

Abstract
Neuronal differentiation and function require extensive stabilization of the microtubule cytoskeleton. Neurons contain a large proportion of microtubules that resist the cold and depolymerizing drugs and exhibit slow subunit turnover. The origin of this stabilization is unclear. Here we have examined the role of STOP, a calmodulin-regulated protein previously isolated from cold-stable brain microtubules. We find that neuronal cells express increasing levels of STOP and of STOP variants during differentiation. These STOP proteins are associated with a large proportion of microtubules in neuronal cells, and are concentrated on cold-stable, drug-resistant, and long-lived polymers. STOP inhibition abolishes microtubule cold and drug stability in established neurites and impairs neurite formation. Thus, STOP proteins are responsible for microtubule stabilization in neurons, and are apparently required for normal neurite formation.
Keywords: neuron, STOP, E-STOP, microtubule-associated protein, calmodulin
Neurons establish connections with other cells over long distances. To do this, the rounded neuronal cell body produces axodendritic extensions. Microtubules play a key role in this remarkable morphogenetic process (Yamada et al., 1970; Drubin et al., 1985; Burgoyne, 1991). A salient feature of neuronal microtubules, thought to be intimately related to their morphogenetic function, is the stability of their assembly state. Neurons contain a high proportion of stable microtubules that resist exposure to cold temperature and to depolymerizing drugs (Webb and Wilson, 1980; Morris and Lasek, 1982; Black et al., 1984; Brady et al., 1984; Baas et al., 1994). As an apparent consequence of polymer stabilization, a large proportion of neuronal microtubules exhibit slow subunit turnover (Lim et al., 1989; Okabe and Hirokawa, 1990; Takeda et al., 1995; Li and Black, 1996). The presence of abundant subsets of long-lived polymers in nerve tissues causes a characteristic accumulation of posttranslationally modified tubulin molecules (for review see Cambray-Deakin, 1991; Paturle-Lafanechère et al., 1994). Stabilized microtubules are apparently required for generating and maintaining neuronal morphology and function (Baas and Heidemann, 1986; Baas and Black, 1990; Baas and Ahmad, 1992). Defects in microtubule stabilization may play a significant role in neuronal pathologies such as Alzheimer's disease (for review see Mandelkow et al., 1995).
Polymer stabilization is an obvious massive property of neuronal microtubules, yet the origin of microtubule stabilization is unclear. Microtubules interact with a multiplicity of cell components, and it is unknown whether microtubule cold stability, microtubule drug resistance, and slow microtubule turnover have a common origin or arise from the action of different factors. Over the past decades, microtubule effectors have mainly been sought among MAPs1 (microtubule-associated proteins; for reviews see Hirokawa, 1994; Mandelkow and Mandelkow, 1995). MAP2 and tau are two well-characterized effectors of microtubule stability in neurons. However, MAP2 and tau do not account for all the stability properties of neuronal microtubules. For instance, these MAPs do not confer cold stability to microtubules (Pirollet et al., 1983; Baas et al., 1994). Therefore, provided that MAPs are indeed the principal factors responsible for the stabilization of brain microtubules, at least one or several other MAPs must be involved in the stabilization of neuronal microtubules. One candidate for such a stabilizing function is STOP (stable tubule-only polypeptide).
STOP was initially isolated from preparations of rat brain cold-stable microtubules (Job et al., 1982), and was found to be a calmodulin-binding protein with potent microtubule-stabilizing capacity in vitro (Job et al., 1981; Job et al., 1982; Margolis et al., 1986). Recently the STOP cDNA has been cloned (Bosc et al., 1996). Expression of the cloned STOP cDNA in human cervical carcinoma cells induced a microtubule stable state (Bosc et al., 1996) reminiscent of that observed in neurons. These results raised the possibility that STOP may be a major factor responsible for microtubule stabilization in neurons, and hence may be important for neuronal morphogenesis. The present work was undertaken to assess the validity of this hypothesis. We tested to see whether STOP was associated with microtubules in neuronal cells, whether such an association occurred preferentially on stable polymers, and whether the inhibition of STOP suppressed microtubule stability. We further assessed the effect of STOP inhibition on neurite formation in cells undergoing neuronal differentiation.
We find that neuronal cells express increasing levels of several STOP variants during differentiation. STOP proteins are associated with a large proportion of axonal microtubules. STOP-associated polymers are also cold-stable, nocodazole-resistant, and contain elevated levels of posttranslationally modified tubulin. STOP inhibition by microinjected STOP antibodies specifically abolishes microtubule cold and drug stability in neurites, providing direct evidence that microtubule association with STOP proteins is the cause of microtubule stabilization. Furthermore, neuronal cells microinjected with STOP antibodies or exposed to STOP antisense oligonucleotides during differentiation show impaired neurite outgrowth.
These data show that microtubule stabilization in neurons is mainly due to the action of STOP proteins, and that STOP proteins are required for normal neurite formation.
Materials and Methods
Cell Culture
Dorsal root ganglia (DRG) were dissected from 14-d-old rat embryos and plated either on glass coverslips or on 35-mm plastic culture dishes coated with 100 μg/ml poly-l-ornithine (Sigma Chemical Co., St Louis, MO) and 10 μg/ml laminin (Sigma Chemcial Co.). DRG were cultured in N2 medium (Bottenstein and Sato, 1979) supplemented with 100 ng/ml NGF (Sigma Chemcial Co.). Undifferentiated PC12 cells were grown in RPMI 1640 (GIBCO BRL, Paisley, Scotland) supplemented with 5% FCS and 5% horse serum, and were subcultured every 3 d. For NGF treatment, PC12 cells were grown on 100 μg/ml poly-l-ornithine, 10 μg/ml laminin– coated glass coverslips, or 35-mm plastic culture dishes in Roswell Park Memorial Institute 1640 supplemented with 1% FCS, 1% horse serum, and 100 ng/ml NGF.
STOP and Tubulin Antibodies
Two rabbit polyclonal antibodies—23N and 23C—were raised against nonoverlapping synthetic peptides corresponding to the NH2-terminal domain (23N) or the COOH-terminal domain (23C) of the rat brain STOP central repeat motif (Bosc et al., 1996). The immunogenic peptides were PAAGKASGADQRDTRRKAG (amino acids [aa] 222–240 of the STOP sequence) and TRTEGHEEKPLPPAQSQTQEGG (aa 246–267 of the STOP sequence) for the 23N and 23C antibodies, respectively. Each peptide was conjugated to keyhole lympet hemocyanin and then injected into rabbits. Sera 23N and 23C were affinity-purified against the corresponding peptide. Monoclonal antibody 296 was a previously described STOP antibody (Pirollet et al., 1989) whose epitope on STOP has recently been localized in the COOH-terminal repeat domain of the protein. Detyrosinated tubulin (Glu-tubulin) antibody (L3) and Δ2-tubulin antibody (L7) were as previously described (Paturle-Lafanechère et al., 1994). A rat monoclonal antibody (YL 1/2) against the tyrosinated form of tubulin (Tyr-tubulin) was kindly provided by Dr. J.V. Kilmartin. Mouse mAb against β-tubulin (clone TUB 2.1) was obtained from Sigma Chemical Co. (St. Louis, MO).
Preparation of Cell and Tissue Extracts
Brains from adult rats or from 14-d-old rat embryos, and cerebella from 2-, 10-, or 20-d-old rats were homogenized in 100 mM MES, 1 mM EGTA, and 1 mM MgCl2 buffer (pH 6.75; 0.75/1, vol/wt) containing protease inhibitors (Complete™ tablets from Boehringer GmbH, Mannheim, Germany). The homogenates were centrifuged at 200,000 g for 10 min at 4°C. Supernatants were then harvested, supplemented with SDS/PAGE sample buffer (Laemmli, 1970), and boiled for 3 min. DRG and PC12 cells were extracted in 1% boiling SDS. After cooling, extracts were centrifuged at 200,000 g for 10 min at 4°C. The supernatants were supplemented with SDS/PAGE sample buffer and then boiled again for 3 min.
SDS-Polyacrylamide Gels and Immunoblotting
SDS-PAGE was performed according to Laemmli (1970). For immunoblotting, proteins were subsequently transferred onto nitrocellulose according to Towbin et al. (1979). Nitrocellulose membranes were processed as previously described (Lieuvin et al., 1994). The 23C, 23N, and mAb 296 primary STOP antibodies were diluted to 1/5,000, 1/10,000 and 1/5,000, respectively. YL1/2, L3, and L7 primary tubulin antibodies were diluted to 1/1,000, 1/100,000 and 1/100,000 respectively. Goat anti–rabbit (Tago Immunologicals, Burlingame, CA), anti–mouse (Cappel Laboratories, Malvern, PA), and donkey anti-rat (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) HRP-coupled secondary antibodies were diluted to 1/5,000.
Immunofluorescence Microscopy
Ganglionic explants or PC12 cells grown on coverslips were fixed in cold methanol and processed for immunofluorescence as in Lieuvin et al. (1994). Rat cerebellum sections were prepared and processed as in Paturle-Lafanechère et al. (1994). The affinity-purified 23C primary STOP antibody was diluted to 1/50. L3 and L7 primary tubulin antibodies were diluted to 1/1,000. TUB 2.1 primary tubulin antibody was diluted to 1/100. Goat anti-rabbit Cy3-coupled secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.) were diluted to 1/1,000. Goat anti– mouse FITC-coupled secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.) were diluted to 1/250. Images were digitalized using an RTE-CCD-1317-K/1 camera (Princeton Instruments Inc., Trenton, NJ) and IPLab Spectrum software (Signal Analytics Co., Vienna, VA).
Electron Microscopy
For control experiments, ganglionic explants on coverslips were washed in PBS buffer and permeabilized by a 3-min immersion in 2 ml of warm 100 mM Pipes, 1 mM EGTA, 1 mM MgCl2, pH 6.65 buffer (PEM buffer) containing 0.1% Triton X100 (vol/vol) and 10% glycerol (vol/vol). Coverslips were rinsed once in PEM buffer containing 10% glycerol (vol/vol), and explants were then fixed for 3 min in the same buffer supplemented with 0.5% 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide (vol/wt; Sigma Chemical Co.) and 0.1 mM GTP. Explants were then further processed for immunoelectron microscopy according to Baas and Black (1990). Explants were successively washed for 3 min at room temperature in PEM, 10% glycerol (vol/vol), and for 1 min in 10 mM Tris, 140 mM NaCl, pH 7.6 (TBS1), 1% BSA (vol/wt). Explants were further incubated for 1 h at room temperature with a primary antibody. Primary antibodies (23C STOP antibody, Glu-tubulin antibody, Tyr-tubulin antibody, and Δ2- tubulin antibody) were diluted to 1/20 in TBS1, 1% BSA (vol/wt). The explants were subsequently washed three times in 20 mM Tris, 140 mM NaCl, pH 8.2 (TBS2), 0.1% BSA (vol/wt), and incubated for 1 h at room temperature with appropriate 5-nm gold-coupled secondary antibodies (British BioCell International, Cardiff, United Kingdom). Secondary antibodies were diluted to 1/2 in TBS2, 0.1% BSA (vol/wt). After three washes in TBS2 buffer, explants were postfixed for 10 min in 0.1 M cacodylate (Taab Laboratories, Aldermaston, United Kingdom), 1% glutaraldehyde (Taab), and 2 mg/ml tannic acid (Mallinckrodt Inc., Paris, KY). Preparations were washed in 0.1 M cacodylate for 3 min and incubated in 0.1 M cacodylate supplemented with 2% osmic acid (vol/vol; Taab) for 10 min. After successive ethanol dehydration, DRG preparations were embedded in Epon (CIPEC, Paris, France) according to Pignot-Paintrand (1992). Frontal ultrathin sections were made using a diamond knife (Diatome Ltd., Biel, Switzerland) on an UltraCut S ultramicrotome (Reichert-Jung, Vienna, Austria) and collected on formvar (Taab)-coated slot grids (ref. 26310; Touzart & Matignon, Vitry-sur-Seine, France). Samples were observed on a 1200 EX II electron microscope (Jeol Ltd., Akishima, Japan).
For nocodazole treatment, explants were exposed for 30 min to 20 μM nocodazole before processing for EM. For cold treatment, explant cells were permeabilized in 4°C PEM-Triton-glycerol buffer and left on ice for 30 min before fixation and processing for EM. Microtubule cold stability tests yielded similar results when performed on intact or permeabilized cells (Lieuvin et al., 1994). For the present study, cell permeabilization was used since (a) it allows nearly instantaneous selection of stabilized polymers without superimposed effects of cell regulations, and (b) minimizes the possibility of STOP redistribution from depolymerizing labile polymers onto surviving stable polymers.
PC12 cells were fixed and processed for electron microscopy examination as in Yu and Baas (1995).
Assay of Microtubule Stability and Antibody Labeling in Electron Microscopy Experiments
For each condition, three independent ganglionic explants on different coverslips were used and processed in parallel for immunoelectron microscopy. Two sets of ten serial ultrathin frontal sections were performed for each embedded sample: one in the proximal region and one in the distal region of DRG cell axons. The proportions of drug- and cold-stable microtubules relative to control microtubules were determined as the ratio of the number of microtubules present per grid mesh in cold- or drug-treated preparations to the corresponding value in control untreated preparations. Section five of each embedded sample was analyzed. The number of polymers present in a mesh randomly selected within the part of the grid containing axons and in two adjacent meshes was determined on each microscopy grid. The variance of the estimated proportions of cold-stable and cold-labile polymers was calculated according to the classical formula:
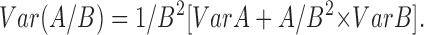 |
For quantification of antibody labeling with STOP or tubulin antibodies, sections 4–6 of each set were analyzed. On each microscopy grid, ten different microscopy fields were examined. Microtubule labeling was quantified by counting the number of gold particles observed along each polymer in the field. Results were expressed in numbers of gold particles per 500 nm of microtubule length. Estimated values of the average number of gold particles present on labile microtubules was determined as follows:
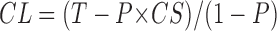 |
where CL, CS, and T represent the average number of gold particles per unit of polymer length in cold-labile, cold-stable, and total microtubules, respectively. P represents the proportion of cold-stable microtubules. In Table I, a P value of 50% corresponding to a conservative estimate of microtubule cold stability (see text) has been used. Using such a P value, CL = 2T − CS and VarCL = 4VarT + VarCS. These estimates rely on the hypothesis that the composition of stable microtubules does not change during labile polymer disassembly. This condition was almost certainly fulfilled during cold stability tests. STOP redistribution was a remote possibility during the cold stability test as designed in the present study (see above). Microtubule detyrosination during concomitant cell lysis and exposure to cold temperature is, in our experience, not detectable. The situation was different in the case of the nocodazole stability test: during the test, STOP proteins could shift from disassembling microtubules to surviving polymers, and in this case the possibility of significant action of the tubulin carboxypeptidase could not be totally excluded.
Table I.
STOP and Tubulin Labeling of Microtubules in DRG Cell Axons
| STOP | Glu-tubulin | Tyr-tubulin | Δ2-tubulin | |||||
|---|---|---|---|---|---|---|---|---|
| T MTs | ||||||||
| m | 12.8 | 11.0 | 21.4 | 19.3 | ||||
| SEM | 0.3 | 0.2 | 0.6 | 0.5 | ||||
| n | 523 | 397 | 333 | 376 | ||||
| CS MTs | ||||||||
| m | 21.4 | 21.3 | 17.0 | 21.9 | ||||
| SEM | 0.6 | 0.5 | 0.5 | 0.6 | ||||
| n | 295 | 261 | 167.0 | 236.0 | ||||
| CL MTs | ||||||||
| m | 4.2 | 0.7 | 25.8 | 16.7 | ||||
| SEM | 0.8 | 0.6 | 1.3 | 1.2 |
DRG cells were either untreated or exposed to cold temperature. Cells were further reacted either with the affinity-purified 23C STOP antibody or with one isoform specific tubulin antibody (Tyr-, Glu- or Δ2-tubulin antibody) and with secondary gold-labeled antibody. The table shows the mean numbers of gold particles observed per 500 nm of microtubule length for total (T MTs) and cold-stable (CS MTs) polymers. The corresponding mean numbers for cold-labile microtubules (CL MTs) were estimated as described under Materials and Methods. m, mean number of gold particle per 500 nm of microtubule length. SEM, standard error of the mean. n, number of microtubules examined.
cDNA Cloning
An oligo(dT)-primed fetal rat brain cDNA library (CLONTECH Laboratories, Inc., Palo Alto, CA) was screened using a 32P-labeled probe corresponding to nucleotides 1138–1935 of the adult rat brain STOP cDNA (Bosc et al., 1996). Screening was performed using standard procedures (Sambrook et al., 1989). A total of 22 overlapping clones was obtained. One clone contained the entire coding sequence corresponding to a novel STOP isoform (see text).
In Vitro Translation of Early (E)-STOP cDNA and Assay of E-STOP Binding to Microtubules
Construction of E-STOP expression vector was done by replacing fragment SacII/ EcoICR I (nucleotides [nt] 997–2843) of STOP cDNA in pSG5-STOP construct (Bosc et al., 1996) by fragment Sac II/Pme I (nt 833–2401) of E-STOP cDNA. The resulting construct (pSG5-E-STOP) was used for in vitro translation of E-STOP and microtubule-binding assays as in Bosc et al. (1996).
Microinjection and Analysis of Fused PC12 Cells
To allow microinjection experiments, PC12 cells were chemically fused: PC12 cells were rinsed in RPMI 1640 supplemented with 20 mM Hepes (GIBCO BRL), centrifuged, and resuspended in 1 ml of polyethylene glycol 1500 (Boehringer GmbH) for 1 min at 37°C. The cell suspension was then diluted in 10 ml of RPMI 1640, 20 mM Hepes, and incubated for 5 min at 37°C. Fused cells were centrifuged for 5 min at 300 g, resuspended in culture medium, and plated on Cellocate coverslips (Eppendorf-Netheler-Hinz GMBH, Hamburg, Germany) coated with 100 μg/ml poly- l-ornithine and 10 μg/ml laminin. Cells were pressure-injected using a 5171 micromanipulator (Eppendorf-Netheler-Hinz GmbH), a 5246 transjector (Eppendorf-Netheler-Hinz GmbH), and an Axiovert 35M microscope (Carl Zeiss, Oberhocken, Germany).
Fused PC12 cells were cultured for 48–72 h in the presence of NGF before assay of antibody effect on microtubule stability. Differentiated fused cells were microinjected with 2 mg/ml affinity-purified 23C or 23N STOP antibodies, or with control IgGs at the same protein concentration. These control IgGs were purified from sera of nonimmunized rabbits. After microinjection and an additional 2–30 h of culture in the presence of NGF, some cells were exposed for 30 min to cold temperature or to 20 μM nocodazole. Subsequent assay of cell microtubule content was as in Lieuvin et al. (1994): after treatment, cells were extracted for 1 min in a large volume of PEM buffer containing 0.1% Triton X100 (vol/vol) and 10% glycerol (vol/vol) to remove soluble tubulin, and were then prepared for immunofluorescence analysis of the remaining polymeric tubulin using mAb TUB 2.1.
To assess STOP antibody effect on neurite outgrowth, fused cells were microinjected either with the 23C antibody or with control IgGs after 1 d of culture in the presence of NGF. After an additional 24–36 h of culture in the presence of NGF, cells were then processed for immunofluorescence analysis. Microinjected IgGs were reacted with a Cy3-coupled IgG antibody, and tubulin was stained with mAb TUB 2.1 and FITC-coupled secondary antibody. Microinjected and control cells were analyzed for the presence of neurite extensions.
Transfection of PC12 Cells with Antisense Oligonucleotides
Five antisense oligonucleotides corresponding to nucleotide sequences located in the central repeat domain of STOP were designed based upon rat brain STOP cDNA sequence (Bosc et al., 1996): ON1as (nt 1172–1189), ON2as (nt 1231–1248), ON3as (nt 1236–1253), ON4as (nt 1536–1553), and ON5as (nt 1843–1860). Control scrambled oligonucleotides were as follows: ON1sc (5′ AGCCGAGCCCTGTGTCTG 3′), ON2sc (5′ AATGCCTCGTCGCTCCGC 3′), ON3sc (5′ CGCGACCGCTTTGGTTCG 3′), ON4sc (5′ GGGGGCTGGGACGGCGAG 3′), and ON5sc (5′ TCTCTCGCCGTCGTTCCC 3′). Control sense oligonucleotides (ON1se, ON2se, ON3se, ON4se, and ON5se) were synthesized as the reverse complement of the antisense oligonucleotides. PC12 cells were transfected with antisense, sense, or scrambled oligonucleotides using DOTAP (Boehringer Mannheim GmbH) as a transfection reagent according to the manufacturer's instructions. For controls, PC12 cells were exposed to DOTAP alone. Cells were observed after 24–36 h of NGF and oligonucleotide treatment, and were analyzed for the presence of neurite extensions using phase contrast microscopy. Cells were scored as positive if they exhibited extensions longer than one cell body diameter. In some experiments, cells were further immunostained with tubulin antibodies for detailed analysis of neurite extensions. Each experiment involved parallel examination of cells treated with DOTAP alone, of cells exposed to one antisense oligonucleotide, and of cells treated with the corresponding sense or scrambled oligonucleotides. For each condition cells grown on three different coverslips were examined. Three microscopy fields were randomly selected on each coverslip. A total of ∼800–1000 cells was screened per condition. Each experiment was run at least in triplicate.
Results
To investigate the distribution of STOP, we used explants of dorsal root ganglia (DRG) from rat embryos. Ganglion explants sprout dense radial arrays of axons during DRG cell differentiation in vitro, and provide a convenient system for ultrastructural studies of statistically significant populations of microtubules (Baas and Black, 1990).
STOP Expression in DRG Cells and Comparison with Pattern of Expression In Vivo
DRG cells were first tested for the pattern of STOP expression on immunoblots of cell extracts (Fig. 1). For this we used three different primary STOP antibodies directed against distinct epitopes of the protein. STOP contains a central repeat domain and a carboxy terminal repeat domain (Fig. 1; Bosc et al., 1996). Two antibodies (23C and 23N) were directed against nonoverlapping peptides derived from the amino acid sequence of STOP central repeat. The third antibody (mAb 296) was directed against epitopes located in the STOP carboxy terminal repeat domain (for details see Materials and Methods). Extracts from adult rat brain and extracts from embryonic rat brain were analyzed in parallel with DRG cell extracts.
Figure 1.

STOP expression in DRG cells. (A) Immunoblot analysis of STOP expression. Proteins from 3-d differentiated DRG cells (lane 1), 10-d differentiated DRG cells (lane 2), adult rat brains (lane 3), and embryonic rat brains (lane 4) were run on 7.5% SDS gels. 20 μg of DRG cell extract proteins were loaded onto the gel. Amounts of loaded brain proteins were adjusted to equilibrate brain and DRG cell STOP signals. Proteins were immunoblotted with polyclonal STOP antibodies 23C and 23N (central repeat antibodies), and with monoclonal STOP antibody 296 (COOH-terminal repeat antibody) as indicated. The bands corresponding to STOP and E-STOP are indicated. Size markers are in kD. (B) Schematic representation of STOP and E-STOP showing domain structure of the two proteins. Both proteins contain a central domain composed of 5 (STOP) or 6 (E-STOP) repeats. STOP also contains a COOH-terminal repeat domain that is lacking in E-STOP. The E-STOP sequence ends at a position corresponding to aa 614 in the STOP sequence (Bosc et al., 1996). The E-STOP sequence data are available from GenBank/EMBL/DDBJ under accession no. AJ002556.
In DRG cell extracts the two central repeat antibodies (23C and 23N) reacted with a major 84-kD polypeptide not recognized by mAb 296 (Fig. 1 A). By contrast, the standard adult rat brain STOP has a higher mass and reacts with all three STOP antibodies (Fig. 1 A). The 84-kD polypeptide was present in cell extracts after 3 d of cell differentiation, and was enhanced after 10 d of cell differentiation. The 84-kD polypeptide was present as a minor band in adult rat brain extracts, but was the dominant STOP antigen in embryonic rat brain (Fig. 1 A). We have chosen to call this 84-kD polypeptide E-STOP to denote its early appearance during development. As a result of screening an embryonic rat brain cDNA library, we have determined the nucleotide sequence of the E-STOP cDNA and the corresponding protein sequence (GenBank/EMBL/DDBJ accession no. AJ002556). E-STOP differs from the standard adult STOP by insertion of an additional central repeat and by deletion of a COOH-terminal amino acid stretch (amino acid [aa] 615–952 of the standard STOP protein; Fig. 1 B). This COOH-terminal domain contains the carboxy terminal STOP repeats recognized by mAb 296. Despite its lack of COOH-terminal repeats, E-STOP shows calmodulin-binding and microtubule-stabilizing activity comparable to adult STOP. As adult STOP (Bosc et al., 1996), E-STOP binds to calmodulin columns in a Ca2+-dependent manner, cosediments with microtubules in microtubule-binding assays, and induces microtubule stabilization when expressed in cells normally devoid of stable microtubules (data not shown).
In addition to E-STOP, 10-d–differentiated DRG cell extracts contained standard adult STOP (Fig. 1 A). DRG cell extracts also contained a 62-kD polypeptide recognized by the two central repeat antibodies (23C and 23N; Fig. 1 A), and by STOP antibodies 22 and 136 directed against epitopes located upstream (aa 160–174) and downstream (aa 485–504) of STOP central repeats, respectively (data not shown). This 62-kD STOP antigen was also present in embryonic brain extracts (Fig. 1 A). It may be a proteolysis product of STOP, or it may be a third STOP isoform that was not further characterized in the present study.
The pattern of STOP expression in DRG cells suggested strong correlation between neuronal cell differentiation and STOP expression. To test whether these conclusions applied to in vivo situations, we assayed STOP expression and distribution in developing rat cerebellum. Immunoblot analysis showed similar patterns of STOP and E-STOP expression during development in cerebellum as in DRG cells (Fig. 2 A). In developing cerebellum, neuronal cells at different stages of differentiation are organized in distinct cell layers. In 2-d-old rats, dividing neuronal cells are still present in the external germinal layer (EGL), whereas cells in the underlying molecular layer (ML) have undergone neuronal differentiation and grown neurite extensions (Burgoyne and Cambray-Deakin, 1988). STOP staining of cerebellum sections from 2-d-old rat cerebellum showed absence of detectable STOP staining in the EGL, while STOP staining was intense in the adjacent ML (Fig. 2, B and C). These results show strong correlation between neuronal differentiation and STOP expression in vivo. At later stages of cerebellum development, patterns of STOP staining were essentially identical to those previously determined in the case of Δ2-tubulin, a known marker of microtubule neuronal differentiation (data not shown; Paturle-Lafanechère et al., 1994).
Figure 2.

STOP expression during postnatal development of the rat cerebellum. (A) Immunoblot analysis of STOP expression in the cerebellum. Proteins from 2-d, 10-d, and 20-d-old rat were run on 7.5% SDS gels. 2 μg of cerebellum extract proteins were loaded onto the gel. Proteins were immunoblotted with polyclonal STOP antibody 23C. The bands corresponding to STOP and E-STOP are indicated. Size markers are in kD. (B and C) Immunofluorescence staining of the cerebellar cortex. Sections of 2-d aged rat cerebellum were stained with polyclonal STOP antibody 23C (B); (C) corresponding DNA staining with Hoechst. EGL, external germinal layer, ML, molecular layer. Bar, 40 μm.
STOP Proteins are Associated with a Large Proportion of Microtubules in DRG Cells Axons
STOP proteins were localized in the axons of DRG cells using both immunofluorescence and immunoelectron microscopy. We used the 23C STOP antibody which only reacts with STOP proteins in DRG cells (see above). An immunofluorescence image of DRG cell axons stained with the 23C STOP antibody is shown in Fig. 3 A. Axons were brightly stained, and the staining was apparently homogeneous along the whole axonal length. Although this showed that STOP proteins were widely distributed in axons, the resolution of immunofluorescence microscopy was not sufficient to assess STOP association with microtubules within the axons. Therefore, we examined the distribution of STOP proteins using immunoelectron microscopy. Processing DRG cells for EM examination involved a cell permeabilization step (see Materials and Methods). We verified by immunoblot assays that no detectable amount of STOP proteins was eliminated during this permeabilization step (data not shown). When stained with the 23C antibody and a gold-coupled secondary antibody, DRG cell axons showed specific microtubule labeling (Fig. 3 B). We determined the number of gold particles per unit of microtubule length in a series of microscopic images, an example of which is shown in Fig. 3 B. This analysis was performed separately for the proximal and distal parts of axons. Results were similar in both parts of axons, and are shown in the case of distal microtubules (Fig. 3 C, black bars). Over 60% of the microtubules were associated with more than 10 gold particles per 500 nm of microtubule length. In parallel experiments in which DRG cell axons were labeled with the secondary antibody alone, we never observed more than 4 gold particles per 500 nm of microtubule length. These data showed that in DRG cells, a large proportion of axonal microtubules had significant STOP staining, and were therefore associated with STOP proteins.
Figure 3.

Immunolocalization of STOP proteins in DRG cell axons. After 10 d of culture, DRG cells were stained using the affinity-purified 23C STOP antibody and a Cy3- (A) or gold-labeled (B) secondary antibody. (A) Immunofluorescence analysis of STOP localization in DRG cell axons. Bar, 25 μm. (B) Immunoelectron microscopy of a DRG cell axon: microtubules were specifically decorated with gold particles. Bar, 50 nm. (C) Gold particle distribution along axonal microtubules in the distal part of DRG cell axons. (Black bars) total microtubules; (dark gray bars) cold stable microtubules; (light gray bars) nocodazole-resistant microtubules. In control experiments with secondary antibody alone, microtubules were never decorated with more than four gold particles.
STOP Proteins are Concentrated on Cold-stable and Drug-resistant Microtubules in DRG Cells
To investigate the relationship between STOP distribution and microtubule stability, differentiated DRG cells treated with cold or nocodazole or untreated were compared for the presence of STOP proteins on microtubules. Treated and untreated DRG cells were processed for immunoelectron microscopy. A comparison between treated or untreated cells for microtubule numbers on grids showed that the distal part of DRG cell axons contained 57 ± 6% (percent ± SD) cold-stable microtubules and 41 ± 6% nocodazole-resistant microtubules (for details see Materials and Methods). Similar proportions of stable microtubules were observed in the proximal part of axons. These estimates were in good agreement with previously published data, reporting 60% cold-stable microtubules and 50% nocodazole-resistant microtubules in differentiated DRG cells (Sekimoto et al., 1995). Microtubule STOP labeling was quantified on distal sections of DRG cell axons. In untreated cells, microtubules (total microtubules) were decorated on average with 12.8 ± 0.3 (mean ± SEM) gold particles per 500 nm of polymer length (Fig. 3 C and Table I). Microtubule labeling was independent of the number of polymers present in the microscopy field (r = 0.08, P > 0.05). In cells exposed to cold temperature, microtubules (cold-stable microtubules) were associated with an average number of 21.4 ± 0.6 gold particles per 500 nm of polymer length (Fig. 3 C and Table I). In cells exposed to nocodazole, microtubules (drug-stable microtubules) were associated on average with 22.0 ± 0.6 gold particles per 500 nm of polymer length (Fig. 3 C). These results showed that cold-stable microtubules and drug-stable microtubules were highly enriched in STOP protein as compared with total microtubules. However, the total microtubule population was heterogeneous, being comprised of stable and labile polymers. Obviously one would like to compare homogeneous populations of stable or labile microtubules. Labile microtubules could not be examined directly, but estimates of the STOP content of labile polymers could be derived from experimental data in the case of cold-labile polymers (see Materials and Methods). The estimated mean number of gold particles per 500 nm of polymer length present on cold-labile microtubules amounted to 4.2 ± 0.8 gold particles (Table I), a value close to that obtained with secondary antibody alone. Such a value indicated that most cold-labile microtubules contained little or no STOP protein, and suggested a very tight relationship between microtubule association with STOP proteins and microtubule cold stability.
In DRG Cells, Microtubule Cold Stability is Tightly Related to Microtubule Detyrosination
It is unknown whether cold stability, a microtubule property tightly related to microtubule association with STOP proteins (see above), is also related to slow microtubule turnover in neurons (Baas et al., 1994). To test whether cold-stable microtubules in DRG cells corresponded to the nondynamic polymers normally present in neuronal cells, we examined cold-stable polymers for their detyrosinated tubulin content. Detyrosinated tubulin differs from normal tubulin by deletion of the COOH-terminal tyrosine residue of α-tubulin, and is known to accumulate in nondynamic polymers (Kreis, 1987; Schulze et al., 1987; Wehland and Weber, 1987; Bulinski and Gundersen, 1991). Detyrosinated tubulin (Glu-tubulin) is generated from normal tyrosinated tubulin (Tyr-tubulin) in microtubules with slow subunit turnover upon long polymer exposure to an ill-defined carboxypeptidase (for reviews see Barra et al., 1988; MacRae, 1997). Neuronal tissues contain large amounts of Glu-tubulin (Fig. 4 A; Baas and Black, 1990; Paturle-Lafanechère et al., 1994). Such an accumulation of Glu-tubulin also occurs in differentiating DRG cells (Fig. 4 A).
Figure 4.

Tubulin modification in DRG cells. (A) Immunoblot analysis of tubulin modification in DRG cells: proteins from DRG cells cultured for 3 d (lane 1) or 10 d (lane 2), adult rat brains (lane 3), and embryonic rat brains (lane 4) were run on 7.5% SDS gels. Amounts of loaded proteins were as indicated in the legend to Fig. 1 A. Proteins were immunoblotted with isoform-specific tubulin antibodies as indicated. (B) Immunofluorescence analysis of Δ2-tubulin localization in 10-d cultured DRG cell axons. Cells were reacted with a specific Δ2-tubulin antibody. Axons showed bright and apparently homogeneous Δ2-tubulin staining along their length. Bar, 25 μm.
Differentiated DRG cells, untreated or exposed to cold temperature, were compared for microtubule content in Glu-tubulin. Cold-treated and control DRG cells were immunostained with a Glu-tubulin antibody and further processed for electron microscopy. Microtubule labeling was quantified on distal sections of DRG cell axons. In control cells, microtubules (total microtubules) were decorated on average with 11.0 ± 0.2 gold particles per 500 nm of polymer length (Table I). In cells exposed to cold temperature, microtubules (cold-stable microtubules) were associated with an average number of 21.3 ± 0.5 gold particles per 500 nm of polymer length (Table I). From these values, the expected mean number of gold particles per 500 nm of polymer length for cold-labile microtubules was estimated to be 0.7 ± 0.6 (Table I). These results indicate a very strong association between microtubule cold stability and microtubule detyrosination in DRG cell axons. This detyrosination of cold-stable microtubules was also evident from the quantification of Tyr-tubulin labeling among the total, cold-stable, and cold-labile polymer populations: cold-stable microtubules contained less Tyr-tubulin than did total or cold-labile microtubules (Table I).
Besides Tyr- and Glu-tubulin, neurons specifically contain a third tubulin variant, Δ2-tubulin (Paturle-Lafanechère et al., 1994). Δ2-tubulin differs from Tyr-tubulin by the deletion of the last two COOH-terminal amino acids of α-tubulin (Paturle-Lafanechère et al., 1991). Δ2-tubulin only appears on very long-lived polymers, and is absent in nonneuronal cells (Paturle-Lafanechère et al., 1994). DRG cells accumulated Δ2-tubulin during differentiation (Fig. 4 A). Δ2-tubulin was present along the whole axon (Fig. 4 B), and only moderately enriched in cold-stable microtubules (Table I).
STOP Expression and Tubulin Modification in PC12 Cells
In the preceding sections it was shown that microtubule association with STOP proteins and microtubule stabilization are two tightly associated polymer properties. STOP may associate with microtubules stabilized by other factors, or STOP may cause microtubule stabilization. To test which of these possibilities was correct, we assessed the effect of STOP inhibition on microtubule stability in neuronal cells. To do this, a neuronal cell system amenable to routine examination of microinjection or antisense experiments was required. We chose PC12 cells, as these cells can be grown indefinitely in vitro in the absence of NGF and undergo neuronal differentiation upon exposure to NGF (Greene and Tischler, 1976). To assess the utility of PC12 cells as a cell model in the context of the present study, they were examined for STOP expression and tubulin modification during differentiation.
As in the case of DRG cells, STOP expression in PC12 cells was analyzed on immunoblots using three different STOP antibodies. Results observed using the 23C STOP antibody are shown in Fig. 5 A. Identical results were observed with the 23N STOP antibody (data not shown). 10 d–differentiated PC12 cells expressed adult STOP, E-STOP, and two additional STOP-reactive polypeptides of 45 kD and 100 kD. Recent work in this laboratory has shown that the 45-kD and 100-kD reactive polypeptides correspond to STOP proteins normally expressed in certain nonneuronal tissues. The complete molecular and functional characterization of nonneuronal STOP proteins will be presented in a forthcoming article (Denarier et al., 1998b ). For the purpose of the present study, the important fact was that differentiated PC12 cells expressed STOP proteins. Differentiated PC12 cells also contained Glu- and Δ2-tubulin (Fig. 5 B). To assess the NGF dependence of STOP and Δ2-tubulin accumulation in PC12 cells, we used immunufluorescence analysis. Mixed populations of differentiated and undifferentiated PC12 cells were stained either with 23C STOP antibody or with Δ2-tubulin antibodies. Differentiated PC12 cells exhibited bright staining of neurite extensions with both antibodies, while undifferentiated cells showed low or background levels of staining (Fig. 5, C and D). These results indicate that, with respect to STOP expression and tubulin modification, the behavior of NGF-treated PC12 cells is similar to that of other neuronal cell models.
Figure 5.

STOP expression and tubulin modification in PC12 cells. (A) Immunoblot analysis of STOP expression in PC12 cells: proteins from PC12 cells after 10 d of differentiation (lane 1), adult rat brains (lane 2), and embryonic rat brains (lane 3) were run on 7.5% SDS gels. 20 μg of PC12 cell proteins were loaded onto the gel. Amounts of loaded brain proteins were adjusted to equilibrate brain and PC12 cell STOP signals. Proteins were immunoblotted with polyclonal STOP antibody 23C. Superimposable results were observed with the 23N STOP antibody (data not shown). The bands corresponding to STOP and E-STOP are indicated. Size markers are in kD. (B) Immunoblot analysis of tubulin modification in PC12 cells: proteins from PC12 cells after 10 d of differentiation (lane 1), adult rat brains (lane 2), and embryonic rat brains (lane 3) were run on 7.5% SDS gels. Amounts of loaded proteins were as indicated in A. Proteins were immunoblotted with isoform-specific tubulin antibodies as indicated. (C and D) Immunofluorescence analysis of mixed populations of differentiated and undifferentiated PC12 cells with 23C STOP antibody (C) or Δ2-tubulin antibody (D). Neurite extensions in differentiated cells showed bright STOP or Δ2-tubulin staining while undifferentiated cells showed low or background levels of staining. Bar, 50 μm.
STOP Proteins are Responsible for Microtubule Cold and Drug Stability in Differentiated PC12 Cells
To test whether the demonstrated STOP association with microtubules was indeed responsible for microtubule stabilization, we examined the effect of STOP inactivation on microtubule stability in PC12 cells. Fused PC12 cells (Okabe and Hirokawa, 1988) were injected either with affinity- purified STOP antibody (23C or 23N) or with control IgGs. STOP central repeat antibodies have the capacity to inhibit STOP binding to microtubules in vitro (Fig. 6). After a recovery period of 6 h, cells were assayed for microtubule resistance to depolymerizing conditions (exposure to cold temperature or to nocodazole). For microtubule content assay, cells were lysed in a large volume of Triton-Pipes buffer to remove free tubulin dimers, and polymeric tubulin was subsequently visualized by immunofluorescence microscopy with tubulin antibody (for details see Materials and Methods and Lieuvin et al., 1994). To identify microinjected cells among other cells, microinjected control IgGs or microinjected STOP antibody were visualized with Cy3-conjugated rabbit IgG antibody (Fig. 7, A–C and G–I, respectively).
Figure 6.

In vitro inhibition of E-STOP binding to microtubules by STOP antibody. In vitro–translated E-STOP (35S-labeled) was sedimented in the presence of bovine brain–polymerized microtubules (for details see Materials and Methods) after preincubation (5 min, 37°C) with polyclonal STOP antibody 23C (0.20 mg/ml) or with naive IgGs at the same concentration, as indicated. Equal amounts of the pellet (P) and supernatant (S) were separated by 7.5% SDS/ PAGE and exposed to autoradiography. In the presence of STOP antibody 23C, extensive inhibition of E-STOP binding to microtubules was observed. Results were similar when E-STOP solutions were preincubated with microtubules before adding STOP antibody 23C, and when STOP antibody 23C was replaced by STOP antibody 23N (data not shown). Size markers are in kD.
Figure 7.

STOP inhibition suppresses microtubule cold and drug stability in fused PC 12 cells Fused PC12 cells injected with control IgGs (A–F) showing IgG (A–C) and microtubule (D–F) staining. (A and D) Untreated cells; (B and E) cold-treated cells; (C and F) nocodazole-treated cells. Fused PC12 cells injected with affinity-purified 23C STOP antibody (G–L) showing STOP antibody (G–I) and microtubule (J–L) staining. (G and J) Untreated cells; (H and K) cold-treated cells; (I and L) nocodazole-treated cells. Note that after cell lysis, enough microinjected IgGs remain associated with insoluble cell structures to allow clear identification of injected cells. STOP antibody injection suppressed microtubule cold and drug stability. Bar, 50 μm.
In the absence of cold or nocodazole treatment, PC12 cells microinjected with nonreactive IgGs showed bright microtubule signal after staining with tubulin antibody (Fig. 7 D). The microtubule signal was not evidently modified when such microinjected cells were exposed to cold temperature or to nocodazole before assay of microtubule content (Fig. 7, E and F, respectively). Nonmicroinjected cells behaved in the same manner as cells injected with nonreactive IgGs (data not shown). These results demonstrate that PC12 cells contain an abundant subset of cold- and drug-resistant microtubules, and that microtubule stability is not modified by the microinjection process.
In the absence of cold or nocodazole treatment, cells microinjected with affinity-purified 23C STOP antibody showed apparently normal microtubule signal when stained with tubulin antibody (Fig. 7 J). However, exposure of such cells to cold temperature or to nocodazole induced complete microtubule disassembly, as shown by absence of detectable microtubule signal in cold- or drug-treated cells (Fig. 7, K and L, respectively). The absence of microtubules in cold-treated STOP antibody–injected cells was also evident in electron micrographs of microinjected cells processed for electron microscopy as in Yu and Baas (1995; data not shown). Thus, injection of the STOP antibody into PC12 cells abolished microtubule cold and drug stability. These striking results were highly reproducible, being observed in all STOP antibody–microinjected cells. Results were identical when affinity-purified 23N antibody was used instead of affinity-purified 23C antibody (data not shown). These results provide compelling evidence that STOP proteins are major determinants of microtubule cold and drug stability in differentiated PC12 cells.
Effect of STOP Inhibition on Neurite Formation in PC12 Cells
There is evidence that cold- and drug-stable polymers are required for neurite integrity (Baas and Heidemann, 1986; Baas and Black, 1990; Baas and Ahmad, 1992). However, such stable polymers may not be required for maintaining preexisting neurites; differentiated PC12 cells microinjected with STOP antibody lack cold- and drug-stable polymers, and nevertheless retain apparently normal neurite extensions (Fig. 7, G–L). We have repeated such experiments with variable delays (2–30 h) between cell microinjection and cell examination. In all STOP antibody– microinjected cells, neurite extensions remained apparently normal, while microtubule cold and drug stability was abolished (data not shown). Therefore, at least within the time duration of our experiments, STOP activity was not required for maintenance of preexisting neurites.
We tested to see whether STOP proteins were required for neurite outgrowth in PC12 cells that were not yet morphologically differentiated. For STOP inhibition, we used both microinjection of STOP antibodies and cell exposure to STOP antisense oligonucleotides. In microinjection experiments, fused PC12 cells were injected either with affinity-purified 23C STOP antibodies or with naive IgGs. Microinjected cells were then grown in the presence of NGF for 36 h and examined for the presence of neurite extensions. Cells were scored as positive if they exhibited extensions longer than one cell body diameter. Pooled data from two independent experiments showed that 33 ± 3% (n = 200) of the cells injected with nonspecific IgGs had significant neurite extensions at the time of examination. Such a yield of cell differentiation was similar to that routinely observed in nonmicroinjected fused PC12 cells. In contrast, only 14 ± 3% (n = 183) of the cells microinjected with the 23C STOP antibody had neurites at the time of examination. This twofold difference between the observed percentages was highly significant (P < 0.001).
Microinjection experiments involve the use of fused PC12 cells. Such cells show a relatively low yield of differentiation. STOP effect on neurite formation was also examined in normal PC12 cells treated with antisense oligonucleotides using DOTAP as transfection reagent. We tested five different antisense oligonucleotides derived from the STOP cDNA sequence coding for the STOP central repeats. In control experiments, the corresponding sense and scrambled oligonucleotides were also tested for their effect on PC12 cell differentiation. In the presence of DOTAP alone, ∼80% of PC12 cells had significant neurite extensions after a 36-h treatment with NGF. In the presence of the different added antisense oligonucleotides, only 18–45% of the cells had significant neurite extensions at the time of examination (Fig. 8, A–C). Such a decrease was never observed in cells treated with sense or scrambled oligonucleotides (Fig. 8 C). Upon immunofluorescence examination, the cells that had grown neurites despite exposure to antisense oligonucleotides exhibited STOP staining in neurites, while other cells showed low or background levels of staining (data not shown). Immunoblot analysis confirmed strong inhibition of STOP expression in the presence of antisense oligonucleotides (Fig. 8 D).
Figure 8.

Inhibition of neurite formation in PC12 cells treated with STOP antisense oligonucleotides. (A and B) Immunofluorescence analysis of PC12 cells after a 36-h NGF treatment in the presence of sense ON4 (A) or antisense ON4 (B). For visualization of neurite extensions, cells were stained with mAb TUB 2.1 tubulin antibody. Bar, 100 μm. (C) Effect of five different STOP antisense oligonucleotides on neurite outgrowth. Each antisense oligonucleotide (as) was tested in parallel with the corresponding sense (se) and scrambled (sc) oligonucleotides. ON1–ON5 designate the five groups of oligonucleotides tested. In each condition, the proportion of differentiated cells among the total cell population was determined. Results are expressed as percent of control values determined in the presence of DOTAP alone (for details see Materials and Methods). (D) Immunoblot analysis of STOP proteins in PC12 cells after a 3-d NGF treatment in the presence of DOTAP alone (c), antisense (as), sense (se), or scrambled (sc) oligonucleotide ON4. 20 μg of PC12 cell proteins were loaded on 7.5% SDS gels. Proteins were immunoblotted with polyclonal STOP antibody 23C. Results marked diminution of STOP expression in cells treated with antisense oligonucleotide ON4. Size markers are in kD.
In conclusion, we find extensive and specific inhibition of neurite outgrowth in PC12 cells upon microinjection of STOP antibody or cell exposure to five different STOP antisense oligonucleotides. These results strongly suggest that STOP activity is required for normal neurite formation.
Discussion
STOP Proteins, Microtubule Stability, and Microtubule Turnover
It has been known for many years that neurons contain abundant subsets of cold-stable microtubules (Webb and Wilson, 1980; Morris and Lasek, 1982; Black et al., 1984; Brady et al., 1984; Baas et al., 1994). Cold stability is a remarkable property of neuronal microtubules, in sharp contrast with the lability of microtubules assembled from pure tubulin in vitro. However, the origin of microtubule cold stabilization in neurons has remained obscure (Baas et al., 1994). Neuronal microtubules interact with many cell components, and it has been uncertain whether cold stability resulted from the action of a single well-defined effector, or had a multiplicity of causes. Among possible factors involved in microtubule cold stabilization, STOP, a neuronal calmodulin- and microtubule-binding protein, was of particular interest because of its capacity to induce microtubule cold stability (Bosc et al., 1996). Here we find that differentiating neuronal cells express several STOP variants. Examination of the STOP distribution at the ultrastructural level in DRG cells shows STOP presence along microtubules and specifically among cold-stable microtubules. Microinjection of STOP antibodies into differentiated PC12 cells causes complete suppression of microtubule cold stability. These results demonstrate that microtubule cold stability in neuronal cells is due to lengthwise association of microtubules with STOP proteins.
Cold stability is not the only remarkable stability property of neuronal microtubules. Although microtubules in cycling cells are universally sensitive to nocodazole, neuronal microtubules are in large part drug-resistant. Before this study it was unclear whether cold stability and drug resistance were independent or related properties of neuronal microtubules (Baas et al., 1994). There is evidence that several MAPs such as MAP2, MAP1B, and tau may be involved in the induction of microtubule drug resistance in neurons (Takemura et al., 1992, Baas et al., 1994), while such MAPs are devoid of cold-stabilizing activity (Pirollet et al., 1983; Baas et al., 1994). As STOP has the capacity to inhibit nocodazole action (Bosc et al., 1996), we have examined STOP contribution to the generation of microtubule resistance to drugs in neurons. We find that drug-resistant microtubules in DRG cells are highly enriched in STOP proteins. Moreover, microinjection of STOP antibodies in PC12 cells abolishes microtubule resistance to nocodazole. This is compelling evidence that STOP proteins are major factors controlling both microtubule cold and drug stability in neurons.
The observation of extensive microtubule drug resistance in neurons has suggested that microtubule turnover may be slow in these cells. Direct measurements of microtubule turnover have shown that a large subset of microtubules requires hours to turn over in neurons instead of a time course of minutes in other cell types (Lim et al., 1989; Okabe and Hirokawa, 1990; Takeda et al., 1995; Li and Black, 1996). Recently, such measurements have also provided a direct demonstration that there is a strong correlation between slow microtubule turnover and microtubule detyrosination in neuronal cells (Li and Black, 1996) as predicted from experiments previously done in other cell types (Kreis, 1987; Schulze et al., 1987; Wehland and Weber, 1987; Bulinski and Gundersen, 1991). In DRG cells, we find that cold-stable (STOP-associated) microtubules are heavily enriched in Glu-tubulin as compared with cold-labile microtubules. We could not test whether STOP inhibition suppresses microtubule detyrosination in PC12 cells. Such a test would have involved microinjection of STOP antibodies followed by Glu-tubulin immunostaining. As both STOP and Glu-tubulin antibodies are rabbit polyclonal antibodies, double-labeling experiments were difficult. However, the tight association between microtubule cold stability and microtubule detyrosination in DRG cells strongly suggests that STOP proteins are major effectors of microtubule dynamics in unperturbed neuronal cells.
When compared with cold-labile microtubules, cold-stable microtubules are much more enriched in Glu-tubulin than in Δ2-tubulin. This finding may seem paradoxical in view of previous evidence that Δ2-tubulin appears only on very long-lived microtubules (Paturle-Lafanechère et al., 1994). The most probable explanation is that in contrast with Glu-tubulin dimers, Δ2-tubulin dimers persist after dissociation from polymers. After polymer dissociation, Glu-tubulin is rapidly converted back to Tyr-tubulin upon action of a tubulin-tyrosine-ligase (MacRae, 1997). In contrast, generation of Δ2-tubulin almost certainly results from an irreversible set of enzymatic reactions (Paturle-Lafanechère et al., 1994). The free Δ2-tubulin dimers can therefore be incorporated into newly forming polymers. Our observation of an almost even distribution of Δ2-tubulin along axons and among axonal microtubules indicates continuous reshuffling of tubulin subunits among axonal polymers. This is in agreement with recent evidence that microtubules continuously exchange their constitutive tubulin subunits with the tubulin molecules of the soluble pool during their migration in axons (Okabe and Hirokawa, 1990; Okabe and Hirokawa, 1992; Funakoshi et al., 1996; Yu et al., 1996).
Taken together, our results show that microtubule stabilization in neurons mainly results from the action of STOP proteins. Apparently, as long as they are associated with active STOP proteins, microtubules are cold-stable, drug-resistant, and become detyrosinated. Over time, STOP activity varies through action of regulatory effectors (Job et al., 1981; Job et al., 1983). Such regulation probably accounts for the lower proportion of nocodazole-resistant microtubules compared with the proportion of cold-stable microtubules in neuronal cells. We surmise that, during the nocodazole test, some initially stable polymers may lose stability as a result of STOP regulation, and will therefore depolymerize.
STOP Isoforms and Neuronal Differentiation
The present study suggests a tight relationship between STOP expression and neuronal differentiation. DRG and PC12 cells, which are of different origins, both show STOP expression during differentiation. Furthermore, STOP expression is also tightly related to neuronal differentiation in developing rat cerebellum. This study also demonstrates the existence of STOP variants that seem to be expressed differentially according to the stage of neuronal differentiation. We have previously described the structure of STOP from adult rat brain. In the present study we have characterized a major STOP variant, E-STOP. E-STOP is the principal STOP protein present in embryonic brain, and differs from adult STOP by the presence of one additional central repeat and deletion of the adult STOP carboxy-terminal repeat domain. Recent work in this laboratory has shown that there are actually multiple developmental and/or cell-specific STOP isoforms. In mice, STOP isoforms are generated from a single gene (Denarier et al., 1998a ) through differential RNA splicing and alternative transcription start sites (Denarier et al., 1998b ). The mouse STOP gene is comprised of four exons (Denarier et al., 1998a ). The coding sequence of the E-STOP cDNA terminates at a location corresponding exactly to the end of exon 3 in the mouse gene. Exon 4, which is lacking in the E-STOP cDNA, encodes the COOH-terminal STOP repeats in adult STOP. E-STOP has calmodulin- and microtubule-binding activity comparable to the complete adult STOP. Therefore, the COOH-terminal repeat domain of STOP is not essential for these aspects of STOP function. This domain may interact with cellular components other than microtubules and calmodulin, and we are currently investigating this possibility.
STOP Proteins and Neurite Formation
We find that in differentiated PC12 cells, STOP inhibition does not induce immediate and obvious neurite retraction. Clearly, neurites can survive in the absence of fully stabilized microtubules, at least within the time duration of our experiments. In contrast, STOP proteins are apparently required for induction of neurite formation in cells that have not differentiated before STOP inhibition. STOP inhibition may either directly affect the mechanics of neurite formation, or may have a more general effect on cell differentiation. Induction of cell differentiation by NGF could depend on adapted regulation of cytoskeletal dynamics. In the absence of STOP activity, such regulation may be perturbed.
Neurons contain several classes of MAPs whose activity is central to neuronal morphogenesis and function (for reviews see Hirokawa, 1994; Mandelkow and Mandelkow, 1995). Antisense oligonucleotides directed against MAP2, MAP1B, and tau have an inhibitory effect similar to STOP antisense oligonucleotides with regard to neurite formation (Hanemaaijer and Ginzburg, 1991; Caceres et al., 1992; Shea et al., 1992; Brugg et al., 1993; Esmaeli-Azad et al., 1994; Sharma et al., 1994). Tau antisense oligonucleotides also trigger neurite retraction (Hanemaaijer and Ginzburg, 1991; Shea et al., 1992). Such an effect on neurite maintenance is absent in the case of MAP1B antisense oligonucleotides (Brugg et al., 1993) and only detectable using refined morphometric analysis in the case of MAP2 antisense oligonucleotides (Sharma et al., 1994).
An integrated view of microtubule regulation in neurons will require an understanding of the cooperative effects of all the various neuronal MAPs. However, complete suppression of microtubule cold stability and drug resistance after STOP inhibition in neuronal cells suggests that STOP proteins are unique with regard to their microtubule-stabilizing effect, and cannot be replaced by other MAPs. Because of this apparent absence of redundancy of the STOP system, the STOP gene knockout should yield clear-cut phenotypes. The suppression of STOP expression in whole animals should determine whether microtubule stabilization by STOP proteins is indispensable to normal neuronal differentiation, as suggested by the results of the present study.
Acknowledgments
We thank Isabelle Pignot-Paintrand for introducing L. Guillaud to EM techniques. We thank Nathalie Scher for preparing this manuscript, and are grateful to Dr. R.L. Margolis for useful advice and review of our manuscript.
This work was supported in part by grants from the French Ministère de l'Enseignement Supérieur et de la Recherche (MESR ACC-SV No. 5), the Commission of the European Communities (No. CHRX CT94.0642), the Ligue Nationale Contre le Cancer, and the Association pour la Recherche sur le Cancer to D. Job.
Abbreviations used in this paper
- aa
amino acid
- DRG
dorsal root ganglia
- E-STOP
early-STOP
- EGL
external germinal layer
- MAPs
microtubule-associated proteins
- ML
molecular layer
- nt
nucleotides, STOP, stable tubule-only polypeptide
Footnotes
Address all correspondence to Didier Job, CEA-Laboratoire du Cytosquelette, INSERM Unité 366, DBMS/CS, CEA-Grenoble, 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France. Tel.: 33-476883801; Fax: 33-476885057; E-mail: job@dsvgre.cea.fr
References
- Baas PW, Ahmad FJ. The plus ends of stable microtubules are the exclusive nucleating structures for microtubules in the axon. J Cell Biol. 1992;116:1231–1241. doi: 10.1083/jcb.116.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Black MM. Individual microtubules in the axon consist of domains that differ in both composition and stability. J Cell Biol. 1990;111:495–509. doi: 10.1083/jcb.111.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Heidemann SR. Microtubule reassembly from nucleating fragments during the regrowth of amputated neurites. J Cell Biol. 1986;103:917–927. doi: 10.1083/jcb.103.3.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Pienkowski TP, Cimbalnik KA, Toyama K, Bakalis S, Ahmad FJ, Kosik KS. Tau confers drug stability but not cold stability to microtubules in living cells. J Cell Sci. 1994;107:135–143. doi: 10.1242/jcs.107.1.135. [DOI] [PubMed] [Google Scholar]
- Barra HS, Arce CA, Argaraña CE. Posttranslational tyrosination/detyrosination of tubulin. Mol Neurobiol. 1988;2:133–153. doi: 10.1007/BF02935343. [DOI] [PubMed] [Google Scholar]
- Black MM, Cochran JM, Kurdyla JT. Solubility properties of neuronal tubulin: evidence for labile and stable microtubules. Brain Res. 1984;295:255–263. doi: 10.1016/0006-8993(84)90974-0. [DOI] [PubMed] [Google Scholar]
- Bosc C, Cronk JD, Pirollet F, Watterson MD, Haiech J, Job D, Margolis RL. Cloning, expression and properties of the microtubule stabilizing protein STOP. Proc Natl Acad Sci USA. 1996;93:2125–2130. doi: 10.1073/pnas.93.5.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci USA. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady ST, Tytell M, Lasek RJ. Axonal tubulin and axonal microtubules: biochemical evidence for cold stability. J Cell Biol. 1984;99:1716–1724. doi: 10.1083/jcb.99.5.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugg B, Reddy D, Matus A. Attenuation of microtubule-associated protein 1B expression by antisense oligodeoxynucleotides inhibits initiation of neurite outgrowth. Neuroscience. 1993;52:489–496. doi: 10.1016/0306-4522(93)90401-z. [DOI] [PubMed] [Google Scholar]
- Bulinski JC, Gundersen GG. Stabilization and post-translational modification of microtubules during cellular morphogenesis. BioEssays. 1991;13:285–293. doi: 10.1002/bies.950130605. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Cambray-Deakin MA. The cellular neurobiology of neuronal development: the cerebellar granule cell. Brain Res Rev. 1988;13:77–101. doi: 10.1016/0165-0173(88)90006-9. [DOI] [PubMed] [Google Scholar]
- Burgoyne, R.D. 1991. Cytoskeleton is a major neuronal organelle. In The Neuronal Cytoskeleton. R.D. Burgoyne, editor. Wiley-Liss, Inc., New York. 1–3.
- Caceres A, Mautino J, Kosik KS. Suppression of MAP2 in cultured cerebellar macroneurons inhibits minor neurite formation. Neuron. 1992;9:607–618. doi: 10.1016/0896-6273(92)90025-9. [DOI] [PubMed] [Google Scholar]
- Cambray-Deakin, M.A. 1991. Cytoskeleton of the growing axon. In The Neuronal Cytoskeleton. R.D. Burgoyne, editor. Wiley-Liss, Inc. New York. 233–255.
- Denarier E, Aguezzoul M, Jolly C, Vourc'h C, Roure A, Andrieux A, Bosc C, Job D. Genomic structure and chromosomal mapping of the mouse STOP gene (Mtap6) . Biochem Biophys Res Commun. 1998a;243:791–796. doi: 10.1006/bbrc.1998.8179. [DOI] [PubMed] [Google Scholar]
- Denarier E, Fourest-Lieuvin A, Bosc C, Pirollet E, Chapel A, Margolis RL, Job D. Nonneuronal isoforms of stop protein are responsible for microtubule cold stability in mammalian fibroblasts. Proc Natl Acad Sci USA. 1998b;75:6055–6060. doi: 10.1073/pnas.95.11.6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Feinstein SC, Shooter EM, Kirschner MW. Nerve growth factor-induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J Cell Biol. 1985;101:1799–1807. doi: 10.1083/jcb.101.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmaeli-Azad B, McCarty JH, Feinstein SC. Sense and antisense transfection analysis of tau function: tau influences net microtubule assembly, neurite outgrowth and neuritic stability. J Cell Sci. 1994;107:869–879. doi: 10.1242/jcs.107.4.869. [DOI] [PubMed] [Google Scholar]
- Funakoshi T, Takeda S, Hirokawa N. Active transport of photoactivated tubulin molecules in growing axons revealed by a new electron microscopic analysis. J Cell Biol. 1996;133:1347–1353. doi: 10.1083/jcb.133.6.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanemaaijer R, Ginzburg I. Involvement of mature tau isoforms in the stabilization of neurites in PC12 cells. J Neurosci Res. 1991;30:163–171. doi: 10.1002/jnr.490300117. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Curr Opin Cell Biol. 1994;6:74–81. doi: 10.1016/0955-0674(94)90119-8. [DOI] [PubMed] [Google Scholar]
- Job D, Fischer EH, Margolis RL. Rapid disassembly of cold-stable microtubules by calmodulin. Proc Natl Acad Sci USA. 1981;78:4679–4682. doi: 10.1073/pnas.78.8.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Job D, Rauch CT, Fischer EH, Margolis RL. Recycling of cold-stable microtubules: evidence that cold stability is due to substoichiometric polymer blocks. Biochemistry. 1982;21:509–515. doi: 10.1021/bi00532a015. [DOI] [PubMed] [Google Scholar]
- Job D, Rauch CT, Fischer EH, Margolis RL. Regulation of microtubule cold stability by calmodulin-dependent and -independent phosphorylation. Proc Natl Acad Sci USA. 1983;80:3894–3898. doi: 10.1073/pnas.80.13.3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis TE. Microtubules containing detyrosinated tubulin are less dynamic. EMBO (Eur Mol Biol Organ) J. 1987;6:2597–2606. doi: 10.1002/j.1460-2075.1987.tb02550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Li Y, Black MM. Microtubule assembly and turnover in growing axons. J Neurosci. 1996;16:531–544. doi: 10.1523/JNEUROSCI.16-02-00531.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieuvin A, Labbé J-C, Dorée M, Job D. Intrinsic microtubule stability in interphase cells. J Cell Biol. 1994;124:985–996. doi: 10.1083/jcb.124.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S-S, Sammak PJ, Borisy GG. Progressive and spatially differentiated stability of microtubules in developing neuronal cells. J Cell Biol. 1989;109:253–263. doi: 10.1083/jcb.109.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae TH. Tubulin post-translational modifications-enzymes and their mechanisms of action. Eur J Biochem. 1997;244:265–278. doi: 10.1111/j.1432-1033.1997.00265.x. [DOI] [PubMed] [Google Scholar]
- Mandelkow E, Mandelkow E-M. Microtubules and microtubule-associated proteins. Curr Opin Cell Biol. 1995;7:72–81. doi: 10.1016/0955-0674(95)80047-6. [DOI] [PubMed] [Google Scholar]
- Mandelkow E-M, Biernat J, Drewes G, Gustke N, Trinczek B, Mandelkow E. Tau domains, phosphorylation, and interactions with microtubules. Neurobiol Aging. 1995;16:355–362. doi: 10.1016/0197-4580(95)00025-a. [DOI] [PubMed] [Google Scholar]
- Margolis RL, Rauch CT, Job D. Purification and assay of a 145-kDa protein (STOP145) with microtubule-stabilizing and motility behavior. Proc Natl Acad Sci USA. 1986;83:639–643. doi: 10.1073/pnas.83.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JR, Lasek RJ. Stable polymers of the axonal cytoskeleton: the axoplasmic ghost. J Cell Biol. 1982;92:192–198. doi: 10.1083/jcb.92.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Hirokawa N. Microtubule dynamics in nerve cells: analysis using microinjection of biotinylated tubulin into PC12 cells. J Cell Biol. 1988;107:651–664. doi: 10.1083/jcb.107.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Hirokawa N. Turnover of fluorescently labeled tubulin and actin in the axon. Nature. 1990;343:479–482. doi: 10.1038/343479a0. [DOI] [PubMed] [Google Scholar]
- Okabe S, Hirokawa N. Differential behaviour of photoactivated microtubules in growing axons of mouse and frog neurons. J Cell Biol. 1992;117:105–120. doi: 10.1083/jcb.117.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paturle-Lafanechère L, Eddé B, Denoulet P, Van Dorsselaer A, Mazarguil H, Le Caer J-P, Wehland J, Job D. Charaterization of a major brain tubulin variant which cannot be tyrosinated. Biochemistry. 1991;30:10523–10528. doi: 10.1021/bi00107a022. [DOI] [PubMed] [Google Scholar]
- Paturle-Lafanechère L, Manier M, Trigault N, Pirollet F, Mazarguil H, Job D. Accumulation of delta 2-tubulin, a major tubulin variant that cannot be tyrosinated, in neuronal tissues and in stable microtubule assemblies. J Cell Sci. 1994;107:1529–1543. doi: 10.1242/jcs.107.6.1529. [DOI] [PubMed] [Google Scholar]
- Pignot-Paintrand I. Orientation of human spermatozoa for electron microscopy: a fast, simple method. Micros Res Tech. 1992;21:75–76. doi: 10.1002/jemt.1070210112. [DOI] [PubMed] [Google Scholar]
- Pirollet F, Job D, Fischer EH, Margolis RL. Purification and characterization of sheep brain cold-stable microtubules. Proc Natl Acad Sci USA. 1983;80:1560–1564. doi: 10.1073/pnas.80.6.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirollet F, Rauch CT, Job D, Margolis RL. Monoclonal antibody to microtubule-associated STOP protein: affinity purification of neuronal STOP activity and comparison of antigen with activity in neuronal and nonneuronal cell extracts. Biochemistry. 1989;28:835–842. doi: 10.1021/bi00428a064. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. 2nd ed. Cold Spring Harbor Laboratory Press, Plainview, New York.
- Schulze E, Asai DJ, Bulinski JC, Kirschner M. Posttranslational modification and microtubule stability. J Cell Biol. 1987;105:2167–2177. doi: 10.1083/jcb.105.5.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekimoto S, Tashiro T, Komiya Y. Two stages in neurite formation distinguished by differences in tubulin metabolism. J Neurochem. 1995;64:354–363. doi: 10.1046/j.1471-4159.1995.64010354.x. [DOI] [PubMed] [Google Scholar]
- Sharma N, Kress Y, Shafit-Zagardo B. Antisense MAP-2 oligonucleotides induce changes in microtubule assembly and neuritic elongation in pre-existing neurites of rat cortical neurons. Cell Motil Cytoskelet. 1994;27:234–247. doi: 10.1002/cm.970270305. [DOI] [PubMed] [Google Scholar]
- Shea TB, Beermann ML, Nixon RA, Fischer I. Microtubule- associated protein tau is required for axonal neurite elaboration by neuroblastoma cells. J Neurosci Res. 1992;32:363–374. doi: 10.1002/jnr.490320308. [DOI] [PubMed] [Google Scholar]
- Takeda S, Funakoshi T, Hirokawa N. Tubulin dynamics in neuronal axons of living zebrafish embryos. Neuron. 1995;14:1257–1264. doi: 10.1016/0896-6273(95)90272-4. [DOI] [PubMed] [Google Scholar]
- Takemura R, Okabe S, Umeyama T, Kanai Y, Cowan NJ, Hirokawa N. Increased microtubule stability and alpha tubulin acetylation in cells transfected with microtubule-associated proteins MAP1B, MAP2 or tau. J Cell Sci. 1992;103:953–964. doi: 10.1242/jcs.103.4.953. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb BC, Wilson L. Cold-stable microtubules from brain. Biochemistry. 1980;19:1993–2001. doi: 10.1021/bi00550a041. [DOI] [PubMed] [Google Scholar]
- Wehland J, Weber K. Turnover of the carboxy-terminal tyrosine of α-tubulin and means of reaching elevated levels of detyrosination in living cells. J Cell Sci. 1987;88:185–203. doi: 10.1242/jcs.88.2.185. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Spooner BS, Wessells NK. Axon growth: roles of microfilaments and microtubules. Proc Natl Acad Sci USA. 1970;66:1206–1212. doi: 10.1073/pnas.66.4.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Baas PW. The growth of the axon is not dependent upon net microtubule assembly at its distal tip. J Neurosci. 1995;15:6827–6833. doi: 10.1523/JNEUROSCI.15-10-06827.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Schwei MJ, Baas PW. Microtubule transport and assembly during axon growth. J Cell Biol. 1996;133:151–157. doi: 10.1083/jcb.133.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]