

Abstract
Ligand-stimulated activation of FGF receptors (FGFRs) in skeletal muscle cells represses terminal myogenic differentiation. Skeletal muscle cell lines and subsets of primary cells are dependent on FGFs to repress myogenesis and maintain growth. To understand the intracellular events that transduce these signals, MM14 skeletal muscle cells were transfected with expression vectors encoding chimeric receptors. The chimeras are comprised of the PDGF β receptor (PDGFβR) extracellular domain, the FGFR-1 intracellular domain, and either the PDGFβR or FGFR-1 transmembrane domain. The chimeric receptors were autophosphorylated upon PDGF-BB stimulation and are capable of stimulating mitogen-activated protein kinase activity. Activation of the tyrosine kinase domain of either chimera repressed myogenesis, suggesting intracellular responses regulating skeletal muscle differentiation are transduced by activation of the FGFR-1 tyrosine kinase. Unexpectedly, we found that activation of either chimeric receptor failed to stimulate cellular proliferation. Thus, it appears that regulation of skeletal muscle differentiation by FGFs requires only activation of the FGFR tyrosine kinase. In contrast, stimulation of proliferation may require additional, as yet unidentified, signals involving the receptor ectodomain, the FGF ligand, and heparan sulfate either alone, or in combination.
Keywords: skeletal muscle, differentiation, fibroblast growth factor, myogenesis, tyrosine kinase
Skeletal muscle differentiation is regulated by FGFs in most skeletal muscle cell lines and in skeletal muscle primary cultures (11, 19, 20, 25). In the developing chick, a population of muscle cells appears to be dependent on FGFs, consistent with a requirement for FGFs in regulating either the growth or differentiation of skeletal muscle cells (unpublished data). As a model system for investigating FGF-dependent skeletal muscle cells, we study FGF signaling in the MM14 skeletal muscle cell line, which is dependent on addition of exogenous FGFs for maintenance of growth and repression of terminal differentiation (20, 24). We have used the MM14 cell line to demonstrate that FGF-mediated signals cannot be replaced by endogenous receptors, other growth factors, or ectopically expressed growth factor receptors (16, 18, 20; our unpublished data). To examine further the intracellular signals transduced upon stimulation of FGF receptor (FGFR)-11 in MM14 cells, we tested the capacity of two receptor chimeras to repress myogenesis and to maintain proliferation.
Numerous studies using receptor chimeras comprised of extracellular and intracellular domains from different receptors have demonstrated that the intracellular and extracellular domains function independently of each other. Intracellular signaling events mediated by orphan receptors have been elucidated by constructing receptor chimeras containing a known ligand binding domains. Thus, chimeric receptors have proven useful for examining the contribution of the intracellular domains of receptor tyrosine kinases to signaling events. Receptor chimeras retain the ligand binding and kinase activity of the extracellular and cytoplasmic domains of the respective receptors from which they were derived. For example, fusion of the EGF receptor extracellular domain to the intracellular domains of either the erbB receptor (30), the HER2 receptor (17), or the insulin receptor (29), results in hybrid receptors with tyrosine kinase activity that is upregulated by EGF. These extensive studies suggest that signals initiated by ligand binding are transduced from the extracellular domain to the intracellular domain by similar mechanisms for all receptor tyrosine kinases (36).
We previously demonstrated that the full-length PDGFβR could not replace the requirement of FGF for repression of skeletal muscle cell differentiation, despite the fact that cells expressing the ectopic receptor activated intracellular signaling pathways in response to PDGF-BB (16). To identify FGFR domains involved in regulating skeletal muscle growth and differentiation, we used two chimeras containing the PDGFβR extracellular domain, the FGFR-1 intracellular domain, and a transmembrane domain from either FGFR-1 or PDGFβR. Although both chimeras repressed skeletal muscle differentiation, we were surprised to find that neither receptor chimera could support proliferation of skeletal muscle cells. These data suggest that signaling pathways required for proliferation cannot be activated by the hybrid receptors and indicate that activation of the intracellular domain of FGFR-1 at least in the context of the hybrid receptors is capable of regulating differentiation of skeletal muscle cells, but is insufficient to support proliferation.
Materials and Methods
Reverse Transcription (RT)-PCR Analysis of FGFRs
Total RNA was isolated from proliferating MM14 cells as previously described (6). 5 μg of total RNA was added to reverse transcriptase buffer (GIBCO-BRL, Gaithersburg, MD) containing 0.025 μg/μl Oligo (dT)12–18 (GIBCO-BRL), 0.01 M DTT (GIBCO-BRL), 0.5 mM dNTP mix (GIBCO-BRL), and 200 U of Superscript II reverse transcriptase (GIBCO-BRL). The reaction was incubated at 42°C for 50 min. Non-reverse transcriptase controls were carried out as described above with the exception of reverse transcriptase addition. Mouse total RNA from an embryonic stem cell mouse tumor was provided by Dr. J. Lee (University of Colorado, Boulder, CO).
PCR amplification was carried out by adding 2 μl of cDNA (or 0.5 μg of plasmid DNA) to PCR buffer containing 0.25 mM each dNTP, 1.5 mM MgCl2, 0.5 μM each of both forward and reverse primers for FGF receptors 1, 2, 3, and 4 as previously described (9), and 5 U of Taq DNA polymerase (GIBCO-BRL). Each reaction was amplified for 35 cycles using the following cycling parameters: denaturation at 94°C for 1 min, annealing at 50°C for 1 min, and elongation at 72°C for 1 min. After amplification, each reaction was resolved on a 0.8% agarose gel containing ethidium bromide and visualized with a UV transilluminator.
Cell Culture and Stable Transfection
Mouse MM14 skeletal muscle cells were cultured as described previously (24). The pcDNA I expression vector (Invitrogen, San Diego, CA) encoding a chimeric receptor consisting of the PDGFβR extracellular and transmembrane domains and the FGFR-1 intracellular domain (pcPRtm/FR1) was obtained from Dr. M. Welsh (Uppsala University, Uppsala, Sweden) (21). pcPRtm/FR1 was co-transfected into MM14 cells with a vector containing the neomycin resistance gene (pKO-neo) by calcium phosphate– mediated transfection (16). Cells expressing the highest level of chimeric receptor were selected from neomycin-resistant cells by fluorescence-activated cell sorting using anti-PDGFβR mAb PR7212 (14), biotinylated goat anti–mouse IgG, and FITC-avidin. The selected cells were subcloned to derive cell lines that proliferated and differentiated in a manner similar to parental MM14 cells.
MM14 cells overexpressing FGFR-1 were generated by co-transfecting with a full-length mouse FGFR-1 cDNA under the control of a Moloney SV-40 promoter and a hygromycin resistance gene under the control of a cytomegalovirus (CMV) promoter (pHyg) at a 20:1 molar ratio (4 pmol/ 0.2 pmol), respectively. After 24 h, 75 μg/ml hygromycin was added and the cells were incubated for 12 d. Colonies resistant to hygromycin were selected and pooled for analysis. The pooled clones were analyzed for growth in FGF-2 and their differentiation capacity.
Transient Transfection Assay
All transient transfections used a calcium phosphate precipitation method (31). Briefly, MM14 cells stably expressing PRtm/FR1 were transiently transfected with 1 μg of CMV-LacZ, 0.5 μg of an α-cardiac actin luciferase reporter construct, and cultured without growth factor or with FGF-2 or PDGF-BB for 36 h. The cells were harvested and assayed for luciferase and β-galactosidase activities using the Tropix Dual Light assay, using an Optocomp luminometer (MGM Instruments, Inc., Hamden, CT) (16). Luciferase values were normalized to β-galactosidase values to correct for variations in transfection efficiencies.
Parental MM14 cells were co-transfected with α-cardiac actin luciferase, CMV-LacZ, and either pcPRtm/FR1, pBJ5PR/FR1tm, or a control vector. pBJ5PR/FR1tm is an expression construct containing the second chimeric receptor gene (PR/FR1tm; provided by Dr. R. Bradshaw [University of California, Irvine, CA]; see Fig. 1 A) that encodes the extracellular domain of the human PDGFβR fused to the transmembrane and intracellular domains of the rat FGFR-1. The coding sequence was cloned into the EcoRI site of pBJ5 in the sense orientation relative to the Sra promoter (32). Cells were cultured with the indicated growth factor for 36 h after osmotic shock, harvested, and then assayed for luciferase and β-galactosidase expression as described above.
Figure 1.


FGFR expression and receptor chimeras. (A) RT-PCR analysis of FGFR expression in MM14 cells. RT-PCR of total RNA from MM14 cells demonstrates that these cells detectably express only FGFR-1. RT-PCR analysis included either RNA prepared from MM14 cells (lanes 2–5); plasmid DNAs for FGFR-1 (lane 6), FGFR-2 (lane 7); total RNA prepared from embryonic stem cell tumors (lanes 8 and 9); and a no DNA control (lanes 10–13). Lanes 1 and 14 contain a 1-kb ladder. Specific primers for FGFR-1, FGFR-2, FGFR-3, and FGFR-4 were used as indicated in the figure. (B) The biological activities of four different receptors were examined in MM14 cells and include FGFR-1, PDGFβR, the PRtm/FR1 chimera containing the PDGFβR extracellular (light gray), transmembrane (white) domains, the FGFR-1 intracellular (dark gray) domain, and the PR/FR1tm chimera containing the PDGFβR extracellular domain (light gray), the FGFR-1 transmembrane (black), and intracellular domain (dark gray).
Immunoblot Analysis
Anti-phosphotyrosine Blots.
Proliferating MM14 cells were washed and incubated in Ham's F10C containing 2 mM CaCl2, supplemented with 2% horse serum without FGF-2 for 3.5 h (proliferating cells) or 57.5 h (differentiated cells). Cells were then stimulated with 2 nM PDGF-BB for 10 min where indicated. Cells were lysed in 25 mM Tris, pH 7.4, 50 mM NaCl, 5 mM EDTA, 2 mM EGTA, 50 mM sodium pyrophosphate, 50 mM NaF, 0.1 mM sodium orthovanadate, 1% Triton X-100, 2 μg/ml leupeptin, 2 μg/ml aprotinin, and 1 mM PMSF. Insoluble material was removed by centrifugation and the protein content of each cell lysate measured using the BCA protein assay (Pierce Chemical Co., Rockford, IL). Samples of lysate containing 18 μg total protein were separated by SDS-PAGE on 7.5% acrylamide gels, and then transferred to an Immobilon membrane (Millipore Corp., Bedford, MA). Tyrosine-phosphorylated proteins were detected using the RC20-conjugated anti-phosphotyrosine mAb coupled to HRP, and then developed for chemiluminescence according to the manufacturer's instructions (Transduction Laboratories, Lexington, KY).
Anti-PDGFβR Blots.
For detection of expressed PRtm/FR1 chimeras, cells were treated as described in the legend and cell lysates containing 60 μg total protein were loaded to each lane. The transferred protein blots were incubated with an affinity-purified anti-PDGFβR mAb (2.5 μg/ml), washed three times for 10 min, and then incubated with an anti–mouse IgG conjugated to HRP. The blots were then developed for chemiluminescent detection as described by the manufacturer (Amersham Corp., Arlington Heights, IL).
Cell Growth and Differentiation Assays
Immunohistochemical Assay for Myosin Heavy Chain (MHC).
MM14 cells stably expressing PRtm/FR1 were grown for 12 h in growth media (contains F10C, 2 mM CaCl2, 15% horse serum) and increasing amounts of FGF-2 (200 pM to 1.5 nM), incubated for 48 h, and then stained for MHC as previously described (16).
DNA Synthesis Assay.
DNA synthesis was assayed by [3H]thymidine incorporation into MM14 cells stably expressing PRtm/FR1 cells as previously described (24). Briefly, cells were plated in 24-well plates at 2,000 cells/well in growth media, with the indicated growth factor(s) for 18 h. The cells were then incubated for 10 h with 2 mCi/ml [3H]thymidine (Dupont-NEN, Boston, MA), and the amount of [3H]thymidine incorporated into DNA was quantitated by liquid scintillation counting.
Immunohistochemical Staining
MM14 cells were transiently transfected as described (13). 24 h after the osmotic shock, 5-bromo-2 deoxyuridine (BrdU; Amersham Corp.) was added to each plate to a final concentration of 10 μM. After 12 h of additional incubation, cells were fixed with 100% methanol and then treated with 2 N HCl for 1 h at 37°C. Fixed cells were blocked in PBS containing 5% goat serum for 1 h at room temperature. The BrdU incorporated into DNA was detected by staining with an anti-BrdU mAb (1:100; Developmental Hybridoma Bank, Iowa City, IA). β-Galactosidase was detected by staining with an anti–β-galactosidase antibody (1:1,000; Boehringer Mannheim Corp., Indianapolis, IN). Bound primary antibodies were visualized with the appropriate immunoglobulin chain–specific goat anti– mouse secondary antibodies (1:500; Southern Biotechnology Associates, Inc., Birmingham, AL) conjugated to either Texas red (for BrdU) or FITC (for β-galactosidase).
MAPK Assays
Immune Complex Kinase Assay.
MM14 cells stably expressing FGFR-1 or the PRtm/FR1 chimera, at a density of 5 × 105 cells per plate, were washed three times with 5 ml of PBS, and then grown for 4 h in F10C containing 2 mM CaCl2, 2% horse serum without FGF-2. Cells were stimulated with either 0.1 nM FGF-2, 2 nM PDGF BB, or 100 nM 12-O-tetra-decnoylphorbol-13-acetate (TPA) for 10 min. Cell lysates were prepared by washing the plates in 4°C TBS, scraping the cells in homogenization buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 5 mM para-nitrophenyl phosphate, 1 mM sodium orthovanadate, 10 mM tetra-sodium pyrophosphate decahydrate, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin) in TBS, sonicating, and then centrifuging at 14,000 g for 15 min at 4°C. Cell lysates were adjusted to 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS. Protein A–Sepharose beads pre-loaded with anti-ERK1 polyclonal antibody (provided by Dr. C. Ashandel, Purdue University, West Lafayette, IN) were incubated in cell lysates for 4 h at 4°C with rotation. The immune complexes were washed three times in homogenization buffer supplemented with 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS and once in 20 mM Tris, pH 7. The immune complexes were then resuspended in 100 μl reaction buffer (12.8 mM Tris, pH 7.4, 5.5 mM para-nitrophenyl phosphate, 51.5 mM MgCl2, 2 μg/ml myelin basic protein, 0.5 mCi/ml [γ-32P]ATP). The in vitro kinase reaction was carried out at 30°C for 30 min. Reaction products were resolved by SDS-PAGE on a 10% polyacrylamide gel. Phosphorylation of myelin basic protein was quantified using a Molecular Dynamics Storm PhosphorImager (Sunnyvale, CA).
elk Reporter Assay.
MAPK (mitogen-activated protein kinase) activity was also determined using Pathdetect ELK1 reporting system (Stratagene, La Jolla, CA) (35). MM14 cells were plated on six-well plates at a density of 40,000 cells/well or on 24-well plates at a density of 8,000 cells/ well and cotransfected with 2.5 μg (500 ng for 24-well plate) pFR-Luc reporter vector, 200 ng (40 ng for 24-well plate) pFA–Elk1 vector, and 1 μg (200 ng for 24-well plate) CMV–LacZ vector per well. 12–16 h after the transfection cells were washed twice with PBS and incubated in media containing 2.5% serum without FGF for 6 h. The cells were then left untreated, or stimulated with 0.1 nM FGF-2 or 1 nM PDGF. Cells were harvested and luciferase activity determined 6 h after mitogen stimulation. Luciferase and β-galactosidase activities were determined using the Tropix Dual Light Assay (Tropix, Bedford, MA). Luciferase activity values were normalized to β-galactosidase activity values to correct for transfection efficiency.
High Level Expression Using a Tetracycline-Repressible System
The DNA fragment encoding a dominant-negative FGFR-1 was subcloned from pcDNFR1 (13) into the EcoRI/XbaI sites of pUHD 10-3 (28) yielding dnFR1-tet. pUHD 10-3 is an expression vector under the control of a tetracycline-repressible hybrid transcriptional activator constitutively expressed from pUHD 15-1 (12). MM14 cells were co-transfected with 1 μg of CMV-LacZ and 10 μg pUHD 15-1 and/or 10 μg of pUHC 13-3 (12) (a luciferase reporter plasmid under the control of the transcriptional activator encoded by pUHD 15-1), and then cultured for 36 h in the presence or absence of 1 μg/ml tetracycline. Cell lysates were prepared and assayed for β-galactosidase and luciferase activities as described previously using a Tropix Dual Light assay (13). To test the effect of a dominant-negative FGFR-1 on the activity of the PR/FR1tm chimera, MM14 cells were transiently transfected with 1 μg of CMV-LacZ, 5 μg of the α-cardiac actin luciferase reporter construct, 10 μg pUHD 15-1, and combinations of pcPR/ FR1tm and dnFR1-tet. After osmotic shock, cells were cultured for 36 h in the absence of tetracycline, with or without the indicated additions of FGF-2 or PDGF-BB. Cell lysates were then prepared and analyzed as previously described (13).
Equilibrium binding of 125I-labeled FGF-2 to intact MM14 and MM14-FR1 cells. MM14 cells (5 × 105) were plated in 35-mm dishes, and 4 h later were incubated with 250 pM 125I-labeled FGF-2 (26) (7 × 103 cpm/fmol) in the presence or absence of 0.96 μM FGF-2 at 4°C. The cells were washed three times with 2 ml F10C containing 25 mM Hepes, pH 7.4, and 0.2% BSA. Cells were then washed twice with 1 ml the same buffer containing 2 M NaCl (low affinity) followed by two 1-ml washes with 20 mM NaAcetate, pH 4.0, containing 2 M NaCl and 0.2% BSA (high affinity). The washes were pooled and counted in a Clinnigamma counter (LKB-Wallac, Turku, Finland).
Results
We wanted to determine if activation of the FGFR tyrosine kinase is sufficient to mediate FGF-dependent biological activities. We chose to use the MM14 skeletal muscle cell line as a model since these cells exhibit two distinct FGF-dependent responses and serve as a model for a population of FGF-dependent myoblasts present in vivo. In the absence of serum, FGFs repress terminal differentiation independent of proliferation, whereas both serum and FGFs appear to be required for stimulating proliferation (7). Moreover, MM14 cells are absolutely dependent on FGFs for repression of terminal differentiation and for growth maintenance, since other growth factors are incapable of replacing FGFs (23).
RT-PCR analysis of FGFRs in MM14 cells indicates that the only detectable FGFR expressed is FGFR-1 (Fig. 1 A). In addition, we have previously demonstrated that MM14 cells do not express the PDGFβR. Therefore, to assess the role of the FGFR-1 tyrosine kinase in repression of myogenesis and stimulation of proliferation in MM14 myoblasts, we examined the activity of two chimeric receptors containing the PDGFβR extracellular domain and the FGFR-1 intracellular domain (Fig. 1 B).
MM14 cells stably expressing the PRtm/FR1 chimera (Fig. 1 B) were selected as described in Materials and Methods, and those clones exhibiting responses to FGF-2 similar to the parental cells were used for further studies. MM14 cells expressing the wild-type PDGFβR (16) and the PRtm/FR1 chimera were examined by Western blotting with an anti-PDGFβR antibody. As expected, the chimeric receptor migrated more rapidly than the wild-type PDGFβR expressed in human fibroblasts or ectopically expressed in MM14 cells, because of the smaller size of the intracellular domain of FGFR-1 (Fig. 2 A). As an initial determinant of the capacity for the receptor chimera to signal, the tyrosine kinase activity was examined by Western blotting using anti-phosphotyrosine antibodies. Immunoblot analysis of proliferating and differentiated PDGF-BB–treated MM14 cells stably expressing the PRtm/FR1 displayed a prominent phosphorylated product migrating at ∼180 kD, the expected mass of the receptor chimera (Fig. 2 B). The tyrosine phosphorylation occurring upon FGF stimulation of the endogenous FGFR-1 is not detectable (data not shown), consistent with our previous observations (16). Presumably, this is due to the low number of receptors per cell (∼700). Tyrosine phosphorylated FGFR-1 is observed only after ectopic overexpression (16).
Figure 2.

Chimeric receptor expression and activity. (A) Western blot analysis of the PDGFβR and the PRtm/FR1 chimera. MM14 cells stably expressing the introduced PDGFβR (lanes 1 and 2), human fibroblasts expressing endogenous PDGFβR (lanes 3 and 4), MM14 cells expressing the PRtm/FR1 chimera (lanes 5 and 6), and untransfected MM14 cells (lanes 7 and 8) were separated by SDS-PAGE, blotted, and then stained with an anti-PDGFβR mAb (lanes 1–8). The solid arrowhead indicates the migration of the PRtm/FR1 chimera and the open arrowhead indicates the migration of the PDGFβR. (B) PDGF-BB–stimulated tyrosine phosphorylation in MM14 cells stably expressing PRtm/FR1. Proliferating (lanes 1 and 2) and differentiated (lanes 3 and 4) cells were left unstimulated (lanes 1 and 3) or were stimulated with 2 nM PDGF-BB (lanes 2 and 4) for 10 min. An anti-phosphotyrosine immunoblot analysis of equal amounts of detergent-solubilized cell extracts is shown. The arrow indicates the position of migration of the tyrosine-phosphorylated chimeric receptor after PDGF-BB stimulation of both proliferating and differentiated cells. The migration of molecular weight markers is indicated on the left.
Next, we tested the ability of the PRtm/FR1 chimera to regulate skeletal muscle differentiation using three independent assays: (1) analysis of muscle-specific reporter gene expression; (2) the presence of muscle-specific MHC protein; and (3) fusion into multinucleated myotubes. Addition of PDGF-BB to MM14 cells stably expressing the PRtm/FR1 chimera inhibited the expression of a muscle-specific reporter gene (Fig. 3 A), and blocked differentiation as judged by either myosin staining (Fig. 3 B) or the relative number of nuclei present in multinucleated myotubes (data not shown). These data were consistent with those observed in one other stable subclone (data not shown).
Figure 3.

Stably expressed PRtm/FR1 chimeric receptors repress differentiation. (A) PDGF-BB represses skeletal muscle-specific gene expression in PRtm/FR1 cells. MM14 cells stably expressing PRtm/FR1 were transiently transfected with a differentiated muscle-specific gene reporter (α-cardiac actin luciferase). Cells transfected with the empty vector (white) or the PRtm/FR1 expression construct (gray) were left untreated (Control), treated with FGF-2, or treated with PDGF-BB. Data are represented as a percent of the control activity. Error bars represent the standard deviation of triplicate analyses. (B) PDGF-BB represses skeletal muscle differentiation in MM14 cells stably expressing the PRtm/FR1 chimeric receptor. MM14 cells transfected with a control vector (the expression plasmid minus the receptor chimera cDNA; white) or the PRtm/FR1 chimera (gray) exhibit a low number of myosin positive cells in the presence of FGF. In the absence of growth factors, cells were maintained in the presence of anti-PDGFβR and anti–FGF-2 antibodies as previously described (16) to reduce stimulation from serum-derived PDGF-BB and residual FGF-2, respectively. Maximum levels of MHC-positive cells are observed in 2% horse serum in the absence of FGF. Each data point is the average of duplicate plates from a representative experiment with an average of 1,500 cells scored for each plate. The number of myosin-positive cells in the presence of FGF (gray bar, 15% HS + FGF) was 0% and thus is too low a value to be observable on the histogram.
It is possible, although unlikely, that the biological activity believed to be mediated by the PRtm/FR1 receptor chimera is an artifact occurring as a result of transfection, selection, or isolation of the two stable subclones. Therefore, we analyzed the ability of the PRtm/FR1 chimera to repress differentiation in transiently transfected cells. In addition, we examined the activity of a similar receptor chimera (PR/FR1tm; see Fig. 1 A), which contains the FGFR-1 transmembrane domain rather than the PDGFβR transmembrane domain. Cells assayed for muscle-specific reporter gene activity after transient transfection with either chimeric receptor construct and a skeletal muscle–specific reporter revealed that both the PRtm/FR1 (Fig. 4 A) and the PR/FR1tm (Fig. 4 B) chimeras similarly inhibited muscle-specific reporter gene transcription in the presence of PDGF-BB. Note that the PR/FR1tm chimera inhibited the endogenous response to FGF-2 (Fig. 4 B).
Figure 4.

Transiently expressed PRtm/FR1 and PR/FR1tm receptor chimeras repress differentiation. (A) PDGF-BB inhibits myogenic differentiation in cells transiently transfected with an expression vector encoding the PRtm/FR1 receptor chimera. MM14 cells were transfected with expression vectors encoding the PRtm/ FR1 receptor chimera (gray) or a control vector (white), a muscle-specific luciferase reporter and a constitutively active LacZ reporter. Cultures were left untreated or fed at 12-h intervals with increasing concentrations (1–2 nM) of FGF-2 or PDGF-BB. At 36 h, the cells were harvested and assayed for luciferase and β-galactosidase activity. (B) PDGF-BB inhibits myogenic differentiation in cells transiently transfected with an expression vector encoding the PR/FR1tm receptor chimera. MM14 cells were transfected with expression vectors encoding the PR/FR1tm receptor chimera (black) or a control vector (white), a muscle-specific luciferase reporter and a constitutively active LacZ reporter. Cells were treated, harvested, and assayed as described for A.
When FGF is removed from MM14 cells in the presence of serum, the cells withdraw from the cell cycle and express muscle-specific gene products (7). Since FGFs may be required for proliferation of MM14 cells and for repression of differentiation, we wanted to determine if activation of the receptor chimeras was sufficient to maintain cell proliferation as well as to repress differentiation. MM14 cells stably expressing the PRtm/FR1 chimera were tested for the ability to synthesize DNA upon addition of PDGF-BB. An analysis of DNA synthesis indicated that these cells could not incorporate [3H]thymidine in the presence of 1 nM PDGF-BB and 15% serum (Fig. 5 A). Higher concentration of PDGF-BB up to 50 nM also failed to elicit DNA synthesis (data not shown). Moreover, the kinetics of cell cycle withdrawal are indistinguishable for PRtm/FR1-expressing cells in the presence or absence of PDGF-BB (Fig. 5 B). Thus, the receptor chimera does not appear to be capable of affecting either DNA synthesis or cell growth. It is possible that the PRtm/ FR1 chimera is defective in a subset of signaling events. However, a different receptor chimera containing the FGFR-1 transmembrane domain would not necessarily be expected to exhibit a similar signaling defect. To test whether or not both receptor chimeras exhibited similar deficiencies in signaling, MM14 cells transiently transfected with an expression vector encoding the PR/FR1tm chimera were examined for their ability to synthesize DNA upon addition of PDGF-BB. Cells transiently co-transfected with PR/FR1tm and LacZ expression vectors were allowed to incorporate BrdU, and were immunostained for BrdU and β-galactosidase. 10–15% of the cells stained positive for β-galactosidase activity, consistent with previously published data on transfection efficiencies (13). In cells ectopically expressing the PR/FR1tm chimera, BrdU incorporation was not seen in the absence of growth factors and in the presence of added PDGF-BB. However, in contrast to the PR tm/FR1 chimera, FGF-2 addition to cells expressing the PR/FR1tm chimera does not promote DNA synthesis (Fig. 6), suggesting that the inclusion of the FGFR-1 transmembrane domain interferes with signaling from the endogenous FGFR-1. The non-transfected cells (β-galactosidase–negative cells) did not incorporate BrdU in the absence of FGF-2 (data not shown). Expression of either receptor chimera appears to be incapable of stimulating DNA synthesis in the presence of PDGF-BB and 15% serum. No significant BrdU incorporation above control levels was seen in PDGF-BB– treated cells negative for β-galactosidase, indicating the absence of PDGFRs and a PDGF response in the parental MM14 cell population. Quantitative analysis of transfected cells revealed that >80% of the PR/FR1tm- and PR tm/FR1-expressing cells failed to synthesize DNA upon addition of PDGF-BB (Fig. 6). Thus, both receptor chimeras act similarly in their ability to transduce only a portion of the FGF signal, suggesting that activation of the intracellular FGFR-1 tyrosine kinase in the context of the chimeric receptor is insufficient to mediate DNA synthesis or cell division.
Figure 5.

Chimeric receptors fail to stimulate DNA synthesis or cell growth. (A) PDGF-BB does not block cell cycle exit in MM14 cells stably expressing the PRtm/FR1 receptor chimera. Cells expressing PRtm/FR1 were cultured in the indicated concentrations of either FGF-2 (□) or PDGF-BB (•) for 18 h. [3H]Thymidine was then added and the amount of [3H]thymidine incorporated was determined after 10 h. Error bars represent the standard deviation of triplicate points. (B) PDGF-BB does not stimulate cell growth in MM14 cells stably expressing the PRtm/ FR1 receptor chimera. Equal numbers of MM14 cells expressing the PRtm/FR1 receptor were grown for 24 h, and then switched to media and 15% horse serum containing no growth factors (⋄), or fed at 12-h intervals with increasing concentrations (0.5–2 nM) of FGF-2 (□), or PDGF-BB (•). At the indicated times the number of cells per 100-mm2 plate were scored.
Figure 6.

Single cell analysis of transiently expressed chimeric receptors. Quantitative analysis of BrdU incorporation. MM14 cells transiently co-transfected with an empty expression vector, or PRtm/FR1, or PR/FR1tm, and a CMV-LacZ expression vector were either left untreated (white), or treated with 200 pM FGF-2 (gray), or 1 nM PDGF-BB (black) after transfection. β-Galactosidase–positive cells were scored for BrdU incorporation and the percent of BrdU-positive cells plotted. A minimum of 200 β-galactosidase–positive cells were scored for each coverslip. One representative experiment is shown. The experiment was repeated with similar results.
It is possible that the receptor chimeras are affecting the mRNA stability, transport, or translation of the muscle-specific reporter gene (see Fig. 4) and thus, the receptor chimeras might not actually be inhibiting skeletal muscle differentiation. Therefore, we confirmed that the chimeric receptors were capable of repressing myogenic differentiation. Skeletal muscle cells transiently transfected with PR/ FR1tm and LacZ expression constructs were examined for the number of nuclei in myotubes. In the presence of PDGF-BB, cells expressing the PR/FR1tm chimera (β-galactosidase–positive cells), were not present in myotubes, similar to that of cells cultured in FGF-2 (data not shown). In co-transfected cells, nuclei were present in myotubes in the absence of PDGF-BB, (data not shown).
We have previously demonstrated that activation of FGF receptors in MM14 cells weakly activates ERKs (16). We thus examined the capacity of the receptor chimeras to activate MAPKs using two independent assays. The PRtm/ FR1 chimera activates ERK1/2 (Fig. 7 A) and both receptor chimeras, as well as the endogenous FGFR-1 are capable of stimulating MAPK activity as determined by an elk reporter gene assay (Fig. 7 B). In this assay, cells expressing the receptor chimeras exhibit high basal activation of the elk reporter (Fig. 7 B), which may be due to receptor activation by residual PDGF-BB present in the serum or auto-activation of receptors resulting from the high level of receptor chimera expressed.
Figure 7.

ERK activation by receptor chimeras in MM14 cells. (A) Proliferating MM14 cells stably expressing PRtm/FR1 were starved and then stimulated for 10 min as indicated. Cell lysates were prepared and ERK1/2 proteins were immunoprecipitated. Immune complexes were subjected to an in vitro kinase assay and the reaction products resolved by SDS-PAGE. Incorporation of 32P was quantified by phosphoimage analysis. The inset illustrates a representative example from the gel analysis (N, F, P, and T correspond to None, FGF-2, PDGF-BB, and TPA, respectively.) Error bars represent the standard deviation of three independent samples. (B) Cells transiently expressing the PR/FR1tm chimera activate an elk reporter gene. MM14 cells transiently co-transfected with the PR/FR1tm chimera, an elk reporter, and a constitutively expressed LacZ construct were either left untreated (black), stimulated with FGF-2 (white), or PDGF-BB (gray).
One potential explanation for the ability of the receptor chimeras to repress differentiation could result from autocrine production and release of endogenous FGFs, a mechanism that we have shown to be able to replace the activity of exogenously applied FGFs (13). To test this hypothesis, we chose to block any potential FGFR-mediated signaling by exploiting a tetracycline-responsive system to induce high level expression of a truncated FGFR-1 (12). The use of this system is required to achieve high level expression of the truncated FGFR-1 necessary to block endogenous FGFR-1 signaling with relatively low amounts of expression vector used for the transient transfection assays. We have shown that ectopic expression of a vector encoding a truncated FGFR1 blocks the activity of exogenously applied FGFs and endogenously expressed FGFs in MM14 cells (13). To test the extent of induction using the tetracycline system, MM14 cells were transiently transfected with expression vectors encoding the luciferase gene under the control of the tetracycline activator, the tetracycline activator plasmid, and a constitutively active LacZ expression vector. Luciferase activity was induced several hundred–fold upon removal of tetracycline (Fig. 8 A). The presence or absence of a wide concentration range of tetracycline had no effect on myogenic differentiation or the ability of FGF to promote proliferation and repress differentiation (data not shown). We then constructed a dominant-negative FGFR expression plasmid (dnFR1-tet) that could be induced using the tetracycline activator. MM14 cells were then transiently transfected with either dnFR1-tet, pBJ5PR/FR1tm, or both along with the tetracycline activator plasmid, the muscle-specific luciferase reporter, and the constitutively active LacZ expression vector. Muscle-specific luciferase reporter assays demonstrate that the action of FGF-2 is blocked upon transfection with the dnFR1-tet construct (Fig. 8 B). However, the PR/ FR1tm chimera is capable of signaling in the presence or absence of the dnFR1-tet construct (Fig. 8 B). Thus, PDGF-dependent signaling mediated by the receptor chimeras is likely to be direct and is not mediated via production and release of endogenous FGFs.
Figure 8.
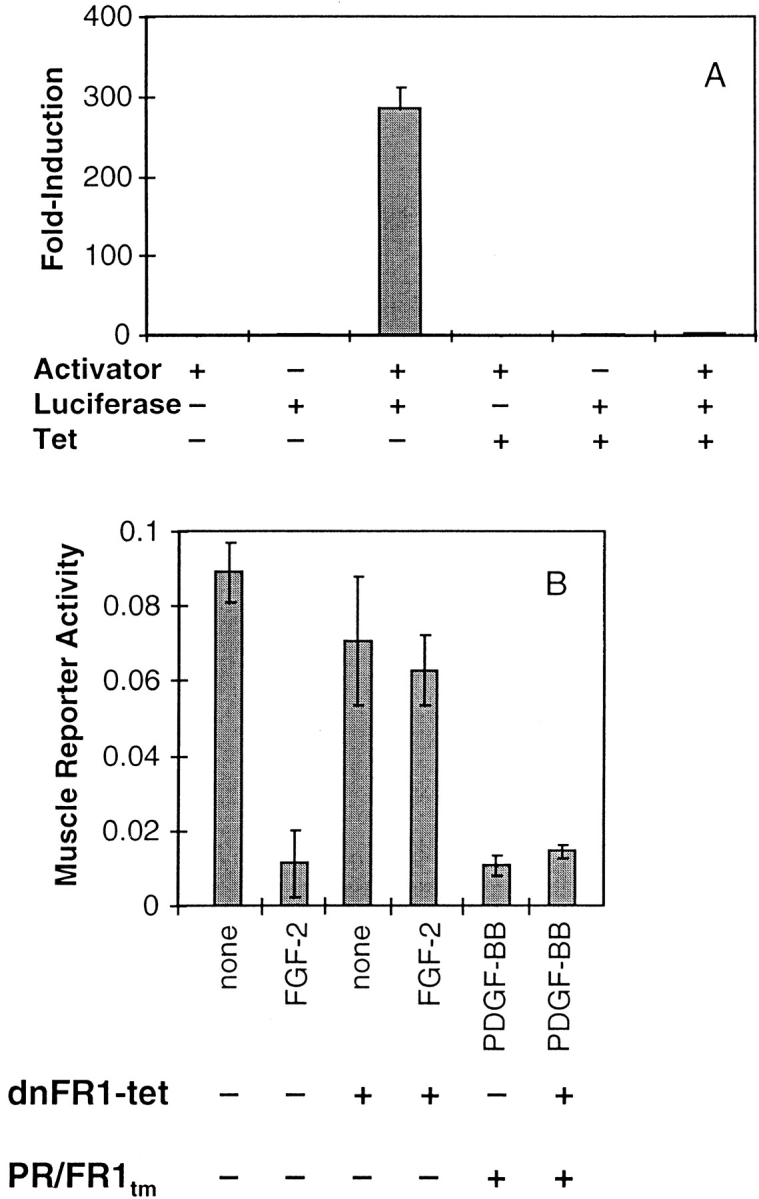
Signaling mediated by the chimeric receptors is not mediated via an indirect autocrine FGF response. (A) Luciferase expression is induced nearly 300-fold in MM14 cells transiently transfected with both the tetracycline activator construct and the tetracycline activator–responsive luciferase reporter construct relative to cells transfected only with the reporter construct. MM14 cells were transiently co-transfected with a LacZ expression construct (CMV-LacZ) along with the tetracycline activator construct (Activator), the luciferase reporter construct under the control of the tetracycline activator (Luciferase), and then cultured in the presence or absence of tetracycline (Tet; 1 μg/ml) for 36 h, harvested and then assayed for luciferase and β-galactosidase activities. (B) MM14 cells were transiently co-transfected with a muscle-specific luciferase reporter gene, CMV-LacZ, the tetracycline activator construct and/or dnFR1-tet, which encodes a dominant-negative FGFR-1 mutant under the control of the tetracycline activator, and where indicated, PR/FR1tm. All cultures were maintained in the absence of tetracycline and received either no treatment, FGF-2 (1 nM), or PDGF-BB (2 nM). After 36 h, the luciferase and β-galactosidase activities were measured to determine muscle-specific promoter activity. Error bars represent the standard deviation of triplicate points. All data shown are from cells incubated in the absence of tetracycline.
A second interpretation that could account for the inability of the receptor chimera to transduce signals for cellular proliferation might be an overstimulation of ERK1/2. The endogenous FGFR-1 weakly stimulates ERK1/2 (see Fig. 7) and ectopic overexpression of the receptor chimeras stimulates ERK1/2 to a greater extent (see Fig. 7). Therefore, we isolated MM14 cells that stably overexpress FGFR-1 (MM14-FR1) at levels 7- to 10-fold higher than parental cells (Fig. 9 A). Consistent with the elevated levels of FGFR-1 in MM14-FR1 cells, activation of ERK1/2 was also significantly increased (Fig. 9 B). However, the dose-responses for FGF-2 stimulation of DNA synthesis in MM14 parental cells and MM14-FR1 were indistinguishable, suggesting that the increase in ERK1/2 activity did not affect signaling for cellular proliferation (Fig. 9 C).
Figure 9.


Overexpression of FGFR-1 enhances ERK1/2 stimulation but fails to affect proliferation. MM14 cells were stably transfected with an FGFR-1 expression vector. MM14 cells overexpressing FGFR-1 (black) were compared with parental cells (white) for FGF-2 binding (A), ERK1/2 activity (B), and DNA synthesis (C). (A) Equilibrium binding was performed using 125I-labeled FGF-2 as described in Materials and Methods. High and low affinity sites were revealed by washing intact cells with high salt or high salt/low pH buffers, respectively. (B) ERK 1/2 activity was determined using myelin basic protein as a substrate for parental (white) and FGFR-1 overexpressing cells (black). Cells were stimulated with DMSO (none), 100 pM FGF-2, or 100 nM TPA for 10 min and then assayed as described in Materials and Methods. The reaction products were resolved by SDS-PAGE and radiolabeled MBP quantified by phosphoimage analysis. The values for DMSO treatment were normalized to 1.0 to compare the two cell lines. Error bars indicate standard deviation. (C) DNA synthesis was examined in parental MM14 cells (□) and MM14-FR1 cells incubated (♦) with increasing concentrations of FGF-2 using a cell proliferation assay as described in Materials and Methods. The results are the representative of a single experiment. Each determination was repeated twice with similar results.
Discussion
To identify specific biological responses mediated by the FGFR-1 tyrosine kinase domain in skeletal muscle cells, we used receptor chimeras comprised of the PDGFβR extracellular and FGFR-1 intracellular domains. We previously demonstrated that PDGFβRs are not present in MM14 cells and that the activated PDGFβR has no detectable effect on skeletal muscle cell proliferation or differentiation (16). Thus, it is an ideal candidate for use in construction of chimeric receptors with FGFR-1 domains and subsequent analysis of receptor domains necessary for signaling. Two distinct receptor chimeras were tested that contained the transmembrane domain of FGFR-1 or the PDGFβR. The genetic defects in skeletal dysplasias and craniosynostosis can result from mutations in the FGFR-3 and FGFR-2 transmembrane domains, respectively (8). These mutations lead to ligand-independent receptor activity or ligand-dependent receptor hyperactivity (8). Thus, it was important to determine if the transmembrane domain replacement would affect chimeric receptor signaling.
Unexpectedly, we found that both PDGFβR/FGFR-1 chimeras were capable of complementing only part of the function of the endogenous FGFR-1. The chimeric receptors were capable of mediating a complex biological response—repression of skeletal muscle differentiation. In contrast to the endogenous FGFR-1 signaling, neither receptor chimera supported cell proliferation. It is unlikely that the extracellular domain of the PDGFβR and binding to PDGF-BB indirectly inhibits FGF signaling since we have shown previously that expression of the full-length PDGFβR had no effect on FGF signaling through endogenous FGFR-1 (16). Interestingly, we noted that the chimera that contained the PDGFRβ transmembrane domain did not affect signaling from the endogenous FGFR-1, whereas the chimera containing the FGFR-1 transmembrane domain acted as a dominant-negative FGFR-1 mutant. While we do not understand the molecular basis for these differences, it is known that mutations in FGFR (8) and neu (2, 3) transmembrane domains result in constitutively active receptors. Thus, alterations in receptor regions juxtaposed to the transmembrane domain may alter or inhibit receptor signaling. Though the chimeric receptors contain different transmembrane domains both yielded identical biological activities, suggesting that FGF regulates skeletal muscle proliferation and differentiation via different signaling systems. We propose that activation of the FGFR-1 receptor tyrosine kinase alone is sufficient to repress myogenesis. This is in contrast to a number of recent reports that implicate FGF and intracellular FGF transport in the biological activity mediated by FGFR-1. In these studies, intracellular transport of FGF ligands was reported to stimulate DNA synthesis independent of FGFR tyrosine kinase activation (33, 34). Our results indicate that regulation of skeletal muscle differentiation appears to require only activation of the FGFR-1 tyrosine kinase and does not appear to require intracellular transport of the FGF ligand nor activation of intracellular events by the ligand.
Since ectopically expressed FGF-1 and FGF-2 repress myogenesis and stimulate proliferation (13), it is possible that the chimeric receptors signal via autocrine-stimulated release of FGFs, that act on the endogenous FGFR-1. To ensure that activation of the FGFR-1 tyrosine kinase was not signaling via autocrine release or production of FGFs, we used a tetracycline-responsive system to overexpress the dominant negative FGFR-1 mutant in cells expressing chimeric receptors. The mutant FGFR-1 blocked all signaling from the endogenous FGFR-1 but did not affect repression of differentiation mediated by the chimeric receptor. Alternatively, the level of activation of MAPKs may be critical for distinguishing between proliferation and differentiation signals. It is possible that overexpression of the receptor chimeras, which led to robust MAPK activation inhibited or altered signals that lead to proliferation. Therefore, we selected cells that overexpressed FGFR-1 and examined FGF-2–induced stimulation of ERK1/2 and FGF-2 stimulation of mitogenesis. Although FGF-2 addition significantly increased ERK1/2 activation, we observed no detectable differences in the mitogenic responses to FGF-2 between the FGFR-1–overexpressing cells and the parental cell line.
Consistent with reports suggesting that signaling events in addition to activation of FGFR kinases are necessary to mediate cellular proliferation by the FGFs, we observed that PRtm/FR1 and PR/FR1tm were incapable of maintaining cellular proliferation or initiating DNA synthesis. It is possible that the extracellular domain of FGFR, FGF, heparan sulfate proteoglycan, or a combination of these is required for DNA synthesis and cell proliferation. We favor a hypothesis that involves FGF, the ectodomain of the FGFR-1, or both, in transducing intracellular signals involved in stimulating cellular proliferation. It is tempting to speculate that the ectodomain of the receptor is required for intracellular transport of the FGF ligand, which then is involved in stimulating DNA synthesis. This hypothesis is consistent with several reports suggesting an alternate signaling role for FGF ligands (1, 4, 5, 15, 33, 34) and for the involvement of FGFR-1 in intracellular transport of the FGF ligand (27).
We hypothesize that it is unlikely that the receptor chimeras are compromised in their ability to activate their respective tyrosine kinase domains. While we cannot compare directly tyrosine phosphorylation of the endogenous FGFR-1 with the ectopically expressed chimeric receptors, we did compare activation of MAPKs and inhibition of skeletal muscle gene expression by both chimeric receptors and the endogenous FGFR-1. We would expect to see defects in signaling that would be distinct for each receptor if the construction of receptor chimeras affected activation of the FGFR-1 tyrosine kinase, yet both chimeric receptors stimulate MAPK activity. However, activation of the receptor chimeras may differ subtly from activation of a wild-type receptor creating a partially defective receptor that is not capable of stimulating cell proliferation. Although we tested to determine if differences in ERK1/2 activation were responsible, differences in activation levels of other signal transducers could be involved. In other experiments, we have constructed a constitutively active FGFR-1 receptor chimera. Preliminary data indicate that the behavior of this receptor is identical to the PDGFβR/ FGFR-1 chimeras as it is capable of repressing myogenesis but cannot support proliferation. Whereas these experiments do not prove an involvement of the ectodomain, ligand, or additional components in FGFR-1 signaling, they do demonstrate that the signaling events required for repression of myogenesis and stimulation of proliferation by FGFR-1 are distinct and separable. Remarkably, both receptor chimeras used in our experiments were fully capable of repressing skeletal muscle differentiation, a complex biological response whose signaling pathways are not understood.
The involvement of intracellular FGFR-1 phosphotyrosines in FGFR-1 signaling has been questioned since mutagenesis of all but two intracellular phosphotyrosines (Y654 and Y655), required for catalytic activity, had no effect on FGFR-1 signaling (22). These data suggest that the molecular mechanisms used by FGFR-1 to transduce intracellular signals may differ significantly from those described for other tyrosine kinase receptors. Recently, a similar conclusion was reached for the muscle-specific receptor tyrosine kinase (MuSK) receptor (10). Chimeric receptors comprised of the trkC extracellular domain and the MuSK intracellular domain, were incapable of acetylcholine receptor clustering, despite the activation of intracellular signaling by the chimera (10).
By determining which biological activities are mediated by the FGFR-1 tyrosine kinase, we have shown that two FGF-dependent cellular activities in a single cell type appear to be independently mediated by distinct signaling mechanisms that result from ligand binding to FGFR-1. Further analysis of the signaling pathways stimulated by the chimeric receptors will provide us with a powerful tool in the delineation if signaling pathways mediating the repression of skeletal muscle differentiation. Domain-swapping experiments with intracellular regions of the PDGFβR and mutagenesis of the intracellular FGFR-1 domains are likely to identify critical regions of the FGFR-1 receptor involved. In addition, we can now use these chimeric receptors for rescue studies to attempt to identify the signaling mechanisms necessary for stimulation of cellular proliferation. Identification of these latter pathways is likely to further our understanding of FGFR signaling and identify the molecular mechanisms involved in the activation of intracellular signaling events that generate specific biological responses.
Acknowledgments
This work was supported by grants from the Public Health Service to B.B. Olwin and by a National Science Foundation predoctoral fellowship to A.J. Kudla.
Abbreviations used in this paper
- BrdU
5-bromo-2 deoxyuridine
- CMV
cytomegalovirus
- ERK
extracellular-regulated kinase
- FGFR
FGF receptor
- MAPK
mitogen-activated protein kinase
- MBP
myelin basic protein
- MHC
myosin heavy chain
- PDGFβR
PDGF β receptor
- PRtm/FR1
PDGFβR/FGFR-1 chimera with PDGFβR transmembrane domain
- PR/ FR1tm
PDGFβR/FGFR-1 chimera with FGFR-1 transmembrane domain
- RT
reverse transcription
- TPA
12-O-tetra-decnoylphorbol-13-acetate
Footnotes
Address all correspondence to Bradley B. Olwin, Department of Molecular, Cellular and Developmental Biology, University of Colorado, Boulder, CO 80309. Tel.: (303) 492-6816. Fax: (303) 492-1587. E-mail: Bradley.Olwin@colorado.edu
References
- 1.Baldin V, Roman AM, Bosc BI, Amalric F, Bouche G. Translocation of bFGF to the nucleus is G1 phase cell cycle specific in bovine aortic endothelial cells. EMBO (Eur Mol Biol Organ) J. 1990;9:1511–1517. doi: 10.1002/j.1460-2075.1990.tb08269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bargmann CI, Hung MC, Weinberg RA. Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell. 1986;45:649–57. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- 3.Bargmann CI, Weinberg RA. Increased tyrosine kinase activity associated with the protein encoded by the activated neu oncogene. Proc Natl Acad Sci USA. 1988;85:5394–5398. doi: 10.1073/pnas.85.15.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouche G, Gas N, Prats H, Baldin V, Tauber JP, Teissie J, Amalric F. Basic fibroblast growth factor enters the nucleolus and stimulates the transcription of ribosomal genes in ABAE cells undergoing G0-G1 transition. Proc Natl Acad Sci USA. 1987;84:6770–6774. doi: 10.1073/pnas.84.19.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgess WH, Shaheen AM, Ravera M, Jaye M, Donohue PJ, Winkles JA. Possible dissociation of the heparin-binding and mitogenic activities of heparin-binding (acidic fibroblast) growth factor-1 from its receptor-binding activities by site-directed mutagenesis of a single lysine residue. J Cell Biol. 1990;111:2129–2138. doi: 10.1083/jcb.111.5.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 7.Clegg CH, Linkhart TA, Olwin BB, Hauschka SD. Growth factor control of skeletal muscle differentiation: commitment to terminal differentiation occurs in G1 phase and is repressed by fibroblast growth factor. J Cell Biol. 1987;105:949–956. doi: 10.1083/jcb.105.2.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Moerlooze L, Dickson C. Skeletal disorders associated with fibroblast growth factor receptor mutations. Curr Opin Genet Dev. 1997;7:378–385. doi: 10.1016/s0959-437x(97)80152-9. [DOI] [PubMed] [Google Scholar]
- 9.DeHamer MK, Guevara JL, Hannon K, Olwin BB, Calof AL. Genesis of olfactory receptor neurons in vitro: regulation of progenitor cell divisions by fibroblast growth factors. Neuron. 1994;13:1083–1097. doi: 10.1016/0896-6273(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 10.Glass DJ, Apel ED, Shah S, Bowen DC, DeChiara TM, Stitt TN, Sanes JR, Yancopoulos GD. Kinase domain of the muscle-specific receptor tyrosine kinase (MuSK) is sufficient for phosphorylation but not clustering of acetylcholine receptors: required role for the MuSK ectodomain? . Proc Natl Acad Sci USA. 1997;94:8848–8853. doi: 10.1073/pnas.94.16.8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gospodarowicz D, Weseman J, Moran J. Presence in brain of a mitogenic agent promoting proliferation of myoblasts in low density culture. Nature. 1975;256:216–219. doi: 10.1038/256216a0. [DOI] [PubMed] [Google Scholar]
- 12.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–51. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hannon K, Kudla AJ, McAvoy MJ, Clase KL, Olwin BB. Differentially expressed fibroblast growth factors regulate skeletal muscle development through autocrine and paracrine mechanisms. J Cell Biol. 1996;132:1151–1159. doi: 10.1083/jcb.132.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hart CE, Seifert RA, Ross R, Bowen-Pope DF. Synthesis, phosphorylation, and degradation of multiple forms of the platelet- derived growth factor receptor studied using a monoclonal antibody. J Biol Chem. 1987;262:10780–10785. [PubMed] [Google Scholar]
- 15.Imamura T, Engleka K, Zhan X, Tokita Y, Forough R, Roeder D, Jackson A, Maier JAM, Hla T, Maciag T. Recovery of mitogenic activity of a growth factor mutant with a nuclear translocation sequence. Science. 1990;249:1567–1570. doi: 10.1126/science.1699274. [DOI] [PubMed] [Google Scholar]
- 16.Kudla AJ, John ML, Bowen-Pope DF, Rainish B, Olwin BB. A requirement for fibroblast growth factor in regulation of skeletal muscle growth and differentiation cannot be replaced by activation of platelet-derived growth factor signaling pathways. Mol Cell Biol. 1995;15:3238–3246. doi: 10.1128/mcb.15.6.3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee J, Dull TJ, Lax I, Schlessinger J, Ullrich A. HER2 cytoplasmic domain generates normal mitogenic and transforming signals in a chimeric receptor. EMBO (Eur Mol Biol Organ) J. 1989;8:167–173. doi: 10.1002/j.1460-2075.1989.tb03361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim RW, Hauschka SD. EGF responsiveness and receptor regulation in normal and differentiation-defective mouse myoblasts. Dev Biol. 1984;105:48–58. doi: 10.1016/0012-1606(84)90260-4. [DOI] [PubMed] [Google Scholar]
- 19.Linkhart TA, Clegg CH, Hauschka SD. Control of mouse myoblast commitment to terminal differentiation by mitogens. J Supramol Structure. 1980;14:483–498. doi: 10.1002/jss.400140407. [DOI] [PubMed] [Google Scholar]
- 20.Linkhart TA, Clegg CH, Hauschka SD. Myogenic differentiation in permanent clonal myoblast cell lines: regulation by macromolecular growth factors in the culture medium. Dev Biol. 1981;86:19–30. doi: 10.1016/0012-1606(81)90311-0. [DOI] [PubMed] [Google Scholar]
- 21.Mares J, Claesson L, Welsh, Welsh M. A chimera between platelet-derived growth factor beta-receptor and fibroblast growth factor receptor-1 stimulates pancreatic beta-cell DNA synthesis in the presence of PDGF-BB. Growth Factors. 1992;6:93–101. doi: 10.3109/08977199209011013. [DOI] [PubMed] [Google Scholar]
- 22.Mohammadi M, Dikic I, Sorokin A, Burgess WH, Jaye M, Schlessinger J. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol Cell Biol. 1996;16:977–989. doi: 10.1128/mcb.16.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olwin BB, Hannon K, Kudla AJ. Are fibroblast growth factors regulators of myogenesis in vivo. . Prog Growth Factor Res. 1994;5:145–158. doi: 10.1016/0955-2235(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 24.Olwin BB, Hauschka SD. Identification of the fibroblast growth factor receptor of Swiss 3T3 cells and mouse skeletal muscle myoblasts. Biochemistry. 1986;25:3487–3492. doi: 10.1021/bi00360a001. [DOI] [PubMed] [Google Scholar]
- 25.Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rapraeger AC, Guimond S, Krufka A, Olwin BB. Regulation by heparan sulfate in fibroblast growth factor signaling. Methods Enzymol. 1994;245:219–240. doi: 10.1016/0076-6879(94)45013-7. [DOI] [PubMed] [Google Scholar]
- 27.Reiland J, Rapraeger AC. Heparan sulfate proteoglycan and FGF receptor target basic FGF to different intracellular destinations. J Cell Sci. 1993;105:1085–1093. doi: 10.1242/jcs.105.4.1085. [DOI] [PubMed] [Google Scholar]
- 28.Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol. 1994;14:1669–1679. doi: 10.1128/mcb.14.3.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riedel H, Dull TJ, Honegger AM, Schlessinger J, Ullrich A. Cytoplasmic domains determine signal specificity, cellular routing characteristics and influence ligand binding of epidermal growth factor and insulin receptors. EMBO (Eur Mol Biol Organ) J. 1989;8:2943–2954. doi: 10.1002/j.1460-2075.1989.tb08444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riedel H, Schlessinger J, Ullrich A. A chimeric, ligand-binding v-erbB/EGF receptor retains transforming potential. Science. 1987;236:197–200. doi: 10.1126/science.3494307. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Second ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. 16.30–16.40.
- 32.Takebe Y, Seiki M, Fujisawa J-I, Hoy P, Yokota K, Arai K-I, Yoshida M, Arai N. SRa promoter: an efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol Cell Biol. 1988;8:466–472. doi: 10.1128/mcb.8.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiedlocha A, Falnes PØ, Madshus IH, Sandvig K, Olsnes S. Dual mode of signal transduction by externally added acidic fibroblast growth factor. Cell. 1994;76:1039–1051. doi: 10.1016/0092-8674(94)90381-6. [DOI] [PubMed] [Google Scholar]
- 34.Wiedlocha A, Falnes PO, Rapak A, Munoz R, Klingenberg O, Olsnes S. Stimulation of proliferation of a human osteosarcoma cell line by exogenous acidic fibroblast growth factor requires both activation of receptor tyrosine kinase and growth factor internalization. Mol Cell Biol. 1996;16:270–280. doi: 10.1128/mcb.16.1.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu L, Sanchez T, Zheng C-F. In vivo signal transduction pathway reporting systems. Strategies. 1997;10:1–3. [Google Scholar]
- 36.Yarden Y, Ullrich A. Growth factor receptor tyrosine kinases. Annu Rev Biochem. 1988;57:443–478. doi: 10.1146/annurev.bi.57.070188.002303. [DOI] [PubMed] [Google Scholar]