

Abstract
NIMA promotes entry into mitosis in late G2 by some mechanism that is after activation of the Aspergillus nidulans G2 cyclin-dependent kinase, NIMXCDC2/NIMECyclin B. Here we present two independent lines of evidence which indicate that this mechanism involves control of NIMXCDC2/NIMECyclin B localization. First, we found that NIMECyclin B localized to the nucleus and the nucleus-associated organelle, the spindle pole body, in a NIMA-dependent manner. Analysis of cells from asynchronous cultures, synchronous cultures, and cultures arrested in S or G2 showed that NIMECyclin B was predominantly nuclear during interphase, with maximal nuclear accumulation in late G2. NIMXCDC2 colocalized with NIMECyclin B in G2 cells. Although inactivation of NIMA using either the nimA1 or nimA5 temperature-sensitive mutations blocked cells in G2, NIMXCDC2/NIMECyclin B localization was predominantly cytoplasmic rather than nuclear. Second, we found that nimA interacts genetically with sonA, which is a homologue of the yeast nucleocytoplasmic transporter GLE2/RAE1. Mutations in sonA were identified as allele-specific suppressors of nimA1. The sonA1 suppressor alleviated the nuclear division and NIMECyclin B localization defects of nimA1 cells without markedly increasing NIMXCDC2 or NIMA kinase activity. These results indicate that NIMA promotes the nuclear localization of the NIMXCDC2/ NIMECyclin B complex, by a process involving SONA. This mechanism may be involved in coordinating the functions of NIMXCDC2 and NIMA in the regulation of mitosis.
Entry into mitosis in Aspergillus nidulans is regulated by the coordinate function of two serine/threonine protein kinases, NIMXCDC2 and NIMA. NIMXCDC2 is an essential histone H1 kinase that is structurally and functionally homologous to fission yeast p34cdc2 (Osmani et al., 1994). NIMA is a β-casein kinase and is structurally distinct from p34cdc2, containing an amino-terminal catalytic domain and a carboxyl-terminal regulatory domain (Osmani et al., 1988b ; Lu et al., 1993; Pu and Osmani, 1995; Pu et al., 1995). Failure to properly activate either of these kinases in G2 prevents the initiation of mitosis, and the combined action of both kinases is critical for coordinating changes in chromosome, microtubule, and nuclear membrane structure during mitosis. For example, mutations preventing the activation of NIMXCDC2 in G2 normally arrests cells in late G2 (Osmani et al., 1991a ; 1994). Although overexpression of NIMA can overcome this interphase arrest, the ensuing mitosis is disorganized such that chromosome condensation occurs but normal spindle assembly does not (O'Connell et al., 1994; Pu and Osmani, 1995). Likewise, nimA mutations normally arrest cells in late G2 (Osmani et al., 1987; 1991a ). Although a bimEAPC1 checkpoint mutation can overcome this G2 arrest, the ensuing mitosis is disorganized and includes aberrant spindle, chromatin, and nuclear membrane structure (Osmani et al., 1988a ; 1991b ).
To ensure the coordinated function of NIMXCDC2 and NIMA, each kinase must somehow be sensitive to the function of the other. Phosphorylation of NIMA by NIMXCDC2 is likely to be involved in making NIMA function sensitive to NIMXCDC2 at the G2 to M transition (Ye et al., 1995). Before activation of NIMXCDC2 in late G2, NIMA is hypophosphorylated and active as a β-casein kinase. Upon activation of NIMXCDC2, NIMA is converted to a hyperphosphorylated, slightly more active form that reacts well with the antiphosphoprotein antibody, MPM2. The finding that NIMXCDC2 is necessary for NIMA hyperphosphorylation and MPM2 reactivity in vivo, and is sufficient for NIMA hyperphosphorylation and MPM2 reactivity in vitro, is consistent with a direct role for NIMXCDC2 in NIMA hyperphosphorylation (Ye et al., 1995).
The role of NIMA hyperphosphorylation in NIMA's function as a mitotic regulator remains to be determined. Hyperphosphorylated NIMA is detectable coincident with the initiation of mitosis in synchronous cultures, consistent with hyperphosphorylation playing a positive role in NIMA's function as a mitotic inducer (Ye et al., 1995). This stimulatory effect could be at the level of NIMA activity, since hyperphosphorylation causes a twofold increase in NIMA's β-casein kinase activity. Hyperphosphorylation could also regulate NIMA function at other levels, for example, by regulating NIMA localization or NIMA proteolysis. The identification of several consensus CDC2 phosphorylation sites in NIMA's carboxyl terminus (Osmani et al., 1988b ; Fry and Nigg, 1995) and the requirement of the carboxyl terminus for NIMA proteolysis (O'Connell et al., 1994; Pu and Osmani, 1995) is suggestive of hyperphosphorylation playing a role in regulating NIMA turnover.
If there is a mechanism making NIMXCDC2 function sensitive to NIMA activity, it does not involve regulation of NIMXCDC2 activation (Osmani et al., 1991a ). Like most eukaryotic cells (Nurse, 1990), activation of CDC2 during G2 in Aspergillus is mediated by its binding to the Cyclin B homologue, NIMECyclin B, which is the principle B-type cyclin associated with activated NIMXCDC2 during G2 (Bergen et al., 1984; Osmani et al., 1994; James et al., 1995). NIMXCDC2/NIMECyclin B is activated by dephosphorylation of tyrosine residue 15 on NIMXCDC2 (O'Connell et al., 1992; Osmani et al., 1994). Tyrosine phosphorylation of NIMXCDC2 requires the function of the p107wee1 homologue, ANKAWEE1, (Ye et al., 1996), and tyrosine dephosphorylation requires the function of the p80cdc25 homologue, NIMTCDC25 (O'Connell et al., 1992). nimA mutations cause a specific cell cycle arrest in G2 very close to the nimTcdc25 mutant arrest point, yet they do not prevent formation of a NIMXCDC2/NIMECyclin B complex, dephosphorylation of NIMXCDC2 on tyrosine 15, or activation of the NIMXCDC2/NIMECyclin B complex as a histone H1 kinase (Osmani et al., 1991a ; Pu et al., 1995). Furthermore, the nimA5 mutation prevents mitosis even in strains expressing a mutant form of NIMXCDC2 which cannot be phosphorylated on threonine 14 or tyrosine 15 (Ye et al., 1996). Thus, loss of NIMA function prevents mitosis by some mechanism other than regulation of the activity of NIMXCDC2/NIMECyclin B.
One way in which NIMXCDC2 function could be affected by NIMA would be if NIMA function was required for proper localization of activated NIMXCDC2. It is known that CDC2/cyclin localization is regulated for certain cyclin-dependent kinase complexes (for example see Pines and Hunter, 1991; Gallant and Nigg, 1992; Ookata et al., 1992; Maridor et al., 1993; Ookata et al., 1995). Here we present evidence from two independent lines of investigation supporting a role for NIMA in the subcellular localization of NIMXCDC2/NIMECyclin B. First, using indirect immunofluorescence analysis of fixed cells, we found that NIMXCDC2 and NIMECyclin B localized to the nucleus and the nuclear-associated organelle, the spindle pole body (SPB)1, in a NIMA-dependent manner. Second, using suppressor analysis, we found that mutations in a homologue of the nucleocytoplasmic transporter GLE2/RAE1 (Brown et al., 1995; Murphy et al., 1996) act as allele-specific suppressors of the nimA1 mutation. Together, these results suggest a role for NIMA in the nuclear localization of the NIMXCDC2/NIMECyclin B and they provide evidence for a mechanism by which NIMXCDC2/NIMECyclin B function is made sensitive to NIMA to coordinate the action of these two mitotic promoting kinases.
Materials and Methods
Strains, Microbiological Techniques, and Genetic Analyses
Aspergillus strains used in this study are listed in Table I. Standard conditions were used for Aspergillus propagation (Morris, 1976; Kafer, 1977), genetics (Pontecorvo et al., 1953), and transformation (Osmani et al., 1987; Gems et al., 1991, 1994). The conditions and procedures used to grow A. nidulans cultures and isolate protein extracts were as described previously (Ye et al., 1995) except where noted in the text. For cytological analyses, A. nidulans cells were grown in liquid YG (Morris, 1976) on coverslips as previously described (Mirabito and Morris, 1993).
Table I.
A. nidulans Strains Used in This Study
| Strain | Genotype | Reference | ||
|---|---|---|---|---|
| A154 | adE20; biA1; wA2, cnxE16; sC12: methG11; nicA2; lacA1; choA1; chaA1 | FGSC* | ||
| A612 | acrA1; riboB2; chaA1 | FGSC | ||
| GR5 | wA2; pryG89; pyroA4 | FGSC | ||
| LPW2 | riboA1; wA2; nimA1; nicA2 | This study | ||
| LPW16 | pyrG89; wA2; sonA1 | This study | ||
| LPW29 | pyrG89, riboB2; wA2; nimA1; sonA1 | This study | ||
| LPW42 | LPW16 transformed with pMTW2 | This study | ||
| PMC645-4 | HA-nimE, wA2, cnxE16; nicA2 | This study | ||
| PMC654-19 | HA-nimE; cnxE16; nimA5; nicA2; chaA1 | This study | ||
| R153 | wA3; pyroA4 | FGSC | ||
| SFC4-21 | pabaA1, yA2; HA-nimE, nimT23; nicA2; choA1 | This study | ||
| SFC403-19 | riboA1, pabaA1; HA-nimE; nimA1; methG11; choA1; chaA1 | This study | ||
| SFC444-1 | pabaA1; HA-nimE; nimA1; nicA2; choA1; sonA1 | This study | ||
| SFC612-5 | riboA1; HA-nimE; nimA5; sonA1 | This study |
FGSC, Fungal Genetics Stock Center, Department of Microbiology, University of Kansas Medical Center, Kansas City, Kansas 66160-7420.
Fluorescence Microscopy
Cells were fixed and stained with 4′,6-diamidino-2-phenylindole (DAPI) to visualize nuclei as previously described (Osmani et al., 1987). Cells were fixed and prepared for indirect immunofluorescence microscopy as previously described (Mirabito and Morris, 1993) with the following exceptions. Cell walls were removed using 40 mg/ml novozyme 234 (NOVO 234) (InterSpex Products, San Mateo, CA), 80 mg/ml Driselase (Interspex Products, Foster City, CA), 1 mM diisopropyl fluorophosphate (DIFP) (Sigma Chemical Co., St. Louis, MO), 2 μg/ml leupeptin, and 40 mg/ml Aprotinin (Sigma Chemical Co.). Lipids were extracted using −20°C methanol for 8 min followed by −20°C acetone for 30 s. Lipid extraction using room temperature methanol or 1% NP-40 yielded similar results. Coverslips were mounted on mounting medium (90% glycerol in TBS containing 1 mg/ml p-phenylenediamine).
Primary Abs used were 12CA5 (Berkeley Antibody Co., Richmond, CA) at 10 mg/ml; rabbit anti-NIMECyclin B serum E8 (Osmani et al., 1994) at 1:1,000; preimmune rabbit serum E8 at 1:1,000; rabbit anti-NIMXCDC2 serum E77 (Osmani et al., 1994) at 1:1,000; preimmune rabbit serum E77 at 1:1,000; MPM2 (gift of J. Kuang, M.D. Anderson Cancer Center, Houston, TX) at 1:1,000; affinity-purified rabbit anti-γ-tubulin (gift of B. Oakley, Ohio State University, Columbus, OH) at 1:100; anti-histone H1 mouse mAb (gift of A. Epstein, University of Southern California, School of Medicine, Los Angeles, CA) at 1:1,000; and DM1A (Sigma Chemical Co.) at 1:100. Secondary Abs (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were CY3-labeled, goat anti–mouse IgG at 1:500, dichlorotriazinylamino fluorescein (DTAF)-labeled, goat anti–mouse IgG at 1:250, CY3-labeled, goat anti–rabbit IgG at 1:500, and DTAF-labeled, goat anti–rabbit IgG at 1:250. Photomicrographs of cells stained with 12CA5 within each figure were produced using identical conditions of fixation, staining, exposure, and enlargement.
For colocalization of NIMECyclin B and SPB or nuclear antigens, some experiments involved double staining using two mouse mAbs (12CA5 and anti-histone H1 or 12CA5 and MPM2). For these experiments, fixed cells were incubated first in 12CA5 and then in a conjugated, anti-mouse secondary antibody, and then in 20 mg/ml of unconjugated, anti-mouse Fab fragments (Jackson ImmunoResearch Laboratories, Inc.) before incubation in the second mouse mAb. This treatment effectively blocked all the anti-mouse IgG sites on the 12CA5 mAb: no further binding to 12CA5 was detectable in control experiments. Colocalization of NIMECyclin B and SPBs was confirmed using 12CA5 and the affinity-purified, rabbit anti– γ-tubulin Ab.
Photomicrographs were captured using either a Photometrics charge-coupled device (CCD) camera (model Sensys; Tucson, AZ) and manipulated using Phase 3 Imaging Systems software (Sterling Heights, MI) (see Fig. 8) or a Diagnostics Instruments Inc., (Media Cybernetics, Silver Spring, MD) CCD camera and then manipulated using Adobe Photoshop 4.0 (Adobe Systems, Inc., San Jose, CA).
Figure 8.

SONA-HA localizes to the nuclear periphery. Cells of the SONA-HA–expressing strain, LPW42, were cultured, fixed, and then prepared for immunocytology as described in Materials and Methods. The image is identified to the left (SONA-HA shows 12CA5 staining). Arrows, example nuclei within the multinucleate cell shown. Digital images were captured using a Sensys Photometrics CCD camera and were merged using Phase 3 Imaging Systems software. Bar, 10 μm.
Culture Conditions for NIME Localization Studies
For analysis of NIMECyclin B and NIMXCDC2 localization in exponentially growing asynchronous cultures, cells were incubated in YG on coverslips at 32°C for 8–12 h. For analysis of NIME localization in cells arrested in S phase, cells were incubated in YG at 32°C for 6 h and then hydroxyurea was added at 50 mM and incubation was continued for an additional 3 h at 32°C. For analysis of NIMECyclin B and NIMXCDC2, localization in cultures shifted to restrictive temperature, cells were incubated in YG liquid at permissive temperature until they contained an average of 2–4 nuclei, and then the cultures were placed in a 44°C incubator for up to 6 h. To generate synchronous cultures, nimTcdc25 or nimA mutants were germinated for 2 h at 32°C, shifted to 44°C for 5 h to arrest cells in G2 of their first cell cycle. The cells were shifted back to either 32°C or room temperature by replacing the medium. Samples were taken before the shift and at the times after the shift as indicated in the legends to Figs. 3 and 5.
Figure 3.

Kinetic analysis of NIMECyclin B localization in cells from a synchronized culture. Cells of the nimTcdc25 mutant, SFC4-21, were cultured, fixed, and then prepared for immunocytology as described in Materials and Methods. The cells were stained for HA-NIMECyclin B, α-tubulin, and DAPI. A and B present data from the same samples plotted to show the fraction of cells with nuclear NIMEHA (▪) relative to nuclear division (▴) in A or relative to spindle formation (▴) in B. Each value was determined by counting more than 300 cells.
Figure 5.
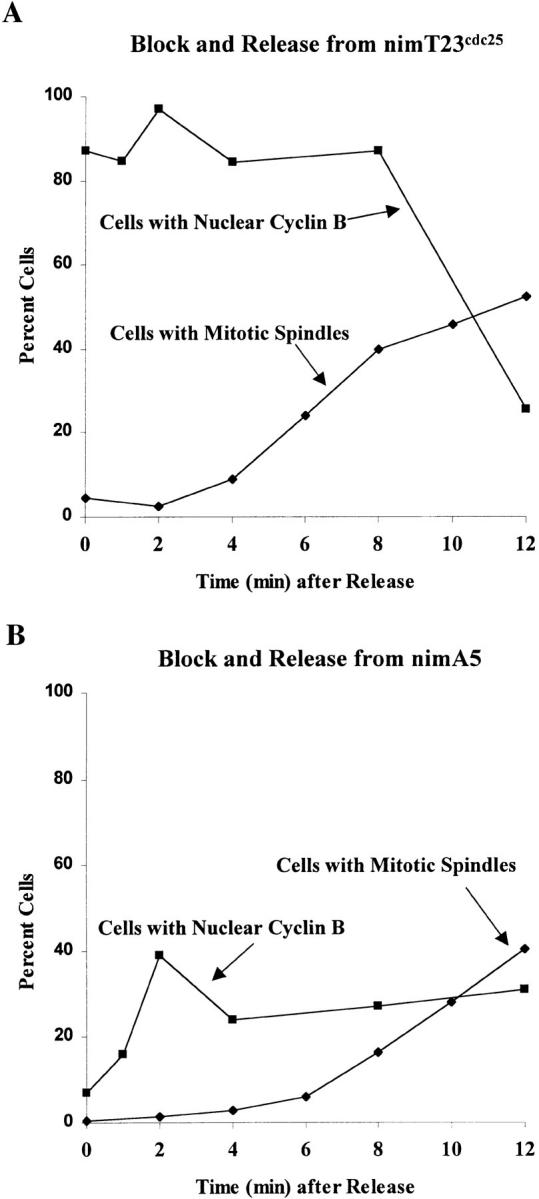
Cells released from the nimA5 G2 arrest accumulate nuclear NIMECyclin B before entering mitosis. Cells of the nimTcdc25 mutant, SFC4-21 (A), and the nimA5 mutant, PMC654-19 (B), were arrested in late G2 by incubation at restrictive temperature and then released from the G2 arrest by shift to permissive temperature (refer to Materials and Methods). The cells were stained for HA-NIMECyclin B to determine the percent cells with nuclear cyclin B (▪) and with anti–α-tubulin to determine the percent cells with mitotic spindles (♦). Each value was determined by counting at least 100 cells.
Isolation and Characterization of Strains Carrying the sonA1 Suppressor Mutation
Spores of the temperature-sensitive (ts) nimA1 strain, LPW2, were mutagenized with 4-nitroquinoline-1-oxide as described (Harris et al., 1994) to achieve a kill rate of 80–95%. The survivors were plated on MAG medium (2% malt extract, 0.2% peptone, 1% dextrose, 2% agar) and incubated at 42°C for 3 d. Revertants were isolated at a frequency of 1,487 revertants out of 4 × 108 survivors plated. Revertants were patched to MAG plates and incubated at 20°C for 8 d to screen for cold sensitivity. Out of 1,487 revertants, 35 were cold sensitive. To screen for extragenic suppressors, each cold-sensitive (cs) revertant was crossed to A612 and random meiotic progeny were selected and scored for the ts and cs phenotypes. Three out of the 35 cs revertants yielded both ts and cs meiotic progeny and demonstrated linkage of the cs and nimA1 suppression phenotypes. This indicated that these revertants contained the nimA1 mutation and a cs, extragenic suppressor of nimA1. Complementation analysis in diploids and genetic crosses indicated that three alleles of a single suppressor gene had been isolated. We named this gene sonA for suppressor of nimA1. Dominance/recessiveness was determined by generating diploids between each revertant and strain A154. The suppressor mutation was assigned to chromosome VIII using standard parasexual analysis (Pontecorvo et al., 1953).
Cloning and Characterization of sonA
Standard procedures were used for DNA preparation and manipulation (Sambrook et al., 1989). Wild-type sonA was cloned by complementation of the cs phenotype of LPW16 using a chromosome VIII–specific cosmid library (Brody et al., 1991) and the autonomously replicating plasmid pAR1-pyr4 (Verdoes et al., 1994) using standard procedures (Gems et al., 1991, 1994). Individual cosmids capable of complementing sonA1 were identified by sib selection. A common 2.6-kb HindIII fragment was the smallest restriction fragment from these cosmids capable of complementing sonA1 in trans. This fragment was sequenced using an Applied Biosystems DNA sequencer (model 373A; Foster City, CA) using procedures recommended by the manufacturer. Open reading frame (ORF) identification and sequence alignments were preformed using the Applied Biosystems Sequence Analysis Program. Database searches were preformed using BLAST (Altschul et al., 1990).
The sonA cDNA was isolated by PCR of A. nidulans total cDNA using Vent polymerase (New England BioLabs, Inc., Beverly, MA). The isolated cDNA covers the entire ORF of SONA, starting at 90 bp upstream from the start codon and ending 32 bp downstream from the stop codon. The sequence for the forward primer was 5′-GCTCTTGATACCCGTCTCTC-3′and for the reverse primer was 5′-CGAATGATGACTAGCCTGGAG-3′.
To determine whether the cloned gene was sonA or a suppressor of sonA1, we integrated the cloned gene at its homologous locus in the sonA1 strain, LPW16, using a two-step gene replacement procedure (O'Connell et al., 1992). In the first step, plasmid pRSW2, containing the sonA1 complementing gene and the A. nidulans pyrG gene, was used to transform LPW16, converting LPW16 from cs to wild-type growth at 20°C. Plasmid pRSW2 was constructed by inserting an EcoRI/BamHI genomic sonA fragment (containing the wild-type sonA gene from 284 bp upstream of the ATG to 140 bp downstream from the stop codon) into pRG3 (Waring et al., 1989). In the second step, a transformant was plated on medium containing uracil and 5-fluoroorotic acid to select for plasmid loss. Both cs and wild-type isolates were obtained as 5-fluoroorotic acid– resistant sectors and plasmid loss was confirmed by Southern blot analysis. Wild-type isolates were confirmed to have lost the sonA1 mutation by standard genetic analysis.
A strain expressing endogenous SONA1 and a recombinant SONA fused to two tandem copies of the hemagglutinin (HA) epitope at its carboxyl terminus (SONA-HA) was created by transforming pMTW2 into LPW16. Plasmid pMTW2, containing sonA fused to two tandem HA epitopes was constructed as follows: a sonA PCR fragment containing 284 bps upstream from the start codon and 140 bps downstream from the stop codon was inserted into pRG3 as a BamHI/EcoRI fragment. A sequence encoding two tandem HA epitopes (5′-TACCCATACGATGTTCCTGACTATGCGGGCTATCCCTATGACGTCCCGGACTATGCGGA-3′) was inserted into the sonA gene immediately before the stop codon by site- directed mutagenesis using the Quick Change™ mutagenesis kit (Strategene, La Jolla, CA). Expression of SONA-HA complemented the cs phenotype of sonA1 and was verified by Western blot analysis using 12CA5.
Analysis of Protein Kinase Levels
To prepare cultures for protein kinase studies, 106 spores/ml were inoculated into flasks containing YG liquid medium and then were shaken at 250 rpm at 32°C for 10 h. For analysis of asynchronous cultures, samples were collected and processed after 10 h at 32°C. For analysis of cultures shifted to restrictive temperature, cultures were brought to 42°C and sampled at the times indicated in Fig. 10. For analysis of cultures shifted back to permissive temperature, 3h, 42°C cultures were brought back to 32°C and sampled at the times indicated in Fig. 10. Procedures for sampling, protein isolation, and protein kinase assays were as previously described (Osmani et al., 1991a ; Ye et al., 1995).
Figure 10.
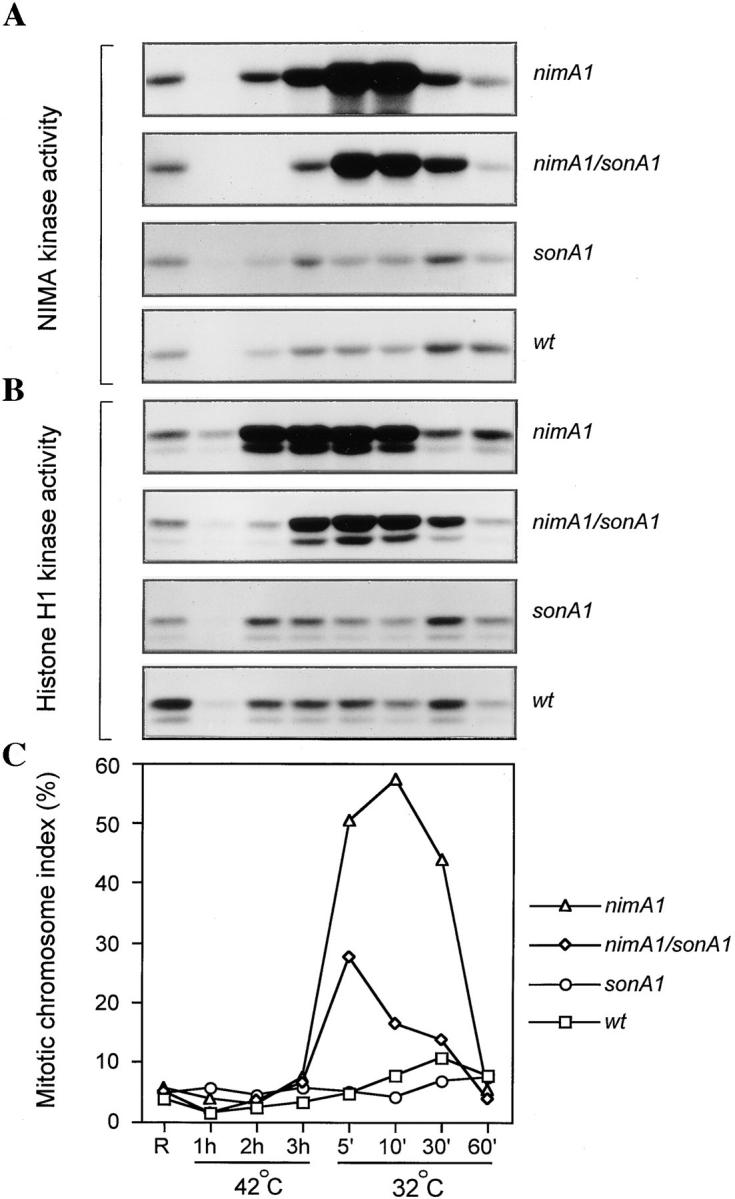
NIMA and NIMXCDC2 kinase activities in wild-type–, nimA1-, and sonA1-containing strains. Cells of a wild-type strain (GR5), a nimA1 containing strain (LPW2), a sonA1-containing strain (LPW16), and a nimA1, sonA1 double mutant strain (LPW29) were cultured and then sampled for kinase activity (A and B) and percentage of mitotic cells (C) as described in Materials and Methods. Samples were taken from exponentially growing, asynchronous cultures (R) or from cultures shifted to 42°C for 1, 2, or 3 h, or from cultures returned to 32°C for 5, 10, 30, or 60 min. A and B show autoradiographs representing NIMA (A) and NIMXCDC2 (B) kinase activities measured in immune complexes isolated from whole cell extracts from the strains indicated at the right.
Results
NIMECyclin B Localized Predominantly to the Nucleus
To determine the subcellular localization of NIMECyclin B, we have created a number of A. nidulans strains (Table I) in which the wild-type NIMECyclin B gene (nimE Cyclin B) has been replaced with an HA-tagged version of nimE Cyclin B (HA-nimE Cyclin B) (Osmani et al., 1994). These strains are phenotypically identical to coisogenic strains containing the untagged version (data not shown), and we can detect HA-NIMECyclin B using the mAb, 12CA5. HA-NIMECyclin B localized predominantly to the nucleus in rapidly growing wild-type cells (Fig. 1, top row). Approximately half of the cells in exponentially growing asynchronous cultures showed detectable nuclear HA-NIMECyclin B staining, whereas essentially every cell showed nuclear staining with anti-histone H1 mAb. The percentage of cells in asynchronous cultures which exhibited nuclear HA-NIMECyclin B staining was consistent in multiple experiments. The nuclear NIMECyclin B staining was punctate and included chromatin, the nucleolus, and the SPB, as judged by staining with 12CA5, DAPI, and either anti–γ-tubulin Ab or MPM2 (Figs. 1 and 2; data not shown). Faint staining of the cytoplasm in cells with bright nuclear staining was also detected. Double staining with 12CA5 and the anti-tubulin Ab, DM1A, showed that all of the cells with nuclear HA-NIMECyclin B contained cytoplasmic microtubules and that cells with mitotic spindles lacked nuclear-specific HA-NIMECyclin B staining (Fig. 1, bottom two rows).
Figure 1.

In situ localization of NIMECyclin B in wild-type cells. Cells of PMC654-4, the strain expressing HA-NIMECyclin B (first, third, and fourth rows) and R153, the no-HA control strain (second row), were cultured, fixed, and then prepared for immunocytology as described in Materials and Methods. Each row shows three images of the same cell. The images corresponding to Ab or DAPI staining are labeled above the panels. Note on cell morphology: A. nidulans is a filamentous fungus that undergoes polar growth and nuclear division without cytokinesis during much of its life cycle. Each elongated, multinucleate cell, or hypha, shown here is the result of the polarized growth and multiple, synchronous nuclear divisions of a uninucleate spore. Hyphal compartments are formed by growth and septation, with all nuclei within one compartment being synchronous. The large bulge at one end of each cell is the spore from which the cell originated. Bar, 10 μm.
Figure 2.

Localization of NIMECyclin B and NIMXCDC2 on nuclei and SPBs in cells arrested in late G2 by a nimTcdc25 mutation. SFC4-21 cells were cultured, fixed, and then prepared for immunocytology as described in Materials and Methods. The cells contained 4–8 elongated, well separated nuclei characteristic of cells arrested in late G2 by a nimTcdc25 mutation (James et al., 1995). The images corresponding to Ab or DAPI staining are labeled above each image. Arrows point to coincident staining of SPBs by anti-NIMECyclin B and MPM2. Bar, 10 μm.
The lack of nuclear NIMECyclin B staining in all cells of an asynchronous culture suggested that nuclear NIMECyclin B localization might be cell cycle stage-specific. To preliminarily investigate this possibility, we localized HA-NIMECyclin B in cells blocked in S phase with hydroxyurea or in late G2 with a ts mutation in nimTcdc25. Both treatments gave essentially the same result: the frequency of nuclear HA-NIMECyclin B staining increased to essentially 100% when cells were blocked in S or G2. Fig. 2 shows examples of the results for cells blocked in G2 by the nimTcdc25 mutation. As cells accumulated at the nimTcdc25 G2 arrest point, nuclear staining became increasingly brighter. The 12CA5 staining result was confirmed by double staining with 12CA5 and polyclonal rabbit anti-NIMECyclin B serum (Osmani et al., 1994). Both Abs stained nuclei, including chromatin, the nucleolus, and SPB-like dots (Fig. 2, top row). To determine if NIMXCDC2 colocalized with NIMECyclin B, we performed double labeling experiments using 12CA5 and polyclonal rabbit anti-NIMXCDC2 specific antisera (Osmani et al., 1994). NIMECyclin B and NIMXCDC2 colocalized on nuclei (Fig. 2, middle row). Preimmune serum controls for the anti-NIME and anti-NIMX antisera showed no nuclear specific staining (data not shown). Nuclear staining was also obtained using rabbit anti-NIMECyclin B and anti-NIMXCDC2 sera on a nimTcdc25 mutant strain which expressed only untagged NIMECyclin B (data not shown).
To determine if the nuclear-associated dots were indeed SPBs, we performed double labeling experiments using polyclonal rabbit anti-NIMECyclin B serum and the mouse mAb, MPM2, which stains the SPB of G2 cells (Martin et al., 1997). Anti-NIMECyclin B and MPM2 staining colocalized on SPBs and the nucleolus in nimTcdc25-arrested cells (Fig. 2, bottom row). Double labeling with 12CA5 and anti– γ-tubulin, or with anti-NIMXCDC2 sera and MPM2 gave similar results (data not shown).
To further characterize the cell cycle dependency of NIMECyclin B localization, we determined the localization of HA-NIMECyclin B in cycling cells from synchronous cultures. We generated synchronous cultures by blocking cells in late G2 using a nimTcdc25 mutation and then releasing the cell cycle arrest. Immediately before release from the G2 arrest, 100% of the cells showed nuclear HA-NIMECyclin B staining (Fig. 3). Within 10 min after release from the G2 arrest, 90% of the cells entered mitosis and showed no nuclear HA-NIMECyclin B staining, whereas the cells in the same sample that had not yet entered mitosis continued to show nuclear HA-NIMECyclin B staining (Fig. 3 B). By 20 min after release, all the cells had finished mitosis (Fig. 3 A) and no cells (out of more than 300 examined) exhibited nuclear HA-NIMECyclin B staining. Cells with nuclear HA-NIMECyclin B began to accumulate 80 min after the release and almost all of the cells exhibited nuclear HA-NIMECyclin B staining (Fig. 3, A and B) just before initiation of the second mitosis. As with the first mitosis, nuclear HA-NIMECyclin B staining was lost coincident with the initiation of mitosis. Out of more than a thousand mitotic cells examined, only two showed very faint nuclear NIMEHA staining. Essentially all cells showed SPB staining at the initial G2 arrest point and in G2 just before the second division (data not shown). SPB staining was not distinguishable from chromatin or nucleolar staining in cells at other time points.
NIMECyclin B Localizes Predominantly to the Cytoplasm in nimA Mutants
To investigate the possibility that NIMA is involved in NIMXCDC2/NIMECyclin B localization, we constructed strains expressing HA-NIMECyclin B and carrying the ts nimA5 or nimA1 mutations. We determined the localization of HA-NIMECyclin B in these strains at restrictive and permissive temperature. At permissive temperature, about half the cells from asynchronous cultures of nimA5 or nimA1 mutants showed nuclear HA-NIMECyclin B localization (data not shown). In contrast, after the shift to restrictive temperature, the nimA5 and nimA1 cells showed little nuclear-specific HA-NIMECyclin B staining (Fig. 4). Instead of being concentrated at nuclei, HA-NIMECyclin B staining was diffuse throughout the cell. Although nimA and nimTcdc25 mutants arrest at essentially the same point in late G2, their NIMECyclin B staining patterns are dramatically different (Fig. 4, compare top row with second and third rows). Antibody access to the nuclei of these cells was confirmed by staining with MPM2 (Fig. 4, middle column) and anti-histone H1 antibodies (data not shown).
Figure 4.

Nuclear-specific NIMECyclin B and NIMXCDC2 localization was prevented by nimA mutations. Cells of the nimTcdc25 mutant, SFC4-21, the nimA5 mutant, PMC654-19, and the nimA1 mutant, SFC403-19, were cultured, fixed, and then prepared for immunocytology as described in Materials and Methods. Left, strain identity.The images corresponding to Ab or DAPI staining are labeled at the top of the panels. Arrows, position of nuclei in the images showing NIMECyclin B and NIMXCDC2 localization. Bar, 10 μm.
It was possible that the above results were caused by masking of the single HA epitope of HA-NIMECyclin B in nimA mutants. To address this possibility, we stained nimA5 cells cultured at restrictive temperature with polyclonal rabbit anti-NIMECyclin B and anti-NIMXCDC2 sera. As with 12CA5 staining, anti-NIMECyclin B staining was diffuse throughout the cells of nimA mutants (Fig. 4, second row). Similar results were obtained with anti-NIMXCDC2 serum (Fig. 4, bottom row).
Like the nimTcdc25 mutation, the nimA5 mutation is readily reversible. A few minutes after return to permissive temperature, nimA5 mutants leave G2 and enter mitosis synchronously (Oakley and Morris, 1983; Osmani et al., 1991a ). If nuclear NIMECyclin B localization is important for mitotic initiation, then one would predict that NIMECyclin B would reaccumulate on nuclei of nimA mutants as cells approach mitosis after return to permissive temperature. To test this, we compared the nuclear HA-NIMECyclin B staining profiles of a nimA5 and a nimTcdc25 mutant at their arrest point in G2 and after release from the G2 arrest into room temperature medium. Release from the G2 arrest under these conditions is relatively slow, allowing for examination of cells as they progress from very late G2 into early mitosis. As above, the percentage of nimTcdc25 cells with nuclear HA-NIMECyclin B decreased after release from the G2 block (Fig. 5 A). In contrast, the percentage of nimA5 cells showing nuclear HA-NIMECyclin B staining increased after release from the block, with 40% of the cells having nuclear HA-NIMECyclin B staining 2 min after the release (Fig 5 B). Under these conditions, reduced but detectable nuclear HA-NIMECyclin B staining remained in some early mitotic cells with very short spindles, although the majority of mitotic cells and all mitotic cells at metaphase or beyond were negative for nuclear HA-NIMECyclin B. All of the nimA5 cells eventually progressed through mitosis and showed no nuclear nimECyclin B staining (data not shown). Based on our observations, it is likely that most if not all nimA5 cells accumulated nuclear HA-NIMECyclin B after return to permissive temperature. The slow, somewhat asynchronous release under these conditions coupled with the loss of nuclear HA-NIMECyclin B staining early in mitosis probably accounts for the shallow slope of nuclear HA-NIMECyclin B curve in Fig. 5 B. We did not observe obvious SPB-like HA-NIMECyclin B staining in nimA5 cells after return to permissive temperature.
nimA Interacts Genetically with sonA, a Gene Related to the Nucleocytoplasmic Transporters GLE2 and RAE1
As part of a search for nimA-interacting genes, we have attempted to identify extragenic suppressors of nimA mutations by isolating induced revertants of the nimA1 heat-sensitive (ts) mutation (refer to Materials and Methods). nimA1 is a tight ts mutation, with colony formation severely inhibited at 42°C, a temperature at which wild-type strains grow well (Fig. 6). We isolated a number of revertants which were simultaneously converted from heat sensitive (ts) to cs, being able to form colonies normally at 42° but not at 20°C (see LPW29 in Fig. 6). For three of these revertants, suppression of nimA1 was unlinked to nimA and tightly linked to the cs phenotype, demonstrating that the original revertants contained a cs, extragenic suppressor of nimA1 (data not shown). These suppressor mutations also conferred a cs phenotype in a nimA1 background (see LPW16 in Fig. 6). All three cs mutations were recessive and in the same complementation group (data not shown), which we designated sonA for suppressor of nimA1. One of these mutations, sonA1, was chosen for further study.
Figure 6.

Colony growth phenotype of the following strains: wild-type (GR5), nimA1 (LPW2), sonA1 (LPW16), and nimA1, sonA1 (LPW29). Strains are identified by their relevant genotype indicated at the top. Each strain was center-point inoculated on MAG medium and then incubated at 42°C for 3 d or at 20°C for 10 d, as indicated.
We cloned the wild-type sonA gene by complementation of the cs defect of a sonA1 mutant (refer to Materials and Methods). A cosmid clone capable of complementing sonA1 was recovered and the complementing sequence was localized to a 2.6-kb genomic fragment. A cDNA clone, generated by PCR, was also sufficient for complementation of sonA1. Sequence of the genomic and cDNA clones indicated that the complementing fragment encoded a 1.2-kb ORF interrupted by a single, 65-bp intron (Fig. 7 B). The cloned gene was sonA and not a suppressor of sonA1 because it was tightly linked to sonA (refer to Materials and Methods) and because the sequence of this gene in the sonA1 strain contained a single C to G mutation resulting in a P to R change at amino acid residue 205 in the ORF (Fig. 7 A).
Figure 7.

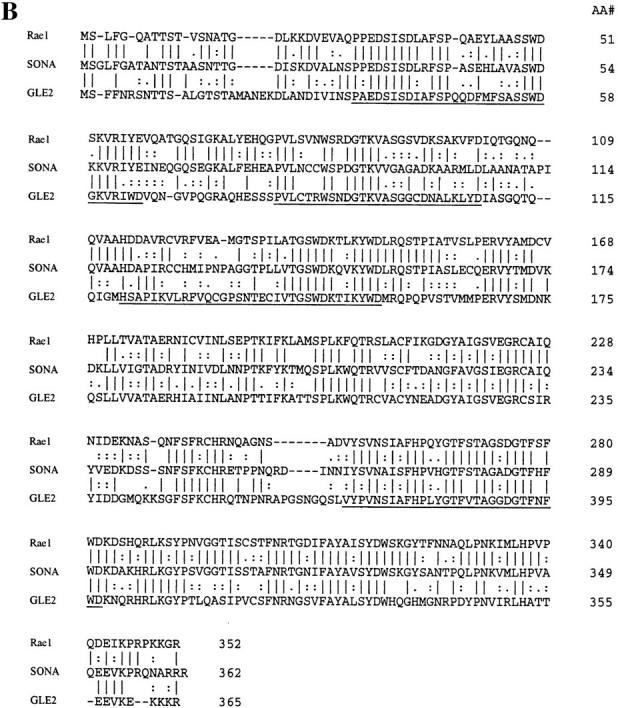
Sequence of sonA and alignment of SONA to GLE2 and RAE1. (A) The nucleotide sequence and predicted ORF in the 2.6-kb genomic sonA clone is shown. Right, amino acid residues of the SONA ORF; underline, predicted WD repeats; boldface at the amino terminus, a single GLFG sequence, found in multiple copies of many nuclear pore proteins; boldface at the carboxyl terminus, a potential nuclear localization sequence. The position of the intron was confirmed by sequencing a cDNA clone. (B) An alignment of SONA to GLE2 and RAE1 is shown. Right, amino acid residue numbers; solid lines, identical residues. Highly conserved and conserved residues, as determined by the Applied Biosystems Sequence Analysis Software, are indicated by the double dots, and single dots, respectively. The sonA cDNA sequence is available from EMBL/GenBank/DDBJ under accession number AF069492.
Conceptual translation of the sonA ORF predicts that the sonA polypeptide (SONA) contains 362 amino acid residues corresponding to a mol wt of ∼39.5 kD. Sequence database searches revealed that SONA is highly similar to Schizosaccharomyces pombe RAE1 (Brown et al., 1995) and Saccharomyces cerevisiae GLE2 (Murphy et al., 1996). SONA is 84.3% similar (58.8% identical) to RAE1 and is 84.3% similar (48.9% identical) to GLE2. Although the four putative β-transducin/WD repeats in RAE1 and GLE2 are well conserved in SONA, the similarity between SONA and RAE1 and GLE2 extends well outside of these repeats along the entire peptide sequences (Fig. 7 B).
To determine the localization of SONA, we constructed strains expressing an HA epitope-tagged version of SONA (SONA-HA). SONA-HA was functional as its expression complemented the cs phenotype of sonA1 (data not shown). Exponentially growing cells expressing SONA-HA showed punctate staining appearing as a ring at the periphery of each nucleus (Fig. 8). A no-HA control strain showed no nuclear ring staining (data not shown; also refer to Fig. 1). SONA-HA staining was not completely coincident with DAPI staining in that it was limited to the nuclear periphery and was evident at the nuclear periphery in regions next to nucleoli, where DAPI staining was excluded. The SONA-HA staining pattern was strikingly similar to the nuclear pore-like staining pattern seen for GLE2 (Murphy et al., 1996).
sonA1 Suppresses the Nuclear Division and NIMECyclin B Localization Defects of nimA1 without Markedly Increasing NIMA or NIMX Kinase Activity
Colonies of sonA1, nimA1 double mutants exhibited slower growth at 42°C than either wild-type or sonA1 single mutant strains (refer to Fig. 6). To examine the suppression of nimA1 by sonA1 in more detail, we followed nuclear division and nuclear morphology during spore germination in strains that were either wild-type, nimA1, sonA1, or nimA1 plus sonA1. Fig. 9 shows examples of cells stained with DAPI to visualize nuclei. At 32°C (permissive growth temperature for nimA1 and sonA1), essentially all of the spores from all the strains examined germinated and underwent nuclear division at approximately wild-type rates. At 42°C, the majority of nimA1 mutant spores failed to undergo a single nuclear division even after 10 h, whereas the majority of spores from wild type had undergone 2–4 divisions. Essentially all the spores of the nimA1, sonA1 double mutant germinated and underwent apparently normal nuclear divisions at 42°C. The rate of nuclear division in this strain was somewhat slower than that in wild type (data not shown). This is consistent with the reduced colony growth rate of sonA1, nimA1 strains, indicating that sonA1 does not suppress nimA1 function to wild-type, nimA1 levels.
Figure 9.

Nuclear division in wild-type–, nimA1-, and sonA1-containing strains. Cells of the wild-type strain, GR5 (a–c), the sonA1 mutant, LPW16 (d–f), the nimA1 mutant, LPW2 (g–i), and the nimA1, sonA1 double mutant, LPW19 (j–l) were stained with DAPI as described in Materials and Methods. Cells shown in panels a, d, g, and j were incubated at 32°C for 8 h. Other panels show cells incubated at 42°C for 8 h (b, e, h, and k) or 10 h (c, f, i, and l). Bar, 10 μm.
It was previously shown that nimA1 mutants arrest in G2 even though they accumulate partially active NIMA and fully activated NIMXCDC2 (Pu et al., 1995). It was possible that sonA1 suppresses nimA1 by somehow increasing the level of NIMA or NIMXCDC2 activity. To investigate these possibilities, we measured the levels of NIMA and NIMXCDC2 kinase activities in a wild-type strain, a nimA1 mutant, a sonA1 mutant, and a nimA1, sonA1 double mutant. Samples were analyzed from asynchronous cultures, from cultures shifted to restrictive temperature, and from cultures shifted to restrictive temperature and then returned to permissive temperature. Fig. 10 shows that 3 h after shift to restrictive temperature (42°C), the nimA1 mutant, LPW2, accumulated NIMA and NIMXCDC2 kinase to levels above that in asynchronous cultures. Return to permissive temperature induced these cells to synchronously enter mitosis, and resulted in a severalfold increase in NIMA activity but no significant increase in NIMXCDC2 activity (Fig. 10 C). These results are in agreement with Pu et al. (1995), and they demonstrate that nimA1 blocks progression into mitosis but does not prevent full activation of NIMXCDC2. The nimA1, sonA1 double mutant, LPW29, did not accumulate NIMA or NIMXCDC2 activity above that of LPW2 either before or after a shift to 42°C (Fig. 10, A and B, compare first and second panels). Based on the peak of mitotic cells after the return to permissive temperature, ∼25% of the LPW29 cells had accumulated in late G2 during incubation at 42°C, consistent with previous data showing that suppression of nimA1 by sonA1 is not complete. The wild-type (GR5) and sonA1 single mutant (LPW16) were essentially identical under these conditions (Fig. 10, A and B, third and fourth panels).
Given that sonA1 did not cause an increase in the levels of NIMA or NIMXCDC2 activity, and that SONA is related to the GLE2/RAE1 nucelocytoplamsic transporter, we considered the possibility that sonA1 may suppress the NIMECyclin B localization defect of nimA1 mutants. We constructed nimA1, sonA1 double mutants and nimA5, sonA1 double mutants which expressed HA-NIMECyclin B as their only NIMECyclin B. Whereas nimA1 cells cultured at restrictive temperature for nimA1 arrested in G2 with HA-NIMECyclin B staining throughout the cell, nimA1, sonA1 double mutants continued to divide and showed nuclear-specific HA-NIMECyclin B staining (Fig. 11). The percentage of nimA1, sonA1 double mutants showing nuclear HA-NIMECyclin B staining was 33% (n = 100) compared with 51% (n = 102) for wild-type cells. These results are consistent with the fact that sonA1 only partially suppresses the growth and nuclear division phenotype of nimA1 mutants (see Figs. 6 and 9). The nimA5, sonA1 double mutant incubated at restrictive temperature arrested in G2 and did not accumulate nuclear-specific HA-NIMECyclin B (data not shown), demonstrating that the ability of sonA1 to suppress the NIMECyclin B localization defect of nimA mutants was allele specific.
Figure 11.

The sonA1 mutation suppresses the nuclear NIMECyclin B localization defect of nimA1. Cells of the nimA1 strain, SFC403-19, and the nimA1, sonA1 double mutant, SFC444-1, were cultured, fixed, and then prepared for immunocytology as described in Materials and Methods. Left, strain identity; top, images corresponding to Ab or DAPI staining; top row arrows, (nimA1 single mutant) position of nuclei in cells showing general cytoplasmic staining; bottom row arrows, position of example nuclei showing NIMECyclin B staining. Bar, 10 μm.
Discussion
Inactivation of the NIMA kinase causes a specific cell cycle arrest in G2 without preventing activation of the H1 kinase activity of NIMXCDC2/NIMECyclin B. NIMA is, therefore, required for mitotic initiation by a mechanism other than activation of NIMXCDC2 as an H1 kinase. Although NIMA itself is probably required for normal mitosis independently of NIMXCDC2 (O'Connell et al., 1994; Lu and Hunter, 1995; Pu and Osmani, 1995), some mechanism must be in place to prevent activated NIMXCDC2 from inappropriately inducing mitosis in the absence of NIMA function. One possibility is that NIMA affects the mitosis-promoting activity of NIMXCDC2 at some level other than activation of enzyme activity. Two lines of evidence presented here, one genetic, the other cytological, lend support to such a hypothesis and indicate a role for NIMA in the nuclear localization of NIMXCDC2/NIMECyclin B.
The NIMXCDC2/NIMECyclin B Complex Localizes to the Nucleus in a NIMA-dependent Manner
We have shown that the major A. nidulans B-type cyclin, NIMECyclin B, localized to the chromatin, nucleolar, and SPB regions of the nucleus in a NIMA-dependent manner. NIMECyclin B localization to the nucleus in S and G2 parallels its accumulation (Ye et al., 1995, 1996), similar in many respects to localization of the S. pombe G2 cyclin, p63cdc13 (Booher et al., 1989; Alfa et al., 1990, 1991; Gallagher et al., 1993; however, also see Audit et al., 1996 for evidence of cytoplasmic p63cdc13 localization during interphase). The localization of NIMXCDC2 to the nucleus correlated with that of NIMECyclin B and was also dependent on NIMA function, suggesting that localization of NIMXCDC2/NIMECyclin B complex itself is perturbed in nimA mutants.
We noted no obvious nuclear NIMECyclin B staining in metaphase cells, even though we have looked at well over a thousand cells from cultures synchronously entering mitosis. We did observe nuclear staining very early in mitosis, before significant SPB separation. These results differ somewhat from cyclin B localization to the mitotic apparatus at metaphase in S. pombe and mammalian cells (for examples see Alfa et al., 1990; Pines and Hunter, 1991; Gallant and Nigg, 1992; Jackman et al., 1995), however, the significance of this difference is not clear. Either we cannot detect whatever NIMECyclin B is present in metaphase nuclei or NIMECyclin B is lost from the nucleus before metaphase. We are currently investigating NIMECyclin B localization in cells overexpressing NIMECyclin B and in cells blocked in mitosis by drugs or by cell cycle mutations to clarify this issue.
NIMA and SONA in the Nucleocytoplasmic Transport of NIMECyclin B
The mechanism by which NIMA functions to promote nuclear NIMECyclin B localization may be indicated by the identification of sonA as an allele-specific suppressor of nimA1. SONA shows high sequence similarity to RAE1 of S. pombe (Brown et al., 1995), GLE2 of S. cerevisiae (Murphy et al., 1996), and the mammalian protein, MRNP41 (Kraemer and Blobel, 1997), all of which have been implicated in nucleocytoplasmic transport. The nuclear mRNA export defect in rae1 −and gle2 −mutants, and the association of MRNP41 with mRNA indicate an important role for these proteins in mRNA export. However, none of these proteins contain RNA binding motifs, suggesting that their interaction with mRNA is indirect. Furthermore, GLE2 and MRNP41 localize predominantly to nuclear pore complexes, gle2−mutations derange nuclear pore complex structure, and GLE2 interacts with SRP1 (importin α) in a two-hybrid assay, suggesting that these proteins play a more general role in nucleocytoplasmic transport (Murphy et al., 1996; Kraemer and Blobel, 1997). The localization of SONA to the nuclear periphery (refer to Fig. 8) is consistent with a role for SONA in nucleocytoplasmic transport.
Given that proper NIMECyclin B localization is dependent on NIMA function (refer to Fig, 4), and that the sonA1 mutation suppresses the mitotic and NIMECyclin B localization defect in nimA1 mutants without causing an increase in NIMA or NIMXCDC2 activity (refer to Fig. 10), we propose that NIMA and SONA are involved in the nucleocytoplasmic transport of NIMECyclin B. Two models consistent with our results (Fig. 12) propose that nuclear localization of NIMECyclin B is a function of its rate of import into the nucleus and its rate of export into the cytoplasm (Fig. 12). These models predict that localization of NIMECyclin B to the SPB is dependent on its accumulation in the nucleus, as if NIMECyclin B accumulates at the nucleoplasmic surface of the SPB. This prediction is consistent with the fact that the SPB is tightly associated with the nucleus throughout the cell cycle in A. nidulans (Oakley and Morris, 1983). SONA is proposed to play a positive role in nuclear export, based on the mRNA export defects of rae1/gle2 mutants, although we cannot formerly exclude a role for SONA in nuclear import. NIMA is proposed to oppose SONA function, either by facilitating nuclear import (Fig. 12 A), or by antagonizing export (Fig. 12 B). In either scenario, loss of NIMA function results in a net decrease in nuclear NIMECyclin B, which can be offset by loss of SONA function.
Figure 12.
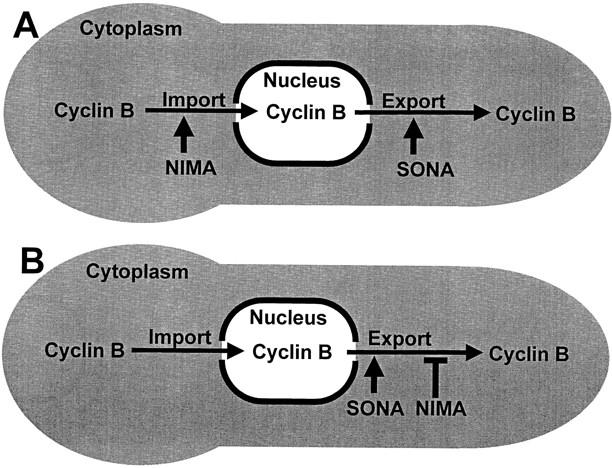
Model for NIMA and SONA function in the nucleocytoplasmic transport of NIMECyclin B.
These models are not meant to exclude additional functions for NIMA beyond that in the nucleocytoplasmic transport of NIMXCDC2/NIMECyclin B. The finding that gain of function mutations in nimA can induce abnormal mitosis in the absence of CDC2 function (O'Connell et al., 1994; Lu and Hunter, 1995; Pu and Osmani, 1995) clearly indicates that NIMA has additional roles in promoting mitosis.
We have proposed a specific (although not necessarily physical) interaction between NIMA and SONA in controlling nuclear NIMECyclin B levels because of the allele-specific suppression of nimA1 (and not nimA5) by sonA1. The mechanism underlying this allele-specific interaction may be explained by quantitative differences in NIMA kinase activity in nimA1 versus nimA5 mutants. For example, extracts from cells arrested in G2 by the nimA1 mutation contain residual NIMA kinase activity (refer to Fig 10; Pu et al., 1995), whereas equivalent extracts from nimA5 cells contain only trace NIMA kinase levels (Ye et al., 1995). Accordingly, the nimA5 mutation may cause too severe a defect in the nuclear accumulation of NIMECyclin B for the sonA1 mutation to suppress. Alternatively, the allele-specific nimA1/sonA1 interaction may be due to a direct interaction between NIMA and SONA. In this regard, we note that SONA contains three consensus NIMA phosphorylation sites (Lu et al., 1994) (refer to Fig. 7; FGAT at 5–8, FYKT at 198–201, and FNRT at 31–316). Regardless of the underlying mechanism, the allele-specific interaction between nimA and sonA indicates a specific interaction and demonstrates that inactivation of sonA does not simply bypass the need for nimA in the promotion of mitosis.
Nucleocytoplasmic Transport and Regulation of Mitosis
This study establishes a role for NIMA in the nuclear localization of NIMXCDC2/NIMECyclin B. One implication of this finding is that nucleocytoplasmic transport and regulation of mitosis are intimately linked. Such transport is particularly relevant to closed mitosis, in which the nuclear envelope remains intact, as occurs in many fungi including A. nidulans, S. pombe, and S. cerevisiae. At the very least, tubulin from disintegrated cytoplasmic microtubules probably needs to be rapidly imported into nuclei to form the intranuclear spindle. Regulators of mitosis may also undergo nuclear or cytoplasmic transport at mitosis, as was proposed for the p107wee1 regulator, p70nim1 in S. pombe (Wu and Russell, 1997). A causal relationship between transport and mitosis is also supported by the G2/M arrest due to a mutation in the S. cerevisiae SRP1 (importin α) gene (Loeb et al., 1995) and by the finding that the S. pombe rae1 − mutation causes a G2 arrest (Brown et al., 1995).
Another significant implication of this work is the identification of a mechanism by which NIMXCDC2/NIMECyclin B function is coordinated with that of NIMA. The requirement for the function of both kinases for the initiation of mitosis can now be explained by a model in which each kinase independently promotes some events of mitosis while also being sensitive to the function of the other. In this model, NIMA is not fully functional until it is hyperphosphorylated by NIMXCDC2/NIMECyclin B and NIMXCDC2/ NIMECyclin B is not properly localized unless NIMA is functional. Given the finding of NIMA-like functions in other organisms (O'Connell et al., 1994; Fry and Nigg, 1995; Gallant et al., 1995; Pu et al., 1995; Pu and Osmani, 1995; Lu and Hunter, 1995), and the evolutionary conservation of SONA/GLE2/RAE1/MNRP41 (refer to Fig. 7; Brown et al., 1995; Murphy et al., 1996; Kraemer and Blobel, 1997), the analysis of NIMA and SONA interactions in A. nidulans should serve as an important model for the elucidation of fundamental mechanisms coordinating nucleocytoplasmic transport and mitosis.
Finally, it is very interesting that mutation of rae1 of S. pombe results in a G2 arrest without preventing full activation of p34cdc2 (Brown et al., 1995; Whalen et al., 1997). Perhaps mutation of rae1 prevents correct localization of p34cdc2/p63cdc13, which would further indicate the conserved nature of this level of mitotic regulation.
Acknowledgments
This work was supported by NIH grants to P.M. Mirabito (GM51931) and S.A.Osmani (GM42564).
Abbreviations used in this paper
- CCD
charge-coupled device
- cs
cold sensitive
- DAPI
4′,6-diamidino-2-phenylindole
- HA
hemagglutinin
- ORF
open reading frame
- SPB
spindle pole body
- ts
temperature sensitive
Footnotes
P.M. Mirabito thanks D.A. Harrison (University of Kentucky, Lexington, KY) for use of his image station and for his skill and patience in helping with imaging and figure preparation. The authors also thank members of the Mirabito and Osmani laboratories, particularly S. Venkatram, for helpful discussions throughout this work and for critical reading of the manuscript.
Address all correspondence to P.M. Mirabito, Molecular and Cellular Biology Section, School of Biological Sciences, University of Kentucky, Lexington, KY 40506-0225. Tel.: (606) 257-7642. Fax: (606) 257-1717. E-mail: pmmira000@pop.uky.edu
References
- Alfa CE, Ducommun B, Beach D, Hyams JS. Distinct nuclear and spindle pole body populations of cyclincdc2in fission yeast. Nature. 1990;347:680–682. doi: 10.1038/347680a0. [DOI] [PubMed] [Google Scholar]
- Alfa CE, Gallagher IM, Hyams JS. Subcellular localization of the p34cdc2/p63cdc13protein kinase in fission yeast. Cold Spring Harbor Symp Quant Biol. 1991;56:489–494. doi: 10.1101/sqb.1991.056.01.055. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Audit M, Barbier M, Soyer-Gobillard MO, Albert M, Geraud ML, Nicolas G, Lenaers G. Cyclin B (p56cdc13) localization in the yeast Schizosaccharomyces pombe: an ultrastructural and immunocytochemical study. Biol Cell. 1996;86:1–10. [PubMed] [Google Scholar]
- Bergen LG, Upshall A, Morris NR. S phase, G2 and nuclear division mutants of Aspergillus nidulans. . J Bact. 1984;159:114–119. doi: 10.1128/jb.159.1.114-119.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher RN, Alfa CE, Hyams JH, Beach DH. The fission yeast cdc2/cdc13/suc1protein kinase: regulation of catalytic activity and nuclear localization. Cell. 1989;58:485–497. doi: 10.1016/0092-8674(89)90429-7. [DOI] [PubMed] [Google Scholar]
- Brody H, Griffith J, Cuticchia AJ, Arnold J, Timberlake WE. Chromosome specific recombinant DNA libraries from the fungus Aspergillus nidulans. . Nucl Acids Res. 1991;19:3105–3109. doi: 10.1093/nar/19.11.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Bharathi A, Ghosh A, Whalen W, Fitzgerald E, Dhar R. A mutation in the Schizosaccharomyces pombe rae1gene causes defects in poly(A)1 RNA export and in the cytoskeleton. J Biol Chem. 1995;270:7411–7419. doi: 10.1074/jbc.270.13.7411. [DOI] [PubMed] [Google Scholar]
- Fry AM, Nigg EA. Cell cycle. The NIMA kinase joins forces with Cdc2. Curr Biol. 1995;5:1122–1125. doi: 10.1016/s0960-9822(95)00227-2. [DOI] [PubMed] [Google Scholar]
- Gallagher IM, Alfa CE, Hyams JS. p63cdc13, a B type cyclin, is associated with both the nucleolar and chromatin domains of the fission yeast nucleus. Mol Biol Cell. 1993;4:1087–1096. doi: 10.1091/mbc.4.11.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant P, Nigg EA. Cyclin B2 undergoes cell cycle dependent nuclear translocation and, when expressed as a nondestructible mutant, causes mitotic arrest in HeLa cells. J Cell Biol. 1992;117:213–224. doi: 10.1083/jcb.117.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant P, Fry AM, Nigg EA. Protein kinases in the control of mitosis: focus on nucleocytoplasmic trafficking. J Cell Sci. 1995;19(Suppl.):21–28. doi: 10.1242/jcs.1995.supplement_19.3. [DOI] [PubMed] [Google Scholar]
- Gems D, Johnstone IL, Clutterbuck JC. An autonomously replicating plasmid transforms Aspergillus nidulansat high frequency. Gene. 1991;98:61–67. doi: 10.1016/0378-1119(91)90104-j. [DOI] [PubMed] [Google Scholar]
- Gems D, Aleksenko A, Belenky L, Robertson S, Ramsden M ,Y. Vinetski, and A.J. Clutterbuck. An “instant gene bank” method for gene cloning by mutant complementation. Mol Gen Genet. 1994;242:467–471. doi: 10.1007/BF00281798. [DOI] [PubMed] [Google Scholar]
- Harris SD, Morrell JL, Hamer JE. Identification and characterization of Aspergillus nidulansmutants defective in cytokinesis. Genetics. 1994;136:517–532. doi: 10.1093/genetics/136.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman M, Firth M, Pines J. Human cyclins B1 and B2 are localized to strikingly different structures: B1 to microtubules, B2 primarily to the Golgi apparatus. EMBO (Eur Mol Biol Organ) J. 1995;14:1646–1654. doi: 10.1002/j.1460-2075.1995.tb07153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SW, Mirabito PM, Scacheri PC, Morris NR. The Aspergillus nidulans bimE(blocked in mitosis) gene encodes multiple cell cycle functions involved in mitotic checkpoint control and mitosis. J Cell Sci. 1995;108:3485–3499. doi: 10.1242/jcs.108.11.3485. [DOI] [PubMed] [Google Scholar]
- Kafer E. Meiotic and mitotic recombination in Aspergillus nidulansand its chromosomal aberrations. Adv Genet. 1977;19:33–131. doi: 10.1016/s0065-2660(08)60245-x. [DOI] [PubMed] [Google Scholar]
- Kraemer D, Blobel G. mRNA binding protein mrnp 41 localizes to both nucleus and cytoplasm. Proc Natl Acad Sci USA. 1997;94:9119–9124. doi: 10.1073/pnas.94.17.9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JD, Schlenstedt G, Pellman D, Kornitzer D, Silver PA, Fink GR. The yeast nuclear import receptor is required for mitosis. Proc Natl Acad Sci USA. 1995;92:7647–7651. doi: 10.1073/pnas.92.17.7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KP, Osmani SA, Means AR. Properties and regulation of the cell cycle-specific NimA protein kinase of Aspergillus nidulans. . J Biol Chem. 1993;268:8769–8776. [PubMed] [Google Scholar]
- Lu KP, Kemp BE, Means AR. Identification of substrate specificity determinants for the cell cycle regulated NIMA protein kinase. J Biol Chem. 1994;269:6603–6607. [PubMed] [Google Scholar]
- Maridor G, Gallant P, Golsteyn R, Nigg EA. Nuclear localization of vertebrate cyclin A correlates with its ability to form complexes with cdk catalytic subunits. J Cell Sci. 1993;106:535–544. doi: 10.1242/jcs.106.2.535. [DOI] [PubMed] [Google Scholar]
- Martin MA, Osmani SA, Oakley BR. The role of gtubulin in mitotic spindle formation and cell cycle progression in Aspergillus nidulans. . J Cell Sci. 1997;110:623–633. doi: 10.1242/jcs.110.5.623. [DOI] [PubMed] [Google Scholar]
- Mirabito PM, Morris NR. BIMA, a TPR-containing protein required for mitosis, localizes to the spindle pole body in Aspergillus nidulans. . J Cell Biol. 1993;120:959–968. doi: 10.1083/jcb.120.4.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris NR. Mitotic mutants of Aspergillus nidulans. . Genet Res. 1976;26:237–254. doi: 10.1017/s0016672300016049. [DOI] [PubMed] [Google Scholar]
- Murphy R, Watkins JL, Wente SR. GLE2, a Saccharomyces cerevisiae homologue of the Schizosaccharomyces pombeexport factor RAE1, is required for nuclear pore complex structure and function. Mol Biol Cell. 1996;7:1921–1937. doi: 10.1091/mbc.7.12.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P. Universal control mechanism regulating onset of M phase. Nature. 1990;344:503–508. doi: 10.1038/344503a0. [DOI] [PubMed] [Google Scholar]
- Oakley BR, Morris NR. A mutation in Aspergillus nidulansthat blocks the transition from interphase to prophase. J Cell Biol. 1983;96:1155–1158. doi: 10.1083/jcb.96.4.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell MJ, Osmani AH, Morris NR, Osmani SA. An extra copy of nimE cyclinB elevates pre-MPF levels and partially suppresses mutation of nimT cdc25 in Aspergillus nidulans. . EMBO (Eur Mol Biol Organ) J. 1992;11:2139–2149. doi: 10.1002/j.1460-2075.1992.tb05273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell MJ, Norbury C, Nurse P. Premature chromatin condensation upon accumulation of NIMA. EMBO (Eur Mol Biol Organ) J. 1994;13:4926–4937. doi: 10.1002/j.1460-2075.1994.tb06820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ookata K, Hisanaga S, Okano T, Tachibana K, Kishimoto T. Relocation and distinct subcellular localization of p34cdc2-cyclin B at meiosis reinitiation in starfish oocytes. EMBO (Eur Mol Biol Organ) J. 1992;11:1763–1772. doi: 10.1002/j.1460-2075.1992.tb05228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ookata K, Hisanaga S, Bulinski JC, Murofushi H, Aizawa H, Itoh TJ, Hotani H, Okumura E, Tachibana K, Kishimoto T. Cyclin B interaction with microtubule-associated protein 4 (MAP4) targets p34cdc2kinase to microtubules and is a potential regulator of M-phase microtubule dynamics. J Cell Biol. 1995;128:849–862. doi: 10.1083/jcb.128.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmani SA, May GS, Morris NR. Regulation of the mRNA levels of nimA, a gene required for the G2M transition in Aspergillus nidulans. . J Cell Biol. 1987;104:1495–1504. doi: 10.1083/jcb.104.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmani SA, Engle DB, Doonan JH, Morris NR. Spindle formation and chromatin condensation in cells blocked at interphase by mutation of a negative cell cycle control gene. Cell. 1988a;52:241–251. doi: 10.1016/0092-8674(88)90513-2. [DOI] [PubMed] [Google Scholar]
- Osmani SA, Pu RT, Morris NR. Mitotic induction and maintenance by overexpression of a G2specific gene that encodes a potential protein kinase. Cell. 1988b;53:237–244. doi: 10.1016/0092-8674(88)90385-6. [DOI] [PubMed] [Google Scholar]
- Osmani A, McGuire SL, Osmani SA. Parallel activation of the NIMA and p34cdc2 cell cycle-regulated protein kinases is required to initiate mitosis in A. nidulans. . Cell. 1991a;67:283–291. doi: 10.1016/0092-8674(91)90180-7. [DOI] [PubMed] [Google Scholar]
- Osmani AH, O'Donnell K, Pu RT, Osmani SA. Activation of the nimA protein kinase plays a unique role during mitosis that cannot be bypassed by absence of the bimE checkpoint. EMBO (Eur Mol Biol Organ) J. 1991b;10:2669–2679. doi: 10.1002/j.1460-2075.1991.tb07810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmani AH, van Peij N, Mischke M, O'Connell MJ, Osmani SA. A single p34cdc2 protein kinase (encoded by nimX cdc2) is required at G1 and G2 in Aspergillus nidulans. . J Cell Sci. 1994;107:1519–1528. doi: 10.1242/jcs.107.6.1519. [DOI] [PubMed] [Google Scholar]
- Pines J, Hunter T. Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport. J Cell Biol. 1991;115:1–17. doi: 10.1083/jcb.115.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontecorvo G, Roper JA, Hemmons LM, MacDonald KD, Buffon AWJ. The genetics of Aspergillus nidulans. . Adv Genet. 1953;5:141–238. doi: 10.1016/s0065-2660(08)60408-3. [DOI] [PubMed] [Google Scholar]
- Pu RT, Osmani SA. Mitotic destruction of the cell cycle regulated NIMA protein kinase of Aspergillus nidulansis required for mitotic exit. EMBO (Eur Mol Biol Organ) J. 1995;14:995–1003. doi: 10.1002/j.1460-2075.1995.tb07080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu RT, Xu G, Wu L, Vierula J, O'Donnell K, Ye XS S.A.Osmani. Isolation of a functional homolog of the cell cycle specific NIMA protein kinase of Aspergillus nidulansand functional analysis of conserved residues. J Biol Chem. 1995;270:18110–18116. doi: 10.1074/jbc.270.30.18110. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 545 pp.
- Verdoes JC, Punt PJ, van der Berg P, Debets F, Stouthamer AH, van den Hondel CAM. Characterization of an efficient gene cloning strategy for Aspergillus niger based on an autonomously replicating plasmid: cloning of the nicB gene of A. niger. . Gene. 1994;146:159–165. doi: 10.1016/0378-1119(94)90288-7. [DOI] [PubMed] [Google Scholar]
- Waring RB, May GS, Morris NR. Characterization of an inducible expression system in Aspergillus nidulansusing alcA and tubulin coding genes. Gene. 1989;79:119–130. doi: 10.1016/0378-1119(89)90097-8. [DOI] [PubMed] [Google Scholar]
- Whalen WA, Bharathi A, Danielewicz D, Dhar R. Advancement through mitosis requires rae1gene function in fission yeast. Yeast. 1997;13:1167–1179. doi: 10.1002/(SICI)1097-0061(19970930)13:12<1167::AID-YEA154>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Wu L, Russell P. Nif1, a novel mitotic inhibitor in Schizosaccharomyces pombe. . EMBO (Eur Mol Biol Organ) J. 1997;16:1342–1350. doi: 10.1093/emboj/16.6.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye XS, Xu G, Pu RT, Fincher RR, McGuire SL, Osmani AH S.A.Osmani. The NIMA protein kinase is hyperphosphorylated and activated downstream of p34cdc2/cyclin B: Coordination of two mitosis promoting kinases. EMBO (Eur Mol Biol Organ) J. 1995;14:986–994. doi: 10.1002/j.1460-2075.1995.tb07079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, X.S., R.R. Fincher, A. Tang, K. O'Donnell, and S.A. Osmani. 1996. Two S phase checkpoint systems, one involving the function of both BIME and Tyr15 phosphorylation of p34cdc2, inhibit NIMA and prevent premature mitosis. EMBO (Eur. Mol. Biol. Organ.) J. 1996. 15: 3599–3610. [PMC free article] [PubMed]