

Abstract
FGF regulates both cell migration and proliferation by receptor-dependent induction of immediate-early gene expression and tyrosine phosphorylation of intracellular polypeptides. Because little is known about the disparate nature of intracellular signaling pathways, which are able to discriminate between cell migration and proliferation, we used a washout strategy to examine the relationship between immediate-early gene expression and tyrosine phosphorylation with respect to the potential of cells either to migrate or to initiate DNA synthesis in response to FGF-1. We demonstrate that transient exposure to FGF-1 results in a significant decrease in Fos transcript expression and a decrease in tyrosine phosphorylation of the FGFR-1, p42mapk, and p44mapk. Consistent with these biochemical effects, we demonstrate that attenuation in the level of DNA synthesis such that a 1.5-h withdrawal is sufficient to return the population to a state similar to quiescence. In contrast, the level of Myc mRNA, the activity of Src, the tyrosine phosphorylation of cortactin, and the FGF-1–induced redistribution of cortactin and F-actin were unaffected by transient FGF-1 stimulation. These biochemical responses are consistent with an implied uncompromised migratory potential of the cells in response to growth factor withdrawal. These results suggest a correlation between Fos expression and the mitogen-activated protein kinase pathway with initiation of DNA synthesis and a correlation between high levels of Myc mRNA and Src kinase activity with the regulation of cell migration.
Polypeptide growth factors are well-described as initiators of both cell migration and growth in vitro through their ability to activate intracellular signaling pathways. A number of polypeptides have been classified as intracellular signaling molecules; these include phospholipase C-γ (6, 50), the GTPase-activating protein for ras (51), the p85 subunit of phosphatidylinositol-3-kinase (17, 55), Src-like tyrosine kinases (45), STAT-kinases (14, 32), mitogen-activated protein (MAP)1 kinases (61), and the Raf proto-oncoprotein (58). However, the biochemical events during G1 that lead to cytoskeleton alterations and specific-gene expression are not well-defined. Moreover, it is not known what mechanism(s) determine whether a cell will migrate and/or proliferate in response to a growth factor.
FGF-1 and FGF-2 are the prototype members of a large family of related genes that regulate such important biological processes as differentiation, embryogenesis, neurogenesis, and angiogenesis, and are potent inducers of cell migration and DNA synthesis in ectoderm- and mesoderm-derived cell types (7, 22). While FGF-1 lacks a classical signal sequence to direct its export through the conventional endoplasmic reticulum–Golgi pathway, it is released in response to stress (38). FGF-1 release is important since it is the interaction between the mitogen and the FGF receptor (R)-1 on the surface of target cells that is essential to induce intracellular signaling and DNA synthesis (18, 69, 73).
Although the mechanism of FGF-induced signal transduction is not well-defined, FGF causes rapid FGFR dimer formation at the cell surface, resulting in phosphorylation of intracellular polypeptides, including phospholipase C-γ (6, 50), p90/FRS2 (19, 44), MAP kinases (65), and Shc (65) as well as autophosphorylation of the FGF receptor (19, 71). Additionally, FGF-1 induces tyrosine phosphorylation of Src (74) during the entire G1 period, which results in tyrosine phosphorylation of the F-actin– binding protein cortactin (72), a protein originally characterized as the major substrate for v-Src (68). Exogenous FGF-1 also upregulates transcription of immediate-early genes (8), and activates a FGFR-1–mediated pathway resulting in FGF-1 translocation from the cell surface to the nucleus during the entire G1 period (71). FGFR-1 also trafficks to a perinuclear locale during this time, and the first immunoglobulin-like loop in FGFR-1 is responsible for this event (57). Furthermore, removing FGF-1 from Balb/c 3T3 cells during the mid-to-late G1 phase (10 h) results in significant attenuation in the level of DNA synthesis as well as in translocation of exogenous FGF-1 and FGFR-1 from the cell surface to nuclear and perinuclear locales (69, 73).
Because the FGF-1 nuclear translocation events correlate with FGF-1–induced tyrosine phosphorylation during the entire G1 period, and because removing exogenous FGF-1 attenuates DNA synthesis induction, we became interested in the plasticity of FGF-1–induced signaling during this period. We examined the effects of transient FGF-1 exposure on FGF-1–induced tyrosine phosphorylation and immediate-early gene expression as they relate to the ability of cells to transition from G0 to the S phase of the cell cycle, or to the ability of cells to migrate. Removal of FGF-1 is accompanied by dephosphorylation of FGFR-1, p90, p42mapk, and p44mapk, all of which are specifically phosphorylated in response to exogenous FGF-1, and by a significant decrease in Fos transcription. Consistent with reversal of these biochemical responses, transient FGF-1 stimulation fails to result in maximal induction of DNA synthesis. In contrast, transient exposure of the cells to FGF-1 results in continuous activation of Src, tyrosine phosphorylation of cortactin, redistribution of F-actin and cortactin to the cell periphery, and expression of both Myc and ODC throughout the withdrawal period, correlating with an uncompromised migratory potential of the cells. The migratory potential of the cells in response to FGF-1 is severely compromised in the presence of a Myc inhibitor. These results suggest that the constant presence of exogenous FGF-1 is required during the entire G1 period in order to sustain Fos expression and tyrosine phosphorylation of FGFR-1, p42mapk, and p44mapk, and to achieve maximal DNA synthesis, whereas sustained activation of the Src pathway, cytoskeletal reorganization, expression of Myc and ODC, and cell migration only require transient exposure to the growth factor.
Materials and Methods
Cell Culture and DNA Synthesis Assays
Swiss 3T3 cells and Balb/c 3T3 cells (American Type Culture Collection, Rockville, MD) were maintained in DMEM (HyClone Laboratories Inc., Logan, UT) supplemented with 10% (vol/vol) calf serum (HyClone Laboratories Inc.) and antibiotics (GIBCO BRL, Gaithersburg, MD). Cellular quiescence was achieved by exposing confluent populations to a serum-free hormone-defined medium (21) containing 10 μg/ml of insulin (DMI) for 48 h as previously described (71). The cells were stimulated by adding 5–10 ng/ml recombinant human FGF-1 (37) and 10 μg/ml heparin (Sigma Chemical Co., St. Louis, MO).
The transient effects of FGF-1 on Swiss 3T3 cells were determined by incubating quiescent monolayers with FGF-1 for 3 h, removing the cell culture medium, and washing the monolayer three times with DMEM containing 10 μg/ml heparin. The monolayer was then supplemented with DMI alone for 0.5, 1.5, or 3 h, and was further restimulated with 5 ng/ml FGF-1 and 10 μg/ml heparin. The level of DNA synthesis was quantitated in four to six independent samples by incorporating [3H]thymidine (0.5 μCi/ml, 20 Ci/mmol; Amersham Corp., Arlington Heights, IL) as previously described (70) by 20 min of pulse labeling 13, 15, 17, 19, 21, and 23 h after restimulation.
Migration Assays
Migration of Balb/c 3T3 cells upon FGF-1 stimulation was determined using an in vitro model of wound repair as previously described (60). Balb/c 3T3 cells were used in this study since Swiss 3T3 cells exhibited a high level of endogenous migration in the absence of FGF-1. In brief, confluent monolayers of Balb/c 3T3 cells were made quiescent in DMI for 48 h. The monolayers were then scraped with a razor blade, and cellular debris was removed by washing with DMEM containing 10 μg/ml heparin. The monolayers were incubated in DMI containing FGF-1 (10 ng/ml) and heparin (10 μg/ml) for the times indicated. In the case of transient conditions, monolayers were incubated in DMI containing FGF-1 and heparin for 3 h, washed with DMEM containing heparin, and then incubated for 1.5 or 3 h in DMI alone before further supplementation with FGF-1 and heparin. For the delayed conditions, monolayers were incubated in DMI for 4.5 or 6 h, respectively, before FGF-1 stimulation. For the short stimulation, monolayers were stimulated with FGF-1 for 3 or 6 h, washed with DMEM containing heparin, and then incubated in DMI for the duration of the experiment. The migration of the transient, delayed, and short-stimulated populations were compared with the migration of cells treated continuously with FGF-1. In all instances, cell migration was halted 22 h after scraping when the cells were fixed with 25% acetic acid/75% methanol and stained with hematoxylin. The number of cells migrating into the denuded area was determined by counting using a light microscope under 100× magnification with a grid. Each condition was examined in duplicate with five representative fields for each plate being counted.
To measure the effect of the c-Myc inhibitor, FR901228 (64) on FGF-1–induced Balb/c migration, confluent monolayers of cells were made quiescent for 24–48 h, and an area of cells was denuded with a razor blade and was stimulated with 10 ng/ml FGF-1 and 10 μg/ml heparin in the presence of 2.5 ng/ml FR901228 as described above. Monolayers were either constantly stimulated with FGF-1 for 22 h, or were transiently stimulated with FGF-1 for 3 h, washed with DMEM containing heparin, and then incubated in DMI for 3 h followed by restimulation with FGF-1 for the duration of the experiment. Unstimulated cells were used as a control. For comparison, each population was stimulated with FGF-1 in the absence of the inhibitor.
Phosphotyrosine Immunoblot Analysis
SDS-PAGE was performed as previously described (71). Confluent monolayers of Swiss 3T3 cells were used to prepare cell lysates for immunoblot analysis. Cells were washed with cold PBS, scraped in cold PBS containing 1 mM sodium vanadate, and collected by centrifugation. Cell pellets were lysed in 0.5 ml of cold lysis buffer (20 mM Tris, pH 7.5, containing 300 mM sucrose, 60 mM KCl, 15 mM NaCl, 5% (vol/vol) glycerol, 2 mM EDTA, 1% (vol/vol) Triton X-100, 1 mM PMSF, 2 mg/ml aprotinin, 2 mg/ml leupeptin, 0.2% (vol/vol) deoxycholate, and 1 mM sodium vanadate, and the lysate was clarified by centrifugation at 4°C. The cytosol was mixed with an equal volume of SDS-PAGE sample buffer, and was heated at 95°C for 10 min. Equal protein concentrations of cytosol lysate were subjected to 7.5% (wt/vol) SDS-PAGE, transferred to nitrocellulose membranes (19), and blotted with an antiphosphotyrosine monoclonal antibody (Upstate Biotechnology Inc., Lake Placid, NY). Phosphorylated proteins were visualized using a horseradish peroxidase–conjugated rabbit antibody against mouse IgG (Bio-Rad Laboratories, Hercules, CA) and the enhanced chemiluminescence detection system (Amersham Corp., Arlington Heights, IL).
Tyrosine phosphorylation of FGFR-1 was determined by immunoprecipitation with affinity-purified polyclonal rabbit antibodies against FGFR-1 that had been raised against a synthetic peptide whose sequence is at the carboxy terminus of FGFR-1 (20, 71). Tyrosine phosphorylation of p44mapk and/or p42mapk was determined by immunoprecipitation with rabbit polyclonal antibodies (aERK-1 and aERK-2, respectively; Santa Cruz Biotechnology, Santa Cruz, CA). Tyrosine phosphorylation of cortactin was determined by immunoprecipitation with polyclonal rabbit antibodies against cortactin that had been raised against a peptide corresponding to residues 343–362 in murine cortactin (72). Cells were lysed as described above, and cytosolic lysates were rotated at 4°C for 1 h with appropriate antibodies followed by the addition of protein A Sepharose (Pharmacia Biotech, Inc., Piscataway, NJ) and further rotation at 4°C for 1 h. The antibody complexes were washed three times with the lysis buffer, and immunoprecipitated proteins were eluted in 50 ml SDS-PAGE sample buffer (46), resolved by 7.5% (wt/vol) or 9% (wt/vol) SDS-PAGE, and immunoblotted with an antiphosphotyrosine monoclonal antibody as described above.
In Vitro Kinase Assays
MAP kinase activity was determined by immunoprecipitation of lysates with aERK-1 and aERK-2 (Santa Cruz Biotechnology) and immobilization of protein–antibody complexes on protein A-Sepharose. Cells were lysed and immunoprecipitated as described above. After immunoprecipitation, the antibody complexes were washed three times with the lysis buffer and once with kinase buffer (40 mM Hepes, pH 7.4, containing 100 mM β-glycerophosphate, 2 mM EDTA, 10 mM MgCl2, 1 mM DTT, 1 mM Na3VO4), and were subsequently incubated in 40 μl of kinase buffer containing 10 μCi of [γ-32P]ATP and 5 μg mylein basic protein at room temperature for 30 min. The phosphorylated proteins were resolved by 12.5% (wt/vol) SDS-PAGE, transferred to nitrocellulose, and visualized by autoradiography. Subsequently, immunoblot analysis was performed using the ERK-1 antibody (Santa Cruz Biotechnology).
The Src kinase activity was determined by immunoprecipitation with monoclonal Src antibody 327 (52) and goat anti–mouse immunoglobulin G antibody (Pierce Chemical Co., Rockford, IL) immobilized on protein A-Sepharose. Cells were lysed and immunoprecipitated as described above. After immunoprecipitation, the antibody complexes were washed three times with the lysis buffer and once with kinase buffer (30 mM Tris, pH 7.4, containing 10 mM MnCl2), and were subsequently incubated in 50 μl of kinase buffer containing 10 μCi of [γ-32P]ATP and 2 μg acid-denatured enolase (Sigma Chemical Co.) at room temperature for 10 min. The phosphorylated proteins were resolved by 9% (wt/vol) SDS-PAGE, transferred to nitrocellulose, and visualized by autoradiography. Subsequently, immunoblot analysis was performed using the Src-2 antibody (Santa Cruz Biotechnology) as previously described (25).
RNA Extraction and Analysis
Total RNA was isolated by the guanidinium isothyocyanate procedure as previously described (24). The RNA was electrophoresed on an 0.8% (wt/ vol) agarose gel containing 2.2 M formaldehyde, capillary-blotted onto nylon membrane filters (Zeta-Probe; Bio-Rad Laboratories), and hybridized to 32P-labeled DNA probes at 65°C for 20 h. Filters were washed at high stringency and exposed to Kodak XAR films (Eastman Kodak Co., Rochester, NY). The Fos probe was an 1.8-kb EcoRI-SalI fragment from the murine Fos genomic clone containing the second, third, and a portion of the fourth Fos exon (49). The ornithine decarboxylase (ODC) probe was a 1.3-kb PstI fragment from the murine ODC cDNA (American Type Culture Collection; reference 3). The Myc probe was a 1.4-kb SstI fragment from the human cDNA (1).
Nuclear Run-on Assay
Nuclei from confluent monolayers of Swiss 3T3 cells were isolated and frozen in two aliquots at −80°C as previously described (29). Nuclear run-on assays were performed by incubating the thawed nuclei for 30 min at 30°C with 100 μCi [32P]UTP, and nascent RNA transcripts were isolated as previously described (33). Plasmid vector (negative control) and plasmid cDNAs (5 μg of each) were linearized, denatured, and applied to nylon membrane filters (Zeta-Probe; Bio-Rad Laboratories) using a slot blot apparatus and cross-linked by UV irradiation. Filters containing DNA probes were prehybridized for at least 2 h at 65°C, and were hybridized in 3 ml of hybridization buffer (33) containing 3 × 107 cpm of [32P]labeled RNA at 65°C for 3 d. Filters were washed at high stringency, dried, and exposed to x-ray film at −80°C. Densitometric analysis was performed using the Lynx 4000 workstation, and values were normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) value. The GAPDH plasmid contained a 1.3-kb human cDNA.
Fluorescence Microscopy
Swiss 3T3 cells were plated on glass coverslips coated with fibronectin, and were then processed for washout experiments. Cell fixation was performed using 3% paraformaldehyde in PBS with incubation at room temperature for 15 min. For cortactin staining, fixed cell monolayers were washed with PBS and blocked with PBS containing 5% BSA, 0.1% Triton X-100, 0.1% Tween 20, and 0.1% NaN3 for 1 h. The monolayer was washed with PBS three times, incubated with anticortactin antiserum (72), diluted 1:50 in blocking buffer for 1 h, washed three times with PBS, and incubated with CY3-conjugated anti–rabbit IgG antibodies (Sigma Chemical Co.) diluted 1:500 in blocking buffer for 0.5 h. The monolayer was washed with PBS three times and embedded in 50% glycerol with phenylenediamine. For F-actin staining, the monolayer was blocked, incubated with 0.1 μg/ml TRITC-conjugated phalloidin (Aldrich Chemical Co., Milwaukee, WI), and embedded as described above. The cells were photographed with a fluorescence microscope (Olympus Corp., Lake Success, NY) at 200× and 600×.
Results
The Kinetics of FGF-1 Removal and Initiation of DNA Synthesis
Prior studies from our laboratory have demonstrated that the presence of exogenous FGF-1 is required during the entire G1 period to achieve maximal levels of DNA synthesis (71). Indeed, exposure of quiescent Swiss 3T3 cells to exogenous FGF-1 followed by FGF-1 withdrawal and supplementation with DMI (short stimulation) results in induction of a low level of DNA synthesis (Fig. 1). To analyze further the effects of transient exposure of exogenous FGF-1 on Swiss 3T3 cells early in G1, we used heparin to remove FGF-1 from the cell culture environment since prior efforts have demonstrated that heparin is able to reduce nonspecific binding of FGF-1 significantly (71). In these experiments, quiescent Swiss 3T3 cell monolayers were incubated with FGF-1 for 3 h, the cell culture medium was removed, and the monolayer was washed with DMEM containing heparin. The monolayer was then supplemented with DMI alone for either 0.5 or 1.5 h. After incubation in the absence of growth factor, the monolayer was restimulated with FGF-1, and at various time points the level of DNA synthesis was determined (Fig. 1 A). This population was designated the transiently stimulated population. For comparison, at the time of restimulation, quiescent Swiss 3T3 cells were exposed to exogenous FGF-1 and designated the delayed population. As shown in Fig. 1 B, the increase in DNA synthesis in cells that were interrupted in their exposure to FGF-1 for 0.5 h was less than the levels of DNA synthesis achieved in the continuously stimulated cultures. However, this brief interruption in exposure of Swiss 3T3 cells to FGF-1 yielded a higher level of DNA synthesis than that observed in the delayed population. In contrast, increasing the interruption time of exposure to exogenous FGF-1 from 0.5 to 1.5 h resulted in a level of DNA synthesis in the transient population that was similar to that observed in the delayed population (Fig. 1 C).
Figure 1.

Dynamics of DNA synthesis in Swiss 3T3 cells restimulated with FGF-1 after removing FGF-1. (A) Schematic diagrams of the experiments using confluent Swiss 3T3 cells that were made quiescent by incubating for 48 h in a serum-free DMI supplemented with 10 μg/ ml insulin. Quiescent cells were stimulated for 3 h with FGF-1 (5 ng/ml) and heparin (10 μg/ml), culture medium was removed, and the monolayer was washed three times with DMEM containing heparin. The cells were incubated for either 0.5 or 1.5 h in DMI alone, and were restimulated with FGF-1 and heparin (transient stimulation). Three types of controls were also performed: (a) constant stimulation, FGF-1 was not removed; (b) short stimulation, cells were stimulated with FGF-1 for 3 h, washed with heparin, and incubated in DMI without FGF-1; and (c) delayed stimulation, FGF-1 was added to quiescent cells simultaneously with restimulation of the cells that were undergoing the transient stimulation. DNA synthesis was monitored using [3H]thymidine incorporation 13–21 h after restimulation. (B) Restimulation with FGF-1 and heparin after a 0.5-h interruption with a heparin wash. [3H]thymidine incorporation (± SD) as a function of time after restimulation with constant stimulation (▪), short stimulation (□), transient stimulation (♦), and delayed stimulation (⋄) of Swiss 3T3 cell populations. (C) Restimulation with FGF-1 and heparin after a 1.5-h interruption with a heparin wash. Description is identical to that described in B.
We have also observed that at the early time points (Fig. 1 C; 13–17 h), the transiently stimulated population demonstrated a small increase in the level of DNA synthesis when compared with the delayed-stimulation population. This relatively small increase may have resulted from the initial 3-h period of exposure to exogenous FGF-1, since a low level of DNA synthesis was also exhibited by the short-stimulated population that is exposed to exogenous FGF-1 for only 3 h (Fig. 1 C; compare filled diamonds with open squares). Thus, we suggest that the difference in the level of DNA synthesis between the transiently-stimulated and delayed-stimulated populations during the early time points (13–17 h) may not be significant since it is similar to the low levels of [3H]thymidine incorporation exhibited by the short-stimulated population (Fig. 1 C). More importantly, however, the levels of DNA synthesis exhibited at the later time points (Fig. 1 C; 19–23 h) by the transiently and delayed-stimulated populations are similar (Fig. 1 C; compare filled diamonds with open diamonds), suggesting that a 3-h period of exposure to exogenous FGF-1 followed by a withdrawal period of 1.5 h is sufficient to generate a population of cells that is similar to a population that has not been exposed to exogenous FGF-1.
Correlation of Gene Expression with FGF-1 Withdrawal in Swiss 3T3 Cells
Because expression of various cell cycle–specific genes has been correlated with DNA synthesis and cell growth, we examined the effects of FGF-1 withdrawal on expression of the immediate-early response genes Myc and Fos, and the early-to-mid response gene ornithine decarboxylase (ODC). RNA was extracted from Swiss 3T3 cells at quiescence after a 1-, 3-, and 6-h exposure to FGF-1, or from populations that were transiently induced with FGF-1 and subsequently subjected to FGF-1 removal by a heparin wash.
Northern blot analysis demonstrated that the steady-state levels of the Fos, Myc, and ODC transcripts were readily induced by FGF-1 after stimulation for 1, 3, or 6 h (Fig. 2). The withdrawal of FGF-1 for 0.5 h did not significantly change the steady-state level of these transcripts. However, removal of exogenous FGF-1 for 1.5 or 3 h resulted in a dramatic decrease in the steady-state level of the Fos transcript, resulting in similar levels to those detected in quiescent cells (Fig. 2 A). Interestingly, restimulation of the cell population with FGF-1 for 1 h after the 3-h withdrawal period resulted in reinduction of the Fos transcript (Fig. 2 A).
Figure 2.
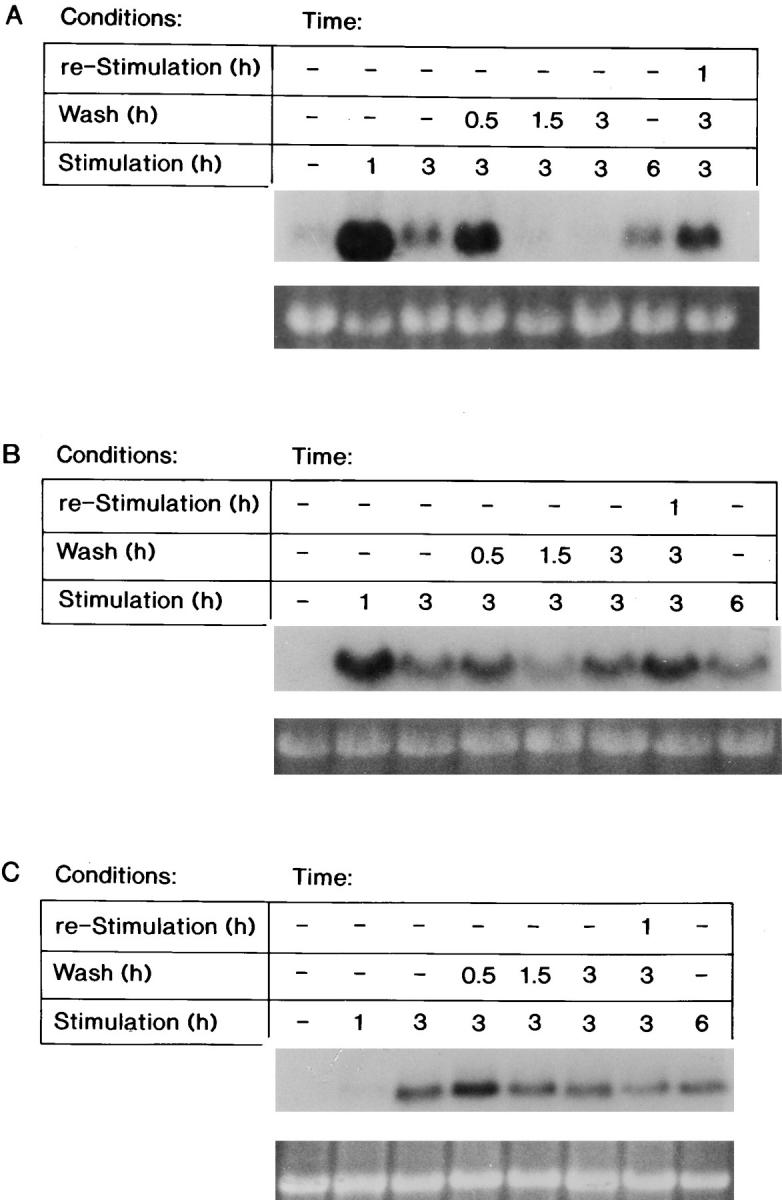
The steady-state mRNA levels of FGF-1–induced genes after FGF-1 removal. RNA was extracted from Swiss 3T3 cells either at quiescence, after a 1-, 3-, or 6-h FGF-1 stimulation, or from populations that were transiently stimulated with FGF-1 (exposed to FGF-1 for 3 h, followed by a heparin wash and supplemented with DMI for 0.5, 1.5, or 3 h, or supplemented with DMI for 3 h and then reexposed to FGF-1 for 1 h). Northern blot analysis was performed as described in experimental procedures. Expression of the (A) Fos transcript (above), of the (B) Myc transcript (above), or of the (C) ODC transcript (above) with corresponding ethidium bromide–stained 18S rRNA (below) is shown.
In contrast, removing FGF-1 for 0.5, 1.5, or 3 h did not significantly decrease the level of either the Myc or ODC transcripts (Fig. 2, B and C). Thus, the decrease in the steady-state level of the Fos transcript after a 1.5-h withdrawal of FGF-1 correlates with a decrease in the level of DNA synthesis (Fig. 1 C), whereas the continual high steady-state levels of Myc and ODC mRNA after a 1.5- or a 3-h withdrawal of FGF-1 are not sufficient to promote progression into the S phase of the cell cycle.
To determine whether downregulation of the Fos transcript in response to FGF-1 removal is regulated at the level of transcriptional initiation, a nuclear run-on analysis was performed. Nuclei were isolated from Swiss 3T3 cells that were exposed to FGF-1 for either 1 or 3 h. An increase in the rate of Fos transcription was observed as compared with the level of Fos transcription from nuclei derived from quiescent cells (Fig. 3). Nuclei isolated from cells that were transiently exposed to FGF-1 for 3 h, followed by a 3-h removal of the mitogen, demonstrated that the level of transcriptional initiation was less than that observed in quiescent cells (Fig. 3). Densitometric measurements determined that a 2.5-fold decrease in Fos transcriptional initiation was observed in cells transiently exposed to FGF-1 as compared with cells that were continuously stimulated with FGF-1 for 6 h. These data suggest that Fos transcript inhibition occurs at least partially at the level of transcriptional initiation. A 1-h restimulation of the cell population transiently exposed to FGF-1 resulted in only a 2.8-fold increase in Fos transcription as compared with quiescent populations, whereas the populations exposed to FGF-1 for 1 h resulted in a 3.4-fold increase. However, the rate of Fos transcription in the 1 h–restimulated populations was similar to the rate of transcription in the nuclei that were continuously exposed to FGF-1 for 6 h. These data argue that the high steady-state level of the Fos transcript after a 1-h restimulation observed by Northern blot analysis (Fig. 2 A) may be due to both transcriptional and posttranscriptional events. In contrast, transient exposure to FGF-1 did not significantly affect ODC transcriptional initiation, where densitometry determined a relative amount of 0.76 for the transiently stimulated population and a relative amount of 0.79 for the 6-h constantly stimulated population.
Figure 3.

Nuclear run-on analysis of transcriptional events after the removal of FGF-1. Nuclei were isolated from quiescent Swiss 3T3 cells that had been stimulated with FGF-1 for 0, 1, 3, and 6 h; had been stimulated with FGF-1 for 3 h, washed with heparin and harvested 3 h after the heparin wash; or had been stimulated with FGF-1 for 3 h, washed with heparin, incubated in DMI without FGF-1 for 3 h, and restimulated with FGF-1 for 1 h. 32P-labeled RNA transcripts (3 × 107 cpm) were hybridized to 5 μg of human Myc, human Fos, murine ODC, and human GAPDH cDNA, and 5 μg of pUC9 DNA that had been linearized and blotted onto nylon membrane filters as described in Materials and Methods.
Interestingly, transcriptional initiation of the Myc transcript did not occur after a 6-h FGF-1 stimulation or after exposure to FGF-1 for 3 h followed by a 3-h withdrawal (Fig. 3). This result suggests that the high level of Myc transcript observed in these populations by Northern blot analysis (Fig. 2 B) was likely due to mRNA stability. Thus, these data suggest that the steady-state level of Fos transcript is sensitive to the continual presence of FGF-1, and that the growth factor is required to activate transcriptional initiation of the Fos transcript. Furthermore, unlike the levels of the Fos transcripts, the steady-state Myc mRNA levels were not affected by transient exposure to FGF-1. Based on the nuclear run-on analysis, this result is most likely due to mRNA stability.
Additionally, to determine the cell cycle progression of the transiently stimulated population, the cells were examined for the presence of cyclin D1, a marker for the cells' progression into the G1 phase of the cell cycle (42, 69). Detectable levels of cyclin D1 were not observed until after the cells were exposed to FGF-1 for 4.5 h. A reduction of cyclin D1 (data not shown) was observed in the population that was stimulated with FGF-1 for 3 h followed by a 3-h withdrawal as compared with the level of cyclin D1 observed after a 6-h FGF-1 stimulation. These results suggest that a 3-h stimulation followed by a 3-h withdrawal period of FGF-1 halts the progression of the cells into the G1 phase of the cell cycle.
The Kinase Activity of MAPK, but Not Src, is Sensitive to FGF-1 Withdrawal
Analysis of Balb/c 3T3 cells has demonstrated FGF-1–dependent tyrosine phosphorylation events throughout the entire G1 transition period (71). To determine the influence of transient exposure to FGF-1 on modification of phosphotyrosine-containing polypeptides, cell lysates were prepared at various times of exposure or removal of FGF-1, and were evaluated by immunoblot analysis. As shown in Fig. 4 A, we observed two distinct patterns of tyrosine phosphorylation. Stimulating cells for 3 or 6 h resulted in the appearance of a 90-kD protein (p90) and the disappearance of a 70-kD protein (p70) as a phosphotyrosyl-containing polypeptide. The cells stimulated for 3 h followed by a 0.5-h withdrawal of FGF-1 did not significantly change the levels of tyrosine phosphorylation, as compared with lysates prepared from cells that were stimulated with FGF-1 for 3 h (Fig. 4 A). However, extending the withdrawal period to 1.5 or 3 h resulted in a dramatic decrease in tyrosine phosphorylation of p90. Moreover, the reappearance of p70 as a phosphotyrosyl-containing polypeptide was also observed (Fig. 4 A). Thus, a 3-h withdrawal of FGF-1 from the cell culture population is sufficient to return the level of tyrosine phosphorylation to a level similar to that observed in unstimulated cells. Furthermore, restimulating the cells with FGF-1 after the 3-h withdrawal results in a pattern of tyrosine phosphorylation similar to that observed in the 3- and 6-h FGF-1–stimulated population.
Figure 4.

(A) Immunoblot analysis of tyrosine phosphorylation in Swiss 3T3 cells after removal of FGF-1. Quiescent Swiss 3T3 cells were exposed to FGF-1 for 0, 3, or 6 h; stimulated with FGF-1 for 3 h, and washed with heparin and harvested 0.5, 1.5, or 3 h after the heparin wash; or were stimulated with FGF-1 for 3 h, washed with heparin, incubated in DMI without FGF-1 for 3 h, and then restimulated with FGF-1 for 1 h. The cells were harvested, and cytosolic lysates were prepared as described in Materials and Methods. Lysates were analyzed by 7.5% (wt/vol) SDS-PAGE, transferred to nitrocellulose membranes, and probed with an antiphosphotyrosine antibody as described (19). The positions of the p90 and p80 bands are indicated by closed arrows, and the p70 band is indicated by an open arrow. (B) Tyrosine phosphorylation of FGFR-1 in response to transient exposure to FGF-1. Quiescent Swiss 3T3 cells were stimulated, washed, and restimulated with FGF-1 in a manner identical to that described above. Cell lysates were immunoprecipitated with a rabbit anti-FGFR-1 polyclonal antibody and subjected to immunoblot analysis (7.5% [wt/vol] SDS-PAGE) with an antiphosphotyrosine antibody as described in Materials and Methods. The position of p130/p145 FGFR-1 polypeptides are indicated by closed arrows. (C) Tyrosine phosphorylation of p44mapk and p42mapk in response to transient exposure to FGF-1. Quiescent Swiss 3T3 cells were stimulated, washed, and restimulated with FGF-1 in a manner identical to that described in Fig. 4 A. Cell lysates were immunoprecipitated with a rabbit anti-ERK-1 polyclonal and rabbit anti-ERK-2 polyclonal antibodies, and were subjected to immunoblot analysis (9% [wt/ vol] SDS-PAGE) with an antiphosphotyrosine antibody as described in Materials and Methods. The positions of p44mapk (ERK-1) and p42mapk (ERK-2) are indicated by closed arrows. (D) In vitro MAP kinase activity in response to transient exposure to FGF-1. Quiescent Swiss 3T3 cells were either stimulated with FGF-1 for 0, 3, 4.5, or 6 h, or were stimulated with FGF-1 for 3 h, washed with heparin, and harvested 1.5 or 3 h after the heparin wash. Cell lysates were immunoprecipitated with anti-Erk-1 and/or anti-Erk-2, and an in vitro kinase assay was performed using MBP as the substrate. The kinase reactions were analyzed by 12.5% (wt/vol) SDS-PAGE, transferred to nitrocellulose membranes, and phosphorylated MBP was visualized by autoradiography. The position of MBP is indicated by a closed arrow. Because the p44mapk band does not resolve away from the antibody band in the immunoblot, we used a different experiment in which a similar trend was observed to quantitate these results: quiescence, 1.0; 3-h stimulation, 14.9; 4.5-h stimulation, 16.4; 3-h stimulation followed by a 1.5-h withdrawal, 3.7; 3-h stimulation followed by a 3-h withdrawal, 2.0; and 6-h stimulation, 24.5. (E) The level of p44mapk (ERK-1) and p42mapk (ERK-2) protein was visualized by immunoblot analysis with p44mapk (ERK-1) polyclonal antibodies. The position of p44mapk and p42mapk is indicated by a closed arrow, and the position of IgG is indicated with an open arrow.
FGF-1 induces tyrosine phosphorylation of FGFR-1 during the entire G1 period (74). To examine the effects of transient exposure of FGF-1 on the levels of the tyrosine phosphorylation of the receptor, immunoprecipitation and immunoblot analysis were used. Quiescent Balb/c 3T3 or Swiss 3T3 cells were either exposed to FGF-1 for 3 and 6 h, or cells were induced with FGF-1 for 3 h, and exogenous FGF-1 was removed with a heparin wash (Fig. 4 B). The level of tyrosine phosphorylation of FGFR-1 is very low in quiescent cells, whereas stimulation of the cells with FGF-1 for either 3 or 6 h resulted in a significant increase in tyrosine phosphorylation of both the p130 and p145 forms of FGFR-1. Removing exogenous FGF-1 for 0.5 h did not influence the level of FGFR-1 tyrosine phosphorylation. However, a 1.5- or a 3-h withdrawal of FGF-1 significantly decreased tyrosine phosphorylation of FGFR-1 to a level that is similar to that observed in quiescent cell lysates. Restimulating the cells for 1 h after the 3-h withdrawal of exogenous FGF-1 induced a prominent tyrosine phosphorylation of FGFR-1 (Fig. 4 B).
Because many of the tyrosine phosphorylated substrates induced by FGF-1 are known, we examined the effect of transient FGF-1 exposure on these substrates. It has been demonstrated that the interaction of the FGFR-1 with FGF-1 leads to activation of the PLC-γ and the Ras pathways; both of these pathways have been shown to contribute to MAP kinase activation (35). MAP kinases are activated rapidly in response to numerous extracellular response systems such as mitogens, neurotransmitters, phorbol esters, and heat shock (56). Persistent activation of MAP kinases throughout the G1 period has been shown to be important for cells to transition into the S phase of the cell cycle (47, 48). To investigate the effects of transient exposure of FGF-1 on activation of p44mapk and p42mapk, we examined the levels of tyrosine phosphorylation by immunoprecipitation and immunoblot analysis (Fig. 4 C). FGF-1 stimulation of cells results in increased levels of tyrosine phosphorylation of both p44mapk and p42mapk after a 3-, 4.5-, and 6-h exposure to FGF-1 (Fig. 4 C). Removing exogenous FGF-1 for 1.5 or 3 h results in a significant decrease in the level of tyrosine phosphorylation of both p44mapk and p42mapk (Fig. 4 C). Consistent with the phosphotyrosyl content of p44mapk and p42mapk, an in vitro kinase assay demonstrated that p44mapk and p42mapk specifically phosphorylated their substrate, MBP, in response to FGF-1 stimulation for 3, 4.5, or 6 h (Fig. 4 D). However, if the cell population was exposed to FGF-1 for 3 h followed by a 1.5- or a 3-h withdrawal period, the MAP kinase activity returns to the levels observed in the quiescent state (Fig. 4 D), while the amount of the p44mapk and p42mapk proteins remain similar (Fig. 4 E). Downregulation of the MAP kinase activity is consistent with downregulation of Fos mRNA in response to growth factor withdrawal, since Fos is a target of the MAP kinase pathway (2), and inhibition of MAP kinase activity results in partial inhibition of Fos (66).
FGF-1 has also been shown to induce tyrosine phosphorylation and activation of Src, which then tyrosine-phosphorylates cortactin (74). Because the Src protein can be phosphorylated on tyrosine residues in an inactive or an active state (13, 54), we used an in vitro kinase assay to determine the effects of transient FGF-1 exposure on Src activity. An in vitro kinase assay in the presence of the Src substrate enolase demonstrates that both the Src protein and enolase are phosphorylated in response to FGF-1 (Fig. 5 A). Surprisingly, the level of Src kinase activity remains the same when the growth factor is removed for 1.5 or 3 h. Densitometric scanning demonstrates that when Src activity, represented by the level of enolase phosphorylation, is normalized for the level of Src protein (Fig. 5 B), Src activity at 4.5 h is 4.0-fold higher than the activity at quiescence, and Src activity after a 3-h stimulation followed by a 1.5-h withdrawal is 3.6-fold higher than at quiescence. Persistent activation of Src, despite withdrawal of growth factor, was observed in both Swiss 3T3 and Balb 3T3 cells (data not shown). Consistent with the Src in vitro kinase data, immunoprecipitation of the Src substrate cortactin and subsequent immunoblot analysis, demonstrates that the level of tyrosine phosphorylation of cortactin is also not sensitive to withdrawal of FGF-1, and increases despite FGF-1 withdrawal (Fig. 5 C).
Figure 5.

(A) In vitro Src kinase activity after the removal of FGF-1. Quiescent Swiss 3T3 cells were either stimulated with FGF-1 for 0, 3, 4.5, or 6 h; or were stimulated with FGF-1 for 3 h, washed with heparin, and harvested 1.5 or 3 h after the heparin wash. Cell lysates were immunoprecipitated with anti-c-Src monoclonal antibody 327, and an in vitro kinase assay was performed using enolase as the substrate. The kinase reactions were analyzed by 9% (wt/vol) SDS-PAGE, transferred to nitrocellulose membranes, and phosphorylated Src and enolase were visualized by autoradiography. The position of Src is indicated by a closed arrow, and the position of enolase is indicated by an open arrow. (B) The level of Src protein was visualized by immunoblot analysis with Src2 polyclonal antibodies. The position of Src is indicated by a closed arrow, and the position of IgG is indicated by an open arrow. (C) Tyrosine phosphorylation of cortactin (p80) in response to transient exposure to FGF-1. Quiescent Balb/c 3T3 cells were stimulated, washed, and restimulated with FGF-1 in a manner identical to that described in Fig. 5 A. Cell lysates were immunoprecipitated with rabbit anticortactin antibody 2719 and subjected to immunoblot analysis (7.5% [wt/vol] SDS-PAGE) with an antiphosphotyrosine antibody as described in Materials and Methods. The position of p80 is indicated by a closed arrow, and the position of IgG is indicated with an open arrow. (D) The level of cortactin protein was visualized by immunoblot analysis with anticortactin polyclonal antibodies (72). The position of cortactin is indicated by a closed arrow, and the position of IgG is indicated by an open arrow.
The FGF-1-Induced Changes of the Actin Cytoskeleton are Maintained After FGF-1 Withdrawal
Src plays a central role in regulating a variety of biological processes that are associated with changes in the cellular architecture. Therefore, we examined the different populations of cells for changes in F-actin and cortactin distribution. We investigated whether the FGF-induced changes in actin redistribution were sensitive to transient FGF-1 stimulation. FGF-1 stimulation for 3 or 6 h results in disappearance of most of the bulky F-actin bundles and redistribution of the F-actin to the cell periphery (compare Fig. 6, A, B, and D). Furthermore, in cells exposed to FGF-1 for 6 h, formation of peripheral polarized stress fibers are readily observed, suggesting that the cells are competent for migration. Since the transiently stimulated population exhibited a similar distribution of F-actin and formation of peripheral polarized stress fibers as observed in the 6-h FGF-1–stimulated population (compare Fig. 6, C and D), we suggest that redistribution of F-actin is insensitive to FGF-1 withdrawal.
Figure 6.
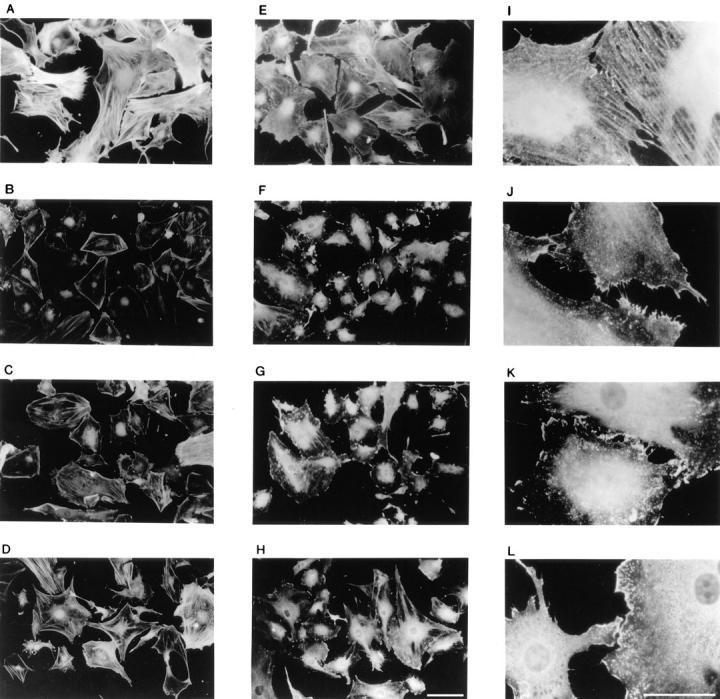
Fluorescence microscopy of actin and cortactin distribution upon differential FGF-1 stimulation. Quiescent and FGF-1–stimulated Swiss 3T3 cell monolayers were processed for fluorescence microscopy as described in Materials and Methods. Quiescent cells (A, E, and I), cells stimulated with FGF-1 for 3 h (B, F, and J), and cells stimulated with FGF-1 for 3 h followed by a 3-h withdrawal period (C, G, and K), and cells stimulated with FGF-1 for 6 h (D, H, and L) were stained with TRITC-phalloidin (A–D) or with anticortactin antibodies (E–L) and photographed with a fluorescence microscope. Bars: (A–H) 20 μm; (I–L) 15 μm.
Cortactin has been demonstrated to be an F-actin–binding protein whose F-actin cross-linking activity is downregulated upon tyrosine phosphorylation (34). Staining quiescent cells with anti-cortactin antibodies revealed that, like F-actin, the protein was distributed throughout the cytoplasm (Fig. 6 E). At a higher magnification, cortactin-positive fibers were readily visible (Fig. 6 I). Exposing the cells to FGF-1 for 3 or 6 h resulted in partial redistribution of cortactin to the cell periphery (Fig. 6, F and H) and disappearance of the cortactin-positive fibers (Fig. 6, J and L). Stimulation with exogenous FGF-1 for 3 h followed by a 3-h withdrawal of the growth factor did not significantly change the peripheral distribution of cortactin (Fig. 6, G and K). Therefore, neither redistribution of F-actin and cortactin nor tyrosine phosphorylation of cortactin require the continual presence of FGF-1.
Withdrawal of FGF-1 Does Not Appear to Result in an Attenuation of Cell Migration
We were surprised to find that Src activation was maintained despite the withdrawal of FGF-1, since Src has been implicated in the regulation of intracellular signaling events that result in proliferation (16, 63). However, activation of Src by dephosphorylating tyrosine 527 results in redistribution of Src from the endosomal membranes to focal adhesion sites (40). In addition, several Src substrates are involved in regulating cellular architecture, and have been implicated in cell attachment and migration (9, 28). Thus, the lack of an effect of transient FGF-1 exposure on Src kinase activity and F-actin and cortactin redistribution prompted us to investigate the migratory potential of the cells by using an in vitro wound repair model (60). In these studies we were unable to use Swiss 3T3 cells since these cells had a high basal level of migration in the absence of FGF-1. Therefore, Balb/c 3T3 cells were used to study the plasticity of FGF-1–induced migration. In this regard, analysis of the phosphotyrosine immunoblots and kinase activity of p44mapk/p42mapk and Src in the Balb/c 3T3 cell population were similar to those obtained with the Swiss 3T3 cell population (data not shown). For the transiently stimulated population, quiescent Balb/c 3T3 cells were wounded and then exposed to FGF-1 for 3 h, the cell culture medium was removed, and the monolayer was washed with DMEM containing heparin. The monolayer was supplemented with DMI alone for either 1.5 or 3 h, and was restimulated with FGF-1 such that the total time from the initial exposure of FGF-1 equaled 22 h. For comparison, cell monolayers were stimulated continuously with FGF-1 for 22 h, and were designated the constantly stimulated population. Furthermore, at the time that FGF-1 was added back into the transiently stimulated populations, cells designated the delayed population were initially exposed to FGF-1. As shown in Fig. 7 A, the constantly stimulated population (22 h) exhibited a marked increase in migration as compared with the population that was not exposed to exogenous FGF-1. The transiently stimulated population demonstrated no significant change in their migratory potential as compared with the constantly-stimulated population. In contrast, the monolayers that were subjected to the delayed stimulation displayed a reduced migratory potential as compared with the transiently stimulated population (Fig. 7 A). These data suggest that the migratory potential of the cell may not be affected by withdrawal of FGF-1 for up to 3 h.
Figure 7.

(A) Migration of Balb/c 3T3 cells upon differential FGF-1 stimulation. Confluent monolayers of Balb/c 3T3 cells were wounded with a razor blade, and then stimulated with FGF-1 as described in Materials and Methods. Unstimulated cells were used as a comparative control (C). The number of cells migrating into the denuded area after a 22-h incubation at 37°C was determined by counting using a microscope with a grid at 100×. Because the total number of migratory cells varied between experiments, these data are the average of two independent experiments, two plates each, and are normalized to the level of total migration in the constantly-stimulated populations. Although the total number of migratory cells varied among experiments, the trends observed for the different populations were indeed consistent. (B) Migration of Balb/c 3T3 cells in the presence or absence of the Myc inhibitor FR901228. Confluent monolayers of quiescent Balb/c 3T3 cells were wounded and stimulated with FGF-1 in the presence of 2.5 ng/ml FR901228 as described in Materials and Methods. The various cell populations were stimulated in the absence of the inhibitor as a control.
The relatively small difference in the migratory potential of the transiently stimulated populations compared with the constantly stimulated populations may be attributed to technical manipulations. If the constantly stimulated and transiently stimulated populations were treated similarly such that the constantly stimulated monolayer is exposed to FGF-1 for 3 h, the cells removed from the incubator, washed with DMEM containing heparin, and then reexposed to FGF-1 for 19 h, we observed a variation in the migration of these cells of 9.25% as compared with the cells constantly stimulated with exogenous FGF-1 for 22 h. Similarly, a variation of 9.20% was observed between migration of the transiently stimulated population and constantly stimulated populations (data not shown), suggesting that these small differences may be due to requisite technical manipulations of the cell culture system. Furthermore, we have also observed by time-lapse photography that migration of the Balb/c 3T3 cell population begins approximately after a 6–8-h stimulation with FGF-1 (data not shown). Consistent with these results, cells in the constant and the transiently stimulated populations started their migration after an 8-h time period (data not shown). Since this in vitro wound repair model may be sensitive to the manipulations required to analyze the migratory phenotype, we suggest that the migration data in combination with the biochemical data correlating the insensitive nature of Src kinase activity and F-actin and cortactin redistribution to FGF-1 withdrawal imply that the migratory potential of the these cells is not sensitive to FGF-1 withdrawal.
Recently, Boyer et al. demonstrated that Src kinases play a role in epithelial cell dispersion in response to EGF that is independent of the Ras pathway and expression of Jun, Fos, Slug, and Myc (5). In contrast, in PDGF-stimulated cells, the target of the Src pathway is Myc (2). Because withdrawal of FGF-1 did not affect either the Src kinase activity or the high steady-state levels of Myc mRNA, we sought to determine whether Myc expression influences the migratory potential of these cells. FR901228 is a fungal metabolite that is used as an antitumor antibiotic (64). The ability of FR901228 to reverse the transformed morphology of Ha-ras-transfected Ras-1 cells can be directly correlated with specific inhibition of the presence of Myc mRNA. FR901228 treatment results in a decrease in Myc protein levels, and neither affects the steady-state levels of Ha-ras, β-actin, or Grb-2 mRNA (64 and Yufang Shi, personal communication) nor macromolecular biosynthesis (64). Thus, quiescent Balb/c 3T3 cells were treated with the Myc inhibitor FR901228 (64) in the presence of FGF-1. Interestingly, we observed a marked decrease in the migratory potential of the constant, transient, and delayed cell populations (Fig. 7 B). The Myc inhibitor also severely decreased the proliferative response of the cells to FGF-1 (data not shown). Thus, the function of Myc appears to be critical for FGF-1 to induce cell migration in vitro.
Discussion
Earlier studies have demonstrated that withdrawal of FGF-1 (71), PDGF, or EGF (67) during the prereplicative period of the cell cycle results in attenuation in the level of DNA synthesis. In this study we have extended this observation by studying the biochemical events following growth factor withdrawal from the cell culture system during the G1 period. We also demonstrated that removal of FGF-1 is sufficient to reverse tyrosine phosphorylation of FGFR-1 and the MAP kinase pathway, and that these biochemical events correlate with an attenuation of FGF-1–induced DNA synthesis. In contrast, transient exposure to exogenous FGF-1 results in continual activation of the FGF-1–induced Src pathway, including tyrosine phosphorylation of cortactin and changes in the cytoskeletal architecture that correlate with an inability of growth factor withdrawal to affect significantly the migratory potential of the cell. The tyrosine kinase receptor–mediated Ras-MAP kinase pathway, which includes activation of the MAP kinases and expression of several nuclear proteins including Fos (27) and Myc (30), has been shown to be essential for cell proliferation. Recently, however, Barone et al. demonstrated that in response to PDGF, the target of the Src pathway is Myc, and the target of the Ras/MAP kinase pathway is Fos in Balb/c 3T3 cells (2). Our results are consistent with these observations, and suggest that in response to FGF-1, Src may lead to high steady-state levels of Myc mRNA. Furthermore, while the MAP kinase pathway may play some role in Myc expression, dephosphorylation of p44mapk and p42mapk in response to FGF-1 withdrawal can be directly correlated with downregulation of Fos expression. In contrast, Boyer et al. demonstrated that activation of Src is required for EGF-induced dispersion of epithelial cells, and that this scattering is independent of the Ras-MAP kinase pathway and immediate– early gene activation, including Myc (5). Additionally, mutation of the Src phosphorylation site in the PDGF receptor-β (Tyr934) results in an increase in PDGF-induced migration and a decrease in the stimulation of DNA synthesis in porcine aortic endothelial cells (31). Although these results appear to contradict the results presented here, it may be possible that these data reflect differences in receptor tyrosine kinase signaling in different cell types, as well as by different growth factors. Indeed, while FGF-1 is able to induce tyrosine phosphorylation of Src and cortactin and increase the steady-state levels of the Fos transcript in presenescent populations of human diploid umbilical vein endothelial cells (25), it is unable to induce phosphorylation of Src and cortactin in senescent populations of human endothelial cells and presenescent and senescent populations of human diploid IMR-90 fibroblasts. However, the levels of Fos mRNA are increased in all cases (26). These data further suggest that FGF-1–induced expression of Fos is not dependent on the Src pathway. Furthermore, while nonproliferative senescent populations of human umbilical vein endothelial cells respond to FGF-1 by inducing both immediate-early (Fos) and early (ODC) transcripts, FGF-1 is unable to signal FGFR-1–mediated tyrosine phosphorylation of Src and cell migration in senescent populations of human endothelial cells (25). In addition, intracellular distribution of Src may also play a role in the ability of Src to regulate cell migration since Src can enhance fibroblast spreading derived from Src homozygous null mice in a manner independent of its intrinsic tyrosine kinase activity, but dependent upon its ability to associate with focal adhesion plaques (41). Since Src is known to associate with the focal adhesion kinase (FAK; 12, 15), and embryonic cells derived from FAK-deficient mice demonstrate an impaired migratory potential (36), it is also possible that the Tyr934 PDGF receptor-β mutant may alter intracellular distribution of Src by an alternative pathway involving PDGF-dependent association of Src and FAK at adhesion sites. However, our data do support a bifurcation of the FGF-1 signal transduction pathway in which disparate intracellular signaling and transcriptional events possibly regulate migratory and mitogenic cell fate. Indeed, the rapid return of Fos expression, but not Myc or ODC, to quiescent levels after FGF-1 withdrawal is consistent with the observation that Myc is the target of the Src pathway, and that Fos is the target of the Ras/MAPK pathway (2) since (a) Src activity, (b) cortactin tyrosine phosphorylation, (c) F-actin and cortactin redistribution, and (d) Myc transcript levels are independent of FGF-1 withdrawal. Furthermore, inhibition of Myc expression severely compromises the cells' migratory potential. Although we do not know the role of ODC expression, both Src (53) and Myc (4, 23, 43) have been demonstrated to be required for migration in other cell types. Recently, Huang et al. demonstrated that Src-induced tyrosine phosphorylation of cortactin results in a decreased ability of cortactin to cross-link actin (34), which may enhance the migratory potential of the cells.
The requirement of extracellular FGF-1 during the G1 phase of the cell cycle for initiation of maximal DNA synthesis may reflect a physiologic safeguard that allows cells to discriminate between a sustained proliferative stimulus and an occasional short-term contact with FGF-1, and thus prevent inopportune proliferation that may lead to hyperplasia and possibly tissue dysfunction. In contrast, sustained proliferative stimulation afforded by FGF-1 may be provided in areas of wound repair and/or inflammation where local conditions such as hypoxia, hyperthermia, and acidosis may result in the long-term presence of extracellular FGF-1, and thus ensure that compensatory cell proliferation is provided in these areas (59). Indeed, the release of FGF-1 as a signal peptide-less mitogen is very tightly regulated (39, 62), and requires extended time periods for accumulation of significant levels of FGF-1 as a functional extracellular protein (39). Therefore, the requirement that cells be stimulated continually with FGF-1 throughout the prereplicative period may be an additional mechanism used to regulate the biological potency of this mitogen.
The FGF prototypes are involved in regulating biologic events in which cell migration is an important component, such as mesoderm formation, angiogenesis, neointima formation, and neurotrophic repair (7, 22). Since many of these biologic responses involve both cell migration and proliferation, it is possible that intermittent exposure of cells to extracellular FGF-1 may be used as a mechanism solely to modify biologic activities associated with cell migration. Interestingly, FGF-1 is a potent inducer of neurite outgrowth and neuronal repair (10). Since the mechanism of neurite formation is not dependent upon cell proliferation and uses Src as an intracellular regulatory agent (11), it is possible that in nonneuronal cells, short-term exposure to FGF-1 may play a role in lamellipodia formation. In confluent populations of cells, this possibility may either lead to reorganization of the cytoskeleton without the initiation of cell migration, or in the case of the endothelial cell, the initiation of sprout formation (75).
Acknowledgments
The authors thank D. Weber and P. Foote for expert secretarial assistance. We are grateful to Drs. Hirotsugu Ueda and Yufang Shi for providing us with the Myc inhibitor FR901228. We also thank Drs. Susan Garfinkel and Bob Friesel for critical reading of the manuscript.
This work was supported in part by National Institutes of Health grants HL32348 and AG7450 to T. Maciag. I.A. Prudovsky was on sabbatical leave from the Engelhardt Institute of Molecular Biology, Moscow, Russia, and T.M. LaVallee was supported by National Institutes of Health Postdoctoral Fellow Training Grant T32 HL07698.
Abbreviations used in this paper
- DMI
defined medium with insulin
- FAK
focal adhesion kinase
- FGFR
FGF receptor
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- MAP
mitogen-activated protein
- MAPK
mitogen-activated protein kinase
- ODC
ornithine decarboxylase
Footnotes
T.M. LaVallee and I.A. Prudovsky contributed equally to the content of this manuscript. The present address of T.M. LaVallee is EntreMed, Inc., 9610 Medical Center Drive, Rockville, MD 20850.
Address all correspondence to Thomas Maciag, Center for Molecular Medicine, Maine Medical Center Research Institute, 125 John Roberts Road, S. Portland, ME 04106. Tel.: 207-761-9783; FAX: 207-828-9071.
References
- 1.Alitalo K, Schwab M, Lin CC, Varmus HE, Bishop JM. Homogeneously staining chromosomal regions contain amplified copies of an abundantly expressed cellular oncogene (cmyc) in malignant neuroendocrine cells from a human colon carcinoma. Proc Natl Acad Sci USA. 1983;80:1707–1711. doi: 10.1073/pnas.80.6.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barone MV, Courtneidge SA. Myc but not Fos rescue of PDGF signaling block caused by kinase-inactive Src. Nature. 1995;378:509–512. doi: 10.1038/378509a0. [DOI] [PubMed] [Google Scholar]
- 3.Berger FG, Gross KW, Watson G. Isolation and characterization of a DNA sequence complementary to an androgen-inducible messenger RNA from mouse kidney. J Biol Chem. 1981;256:7006–7013. [PubMed] [Google Scholar]
- 4.Biro S, Fu YM, Yu ZX, Epstein SE. Inhibitory effects of antisense oligodeoxynucleotides targeting cmyc mRNA on smooth muscle cell proliferation and migration. Proc Natl Acad Sci USA. 1993;90:654–658. doi: 10.1073/pnas.90.2.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyer B, Roche S, Denoyelle M, Thiery JP. Src and Ras are involved in separate pathways in epithelial cell scattering. EMBO (Eur Mol Biol Organ) J. 1997;16:5904–5913. doi: 10.1093/emboj/16.19.5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burgess WH, Dionne CA, Kaplow J, Mudd R, Friesel R, Zilberstein A, Schlessinger J, Jaye M. Characterization and cDNA cloning of phospholipase C-gamma, a major substrate for heparin-binding growth factor 1 (acidic fibroblast growth factor) activated tyrosine kinase. Mol Cell Biol. 1990;10:4770–4777. doi: 10.1128/mcb.10.9.4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgess WH, Maciag T. The heparin-binding (fibroblast) growth factor family of proteins. Annu Rev Biochem. 1989;58:575–606. doi: 10.1146/annurev.bi.58.070189.003043. [DOI] [PubMed] [Google Scholar]
- 8.Burgess WH, Shaheen AM, Ravera M, Jaye M, Donohue PJ, Winkles JA. Possible dissociation of the heparin-binding and mitogenic activities of heparin-binding (acidic fibroblast) growth factor1 from its receptor-binding activities by site-directed mutagenesis of a single lysine residue. J Cell Biol. 1990;111:2129–2138. doi: 10.1083/jcb.111.5.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cary LA, Chang JF, Guan J. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J Cell Sci. 1996;109:1787–1794. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- 10.Cheng H, Cao Y, Olson L. Spinal cord repair in adult paraplegic rats: Partial restoration of hind limb function. Science. 1996;273:510–513. doi: 10.1126/science.273.5274.510. [DOI] [PubMed] [Google Scholar]
- 11.Clark EA, Brugge JS. Integrins and signal transduction pathways: The road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 12.Cobb BS, Schaller MD, Leu TH, Parsons JT. Stable association of pp60src and pp59fyn with the focal adhesion–associated protein tyrosine kinase. Mol Cell Biol. 1994;14:147–155. doi: 10.1128/mcb.14.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper JA, Howell B. The when and how of src regulation. Cell. 1993;73:1051–1054. doi: 10.1016/0092-8674(93)90634-3. [DOI] [PubMed] [Google Scholar]
- 14.Darnell JE, Jr, Kerr IM, Stark GR. JakSTAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 15.Eide BL, Turck CW, Escobedo JA. Identification of Tyr319 as the primary site of tyrosine phosphorylation and pp60src association in the focal adhesion kinase, pp125FAK. Mol Cell Biol. 1995;15:2819–2827. doi: 10.1128/mcb.15.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erpel T, Courtneidge SA. Src family protein tyrosine kinases and cellular signal transduction pathways. Curr Opin Cell Biol. 1995;7:176–182. doi: 10.1016/0955-0674(95)80025-5. [DOI] [PubMed] [Google Scholar]
- 17.Escobedo JA, Navankasattusas S, Kavanaugh WM, Milfay D, Fried VA, Williams LT. cDNA cloning of a novel 85kd protein that has SH2 domains and regulates binding of PI3kinase to the PDGF beta receptor. Cell. 1991;65:75–82. doi: 10.1016/0092-8674(91)90409-r. [DOI] [PubMed] [Google Scholar]
- 18.Forough R, Zhan X, MacPhee M, Friedman S, Engleka KA, Sayers T, Wiltrout RH, Maciag T. Differential transforming abilities of nonsecreted and secreted forms of human fibroblast growth factor1. J Biol Chem. 1993;268:2960–2968. [PubMed] [Google Scholar]
- 19.Friesel R, Burgess WH, Maciag T. Heparin-binding growth factor 1 stimulates tyrosine phosphorylation in NIH 3T3 cells. Mol Cell Biol. 1989;9:1857–1865. doi: 10.1128/mcb.9.5.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friesel R, Dawid IB. cDNA cloning and developmental expression of fibroblast growth factor receptors from Xenopus laevis. . Mol Cell Biol. 1991;11:2481–2488. doi: 10.1128/mcb.11.5.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friesel R, Maciag T. Internalization and degradation of heparin binding growth factorI by endothelial cells. Biochem Biophys Res Commun. 1988;151:957–964. doi: 10.1016/s0006-291x(88)80459-5. [DOI] [PubMed] [Google Scholar]
- 22.Friesel RE, Maciag T. Molecular mechanisms of angiogenesis: Fibroblast growth factor signal transduction. FASEB J. 1995;9:919–925. doi: 10.1096/fasebj.9.10.7542215. [DOI] [PubMed] [Google Scholar]
- 23.Fukuyama J, Miyazawa K, Hamano S, Ujiie A. Inhibitory effects of tranilast on proliferation, migration, and collagen synthesis of human vascular smooth muscle cells. Can J Physiol Pharmacol. 1996;74:80–84. [PubMed] [Google Scholar]
- 24.Garfinkel S, Haines DS, Brown S, Wessendorf J, Gillespie DH, Maciag T. Interleukin1α mediates an alternative pathway for the antiproliferative action of poly(I.C) on human endothelial cells. J Biol Chem. 1992;267:24375–24378. [PubMed] [Google Scholar]
- 25.Garfinkel S, Hu X, Prudovsky IA, McMahon GA, Kapnik EM, McDowell SD, Maciag T. FGF1-dependent proliferative and migratory responses are impaired in senescent HUVEC, and correlate with the inability to signal tyrosine phosphorylation of FGFR1 substrates. J Cell Biol. 1996;134:783–791. doi: 10.1083/jcb.134.3.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garfinkel S, Wessendorf JHM, Hu X, Maciag T. The human diploid fibroblast senescence pathway is independent of interleukin-1α mRNA levels and tyrosine phosphorylation of FGFR-1 substrates. Biochim Biophys Acta. 1996;1314:109–119. doi: 10.1016/s0167-4889(96)00105-x. [DOI] [PubMed] [Google Scholar]
- 27.Gille H, Sharrocks AD, Shaw PE. Phosphorylation of transcription factor p62(TCF) by MAP kinase stimulates ternary complex formation at cfos promoter. Nature. 1992;358:414–417. doi: 10.1038/358414a0. [DOI] [PubMed] [Google Scholar]
- 28.Gilmore AP, Romer LH. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol Biol Cell. 1996;7:1209–1224. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenberg ME, Ziff EB. Stimulation of 3T3 cells induces transcription of the cfos protooncogene. Nature. 1984;311:433–438. doi: 10.1038/311433a0. [DOI] [PubMed] [Google Scholar]
- 30.Gupta S, Davis RJ. MAP kinase binds to the NH2terminal activation domain of cMyc. FEBS Lett. 1994;353:281–285. doi: 10.1016/0014-5793(94)01052-8. [DOI] [PubMed] [Google Scholar]
- 31.Hansen K, Johnell M, Siegbahn A, Rorsman C, Engstrom U, Wernstedt C, Heldin CH, Ronnstrand L. Mutation of a Src phosphorylation site in the PDGF β receptor leads to increased PDGF-stimulated chemotaxis but decreased mitogenesis. EMBO (Eur Mol Biol Organ) J. 1996;15:5299–5313. [PMC free article] [PubMed] [Google Scholar]
- 32.Heim MH, Kerr IM, Stark GR, Darnell JE., Jr Contribution of STAT SH2 groups to specific interferon signaling by the JakSTAT pathway. Science. 1995;267:1347–1349. doi: 10.1126/science.7871432. [DOI] [PubMed] [Google Scholar]
- 33.Celano P, Berchtold C, Gasero RA. A simplification of the nuclear runoff transcription assay. Biotechniques. 1989;7:942–944. [PubMed] [Google Scholar]
- 34.Huang C, Ni Y, Wang T, Gao Y, Haudenschild CC, Zhan X. Downregulation of the filamentous actin cross-linking activity of cortactin by Src-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:13911–13915. doi: 10.1074/jbc.272.21.13911. [DOI] [PubMed] [Google Scholar]
- 35.Huang J, Mohammadi M, Rodrigues GA, Schlessinger J. Reduced activation of RAF1 and MAP kinase by a fibroblast growth factor receptor mutant deficient in stimulation of phosphatidylinositol hydrolysis. J Biol Chem. 1995;270:5065–5072. doi: 10.1074/jbc.270.10.5065. [DOI] [PubMed] [Google Scholar]
- 36.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Aizawa S. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAKdeficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 37.Imamura T, Engleka K, Zhan X, Tokita Y, Forough R, Roeder D, Jackson A, Maier JAM, Hla T, Maciag T. Recovery of mitogenic activity of a growth factor mutant with a nuclear translocation sequence. Science. 1990;249:1567–1570. doi: 10.1126/science.1699274. [DOI] [PubMed] [Google Scholar]
- 38.Jackson A, Friedman S, Zhan X, Engleka KA, Forough R, Maciag T. Heat shock induces the release of fibroblast growth factor1 from NIH3T3 cells. Proc Natl Acad Sci USA. 1992;89:10691–10695. doi: 10.1073/pnas.89.22.10691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jackson A, Tarantini F, Gamble S, Friedman S, Maciag T. The release of fibroblast growth factor1 from NIH 3T3 cells in response to temperature involves the function of cysteine residues. J Biol Chem. 1995;270:33–36. doi: 10.1074/jbc.270.1.33. [DOI] [PubMed] [Google Scholar]
- 40.Kaplan KB, Bibbins KB, Swedlow JR, Arnaud M, Morgan DO, Varmus HE. Association of the aminoterminal half of csrc with focal adhesions alters their properties and is regulated by phosphorylation of tyrosine 527. EMBO (Eur Mol Biol Organ) J. 1994;13:4745–4756. doi: 10.1002/j.1460-2075.1994.tb06800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaplan KB, Swedlow JR, Morgan DO, Varmus HE. CSrc enhances the spreading of Src/fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev. 1995;9:1505–1517. doi: 10.1101/gad.9.12.1505. [DOI] [PubMed] [Google Scholar]
- 42.Kato JY, Matsuoka M, Strom DK, Sherr CJ. Regulation of cyclin D–dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol Biol Cell. 1994;14:2713–2721. doi: 10.1128/mcb.14.4.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koster R, Blatt LM, Streubert M, Zietz C, Hermeking H, Brysch W, Sturzl M. Consensus interferon and plateletderived growth factor adversely regulate proliferation and migration of kaposi's sarcoma cells by control of cmyc expression. Am J Pathol. 1996;149:1871–1885. [PMC free article] [PubMed] [Google Scholar]
- 44.Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. A lipid-anchored Grb2-binding protein that links fgf receptor activation to the ras/MAPK signaling pathway. Cell. 1997;89:693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- 45.Kypta RM, Goldberg Y, Ulug ET, Courtneidge SA. Association between the PDGF receptor and members of the SRC family of tyrosine kinases. Cell. 1990;62:481–492. doi: 10.1016/0092-8674(90)90013-5. [DOI] [PubMed] [Google Scholar]
- 46.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 47.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 48.Meloche S, Seuwen K, Pages G, Pouyssegur J. Biphasic and synergistic activation of p44mapk (ERK1) by growth factors: correlation between late phase activation and mitogenicity. Mol Endocrinol. 1992;6:845–854. doi: 10.1210/mend.6.5.1603090. [DOI] [PubMed] [Google Scholar]
- 49.Miller AD, Curran T, Verma IM. Cfos protein can induce cellular transformation: A novel mechanism of activation of a cellular oncogene. Cell. 1984;36:51–60. doi: 10.1016/0092-8674(84)90073-4. [DOI] [PubMed] [Google Scholar]
- 50.Mohammadi M, Honegger AM, Rotin D, Fischer R, Bellot F, Li W, Dionne CA, Jaye M, Rubinstein M, Schlessinger J. A tyrosine-phosphorylated carboxy terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase Cgamma 1. Mol Cell Biol. 1991;11:5068–5078. doi: 10.1128/mcb.11.10.5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Molloy CJ, Bottaro DP, Fleming TP, Marshall MS, Gibbs JB, Aaronson SA. PDGF induction of tyrosine phosphorylation of GTPase activating protein. Nature. 1989;342:711–714. doi: 10.1038/342711a0. [DOI] [PubMed] [Google Scholar]
- 52.Mukhopadhyay D, Tsiokas L, Zhou XM, Foster D, Brugge JS, Sukhatme VP. Hypoxic induction of human vascular endothelial growth factor expression through Src activation. Nature. 1995;375:577–581. doi: 10.1038/375577a0. [DOI] [PubMed] [Google Scholar]
- 53.Mureebe L, Nelson PR, Yamamura S, Lawitts J, Kent KC. Activation of pp60csrc is necessary for human vascular smooth muscle cell migration. Surgery. 1997;122:138–144. doi: 10.1016/s0039-6060(97)90002-7. [DOI] [PubMed] [Google Scholar]
- 54.Nada S, Okada M, Macauley A, Cooper JA, Nakagawa H. Cloning of a complementary DNA for a protein tyrosine kinase that specifically phosphorylates a negative regulatory site of P60CSRC. Nature. 1991;351:69–72. doi: 10.1038/351069a0. [DOI] [PubMed] [Google Scholar]
- 55.Otsu M, Hiles I, Gout I, Fry MJ, Ruizlarrea F, Panayotou G, Thompson A, Dhand R, Hsuan J, Totty N, et al. Characterization of Two 85kd proteins that associate with receptor tyrosine kinases, middleT/pp60csrc complexes, and PI3 kinase. Cell. 1991;65:91–104. doi: 10.1016/0092-8674(91)90411-q. [DOI] [PubMed] [Google Scholar]
- 56.Pelech SL, Sanghera JS. Mitogen-activated protein kinases versatile transducers for cell signaling. Trends Biochem Sci. 1992;17:233–238. doi: 10.1016/s0968-0004(00)80005-5. [DOI] [PubMed] [Google Scholar]
- 57.Prudovsky I, Savion N, LaVallee TM, Maciag T. The nuclear trafficking of extracellular FGF1 correlates with the perinuclear association of the FGFR1α isoforms but not the FGFR1β isoforms. J Biol Chem. 1996;271:14198–14205. doi: 10.1074/jbc.271.24.14198. [DOI] [PubMed] [Google Scholar]
- 58.Rapp UR. Role of Raf1 serine/threonine protein kinase in growth factor signal transduction. Oncogene. 1991;6:495–500. [PubMed] [Google Scholar]
- 59.Sano H, Forough R, Maier JA, Case JP, Jackson A, Engleka K, Maciag T, Wilder RL. Detection of high levels of heparin binding growth factor1 (acidic fibroblast growth factor) in inflammatory arthritic joints. J Cell Biol. 1990;110:1417–1426. doi: 10.1083/jcb.110.4.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sato Y, Rifkin DB. Autocrine activities of basic fibroblast growth factor: regulation of endothelial cell movement, plasminogen activator synthesis, and DNA synthesis. J Cell Biol. 1988;107:1199–1205. doi: 10.1083/jcb.107.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sturgill TW, Wu J. Recent progress in characterization protein kinase cascades for phosphorylation of ribosomal protein S6. Biochim Biophys Acta. 1991;1092:350–357. doi: 10.1016/s0167-4889(97)90012-4. [DOI] [PubMed] [Google Scholar]
- 62.Tarantini F, Gamble S, Jackson A, Maciag T. The cysteine residue responsible for the release of fibroblast growth factor-1 resides in a domain independent of the domain for phosphatidylserine binding. J Biol Chem. 1995;270:29039–29042. doi: 10.1074/jbc.270.49.29039. [DOI] [PubMed] [Google Scholar]
- 63.Taylor SJ, Shalloway D. Src and the control of cell division. Bioessays. 1996;18:9–11. doi: 10.1002/bies.950180105. [DOI] [PubMed] [Google Scholar]
- 64.Ueda H, Nakajima H, Hori Y, Goto T, Okuhara M. Action of FR901228, a novel antitumor bicyclic depsipeptide produced by chromobacterium violaceum no. 968, on Haras transformed NIH3T3 cells. Biosci Biotech Biochem. 1994;58:1579–1583. doi: 10.1271/bbb.58.1579. [DOI] [PubMed] [Google Scholar]
- 65.Wang JK, Gao G, Goldfarb M. Fibroblast growth factor receptors have different signaling and mitogenic potentials. Mol Cell Biol. 1994;14:181–188. doi: 10.1128/mcb.14.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Z, Templeton DM. Induction of c-fos proto-oncogene in mesangial cells by cadium. J Biol Chem. 1998;273:73–79. doi: 10.1074/jbc.273.1.73. [DOI] [PubMed] [Google Scholar]
- 67.Westermark B, Heldin C-H. Similar action of platelet-derived growth factor and epidermal growth factor in the prereplicative phase of human fibroblasts suggests a common intracellular pathway. J Cell Physiol. 1985;124:43–48. doi: 10.1002/jcp.1041240108. [DOI] [PubMed] [Google Scholar]
- 68.Wu H, Parsons JT. Cortactin, an 80/85 kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J Cell Biol. 1993;120:1417–1426. doi: 10.1083/jcb.120.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiong Y, Connolly T, Futcher B, Beach D. Human Dtype cyclin. Cell. 1991;65:691–699. doi: 10.1016/0092-8674(91)90100-d. [DOI] [PubMed] [Google Scholar]
- 70.Zhan X, Goldfarb M. Growth factor requirements of oncogene transformed NIH 3T3 and BALB/c 3T3 cells cultured in defined media. Mol Cell Biol. 1986;6:3541–3544. doi: 10.1128/mcb.6.10.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhan X, Hu X, Friesel R, Maciag T. Long term growth factor exposure and differential tyrosine phosphorylation are required for DNA synthesis in BALB/c 3T3 cells. J Biol Chem. 1993;268:9611–9620. [PubMed] [Google Scholar]
- 72.Zhan X, Hu X, Hampton B, Burgess WH, Friesel R, Maciag T. Murine cortactin is phosphorylated in response to fibroblast growth factor1 on tyrosine residues late in the G1 phase of the BALB/c 3T3 cell cycle. J Biol Chem. 1993;268:24427–24431. [PubMed] [Google Scholar]
- 73.Zhan X, Hu XG, Friedman S, Maciag T. Analysis of endogenous and exogenous nuclear translocation of fibroblast growth factor1 in NIH 3T3 cells. Biochem Biophys Res Commun. 1992;188:982–991. doi: 10.1016/0006-291x(92)91328-n. [DOI] [PubMed] [Google Scholar]
- 74.Zhan X, Plourde C, Hu X, Friesel R, Maciag T. Association of fibroblast growth factor receptor1 with cSrc correlates with association between cSrc and cortactin. J Biol Chem. 1994;269:20221–20224. [PubMed] [Google Scholar]
- 75.Zimrin AB, Pepper MS, McMahon GA, Nguyen F, Montesano R, Maciag T. An antisense oligonucleotide to the Notch ligand Jagged enhances fibroblast growth factorinduced angiogenesis in vitro. J Biol Chem. 1996;271:32499–32502. doi: 10.1074/jbc.271.51.32499. [DOI] [PubMed] [Google Scholar]