Oxygen tension plays a major role in the regulation of VEGF expression in a variety of cell types, including endothelial cells. Several findings have implicated VEGF as the major mediator of the angiogenic effect of hypoxia (Shweiki et al., 1992; Goldberg and Schneider, 1994; Minchencko et al., 1994; Liu et al., 1995; Shima et al., 1995; Forsythe et al., 1996). In addition, several cytokines and growth factors, as well as tumor promoters upregulate VEGF expression in many cell types (Garrido et al., 1993; Goldman et al., 1993; Pertovaara et al., 1994; Li et al., 1995; Tsai et al., 1995; Ryuto et al., 1996). It has been proposed that VEGF may act as a paracrine mediator for indirect-acting angiogenic factors, such as transforming growth factor beta (Brogi et al., 1994). However, the effect of cytokines and growth factors on VEGF expression in vascular endothelial cells has not been investigated.
Numerous lines of evidence implicate VEGF as a pivotal factor in the regulation of normal and pathological vasculogenesis and angiogenesis. VEGF promotes angiogenesis in different experimental models in vitro and in vivo (Leung et al., 1989; Plouet et al., 1989; Pepper et al., 1992; Nicosia et al., 1994; Phillips et al., 1995). The loss of even a single VEGF allele results in embryonic lethality, showing the irreplaceable role of this factor in the development of the vascular system (Carmeliet et al., 1996; Ferrara et al., 1996). In addition, VEGF has been shown to have therapeutic effects on coronary and limb ischemia (Takeshita et al., 1994; Pearlman et al., 1995; Harada et al., 1996; Isner et al., 1996). VEGF mRNA is markedly upregulated in the majority of human tumors so far examined (for review see Ferrara and Davis-Smyth, 1997). Its expression in several tumors has been correlated with high vascularity, lymph node metastasis, and liver metastasis, and a poorer prognosis than VEGF-negative tumors (Toi et al., 1994; Maeda et al., 1996). Antibodies to VEGF or expression of a dominant-negative VEGF receptor inhibit tumor growth in vivo without affecting tumor cell proliferation in vitro. In animals treated with anti-VEGF antibodies the density of blood vessels in tumor sections is lower than in the tumors of control animals, showing that the inhibitory effect of the antibody on tumor growth is mediated by a blockage of the angiogenic activity of VEGF (Kim et al., 1993; Millauer et al., 1994; Warren et al., 1995; Borgstrom et al., 1996). These findings implicate VEGF as the major tumor angiogenesis factor so far identified.
In addition to promoting vasculogenesis and angiogenesis, VEGF enhances vascular permeability and induces fenestrations in the endothelium of small capillaries and venules (Senger et al., 1983; Roberts and Palade, 1995). An increase in vascular permeability may be required for angiogenesis during tumor growth and wound healing (Dvorak, 1986). The observation of VEGF expression around microvessels whose endothelium is normally quiescent has generated the hypothesis that VEGF is also required for the maintenance of the differentiated state of blood vessels (Ferrara et al., 1992; Alon et al., 1995).
The findings that FGF-2 can modulate angiogenesis with an autocrine mechanism (Sato and Rifkin, 1988; Mignatti et al., 1991; Sato et al., 1991; Peverali et al., 1994; Bikfalvi et al., 1995; Klein et al., 1996) and that FGF-2 and VEGF have synergistic effects on angiogenesis (Pepper et al., 1992) prompted us to investigate potential interactions between these two potent angiogenic factors in vascular endothelial cells. Here we report that FGF-2 modulates endothelial cell expression of VEGF through both autocrine and paracrine mechanisms of action. In vivo the endothelial cells of quiescent vessels do not express VEGF; upon stimulation with FGF-2 the endothelium of newly forming capillaries produces VEGF. The systemic administration of VEGF antibody to mice that received corneal implants of FGF-2 pellets results in a dramatic decrease in angiogenesis, implicating endothelial cell VEGF as a major mediator of the angiogenic activity of FGF-2.
Materials and Methods
Materials
Human recombinant VEGF165 and mouse recombinant VEGF165 were purchased from R & D Systems, Inc. (Minneapolis, MN); anti-human VEGF antibody (αVEGF sc507) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); the neutralizing monoclonal antibody to human VEGF, mAb #577B11, was a gift of Texas Biotechnology, Inc. (Houston, TX); mouse and rabbit nonimmune (n.i.) IgG were purchased from Sigma Chemical Co. (St. Louis, MO); human recombinant FGF-2 was obtained from Scios Nova and Synergen, Inc. (Boulder, CO); anti– rFGF-2 antibody has been described (R58; Pintucci et al., 1996); anti-von Willebrand factor antibody (vWF A0082) was purchased from Dako Corp. (Carpinteria, CA), and doxycycline from Sigma Chemical Co. protein concentrations were measured by the Bio-Rad DC protein assay reagent (Bio-Rad Laboratories, Hercules, CA) using BSA as a standard.
Cells and Media
Bovine aortic endothelial cells (BAE) and bovine capillary endothelial cells (BCE) were isolated as described (Folkman et al., 1979) and grown in alpha modified minimum essential medium (αMEM; Fisher Scientific Co., Pittsburgh, PA) supplemented with 5% calf serum and 2mM l-glutamine (GIBCO BRL, Gaithersburg, MD). These cells were used between passages 6 and 15 in culture. Human umbilical vein endothelial (HUVE) cells and human aortic endothelial (HAE) cells were purchased from Clonetics (San Diego, CA). These cells were grown in endothelial cell basal medium-2 (Clonetics) containing 2% FCS and the endothelial cell growth supplements provided by the company. Both HUVE and HAE cells were used between passages 3 and 5 in culture. NIH 3T3 fibroblasts transfected with the cDNAs for human low M r FGF-2 (LMW FGF-2), high M r FGF-2 (HMW FGF-2), or all FGF-2 isoforms (wt FGF-2), and NIH 3T3 cells transfected with the vector alone (ZipNeo) have been described (Quarto et al., 1991a ,b; Bikfalvi at al., 1995). These cells were grown in DME supplemented with 10% FBS, 2 mM l-glutamine, and 500 μg/ml geneticin (G418; Sigma Chemical Co.).
Cell Proliferation Assay
HUVE or HAE cells were seeded into 96-well plates (2.5 × 103 cells/well) in 50 μl of endothelial cell basal medium-2 supplemented with heparin and 1% FCS. After overnight incubation, FGF-2 or VEGF were added to a final concentration of 10 or 30 ng/ml, respectively, in the presence or absence of 10 μg/ml of either anti-human VEGF monoclonal antibody or anti–FGF-2 antibody or mouse n.i. IgG. Control cultures received equivalent volumes of medium with no addition. The medium was replaced every third day with fresh medium with or without addition of FGF-2, VEGF, and the IgGs. Triplicate samples were trypsinized every 24 h and counted with a hemocytometer.
Transfection of NIH 3T3 Cells with FGF-2 cDNA under Control by the Tetracycline-dependent Transactivator
EcoRI–EcoRI cDNAs for LMW and HMW FGF-2 were cloned into the EcoRI site of the plasmid pUHD10-3 (provided by H. Bujard, Zentrum für Molekulare Biologie der Universität, Heidelberg, Germany), which contains the binding sequence for the tetracycline-controlled transactivator (rtTA) in front of a multiple cloning site (Gossen et al., 1995). NIH 3T3 cells were transfected with the plasmid pUHD172-1Neo (also provided by H. Bujard), which contains a mutagenized form of rtTA spliced to a nuclear translocation sequence and the neomycin resistance gene (Gossen et al., 1995). Transfected cells were selected in medium containing 500 μg/ml of G418. One clone of G418-resistant cells that constitutively expressed rtTA was cotransfected with the plasmids pUHD–LMW FGF-2 or pUHD–HMW FGF-2 and the plasmid pCEP4 containing the hygromycin resistance gene (Invitrogen, Carlsbad, CA). Stable cotransfectants were selected in medium containing 500 μg/ml of both G418 and hygromycin (Calbiochem-Novabiochem, San Diego, CA). Hygromycin/ G418-resistant cells were characterized for FGF-2 expression by Western blotting of extracts of cells grown in serum-free medium with or without 1 μg/ml of doxycycline for 24 h.
Preparation of SK-Hep1 Cell-conditioned Medium
Confluent cultures of SK-Hep1 cells in 150 cm2 culture flasks were washed twice with PBS and incubated for 17 h in the presence of 15 ml of serum-free DME. The culture supernatant was centrifuged at 1,000 g for 15 min and either used immediately or stored at −20°C.
Northern Blotting
Total RNA was extracted from cells with the Trizol reagent (GIBCO BRL) according to the manufacturer's instructions. The RNA (20 μg) was run in a 1% formaldehyde–agarose gel and transferred to a positively charged nylon membrane (Boehringer Mannheim Biochemicals, Indianapolis, IN). The blot was prehybridized in digoxygenin (DIG) Easy Hyb (Boehringer Mannheim Biochemicals) for 2 h at 42°C and then hybridized overnight at 42°C with a 403-bp DIG-labeled cDNA probe to human VEGF121 obtained by reverse transcriptase–PCR using the primers described in Houk et al. (1991). The cDNA was labeled by running the PCR reaction in the presence of DIG-labeled nucleotides (Boehringer Mannheim Biochemicals). After hybridization, the membrane was washed twice in 2× SSC, 0.1% SDS at room temperature for 15 min and twice in 1× SSC, 0.1% SDS at 50°C for 15 min. The detection of the probe was performed using the Genius 7 kit (Boehringer Mannheim Biochemicals) according to the manufacturer's instructions. The membranes were exposed to autoradiographic films (Hyperfilm MP; Amersham Life Technologies, Arlington Heights, IL) for 1 min.
Western Blotting
Confluent endothelial cells or NIH 3T3 cells in 100-mm dishes were washed with PBS and incubated overnight in serum-free medium. The conditioned medium was concentrated 80-fold by ultrafiltration in Centricon tubes (Amicon, Inc., Beverly, MA). The cells were lysed in 100 mM Tris-HCl, pH 8.1, containing 0.5% Triton X-100, 10 μg/ml leupeptin (Sigma Chemical Co.), and 400 μM Pefablock (Boehringer Mannheim Biochemicals). Cell extract protein (200 μg from endothelial cells, 50 μg from NIH 3T3 cells) or concentrated conditioned medium was electrophoresed in SDS–12% polyacrylamide gel under reducing conditions and then blotted to a polyvinylidene difluoride membrane (Immobilon P; Millipore Corp., Waters Chromatography, Bedford, MA) for 4 h at 45 V. The membrane was incubated with 5% skim milk (Carnation; Nestlé Food Co., Glendale, CA) in 20 mM Tris buffer, pH 7.4, overnight at 4°C to block nonspecific binding. VEGF or FGF-2 were detected by incubating the membrane with 0.2 μg/ml of antibody to human VEGF (αVEGF sc507; Santa Cruz Biotechnology, Inc.) or to human rFGF-2 (R58; Pintucci et al., 1996) for 1 h at room temperature. After incubation with horseradish peroxidase-conjugated donkey anti–rabbit IgG (1:5,000) for 1 h (Amersham Life Technologies), immune complexes were detected with the enhanced chemiluminescence ECL™ detection system (Amersham Life Technologies). The membranes were exposed to autoradiographic films (Hyperfilm MP; Amersham Life Technologies) for 10 s–1 min.
Metabolic Labeling and Immunoprecipitation
Confluent endothelial cells were incubated for 2 h with SK-Hep1 cell-conditioned medium or with control medium. After incubation with methionine/ cysteine-free DME for 1 h, the medium was replaced with methionine/cysteine-free DME containing 150 μCi/ml of 35S-methionine/cysteine (ICN Biomedicals, Inc., Costa Mesa, CA) and the incubation was continued for 4 h. The medium was collected and centrifuged at 1,000 g for 5 min, diluted 1:1 with RIPA buffer (PBS, 1% Tergitol, 0.5% deoxycholic acid, 0.1% SDS) and incubated with 1 μg/ml of anti-human VEGF IgG (αVEGF sc507; Santa Cruz Biotechnology, Inc.) for 1 h followed by 1 h of incubation with protein A–Agarose (Boehringer Mannheim Biochemicals). After washing three times with RIPA buffer, the resin was loaded onto a SDS–12% polyacrylamide gel and run under reducing conditions. The gel was fixed in 30% methanol and 10% acetic acid for 30 min and then incubated for 30 min in Amplify (Amersham Life Technologies). Dried gels were exposed to autoradiographic films (Hyperfilm βmax; Amersham Life Technologies) for 7 d at −80°C.
Densitometry
Northern blot and Western blot bands were analyzed with a Shimadzu scanning densitometer (model C6-93-01PC; Shimadzu Scientific Instruments, Inc., Columbia, MD) using dedicated software.
In Vivo Angiogenesis Assay
Angiogenesis assays in the mouse cornea were performed as described (Chen et al., 1995). Briefly, a corneal pocket was created with a modified von Graefe cataract knife in both eyes of 4–6-wk-old C57B mice (The Jackson Laboratory, Bar Harbor, ME). One 0.34 × 0.34-mm sucrose aluminum sulfate (Bukh Meditec, Copenhagen, Denmark) pellet coated with hydron polymer type NCC (IFN Sciences, New Brunswick, NJ), containing 50 ng of rFGF-2 was implanted into each pocket. Pellets without rFGF-2 were used as negative controls. The corneas were routinely examined by slit-lamp biomicroscopy on postoperative days 5–7. After examination, the mice were killed and the eyes excised, embedded in tissue-freezing medium (Triangle Biomedical Sciences, Durham, NC), and then frozen in a mixture of dry ice and 2-methylbutane (Fisher Scientific Co.). Frozen eyes were stored at −80°C.
Immunohistochemistry
30-μm frozen sagittal sections of mouse eyes were fixed in 4% paraformaldehyde in PBS for 15 min and then incubated with 1 μg/ml goat n.i. IgG for 1 h to block nonspecific binding. To detect VEGF or von Willebrand factor (vWF), the specimens were incubated overnight at 4°C with 2 μg/ml of rabbit antibody to murine VEGF (αVEGF sc507) or with 2 μg/ml of rabbit antibody to human vWF that cross-reacts with mouse vWF. Rabbit n.i. IgG was used as a negative control. The sections were washed in 20 mM Tris buffer, pH 7.4, incubated with biotinylated goat anti–rabbit antiserum (Vectastain ABC Kit; Burlingame, CA) for 45 min and subsequently in 1% Triton X-100 (Sigma Chemical Co.) for 30 min. After washing in 20 mM Tris buffer, pH 7.4, avidin–biotin amplification was performed according to the manufacturer's instructions (Vectastain ABC Kit). Antigen–antibody complexes were detected by incubation with 3,3 diaminobenzamine (Sigma Chemical Co.) at room temperature for 10 min. The slides were counterstained with Gil's hematoxylin 1 (Sigma Chemical Co.).
In Situ Hybridization
Single-stranded, sense (S) and antisense (AS) RNA probes were generated by run-off in vitro transcription in the presence of DIG-labeled nucleotides (Transcription kit; Boehringer Mannheim Biochemicals) of a linearized pCR™II plasmid (TA cloning kit; Invitrogen) containing the 403-bp VEGF cDNA insert used for Northern blotting. 30-μm frozen sections of mouse eyes were fixed in 4% paraformaldehyde in PBS for 15 min, digested with proteinase K (1 μg/ml; Boehringer Mannheim Biochemicals) at room temperature for 10 min and then acetylated with 0.25% acetic anhydride in 100 mM triethanolamine. Hybridization with 0.2 μg/ml of the DIG-labeled S or AS VEGF riboprobe was performed in deionized 40% formamide, 10% dextran sulphate, 1× Denhardt's solution, 4× SSC, 10 mM DTT, and 1 mg/ml sheared salmon sperm DNA overnight at 42°C in a moist chamber. The slides were washed three times in 0.1× SSC for 20 min at 55°C and digested with RNase A (20 μg/ml; Boehringer Mannheim Biochemicals). The immunological detection was performed using sheep anti-DIG IgG conjugated with alkaline phosphatase 0.5 U/ml (anti–DIG-AP Fab fragment; Boehringer Mannheim Biochemicals) according to the manufacturer's instructions. The chromogenic reaction with NBT/BCIP (Boehringer Mannheim Biochemicals) was developed in 100 mM Tris buffer, 50 Mm NaCl, 50 mM MgCl2, and 1 mM levamisole, pH 9.5, for 4 h at room temperature.
Results
FGF-2 Stimulates Endothelial Cell Expression of VEGF through Both Autocrine and Paracrine Mechanisms
To test the effect of FGF-2 on VEGF expression by endothelial cells, confluent BCE or BAE cells were incubated for 4 h with serum-free medium or with medium containing 10 ng/ml of human recombinant FGF-2 (rFGF-2). Northern blotting with a cDNA probe to human VEGF showed a band of 3.9 kb, consistent with the expected size of VEGF mRNA (Tischer et al., 1991; Shima et al., 1995) (Fig. 1 A). The intensity of this band was ten- and fivefold higher in rFGF-2–treated BCE and BAE cells, respectively, than in control cells. Western blotting under nonreducing conditions with antibody to human VEGF showed a band of 46 kD that comigrated with human recombinant VEGF165 (R & D Systems) in medium conditioned by FGF-2–treated endothelial cells. This band was very faint or absent in the conditioned medium or extracts of control BCE or BAE cells (Fig. 1 B). Reverse transcriptase–PCR with primers for VEGF (Houk et al., 1991) showed two transcripts whose sequences corresponded to VEGF165 and VEGF121 and a minor transcript corresponding to VEGF189 (data not shown). However, Western blotting of cell-conditioned medium or extract showed no immunoreactive proteins other than VEGF165. These data showed that exogenous FGF-2 upregulates the expression of VEGF165 in cultured endothelial cells.
Figure 1.
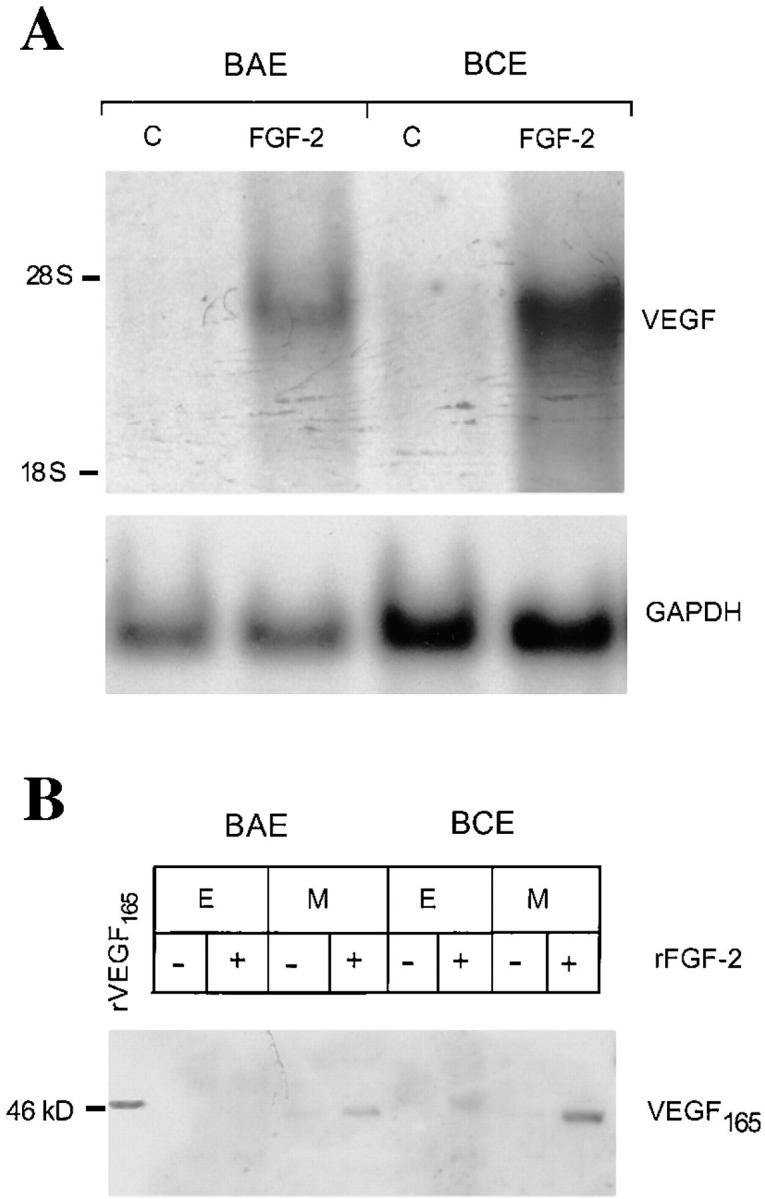
FGF-2 induces VEGF expression in endothelial cells. (A) Northern blotting analysis of total RNA (20 μg) from BAE or BCE cells treated with rFGF-2 (10 ng/ml) or with control medium (C) for 4 h. The RNA blot was hybridized with a DIG-labeled cDNA probe for human VEGF as described in Materials and Methods. GAPDH mRNA is shown as a control. The position of 28 S and 18 S ribosomal RNA is shown on the left. This experiment was repeated four times with comparable results. (B) Western blotting analysis of Triton X-100 extracts (E; 200 μg) or conditioned medium (M) from BAE or BCE cells treated with 10 ng/ml of rFGF-2 (+) or with control medium (−) for 17 h. The samples were electrophoresed under nonreducing conditions. The protein blot was hybridized with human VEGF IgG; antigen–antibody complexes were detected as described in Materials and Methods. Recombinant VEGF165 (10 ng) was run as a control in the leftmost lane. Molecular masses are shown in kD on the left. This experiment was repeated three times with comparable results.
The endothelial cells we used had been selected in our laboratory for low expression of endogenous FGF-2, relative to other strains of BCE or BAE cells. The cells expressed 18, 22, and 24 kD FGF-2, as assessed by Western blotting with antibody to human rFGF-2 (Fig. 2 A). Because in the absence of exogenous rFGF-2 BCE or BAE cells did not express VEGF (Fig. 1), this finding indicated that either VEGF expression is regulated only by exogenous FGF-2 or that the levels of endogenous FGF-2 in our endothelial cells were too low to induce VEGF expression. To test these hypotheses, BCE cells were incubated with SK-Hep1 hepatoma cell-conditioned medium, which upregulates endothelial cell expression of FGF-2 mRNA (Peverali et al., 1994). Consistent with previous findings (Moscatelli et al., 1986b ; Peverali et al., 1994), Western blotting of concentrated SK-Hep1 cell-conditioned medium showed no detectable FGF-2 (<0.25 ng/ml) (Fig. 2 A). Incubation with SK-Hep1 cell-conditioned medium resulted in a five- and twofold increase in LMW FGF-2 and HMW FGF-2, respectively (Fig. 2 A), and in a fourfold increase in VEGF mRNA (Fig. 2 B). Addition of neutralizing rFGF-2 IgG (10 μg/ml) decreased VEGF mRNA expression to control levels. In contrast, n.i. IgG was ineffective (Fig 2 B). Similarly, immunoprecipitation experiments showed that 2.5-fold higher levels of VEGF165 were produced by endothelial cells exposed to the conditioned medium relative to cells incubated with control medium. Addition of anti–rFGF-2 antibody abolished this effect, whereas n.i. serum was ineffective (data not shown). Because SK-Hep1 cell-conditioned medium contains no FGF-2, these results showed that upregulation of endogenous FGF-2 results in increased expression of endothelial cell VEGF165.
Figure 2.

SK-Hep1–conditioned medium induces FGF-2 and VEGF expression in endothelial cells. (A) Right, Western blotting analysis of Triton X-100 extracts (200 μg) from BCE cells incubated for 17 h with SK-Hep1 cell-conditioned medium (SK-Hep1 c.m.) or with control medium (C). Recombinant LMW FGF-2 (rFGF-2; 10 ng) was run as a control in the leftmost lane. Left, 4 ml of SK-Hep1 cell-conditioned medium was concentrated 80-fold and analyzed by Western blotting as described in Materials and Methods. 1 ng of rFGF-2 was run in the same blot as a control. Antigen–antibody complexes were detected as described in Materials and Methods. Molecular masses are shown in kD between the two panels. The positions of LMW and HMW FGF-2 bands are shown on the right. This experiment was repeated three times with similar results. (B) Northern blotting analysis of total RNA (20 μg) from BCE cells incubated for 2 h with either control medium (C) or with SK-Hep1 cell-conditioned medium (SK-Hep1 c.m.) or 10 ng/ ml of rFGF-2 in the presence or absence of 10 μg/ml of neutralizing antibody to rFGF-2 (αFGF-2 IgG) or n.i. IgG (n.i. IgG). The RNA blots were hybridized with a DIG-labeled cDNA probe to human VEGF as described in Materials and Methods. GAPDH mRNA is shown as a control. This experiment was repeated three times with similar results.
VEGF Mediates FGF-2–induced Proliferation of Endothelial Cells
To test the functional significance of VEGF produced by endothelial cells stimulated with FGF-2, we characterized the effect of neutralizing monoclonal antibody to VEGF on the proliferation of endothelial cells treated with FGF-2. Because the monoclonal antibody we used was raised against human VEGF, HUVE or HAE cells were used for these experiments. As shown in Fig. 3 (A and C), addition of VEGF antibody to FGF-2–treated HUVE or HAE cells inhibited cell proliferation by 100 and 50%, respectively. In control cultures, the VEGF antibody completely neutralized the activity of recombinant VEGF on HUVE or HAE cell proliferation (Fig. 3, B and D). Thus, endothelial cell-derived VEGF is an important downstream mediator of the mitogenic activity of FGF-2.
Figure 3.

Antibody to VEGF inhibits FGF-2–induced endothelial cell proliferation. Growth curves of HUVE (A and B) or HAE (C and D) cells in the absence (□) or in the presence of 10 ng/ml of FGF-2 (⋄) (A and C) or 30 ng/ml of VEGF (▴) (B and D) without or with addition of either 10 μg/ml of anti-VEGF (▴) or anti–FGF-2 (○) or n.i. IgG (▪). The cells were grown and counted as described in Materials and Methods. Each point represents mean and standard deviation of triplicate samples from a representative experiment.
Endogenous LMW FGF-2 and HMW FGF-2 Modulate VEGF Expression through Different Mechanisms of Action
Our endothelial cells expressed both LMW FGF-2 and HMW FGF-2 (Fig. 2 A). To characterize possible differential roles of the FGF-2 isoforms in VEGF upregulation we used a panel of NIH 3T3 cell clones transfected with the cDNAs for either LMW FGF-2 (LMW FGF-2), HMW FGF-2 (HMW FGF-2), or all FGF-2 isoforms (wt FGF-2). NIH 3T3 cells transfected with the vector alone (ZipNeo) were used as control. By Western blotting, all the clones of FGF-2 transfectants used in our experiments expressed comparable levels of FGF-2 (Fig 4 A). Control cells transfected with the vector alone produced no FGF-2.
Figure 4.


FGF-2 and VEGF expression in NIH 3T3 cells transfected with FGF-2 cDNA. (A) FGF-2 expression. Triton X-100 extracts (50 μg) of clones of NIH 3T3 cells transfected with the cDNAs for HMW (clones 1 and 2), LMW (clones 3 and 4) or WT FGF-2, or from control cells transfected with the vector alone (ZIPNEO) were analyzed by Western blotting with antibody to human FGF-2. Recombinant LMW FGF-2 (rFGF-2; 10 ng) was run as a control in the leftmost lane. (B) VEGF expression. Medium conditioned by the clones of FGF-2 transfectants shown in A was analyzed by Western blotting with antibody to VEGF. Recombinant VEGF165 (10 ng) was run as a control in the leftmost lane. Western blotting was carried out under reducing conditions as described in Materials and Methods. Molecular masses are shown in kD on the left. This experiment was repeated three times with similar results. (C) Northern blotting analysis of total RNA (20 μg) from clones of NIH 3T3 cells transfected with the cDNAs for HMW (clone 1), LMW (clone 3) or WT FGF-2, or from control cells transfected with the vector alone (ZIPNEO). The RNA blot was hybridized with a DIG-labeled cDNA probe to human VEGF as described in Materials and Methods. The position of 28 S and 18 S ribosomal RNA is shown on the left. 18S ribosomal RNA is shown as a control. This experiment was repeated three times with comparable results.
Western blotting of conditioned medium under reducing conditions showed two bands of 23 and 21 kD, respectively, consistent with two glycosylation variants of VEGF165 (Houck et al., 1991) in medium conditioned by the FGF-2 transfectants (Fig. 4 B). Medium conditioned by LMW FGF-2 or HMW FGF-2 transfectants contained comparable levels of VEGF165; wt FGF-2 cell clones expressed higher levels of VEGF165. In contrast, control cells transfected with the vector alone produced no VEGF. No other isoforms of VEGF were detected in the conditioned medium or cell extracts (data not shown) of any of the transfected cells (Fig. 4 B). Northern blotting of RNA from the FGF-2 transfectants with a cDNA probe to human VEGF showed a band of 3.9 kb, consistent with the expected size of murine VEGF mRNA (Shima et al., 1995). This band was absent in control, vector-transfected cells (Fig. 4 C).
To rule out the possibility that the differences in VEGF expression between the FGF-2 transfectants and the control cells reflected clonal variability, we used clones of NIH 3T3 cells transfected with the cDNAs for either LMW FGF-2 or HMW FGF-2 whose expression was controlled by the tetracycline-dependent transactivator (Gossen et al., 1995). These transfectants had comparable growth rates in the absence or in the presence of doxycycline, a tetracycline analogue (data not shown). However, HMW FGF-2 transfectants had a higher growth rate than LMW FGF-2 transfectants. Addition of doxycycline to the culture medium resulted in a dramatic upregulation of both FGF-2 and VEGF165 expression by the LMW FGF-2 and the HMW FGF-2 transfectants (Fig. 5) but did not affect FGF-2 or VEGF expression in control, vector-transfected cells (data not shown).
Figure 5.

FGF-2 and VEGF expression in NIH 3T3 cells transfected with FGF-2 cDNA under control by the tetracycline- dependent transactivator. NIH 3T3 cells transfected with the cDNAs for either LMW FGF-2 or HMW FGF-2 cDNA under control by the tetracycline-dependent transactivator were grown for 24 h in serum-free medium with (+) or without (−) addition of 1 μg/ml of doxycycline. The conditioned medium was collected and the cells were lysed with Triton X-100. Cell extracts and concentrated media were analyzed by Western blotting under nonreducing conditions as described in Materials and Methods. (A) Western blotting analysis of cell extracts (50 μg) with antibody to FGF-2. Extract of NIH 3T3 cells transfected with wt FGF-2 cDNA (WT FGF-2) was run as a control in the rightmost lane. (B) Western blotting analysis of conditioned media with antibody to VEGF. The samples were electrophoresed under nonreducing conditions. Recombinant VEGF165 (10 ng) was run as a control in the rightmost lane. Molecular masses are shown in kD on the left of each panel. This experiment was repeated twice with comparable results.
To confirm that VEGF expression in NIH 3T3 cells was mediated by FGF-2, HMW FGF-2 and LMW FGF-2 transfectants were grown for 9 h in the presence of neutralizing antibody to FGF-2. VEGF expression was characterized by Northern blotting (data not shown) and by Western blotting (Fig. 6). Addition of the antibody abolished VEGF165 expression in a dose-dependent manner in LMW FGF-2 transfectants but had no effect on HMW FGF-2 transfectants. This result is consistent with previous findings of different cell compartmentalization and mechanisms of action of LMW FGF-2 and HMW FGF-2 (Quarto et al., 1991b ; Bikfalvi et al., 1995; Klein et al., 1996).
Figure 6.

Effect of anti–FGF-2 antibody on VEGF expression by NIH 3T3 cells transfected with LMW FGF-2 or HMW FGF-2 cDNA. Western blotting analysis of medium conditioned by LMW FGF-2 or HMW FGF-2 trans-fectants (clones 1 and 3 shown in Fig. 4) in the absence or in the presence of the indicated concentrations of anti-FGF-2 antibody (αFGF-2) or n.i. (n.i.) IgG (50 μg/ml). (A) LMW FGF-2 transfectants (clone 3). (B) HMW FGF-2 transfectants (clone 1). FGF-2 expression by these clones is shown in Fig. 4 A. The cells were preincubated for 3 h in serum-free medium with or without the indicated concentrations of anti– FGF-2 antibody or n.i. IgG; the medium was replaced with fresh, serum-free medium with or without anti–FGF-2 antibody or n.i. IgG and the incubation was continued for 9 h. The conditioned medium was analyzed by Western blotting under reducing conditions with anti-VEGF antibody as described in Materials and Methods. Recombinant VEGF165 (10 ng) was run as a control in the leftmost lane of the gels shown in A and B. This experiment was repeated three times with similar results.
These data showed that VEGF expression is modulated by endogenous FGF-2 as well as by exogenous FGF-2. VEGF upregulation by endogenous LMW FGF-2 requires release of FGF-2 from the cells and interaction with cell membrane receptors. In contrast, endogenous HMW FGF-2 acts through an intracellular mechanism.
FGF-2 Induces VEGF Expression in the Endothelial Cells of Growing Capillaries
To test the hypothesis that FGF-2 can upregulate endothelial cell expression of VEGF in vivo, we used the angiogenesis assay in the mouse cornea. Hydron pellets containing either 50 ng of rFGF-2 or no rFGF-2 were implanted in corneal pockets of C57B mice 1 mm away from the limbic vessels. 6 d after implantation, when maximum capillary formation was observed in eyes with the rFGF-2 pellets, 30-μm sagittal sections of the entire eye were prepared and analyzed both by in situ hybridization and by immunohistochemistry.
In situ hybridization with a DIG-labeled antisense VEGF riboprobe of sections of eyes that received sham pellets showed that the endothelial cells of the limbic vessels expressed no detectable VEGF mRNA (Fig. 7 A). In sections of eyes that received rFGF-2 pellets, the VEGF riboprobe hybridized to VEGF mRNA present in the endothelial cells of the newly formed capillaries in the limbic region (Fig. 7 B). Photomicrographs taken in a through-focus series at high magnification (1,500×) showed that the VEGF-expressing endothelial cells were predominantly located in branching vessels (Fig. 8). The probe did not hybridize significantly to occasional fibroblasts or other cell types present in the corneal stroma (Figs. 7 and 8).
Figure 7.
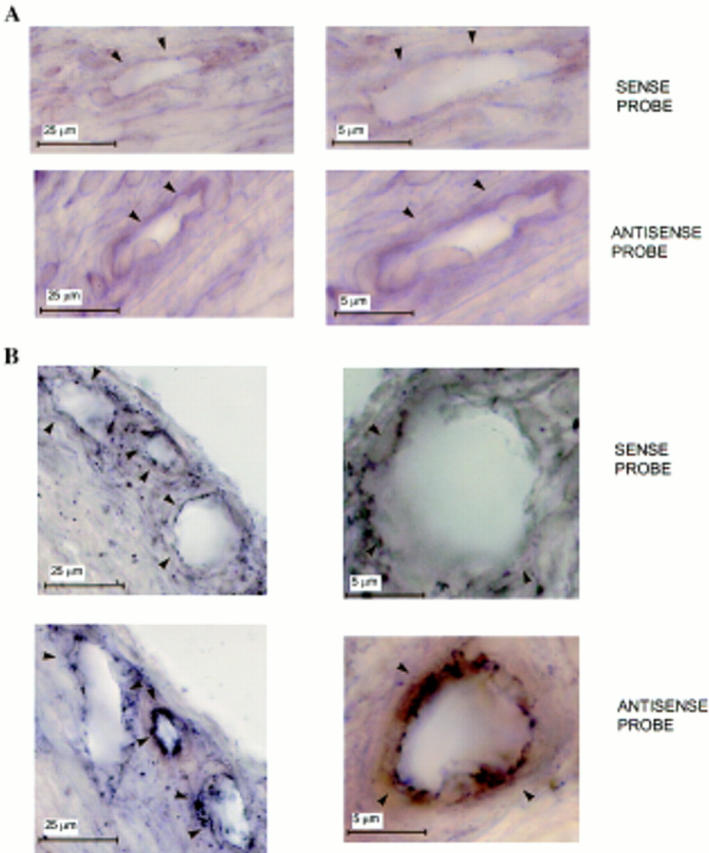
Expression of VEGF mRNA by vascular endothelium in vivo.In situ hybridization with DIG-labeled sense or antisense VEGF riboprobes of adjacent 30-μm sections of mouse corneas that received either (A) sham pellets or (B) pellets containing 50 ng of rFGF-2. Implantation of pellets in the cornea, preparation of the probes, and in situ hybridization were carried out as described in Materials and Methods. Contrast was enhanced by computer to increase the appearance of the reaction product. Arrowheads, vessel's wall. (A) Sections of limbic vessels show no hybridization with the probes. (B) Sections of newly forming capillaries in the stroma of the cornea show hybridization of the endothelium with the antisense but not with the sense probe. Hybridization signals (brown-black staining) are present only in the endothelium of newly formed vessels in FGF-2–treated eyes.
Figure 8.

Expression of VEGF mRNA by the endothelial cells of branching capillaries. Photomicrographs taken in a through-focus series (1-μm steps) of a 30 μm-thick section of mouse cornea hybridized in situ with a DIG-labeled antisense riboprobe to VEGF. A hydron pellet containing 50 ng of rFGF-2 was implanted in the cornea 5 d before sectioning. Implantation of the pellet in the cornea, preparation of the probes, and in situ hybridization were carried out as described in Materials and Methods. Contrast was enhanced by computer to increase the appearance of the reaction product. Arrowheads, the wall of a capillary branching out of a larger vessel (top left corner of each panel). A homogenous hybridization signal is associated with the endothelium of the branching capillary (arrowheads, panels 2–5) but not with the endothelium of the larger vessel. In panels 1 and 5 the capillary is below and above the focus plane, respectively.
Immunostaining with antibody to VEGF showed that the quiescent vessels of the limbic region of corneas that received sham pellets had no VEGF (Fig 9). In sections of eyes that received rFGF-2 pellets, the endothelium of newly formed capillaries stained intensely with VEGF IgG (Fig. 10). No other cell types present in the corneal stroma, except occasional cells adjacent to the vessels' lumina, were stained. These cells may represent accessory vascular cells (e.g., pericytes), acute inflammatory infiltrates or endothelial cells of adjacent, collapsed capillaries. It is noteworthy that significant hybridization of the VEGF riboprobe occurred only to cells surrounding the lumina (Figs. 7 and 8). All the periluminal cells stained with VEGF antibody also stained positively with antibody to vWF, an endothelial cell marker, confirming that the VEGF mRNA-producing cells were indeed endothelial cells (Figs. 9 and 10). Thus, although no VEGF is expressed by quiescent vascular endothelium, induction of angiogenesis with rFGF-2 results in VEGF expression by the endothelial cells of forming capillaries.
Figure 9.
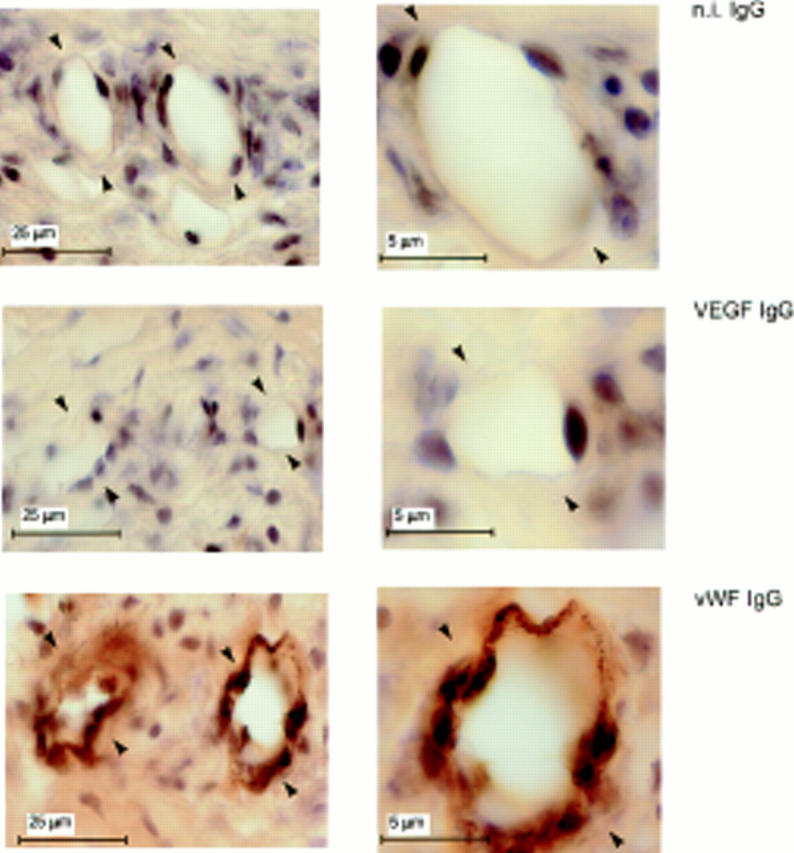
Lack of VEGF expression in quiescent endothelium. Adjacent 30-μm sections of mouse corneas that received pellets with no FGF-2 were immunostained with antibody to mouse VEGF (VEGF IgG) or to vWF (vWF IgG) or with n.i. IgG (n.i. IgG) as described in Materials and Methods. The endothelium of the limbic vessels (arrowheads) stains positively for vWF but not for VEGF.
Figure 10.

VEGF expression in the endothelium of newly formed capillaries. Adjacent 30-μm sections of mouse corneas that received pellets containing 50 ng of rFGF-2 were immunostained with antibody to mouse VEGF (VEGF IgG) or to vWF (vWF IgG) or with n.i. IgG (n.i. IgG) as described in Materials and Methods. The endothelium of newly formed capillaries (arrowheads) in the stroma of the cornea stains positively both for vWF and for VEGF. Some paraluminal cells also stain positively with both VEGF and vWF antibody.
Endothelial Cell VEGF Mediates FGF-2–induced Angiogenesis
To investigate the functional significance of endothelial cell expression of VEGF during FGF-2–induced angiogenesis, 18 mice that received corneal implants of pellets containing rFGF-2 (50 ng) in both eyes were randomized into three groups of six animals and given i.v. injections of either PBS, PBS containing n.i. IgG (100 μg), or neutralizing anti-human VEGF monoclonal antibody (100 μg). This antibody neutralizes mouse VEGF in in vitro proliferation assay almost as efficiently as human VEGF (Fig. 11; see Fig. 3 for comparison). The mice were injected 1 d before pellet implantation and on postoperative days 1 and 3. The corneas were photographed on day 5.
Figure 11.

Effect of monoclonal antibody to human VEGF on endothelial cell proliferation induced by mouse VEGF. Growth curves of HUVE cells in the absence (□) or in the presence of 30 ng/ml of mouse VEGF (♦) and of either monoclonal anti-human VEGF antibody (▴, 10 μg/ml; ▵, 30 μg/ml) or n.i. IgG (□, 10 μg/ml; ▪, 30 μg/ml). The cells were grown and counted as described in Materials and Methods. Each point represents mean and standard deviation of triplicate samples from a representative experiment.
The animals injected with anti-VEGF antibody consistently showed markedly reduced angiogenesis relative to control mice injected with PBS or n.i. IgG. As shown in Fig. 12, the length of the vessels was the same in the three groups. However, the capillaries of mice treated with anti-VEGF antibody appeared to be consistently reduced in number, and were thinner and less branched than in control animals. Similar results were obtained when the VEGF antibody (1 μg) or n.i. IgG were added in the same pellets containing rFGF-2 (Fig. 13).
Figure 12.
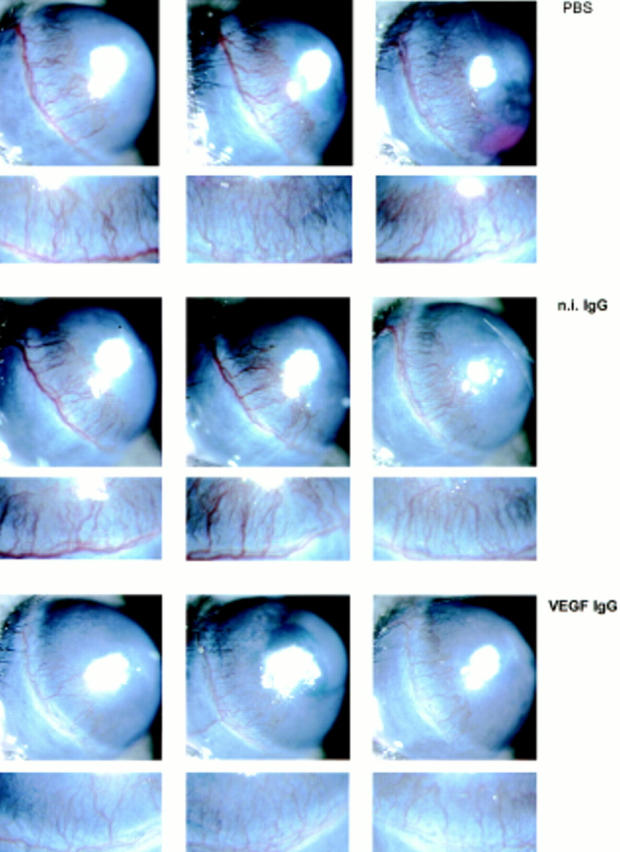
Effect of anti-VEGF antibody on FGF-2–induced angiogenesis. Hydron pellets containing 50 ng of rFGF-2 were implanted in the cornea of both eyes of 18 Swiss Webster mice as described in Materials and Methods. The animals were randomized into three groups of six mice and given i.v. injections of either PBS or PBS containing n.i. IgG (100 μg) or neutralizing anti-human VEGF monoclonal antibody (VEGF IgG; 100 μg) 1 d before pellet implantation and on postoperative days 1 and 3. The corneas were photographed by slit-lamp biomicroscopy on day 5 after implantation of the pellet. The eyes of the animals injected with VEGF antibody have fewer and thinner corneal limbic capillaries than those of animals injected with PBS or n.i IgG. An enlargement of the limbic area containing the newly formed vessels is shown below each photograph of the corresponding eye. This experiment was repeated twice with comparable results. In control mice that received corneal implants of pellets containing 200 ng of human recombinant VEGF, the same treatment with the VEGF antibody abolished the angiogenic response almost completely (data not shown).
Figure 13.
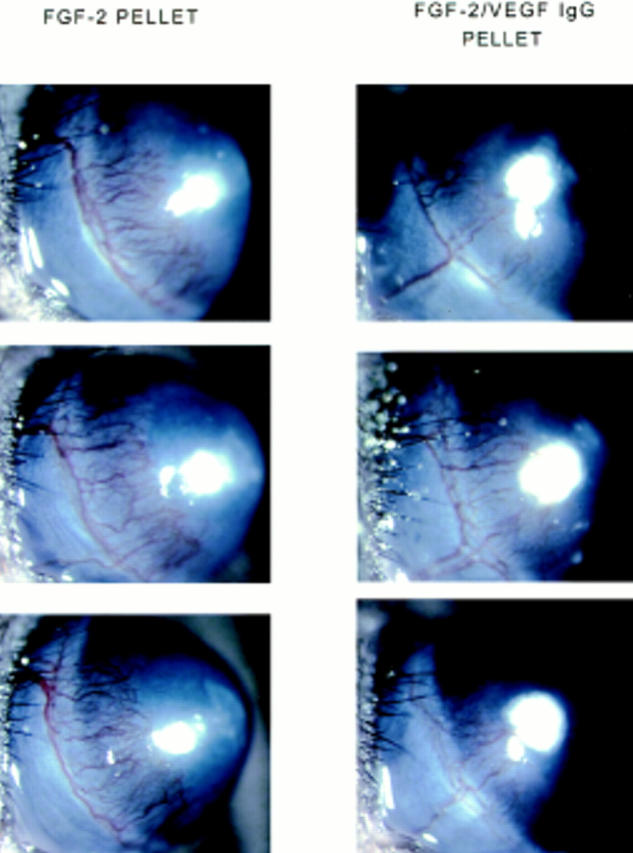
Effect of anti-VEGF antibody on FGF-2–induced angiogenesis. Hydron pellets containing 50 ng of rFGF-2 with or without 1 μg of anti-VEGF monoclonal antibody were implanted in the cornea of both eyes of Swiss Webster mice as described in Materials and Methods. Each group consisted of six animals. The corneas were photographed by slit-lamp biomicroscopy on day 5 after implantation of the pellet. FGF-2 pellet: eyes that received corneal implantation of pellets containing FGF-2 alone. FGF-2/ VEGF IgG pellet: eyes that received corneal implantation of pellets containing FGF-2 and antibody to VEGF. The eyes that received corneal implants of pellets containing FGF-2 and VEGF antibody have fewer and thinner corneal limbic capillaries than eyes that received corneal implants of pellets containing FGF-2 alone.
To quantitate these effects, the number of vessels was measured on enlarged photographs of all the eyes. Because vessel branching also appeared to be reduced in antibody-treated animals, the vessels were counted both proximally to and distally from the limbus (i.e., close to the pellet). As shown in Table I, in control mice the number of vessels distal from the limbus was 2.0- to 2.5-fold higher than in the paralimbic region. In mice receiving VEGF antibody either by intravenous injection or by corneal implant the number of vessels proximal to the limbic region showed only 12 and 26% decrease, respectively. However, distally from the limbus, the systemic and the topical antibody treatments resulted in 40 and 50% decrease in vessel number, respectively, showing that vessel branching was considerably reduced or abolished by the VEGF antibody. Because only the endothelial cells of forming capillaries in the cornea expressed VEGF mRNA, the effect of the antibody must be mediated by the neutralization of the endothelial cell-derived growth factor. Therefore, endothelial cell VEGF represents a major autocrine effector of the angiogenic response induced by FGF-2.
Table I.
Number of Capillaries Proximal to and Distal from the Limbic Region of the Cornea of Mice Receiving mAb to VEGF by i.v. Injection or by Corneal Implant in the FGF-2–Containing Pellet
| i.v. injection | Pellet | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Proximal | Distal | Proximal | Distal | |||||||||||||
| vessel | vessel | vessel | vessel | |||||||||||||
| (n)* | (%) | (n)* | (%) | (n)* | (%) | (n)* | (%) | |||||||||
| PBS | 15.7 ± 1.1 | 100.0 | 41 ± 3.1 | 97.2 | — | — | — | — | ||||||||
| n.i. IgG | 15.7 ± 1.1 | 100.0 | 42.3 ± 1.5 | 100.0 | 15.3 ± 1.0 | 100.0 | 30.0 ± 1.3 | 100.0 | ||||||||
| VEGF Ab | 13.8 ± 1.5 | 87.9‡ | 25.2 ± 0.9 | 59.6§ | 11.3 ± 0.9 | 73.8‡ | 15.2 ± 0.9 | 50.7§ | ||||||||
Mean of 12 eyes per group ± SEM.
P < 0.01.
P < 0.01. Statistical analysis performed by analysis of variance.
Discussion
Experimental evidence has shown that angiogenesis is regulated by a variety of growth factors and cytokines with a paracrine mode of action. FGF-2 and VEGF are the most potent angiogenesis inducers and have a synergistic effect on angiogenesis (Pepper et al., 1992; Asahara et al., 1995). In this work we provide evidence that vascular endothelial cell-derived FGF-2 and VEGF are involved in a cascade of growth factor activities that modulate angiogenesis with an autocrine mechanism of action. In this cascade, endogenous or exogenous FGF-2 regulates endothelial cell expression of VEGF, which is a major autocrine mediator of FGF-2-induced angiogenesis. These conclusions are based on the following observations: (a) although quiescent endothelial cells in vitro do not express VEGF, addition of exogenous FGF-2 or upregulation of endogenous FGF-2 expression result in increased VEGF synthesis; (b) antibody to VEGF inhibits FGF-2-induced proliferation of endothelial cells; (c) in cultured cells endogenous FGF-2 upregulates VEGF expression through both extracellular and intracellular mechanisms: VEGF upregulation by LMW FGF-2 requires FGF-2 release from the cells and interaction with cell membrane receptors; HMW FGF-2 acts through an intracellular mechanism; (d) although the quiescent endothelial cells of the limbic vessels of mouse cornea express no VEGF, during angiogenesis induced by implantation of FGF-2 pellets the endothelial cells of newly formed capillaries express both VEGF mRNA and protein; VEGF mRNA is located primarily to the endothelium of branching vessels; and (e) systemic administration of neutralizing VEGF antibody inhibits angiogenesis induced by FGF-2 in the mouse cornea.
Our cultured endothelial cells expressed significant amounts of FGF-2 but no VEGF. Upregulation of VEGF mRNA and protein required addition of exogenous FGF-2 or upregulation of endogenous FGF-2, indicating that the endogenous levels of FGF-2 were not sufficient to induce VEGF expression. This conclusion is also supported by our experiments with NIH 3T3 fibroblasts transfected with FGF-2 cDNA and the tetracycline resistance transactivator. Because the tetracycline-controlled transactivator was leaky, in the absence of doxycycline LMW FGF-2 and HMW FGF-2 transfectants expressed detectable amounts of FGF-2. However, the expression of VEGF was very low or below detection limit (refer to Fig. 5). Upregulation of FGF-2 synthesis by addition of doxycycline resulted in a dramatic increase in VEGF expression. Therefore, the level of endogenous FGF-2 or the presence of exogenous FGF-2 are important factors in the regulation of VEGF expression in endothelial cells.
Our characterization of NIH 3T3 cells transfected with LMW FGF-2 or HMW FGF-2 cDNA showed that both HMW FGF-2 and LMW FGF-2 upregulate VEGF expression. Transfectants that constitutively express high levels of LMW FGF-2 or HMW FGF-2 also produce high levels of VEGF, whereas control, vector-transfected cells express no VEGF. LMW FGF-2 and HMW FGF-2 transfectants produce comparable levels of VEGF. However, the high amounts of FGF-2 produced by these cells may mask differences in the efficiency of VEGF regulation between LMW FGF-2 and HMW FGF-2. Two observations suggest that LMW FGF-2 may upregulate VEGF expression more efficiently than HMW FGF-2. Treatment of endothelial cells with SK-Hep1 cell-conditioned medium resulted in five- and twofold increased synthesis of LMW FGF-2 and HMW FGF-2, respectively. Concomitantly, VEGF mRNA and VEGF165 levels were also upregulated. Addition of neutralizing antibody to FGF-2 blocked the upregulation of VEGF mRNA and protein. Because HMW FGF-2 is primarily intracellular and acts through an intracrine mechanism(s) (Bugler et al., 1991; Florkiewicz et al., 1991; Quarto et al., 1991a ,b; Renko et al., 1991; Bikfalvi et al., 1995), the antibody did not neutralize HMW FGF-2 activity. However, the upregulation of HMW FGF-2 did not result in increased VEGF levels. This observation is consistent with the results of our analysis of VEGF expression in clones of NIH 3T3 cells transfected with FGF-2 cDNA and the tetracycline resistance transactivator. In the absence of doxycycline, LMW FGF-2 transfectants expressed low amounts of both LMW FGF-2 and VEGF. In contrast, the HMW FGF-2 transfectants had higher levels of HMW FGF-2 but expressed no VEGF (refer to Fig. 5).
Neutralizing antibody to FGF-2 abolished VEGF expression in NIH 3T3 cells that produce LMW FGF-2 but had no effect on cells that express HMW FGF-2, showing that LMW FGF-2 and HMW FGF-2 modulate VEGF expression with different mechanisms of action. This finding is consistent with previous observations that HMW FGF-2 is exclusively found intracellularly and accumulates in the nucleus, whereas LMW FGF-2 is released by the cells and binds to surface receptors (Bugler et al., 1991; Mignatti et al., 1991; Florkiewicz et al., 1991; Quarto et al., 1991a ,b; Renko et al., 1991; Bikfalvi et al., 1995). LMW FGF-2 and HMW FGF-2 modulate different cell functions, including migration, proliferation and FGF receptor downregulation (Bikfalvi et al., 1995). It has not been established whether these cell functions are also mediated through VEGF expression.
Many tumor or transformed cells express high levels of VEGF (Ferrara and Davis-Smyth, 1997). The upregulation of VEGF expression in endothelial cells or in FGF-2–transfected NIH 3T3 cells might result from the growth stimulatory effect of FGF-2 or from nonspecific effects derived from cell transformation. However, our analysis of NIH 3T3 cells transfected with FGF-2 cDNA and the tetracycline resistance transactivator rules out this hypothesis. Whereas expression of both FGF-2 and VEGF was dramatically upregulated in the presence of doxycycline, the cells had comparable growth rates in the presence or in the absence of the antibiotic (data not shown). In the presence of doxycycline, LMW FGF-2 transfectants had lower growth rates than HMW FGF-2 transfectants (data not shown) but expressed comparable amounts of VEGF.
VEGF expression is regulated by a variety of factors, including hypoxia, cytokines, and tumor promoters. FGF-2 upregulates VEGF levels in vascular smooth muscle cells (Stavri et al., 1995). However, only hypoxia and advanced glycation end products have been shown to regulate VEGF expression in vascular endothelial cells (Liu et al., 1995; Yamagishi et al., 1997). Our study is the first demonstration that VEGF expression in endothelial cells is modulated by FGF-2, a potent angiogenesis inducer. This finding has several important implications. First, VEGF is an important downstream mediator of the angiogenic activity of FGF-2, as shown by the inhibitory effect of VEGF antibody in vitro and in vivo. Second, although other cell types, including pericytes, smooth muscle cells, and inflammatory cells may participate in angiogenesis, capillary formation can be mediated by endothelial cell-derived growth factors through an autocrine mechanism independent of accessory cells. Third, FGF-2 and VEGF have synergistic effects on angiogenesis (Pepper et al., 1992; Asahara et al., 1995). Thus, a cascade of autocrine events in which one angiogenic factor upregulates the other appears to represent a very efficient mechanism for angiogenesis.
Recently, FGF-2 antibodies have been shown to block VEGF-induced angiogenesis in vitro (Mandriota and Pepper, 1997). Although this effect is opposite to the one we report here, it is consistent with our observation of an autocrine mechanism of angiogenesis mediated by FGF-2 and points to complex interactions between FGF-2 and VEGF. It is possible that endogenous endothelial cell FGF-2 is required for efficient angiogenesis and/or that the simultaneous presence of both FGF-2 and VEGF in vascular endothelial cells is required for angiogenesis in the adult organism. Mice genetically deficient in FGF-2 (FGF-2 knockout) have a normal vasculature and no apparent defects related to impaired angiogenesis (Ortega et al., 1998; Zhou et al., 1998). However, whereas VEGF expression during development may be modulated by factors other than FGF-2, the levels of VEGF in the endothelium of forming capillaries in adult FGF-2 knockout mice have not yet been characterized. More important, whether FGF-2 knockout mice have normal VEGF levels and angiogenesis in pathological settings such as tumor growth also remains to be investigated.
Our in vivo angiogenesis experiments with the mouse cornea show that FGF-2 also modulates endothelial cell expression of VEGF in vivo, and that endothelial cell VEGF is an important, autocrine effector of FGF-2–induced angiogenesis.
Consistent with previous findings, our in situ hybridization and immunohistochemistry experiments showed that the endothelium of the quiescent limbic vessels of the mouse cornea does not express VEGF. However, after stimulation with FGF-2, the endothelial cells of newly formed capillaries produce both VEGF mRNA and protein. The analysis of VEGF mRNA in several tumors has shown expression in the tumor cells but not in the endothelium of the tumor vasculature (Ferrara and Davis-Smyth, 1997). Conversely, immunohistochemical studies have shown VEGF associated with the endothelium of tumor capillaries, indicating that VEGF secreted by tumor cells is bound by its receptors present exclusively on endothelial cells (Plate et al., 1992, 1994; Brown et al., 1993; Qu-Hong et al., 1995). In these studies the relative abundance of VEGF transcript in the tumor cells might mask the VEGF mRNA of the endothelial cells. Our experimental model of angiogenesis in the mouse cornea obviates the problem of identifying the source of endothelium-associated VEGF, since only occasional fibroblasts are normally present in the stroma of the cornea. Inflammatory cells may be present in the cornea during angiogenesis. However, in our in situ hybridization experiments, the VEGF mRNA probe hybridized significantly only to vWF–positive cells of newly formed capillaries, indicating endothelial cells as the major source of VEGF.
On the basis of our data, we cannot exclude that in the cornea assay VEGF from cell types other than endothelial cells, including smooth muscle cells, macrophages, or T cells (Freeman et al., 1995; Stavri et al., 1995; McLaren et al., 1996), may contribute to early stages of angiogenesis, e.g., capillary sprouting from the limbic vessel. However, our finding that VEGF antibody did not significantly reduce the number of capillaries sprouting from the limbic vessel but did considerably decrease subsequent branching indicates that the contribution of cell types other than endothelial cells may be independent of VEGF. In contrast, VEGF derived from endothelial cells may play a role at later stages of angiogenesis. Whatever the contribution of other cell types to angiogenesis, our finding that in vitro FGF-2–stimulated endothelial cells rapidly (2–4 h) upregulate VEGF mRNA expression suggests that in vivo VEGF receptors can be more promptly saturated by the endothelial cell–derived ligand with an autocrine mechanism than with a paracrine mechanism by VEGF derived from other sources.
VEGF mRNA was located predominantly to sites of vessel branching. Interestingly, FGF-2 has also been found associated with the basal lamina of branching capillaries (Cordon et al., 1990), indicating that FGF-2 and VEGF may be primarily expressed by the endothelium of growing capillaries and play a role in vessel formation. This hypothesis is confirmed by our finding that neutralizing VEGF antibody dramatically inhibits FGF-2 induced angiogenesis in the mouse cornea. It is noteworthy that the antibody appears to predominantly reduce branching of the newly formed vessels, an observation that correlates with the location of FGF-2 and VEGF.
A number of experimental and clinical studies have implicated VEGF as a paracrine angiogenesis factor in different physiological and pathological settings. The levels of VEGF in several tumors have been shown to correlate with tumor vascularity, a negative prognostic indicator for a variety of malignancies (Toi et al., 1994; Maeda et al., 1996). However, other studies have failed to demonstrate such correlation (Dvorak et al., 1995). The autocrine or paracrine regulation of endothelial cell VEGF by FGF-2 may represent an important mechanism of angiogenesis under conditions in which insufficient or no VEGF is derived from other cell types. This autocrine mechanism may be triggered by normal or tumor cell-derived FGF-2 or by upregulation of endothelial cell FGF-2. Factors that regulate endothelial cell expression of FGF-2 are essentially unknown. Some tumor cell lines secrete a peptide(s) that upregulates endothelial cell expression of FGF-2 and induces in vitro angiogenesis (Peverali et al., 1994). FGF-2 upregulation results in increased expression of urokinase, a proteinase implicated in angiogenesis. Urokinase upregulation and in vitro angiogenesis are mediated by endothelial cell FGF-2 with an autocrine mechanism of action (Peverali et al., 1994). The purification and characterization of this and other potential factors that upregulate endothelial cell FGF-2 will be important for our comprehension of the paracrine and autocrine mechanisms of angiogenesis.
In all the experiments we performed, i.v. or local administration of VEGF antibody consistently inhibited angiogenesis. However, our antibody did not totally abrogate FGF-2–induced capillary formation. This result can be explained by two hypotheses: (a) the monoclonal antibody we used was raised against human VEGF: although we found that in vitro it can completely neutralize mouse VEGF almost as efficiently as human VEGF, it is possible that in vivo this antibody does not completely neutralize or the doses we administered did not afford complete neutralization of mouse VEGF; and (b) FGF-2 may not only act by inducing endothelial cell expression of VEGF but also induce angiogenesis through VEGF-independent mechanisms. Mouse embryos lacking a single VEGF allele have several abnormalities in their vasculature and die in utero (Carmeliet et al., 1996; Ferrara et al., 1996), showing that FGF-2 alone is not sufficient for normal vasculogenesis and angiogenesis during development. However, in the adult organism FGF-2 might at least in part complement the lack of VEGF. Although these hypotheses await confirmation, our experiments clearly show endothelial cell VEGF as a major downstream mediator of the angiogenic activity of FGF-2.
Acknowledgments
The authors thank B. Merhara (New York University Medical Center) for his expert help with the histology work, T. Broke (Texas Biotechnology, Inc.) for the generous gift of neutralizing monoclonal anti-VEGF IgG #577B11, and R. Crowe, E. Anglade, and H. Logan for their valuable assistance. We are particularly indebted to E. Grossi and J. Weider (all five from New York University Medical Center) for their precious help with statistical analysis and computer imaging.
This work was supported by funds from the S.A. Localio Laboratory for General Surgery Research and from the Laboratory for Cardiovascular Surgery Research, and in part by grant CA 34282 from the National Institutes of Health to D.B. Rifkin. P. Mignatti was partially supported by an American Cancer Society International Cancer Research Fellowship through the International Union Against Cancer.
Abbreviations used in this paper
- BAE
bovine aortic endothelial cells
- BCE
bovine capillary endothelial cells
- DIG
digoxygenin
- FGF-2
fibroblast growth factor-2
- HAE
human aortic endothelial cells
- HMW FGF-2
high molecular weight fibroblast growth factor-2
- HUVE
human umbilical vein endothelial cells
- LMW FGF-2
low molecular weight fibroblast growth factor-2
- rFGF-2
recombinant fibroblast growth factor-2
- VEGF
vascular endothelial growth factor
- vWF
von Willebrand factor
Footnotes
Address all correspondence to Paolo Mignatti, Department of Cell Biology, New York University Medical Center, 550 First Avenue, New York, NY 10016. Tel.: (212) 263-1478. Fax: (212) 263-0147. E-mail: mignap01 @http://mcrcr.med.nyu.edu
References
- Abraham JA, Mergia A, Whang JL, Tumolo A, Friedman J, Hjerrild KA, Gospodarowicz D, Fiddes JC. Nucleotide sequence of a bovine clone encoding the angiogenic protein, basic fibroblast growth factor. Science. 1986;233:545–548. doi: 10.1126/science.2425435. [DOI] [PubMed] [Google Scholar]
- Alon T, Hemo I, Itin A, Pe'er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nature Med. 1995;1:1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- Asahara T, Bauters C, Zheng LP, Takeshita S, Bunting S, Ferrara N, Symes JF, Isner JM. Synergistic effect of vascular endothelial growth factor and basic fibroblast growth factor on angiogenesis in vivo. Circulation. 1995;92(Suppl.):365–371. doi: 10.1161/01.cir.92.9.365. [DOI] [PubMed] [Google Scholar]
- Bashkin P, Doctrow S, Klagsbrun M, Svahn CM, Folkman J, Vlodavski I. Basic fibroblast growth factor binds to subendothelial extracellular matrix and is released by heparitinase and heparin-like molecules. Biochemistry. 1989;28:1737–1743. doi: 10.1021/bi00430a047. [DOI] [PubMed] [Google Scholar]
- Basilico C, Moscatelli D. The FGF family of growth factors and oncogenes. Adv Cancer Res. 1992;59:115–165. doi: 10.1016/s0065-230x(08)60305-x. [DOI] [PubMed] [Google Scholar]
- Bikfalvi A, Klein S, Pintucci G, Quarto N, Mignatti P, Rifkin DB. Differential modulation of cell phenotype by different molecular weight forms of basic fibroblast growth factor: possible intracellular signaling by the high molecular weight forms. J Cell Biol. 1995;129:233–243. doi: 10.1083/jcb.129.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikfalvi A, Klein S, Pintucci G, Rifkin DB. Biological roles of fibroblast growth factor-2. Endocr Rev. 1997;18:26–45. doi: 10.1210/edrv.18.1.0292. [DOI] [PubMed] [Google Scholar]
- Borgstrom P, Hillan KJ, Sriramarao P, Ferrara N. Complete inhibition of angiogenesis and growth of microtumors by anti-vascular endothelial growth factor neutralizing antibodies, novel concepts of angiostatic therapy from intravital videomicroscopy. Cancer Res. 1996;56:4032–4039. [PubMed] [Google Scholar]
- Breier G, Damert A, Plate KH, Risau W. Angiogenesis in embryos and ischemic diseases. Thromb Haemost. 1997;78:678–683. [PubMed] [Google Scholar]
- Brogi E, Wu T, Namiki A, Isner JM. Indirect angiogenic cytokines upregulate VEGF and bFGF expression in vascular smooth muscle cells, whereas hypoxia upregulates VEGF expression only. Circulation. 1994;90:649–652. doi: 10.1161/01.cir.90.2.649. [DOI] [PubMed] [Google Scholar]
- Brown LF, Berse B, Jackman RW, Tognazzi K, Manseau EJ, Senger DR, Svorak HF. Expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in adenocarcinomas of the gastrointestinal tract. Cancer Res. 1993;53:4727–4735. [PubMed] [Google Scholar]
- Bugler B, Amalric F, Prats H. Alternative initiation of translation determines cytoplasmic or nuclear localization of basic fibroblast growth factor. Mol Cell Biol. 1991;11:573–577. doi: 10.1128/mcb.11.1.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsestein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;38:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Chen C, Parangi S, Tolentino MJ, Folkman J. A strategy to discover circulating angiogenesis inhibitors generated by human tumors. Cancer Res. 1995;55:4230–4233. [PubMed] [Google Scholar]
- Cordon CC, Vlodavski I, Haimovitz FA, Hicklin D, Fuks Z. Expression of basic fibroblast growth factor in human normal tissues. Lab Invest. 1990;63:832–840. [PubMed] [Google Scholar]
- DiMario J, Buffinger N, Yamada S, Strohman RC. Fibroblast growth factor in the extracellular matrix of dystrophic (mdx) mouse muscle. Science. 1989;244:688–690. doi: 10.1126/science.2717945. [DOI] [PubMed] [Google Scholar]
- Dionne CA, Crumley G, Bellot F, Kaplow JM, Searfoss G, Ruta M, Burgess WH, Jaye M, Schlessinger J. Cloning and expression of two distinct high-affinity receptors cross-reacting with acidic and basic fibroblast growth factor. EMBO (Eur Mol Biol Organ) J. 1990;9:2685–2692. doi: 10.1002/j.1460-2075.1990.tb07454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak HF, Tumors : wounds that do not heal. Similarity between tumor stroma generation and wound healing N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular permeability and angiogenesis. Am J Pathol. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Houck K, Jakeman L, Leung DW. Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr Rev. 1992;13:18–32. doi: 10.1210/edrv-13-1-18. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KL, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- Flaumenhaft R, Moscatelli D, Saksela O, Rifkin DB. Role of extracellular matrix in the action of basic fibroblast growth factor: matrix as a source of growth factor for long-term stimulation of plasminogen activator production and DNA synthesis. J Cell Physiol. 1989;140:75–81. doi: 10.1002/jcp.1041400110. [DOI] [PubMed] [Google Scholar]
- Florkiewicz RZ, Sommer A. Human basic fibroblast growth factor gene encodes four polypeptides: three initiate translation from non-AUG codons. Proc Natl Acad Sci USA. 1989;86:3978–3981. doi: 10.1073/pnas.86.11.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florkiewicz RZ, Baird A, Gonzalez AM. Multiple forms of bFGF: differential nuclear and cell surface localization. Growth Factors. 1991;4:265–275. doi: 10.3109/08977199109043912. [DOI] [PubMed] [Google Scholar]
- Folkman J. What is the evidence that tumors are angiogenesis-dependent? . J Natl Cancer Inst. 1991;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- Folkman J. The role of angiogenesis in tumor growth. Semin Cancer Biology. 1992;3:65–71. [PubMed] [Google Scholar]
- Folkman J, Klagsbrun M. Angiogenic factors. Science. 1987;235:442–447. doi: 10.1126/science.2432664. [DOI] [PubMed] [Google Scholar]
- Folkman J, Haudenschild CC, Zetter BR. Long-term culture of capillary endothelial cells. Proc Natl Acad Sci USA. 1979;76:5217–5221. doi: 10.1073/pnas.76.10.5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J, Klagsbrun M, Sasse J, Wadzinski M, Ingber D, Vlodavski I. A heparin-binding angiogenic protein—basic fibroblast growth factor—is stored within basement membrane. Am J Pathol. 1988;130:393–400. [PMC free article] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL , Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR, Schneck FX, Gagnon ML, Corless C, Soker S, Niknejad K, Peoples GE, Klagsbrun M. Peripheral blood T lymphocytes and lymphocytes infiltrating human cancers express vascular endothelial growth factor: a potential role for T cells in angiogenesis. Cancer Res. 1995;55:4140–4145. [PubMed] [Google Scholar]
- Garrido C, Saule S, Gospodarowicz D. Transcriptional regulation of vascular endothelial growth factor expression in ovarian bovine granulosa cells. Growth Factors. 1993;8:109–117. doi: 10.3109/08977199309046931. [DOI] [PubMed] [Google Scholar]
- Goldberg MA, Schneider TJ. Similarities between the oxygen-sensing mechanisms regulating the expression of vascular endothelial growth factor and erythropoietin. J Biol Chem. 1994;269:4355–4359. [PubMed] [Google Scholar]
- Goldman CK, Kim J, Wong WL, King V, Brock T, Gillespie GY. Epidermal growth factor stimulates vascular endothelial growth factor production by human malignant glioma cells: a model of glioblastoma multiform pathophysiology. Mol Cell Biol. 1993;4:121–133. doi: 10.1091/mbc.4.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- Harada K, Friedman M, Lopez J, Wang S, Li J, Prasad PV, Pearlman JD, Edelman E, Sellke FW, Simons M. Vascular endothelial growth factor in chronic myocardial ischemia. Am J Physiol. 1996;270:H1791–H1802. doi: 10.1152/ajpheart.1996.270.5.H1791. [DOI] [PubMed] [Google Scholar]
- Horak ER, Leek R, Klenk N, LeJeune S, Smith K, Stuart N, Greenall M, Stepniewska K, Harris AL. Angiogenesis, assessed by platelet/ endothelial cell adhesion molecule antibodies, as indicator of node metastases and survival in breast cancer. Lancet. 1992;340:1120–1124. doi: 10.1016/0140-6736(92)93150-l. [DOI] [PubMed] [Google Scholar]
- Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocr. 1991;5:1806–1814. doi: 10.1210/mend-5-12-1806. [DOI] [PubMed] [Google Scholar]
- Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 1992;267:26031–26037. [PubMed] [Google Scholar]
- Isner JM, Pieczek A, Schainfeld R, Blair R, Haley L, Asahara T, Rosenfield K, Razvi S, Walsh K, Symes JF. Clinical evidence of angiogenesis following arterial gene transfer of phVEGF165 in patient with ischaemic limb. Lancet. 1996;348:370–374. doi: 10.1016/s0140-6736(96)03361-2. [DOI] [PubMed] [Google Scholar]
- Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, Connolly DT. Vascular permeability factor, an endothelial cell mitogen related to platelet derived growth factor. Science. 1989;246:1309–1312. doi: 10.1126/science.2479987. [DOI] [PubMed] [Google Scholar]
- Keegan K, Johnson DE, Williams LT, Hayman MJ. Isolation of an additional member of the fibroblast growth factor receptor family, FGFR-3. Proc Natl Acad Sci USA. 1991;88:1095–1099. doi: 10.1073/pnas.88.4.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Winer J, Armanini M, Gillet N, Phillips HS, Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumor growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- Klein S, Giancotti FG, Presta M, Albelda SM, Buck CA, Rifkin DB. Basic fibroblast growth factor modulates integrin expression in microvascular endothelial cells. Mol Biol Cell. 1993;4:973–982. doi: 10.1091/mbc.4.10.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S, Bikfalvi A, Birkenmeier TM, Giancotti FG, Rifkin DB. Integrin regulation by endogenous expression of 18-kDa fibroblast growth factor-2. J Biol Chem. 1996;271:22583–22590. doi: 10.1074/jbc.271.37.22583. [DOI] [PubMed] [Google Scholar]
- Lee PL, Johnson DE, Cousens LS, Fried VA, Williams LT. Purification and complementary DNA cloning of a receptor for basic fibroblast growth factor. Science. 1989;245:57–60. doi: 10.1126/science.2544996. [DOI] [PubMed] [Google Scholar]
- Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem. 1995;270:13333–13340. doi: 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
- Li J, Perrella MA, Tsai JC, Yet SF, Hsieh CM, Yoshizumi M, Patterson C, Endego WO, Zhou F, Lee M. Induction of vascular endothelial growth factor gene expression by interleukin-1 beta in rat aortic smooth muscle cells. J Biol Chem. 1995;270:308–312. doi: 10.1074/jbc.270.1.308. [DOI] [PubMed] [Google Scholar]
- Liu Y, Cox SR, Morita T, Kourembanas S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5′ enhancer. Circ Res. 1995;77:638–643. doi: 10.1161/01.res.77.3.638. [DOI] [PubMed] [Google Scholar]
- Macchiarini P, Fontanini G, Hardin MJ, Squartini F, Angeletti CA. Relation of neovascularization to metastasis of non-small-cell lung cancer. Lancet. 1992;340:145–146. doi: 10.1016/0140-6736(92)93217-b. [DOI] [PubMed] [Google Scholar]
- Maeda K, Chung YS, Ogawa Y, Takatsuka S, Kang SM, Ogawa M, Sawada T, Sowa M. Prognostic value of vascular endothelial growth factor expression in gastric carcinoma. Cancer. 1996;77:858–863. doi: 10.1002/(sici)1097-0142(19960301)77:5<858::aid-cncr8>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Mandriota SJ, Pepper MS. Vascular endothelial growth factor-induced in vitro angiogenesis and plasminogen activator expression are dependent on endogenous basic fibroblast growth factor. J Cell Sci. 1997;110:2293–2302. doi: 10.1242/jcs.110.18.2293. [DOI] [PubMed] [Google Scholar]
- McLaren J, Prentice A, Charnock-Jones DS, Millican SA, Muller KH, Sharkey AM, Smith SK. Vascular endothelial growth factor is produced by peritoneal fluid macrophages in endometriosis and is regulated by ovarian steroids. J Clin Invest. 1996;98:482–489. doi: 10.1172/JCI118815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignatti P, Rifkin DB. Release of basic fibroblast growth factor, an angiogenic factor devoid of secretory signal sequence: a trivial phenomenon or a novel secretion mechanism? . J Cell Biochem. 1991;47:201–207. doi: 10.1002/jcb.240470303. [DOI] [PubMed] [Google Scholar]
- Mignatti P, Morimoto T, Rifkin DB. Basic fibroblast growth factor released by single, isolated cells stimulates their migration in an autocrine manner. Proc Natl Acad Sci USA. 1991;88:11007–11011. doi: 10.1073/pnas.88.24.11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignatti P, Morimoto T, Rifkin DB. Basic fibroblast growth factor, a protein devoid of secretory signal sequence, is released by cells via a pathway independent of the endoplasmic reticulum-Golgi complex. J Cell Physiol. 1992;151:81–93. doi: 10.1002/jcp.1041510113. [DOI] [PubMed] [Google Scholar]
- Millauer B, Shawver LK, Plate KH, Risau W, Ullrich A. Glioblastoma growth is inhibited in vivo by a negative dominant Flk-1 mutant. Nature. 1994;367:576–579. doi: 10.1038/367576a0. [DOI] [PubMed] [Google Scholar]
- Minchenko A, Bauer T, Salceda S, Caro J. Hypoxic stimulation of vascular endothelial growth factor expression in vivo and in vitro. Lab Invest. 1994;71:374–379. [PubMed] [Google Scholar]
- Moscatelli D, Presta M, Rifkin DB. Purification of a factor from human placenta that stimulates endothelial cell protease production, DNA synthesis and migration. Proc Natl Acad Sci USA. 1986a;83:2091–2095. doi: 10.1073/pnas.83.7.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscatelli D, Presta M, Joseph-Silverstein J, Rifkin DB. Both normal and tumor cells produce basic fibroblast growth factor. J Cell Physiol. 1986b;129:273–276. doi: 10.1002/jcp.1041290220. [DOI] [PubMed] [Google Scholar]
- Moscatelli D. High and low affinity binding sites for basic fibroblast growth factor on cultured cells: absence of a role for low affinity binding in the stimulation of plasminogen activator production by bovine capillary endothelial cells. J Cell Physiol. 1987;131:123–130. doi: 10.1002/jcp.1041310118. [DOI] [PubMed] [Google Scholar]
- Moscatelli D, Joseph-Silverstein J, Manejias R, Rifkin DB. M r25,000 heparin-binding protein from guinea pig brain is a high molecular weight form of basic fibroblast growth factor. Proc Natl Acad Sci USA. 1987;84:5778–5782. doi: 10.1073/pnas.84.16.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicosia RF, Nicosia SV, Smith M. Vascular endothelial growth factor, platelet-derived growth factor and insulin-like growth factor-1 promote rat aortic angiogenesis in vitro. Am J Pathol. 1994;145:1023–1029. [PMC free article] [PubMed] [Google Scholar]
- Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci USA. 1998;95:5672–5677. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: deposition into the subepithelial extracellular matrix and bioactivity of ECM-bound VEGF. Mol Biol Cell. 1993;4:1317–1326. doi: 10.1091/mbc.4.12.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen J, Makela TP, Eerola E, Korhonen J, Hirvonen H, Claesson-Welsh L, Alitalo K. FGFR-4, a novel acidic fibroblast growth factor receptor with a distinct expression pattern. EMBO (Eur Mol Biol Organ) J. 1991;10:1347–1354. doi: 10.1002/j.1460-2075.1991.tb07654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman JD, Hibberd MG, Chuang ML, Harada K, Lopez JJ, Gladstone SR, Friedman SR, Sellke FW, Symons M. Magnetic resonance mapping demonstrates benefits of VEGF-induced myocardial angiogenesis. Nature Med. 1995;1:1085–1089. doi: 10.1038/nm1095-1085. [DOI] [PubMed] [Google Scholar]
- Pepper MS, Ferrara N, Orci L, Montesano R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem Biophys Res Comm. 1992;189:824–831. doi: 10.1016/0006-291x(92)92277-5. [DOI] [PubMed] [Google Scholar]
- Pertovaara L, Kaipainen A, Mustonen T, Orpana A, Ferrara N, Saksela O, Alitalo K. Vascular endothelial growth factor is induced in response to transforming growth factor-β in fibroblastic and epithelial cells. J Biol Chem. 1994;269:6271–6274. [PubMed] [Google Scholar]
- Peverali FA, Mandriota SJ, Ciana P, Marelli R, Quax P, Rifkin DB, Della G, Valle, Mignatti P. Tumor cells secrete an angiogenic factor that stimulates basic fibroblast growth factor and urokinase expression in vascular endothelial cells. J Cell Physiol. 1994;161:1–14. doi: 10.1002/jcp.1041610102. [DOI] [PubMed] [Google Scholar]
- Phillips GD, Stone AM, Jones BD, Schultz JC, Whitehead RA, Knighton DR. Vascular endothelial growth factor (rhVEGF165) stimulates angiogenesis in the rabbit cornea. In Vivo. 1995;8:961–965. [PubMed] [Google Scholar]
- Pintucci G, Quarto N, Rifkin DB. Methylation of high molecular weight fibroblast growth factor-2 determines post-translational increases in molecular weight and affects its intracellular distribution. Mol Biol Cell. 1996;7:1249–1258. doi: 10.1091/mbc.7.8.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumor angiogenesis factor in vivo. Nature. 1992;359:845–847. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- Plate KH, Breier G, Weich HA, Mennel HD, Risau W. Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer. 1994;59:520–529. doi: 10.1002/ijc.2910590415. [DOI] [PubMed] [Google Scholar]
- Plouet J, Schilling J, Gospodarowicz D. Isolation and characterization of a newly identified endothelial cell mitogen produced by AtT20 cell. EMBO (Eur Mol Biol Organ) J. 1989;8:8301–8306. doi: 10.1002/j.1460-2075.1989.tb08557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak Z, Cohen T, Sivan R, Kandelis Y, Spira G, Vlodavsky I, Keshet E, Neufeld G. VEGF145, a secreted vascular endothelial growth factor isoform that binds to extracellular matrix. J Biol Chem. 1997;272:7151–7158. doi: 10.1074/jbc.272.11.7151. [DOI] [PubMed] [Google Scholar]
- Prats H, Kaghad M, Prats AC, Klagsbrun M, Lellias JM, Liauzun P, Chalon P, Tauber JP, Amalric F, Smith JA, Caput D. High molecular mass forms of basic fibroblast growth factor are initiated by alternative CUG codons. Proc Natl Acad Sci USA. 1989;86:1836–1840. doi: 10.1073/pnas.86.6.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarto N, Finger FP, Rifkin DB. The NH2-terminal extension of high molecular weight bFGF is a nuclear targeting signal. J Cell Physiol. 1991a;147:311–318. doi: 10.1002/jcp.1041470217. [DOI] [PubMed] [Google Scholar]
- Quarto N, Talarico D, Florkiewicz R, Rifkin DB. Selective expression of high molecular weight basic fibroblast growth factor confers a unique phenotype to NIH 3T3 cells. Cell Regul. 1991b;2:699–708. doi: 10.1091/mbc.2.9.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu-Hong J, Nagy JA, Senger DR, Dvorak HF, Dvorak AM. Ultrastructural localization of VEF/VEGF to the albuminal plasma membrane and vesiculovacuolar organelles of tumor microvascular endothelium. J Histochem Cytochem. 1995;43:381–389. doi: 10.1177/43.4.7534783. [DOI] [PubMed] [Google Scholar]
- Renko M, Quarto N, Morimoto T, Rifkin DB. Nuclear and cytoplasmic localization of different basic fibroblast growth factor species. J Cell Physiol. 1991;144:108–114. doi: 10.1002/jcp.1041440114. [DOI] [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Roberts WG, Palade G. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci. 1995;108:2369–2379. doi: 10.1242/jcs.108.6.2369. [DOI] [PubMed] [Google Scholar]
- Rojeli S, Klagsbrun M, Atzmon R, Kurokawa M, Haimovitz A, Fuks Z, Vlodavski I. Basic fibroblast growth factor is an extracellular matrix component required for supporting the proliferation of vascular endothelial cells. J Cell Biol. 1989;109:823–831. doi: 10.1083/jcb.109.2.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryuto M, Ono M, Izumi H, Yoshida S, Weich HA, Kohno K, Kuwano M. Induction of vascular endothelial growth factor by tumor necrosis factor alpha in human glioma cells. Possible roles of SP-1. J Biol Chem. 1996;271:28220–28228. doi: 10.1074/jbc.271.45.28220. [DOI] [PubMed] [Google Scholar]
- Sato Y, Rifkin DB. Autocrine activities of basic fibroblast growth factor: regulation of endothelial cell movement, plasminogen activator synthesis and DNA synthesis. J Cell Biol. 1988;107:1199–1205. doi: 10.1083/jcb.107.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Shimada T, Takaki R. Autocrinological role of basic fibroblast growth factor on tube formation of vascular endothelial cells in vitro. Biochem Biophys Res Comm. 1991;180:1098–1102. doi: 10.1016/s0006-291x(05)81179-9. [DOI] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Risau W, Vollmer E, Sorg C. In situ detection of basic fibroblast growth factor by highly specific antibodies. Am J Pathol. 1990;137:85–92. [PMC free article] [PubMed] [Google Scholar]
- Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- Senger DR, Van Der Water L, Brown LF, Nagy JA, Dvorak HF. Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Met Rev. 1993;12:303–324. doi: 10.1007/BF00665960. [DOI] [PubMed] [Google Scholar]
- Shima DT, Adamis AP, Ferrara N, Yeo KT, Yeo TK, Allende M, Folkman J, D'Amore P. Hypoxic induction of vascular endothelial cell growth factors in the retina: identification and characterization of vascular endothelial growth factor (VEGF) as the sole mitogen. Mol Med. 1995;2:64–71. [PMC free article] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Sommer A, Brewer MT, Thompson RC, Moscatelli D, Presta M, Rifkin DB. A form of human fibroblast growth factor with an extended amino terminus. Biochem Biophys Res Comm. 1987;144:543–550. doi: 10.1016/s0006-291x(87)80001-3. [DOI] [PubMed] [Google Scholar]
- Stavri GT, Zachary IC, Martin JF, Erusalimsky JD. Basic fibroblast growth factor upregulates the expression of vascular endothelial growth factor in vascular smooth muscle cells: synergistic interaction with hypoxia. Circulation. 1995;92:11–14. doi: 10.1161/01.cir.92.1.11. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Zhung L, Brogi E, Kearney M, Pu L-Q, Bunting S, Ferrara N, Symes JF, Isner JM. Therapeutic angiogenesis: a single intra-arterial bolus of vascular endothelial growth factor augments collateral vessel formation in a rabbit ischemic hind limb model. J Clin Invest. 1994;93:662–670. doi: 10.1172/JCI117018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, Fiddes J, Abraham J. The human gene for vascular endothelial growth factor. J Biol Chem. 1991;266:11947–11954. [PubMed] [Google Scholar]
- Toi M, Hoshima S, Takayanagi T, Tominaga T. Association of vascular endothelial growth factor expression with tumor angiogenesis and with early relapse in primary breast cancer. Jpn J Cancer Res. 1994;85:1045–1049. doi: 10.1111/j.1349-7006.1994.tb02904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JC, Goldman CK, Gillespie Y. Vascular endothelial growth factor in human glioma cell lines: induced secretion by EGF, PDGF-BB and bFGF. J Neurosurg. 1995;82:864–873. doi: 10.3171/jns.1995.82.5.0864. [DOI] [PubMed] [Google Scholar]
- Vartanian RK, Weidner N. Correlation of intratumoral endothelial cell proliferation with microvessel density (tumor angiogenesis) and tumor proliferation in breast carcinoma. Am J Pathol. 1994;144:1188–1194. [PMC free article] [PubMed] [Google Scholar]
- Vlodavski I, Folkman J, Sullivan R, Fridman R, Ishai-Michaeli R, Sasse J, Klagsbrun M. Endothelial cell-derived basic fibroblast growth factor. Synthesis and deposition into subendothelial extracellular matrix. Proc Natl Acad Sci USA. 1987;84:2292–2296. doi: 10.1073/pnas.84.8.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakui S, Furusato M, Itoh T, Sasaki H, Akiyama A, Kinoshita I, Asano K, Tokuda T, Aizawa S, Ushigome S. Tumour angiogenesis in prostatic carcinoma with and without bone marrow metastasis: a morphometric study. J Pathol. 1992;168:257–262. doi: 10.1002/path.1711680303. [DOI] [PubMed] [Google Scholar]
- Warren RS, Yuan H, Matli MR, Gillet NA, Ferrara N. Regulation by vascular endothelial growth factor of human colon cancer tumorigenesis in a mouse model of experimental liver metastasis. J Clin Invest. 1995;95:1789–1797. doi: 10.1172/JCI117857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner N, Semple JP, Welch W, Folkman J. Tumor angiogenesis and metastasis. Correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- Yamagishi S, Yonekura H, Yamamoto Y, Katsuno K, Sato F, Mita I, Ooka H, Satozawa N, Kawakami T, Nomura M, Yamamoto H. Advanced glycation end products-driven angiogenesis in vitro. Induction of the growth and tube formation of human microvascular endothelial cells through autocrine vascular endothelial growth factor. J Biol Chem. 1997;272:8723–8730. doi: 10.1074/jbc.272.13.8723. [DOI] [PubMed] [Google Scholar]
- Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, Luo W, et al. Fibroblast growth factor 2 control of vascular tone. Nature Med. 1998;4:201–207. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]