

Abstract
Recent studies have established cell type– specific, proapoptotic, or antiapoptotic functions for the transcription factor NF-κB. In each of these studies, inhibitors of NF-κB activity have been present before the apoptotic stimulus, and so the role of stimulus- induced NF-κB activation in enhancing or inhibiting survival could not be directly assessed. Sindbis virus, an alphavirus, induces NF-κB activation and apoptosis in cultured cell lines. To address whether Sindbis virus– induced NF-κB activation is required for apoptosis, we used a chimeric Sindbis virus that expresses a superrepressor of NF-κB activity. Complete suppression of virus-induced NF-κB activity neither prevents nor potentiates Sindbis virus–induced apoptosis. In contrast, inhibition of NF-κB activity before infection inhibits Sindbis virus–induced apoptosis. Our results demonstrate that suppression of steady-state, but not stimulus-induced NF-κB activity, regulates expression of gene products required for Sindbis virus–induced death. Furthermore, we show that in the same cell line, NF-κB can be proapoptotic or antiapoptotic depending on the death stimulus. We propose that the role of NF-κB in regulating apoptosis is determined by the death stimulus and by the timing of modulating NF-κB activity relative to the death stimulus.
Multicellular organisms use a programmed process known as apoptosis to eliminate damaged or unwanted cells during development, in adulthood, or in disease states. In some instances, cells destined to die by apoptosis must synthesize new mRNAs encoding putative death proteins for ordered self-destruction to occur. Indeed, general inhibitors of transcription and translation inhibit apoptotic death in response to a host of stimuli, even when added many hours after the onset of the death stimulus (Martin et al., 1988; Oppenheim et al., 1990; Ratan et al., 1994; Jacobson et al., 1997). A requirement for transcription in the proper execution of some types of apoptosis has stimulated a search for DNA-binding proteins known as transcription factors that are activated by apoptotic stimuli, and that govern expression of putative death proteins (Jehn and Osborne, 1997). Several apoptosis-associated transcription factors have been identified, including p53 (Lowe et al., 1993), c-jun (Estus et al., 1994; Ham et al., 1995), Nurr-77/Nor-1 (Liu et al., 1994; Woronicz et al., 1994), and interferon regulatory factor-1 (Tanaka et al., 1994).
Recent data suggests that another transcription factor, NF-κB, though classically associated with immune and inflammatory responses (Baeuerle and Henkel, 1994), can also be added to the list of apoptosis-associated transcription factors. The NF-κB family of transcription factors is composed of five different subunits, including p50, p52, p65/Rel A, c-Rel, and Rel-B, which can form either homodimers or heterodimers (Baeuerle and Baltimore, 1996). Overexpression of one of these NF-κB subunits, c-rel, in chick bone marrow cells leads to apoptosis (Abbadie et al., 1993). These observations suggest that in some cell types, NF-κB activation is sufficient to activate the apoptotic process. Additionally, inhibition of NF-κB activity using κB-specific transcription factor decoys inhibits Sindbis virus–induced cell death in AT-3 prostate carcinoma cells (Lin et al., 1995). Moreover, overexpression of a truncated dominant-negative form of p65/rel A inhibits serum deprivation–induced NF-κB–dependent gene activation and apoptosis in human embryonic kidney cells (Grimm et al., 1996). These results identify apoptotic paradigms where NF-κB activation is necessary for cell death initiation, and confirm that NF-κB can act as a proapoptotic transcription factor. Indeed, several proapoptotic genes, including c-Myc (La Rosa et al., 1994), p53 (Wu and Lozano, 1994), Fas ligand (Takahashi et al., 1994), and interleukin-1 β–converting enzyme (caspase-1; Casano et al., 1994) have NF-κB–binding sequences in their promoter regions.
In contrast to the aforementioned observations, NF-κB activity appears to regulate genes that suppress some types of apoptosis. For example, suppression of NF-κB activity in immature B cells after adding anti-IgM (Wu et al., 1996); in cells stimulated with TNF-α, radiation, or daunorubicin (Liu et al., 1996; Beg and Baltimore, 1996; Wang et al., 1996; van Antwerp et al., 1996); or in liver during development (Beg et al., 1995); dramatically enhances cell death. Putative NF-κB–regulated antiapoptotic genes include manganese superoxide dismutase (Wong et al., 1989) and the zinc finger protein A20 (Opipari et al., 1992). Thus, NF-κB activity can play a pivotal role in triggering antiapoptotic or proapoptotic pathways; however, the precise factors that determine NF-κB's ability to regulate these divergent biological outcomes remain unclear.
Under basal nonstress conditions, NF-κB is sequestered in most cells in the cytoplasm by a member of the IκB family of proteins: IκBα, IκBβ, or IκBε (Baeuerle and Baltimore, 1988; Baeuerle and Baltimore, 1996). Binding of IκB to NF-κB masks nuclear localization signals on NF-κB and prevents its translocation to the nucleus (Beg et al., 1992). In response to a host of stimuli, including TNF-α, virus infection, lipopolysaccharide, and UV light, IκBα is phosphorylated, followed by its degradation via the ubiquitin–proteasome pathway (Henkel et al., 1993; Palombella et al., 1994; Thanos and Maniatis, 1995; Chen et al., 1996; DiDonato et al., 1995; DiDonato et al., 1996; DiDonato et al., 1997; Régnier et al., 1997). Degradation of IκB α allows free NF-κB to translocate to the nucleus to influence expression of its target genes. Degradation of IκB α can be inhibited by mutating either serine 32 or 36 in the NH2-terminal region of IκB α to alanines. (Brown et al., 1995; Traenckner et al., 1995; DiDonato et al., 1996). Overexpression of the 32A/36A NH2-terminal mutant (IκB M) is an effective molecular strategy for suppressing NF-κB activation in cultured cells (Wang et al., 1996). Indeed, stable cell lines containing the superrepressor of NF-κB, IκB M have been generated and used to demonstrate an antiapoptotic role for NF-κB activity in TNF α, daunorubicin, or UV irradiation–induced apoptosis (Wang et al., 1996; van Antwerp et al., 1996). However, this approach does not distinguish whether suppression of NF-κB activity must be initiated before or after the apoptotic stimulus to regulate apoptosis.
To begin to address this question, we used the alphavirus Sindbis virus, which induces apoptosis in cultured cells (Levine et al., 1993) and in the brains of newborn mice (Lewis et al., 1996). Like many other inducers of apoptosis, the precise mechanisms by which Sindbis virus (SV)1 induces apoptosis is unknown, but many general antagonists of apoptosis, including Bcl-2 (Levine et al., 1993; Levine et al., 1996), Bcl-XL (Cheng et al., 1996), or the antioxidant N-acetylcysteine (NAC; Lin et al., 1995) can block SV-induced death. Moreover, SV induces activation of NF-κB via a classical ubiquitin–proteasome pathway many hours in advance of morphological evidence of apoptosis (Lin et al., 1998). Previous studies from our laboratory demonstrated that NF-κB activity is required for cell death in AT-3 prostate carcinoma cells (Lin et al., 1995), but as with other studies, the NF-κB inhibitors were present before the apoptotic stimulus was applied, and so the role of SV-induced NF-κB activation in regulating apoptosis could not be directly assessed.
Recently, a recombinant form of SV has been developed as a vector to express heterologous proteins after infection, allowing their roles in SV-induced death to be examined (Cheng et al., 1996). We used this vector to generate a recombinant SV that carries the superrepressor form of IκB α. Such a vector can not only elicit apoptosis, but can also inhibit nuclear NF-κB activity after, but not before infection. We report here that complete suppression of NF-κB activation after SV infection neither suppresses nor potentiates cell death. In contrast, stable expression of a superrepressor form of IκB α in cell lines resulting in constitutive suppression of NF-κB activation suppresses SV-induced death. These data suggest that NF-κB activity induced by an apoptotic stimulus such as SV infection does not influence cell viability, while NF-κB activity before SV infection is critical in regulating a proapoptotic response.
Materials and Methods
Culture and Viability Studies
AT-3 rat prostate carcinoma cells were cultured as previously described (Lin et al., 1995) in RPMI 1640 medium containing 10% FCS, 2 mM l-glutamine, penicillin (50 U/ml), and streptomcyin (50 μg/ml). Wild-type, p65 knockout (p65−/−), and p50 knockout (p50−/−) 3T3 fibroblasts or EFs (mouse embryonic fibroblasts) were cultured as previously described (Beg and Baltimore, 1996). For viability studies, 103 AT-3 cell/well or 104 mouse embryo fibroblasts/well were plated in 96-well dishes. One day after plating, SV (strain AR339) or recombinant SV (wild-type, M, or R) were added at a multiplicity of infection of 10 plaque-forming units per cell. Viability was assessed using a viability index generated by measuring the cell-associated LDH activity (Promega Corp., Madison, WI) in SV- infected cells and dividing this value by the cell-associated LDH activity in mock-infected cells. In parallel, viability was also assessed by using the metabolic label alamarBlue (Accu Med International, Chicago, IL) and by making morphological observations of cell death using phase contrast microscopy. Apoptotic cells were identified by the presence of phase bright apoptotic bodies, or by pyknosis associated with membrane blebbing. Previous studies have verified that these morphological changes induced by SV in AT-3 cells are correlated with DNA laddering in multimers of 185 bp, and hypercondensation and fragmentation of chromatin characteristic of apoptosis (Lin et al., 1995).
For the cytotoxicity experiments involving TNF α (mouse; Boehringer Mannheim Corp., Indianapolis, IN), 104 AT-3 Zeo or AT-3 IκB Zeo cells were seeded/well in a 96-well plate; for cytotoxicity experiments involving H2O2, staurosporine 103 AT-3 Zeo or AT-3 IκB Zeo cells were seeded/ well in a 96-well plate. In each of these death paradigms, viability was assessed as described above.
Generation of Recombinant Sindbis Viruses Containing IκB wild-type (wt), IκB M, and IκB R
A fragment of hemagglutinin (HA; 3×)-tagged IκB α wt or an HA (3×) tagged-IκB α with serines 32 and 36 mutated to alanines (M) was cut from the HindIII/NotI sites of the corresponding Bluescript II Ks+ plasmids (DiDonato et al., 1996). Each fragment containing IκB wt or IκB M was blunt end–ligated into the BstEII site of a recombinant SV vector (ds TE12Q) in forward and reverse orientations. The SV vectors containing wild-type and mutated forms of HA-IκB α were linearized with PvuI and transcribed in vitro with SP6 RNA polymerase (GIBCO BRL, Gaithersburg, MD). The resultant RNA transcripts were then transfected into BHK cells using 10 μg of lipofectamine (GIBCO BRL) according to the manufacturer's instructions. 24 h after transfection, recombinant viruses were harvested from the supernatant of the culture media of the transfected BHK cells. Viral titers were determined by standard plaque assays as previously described.
Electrophoretic Mobility Shift Assays (EMSAs)
Cytoplasmic and nuclear extracts were generated as previously described (Lin et al., 1995). The protein concentration was determined by BCA assay (Pierce Chemical Co., Rockford, IL). Binding reactions for EMSA were performed at room temperature for 15 min using 6–8 μg of nuclear protein and 40,000 cpm (NF-κB) or 25,000 cpm (AP-1) of radiolabeled oligonucleotide. DNA–protein complexes were separated from unbound probe on native 5.3% polyacrylamide gels at 200–250 V for 2–3 h. The resultant gel was vacuum-dried and exposed on film (Eastman Kodak Co., Rochester, NY) overnight at −80°C.
Immunoblot Analysis
20 μg of protein from cytoplasmic extracts were boiled in Laemmeli buffer and electrophoresed under reducing conditions on 12% polyacrylamide gels. Proteins were then transferred to a polyvinylidene difluoride membrane (Bio-Rad Laboratories, Hercules, CA). Nonspecific binding was inhibited by incubation in Tris-buffered saline (TBST: 50 mM Tris-HCl, pH 8.0, 0.9% NaCl, 0.1% Tween 20) containing 5% nonfat dried milk for 2 h. Rabbit anti-IκB α antibody (C-21; Santa Cruz Biotechnology, Santa Cruz, CA) and a polyclonal antibody recognizing Sindbis virus structural proteins (a gift from Dr. Diane Griffin, The Johns Hopkins University School of Hygiene and Public Health) were diluted in 1% milk-TBST at 1:1,000. Mouse anti-HA (12 CA5, Boehringer Mannheim) monoclonal antibody was diluted 1:1,000 in 1% milk TBST. After exposure of membranes to HRP-conjugated anti-rabbit secondary antibody or HRP-linked anti-mouse secondary antibody for 1.5 h, immunoreactive proteins were detected according to the enhanced chemiluminescent protocol (Amersham Corp., Arlington Heights, IL). For quantitiave analysis of IκB α degradation after SV infection, secondary antibody incubation and immunocomplexes detection were performed according to the enhanced chemiluminescent fluorescence protocol (Amersham Corp.).
Generation of AT-3 Cell Lines with Enforced Expression of IκBs
A HindIII/Not I fragment containing HA-tagged IκB M was blunt-ended and ligated into the NheI site of an expression vector, pcDNA 3.1/Zeo (Invitrogen, San Diego, CA). 2 μg of pcDNA 3.1/HA-IκB Zeo or empty vector pcDNA 3.1/Zeo was transfected into AT-3 cells (2 × 105) using lipofectamine as described above. Resistant pools of clones containing the transfected plasmids were selected with Zeocin (Invitrogen Corp.) at 350 μg/ml. Approximately 30 pools of clones (AT-3 IκB Zeo or AT-3 Zeo) were isolated and expanded. Expression of IκB M in two independent pools of clones was verified by immunoblotting using an anti-IκB α antibody as described above.
Luciferase Assays
105 AT-3 Zeo or AT-3 IκB Zeo cells were transfected using the lipofectamine method, with 1.5 μg of SV40κB-luc reporter constructs (Ting et al., 1996) along with 500 ng of pCMV-βgal plasmids. Luciferase activity was determined 30 h after transfection, and was normalized on the basis of β-galactosidase expression level. Both the luciferase assay system and β-galactosidase assay system were purchased from Promega Corp.
Results
Inhibition of SV-induced NF-κB Activity in AT-3 Cells by Expression of IκB α in a Sindbis Virus Vector
To test whether SV-induced NF-κB activity is required for SV-induced apoptosis, we generated a recombinant SV that carries the wild-type IκB α gene (SV-IκB wt), or a more stable mutant form of IκB α (SV-IκB M), each containing three influenza virus HA epitope tags. As a control, we inserted the HA-tagged IκB wt in the reverse orientation into the SV vector. Infection of AT-3 cells with SV-IκB wt or SV-IκB M revealed the expression of HA-tagged IκB 8 h after infection (Fig. 1, A and B). Using an anti-HA antibody, we detected two bands reflecting HA-IκBs that likely are translated from different initiation sites within the HA-tagged region on the virus vector (Fig. 1 B). As expected, quantitative analysis of immunoblots using ECF and phosphorimaging showed 20% greater levels of HA-IκB in extracts from SV-IκB M–infected cells as compared with extracts from SV-IκB wt-infected cells.
Figure 1.


Inhibition of SV-induced NF-κB activity in AT-3 cells by heterologous expression of the inhibitory subunit of NF-κB, IκB α in a SV vector. (A–C) Cytoplasmic extracts were prepared from AT-3 cells infected with SV-IκB wt, SV-IκB M, or SV-IκB R at the time points indicated. A shows a Western blot analysis against IκBα. HA-IκB corresponds to the heterologous HA-tagged human IκB α; IκB corresponds to the endogenous IκB α; endogenous nonphosphorylated IκB α and nonspecific bands are denoted by an asterisk and N.S., respectively. (B) The blot shown in A was stripped, and anti-HA immunoreactive proteins are shown. C shows the Western blot analysis against the sindbis virus structural proteins. The upper arrow points to the viral envelope glycoproteins E1 and E2; the lower arrow points to the viral capsid protein. (D) EMSA performed with 6–8 μg of nuclear extracts corresponding to cytoplasmic extracts used in A–C using a 32P-labeled oligonucleotide with a consensus NF-κB sequence from the MHC class I promoter. Upper arrow points to the predominant complex induced by SV, p50-p65 heterodimer; the lower arrow points to p50–p50 homodimers. The composition of the induced NF-κB complexes were identified in a previous study (Lin et al., 1995).
Additionally, overexpression of IκB by the vector SV appeared to slow degradation of endogenous IκB α because we were able to detect modified, likely phosphorylated higher molecular weight forms of endogenous IκB α in cytoplasmic extracts from cells infected with SV-IκB wt and SV-IκB M, but not SV-IκB R-infected cells (Fig. 1 A). Moreover 39, 25, and 37% of endogenous IκB α from AT-3 cells infected with SV-IκB wt, SV-IκB M, and SV-IκB R, respectively, was degraded or converted to a modified form 16 h after infection as compared with IκB α in mock-infected cells (corresponding band is labeled by a star in Fig. 1 A). Altogether, these data demonstrate that we are able to express heterologous IκB wt and IκB M in AT-3 cells using the SV vector, and that, like the wild-type SV (Lin et al., 1998), the recombinant SVs result in degradation of endogenous IκB α.
To determine whether the expressed IκBs are able to suppress SV-induced NF-κB activity, we performed EMSAs on nuclear extracts from infected and mock-infected cells using a 32P-labeled probe with a consensus NF-κB binding site. As shown in Fig. 1 D, 8 h after infection, SV-IκB R elicited a dramatic increase in two NF-κB complexes, previously defined as p50–p65 and p50–p50 (Lin et al., 1995); SV-IκB M completely suppressed p50–p65 activity and diminished p50–p50 activity; and SV-IκB wt induced a level of p50–p65 activity intermediate between the SV-IκB M and the SV-IκB R. The differences in NF-κB activity among recombinant SV-IκBs was not due to their different replication rates in AT-3 cells, because levels of SV structural proteins (envelope glycoproteins, E1/E2, and capsid proteins) were similar in cells infected with each recombinant virus (Fig. 1 C).
Complete Suppression of SV-induced NF-κB Activity Does Not Inhibit SV-induced Apoptosis in AT-3 Cells
We demonstrated that SV-induced NF-κB activity could be suppressed using a recombinant SV containing a wt or mutant form of IκB α. To determine the effects of suppressing SV-induced NF-κB activity on apoptosis, we measured viability of cells infected with the different recombinant viruses at several time points after infection. As shown in Fig. 2, we found that SV-IκB wt, SV-IκB M, and SV-IκB R triggered cell death with similar kinetics in AT-3 cells. Similar results were observed in BHK cells (data not shown). These results suggest that SV-induced NF-κB activity is not required for SV-induced apoptosis. However, in a previous study we established that NF-κB is required for SV-induced death by pretreating AT-3 cells with NF-κB–specific transcription factor decoys that competitively inhibit binding of NF-κB to its target DNA sites. To reconcile these apparently conflicting observations, we hypothesized that NF-κB activity before infection is necessary for SV-induced apoptosis; that is, steady-state levels of NF-κB may regulate expression of latent death genes before SV infection.
Figure 2.

Cell viability of AT-3 cells infected with SV-IκB wt, SV-IκB M, and SV-IκB R. Percent viability was determined 24 and 48 h after infection by the alamar blue assay. Results are expressed as the mean ± SEM for experiments performed in triplicate (P > 0.05). Error bars that did not show up are hidden by the symbols.
Stable Overexpression of IκB M in AT-3 Cells Delays SV-induced Apoptosis
AT-3 cells that overexpress IκB M were established to examine whether inhibition of NF-κB activity beginning before SV infection inhibits SV-induced death. Two pools of cell clones stably expressing HA-tagged IκB M (AT-3 IκB Zeo) were generated along with a pool of control clones expressing a zeocin resistance gene product alone (AT-3 Zeo; Fig. 3 A). A pool of cell clones represents a population of one or more individual clones. EMSA indicated that IκB M overexpression in AT-3 cells functions as a superrepressor because there was significant suppression of NF-κB binding activity after SV infection in AT3-IκB Zeo clones compared with the AT-3 Zeo cells (Fig. 3 B). A slight induction of NF-κB activity 6 h after infection in the AT-3 IκB cells was observed, likely due to the heterogeneous levels of HA-IκB M expression in our pool of AT-3 IκB clones.
Figure 3.




The effect of stable overexpression of IκB α M on SV-induced apoptosis in AT-3 cells. (A) Western blot analysis demonstrating expression of HA-tagged IκB M in two pools of AT-3 IκB Zeo cell clones, but not in the corresponding AT-3 cells containing the zeocin resistance gene alone. (B) EMSA was performed using nuclear extracts obtained in parallel from SV-infected AT-3 Zeo or AT-3 IκB Zeo cells O, 4, and 6 h after infection. The upper arrow points to the p50–p65 complex, and the lower arrow points to p50–p50 homodimers. (C) Cell viability of AT-3 Zeo and AT-3 IKB Zeo cells 40 h after SV infection. Viability was determined by LDH assay as described in Materials and Methods. and is expressed as mean ± SEM for three separate experiments (P < 0.005). (D) Basal level of NF-κB transcriptional activity was nearly completely suppressed in AT-3 IκB Zeo cells compared with AT-3 Zeo cells. Basal level of NF-κB activity was determined by κB luciferase reporter system (P < 0.005).
AT-3 IκB Zeo cells that constitutively suppress NF-κB activity are more resistant to SV-induced death than are AT-3 Zeo cells (Fig. 3 C). The protection conferred by stable overexpression of IκB M could not be attributed to decreased viral entry or viral replication in these cells, because viral production measured 4 and 8 h after infection was similar (P > 0.5) in AT-3 IκB Zeo and AT-3 Zeo cells (data not shown). These results suggest that inhibition of NF-κB activity beginning before, but not after SV infection prevents SV-induced death. Indeed, using a luciferase reporter regulated by eight NF-κB binding sites, we found nearly complete suppression of constitutive NF-κB activity in AT-3 IκB Zeo cells compared with AT-3 Zeo cells (Fig. 3 D) or parental AT-3 cells (not shown).
Short-term Treatment of AT-3 Cells with N-acetyl-l-cysteine Combined with Infection of AT-3 Cells with SV-IκB M, but not SV-IκB R, Prevents Cell Death
We next used a different approach to confirm that inhibition of NF-κB activity must be initiated in advance of the apoptotic stimulus to prevent SV-induced death. In previous studies, we demonstrated that NAC at 30 mM prevents SV-induced apoptosis without interfering with viral replication (Lin et al., 1995). Therefore, we infected AT-3 cells with recombinant SV-IκBs in the presence of NAC. In this way, we could suppress activation of apoptosis induced by SV, but allow the recombinant SV to replicate and concomitantly express the exogenous HA–tagged IκB M. As expected, nearly complete survival was maintained in SV-IκB M and SV-IκB R–infected cells in the presence of 30 mM NAC (Fig. 4 A), and expression of HA-tagged IκB M in the SV-IκB M–infected cells, but not the SV-IκB R–infected cells, was verified by immunoblotting (data not shown). After 19 h, the NAC was removed. The cells infected with SV-IκB M were significantly protected from cell death as compared with cells infected with SV-IκB R 24 and 48 h after NAC withdrawal (Fig. 4 A). AT-3 cells that have been treated with NAC for 19 h appear to have no defects in NF-κB activation, because 4 h after NAC withdrawal, cells infected with SV-IκB R, but not the SV-IκB M, elicited increased nuclear NF-κB activity (Fig. 4 B).
Figure 4.


Short-term treatment of AT-3 cells with N-acetyl-l-cysteine (NAC) combined with infection with SV-IKB M, but not SV-IKB R, prevents cell death. (A) 2.5 × 103 cells/well in 96-well plates were infected with SV-IκB M or SV-IκB R, and were treated with 30 mM NAC for 19 h. The cells were washed twice with PBS, and fresh media without NAC was added. Cell viability was determined by the alamar blue assay 0, 24, and 48 h after NAC withdrawal. Results are expressed as the mean ± SEM for three experiments (P < 0.005) 24 and 48 hours after NAC withdrawal. (B) EMSAs were performed on nuclear extracts from SV-IκB M and SV-IκB R–infected cells 4 h after NAC withdrawal using 6–8 μg of protein and radiolabeled NF-κB or AP-1 probes. The quality of the nuclear exatracts from the treated and infected cells was confirmed by analysis of AP-1 DNA activity, indicating that NAC does not interfere with global nuclear protein activity. The arrow points to the NF-κB p50–p65 complex; the lower band is the NF-κB p50–p50 complex.
Embryonic Fibroblasts or 3T3 Cells Derived from p65−/− Mice, but Not Embryonic Fibroblasts Derived from p50−/− Mice, are Resistant to SV-induced Apoptosis
We next investigated whether inhibition of NF-κB activity must be initiated before the apoptotic stimulus to observe protection from SV-induced apoptosis in other cell types. Cell survival was examined in primary embryonic fibroblasts (EFs) or immortalized fibroblasts (3T3) from p65 knockout (p65−/−), p50 knockout (p50−/−), and wt mice infected with SV. Similar to our observations in AT-3 cells, SV-IκB wt, SV-IκB M, and SV-IκB R induced death in 3T3 cells with identical kinetics (Fig. 5 A). In contrast, p65−/− 3T3 cells, but not p50−/− 3T3 cells, were more resistant to SV-induced apoptosis than the wt 3T3 cells. These effects are unlikely to be due to secondary changes resulting from immortalization, because EFs from p65−/− mice, but not p50−/− mice, showed significant protection from SV-induced cell death as well (Fig. 5 B). Viral titers determined from the infected cell supernatant of wt 3T3 cells and p65−/− 3T3 cells 4, 8, and 12 h after infection indicated that SV was replicating equally well in both cells (P > .05; data not shown).
Figure 5.
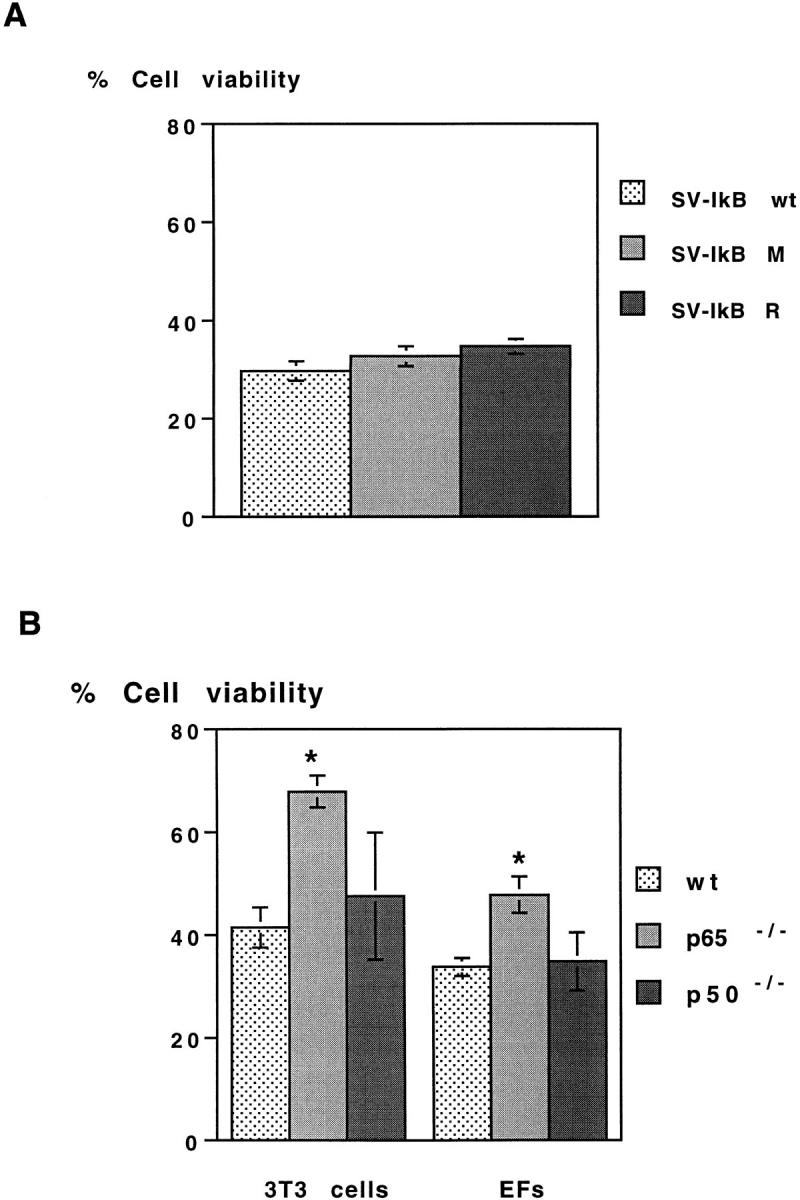
Cell viability of wt, p65 knockout, and p50 knockout embryo fibroblasts. (A) Viability of wt 3T3 cells infected with SV-IκB wt, SV-IκB M, and SV-IκB R 24 h after infection (P > 0.05). (B) Viability of wt, p65−/−, and p50−/− 3T3 cells and EFs infected with wt SV 24 h after infection. 3T3 refers to immortalized fibroblasts, while EFs refers to primary fibroblasts. Viability was determined by LDH assay as described in Materials and Methods and normalized to identical cell densities among the three cell types. Results are expressed as mean ± SEM for 3–5 experiments (P < 0.05).
Altogether, data from AT-3 prostate carcinoma cells and mouse embryo fibroblasts experiments argue that inhibition of NF-κB activity must be initiated before SV infection to suppress cell death, while inhibition of SV-induced NF-κB activity alone neither potentiates nor inhibits SV-induced death.
The Proapoptotic and Antiapoptotic Roles of NF-κB are Determined by the Death Stimulus
Our observations that fibroblasts from p65−/− mice are more resistant to SV–induced apoptosis are in contrast to previous observations that TNF α–induced apoptosis is potentiated in fibroblasts from p65−/− mice (Beg and Baltimore, 1996). These results suggest that the proapoptotic or antiapoptotic roles of NF-κB are determined by the death stimulus, and are not necessarily dependent on the cell type. To examine this possibility further, we compared viabilities in populations of AT-3 Zeo and AT-3 IκB M clones in response to a broad range of cytotoxic stimuli, including hydrogen peroxide (H2O2), TNF α, the general protein kinase inhibitor staurosporine (STS), and serum deprivation. Similar to SV infection, we found that cell death induced by 100 μM (not shown) or 500 μM peroxide was delayed in two populations of IκB M overexpressing cell clones as compared with control cells (Fig. 6 A); thus, in addition to SV infection, NF-κB also appears to exhibit prodeath activity after peroxide treatment. In contrast (and in agreement with observations from others; Liu et al., 1996; Beg and Baltimore, 1996; Wang et al., 1996; van Antwerp et al., 1996), exposure of AT3-IκB M cells to TNF α (10 ng/ml) or staurosporine (STS at 100 nM or 2 μM) results in significantly greater levels of cell death than AT3-Zeo cells exposed to each of these stimuli (Fig. 6, B and C). Finally, AT-3 Zeo or AT-3 IκB cells died at similar rates (P > 0.1) in response to 48 h of serum deprivation (Table I). These observations support the notion that the death stimulus is critically important in determining whether NF-κB regulates prodeath, antideath proteins, or is an innocent bystander.
Figure 6.

The effect of overexpression of IκB M on H2O2-, TNF α-, and STS-induced death in AT-3 cells. Cell viability was determined by LDH assay as described in Materials and Methods, and results are expressed as mean ± SEM for three separate experiments. AT-3 Zeo or AT-3 IκB Zeo cells exposed to: (A) 500 μM peroxide for 4 h (P < 0.005), (B) TNF-α (10 ng/ml) for 24 h (P < 0.005), and (C) 100 nM or 2 μM STS for 24 or 48 h (P < 0.005).
Table I.
The Role of NF-κB in Regulating Apoptosis in AT-3 Cells Depends on the Death Stimulus
| Stimulus | Proapoptotic | Antiapoptotic | ||
|---|---|---|---|---|
| SV | + | − | ||
| H2O2 (100 μM and 500 μM) | + | − | ||
| TNF (10 ng/ml) | − | + | ||
| STS (100 nM and 2 μM) | − | + | ||
| Serum deprivation (48 h) | − | − |
Discussion
Elucidation of the signaling pathways leading to activation of NF-κB, the precise identity of NF-κB family members, and definition of consensus DNA-binding sequences of this family of DNA-binding proteins have provided powerful molecular tools for suppressing nuclear NF-κB activity in intact cells, and in evaluating the role of NF-κB in apoptosis (Abbadie et al., 1993; Barger et al., 1995; Lin et al., 1995; Beg and Baltimore, 1996; Wang et al., 1996; van Antwerp et al., 1996). In each of these studies, deletion of individual NF-κB subunits or expression of inhibitors of NF-κB nuclear activity were initiated before the apoptotic stimulus, so it could not be determined if the critical cell death regulatory function(s) of NF-κB activity occur before or after the death stimulus. Indeed, in previous studies we showed that suppression of SV-induced NF-κB activity by pretreatment with NF-κB–specific transcription factor decoys inhibits SV-induced apoptosis (Lin et al., 1995). Our previous results could not distinguish between two likely models (Fig. 7). In the first model (see Fig. 7 A), SV induces degradation of IκB, leading to translocation of NF-κB to the nucleus and expression of one or more κB- dependent death proteins. In the second model (see Fig. 7 B), nuclear NF-κB activity occurring at some time before the SV infection is required for expression of a protein involved in apoptosis that is directly or indirectly activated by SV infection. In the second scheme, SV-induced NF-κB activity could be involved in regulating responses not directly related to cell death, such as the inflammatory response. Herein, we show that a recombinant SV carrying a superrepressor form of IκB α suppresses SV-induced NF-κB nuclear activity (Fig. 1), but does not inhibit or potentiate apoptosis in AT-3 prostate carcinoma cells (Fig. 2) or 3T3 embryo fibroblasts (Fig. 5 A). These results, combined with our observations that AT-3 cells stably overexpressing the superrepressor of NF-κB (Fig. 3) or mouse embryo fibroblasts deleted in the p65 subunit of NF-κB (Fig. 4) are more resistant to SV-induced death, are consistent with the second model. Furthermore, these data imply a requirement for a p65-containing NF-κB complex to drive expression of an essential gene for SV-induced cell death.
Figure 7.

Proposed models by which NF-κB regulates SV-induced apoptosis. (A) SV induces degradation of IκB, allowing translocation of an NF-κB heterodimer from the cytoplasm to the nucleus. NF-κB activates transcription of one or more unknown death genes required for cell death. (B) Before infection, steady state NF-κB activity regulates expression of an inactive form of a death protein that is posttranscriptionally or posttranslationally activated by SV, leading to apoptosis. In this scheme, SV-induced NF-κB activity is not involved in regulating SV-induced apoptosis (dashed line).
If Sindbis-virus–induced NF-κB activation is not an important determinant in cell viability, what is the stimulus before infection leading to NF-κB–dependent synthesis of prodeath proteins? Several lines of evidence have demonstrated that NF-κB activity can be modulated during cell cycle progression (Baldwin et al., 1991; Li et al., 1993; Evans et al., 1993; Duckett et al., 1995; Bash et al., 1997). In quiescent 3T3 fibroblasts, NF-κB is quickly induced by stimulation with serum or growth factors as cells transition from G0 to G1 stages of the cell cycle (Duckett et al., 1995). Moreover, specific cyclin-dependent kinases were shown to regulate transcriptional activation by NF-κB through the interaction with the coactivator p300 (Perkins et al., 1997). Thus, a cell cycle–dependent NF-κB activity could be activated before infection. Such an activity may be undetectable by EMSA (Fig. 1 D and Fig. 3 B), but detectable by NF-κB reporter gene assay (Fig. 3 D), which is more sensitive. The inability to detect NF-κB DNA binding may be due to the unsynchronized nature of the population of cells we study, or because cell cycle–dependent activation of NF-κB– dependent genes occurs as a result of phosphorylation of nuclear p65-containing complexes without enhanced nuclear translocation of NF-κB (Finco et al., 1997). We speculate that the cell cycle–dependent activation of NF-κB may regulate expression of inactive forms of one or more apoptotic effectors or regulators before the apoptotic stimulus. Of note, previous studies have shown that inducible expression of a dominant negative Ras inhibits cell cycle progression and SV-induced apoptosis, while infection of PC12 cells with recombinant SV carrying a dominant negative Ras has no effect on viability (Joe et al., 1996). These findings, along with data presented herein and recent observations demonstrating that oncogenic Ras activates NF-κB–dependent gene expression, are consistent with the following model: a cell cycle– dependent Ras-induced signal transduction pathway activates NF-κB that regulates the expression of proapoptotic genes before SV infection.
How can the proapoptotic role of NF-κB in the setting of SV infection be reconciled with the established antiapoptotic role of NF-κB in response to other apoptotic stimuli? The stimulus, the cell type, and the subunit of NF-κB activated have all been proposed as critical factors that determine whether this transcription factor promotes or suppresses apoptosis. We provide evidence suggesting that the stimulus may be the main factor in determining NF-κB's ability to regulate divergent biological outcomes. First, overexpression of IκB M in AT-3 cells protects from SV and H202-induced cell death, but potentiates TNF-α and staurosporine-induced cell death, indicating that suppression of NF-κB activity can be proapoptotic or antiapoptotic in the same cell type, depending on the stimulus. Second, fibroblasts from p65−/−mice, but not p50−/− mice, are resistant to SV-induced death, but more sensitive to TNF-α–induced death. These observations show that the deleted subunit of NF-κB is required for specific target gene activation leading to either a pro- or antiapoptotic response, but is not a necessary determinant in the proapoptotic or antiapoptotic actions of NF-κB. Finally, our observation that serum deprivation–induced death occurs with similar kinetics in AT-3 Zeo and AT-3 IκB Zeo cells suggests that NF-κB is not part of this common pathway of apoptotic death, and that other stimulus-specific pathways may need to be simultaneously activated to determine NF-κB's role in determining cell survival or death.
Our results are congruent with the emerging concept of private cell death pathways (Smith and Osborne, 1997). This notion acknowledges the existence of a common effector phase to apoptotic cell death, but states that within a given cell, many stimuli are capable of inducing apoptosis, and that each stimulus may do so by inducing a unique set of genes. These genes in turn may positively or negatively regulate activation of the common effector pathway. Heretofore, the best-studied examples of stimulus-specific cell death pathways have been in lymphocytes, where ionizing radiation (Lowe et al., 1993), glucocorticoids (Liu et al., 1994), and T cell activation (Woronicz et al., 1994) each activate unique transcription factor responses that converge on a common final effector pathway to apoptosis. Our studies carry this concept a step further, and suggest that a single transcription factor, NF-κB, can mediate activation or suppression of cell death signals in the same cell type depending on the stimulus (Fig. 6; Table I). Furthermore, we identify that timing of NF-κB activation is another important variable in determining whether this transcription factor regulates expression of prodeath proteins, or is an innocent bystander (Figs. 2, 3 C, and 5). The demonstration herein that steady-state NF-κB activity is required to promote SV-induced cell death in AT-3 prostate carcinoma cells and 3T3 fibroblasts will focus future studies aimed at defining one or more NF-κB–regulated genes required for SV-induced apoptosis.
Accumulating evidence indicates that apoptosis results from activation of a constitutively expressed suicide program that is conserved in evolution from nematodes to humans (Steller, 1995; Martin and Green, 1995). Interestingly, in prokaryotes such as Escherichia coli, a protease whose specific substrate is the translational elongation factor EF-Tu, is constitutively expressed. During infection with bacteria phage, this protease is activated and cleaves EF-Tu, leading to death of the cells (Snyder, 1995; Jacobson et al., 1997). Our findings suggest a parallel example in mammalian cells where SV infection posttranscriptionally or posttranslationally activates a constitutively expressed suicide program that is regulated in part, by NF-κB before infection.
Acknowledgments
We would like to thank Dr. Diane Griffin for helpful comments, and Dr. Brian Seed for providing the NF-κB luciferase reporter construct. This work was supported by grants from the National Institutes of Health (NINDS R29 NS34943 and KO8 NS01951 R.R. Ratan).
Footnotes
1. Abbreviations in this paper: EMSA, electrophoretic mobility shift assay; HA, hemagglutinin; M, mutant; NAC, N-acetylcysteine; R, reverse; SV, Sindbis virus; STS, staurosporine; wt, wild-type.
Address all correspondence to Rajiv R. Ratan, M.D., Ph.D., Neurology Laboratories at Beth Israel Deaconess Medical Center, Harvard Institutes of Medicine, Rm. 857; 77 Avenue Louis Pasteur, Boston, MA 02115.
References
- Abaddie CN, Kabrun F, Bouali F, Vandenbunder B, Enrietto P. High levels of c-rel expression are associated with programmed cell death in the developing avian embryo and in bone marrow cells in vitro. Cell. 1993;75:899–912. doi: 10.1016/0092-8674(93)90534-w. [DOI] [PubMed] [Google Scholar]
- Barger SW, Horster D, Furukawa K, Goodman Y, Kriegelstein J, Mattson MP. Tumor necrosis factor alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+accumulation. Proc Natl Acad Sci USA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D. IκB: a specific inhibitor of the NF-κB transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Baldwin AS, Jr, Azizkhan JC, Jensegn DE, Beg AA, Coodly LR. Induction of NF-κB DNA binding activity during the G0-to-G1transition in mouse fibroblasts. Mol Cell Biol. 1991;11:4843–4951. doi: 10.1128/mcb.11.10.4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bash J, Zong W-X, Gélinas C. c-Rel arrests the proliferation of HeLa cells and affects critical regulators of the G1/S-phase transition. Mol Cell Biol. 1997;17:6526–6536. doi: 10.1128/mcb.17.11.6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α–induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Beg AA, Ruben SM, Scheinman RI, Haskill S, Rosen CA, Baldwin AS., Jr IκB interacts with the nuclear localization sequences of the subunits of NF-κB: a mechanism for cytoplasmic retention. Gene Dev. 1992;6:1899–1913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the Rel A component of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of IκB-α proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- Casano FJ, Rolando AM, Nudgett JS, Molineaux SM. The structure and complete nucleotide sequence of the murine gene encoding interleukin-1 β converting enzyme (ICE) Genomics. 1994;20:474–481. doi: 10.1006/geno.1994.1203. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IκBα by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- Cheng EHY, Levine B, Boise LH, Thompson CB, Hardwick JM. Bax-independent inhibition of apoptosis by Bcl-XL . Nature. 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- DiDonato JA, Mercurio F, Rosette C, Jian W-L, Suyang H, Ghosh S, Karin M. Mapping of the inducible IκB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1995;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato JA, Mercurio F, Karin M. Phosphorylation of IκBα precedes but is not sufficient for its dissociation from NF-κB. Mol Cell Biol. 1996;15:1302–1311. doi: 10.1128/mcb.15.3.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Duckett CS, Perkins ND, Leung K, Agranoff AB, Nabel GJ. Cytokine induction of nuclear factor κB in cycling and growth-arrested cells. J Biol Chem. 1995;270:18836–18840. doi: 10.1074/jbc.270.32.18836. [DOI] [PubMed] [Google Scholar]
- Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-junas necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RB, Gottlieb PD, Bose HR., Jr Identification of a Rel- related protein in the nucleus during the S phase of the cell cycle. Mol Cell Biol. 1993;13:6147–6156. doi: 10.1128/mcb.13.10.6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finco TS, Westwick JK, Norris JL, Beg AA, Der CJ, Baldwin AS., Jr Oncogenic Ha-Ras-induced signaling activates NF-κB transcriptional activity, which is required for cellular transformation. J Biol Chem. 1997;272:24113–24116. doi: 10.1074/jbc.272.39.24113. [DOI] [PubMed] [Google Scholar]
- Grimm S, Bauer MKA, Baeuerle PA, Schulze-Osthoff K. Bcl-2 down-regulates the activity of transcription factor NF-κB induced upon apoptosis. J Cell Biol. 1996;134:13–23. doi: 10.1083/jcb.134.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Henkel T, Machleidt T, Alkalay I, Krönke M, Ben-Neriah Y, Baeuerle PA. Rapid proteolysis of IκB-α is necessary for activation of transcription factor NF-κB. Science. 1993;365:182–185. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Jenh BM, Osborne BA. Gene regulation associated with apoptosis. Crit Rev Eukaryot Gene Expr. 1997;7:179–193. doi: 10.1615/critreveukargeneexpr.v7.i1-2.100. [DOI] [PubMed] [Google Scholar]
- Joe AK, Ferrare G, Jiang HH, Liang XH, Levine B. Dominant inhibitory Ras delays Sindbis-virus induced apoptosis in neuronal cells. J Virol. 1996;70:7744–7751. doi: 10.1128/jvi.70.11.7744-7751.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa FA, Pierce JP, Sonenshein GE. Differential regulation of the c-myc oncogene promoter by the NF-κB Rel family of transcription factors. Mol Cell Biol. 1994;14:1039–1044. doi: 10.1128/mcb.14.2.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Huang Q, Isaacs JT, Reed JC, Griffin DE, Hardwick JM. Conversion of lytic persistent alphavirus infection by the bcl-2cellular oncogene. Nature. 1993;361:739–742. doi: 10.1038/361739a0. [DOI] [PubMed] [Google Scholar]
- Levine B, Goldman JE, Jiang HH, Griffin DE, Hardwick JM. Bcl-2 protect mice against alphavirus encephalitis. Proc Natl Acad Sci USA. 1996;93:4810–4815. doi: 10.1073/pnas.93.10.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, Wesselingh SL, Griffin DE, Hardwick JM. Alphavirus-induced apoptosis in mouse brains correlates with neurovirulence. J Virol. 1996;70:1828–1835. doi: 10.1128/jvi.70.3.1828-1835.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Sedivy JM. Raf-1 protein kinase activates the NF-κB transcription factor by dissociating the cytoplasmic NF–κB–IκB complex. Proc Natl Acad Sci USA. 1993;90:9247–9251. doi: 10.1073/pnas.90.20.9247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K-I, Lee S-H, Narayanan R, Baraban JM, Hardwick JM, Ratan RR. Thiol agents and Bcl-2 identify an alphavirus-induced pathway that requires activation of the transcription factor NF-κB. J Cell Biol. 1995;131:1149–1161. doi: 10.1083/jcb.131.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, K.-I., J.M. Baraban, and R.R. Ratan. 1998. Inhibition versus induction of apoptosis by proteasome inhibitors depends on concentration. Cell Death and Differentiation. In press. [DOI] [PubMed]
- Liu Z-G, Smith SW, McLaughlin KA, Schwartz LM, Osborne BA. Apoptotic signals delivered through the T-cell receptor of a T-cell hybrid require the immediate-early gene nur77. Nature. 1994;367:281–284. doi: 10.1038/367281a0. [DOI] [PubMed] [Google Scholar]
- Liu Z-G, Hsu H, Goeddel D, Karin M. Disection of TNF receptor 1 effector function: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? . Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JE, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opipari AW, Jr, Hu HM, Yabkowitz R, Dixit VM. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J Biol Chem. 1992;267:12424–12427. [PubMed] [Google Scholar]
- Oppenheim RW, Prevette D, Tytell M, Homma S. Naturally occurring and induced neuronal death in the chick embryo in vivo requires protein and RNA synthesis. Dev Biol. 1990;138:104–113. doi: 10.1016/0012-1606(90)90180-q. [DOI] [PubMed] [Google Scholar]
- Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- Perkins ND, Felzein LK, Betts JC, Leung K, Beach DH, Nabel G. Regulation of NF-κB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Régnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- Smith SW, Osborne BA. Private pathways to a common death. J NIH Res. 1997;9:33–37. [Google Scholar]
- Snyder L. Phage-exclusion enzymes: a bonanza of biochemical and cell biology reagents? . Mol Microbiol. 1995;15:415–420. doi: 10.1111/j.1365-2958.1995.tb02255.x. [DOI] [PubMed] [Google Scholar]
- Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Tanaka M, Inazawa J, Abe T, Suda T, Nagata S. Human Fas ligand: gene structure, chromosomal localization and species specificity. Int Immnuol. 1994;6:1567–1574. doi: 10.1093/intimm/6.10.1567. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Ishihara M, Harada H, Kimura T, Matsuyama T, Lamphier MS, Aizawa S, Mak TW, Taniguchi T. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell. 1994;77:829–839. doi: 10.1016/0092-8674(94)90132-5. [DOI] [PubMed] [Google Scholar]
- Ting AT, Pimentel-Muiños FX, Seed B. RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO (Eur Mol Biol Organ) J. 1996;15:6189–6196. [PMC free article] [PubMed] [Google Scholar]
- Thanos D, Maniatis T. NF-κB: A lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- Traenckner EB-M, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human IκB-α on serines 32 and 36 controls IκB-α proteolysis and NF-κB activation in response to diverse stimuli. EMBO (Eur Mol Biol Organ) J. 1995;14:2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Wang C-Y, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy–induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Wong GH, Elwell JH, Oberley LW, Goeddel DV. Manganous superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell. 1989;58:923–931. doi: 10.1016/0092-8674(89)90944-6. [DOI] [PubMed] [Google Scholar]
- Woronicz JD, Calnan B, Ngo V, Winoto A. Requirement for the orphan steroid receptor Nur77 in apoptosis of T-cell hybridomas. Nature. 1994;367:277–280. doi: 10.1038/367277a0. [DOI] [PubMed] [Google Scholar]
- Wu H, Lozano G. NF-κB activation of p53. A potential mechanism for suppressing cell growth in response to stress. J Biol Chem. 1994;269:20067–20074. [PubMed] [Google Scholar]
- Wu M, Lee H, Bellas RE, Schauer SL, Arsura M, Katz D, Fitzgerald MJ, Rothstein TL, Sherr DH, Sonenshein GE. Inhibition of NF-κB/Rel induces apoptosis of murine B cells. EMBO (Eur Mol Biol Organ) J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]