

Abstract
Integrin αIIbβ3 mediates platelet aggregation and “outside-in” signaling. It is regulated by changes in receptor conformation and affinity and/or by lateral diffusion and receptor clustering. To document the relative contributions of conformation and clustering to αIIbβ3 function, αIIb was fused at its cytoplasmic tail to one or two FKBP12 repeats (FKBP). These modified αIIb subunits were expressed with β3 in CHO cells, and the heterodimers could be clustered into morphologically detectable oligomers upon addition of AP1510, a membrane-permeable, bivalent FKBP ligand. Integrin clustering by AP1510 caused binding of fibrinogen and a multivalent (but not monovalent) fibrinogen-mimetic antibody. However, ligand binding due to clustering was only 25–50% of that observed when αIIbβ3 affinity was increased by an activating antibody or an activating mutation. The effects of integrin clustering and affinity modulation were additive, and clustering promoted irreversible ligand binding. Clustering of αIIbβ3 also promoted cell adhesion to fibrinogen or von Willebrand factor, but not as effectively as affinity modulation. However, clustering was sufficient to trigger fibrinogen-independent tyrosine phosphorylation of pp72Syk and fibrinogen-dependent phosphorylation of pp125FAK, even in non-adherent cells. Thus, receptor clustering and affinity modulation play complementary roles in αIIbβ3 function. Affinity modulation is the predominant regulator of ligand binding and cell adhesion, but clustering increases these responses further and triggers protein tyrosine phosphorylation, even in the absence of affinity modulation. Both affinity modulation and clustering may be needed for optimal function of αIIbβ3 in platelets.
Integrins are type I transmembrane αβ heterodimers that mediate cell adhesion and signaling in a highly regulated manner (Clark and Brugge, 1995). Several modes of integrin regulation have been demonstrated or postulated, including control of expression on the cell surface by coordinate subunit biosynthesis and recycling (Bennett, 1990; Bretscher, 1992), modulation of receptor affinity by conformational changes in the heterodimer (Sims et al., 1991; Shattil et al., 1998), and modulation of receptor avidity by lateral diffusion of heterodimers to form higher order multimers or clusters (Detmers et al., 1987; van Kooyk et al., 1994; Kucik et al., 1996; Bazzoni and Hemler, 1998). The latter process may be promoted by interactions of integrins with multivalent, extracellular ligands (Peerschke, 1995b ; Simmons et al., 1997), and with components of the dynamic actin cytoskeleton (Sastry and Horwitz, 1993; Fox et al., 1996; Kucik et al., 1996). Integrin function must sometimes be regulated acutely over seconds to minutes to enable the kinds of rapid changes in cell adhesion and migration that are required during immune responses, inflammation, and hemostasis. Several integrins in blood cells are targets of this type of activation or “inside-out” signaling, including α4β1, αLβ2, and αMβ2 in leukocytes, and αIIbβ3 and αVβ3 in platelets (Bennett et al., 1997; Bazzoni and Hemler, 1998; Shattil et al., 1998). It seems likely that some combination of conformational change and receptor clustering is involved in activating the ligand-binding function of these integrins. However, evidence to support one or the other mechanism has been largely indirect, and it has been difficult to determine the relative contributions of each in intact cells. The distinction between integrin affinity and avidity modulation is not academic because the underlying mechanisms may be different, with implications for therapeutic strategies to block or promote integrin functions in pathological conditions (Coller, 1997; Bazzoni and Hemler, 1998).
One of the best-studied integrins from the standpoint of acute regulation is αIIbβ3, which interacts with Arg-Gly-Asp–containing ligands, such as fibrinogen and von Willebrand factor (vWf),1 to effect platelet aggregation and spreading on vascular surfaces. Platelet agonists, such as thrombin and ADP, cause rapid changes in the adhesive function of αIIbβ3, as evidenced by increases in the binding of soluble fibrinogen, vWf, and ligand-mimetic mAbs, including PAC1 (Shattil et al., 1985). Antagonists, such as prostacyclin and nitric oxide can inhibit and, under some conditions reverse these acute changes (Graber and Hawiger, 1982; Freedman et al., 1997). Coupled with evidence from fluorescence resonance energy transfer studies showing that the αIIb and β3 subunits undergo changes in relative orientation during platelet activation (Sims et al., 1991), a current hypothesis is that ligand binding to αIIbβ3 is controlled, at least in part, by changes in heterodimer conformation that affect access of the ligand to recognition sites in the receptor (Loftus and Liddington, 1997; Shattil et al., 1998). On the other hand, it is entirely possible that clustering of αIIbβ3 also occurs during the process of platelet activation. Indeed, clustering of αIIbβ3 on the platelet surface has been detected by electron microscopy after ligand binding (Isenberg et al., 1987; Simmons et al., 1997). Were clustering to occur directly in response to platelet agonists, it could enhance ligand binding through chelate and rebinding effects. Furthermore, “outside-in” signaling through αIIbβ3, manifested by activation of specific protein tyrosine kinases, lipid kinases, and cytoskeletal reorganization (Fox et al., 1993; Banfic et al., 1998; Shattil et al., 1998), seems to require the binding of multivalent ligands (Huang et al., 1993), indirectly suggesting a functional role for oligomerization of αIIbβ3.
Since platelets are not amenable to genetic manipulations ex vivo, heterologous expression systems have been used to study the structure and function of αIIbβ3 (O'Toole et al., 1994; Loh et al., 1996). For example, human αIIbβ3 expressed in CHO cells binds little or no fibrinogen or PAC1 and is therefore, in a constitutively low affinity/ avidity state, just as it is in resting platelets. However, the affinity of αIIbβ3 can be increased by incubation of the cells with particular “LIBS” mAb Fab fragments that bind to the α or β integrin subunit and induce a conformational change in the extracellular portion of the receptor to expose ligand binding sites (O'Toole et al., 1994). Under these experimental conditions, monovalent LIBS Fab fragments by themselves would not be expected to induce receptor clustering. In the present study, we have used new modifications of this experimental system to establish the separate contributions of affinity modulation and receptor clustering to the functions of αIIbβ3. Specifically, single or tandem repeats of the FK506-binding protein, FKBP12 (FKBP), have been fused to the cytoplasmic tail of αIIb to conditionally cluster heterodimers into oligomers from inside the cell using AP1510, a synthetic, bivalent, and membrane-permeable FKBP dimerizer (Amara et al., 1997). The results establish that affinity modulation and receptor clustering can play complementary roles in the adhesive and signaling functions of this prototypic integrin. Whereas affinity modulation is the predominant mechanism for regulating ligand binding to αIIbβ3, receptor clustering facilitates this process and promotes outside-in signaling, even in the absence of affinity modulation.
Materials and Methods
Plasmid Constructions and Expression of Recombinant Forms of αIIbβ3 in CHO Cells
A pCDM8 template containing full-length αIIb (O'Toole et al., 1994) was subjected to PCR with Pfu polymerase (Stratagene, La Jolla, CA) to place XbaI and SpeI restriction sites at the 5′ and 3′ ends of αIIb, respectively. The PCR product was cut with XbaI and SpeI and ligated into an XbaI-cut, CMV-based mammalian expression vector, pCF1E (ARIAD Pharmaceutical, Inc., Cambridge, MA). Plasmids with inserts in the correct orientation were amplified and purified for CHO cell transfections (Maxi-Prep; QIAGEN Inc., Chatsworth, CA). The resulting αIIb(FKBP)/pCF1E plasmid encoded αIIb fused in-frame to FKBP, which in turn was fused in-frame to a hemagglutinin epitope tag (see Fig. 1). To construct αIIb fused to two tandem FKBP repeats (αIIb(FKBP)2), a single FKBP was removed from pCF1E with XbaI/SpeI and ligated into SpeI-cut αIIb(FKBP)/pCF1E. The remaining αIIb and β3 cDNAs depicted in Fig. 1 were in pCDM8 (O'Toole et al., 1994). cDNA coding full-length human Syk was in EMCV (Gao et al., 1997). Plasmid inserts were analyzed by automated sequencing to confirm authenticity.
Figure 1.

Integrin constructs used in this study. The vertical bar represents the cell membrane. Integrin extracellular domains are to the left of the bar and intracellular domains to the right. The relative sizes of the various domains are not drawn to scale. For example, the cytoplasmic tail of αIIb contains 20 amino acid residues and a single FKBP repeat contains ∼100 residues. The asterisk in β3(S752P) marks the site of the point mutation.
cDNAs were transfected into CHO-K1 cells with lipofectamine according to the manufacturer's instructions (GIBCO BRL, Gaithersburg, MD). Typically, 0.5–2 μg of each plasmid was used, supplemented when necessary with empty vector DNA (pCDNA3; Invitrogen, San Diego, CA) for a total of 4 μg per dish. Cells were maintained for 48 h for transient expression or subjected to antibiotic selection for stable expression. Stable cell lines were selected further for high integrin expression by single cell FACS® sorting using an αIIbβ3-specific murine mAb, D57 (O'Toole et al., 1994).
Characterization of Recombinant Integrins in CHO Cells
Cell surface expression of recombinant αIIbβ3 was assessed by flow cytometry using biotin-D57 and FITC-streptavidin (Leong et al., 1995). αIIb expression was also evaluated by Western blotting. 48 h after transfection, the cells were lysed in 66 mM Tris-HCl, pH 7.4, containing 2% SDS and 30 μg of protein were electrophoresed in SDS–7.5% polyacrylamide gels under nonreducing conditions, transferred to nitrocellulose, and then subjected to Western blotting with murine mAb B1B5 specific for αIIb or antibody 12CA5 specific for the hemagglutinin epitope tag (Abrams et al., 1992). After addition of affinity-purified, HRP-conjugated goat anti– mouse IgG (Biosource International, Camarillo, CA), blots were developed for 0.1–1 min by enhanced chemiluminescence (ECL; Amersham, Arlington Heights, IL).
Confocal Microscopy
To establish whether AP1510 could induce clustering of αIIb(FKBP)2β3 that was detectable morphologically, cells stably expressing αIIb(FKBP)2β3 were incubated in the presence of 1,000 nM AP1510 (or 0.5% EtOH as a vehicle control) for 30 min at room temperature. Then, analogous to the method used by Yauch and co-workers to detect antibody-induced integrin clustering (Yauch et al., 1997), the cells were incubated with 10% goat serum for 30 min at room temperature, followed by 10 μM FITC-D57 or unlabeled D57 for 30 min on ice. After washing, the sample containing unlabeled D57 was incubated for 30 min with FITC-labeled goat anti–mouse heavy and light chains (1:500; Biosource International) to deliberately cluster the integrin as a positive control. All samples were fixed in 4% paraformaldehyde, resuspended in mounting medium (Fluorosave; Calbiochem-Novabiochem, San Diego, CA), and analyzed on glass slides with an MRC 1024 laser scanning confocal imaging system (Bio-Rad Laboratories, Hercules, CA).
Measurements of Ligand Binding Due to Clustering and Affinity Modulation of αIIbβ3
Ligand binding to αIIbβ3 in CHO cells was assessed by flow cytometry using a saturating amount of the fibrinogen-mimetic, murine monoclonal IgMκ antibody, PAC1 (Kashiwagi et al., 1997). CHO cells were resuspended to 107 cells/ml in Tyrode's buffer supplemented with 2 mM CaCl2 and MgCl2 (O'Toole et al., 1994). For most experiments, 4 × 105 cells were added to tubes containing a final concentration of 0.4% PAC1 ascites or 40 nM purified PAC1 in a final vol of 50 μl, and then incubated for 30 min at room temperature. In some experiments, monovalent recombinant PAC1 Fab produced in insect cells and purified by nickel-agarose chromatography was used instead of PAC1 IgM at a final concentration of 30 nM (Abrams et al., 1994). As indicated for each experiment, cell incubations were also carried out in the presence of one or more of the following reagents: 10–5,000 nM AP1510 (or vehicle buffer) to cluster αIIb(FKBP)2β3 or αIIb(FKBP)β3 (Amara et al., 1997), 150 μg/ml anti-LIBS6 Fab to convert αIIbβ3 into a high affinity form through conformational changes (Du et al., 1993; Kashiwagi et al., 1997), and 10 μM integrilin, an αIIbβ3 antagonist to specifically block PAC1 binding (Scarborough et al., 1993). Preliminary experiments with AP1510 and anti–LIBS6 Fab indicated that a 10– 30-min incubation of cells with these reagents was sufficient to achieve their maximum effects. Cells were then washed and incubated on ice for 30 min with biotin-D57, followed by phycoerythrin-streptavidin and either FITC-labeled goat anti–mouse μ heavy chain antibody (to label PAC1 IgM) or FITC-labeled goat anti–mouse heavy and light chain antibody (to label PAC1 Fab) (both from Biosource International). Samples were diluted with 0.5 ml Tyrode's buffer containing 2 μg/ml propidium iodide (Sigma Chemical Co., St. Louis, MO) and analyzed on a FACSCalibur® flow cytometer (Becton Dickinson Co., Mountain View, CA). After electronic compensation, PAC1 binding (FL1 channel) was analyzed on the gated subset of live cells (propidium iodide–negative, FL3) that was positive for αIIbβ3 expression (FL2). To control for variations in integrin expression from transfection to transfection, PAC1 binding, measured as mean fluorescence intensity in arbitrary units, was expressed relative to the levels of αIIbβ3, measured simultaneously with biotin-D57.
Adhesion of CHO Cells to Fibrinogen and vWF
Immulon-2 microtiter wells (Dynex Laboratories, Chantilly, VA) were coated with purified fibrinogen (Enzyme Research Laboratories, South Bend, IN) or vWf (Ruggeri et al., 1983) overnight at 4°C at coating concentrations ranging from 0.01–2 μg/well, followed by blocking with 20 mg/ml BSA. CHO cells stably expressing αIIb(FKBP)2β3 were labeled for 30 min at 37°C with 2 μM BCECF-AM (Molecular Probes, Eugene, OR). After washing, the cells were resuspended to 106/ml, incubated for 30 min in the presence of 1,000 nM AP1510 and/or 150 μg/ml anti–LIBS6 Fab, and then 100-μl aliquots were added to the coated microtitre wells for 90 min at 37°C. After washing three times with 150 μl of PBS, cell adhesion was quantitated by cytofluorimetry (Leng et al., 1998).
Protein Tyrosine Phosphorylation in CHO Cells
Stable cell lines expressing αIIb(FKBP)2β3 were transiently transfected with EMCV-Syk and placed into complete DME with 10% FBS. 24 h after transfection, the amount of serum was reduced to 0.5%, and 48 h after transfection, the cells were resuspended to 3 × 106/ml in DME and slowly rotated at 37°C for 45 min in the presence of 20 μM cycloheximide. Then cells were incubated for 10 min with one or more of the following: 1,000 nM AP1510 to stimulate receptor clustering, 150 μg/ml anti–LIBS6 Fab to increase integrin affinity, or 250 μg/ml fibrinogen to achieve ligand binding. As a positive tyrosine phosphorylation control, one aliquot of cells was allowed to attach for 60 min to a dish coated with αIIbβ3 antibody D57. Cells were lysed in RIPA buffer containing 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 158 mM NaCl, 10 mM Tris, pH 7.4, 1 mM Na2EGTA, 0.5 mM leupeptin, 0.25 mg/ml Pefabloc, 5 μg/ml aprotinin, 20 mM NaF, 3 mM β-glycerophosphate, 10 mM sodium pyrophosphate, and 5 mM sodium vanadate. After clarification, 200 μg of protein were immunoprecipitated with rabbit antiserum specific for Syk or FAK (Gao et al., 1997). Immunoprecipitates were subjected to Western blotting with anti-phosphotyrosine mAbs, 4G10, and PY20 (Upstate Biotechnology Inc., Lake Placid, NY and Transduction Laboratories, Lexington, KY, respectively), followed by stripping and reprobing with mAb 4D10 to Syk or antiserum to FAK (Gao et al., 1997). Immunoreactive bands detected by ECL were quantitated by calibrated densitometry using a flatbed scanner, Power Center Pro 240 computer, and NIH Image software.
Results
Heterologous Expression of αIIbβ3 Containing Dimerization Motifs
The purpose of these studies was to evaluate the possible contributions of integrin clustering and affinity modulation to the adhesive and signaling functions of αIIbβ3. A CHO cell model system, previously used to study factors that influence the ligand-binding affinity of human αIIbβ3 (O'Toole et al., 1994; Hughes et al., 1996; Zhang et al., 1996; Kashiwagi et al., 1997), has now been adapted to study integrin clustering. Full-length αIIb was engineered to contain one or two FKBP repeats and a hemagglutinin epitope tag at the extreme COOH terminus of the cytoplasmic tail (Fig. 1). In theory, a protein containing a single FKBP may dimerize upon addition of a membrane-permeable, bivalent FKBP ligand, such as AP1510, and a protein with two or more FKBP repeats may form larger oligomers (Amara et al., 1997; Yap et al., 1997; Yang et al., 1998). Consequently, we reasoned that if αIIb(FKBP) or αIIb(FKBP)2 were successfully coexpressed on the surface of CHO cells along with β3, then αIIbβ3 heterodimers might be converted into a dimer of dimers ((αIIbβ3)2) or into even larger oligomers in response to AP1510.
After transient or stable expression in CHO cells, both αIIb(FKBP)β3 and αIIb(FKBP)2β3 were found to be expressed to the same extent as wild-type αIIbβ3, as determined by the binding of D57, an αIIbβ3-specific antibody (Fig. 2 A). In addition, Western blotting of the cell lysates with an antibody specific for the extracellular domain of αIIb (B1B5) showed that αIIb(FKBP) and αIIb(FKBP)2 exhibited the predicted slower electrophoretic mobilities compared with the smaller wild-type αIIb subunit (Fig. 2 B). The αIIb(FKBP) and αIIb(FKBP)2 fusion proteins also reacted on Western blots with an antibody to the COOH-terminal epitope tag, further suggesting that they represented full-length proteins (Fig. 2 B). Thus, the fusion of one or two FKBP repeats to the cytoplasmic tail of αIIb does not interfere with the transient or stable expression of this subunit in CHO cells to form an αIIbβ3 complex. Therefore, in the following studies transient and stable cell lines were used as indicated, depending on the experimental protocol.
Figure 2.
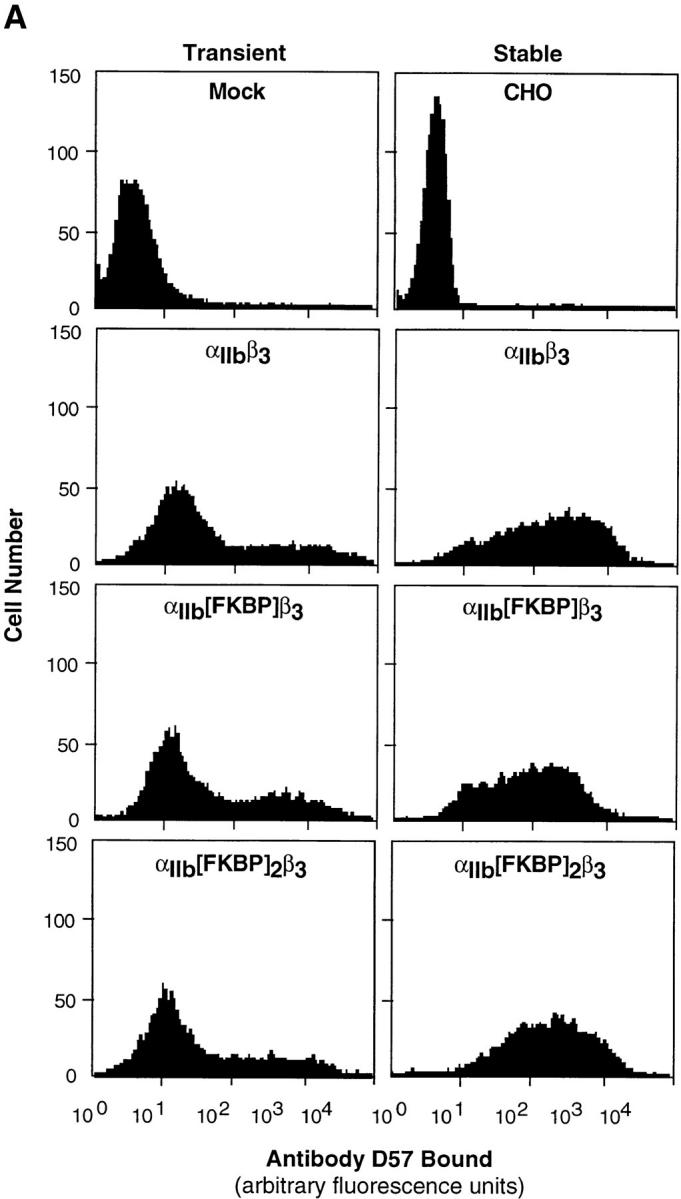

Expression of αIIb(FKBP) fusion constructs with β3 in CHO cells. In A, cells were transiently or stably transfected with the indicated integrin subunits and stained with a combination of the anti-αIIbβ3 mAb, biotin-D57, and FITC-streptavidin for assessment of integrin surface expression by flow cytometry. One sample of cells was mock transfected to serve as a negative control in the transient transfections, and untransfected CHO cells served as a negative control for the stable transfectants. In B, cells were transiently transfected with the indicated integrin constructs. 48 h later, the cells were lysed with SDS sample buffer and 30 μg of protein were subjected to Western blotting with an mAb to αIIb (B1B5) or antibody 12CA5 to the hemagglutinin epitope tag located at the COOH terminus of αIIb(FKBP) and αIIb(FKBP)2. Brackets indicate the region on each blot where the different forms of αIIb are located. The very light band seen in the CHO cell lane on the B1B5 blot was neither consistent nor specific. In this particular experiment, the level of expression of αIIb(FKBP)2β3 was less than that of αIIb(FKBP)β3, accounting for the difference in band intensities between the two samples.
Conditional Clustering of αIIbβ3 in CHO Cells
Large integrin oligomers might be detectable in CHO cells at the level of light microscopy (van Kooyk et al., 1994; Yauch et al., 1997). To determine if clusters of αIIb(FKBP)2β3 could be detected morphologically, CHO cells stably expressing †this integrin were incubated for 30 min at room temperature with 1,000 nM AP1510 (or vehicle buffer as a control), stained with FITC-D57 on ice, fixed, and then examined by confocal microscopy. D57 staining was entirely surface associated, and cells that had been treated with buffer instead of AP1510 exhibited a finely patchy distribution of αIIb(FKBP)2β3 (Fig. 3, A–C). In contrast, αIIb(FKBP)2β3 in cells treated with AP1510 exhibited a coarse patchiness (Fig. 3, E–G), similar to that observed when the D57 antibody was deliberately cross-linked with a secondary antibody before cell fixation (Fig. 3 H). The same results with AP1510 were obtained with another independent αIIb(FKBP)2β3 clone; in contrast, AP1510 caused no discernible clustering of wild-type αIIbβ3 in CHO cells (not shown). These results are consistent with the conclusion that oligomerization of αIIb(FKBP)2β3 can be induced conditionally from within the cell using AP1510, enabling us to conduct a detailed study of the functional consequences of integrin clustering.
Figure 3.

Distribution of αIIb(FKBP)2β3 in CHO cells. CHO cells stably expressing αIIb(FKBP)2β3 were incubated in suspension for 30 min in the absence (A–D, and H) or presence (E–G) of 1,000 nM AP1510. They were then incubated at 4°C with FITC-D57 to stain the integrin (A–C, and E–G), fixed, and then deposited onto glass slides for confocal microscopy. As a negative control, cells in D were incubated only with FITC goat anti–mouse immunoglobulin. As a positive control, cells in H were incubated with unlabeled D57, followed by FITC goat anti–mouse immunoglobulin to deliberately cross-link the integrin before fixation. Panels represent single images collected from the entire series of 0.5-μm focal planes. Images are from a single experiment representative of four so performed. Bar, 10 μm.
Receptor Clustering in the Regulation of Ligand Binding to αIIbβ3
Activation of αIIbβ3 is required for the binding of soluble, macromolecular Arg-Gly-Asp–containing ligands, such as fibrinogen, vWf, and fibrinogen-mimetic antibodies, such as PAC1. To evaluate the contribution of clustering to αIIbβ3 activation, flow cytometry was used to quantitate the specific binding of PAC1 to transiently transfected CHO cells. Specific binding was defined as that inhibitable by 10 μM integrilin, an αIIbβ3-selective antagonist, and it was expressed relative to the amount of αIIbβ3 on the cell surface, determined simultaneously with antibody D57. In cells expressing αIIb(FKBP)2β3, there was little binding of PAC1, indicating that, like αIIbβ3, this integrin is in a constitutive low affinity/avidity state. AP1510 caused a dose-dependent increase in PAC1 binding to αIIb(FKBP)2β3 cells (Fig. 4, closed circles), without affecting the levels of surface expression of this integrin. However, PAC1 binding due to AP1510 appeared modest compared with binding in response to upregulation of integrin affinity by an activating antibody Fab fragment, anti–LIBS6 Fab (Fig. 4, open circles).
Figure 4.

Effect of the dimerizer, AP1510, and the activating antibody, anti–LIBS6 Fab, on PAC1 binding to CHO cells expressing αIIb(FKBP)2β3. Cells were transfected with αIIb(FKBP)2 and β3. After 48 h, they were incubated for 30 min with PAC1 along with AP1510 and/or 150 μg/ml anti-LIBS6. Then PAC1 binding to αIIb(FKBP)2β3-expressing cells was quantitated by flow cytometry as described in Materials and Methods. Specific PAC1 binding was defined as binding inhibitable by 10 μM integrilin, a selective αIIbβ3 antagonist. It was expressed relative to the amount of integrin per cell determined simultaneously with antibody D57.
To evaluate possible mechanistic differences between integrin clustering and affinity modulation in the control of ligand binding, additional experiments were performed with cells expressing αIIb(FKBP)2β3. First, we considered the possibility that AP1510 caused PAC1 binding by increasing integrin affinity rather than (or in addition to) avidity. However, AP1510 failed to stimulate the binding of a monovalent PAC1 Fab fragment to αIIb(FKBP)2β3, although this form of PAC1 bound normally in response to anti–LIBS6 Fab (Fig. 5). Since a monovalent ligand might be expected to be sensitive to affinity modulation but less sensitive than a multivalent ligand to avidity modulation, this result suggests that AP1510 was indeed working by clustering the integrin. Second, PAC1 binding in response to AP1510 was completely prevented if the cells were preincubated for 30 min with 4 mg/ml of 2-deoxy-d-glucose and 0.2% sodium azide to deplete metabolic ATP (two separate experiments). Since oligomerization by AP1510 should be energy independent, this suggests that metabolic energy is needed to maintain the receptor in a proper conformation, even when ligand binding is triggered by receptor clustering. Third, the effect of a specific point mutation (β3(S752P)) or a truncation (β3(Δ724)) of the β3 cytoplasmic tail were studied because both have been shown to disrupt affinity modulation of αIIbβ3 in platelets and CHO cells (Chen et al., 1992, 1994; O'Toole et al., 1994; Wang et al., 1997). Whereas β3(S752P) abolished PAC1 binding in response to AP1510, β3(Δ724) had no such effect (Fig. 6). Thus the β3 cytoplasmic tail plays a role in ligand binding triggered by integrin clustering, but there must be differences in the structural features of β3 required for affinity and avidity modulation.
Figure 5.

Relative effects of αIIbβ3 clustering and affinity modulation on the binding of multivalent PAC1 IgM and monovalent PAC1 Fab to CHO cells. Cells stably expressing αIIb(FKBP)2β3 were incubated with a saturating concentration of PAC1 IgM (40 nM) or recombinant PAC1 Fab (30 nM) for 30 min in the absence or presence of AP1510, and specific binding of PAC1 was quantitated by flow cytometry. Unlike most other experiments, PAC1 was expressed here simply as mean fluorescence intensity in arbitrary units since correction for the degree of integrin expression was not necessary. Data represent the means ± SD of triplicate values from one experiment representative of two so performed.
Figure 6.

Effects of β3 cytoplasmic tail mutations on PAC1 binding caused by receptor clustering and affinity modulation. CHO cells were transiently transfected with the indicated αIIb and β3 subunits. 48 h later, they were incubated for 30 min with PAC1 along with AP1510 and/or 150 μg/ml anti–LIBS6 Fab, and specific PAC1 binding was quantitated by flow cytometry. Note the almost 10-fold difference in scales of the y axes. Data represent the means ± SEM of three experiments.
Thus far, the results support the validity of this model system to study integrin clustering, and they suggest that both clustering and affinity modulation can regulate ligand binding to αIIbβ3. Further studies were performed to determine the relative contributions of clustering and affinity modulation to ligand binding under conditions in which the effects of AP1510 and anti–LIB6 Fab were maximal (1,000 nM and 150 μg/ml, respectively). CHO cells were transiently transfected with either αIIbβ3, αIIb(FKBP)β3, αIIb(FKBP)2β3, or αIIb/α6Aβ3 (a constitutive, high affinity mutant [O'Toole et al., 1994]), and ligand binding was evaluated 48 h later. Whereas AP1510 had no effect on PAC1 binding to wild-type αIIbβ3, it increased binding to both αIIb(FKBP)β3 and αIIb(FKBP)2β3 such that specific PAC1 binding was increased approximately twofold (P < 0.001) (Fig. 7). However, PAC1 binding induced by AP1510 amounted to only 50% of the binding observed with the high affinity αIIb/α6Aβ3 chimera, and only 25% of the binding induced by anti–LIBS6 Fab (Fig. 7). Nevertheless, the PAC1 binding caused by clustering was statistically significant (P < 0.03) and approximately additive to the binding caused by affinity modulation (Fig. 7).
Figure 7.

Relative effects of receptor clustering and affinity modulation on PAC1 binding to αIIbβ3. CHO cells were transiently transfected with the indicated integrin constructs. 48 h later, they were incubated for 30 min with PAC1 along with AP1510 and/or 150 μg/ml anti–LIBS6 Fab, and specific PAC1 binding was quantitated by flow cytometry. Note the difference in scales of the y axes. Data represent the means ± SEM of three to five experiments.
Fibrinogen and PAC1 binding to activated platelets is initially reversible by the addition of EDTA, but binding becomes progressively irreversible over 15–60 min (Peerschke, 1995a ; Fox et al., 1996). In CHO cells that expressed αIIb(FKBP)2β3 and were treated with both anti–LIBS6 Fab and AP1510 to achieve maximal PAC1 binding, the added component of ligand binding resulting from AP1510 was fully reversible at 10 min but irreversible at 30 min (Fig. 8). Similar results were obtained when FITC-fibrinogen was used instead of PAC1 to monitor ligand binding (not shown). This series of experiments demonstrates that affinity modulation is the predominant regulator of ligand binding to αIIbβ3. However, receptor clustering plays an additive role in promoting eventual irreversible binding of the ligand.
Figure 8.

Effect of receptor clustering on reversible and irreversible ligand binding to αIIb(FKBP)2β3. Binding of PAC1 in response to anti–LIBS6 Fab was initiated in CHO cells stably- expressing αIIb(FKBP)2β3, either in the absence or presence of AP1510. After 10 or 30 min, half of each sample was treated with 5 mM EDTA to displace reversibly bound PAC1 and half was not. Then specific PAC1 binding was determined. Reversible PAC1 binding was defined as specific binding displaced by EDTA, and irreversible binding was defined as specific binding that was not displaced by EDTA. Data represent the means ± SEM of three experiments.
αIIbβ3 Clustering in the Regulation of Cell Adhesion
Activation of platelets by agonists leads to increased cell adhesion to the αIIbβ3 ligands, fibrinogen, and vWf (Savage et al., 1992). To determine the relative contributions of αIIbβ3 clustering and affinity modulation to cell adhesion, CHO cells that stably expressed αIIb(FKBP)2β3 were loaded with BCECF as a fluorescent marker and incubated for 90 min in microtiter wells coated with fibrinogen or vWf. Cell adhesion was dependent on the coating concentration of fibrinogen (Fig. 9, left panel ) and vWf (Fig. 9, right panel ), as well as on the presence of αIIb(FKBP)2β3, since it was blocked by 10 μM integrilin. AP1510 (1,000 nM) increased the extent of cell adhesion, but only very modestly and only at the higher coating concentrations of fibrinogen and vWf. On the other hand, increasing integrin affinity with anti–LIBS6 Fab (150 μg/ml) caused a more marked increase in cell adhesion, even at the lower ligand concentrations (Fig. 9, left and right panels). Thus under these assay conditions, receptor clustering is not as effective as affinity modulation in regulating cell adhesion via αIIbβ3.
Figure 9.

Relative effects of receptor clustering and affinity modulation on CHO cell adhesion to fibrinogen or vWF. As described in Materials and Methods, CHO cells stably expressing αIIb(FKBP)2β3 were fluorescently labeled with BCECF, and then incubated for 90 min in microtiter wells coated with fibrinogen (left panel ) or vWf (right panel ) in the presence of 1,000 nM AP1510 and/or 150 μg/ml anti–LIBS6 Fab. After washing, cell adhesion was quantitated by cytofluorimetry. Adhesion was expressed as a percentage of total cells added. This experiment is representative of three so performed. Not shown is the fact that in the absence of AP1510, the adhesion of αIIb(FKBP)2β3 cells was the same as for cells expressing wild-type αIIbβ3.
αIIbβ3 Clustering in the Regulation of Outside-In Signaling
In platelets and CHO cell transfectants, fibrinogen binding to αIIbβ3 leads to tyrosine phosphorylation and activation of Syk and FAK. The binding of soluble fibrinogen is sufficient to activate Syk, but additional post–ligand binding events, such as cytoskeletal reorganization, are necessary for activation of FAK (Huang et al., 1993; Clark et al., 1994; Gao et al., 1997). To study the role of integrin clustering in these events, CHO cells stably expressing αIIb(FKBP)2β3 were transiently-transfected with human Syk, and tyrosine phosphorylation of Syk and endogenous hamster FAK was examined. Fig. 10 A shows the raw data for a single experiment and Fig. 10 B shows a summary of three separate experiments. Cells maintained in suspension for 10 min in the absence or presence of fibrinogen exhibited a low level of tyrosine phosphorylation of Syk and FAK. However, addition of 1,000 nM AP1510 caused an average 2.8-fold increase in tyrosine phosphorylation of Syk, even in the absence of fibrinogen (P < 0.05), and this response was greater still in the presence of fibrinogen (5.4-fold; P < 0.02). In contrast, in the absence of fibrinogen integrin clustering by AP1510 did not stimulate an increase in FAK tyrosine phosphorylation, but increased FAK phosphorylation was observed in the presence of fibrinogen (3.5-fold; P < 0.001). Thus, integrin clustering can trigger tyrosine phosphorylation of Syk even in the absence of fibrinogen binding, whereas phosphorylation of FAK requires both receptor clustering and fibrinogen binding. Affinity modulation by anti-LIBS6 is not necessary in either case.
Figure 10.


Effect of integrin clustering on tyrosine phosphorylation of Syk and FAK. In A, CHO cells stably-expressing αIIb(FKBP)2β3 were transiently transfected with Syk. 48 h later, they were incubated in suspension for 10 min as indicated with 1,000 nM AP1510, 150 μg/ml anti–LIBS6 Fab, and/or 250 μg/ml fibrinogen. Lysates were prepared and immunoprecipitated with rabbit antiserum to Syk (top) or FAK (bottom), and samples were subjected to Western blotting with antibodies 4G10 and PY20 to phosphotyrosine (anti–P-Tyr). Finally, blots were stripped and reprobed with antibodies to Syk or FAK to assess gel loading. The first lane in each blot is a negative control using normal rabbit serum (NRS) for immunoprecipitation. The last lane (Adh) is a “positive control” in which cells were allowed to become adherent over 60 min to immobilized antibody D57 before lysis. In B, the data represent the means ± SEM of three such experiments. The intensities of the phosphotyrosine bands are expressed relative to the intensities of the matching Syk and FAK immunoreactive bands. Asterisks denote statistically significant differences (P ≤ 0.05) from the cells incubated in suspension without additives (black bar labeled –).
Discussion
In this study, engraftment of one or two FKBP repeats onto the COOH terminus of the αIIb subunit enabled us to cluster integrin αIIbβ3 in a conditional fashion by treating CHO cells with a synthetic, bivalent FKBP ligand, AP1510. This permitted us for the first time to conduct a detailed comparison of the functional effects of receptor clustering, initiated from within the cell, with the effects of increasing integrin affinity through conformational changes. The major conclusions of this work are: (a) Conformational changes play a predominant role in αIIbβ3 activation in CHO cells, as monitored by ligand binding and cell adhesion assays. (b) Clustering causes a modest increment in reversible and ultimately irreversible binding of multivalent ligands to αIIbβ3, and this binding is additive to that caused by affinity modulation. (c) Ligand binding resulting from receptor clustering is dependent on cellular metabolic energy and is sensitive to some, but not all, of the mutations or deletions in the β3 cytoplasmic tail that block affinity modulation of the receptor. (d) Integrin clustering promotes ligand-independent tyrosine phosphorylation of Syk, and ligand- dependent phosphorylation of FAK, even when cells are maintained in suspension and even in the absence of deliberate affinity modulation. Thus, by being able to manipulate integrin clustering and affinity separately and in a controlled manner, we conclude that these two processes play complementary roles in the function of αIIbβ3.
Integrin Clustering and Inside-Out Signaling
Inside-out signaling is responsible for acute regulation of the ligand binding function of integrins. In the case of integrins that normally engage soluble adhesive ligands in vivo, such as αIIbβ3, inside-out signaling can be monitored directly using labeled ligands or ligand-mimetic antibodies, such as PAC1. Alternatively, it can be assessed by cell adhesion assays. Although physiologically relevant, cell adhesion is a more indirect measure of integrin activation because it can be influenced by factors, such as actin polymerization, cell spreading, and focal adhesion turnover, that may affect the overall strength of the adhesion process through mechanisms other than regulation of ligand binding (Burridge and Chrzanowska-Wodnicka, 1996; Yamada and Geiger, 1997; Hall, 1998). Thus, when possible, it is preferable to monitor inside-out signaling by ligand binding assays, as in the current study.
Ligand binding to integrins is thought to be regulated by a combination of affinity and avidity modulation (van Kooyk and Figdor, 1993; Diamond and Springer, 1994; Bazzoni and Hemler, 1998). In the case of αIIbβ3, platelet activation is believed to cause a modification of the conformation or orientation of the integrin cytoplasmic tails that is transmitted across the membrane, leading to increased access of the ligand to binding sites in the receptor (Loftus and Liddington, 1997; Shattil et al., 1998). However, cell activation appears to promote ligand binding to certain β1 and β2 integrins by also stimulating the lateral diffusion and clustering of these receptors (van Kooyk and Figdor, 1993; Kucik et al., 1996; Bazzoni and Hemler, 1998; Shattil et al., 1998), and the same might be true for αIIbβ3. Several experimental approaches have been used to cluster integrins, including treatment of cells with multivalent antibodies or chemical cross-linkers, incubation of cells with ligand-coated beads, and promotion of cell spreading (Kornberg et al., 1991; Dorahy et al., 1995; Hotchin and Hall, 1995; Miyamoto et al., 1995). Whereas each of these has provided important information about outside-in signaling, none is entirely suitable for studies of soluble ligand binding to αIIbβ3. The use here of AP1510 to cluster αIIb(FKBP)β3 or αIIb(FKBP)2β3, while CHO cells were maintained in suspension demonstrates unambiguously that affinity and avidity modulation can complement one another with respect to the control of ligand binding. Given the wide variety of soluble, matrix- and cell-associated ligands that integrins must contend with, it is likely that the relative contributions of affinity and avidity modulation will vary with the integrin and the cell type.
Fortunately, the fusion of single or tandem FKBP repeats to the αIIb cytoplasmic tail did not interfere with αIIbβ3 expression or function in CHO cells. Perhaps this means that the very COOH terminus of the α subunit is dispensable for the integrin functions that were assessed. On the other hand, direct attachment of FKBP to the β3 tail interferes with energy-dependent affinity modulation of αIIbβ3, possibly by disrupting necessary interactions of the β3 tail with regulatory proteins (Hato, T., and Shattil, S.J., unpublished observations). We ascribe any functional effects of AP1510 on αIIb(FKBP)β3 and αIIb(FKBP)2β3 to receptor clustering. Although the evidence for this is strong, it is largely indirect. First, AP1510 only affected those forms of αIIbβ3 that contained FKBP repeats (Figs. 6 and 7). Second, confocal microscopy showed that AP1510 treatment was associated with the appearance of coarse patches of integrin staining in the surface membrane (Fig. 3). Finally, AP1510 caused binding of a multivalent but not monovalent form of PAC1, precisely what might be expected in the case of integrin clustering (Fig. 5). Interestingly, the effect of AP1510 on PAC1 binding to αIIb(FKBP)β3 and αIIb(FKBP)2β3 was nearly equivalent (Fig. 7). Assuming that AP1510 can only dimerize αIIb(FKBP)β3, this suggests that formation of a dimer of dimers, e.g., (αIIb(FKBP)β3)2, may be sufficient to initiate some ligand binding to αIIbβ3.
What are the biological implications of αIIbβ3 clustering during inside-out signaling? Ligand binding resulting from clustering of αIIb(FKBP)2β3 required a normal β3 cytoplasmic tail since a tail point mutation (S752P) disrupted this process (Fig. 6). This same mutation disrupts energy- dependent affinity modulation of αIIbβ3 in CHO cells and in human platelets, where it is responsible for a bleeding diathesis (Chen et al., 1992, 1994). In contrast, another β3 cytoplasmic modification, a truncation starting at residue Arg 724, did not disrupt ligand binding induced by clustering of αIIb(FKBP)2β3, although it does abolish affinity modulation of αIIbβ3 in CHO cells and platelets (O'Toole et al., 1994; Wang et al., 1997). This suggests that there are differences in the structural elements of the β3 tail that are needed for affinity and avidity modulation. A growing number of proteins have been shown to interact directly with the cytoplasmic tails of α or β integrin subunits, at least in vitro, and overexpression of some of them, including calreticulin (α tails) (Coppolino et al., 1997), cytohesin-1 (β2 tail) (Kolanus et al., 1996), and β3-endonexin (β3 tail) (Kashiwagi et al., 1997) can affect ligand binding and cell adhesion. When overexpressed in CHO cells, several other signaling proteins, including H-ras (Hughes et al., 1996), R-ras (Zhang et al., 1996), and CD98 (Fenczik et al., 1997) have been shown to modulate the ligand binding properties of αIIbβ3; however, it is not known if any of these proteins interact directly with the integrin. The relative effects of potential regulatory molecules such as these on receptor clustering and receptor affinity remain to be determined.
The extent of PAC1 binding observed in response to integrin clustering was only a fraction of that observed with affinity modulation. Yet ligand binding resulting from clustering and affinity modulation was additive (Fig. 7). Clustering also facilitated CHO cell adhesion to immobilized fibrinogen and vWf, but once again, this effect was relatively minor compared with the effect of affinity modulation and it was apparent only at the higher coating concentrations of the ligands (Fig. 9). On the other hand, if clustering of αIIbβ3 were to cause increased ligand binding in platelets as it does in CHO cells, it could affect the ultimate size of a platelet aggregate and hence the delicate balance between adequate and inadequate hemostasis or the difference between partial and total arterial occlusion by a platelet-rich thrombus. Conceivably, the effects of integrin clustering on ligand binding may be even more pronounced in platelets than in the CHO cell model system because receptor density may be higher in platelets and clustering may be stimulated by agonists that trigger integrin interactions with multivalent, polymerizing ligands on both sides of the plasma membrane (Hartwig, 1992; Fox et al., 1996; Simmons and Albrecht, 1996). Furthermore, any increase in adhesive strength promoted by αIIbβ3 clustering in vivo might help platelets resist detachment from sites of vascular injury in response to hemodynamic forces (Savage et al., 1996).
Integrin Clustering and Outside-In Signaling
A potential limitation of the chemical dimerization approach used here is that it may not reflect or trigger the types of interactions between αIIbβ3, cytoskeletal proteins, and signaling molecules that take place normally during outside-in signaling. For example, in platelets, the binding of fibrinogen to αIIbβ3 is sufficient to trigger tyrosine phosphorylation and activation of Syk, whereas tyrosine phosphorylation of FAK requires additional post–ligand binding events that occur during platelet aggregation or spreading (Haimovich et al., 1993; Huang et al., 1993). In this regard, clustering of αIIb(FKBP)2β3 in CHO cells by AP1510 caused significant tyrosine phosphorylation of Syk, even when the cells were maintained in suspension without fibrinogen (Fig. 10). Since integrin-dependent tyrosine phosphorylation of Syk correlates with induction of Syk kinase activity in both platelets and CHO cells (Clark et al., 1994; Gao et al., 1997), these results suggest that the binding of multivalent fibrinogen to αIIbβ3 triggers Syk activation, at least in part, by inducing integrin clustering.
In contrast to the results for Syk, clustering of αIIb(FKBP)2β3 by AP1510 was not sufficient to cause tyrosine phosphorylation of FAK in cells maintained in suspension. However, fibrinogen binding together with receptor clustering were sufficient to induce the response (Fig. 10). These results highlight the apparent differences in coupling mechanisms between αIIbβ3 and Syk and αIIbβ3 and FAK (Gao et al., 1997). At the same time, they demonstrate unambiguously that conditional clustering of αIIbβ3 in CHO cells can recapitulate a pattern of outside-in signaling that is characteristic of platelets. In nucleated cells, integrin and growth factor signaling pathways collaborate to regulate gene expression, cell adhesion, and motility (Schwartz et al., 1995; Juliano, 1996; Sastry and Horwitz, 1996; Yamada and Geiger, 1997). One hallmark of integrated signaling networks is tight control of enzyme activity and protein subcellular localization though regulated protein–protein interactions (Pawson and Scott, 1997). Chemical inducers of dimerization can be used to promote controlled homodimerization and heterodimerization of proteins in vivo as well as ex vivo (Rivera et al., 1996; Spencer et al., 1996; Clackson, 1997; Yang et al., 1998). Consequently, they should prove useful in evaluating diverse aspects of integrin signaling.
Acknowledgments
The authors are grateful to J. Amara and V. Rivera (ARIAD Pharmaceuticals, Inc., Cambridge, MA) for supplying pCF1E and AP1510; and to several colleagues for their gifts of other reagents: J. Brugge (Harvard Medical School, Boston, MA [EMCV-Syk]); M. Ginsberg (Scripps Research Institute [antibodies D57 and No. 2308, αIIb and β3 pCDM8 plasmids]); D. Phillips (Cor Therapeutics, Inc., South San Francisco, CA [Integrilin]); and Z. Ruggeri (Scripps Research Institute [von Willebrand factor]).
Abbreviations used in this paper
- ECL
enhanced chemiluminescence
- FKBP
FK506 binding protein, or FKBP12
- vWf
von Willebrand factor
Footnotes
These studies were supported, in part by grants from the National Institutes of Health (HL56595, HL57900).
Address all correspondence to Dr. Shattil, Department of Vascular Biology, The Scripps Research Institute, 10550 North Torrey Pines Rd., VB-5, La Jolla, CA 92037. Tel.: (619) 784-7148. Fax: (619) 784-7422. E-mail: shattil@scripps.edu
References
- Abrams C, Deng J, Steiner B, Shattil SJ. Determinants of specificity of a baculovirus-expressed antibody Fab fragment that binds selectively to the activated form of integrin αIIbβ3 . J Biol Chem. 1994;269:18781–18788. [PubMed] [Google Scholar]
- Abrams CS, Ruggeri ZM, Taub R, Hoxie JA, Nagaswami C, Weisel W, Shattil SJ. Anti-idiotypic antibodies against an antibody to the platelet glycoprotein (GP) IIb-IIIa complex mimic GP IIb-IIIa by recognizing fibrinogen. J Biol Chem. 1992;267:2775–2785. [PubMed] [Google Scholar]
- Amara JF, Clackson T, Rivera VM, Guo T, Keenan T, Natesan S, Pollock R, Yang W, Courage NL, Holt DA, Gilman M. A versatile synthetic dimerizer for the regulation of protein–protein interactions. Proc Natl Acad Sci USA. 1997;94:10618–10623. doi: 10.1073/pnas.94.20.10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfic H, Tang XW, Batty IH, Downes CP, Chen CS, Rittenhouse SE. A novel integrin-activated pathway forms PKB/Akt-stimulatory phosphatidylinositol 3,4-bisphosphate via phosphatidylinositol 3-phosphate in platelets. J Biol Chem. 1998;273:13–16. doi: 10.1074/jbc.273.1.13. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Hemler ME. Are changes in integrin affinity and conformation overemphasized? . Trends Biochem Sci. 1998;23:30–34. doi: 10.1016/s0968-0004(97)01141-9. [DOI] [PubMed] [Google Scholar]
- Bennett JS. The molecular biology of platelet membrane proteins. Semin Hematol. 1990;27:186–204. [PubMed] [Google Scholar]
- Bennett JS, Chan C, Vilaire G, Mousa SA, DeGrado WF. Agonist-activated αVβ3on platelets and lymphocytes binds to the matrix protein osteopontin. J Biol Chem. 1997;272:8137–8140. doi: 10.1074/jbc.272.13.8137. [DOI] [PubMed] [Google Scholar]
- Bretscher MS. Circulating integrins: α5β1, α6β4 and Mac-1, but not α3β1, α4β1or LFA-1. EMBO (Eur Mol Biol Organ) J. 1992;11:405–410. doi: 10.1002/j.1460-2075.1992.tb05068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Biol. 1996;12:463–519. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Chen Y-P, Djaffar I, Pidard D, Steiner B, Cieutat AM, Caen JP, Rosa J-P. Ser-752 → Pro mutation in the cytoplasmic domain of integrin β3 subunit and defective activation of platelet integrin αIIbβ3(glycoprotein IIb-IIIa) in a variant of Glanzmann thrombasthenia. Proc Natl Acad Sci USA. 1992;89:10169–10173. doi: 10.1073/pnas.89.21.10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-P, O'Toole TE, Ylänne J, Rosa J-P, Ginsberg MH. A point mutation in the integrin β3 cytoplasmic domain (S752→ P) impairs bidirectional signaling through αIIbβ3(platelet glycoprotein IIb-IIIa) Blood. 1994;84:1857–1865. [PubMed] [Google Scholar]
- Clackson T. Controlling mammalian gene expression with small molecules. Curr Opin Chem Biol. 1997;1:210–218. doi: 10.1016/s1367-5931(97)80012-9. [DOI] [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Integrins and signal transduction pathways. The road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Clark EA, Shattil SJ, Ginsberg MH, Bolen J, Brugge JS. Regulation of the protein tyrosine kinase, pp72syk, by platelet agonists and the integrin, αIIbβ3 . J Biol Chem. 1994;46:28859–28864. [PubMed] [Google Scholar]
- Coller BS. Platelet GPIIb/IIIa antagonists: the first anti-integrin receptor therapeutics. J Clin Invest. 1997;99:1467–1471. doi: 10.1172/JCI119307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppolino MG, Woodside MJ, Demaurex N, Grinstein S, St-Arnaud R, Dedhar S. Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nature. 1997;386:843–847. doi: 10.1038/386843a0. [DOI] [PubMed] [Google Scholar]
- Detmers PA, Wright SD, Olsen E, Kimball B, Cohn ZA. Aggregation of complement receptors on human neutrophils in the absence of ligand. J Cell Biol. 1987;105:1137–1145. doi: 10.1083/jcb.105.3.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Springer TA. The dynamic regulation of integrin adhesiveness. Curr Biol. 1994;4:506–517. doi: 10.1016/s0960-9822(00)00111-1. [DOI] [PubMed] [Google Scholar]
- Dorahy DJ, Berndt MC, Burns GF. Capture by chemical crosslinkers provides evidence that integrin αIIbβ3forms a complex with protein tyrosine kinases in intact platelets. Biochem J. 1995;309:481–490. doi: 10.1042/bj3090481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Gu M, Weisel J, Nagaswami C, Bennett JS, Bowditch R, Ginsberg MH. Long range propagation of conformational changes in integrin αIIbβ3 . J Biol Chem. 1993;268:23087–23092. [PubMed] [Google Scholar]
- Fenczik CA, Sethi T, Ramos JW, Hughes PE, Ginsberg MH. Complementation of dominant suppression implicates CD98 in integrin activation. Nature. 1997;390:81–85. doi: 10.1038/36349. [DOI] [PubMed] [Google Scholar]
- Fox J, Shattil SJ, Kinlough-Rathbone R, Richardson M, Packham MA, Sanan DA. The platelet cytoskeleton stabilizes the interaction between αIIbβ3and its ligand and induces selective movements of ligand-occupied integrin. J Biol Chem. 1996;271:7004–7011. doi: 10.1074/jbc.271.12.7004. [DOI] [PubMed] [Google Scholar]
- Fox JEB, Lipfert L, Clark EA, Reynolds CC, Austin CD, Brugge JS. On the role of the platelet membrane skeleton in mediating signal transduction. Association of GP IIb-IIIa, pp60c-src, pp62c-yes, and the p21rasGTPase-activating protein with the membrane skeleton. J Biol Chem. 1993;268:25973–25984. [PubMed] [Google Scholar]
- Freedman JE, Loscalzo J, Barnard MR, Alpert C, Keaney JF, Jr, Michelson AD. Nitric oxide released from activated platelets inhibits platelet recruitment. J Clin Invest. 1997;100:350–356. doi: 10.1172/JCI119540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Zoller K, Ginsberg MH, Brugge JS, Shattil SJ. Regulation of the pp72Syk protein tyrosine kinase by platelet integrin αIIbβ3 . EMBO (Eur Mol Biol Organ) J. 1997;16:6414–6425. doi: 10.1093/emboj/16.21.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber SE, Hawiger J. Evidence that changes in platelet cyclic AMP levels regulate the fibrinogen receptor on human platelets. J Biol Chem. 1982;257:14606–14609. [PubMed] [Google Scholar]
- Haimovich B, Lipfert L, Brugge JS, Shattil SJ. Tyrosine phosphorylation and cytoskeletal reorganization in platelets are triggered by interaction of integrin receptors with their immobilized ligands. J Biol Chem. 1993;268:15868–15877. [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hartwig JH. Mechanisms of actin rearrangements mediating platelet activation. J Cell Biol. 1992;118:1421–1442. doi: 10.1083/jcb.118.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchin NA, Hall A. The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J Cell Biol. 1995;131:1857–1865. doi: 10.1083/jcb.131.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M-M, Lipfert L, Cunningham M, Brugge JS, Ginsberg MH, Shattil SJ. Adhesive ligand binding to integrin αIIbβ3 stimulates tyrosine phosphorylation of novel protein substrates before phosphorylation of pp125FAK . J Cell Biol. 1993;122:473–483. doi: 10.1083/jcb.122.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PE, Renshaw MW, Pfaff M, Forsyth J, Keivens VM, Schwartz MA, Ginsberg MH. Suppression of integrin activation: a novel function of a Ras/Raf-initiated MAP-kinase pathway. Cell. 1996;88:521–530. doi: 10.1016/s0092-8674(00)81892-9. [DOI] [PubMed] [Google Scholar]
- Isenberg WM, McEver RP, Phillips DR, Shuman MA, Bainton DF. The platelet fibrinogen receptor: an immunogold-surface replica study of agonist-induced ligand binding and receptor clustering. J Cell Biol. 1987;104:1655–1663. doi: 10.1083/jcb.104.6.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano R. Cooperation between soluble factors and integrin-mediated cell anchorage in the control of cell growth and differentiation. Bioessays. 1996;18:911–917. doi: 10.1002/bies.950181110. [DOI] [PubMed] [Google Scholar]
- Kashiwagi H, Schwartz MA, Eigenthaler MA, Davis KA, Ginsberg MH, Shattil SJ. Affinity modulation of platelet integrin αIIbβ3 by β3-endonexin, a selective binding partner of the β3 integrin cytoplasmic tail. J Cell Biol. 1997;137:1433–1443. doi: 10.1083/jcb.137.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolanus W, Nagel W, Schiller B, Zeitlmann L, Godar S, Stockinger H, Seed B. α-L-β-2 integrin/LFA-1 binding to ICAM-1 induced by cytohesin-1, a cytoplasmic regulatory molecule. Cell. 1996;86:233–242. doi: 10.1016/s0092-8674(00)80095-1. [DOI] [PubMed] [Google Scholar]
- Kornberg LJ, Earp HS, Turner CE, Prockop C, Juliano RL. Signal transduction by integrins: increased protein tyrosine phosphorylation caused by clustering of β1integrins. Proc Natl Acad Sci USA. 1991;88:8392–8396. doi: 10.1073/pnas.88.19.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucik DF, Dustin ML, Miller JM, Brown EJ. Adhesion-activating phorbol ester increases the mobility of leukocyte integrin LFA-1 in cultured lymphocytes. J Clin Invest. 1996;97:2139–2144. doi: 10.1172/JCI118651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng L, Kashiwagi H, Ren X-D, Shattil SJ. RhoA and the function of platelet integrin αIIbβ3 . Blood. 1998;91:4206–4215. [PubMed] [Google Scholar]
- Leong L, Hughes PE, Schwartz MA, Ginsberg MH, Shattil SJ. Integrin signaling: Roles for the cytoplasmic tails of αIIbβ3 in the tyrosine phosphorylation of pp125FAK . J Cell Sci. 1995;108:3817–3825. doi: 10.1242/jcs.108.12.3817. [DOI] [PubMed] [Google Scholar]
- Loftus JC, Liddington RC. Cell adhesion in vascular biology. New insights into integrin–ligand interaction. J Clin Invest. 1997;99:2302–2306. doi: 10.1172/JCI119408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh E, Qi WW, Vilaire G, Bennett JS. Effect of cytoplasmic domain mutations on the agonist-stimulated ligand binding activity of the platelet integrin αIIbβ3 . J Biol Chem. 1996;271:30233–30241. doi: 10.1074/jbc.271.47.30233. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Teramoto H, Coso OA, Gutkind JS, Burbelo PD, Akiyama SK, Yamada KM. Integrin function: Molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol. 1995;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole TE, Katagiri Y, Faull RJ, Peter K, Tamura R, Quaranta V, Loftus JC, Shattil SJ, Ginsberg MH. Integrin cytoplasmic domains mediate inside-out signaling. J Cell Biol. 1994;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Peerschke EI. Regulation of platelet aggregation by post-fibrinogen binding events. Thromb Haemost. 1995a;73:862–867. [PubMed] [Google Scholar]
- Peerschke EIB. Bound fibrinogen distribution on stimulated platelets—examination by confocal scanning laser microscopy. Am J Pathol. 1995b;147:678–687. [PMC free article] [PubMed] [Google Scholar]
- Rivera VM, Clackson T, Natesan S, Pollock R, Amara JF, Keenan T, Magari SR, Phillips T, Courage NL, Cerasoli F, Jr, et al. A humanized system for pharmacologic control of gene expression. Nat Med. 1996;2:1028–1032. doi: 10.1038/nm0996-1028. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM, De Marco L, Gatti L, Bader R, Montgomery RR. Platelets have more than one binding site for von Willebrand factor. J Clin Invest. 1983;72:1–12. doi: 10.1172/JCI110946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry SK, Horwitz AF. Integrin cytoplasmic domains: mediators of cytoskeletal linkages and extra- and intracellular initiated transmembrane signaling. Curr Opin Cell Biol. 1993;5:819–831. doi: 10.1016/0955-0674(93)90031-k. [DOI] [PubMed] [Google Scholar]
- Sastry SK, Horwitz AF. Adhesion-growth factor interactions during differentiation: an integrated biological response. Dev Biol. 1996;180:455–467. doi: 10.1006/dbio.1996.0319. [DOI] [PubMed] [Google Scholar]
- Savage B, Shattil SJ, Ruggeri ZM. Modulation of platelet function through adhesion receptors. A dual role for glycoprotein IIb–IIIa (integrin αIIbβ3) mediated by fibrinogen and glycoprotein Ib-von Willebrand factor. J Biol Chem. 1992;267:11300–11306. [PubMed] [Google Scholar]
- Savage B, Saldívar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84:289–297. doi: 10.1016/s0092-8674(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Scarborough RM, Naughton MA, Teng W, Rose JW, Phillips DR, Nannizzi L, Arfsten A, Campbell AM, Charo IF. Design of potent and specific integrin antagonists. Peptide antagonists with high specificity for glycoprotein IIb-IIIa. J Biol Chem. 1993;268:1066–1073. [PubMed] [Google Scholar]
- Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- Shattil SJ, Hoxie JA, Cunningham M, Brass LF. Changes in the platelet membrane glycoprotein IIb-IIIa complex during platelet activation. J Biol Chem. 1985;260:11107–11114. [PubMed] [Google Scholar]
- Shattil SJ, Kashiwagi H, Pampori N. Integrin signaling: the platelet paradigm. Blood. 1998;91:2645–2657. [PubMed] [Google Scholar]
- Simmons SR, Albrecht RM. Self-association of bound fibrinogen on platelet surfaces. J Lab Clin Med. 1996;128:39–50. doi: 10.1016/s0022-2143(96)90112-2. [DOI] [PubMed] [Google Scholar]
- Simmons SR, Sims PA, Albrecht RM. αIIbβ3 redistribution triggered by receptor cross-linking. Arterioscler Thromb Vasc Biol. 1997;17:3311–3320. doi: 10.1161/01.atv.17.11.3311. [DOI] [PubMed] [Google Scholar]
- Sims PJ, Ginsberg MH, Plow EF, Shattil SJ. Effect of platelet activation on the conformation of the plasma membrane glycoprotein IIb-IIIa complex. J Biol Chem. 1991;266:7345–7352. [PubMed] [Google Scholar]
- Spencer DM, Belshaw PJ, Chen L, Ho SN, Randazzo F, Crabtree GR, Schreiber SL. Functional analysis of Fas signaling in vivo using synthetic inducers of dimerization. Curr Biol. 1996;6:839–847. doi: 10.1016/s0960-9822(02)00607-3. [DOI] [PubMed] [Google Scholar]
- van Kooyk Y, Figdor CG. Activation and inactivation of adhesion molecules. Curr Top Micro Immunol. 1993;184:235–248. doi: 10.1007/978-3-642-78253-4_19. [DOI] [PubMed] [Google Scholar]
- van Kooyk Y, Weder P, Heije K, Figdor CG. Extracellular Ca2+modulates leukocyte function-associated antigen-1 cell surface distribution on T lymphocytes and consequently affects cell adhesion. J Cell Biol. 1994;124:1061–1070. doi: 10.1083/jcb.124.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Shattil SJ, Ambruso DR, Newman PJ. Truncation of the cytoplasmic domain of β3 in a variant form of Glanzmann thrombasthenia abrogates signaling through the integrin αIIbβ3 complex. J Clin Invest. 1997;100:2393–2403. doi: 10.1172/JCI119780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, Geiger B. Molecular interactions in cell adhesion complexes. Curr Opin Cell Biol. 1997;9:76–85. doi: 10.1016/s0955-0674(97)80155-x. [DOI] [PubMed] [Google Scholar]
- Yang J, Symes K, Mercola M, Schreiber SL. Small-molecule control of insulin and PDGF receptor signaling and the role of membrane attachment. Curr Biol. 1998;8:11–18. doi: 10.1016/s0960-9822(98)70015-6. [DOI] [PubMed] [Google Scholar]
- Yap AS, Brieher WM, Pruschy M, Gumbiner BM. Lateral clustering of the adhesive ectodomain: a fundamental determinant of cadherin function. Curr Biol. 1997;7:308–315. doi: 10.1016/s0960-9822(06)00154-0. [DOI] [PubMed] [Google Scholar]
- Yauch RL, Felsenfeld DP, Kraeft SK, Chen LB, Sheetz MP, Hemler ME. Mutational evidence for control of cell adhesion through integrin diffusion/clustering, independent of ligand binding. J Exp Med. 1997;186:1347–1355. doi: 10.1084/jem.186.8.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Vuori K, Wang H-G, Reed JC, Ruoshlati E. Integrin activation by R-ras. Cell. 1996;85:61–69. doi: 10.1016/s0092-8674(00)81082-x. [DOI] [PubMed] [Google Scholar]