

Abstract
Three previously identified genes from Saccharomyces cerevisiae, VMA12, VMA21, and VMA22, encode proteins localized to the endoplasmic reticulum (ER). These three proteins are required for the biogenesis of a functional vacuolar ATPase (V-ATPase), but are not part of the final enzyme complex. Subcellular fractionation and chemical cross-linking studies have revealed that Vma12p and Vma22p form a stable membrane associated complex. Cross-linking analysis also revealed a direct physical interaction between the Vma12p/Vma22p assembly complex and Vph1p, the 100-kD integral membrane subunit of the V-ATPase. The interaction of the Vma12p/Vma22p complex with Vph1p was transient (half-life of ∼5 min), reflecting trafficking of this V-ATPase subunit through the ER en route to the vacuolar membrane. Analysis of these protein–protein interactions in ER-blocked sec12 mutant cells indicated that the Vph1p-Vma12p/Vma22p interactions are quite stable when transport of the V-ATPase out of the ER is blocked. Fractionation of solubilized membrane proteins on a density gradient revealed comigration of Vma22p and Vma12p, indicating that they form a complex even in the absence of cross-linker. Vma12p and Vma22p migrated to fractions separate from Vma21p. Loss of Vph1p caused the Vma12p/Vma22p complex to sediment to less dense fractions, consistent with association of Vma12p/ Vma22p with nascent Vph1p in ER membranes. This is the first evidence for a dedicated assembly complex in the ER required for the assembly of an integral membrane protein complex (V-ATPase) as it is transported through the secretory pathway.
Keywords: vacuoles, vacuolar ATPase, endoplasmic reticulum, molecular chaperones, membrane proteins
The Saccharomyces cerevisiae vacuolar proton-translocating ATPase (yeast V-ATPase)1 is the best characterized member of the V-type ATPase family of membrane complexes found in all eukaryotic cells, and thus provides an excellent model to study the assembly of a multi-subunit enzyme complex (Forgac, 1989; Stevens and Forgac, 1997). Biochemical and genetic analyses have allowed the identification of a large number of yeast VMA (vacuolar membrane ATPase) genes required for the assembly of the V-ATPase enzyme complex (Stevens and Forgac, 1997). The V-ATPase is composed of a V1 catalytic sector of peripherally associated proteins facing the cytosol, assembled onto the Vo sector of integral membrane proteins. At least 13 genes encode subunits of the V-ATPase enzyme complex (Hirata et al., 1997). Assembly of the multisubunit V-ATPase is a complex process requiring the coordinated association of subunits synthesized in the cytosol with subunits targeted to the vacuole via the endomembrane network comprising the secretory pathway.
Examination of various V-ATPase mutants, lacking either a V1 or Vo subunit, has revealed two assembly phenotypes describing the fate of the remaining subunits in these mutant yeast cells. Loss of a V1 subunit (69, 60, 42, 32, 27, 14, and 13 kD) does not affect the stability of any of the remaining subunits, but does prevent the assembly of the V1 subunits onto the vacuolar membrane. The exception being the loss of the 54-kD V1 subunit (Vma13p), which does not prevent the assembly of the remaining V1 subunits onto the vacuolar membrane, but the V-ATPase complex that does assemble is inactive (Ho et al., 1993 b). Thus, it appears that the stability of the individual V1 subunits is not dependent on the other V1 subunits being present in the cell, but the ability to assemble onto the vacuolar membrane requires the presence of all the V1 subunits (except the 54-kD subunit). The Vo subcomplex is assembled and targeted to the vacuolar membrane in cells lacking any V1 subunit.
In contrast, the loss of a Vo subunit affects both the assembly and stability of the remaining Vo subunits. In addition, loss of a Vo subunit prevents V-ATPase assembly, however, the V1 subunits associate as a V1 subcomplex in the cytosol (Doherty and Kane, 1993; Tomashek et al., 1996). The Vo subcomplex of the yeast V-ATPase is composed of at least five different subunits of molecular mass 100, 36, 23, 17, and 16.5 kD. In cells lacking a Vo subunit, the 36-kD (Vma6p) peripherally associated protein fails to assemble onto the membrane, but remains stable in the cytosol (Bauerle et al., 1993; Graham, L.A., unpublished results). The effect on the 23-kD (Vma16p), 17-kD (Vma11p), and 16-kD (Vma3p) proteins has been harder to assess due to their extreme hydrophobicity (Hirata et al., 1997). Loss of a Vo subunit, such as the 36-kD subunit (Vma6p) or the 16.5-kD subunit (Vma3p), resulted in decreased steady state levels of the 100-kD V-ATPase subunit present on vacuolar membranes as determined by immunoblot analysis (Bauerle et al., 1993).
The 100-kD V-ATPase subunit, encoded by the VPH1 gene, is unique to V-ATPase complexes and has no homologous counterpart in the structurally and functionally similar F1Fo-ATPase complex (Manolson et al., 1992; Stevens and Forgac, 1997). The 100-kD protein can be divided into a hydrophilic NH2-terminal half and a hydrophobic COOH terminus predicted to form six to seven membrane-spanning domains. Similar to other Vo proteins in wild-type cells, Vph1p is inserted into the membranes of the endoplasmic reticulum (ER) upon synthesis and targeted to the vacuole by the secretory pathway. Loss of Vph1p prevents assembly of the Vo subcomplex and thus the V-ATPase never assembles on vacuolar membranes.
Genetic analysis also led to the identification of three VMA genes (VMA12, VMA21, and VMA22) encoding proteins that are not subunits of the final complex, but are required for the assembly of a functional V-ATPase (Hirata et al., 1993; Ho et al., 1993a ; Hill and Stevens, 1994, 1995; Jackson and Stevens, 1997). Loss of either Vma12p, Vma21p, or Vma22p results in Vma− phenotypes identical to those observed for mutant cells that lack a V-ATPase subunit. The mutant cells display loss of V-ATPase activity, increased sensitivity to calcium, the inability to grow in media buffered to neutral pH, and cannot use nonfermentable carbon sources. The loss of either Vma12p, Vma21p, or Vma22p thus appears to block the assembly of a functional V-ATPase enzyme complex. It has been shown previously that loss of either Vma12p, Vma21p, or Vma22p, the nonsubunit assembly factors, also reduced the steady state cellular levels of Vph1p (Hill and Stevens, 1994, 1995; Hirata et al., 1993). Whereas Vph1p has a half-life of >400 min in wild-type cells, vma12Δ, vma21Δ, and vma22Δ cells demonstrated an increase in the rate of degradation of Vph1p, resulting in a half-life of ∼30 min (Hill and Stevens, 1994, 1995; Jackson and Stevens, 1997). Cells lacking either Vma12p, Vma21p, or Vma22p do not display general defects in the processing, targeting or assembly of other vacuolar membrane or plasma membrane proteins (Bachhawat et al., 1993; Hill and Stevens, 1994). Finally, protease protection studies revealed that Vph1p was inserted normally into the ER membranes of vma12Δ cells, indicating that Vma12p, Vma21p, and Vma22p are involved in assembly of the V-ATPase and not translocation of Vph1p (Jackson and Stevens, 1997).
All three proteins, Vma12p, Vma21p, and Vma22p, have been immunolocalized to the membranes of the ER and not the vacuole (Hill and Stevens, 1994, 1995; Jackson and Stevens, 1997). Vma21p is an extremely hydrophobic protein of 8.5 kD possessing a COOH-terminal di-lysine motif (KKED), which functions to retain the protein in the ER (Hill and Stevens, 1994). Neither Vma12p or Vma22p contains an obvious ER retention signal and only Vma12p (25 kD) is predicted to be an integral membrane protein. Since the loss of any one of these three proteins results in an identical phenotype, we set out to determine whether these proteins function together in aiding the assembly of the V-ATPase complex transiting the secretory pathway on its way to the vacuole.
The first indication that the V-ATPase assembly factors may interact came from subcellular fractionation analysis of Vma22p. Vma22p is predicted to be a hydrophilic protein (21 kD), yet in wild-type cells this protein is found associated with ER membranes. The fractionation profile of Vma22p in vma12Δ cells was strikingly different than that seen in wild-type cells; all the Vma22p in the mutant cells now fractionated with cytosolic proteins in the supernatant (Hill and Stevens, 1995). The ability of Vma22p to associate stably with ER membranes was thus found to depend on the presence of Vma12p.
To further characterize the V-ATPase assembly process, we investigated the interactions between the assembly factors as well as between the assembly factors and a V-ATPase subunit, Vph1p, as it assembles into the Vo subcomplex in the ER. Vma12p and Vma22p were found to interact directly as determined by chemical cross-linking analysis and cofractionation under conditions of gentle detergent solubilization. These studies revealed a transient interaction (t1/2 = ∼5 min) between the ER localized Vma12p/Vma22p complex and the vacuolar ATPase subunit Vph1p. Vma21p does not appear to be a component of the Vma12p/Vma22p complex as determined by fractionation studies nor does it appear to interact directly with the 100-kD subunit. We propose that Vma12p together with Vma22p forms an assembly complex that interacts with Vo subunits in the ER as they are being assembled before transport of the V-ATPase complex to the vacuole. Vma12p and Vma22p provide the first example of an hetero-oligomeric integral membrane assembly complex apparently dedicated to the biogenesis of a single multisubunit enzyme complex.
Materials and Methods
Materials
Monoclonal antibodies recognizing Vph1p (10D7A) was purchased from Molecular Probes Inc. (Eugene, OR) and used for immunoblot analysis. 35S-Express label was purchased from DuPont NEN (Boston, MA). Fixed Staphylococcus aureus cells (IgGSorb) used to collect immune complexes was purchased from the Enzyme Center (Malden, MA). Zymolyase 100T prepared from Arthrobacter luteus was purchased from ICN Biochemicals, Inc. (Costa Mesa, CA). All other reagents were purchased from Sigma Chemical Co. (St. Louis, MO).
Yeast Strains and Media
Yeast strains and plasmids used in this study are listed in Table I. All yeast strains were cultured either in SD minimal media (0.67% yeast nitrogen base, 2% dextrose) supplemented with the appropriate amino acids or rich media (YEPD buffered to pH 5.0; Yamashiro et al., 1990) using standard techniques. All yeast strains were cultured in SD media lacking methionine before whole cell labeling using [35S]methionine.
Table I.
Yeast Strains and Plasmids Used in This Study
| Genotype | Source | |||
|---|---|---|---|---|
| Strain | ||||
| SF838-1D | MATα ura3-52 leu2-3,112 his4-519 ade6 pep4-3 | Rothman and Stevens, 1986 | ||
| KHY31 | MATα ura3-52 leu2-3,112 his4-519 ade6 pep4-3 vph1Δ::LEU2 | This study | ||
| CRY6 | MATα ura3-52 leu2-3,112 his4-519 ade6 pep4-3 vma2Δ::LEU2 | Yamashiro et al., 1990 | ||
| MKY2 | MATα ura3-52 leu2-3,112 his4-519 ade6 pep4-3 vma3Δ::LEU2 | Kane et al., 1992 | ||
| CBY1 | MATα ura3-52 leu2-3,112 his4-519 ade6 pep4-3 vma6Δ::LEU2 | Bauerle et al., 1993 | ||
| RHA107 | MATα ura3-52 leu2-3,112 his4-519 ade6 pep4-3 vm11Δ::LEU2 | Gift from R. Hirata | ||
| LGY9 | MATα ura3-52 leu2-3,112 his4-519 ade6 pep4-3 vma16Δ::LEU2 | This study | ||
| DJY102 | MATα ura3-52 leu2-3,112 his4-519 ade6 pep4-3 vma12Δ::LEU2 | Jackson and Stevens, 1997 | ||
| KHY3 | MATα ura3-52 leu2-3,112 his4-519 ade6 pep4-3 vma21Δ::LEU2 | Hill and Stevens, 1994 | ||
| KHY34 | MATα ura3-52 leu2-3,112 his4-519 ade6 pep4-3 vma22Δ::LEU2 | Hill and Stevens, 1995 | ||
| Plasmid | ||||
| pKH28 | pRS316 VMA21::HA | Hill and Stevens, 1994 |
Vph1p Immunoprecipitations
Denaturing immunoprecipitation from [35S]methionine labeled cells of the 100-kD V-ATPase subunits using anti-Vph1p polyclonal sera was performed as previously described (Hill and Stevens, 1994). In brief, cells were radiolabeled with 100 μCi per 0.5 OD600 nm of cells for 10 min, 50 mM excess unlabeled methionine and cysteine was added, and samples were removed at various times. Cells were converted to spheroplasts, lysed in 0.6% SDS, heated to 65°C for 5 min, and the 100-kD subunit was quantitatively immunoprecipitated from the cell extracts.
Subcellular Fractionation
Fractionation of organelles from wild-type and vma mutant cells was performed by differential centrifugation of cell lysates as described by Horazdovsky and Emr (1993). 10 OD600 of exponentially growing cells (1–2 × 108 cells/ml) were spheroplasted, lysed and centrifuged for 5 min at 500 g to remove unbroken cells. The supernatant was centrifuged for 10 min at 13,000 g to generate the membrane pellet fraction (P13), and the supernatant (S13) was centrifuged at 100,000 g for 30 min generating the P100 pellet fraction and S100 supernatant. The membrane pellets were resuspended in 300 μl sample buffer. Proteins in the S100 fraction were precipitated by the addition of TCA to a final concentration of 5%, collected by centrifugation for 5 min at 13,000 g, washed twice with acetone, air dried and resuspended in 300 μl sample buffer. The proteins in 20 μl of each fraction were separated by 15% SDS-PAGE, transferred to nitrocellulose, and probed with anti-Vma22p polyclonal antibodies (Hill and Stevens, 1995). Secondary antibodies conjugated to horseradish peroxidase were from Amersham and were detected using chemiluminescent reagent (DuPont NEN).
Cross-linking Radiolabeled Proteins and Double Immunoprecipitation
Wild-type cells (SF838-1Dα) were labeled with [35S]methionine for 30 min and then pulsed with an additional equivalent aliquot of radiolabel for 5 min to observe steady state interactions. Cells for pulse–chase experiments were labeled for 5 min and then chased with the addition of 50 mM excess cold methionine. Samples were removed at various times during the chase, treated with 10 mM sodium azide, and incubated on ice 5 min before collection by centrifugation. Cells were treated with 10 mM DTT for 5 min, spheroplasted using 250 μg/ml zymolyase, lysed in PBS, 100 mM sorbitol and treated with 250 μM of the reversible cross-linking reagent dithio-bis(succinimidylpropionate) (DSP) for 30 min at room temperature according to the protocol of Stack et al. (1993). The cross-linker was quenched by the addition of 50 mM Tris, pH 7.5. Proteins were precipitated from each sample by the addition of TCA to a final concentration of 5% as before, protein pellets were washed with acetone and resuspended in 100 μl of 4 M urea, 1% SDS and heated for 5 min at 65°C. Primary immunoprecipitations were carried out under nonreducing denaturing conditions using rabbit polyclonal antibodies against Vma22p. Samples from the first immunoprecipitation were resuspended in 100 μl of 4 M urea, 1% SDS, and 5% β-mercaptoethanol (to disrupt the disulfide cross-link), heated as before and a second immunoprecipitation with either anti-Vph1p sera (Hill and Stevens, 1994), anti-Vma22p sera (Hill and Stevens, 1995), anti-Vma12p sera (Hirata et al., 1993), or anti-ALP sera (Nothwehr et al., 1995) was performed. Proteins from the second immunoprecipitation were separated by SDS-PAGE, the dried gels were exposed to a phosphor screen and analyzed using a STORM PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
Sucrose Density Gradient Fractionation
Exponentially growing cells (500 OD600 total) were treated with 10 mM DTT and converted to spheroplasts by incubation with 50 μg/ml zymolyase in 1.2 M sorbitol, 20 mM potassium phosphate buffer, pH 7.6, and 1 mM MgCl2 for 1 h at 30°C. Spheroplasts were washed once with 1.2 M sorbitol and lysed by resuspension in 40 ml of ice cold PBS, 100 mM sorbitol containing 2× protease inhibitor cocktail (2 mM PMSF, 2 mg/ml leupeptin, and 2 mg/ml pepstatin). P13 membranes were collected by centrifugation of lysed spheroplasts for 15 min at 13,000 g, after a 500-g clearing spin for 5 min, and resuspended in PBS. Membranes were solubilized in 1% Triton X-100 at a final protein concentration of 1 mg/ml by incubation for 30 min at 4°C. Protein concentration of the membrane fraction was determined according to the method of Markwell et al. (1978). Solubilized membranes were centrifuged for 15 min at 13,000 g at 4°C and the supernatant was transferred to a fresh tube. A total of 600 μg of detergent solubilized P13 membrane protein were loaded onto a 12 ml 5–15% sucrose density gradient prepared according to Chapman et al. (1996). Fractionation was achieved by centrifugation at 150,000 g for 16 h at 4°C. Twelve 1-ml fractions were collected and proteins precipitated by the addition of TCA (5% final concentration). The protein pellets were dissolved in 200 μl sample buffer and 20 μl of each sample were subjected to SDS-PAGE. Immunoblot analysis of the proteins was performed according to previously described protocols (Bauerle et al., 1993).
Results
Instability of the 100 kD V-ATPase Subunit Reflects Failure to Assemble in the ER
Assembly of a functional V-ATPase requires the biosynthesis of all the subunits plus Vma12p, Vma21p, and Vma22p (Stevens and Forgac, 1997). Determining the stability of pulse radiolabeled 100-kD Vo subunit (Vph1p) in various vma mutants serves as a means to follow the assembly status of the Vo subcomplex. Kinetic analysis of newly synthesized Vph1p in wild-type cells revealed no detectable change in the level of the protein over 80 min and an estimated half-life in excess of 400 min or the equivalent of 3–4 cell divisions (Fig. 1; Hill and Stevens, 1995). The stability of Vph1p in mutant cells lacking a V1 subunit (vma2Δ) was the same as wild-type, consistent with the ability of these cells to assemble and target the Vo subcomplex to the vacuole (Kane et al., 1992). In contrast, newly synthesized Vph1p immunoprecipitated from mutant cells lacking a Vo subunit, vma3Δ or vma6Δ, revealed rapid degradation of Vph1p with an estimated half-life of ∼30 min (Fig. 1). These data are consistent with the decreased steady state levels observed previously in these mutants by immunoblot analysis (Bauerle et al., 1993).
Figure 1.

Stability of the 100-kD V-ATPase subunit (Vph1p) in wild-type and vma mutant cells. Cells were pulse labeled with [35S]methionine for 10 min and chased in the presence of 50 mM unlabeled methionine. Samples were removed at 0, 30, 60, and 90 min during the chase, denaturing cell extracts prepared, and Vph1p was immunoprecipitated using polyclonal sera. Immunoprecipitated proteins were separated by SDS-PAGE, dried gels were exposed to a phosphor screen, and data was collected using a phosphorimaging system. WT, wild-type.
Table II summarizes the stability of newly synthesized Vph1p in a number of vma mutant strains. Also reported in Table II is the localization of Vph1p in vma mutant backgrounds, as determined by indirect immunofluorescence using affinity-purified anti-Vph1p polyclonal antibodies. The stability of Vph1p was observed to be sensitive to the loss of any other Vo subunit. Similarly, the stability of Vph1p was greatly decreased in cells lacking any one of the assembly factors. In mutants lacking a Vo subunit or assembly factor, Vph1p immunolocalized to the ER, consistent with previous reports (Kane et al., 1992; Bauerle et al., 1993; Hill and Stevens, 1994, 1995; Jackson and Stevens, 1997). The data reported here for vma2Δ mutant cells, which lack the 60-kD V1 subunit, are similar to those obtained for a vma mutant lacking any other V1 subunit (Table II).
Table II.
Turnover of Vph1p in Various vma Mutant Strains
| Strain | Defect | Half-life* | Localization‡ | |||
|---|---|---|---|---|---|---|
| Wild-type | None | >5 h | Vacuole | |||
| vma2Δ | V1 subunit | >5 h | Vacuole | |||
| vma3Δ | V0 subunit | ∼30 min | ER | |||
| vma11Δ | V0 subunit | ∼30 min | ER | |||
| vma16Δ | V0 subunit | ∼30 min | ER | |||
| vma6Δ | V0 subunit | ∼30 min | ER | |||
| vma12Δ | Assembly factor | ∼30 min | ER | |||
| vma21Δ | Assembly factor | ∼30 min | ER | |||
| vma22Δ | Assembly factor | ∼30 min | ER |
Observed half-life between 25–35 min for all mutants lacking either a V0 subunit or an assembly factor.
Localization of Vph1p was determined by indirect immunofluorescence using anti-Vph1p affinity-purified polyclonal antibodies.
Membrane Association of Vma22p Requires only Vma12p
Both Vma12p and Vma21p are proteins with predicted transmembrane domains and each has been demonstrated to associate with cellular membrane fractions (Hirata et al., 1993; Hill and Stevens, 1994). Vma12p and Vma21p have been localized using indirect immunofluorescence to perinuclear membranes consistent with ER localization (Hill and Stevens, 1994; Jackson and Stevens, 1997). In contrast, Vma22p is predicted to be a hydrophilic protein lacking any membrane spanning domains, yet it too is found associated with ER membranes (Hill and Stevens, 1995). It has been shown previously that Vma22p does not associate with membranes in vma12Δ mutant cells (Hill and Stevens, 1995). Since the disruption of either vma12Δ, vma21Δ, or vma22Δ results in a phenotype similar to the complete loss of any Vo subunit (vma3Δ, vma6Δ, vma11Δ, or vma16Δ), it is possible that the membrane association of Vma22p is dependent on both of the other assembly factors and/or on the assembly substrates (the Vo subunits).
We examined the effect of the loss of various Vo subunits on the membrane association of Vma22p (Fig. 2). Cells were converted to spheroplasts, lysed, and then centrifuged at low speed (2000 g) to remove unbroken cells generating S2 (total cell lysate fraction). Proteins were fractionated by differential centrifugation into P13 proteins that sedimented at 13,000 g (ER, vacuoles, and plasma membranes), P100 proteins that sedimented at 100,000 g (Golgi, endosomes, vesicles), and the remaining S100 supernatant. In wild-type cells Vma22p fractionated between the P13 membrane fraction and the cytosolic fraction (S100) with a trace found in the P100 membranes. The percentage of Vma22p present in the S100 fraction varied somewhat between preparations and most likely represents Vma22p released from the membranes during the generation of the subcellular fractionations. The fractionation profile of Vma22p in mutants lacking any Vo subunit or vma21Δ was identical to that of wild-type cells, with ∼50% of the protein associated with the ER membranes of the P13 pellet. In cells lacking Vma12p, over 90% of the Vma22p in the cell was found in the cytosol fraction, consistent with previously published results (Hill and Stevens, 1995). Only the loss of Vma12p prevented Vma22p from fractionating with the P13 membranes. The association of Vma22p with ER membranes was not dependent on the presence of any Vo subunit. These results are consistent with a model in which Vma22p associates with the ER membrane through a direct interaction with Vma12p, and that this complex forms independently of the availability of assembly substrates (the Vo subunits).
Figure 2.

Subcellular fractionation of Vma22p in wild-type and vma mutant cells. Cells were converted to spheroplasts, lysed osmotically, and fractionated by differential centrifugation generating S2 (total lysate), P13 (13,000-g membrane pellet), P100 (100,000-g membrane pellet), and S100 (100,000-g supernatant). Proteins from equal proportions of each fraction were separated by SDS-PAGE, and immunoblots probed with anti-Vma22p polyclonal sera. WT, wild-type.
Vma12p and Vma22p Interact with the 100 kD V-ATPase Subunit in the ER
To determine whether Vma12p and Vma22p are part of a V-ATPase assembly complex, we used the reversible cross-linking reagent DSP to form a stable association between Vma22p and any interacting proteins. DSP reacts with primary amines available within a protein and when used at low concentrations only nearest neighbor proteins are cross-linked since the spacer arm length between the reactive groups is only 1.2 nm. Wild-type cells were radiolabeled, converted to spheroplasts, osmotically lysed, and then treated with 250 μM DSP following the protocol of Stack et al. (1993). Various concentrations of DSP were tested (10–800 μM) and 250 μM was found to be the optimal concentration. All noncross-linked protein interactions were disrupted using urea and SDS before immunoprecipitation under nonreducing conditions with antibodies specific for Vma22p. Vma22p and proteins cross-linked to Vma22p were treated with reducing agent to disrupt the disulfide cross-link and a second round of denaturing immunoprecipitation was performed with anti-Vma12p, anti-Vph1p, or anti-ALP antibodies to determine whether these specific proteins had cross-linked to Vma22p.
Specific proteins identified as cross-linked to Vma22p in wild-type yeast radioactively labeled for 30 min are shown in the left panel of Fig. 3 (lanes 1–3). The position of Vma22p is shown in lane 3, where anti-Vma22p antibody was used in both rounds of immunoprecipitation. Vma22p did not cross-link to Vma12p or Vph1p in the absence of DSP or when preimmune sera was used in the denaturing immunoprecipitation (Fig. 3, lanes 6–10). Fig. 3 (lane 2) also shows that Vma12p associated with Vma22p and was immunoprecipitated using anti-Vma12p antibodies in the second round of immunoprecipitation. The predicted molecular mass of Vma12p is 25.2 kD and that of Vma22p is 21 kD, consistent with the migration of these proteins subjected to SDS-PAGE. These data indicate that Vma22p and Vma12p interact directly in wild-type cells, and that this interaction is specific.
Figure 3.

Cross-linking radiolabeled assembly factor proteins and the 100-kD V-ATPase subunit. Wild-type cells were labeled with [35S]methionine for 30 min followed by an equivalent 5-min pulse of label. Cells were converted to spheroplasts, osmotically lysed, and then treated with or without 250 μM DSP at room temperature for 30 min. The cross-linker was quenched with the addition of 50 mM Tris, pH 7.5, and cross-linked proteins were TCA precipitated, acetone washed, and then resuspended in denaturing buffer without reducing agent. Proteins were immunoprecipitated with anti-Vma22p polyclonal sera or preimmune sera and resuspended in buffer plus 5% β-mercaptoethanol and reimmunoprecipitated using anti-Vma22p, anti-Vph1p, anti-Vma12p, or anti-ALP polyclonal sera. Proteins from the second immunoprecipitation were resuspended in sample buffer, separated by SDS-PAGE, and exposed to a phosphor screen.
Loss of either Vma12p or Vma22p prevents the assembly of the Vo subcomplex by affecting the stability of Vph1p (the 100-kD Vo subunit), and since Vma12p and Vma22p interact directly, we propose that Vma12p and Vma22p form an ER-localized assembly complex required for the biosynthesis of a functional V-ATPase. We set out to test this model by investigating whether Vma12p/ Vma22p and the V-ATPase subunit Vph1p interact directly. Using the same type of approach outlined above, Vma22p and proteins associated with Vma22p were first immunoprecipitated from cross-linked radiolabeled cell extracts with a subsequent immunoprecipitation using anti-Vph1p antibodies after disrupting the cross-links. These data revealed that Vph1p was cross-linked to Vma22p (Fig. 3, lane 1).
Failure to immunoprecipitate newly synthesized alkaline phosphatase (ALP, another vacuolar membrane protein in transit through the ER; Klionsky and Emr, 1989) from cross-linked extracts using anti-Vma22p antibodies (Fig. 3, lane 4) demonstrates that the cross-linking and double immunoprecipitations have identified specific interactions between the assembly factors and the 100-kD V-ATPase subunit occurring early in the assembly pathway. The expected position of ALP on the gels was determined using anti-ALP antibodies for both rounds of immunoprecipitations (Fig. 3, lane 5).
Examination of the proteins immunoprecipitated by anti-Vma22p sera in the presence of cross-linking reagent and before the second immunoprecipitation revealed a complex pattern of protein bands of various sizes and intensity (data not shown). When anti-Vma12p antibodies were used in the initial immunoprecipitation (in place of the anti-Vma22p antibodies); both Vma22p and Vph1p were cross-linked to Vma12p (data not shown). However, we did observe that Vma22p was recovered less efficiently as a cross-linked complex when Vma12p was used for the first immunoprecipitation compared with the amount of Vma12p isolated when Vma22p was used as the first immunoprecipitation. Therefore, in all subsequent cross-linking experiments we routinely immunoprecipitated Vma22p first.
Vph1p Transiently Interacts with the Vma12p/Vma22p Assembly Complex
Both Vma12p and Vma22p have been localized to the membranes of the ER (Hill and Stevens, 1995; Jackson and Stevens, 1997), yet Vph1p localizes with the V-ATPase to the vacuolar membrane (Manolson et al., 1992). To address the nature of the interaction between Vma12p, Vma22p and Vph1p we first performed a kinetic analysis of the interactions between these three proteins. Using radiolabeled wild-type cell extract treated with DSP, we examined the interaction of the assembly complex with Vph1p. Cells were pulse labeled for a short time (5 min) then samples were removed at various times during the chase. Cells were converted to spheroplasts, osmotically lysed, treated with DSP and these proteins were immunoprecipitated with anti-Vma22p antibodies as described above. The immunoprecipitated proteins from each timepoint sample were divided into aliquots, the cross-links were broken using a reducing agent, and a second round of immunoprecipitation was performed using anti-Vma22p, anti-Vma12p, and anti-Vph1p antibodies.
Immediately after the short radiolabel pulse, we observed the interaction of newly synthesized Vph1p and Vma12p with Vma22p (Fig. 4, 0 min chase), similar to what was observed previously (Fig. 3). The interaction of Vma12p with Vma22p remained constant over the entire 160-min chase period, as revealed by the constant amount of Vma12p detected in the second immunoprecipitation. These data are consistent with the model that Vma12p and Vma22p form a stable complex in the ER membrane. Unfortunately, we were unable to detect Vma21p (the third ER-localized V-ATPase assembly factor; Hill and Stevens, 1994) cross-linked to either Vma12p or Vma22p in wild-type cells expressing epitope-tagged Vma21p. We were able to immunoprecipitate Vma21p from denatured extracts in the absence of DSP but apparently, treatment with the cross-linker rendered Vma21p nonimmunoprecipitable, even after the cross-links were reduced (Graham, L. A., unpublished results).
Figure 4.

Time course of interaction between Vma12p/Vma22p and Vph1p. Wild-type cells were labeled with [35S]methionine for 5 min and chased in the presence of excess unlabeled methionine. Samples were removed at the indicated times and cell lysates treated with cross-linking agent DSP as described in Fig. 3. Two rounds of immunoprecipitations were performed first using anti-Vma22p then followed by either anti-Vma22p, anti-Vma12p, or anti-Vph1p. The proteins of the immunoprecipitated complexes were separated by SDS-PAGE and gels exposed to a phosphor screen.
The amount of Vph1p interacting with Vma22p was at its highest in the early timepoint (Fig. 4, 0 min chase), and based on comparison of radioactively labeled and cross-linked Vph1p after immunoprecipitation with anti-Vph1p sera alone, we estimate that as much as 20% of radiolabeled Vph1p is recovered in a cross-linked complex with Vma22p at this early timepoint. The level of Vph1p that could be cross-linked to Vma22p decreased quickly during the subsequent chase. These results suggest that Vph1p interacts transiently with the Vma12p/Vma22p assembly complex on its way to the vacuolar membrane. The interaction between the Vma12p/Vma22p assembly complex and Vph1p is estimated to occur with a half-life of less than 5 min. It is important to note that the decay of the Vph1p band with time in Fig. 4 does not reflect degradation, since Vph1p had a half-life of >400 min in these cells. The very short half-life of this interaction argues against the possibility of the Vma12p/Vma22p assembly complex remaining associated with the assembled V-ATPase complex until it reaches the vacuole. Rather, the transient nature of the interaction supports the idea that these are specific interactions observed between the assembly factors and Vph1p during assembly of the V-ATPase in the ER.
The kinetics of the interaction between the V-ATPase subunit (Vph1p) and the Vma12p/Vma22p assembly complex suggest that the interaction may occur only for the duration of the assembly of the membrane sector of the V-ATPase (Vo). According to this model, the Vma12p/ Vma22p assembly complex would release the assembled Vo sector, and the Vo sector and/or assembled V-ATPase complex would be loaded into ER-derived vesicles bound for the Golgi complex. To determine whether Vph1p would continue to associate with the assembly complex in the absence of membrane traffic out of the ER, we used a temperature sensitive allele, sec12-4, to block protein exit from the ER at the restrictive temperature (Nakano et al., 1988). The sec12-4 cells were grown at 23°C before the addition of [35S]methionine, and an aliquot of the cells was shifted to the nonpermissive temperature (37°C for 15 min) to induce the temperature-sensitive block. Cells were radiolabeled for 5 min and chased for various times after the addition of excess unlabeled methionine, treated with DSP, and then immunoprecipitated first using anti-Vma22p antibodies, and subsequently using anti-Vph1p antibodies, as described for the previous experiment (see Materials and Methods). The lower panel in Fig. 5, shows the same transient interaction between Vma22p and Vph1p in the sec12-4 cells at the permissive temperature of 23°C as observed for wild-type cells (Fig. 4). By contrast, the interaction between Vma22p and Vph1p in sec12-4 cells at the nonpermissive temperature, when exit from the ER is blocked, did not change significantly during the 160-min chase time (Fig. 5, top). Induction of the sec block was confirmed by parallel immunoprecipitation of CPY from samples removed at the 0 and 80 min timepoint. The sec12-4 cells radiolabeled and chased at 23°C showed wild-type processing of CPY whereas cells labeled and chased at 37°C failed to process CPY, it remained in the ER precursor form (data not shown), confirming protein exit from the ER was blocked. These data indicate that the interaction between the Vma12p/Vma22p assembly complex and newly synthesized Vph1p is initiated in the ER, but that release of Vph1p from the complex is dependent on exit of the Vo sector or V-ATPase from the ER.
Figure 5.

Effect of blocking protein exit from the ER on the interaction between Vma12p/Vma22p and Vph1p. sec12-4 cells were grown at 23°C and shifted to 37°C for 15 min before labeling with [35S]methionine. Cells were labeled for 5 min, chased, lysates treated with DSP and double immunoprecipitated as described in Fig. 3, first using anti-Vma22p followed by anti-Vph1p polyclonal sera.
Vma12p and Vma22p Form a Stable Assembly Complex
The results presented thus far suggest that it may be possible to identify a stable Vma12p/Vma22p assembly complex without the use of a chemical cross-linker by density gradient fractionation. Microsomal (P13) membranes were prepared from wild-type yeast cells expressing a fully functional epitope-tagged form of Vma21p (Vma21p-HA, pKH28; Hill and Stevens, 1994). The membranes were diluted to give a protein concentration of 1 mg/ml and the membrane proteins solubilized in 1% Triton X-100. The solubilized membrane proteins were separated by centrifugation (16 h at 150,000 g) through a 5–15% sucrose density gradient (Chapman et al., 1996). The migration of the solubilized membrane proteins in the gradient was largely dependent on shape and molecular mass. Density gradient fractions were examined by immunoblot analysis for the presence of the assembly factors, Vma12p, Vma22p, and Vma21p, and the 100-kD V-ATPase subunit.
Immunoblot analysis revealed that the majority of Vma12p and Vma22p migrated to the same fractions in the sucrose density gradient fractionation of wild-type membranes (Fig. 6 A, fractions 6 and 7). We propose that the comigration of Vma12p and Vma22p represents the stable association of at least these two proteins, under conditions of gentle detergent solubilization, forming the assembly complex observed by cross-linking analysis (Figs. 3 and 4). Immunoblot analysis of the sucrose gradient fractions revealed the Vma21p was present only in fractions 1 and 2 (Fig. 6 A), well separated from the fractions containing Vma12p and Vma22p (fractions 6 and 7). It is possible that under the conditions used for solubilization (1% Triton X-100) and fractionation that Vma21p disassociated from the Vma21p/Vma22p assembly complex. Alternatively, Vma21p may not be part of the Vma12p/Vma22p assembly complex, and could function independently in the assembly of V-ATPase integral membrane subunits. Vma21p could assist the assembly of the V-ATPase either upstream or downstream of Vma12p/Vma22p or even possibly on a parallel pathway interacting with other Vo subunits.
Figure 6.
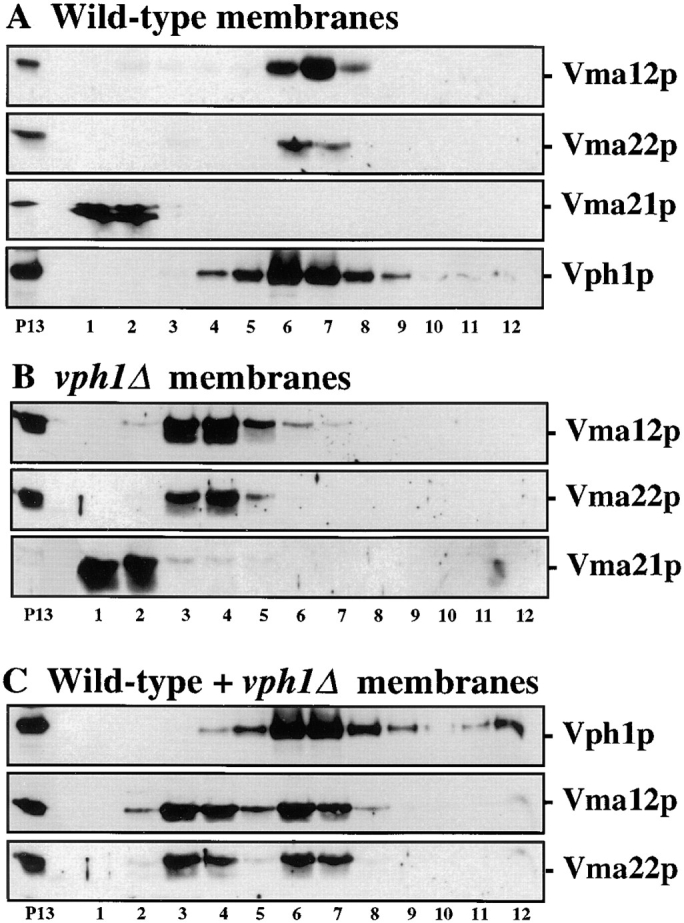
Sucrose gradient fractionation of detergent solubilized P13 membranes probed for assembly factors and Vph1p. P13 membranes prepared from wild-type and vph1Δ cells transformed with pKH28 (Vma21p::HA) were solubilized with 1% Triton X-100 and layered on top of a 5–15% linear sucrose density gradient. After centrifugation, twelve 1-ml fractions were collected from the top of the gradient and the proteins were precipitated by the addition of TCA. Equal proportions of each fraction were subjected to SDS-PAGE and immunoblot analysis. A portion of the solubilized membrane proteins was removed before gradient separation and analyzed with the gradient fractions (lane labeled P13) to aid in the identification of each band. (A) Wild-type fractions probed with either anti-Vma22p, anti-Vma12p, or anti-HA polyclonal sera and anti-Vph1p monoclonal antibodies. (B) Fractionated membranes from vph1Δ cells probed with either anti-Vma12p, anti-Vma22p or anti-HA antibodies. (C) Equal amounts of wild-type and vph1Δ P13 membranes were mixed, detergent solubilized (1% Triton X-100), and fractionated on a sucrose density gradient. Gradient fractions were probed with anti-Vma12p, anti-Vma22p, and anti-Vph1p antibodies.
Immunoblot analysis of the gradient fractionation of wild-type solubilized membranes with antibodies against the 100-kD V-ATPase subunit (Vph1p) is shown in the bottom panel of Fig. 6 A. Vph1p was present in several fractions of the gradient (fractions 3–12) with a peak of the protein present in fractions 6 and 7. Under steady state conditions, Vph1p is localized predominantly to the membranes of the vacuole as observed by immunofluorescence, but would be present in several types of complexes such as a Vo subcomplex, lacking the V1 catalytic subunits (Kane et al., 1992; Ho et al., 1993a ; Graham et al., 1995), as well as the fully assembled V-ATPase complex. Detergent solubilization of vacuolar membranes has been observed to dissociate the V1 subcomplex from a portion of the fully assembled V-ATPase complexes. In addition, the sucrose density gradients used in this work did not separate intact V-ATPase from the Vo subcomplex, thus explaining the broad sedimentation profile of Vph1p throughout the gradient.
The migration of the Vma12p/Vma22p assembly complex in the density gradients overlapped with fractions containing Vph1p either as a component of the V-ATPase complex or Vo subcomplex. Our cross-linking analysis demonstrated direct interaction between the Vma12p/ Vma22p assembly complex and the Vph1p V-ATPase subunit. Taken together, the migration of the Vma12p/ Vma22p complex appears to be due to its association with the V-ATPase subunit, Vph1p. Using pulse-label conditions we showed that a small radioactively labeled pool of newly synthesized Vph1p interacts with the Vma12p/ Vma22p assembly complex at very early time points and that the interaction is transient. The gradients reflect steady state cellular conditions in which Vph1p is continually being synthesized and inserted into the membranes of the ER where it interacts directly with the ER localized Vma12p/Vma22p assembly complex.
If the proteins present in the more dense gradient fractions (Fig. 6 A) represent a complex between the Vma12p/ Vma22p assembly factors and Vph1p, then the migration of these proteins should be altered in cells lacking Vph1p. To examine this possibility we fractionated detergent solubilized membranes prepared from yeast cells lacking Vph1p (KHY31; vph1Δ). Gradient fractions were probed with antibodies against Vma12p or Vma22p and the results are shown in Fig. 6 B. The proteins continued to cosediment, suggesting that Vma12p and Vma22p remain associated in a complex even in the absence of substrate (Vph1p). Although, in the absence of Vph1p, Vma12p, and Vma22p did not migrate as far into the density gradient (Fig. 6 B) compared with the fractionation profile of Vma12p and Vma22p in solubilized membranes prepared from wild-type cells (Fig. 6 A). Instead of migrating to fractions 6 and 7, the majority of the Vma12 and Vma22 proteins were found in fractions 3 and 4 in the absence of Vph1p. These data are consistent with the subcellular fractionation results since the membrane association of Vma22p, via Vma12p, was not altered in vph1Δ cells (Fig. 2). Even in the absence of Vph1p, Vma12p, and Vma22p continued to cosediment to fractions discrete from the fractions containing Vma21p (Fig. 6 B, fractions 3 and 4 vs fractions 1 and 2).
The differing fractionation profiles of the solubilized Vma12p/Vma22p assembly complex in samples prepared from wild-type or vph1Δ mutant membranes suggests that in steady state the majority of the ER localized assembly complex in wild-type cells is associated with newly synthesized Vph1p as it transits the ER enroute to the vacuole. Alternatively, the association between the Vma12p/Vma22p assembly complex and Vph1p could simply be the result of cell lysis and mixing of membrane compartments during detergent solubilization. To rule out the second possibility, membranes from wild-type and vph1Δ cells were isolated separately, diluted to a protein concentration of 1 mg/ml, equal volumes mixed together before detergent solubilization, then the solubilized membranes were fractionated on the density gradient. The gradient fractions were probed with antibodies against Vph1p, Vma12p, and Vma22p and the results are shown in Fig. 6 C.
The fractionation profile of the Vph1p in the mixed and solubilized membranes is similar to that seen for wild-type membranes alone, since vph1Δ membranes lack any Vph1 protein and addition of these membranes does not affect the migration of the wild-type Vph1p-containing complexes. If the fractionation profile of the Vma12p/Vma22p assembly complex from wild-type solubilized membranes is solely the result of associations formed after solubilization then we would expect the Vma12p/Vma22p from vph1Δ membranes (Fig. 6 B) to associate with the 100-kD from wild-type cells and shift to the more dense fractions. If the fractionation of Vma12p/Vma22p reflects interactions occurring presolubilization then we would predict a biphasic fractionation profile for these proteins representing their independent behavior in wild-type and vph1Δ cells. Probing gradient fractions from the mixed and solubilized membranes with antibodies against Vma12p and Vma22p revealed that these proteins were present in both the less dense fractions (fractions 3 and 4) similar to vph1Δ membranes and in the more dense fractions (fractions 6 and 7) similar to the wild-type membranes. As a further control, purified vacuolar membranes from wild-type cells and P13 membranes from vph1Δ cells were mixed and detergent solubilized, and subjected to density gradient centrifugation. This mixing control revealed that the Vma12 and Vma22 proteins continued to fractionate only to the less dense fractions whereas Vph1p sedimented to the more dense fractions (data not shown). Thus, the migration of the Vma12p/Vma22p assembly complex from wild-type membranes is the result of the in vivo association with the Vph1p assembling into the Vo subcomplex the membranes of the ER.
Vma21p Is Not Part of the Vma12p/Vma22p ER Assembly Complex
Phenotypic analysis of yeast cells lacking either Vma12p, Vma21p, or Vma22p, revealed that all three of these ER localized proteins are required for assembly of the yeast V-ATPase. Even though Vma21p is a small (77 amino acids) hydrophobic protein, we did succeed in constructing a COOH-terminal epitope-tagged Vma21p (Vma21p-HA, pKH28) that was able to fully complement the vma21Δ disruption strain (Hill and Stevens, 1994). As seen in Fig. 6, sucrose density gradient fractions probed with anti–HA antibodies revealed that Vma21p did not cosediment with Vma12p/Vma22p or the 100-kD V-ATPase subunit.
To investigate the role of Vma21p from another angle, we determined whether the loss of Vma21p affected the association between Vma12p, Vma22p, and Vph1p that we detected in wild-type cells. Using the same type of analysis as shown in Figs. 4 and 5, we examined the kinetics of the interaction between these proteins in vma21Δ cells by following the fate of newly synthesized, radiolabeled proteins. Fig. 7 shows that newly synthesized Vma12p and Vma22p interacted stably even when Vma21p was absent from the cells. The loss of Vma21p did not prevent the formation of the Vma12p/Vma22p assembly complex nor did it appear to destabilize the complex. It was possible to follow the fate of Vph1p in vma21Δ cells because although the loss of Vma21p destabilized Vph1p (Table II; Hill and Stevens, 1994), vma21Δ yeast cells synthesized Vph1p at the same rate as wild-type cells. We found that the loss of Vma21p did not affect the association of Vma12p, Vma22p, and Vph1p, indicating that Vma21p was not required for the association of Vma22p with newly synthesized Vph1p (Fig. 7). We have also observed that Vph1p associates with Vma12p/Vma22p in cells lacking any other integral membrane Vo subunit, such as the proteolipids Vma3p, Vma11p, and Vma16p (data not shown). These data suggest that Vma21p functions in an assembly role either on a separate but parallel pathway (e.g., with the proteolipids) to the Vma12p/Vma22p assembly complex or may even function downstream of Vma12p/Vma22p (but still in the ER).
Figure 7.

Cross-linking Vma12p to Vma22p and to Vph1p in vma21Δ mutant cells. Proteins in vma21Δ cells were pulse labeled for 5 min with [35S]methionine, cells extracts were treated with DSP and two rounds of immunoprecipitations were performed using anti-Vma22p followed by anti-Vma22p, anti-Vma12p, or anti-Vph1p polyclonal sera as described in Fig. 4.
Discussion
Resident ER proteins play a variety of roles including aiding translocation, folding, assembly, and targeting of proteins destined for other cellular compartments. In yeast, ER proteins such as Kar2p and calnexin function as general molecular chaperones for a range of proteins in transit through the ER (Gething and Sambrook, 1992; Brodsky and McCracken, 1997). In this work, we examined the role of two ER-localized proteins, Vma12p and Vma22p, which are required for the assembly of the integral membrane subunits of the V-ATPase. We have shown that Vma12p and Vma22p associate to form a stable ER-localized assembly complex that interacts transiently with Vph1p (the 100-kD integral membrane V-ATPase subunit). We also found that Vma12p/Vma22p remain associated with Vph1p as long as the newly assembled V-ATPase remains in the ER. Finally, we report that whereas Vma21p is a third assembly factor absolutely required for V-ATPase assembly, this protein appears not to be a component of the Vma12p/Vma22p complex.
ER Localized Vma12p/Vma22p Assembly Complex
Vma12p, Vma21p, and Vma22p, have been shown previously to reside in or associate with the ER (Hirata et al., 1993; Hill and Stevens, 1994, 1995; Jackson and Stevens, 1997). Whereas Vma12p and Vma21p each possess two potential transmembrane domains, Vma22p is a hydrophilic protein lacking any obvious transmembrane domains. Preliminary subcellular fractionation analysis suggested a role for Vma12p in mediating the membrane association of Vma22p (Hill and Stevens, 1995). We have extended the subcellular fractionation analysis and found that membrane association of Vma22p was only dependent on Vma12p, and not on either Vma21p or any Vo subunit.
We used the reversible cross-linking reagent DSP to determine that Vma12p and Vma22p interact directly within the membranes of the ER. Vma12p and Vma22p associated shortly after synthesis and this association was stable (i.e., over several cell divisions). We observed that the Vma12p/Vma22p complex was stable even after solubilization of ER membranes with 1% Triton X-100 and fractionation on a sucrose density gradient in the absence of cross-linking reagent. The loss of Vma21p, the third assembly factor, did not affect either the formation or the stability of the Vma12p/Vma22p complex as determined by subcellular fractionation, chemical cross-linking, and density gradient fractionation. These data indicate that Vma12p and Vma22p form a complex in the ER whose formation and stability requires neither Vma21p nor assembly substrates (Vo subunits).
Direct Interaction of Vma12p/Vma22p With Vph1p
In wild-type cells the 100-kD subunit of the V-ATPase (Vph1p) is a very stable protein with a half-life spanning several cell generations. In contrast, yeast cells lacking any one of the three assembly factors or any other Vo subunit rapidly degrade Vph1p in the ER. Our cross-linking analysis revealed a direct interaction between the Vma12p/ Vma22p assembly complex in the ER and newly synthesized Vph1p. We conclude that the interaction of Vph1p with the assembly complex stabilizes this Vo subunit in the ER allowing it to assemble into the Vo subcomplex.
Our kinetic analysis of the interaction between the Vma12p/Vma22p assembly complex and the 100-kD subunit in wild-type cells revealed that the interaction was transient, with a half-life of ∼5 min. This transient interaction presumably reflects the length of time required to assemble the Vo subcomplex and transport it from the ER. When exit from the ER was blocked with a conditional sec mutation (sec12-4), the interaction between Vma12p/ Vma22p and Vph1p was significantly stabilized. Therefore, we conclude that release of Vph1p from the assembly complex requires exit from the ER, and exit from the ER is possible only if the Vo subcomplex is fully assembled.
The interaction between Vph1p and the Vma12p/ Vma22p assembly complex was confirmed by fractionation analysis of detergent solubilized membranes. Fractionation of proteins from wild-type cells revealed the cosedimentation of Vma12p/Vma22p and Vph1p. Almost all the Vma12p/Vma22p was present in the denser fractions due to the association of this complex with Vph1p. In the absence of Vph1p, the Vma12p/Vma22p complex migrated to less dense fractions. From these data we conclude that Vma12p/Vma22p and Vph1p associate in the ER during assembly of the V-ATPase complex.
Determining the role of the Vma21p in the assembly of the V-ATPase has been difficult. Vma21p is a small, extremely hydrophobic protein localized to the membranes of the ER and required for V-ATPase assembly (Hill and Stevens, 1994). Whereas it was possible to immunoprecipitate Vma21p::HA with anti-HA antibodies, we never observed cross-linking of Vma21p::HA to either Vma12p or Vma22p. One possibility is that Vma21p does in fact associate with Vma12p/Vma22p, but we are unable to detect this interaction. Consistent with this interpretation is the observation that reaction of DSP with Vma21p-HA was found to mask the HA epitope (Graham, L.A., unpublished results), making it impossible to immunoprecipitate the cross-linked product (even after breaking the cross-links with reducing agent). In fact, three of the four lysine residues (the primary target for DSP) in Vma21p are immediately adjacent to the HA-epitope tag. An alternative interpretation of the cross-linking results is that Vma21p does not actually associate directly with the Vma12p/ Vma22p complex, but instead functions either before or after the Vma12p/Vma22p complex in aiding the assembly of the V-ATPase.
We found that Vma21p fractionated away from the Vma12p/Vma22p complex when detergent solubilized proteins were separated on a density gradient. Although we cannot rule out the possibility that the solubilization and fractionation conditions used disrupted the tenuous interaction of Vma21p with the Vma12p/Vma22p assembly complex, we propose that Vma21p functions in a pathway parallel to the Vma12p/Vma22p assembly complex as described below, and may not interact directly with Vph1p. It is also possible that Vma21p functions to escort the V-ATPase complex from the ER to the Golgi, and is then retrieved back to the ER by virtue of its COOH-terminal di-lysine motif (Hill and Stevens, 1994).
Unlike general molecular chaperones such as Kar2p/BiP (Gething and Sambrook, 1992), Vma12p, Vma21p, and Vma22p represent a class of ER resident proteins dedicated to the assembly of a specific enzyme complex, the V-ATPase. These proteins are not required for the localization or stability of other membrane proteins transiting the secretory pathway (Hill and Stevens, 1994, 1995). Several other members of this class of dedicated ER proteins have been identified in yeast; Shr3p is required specifically for the exit of the plasma membrane amino acid permeases from the ER (Ljungdahl et al., 1992; Kuehn et al., 1996) and correct localization of the Golgi protein α1,2-mannosyltransferase requires the function of Anp1p in the ER (Chapman and Munro, 1994).
Model For V-ATPase Assembly in the ER
The identification of three ER localized proteins dedicated to the assembly of the yeast V-ATPase indicates that the assembly process is carefully orchestrated. Failure to properly assemble the Vo subcomplex in the ER targets at least one Vo subunit, Vph1p, for rapid degradation. The proposed model for the assembly of the V-ATPase involves two parallel pathways describing Vo sector assembly occurring in the ER (Fig. 8). The first step in the assembly pathway would involve the association of the fully translocated and folded Vph1p with the Vma12p/Vma22p assembly complex in the ER membrane. Based on the finding that Vph1p associates with Vma12p/Vma22p independent of Vma21p or any other Vo subunit, we propose a second parallel (or downstream) step involving the third assembly factor Vma21p in the association of Vma3p, Vma11p, and Vma16p to form the proton-translocating channel. The proton pore of the V-ATPase is a hetero-oligomeric complex composed of three different, but related, highly hydrophobic subunits (the proteolipids Vma3p, Vma11p, and Vma16p; Hirata et al., 1997). Because the proteolipids are so difficult to work with experimentally, it remains unclear what role Vma12p/Vma22p or Vma21p play in the assembly of the proteolipid proton pore complex.
Figure 8.

Model of V-ATPase assembly in the ER. Vma12p and Vma22p associate to form a stable assembly complex that interacts directly with the 100-kD V-ATPase subunit (Vph1p) in the ER membrane. The three proteolipids (Vma3p, Vma11p, and Vma16p), components of the proton pore, must assemble together with Vph1p in the ER. Release of Vph1p by the assembly complex requires exit of the V-ATPase subunit from the ER. Since Vph1p is unable to exit the ER in the absence of the other Vo subunits, we propose the Vo complex must assemble completely before it can exit the ER and proceed to the vacuole. A precise role for the assembly factor Vma21p is not yet clear.
After the Vo subunits have associated with the appropriate assembly factors, we propose that Vph1p associates with Vma3p, Vma11p, and Vma16p in the ER membrane. In support of this model are the findings that cells unable to assemble the proton pore (due to the absence of Vma3p, Vma11p, or Vma16p) also rapidly degrade Vph1p in the ER (Table II). Sometime during or after the step in which Vph1p associates with the proton pore subunits, we propose that the fifth Vo subunit, Vma6p, joins the complex. The fully assembled Vo subcomplex, regardless of whether the V1 sector is associated or not (Kane et al., 1992), would then be competent to exit the ER. Our future efforts will be focused on the role of Vma21p in V-ATPase assembly and on determining the interactions between the various Vo subunits in the ER membrane.
Acknowledgments
We thank Nia Bryant for critical reading of this manuscript.
This work was supported by a grant from the National Institutes of Health (38006) to T.H. Stevens and Human Frontier Science Program Organization Grant RG-389/94M to T.H. Stevens.
Abbreviations used in this paper
- ALP
alkaline phosphatase
- CPY
carboxypeptidase Y
- DAPI
4′,6′-diamidino-2-phenylindole
- DSP
dithio-bis(succinimidylpropionate)
- V-ATPase
vacuolar proton-translocating ATPase
- V1
peripheral catalytic sector of the V-ATPase
- Vo
integral membrane sector of the V-ATPase
- VMA
vacuolar membrane ATPase
- VPH
vacuolar pH
- YEPD
1% yeast extract, 2% peptone, and 2% dextrose
Footnotes
Dedicated to the memory of Wesley C. Graham.
References
- Bachhawat AK, Manolson MF, Murdock DG, Garman JD, Jones EW. The VPH2 gene encodes a 25 kD protein required for activity of the yeast vacuolar H+-ATPase. Yeast. 1993;9:175–184. doi: 10.1002/yea.320090208. [DOI] [PubMed] [Google Scholar]
- Bauerle C, Ho MN, Lindorfer MA, Stevens TH. The Saccharomyces cerevisiae VMA6 gene encodes the 36-kD subunit of the vacuolar H+-ATPase membrane sector. J Biol Chem. 1993;268:12749–12757. [PubMed] [Google Scholar]
- Brodsky JL, McCracken AA. ER-associated and proteosome-mediated protein degradation: how two topologically restricted events came together. Trends Cell Biol. 1997;7:151–156. doi: 10.1016/S0962-8924(97)01020-9. [DOI] [PubMed] [Google Scholar]
- Chapman RE, An S, Edwardson JM, Jahn R. A novel function for the second C2 domain of synaptotagamin; Ca2+-triggered dimerization. J Biol Chem. 1996;271:5844–5849. doi: 10.1074/jbc.271.10.5844. [DOI] [PubMed] [Google Scholar]
- Chapman RE, Munro S. The functioning of the yeast Golgi apparatus requires an ER protein encoded by ANP1, a member of a new family of genes affecting the secretory pathway. EMBO (Eur Mol Biol Organ) J. 1994;13:4896–4907. doi: 10.1002/j.1460-2075.1994.tb06817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty RD, Kane PM. Partial assembly of the yeast vacuolar H+-ATPase in mutants lacking one subunit of the enzyme. J Biol Chem. 1993;268:16845–16851. [PubMed] [Google Scholar]
- Forgac M. Structure and function of vacuolar class of ATP-driven proton pumps. Physiol Reviews. 1989;69:765–796. doi: 10.1152/physrev.1989.69.3.765. [DOI] [PubMed] [Google Scholar]
- Gething MJ, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Graham LA, Hill KJ, Stevens TH. VMA8 encodes a 32-kD V1 subunit of the Saccharomyces cerevisiae vacuolar H+-ATPase required for the function and assembly of the enzyme complex. J Biol Chem. 1995;270:15037–15044. doi: 10.1074/jbc.270.25.15037. [DOI] [PubMed] [Google Scholar]
- Hill KJ, Stevens TH. Vma21p is a yeast membrane protein retained in the endoplasmic reticulum by a di-lysine motif and is required for the assembly of the vacuolar H+-ATPase complex. Mol Biol Cell. 1994;5:1039–1050. doi: 10.1091/mbc.5.9.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill KJ, Stevens TH. Vma22p is a novel endoplasmic reticulum associated protein required for assembly of the yeast vacuolar H+-ATPase complex. J Biol Chem. 1995;270:22329–22336. doi: 10.1074/jbc.270.38.22329. [DOI] [PubMed] [Google Scholar]
- Hirata R, Graham LA, Takatsuki A, Stevens TH, Anraku Y. VMA11 and VMA16 encode second and third proteolipid subunits of the Saccharomyces cerevisiae vacuolar membrane H+-ATPase. J Biol Chem. 1997;272:4795–4803. doi: 10.1074/jbc.272.8.4795. [DOI] [PubMed] [Google Scholar]
- Hirata R, Umemoto N, Ho MN, Ohya Y, Stevens TH, Anraku Y. VMA12 is essential for assembly of the vacuolar H+-ATPase subunits onto the vacuolar membrane in Saccharomyces cerevisiae. . J Biol Chem. 1993;268:961–967. [PubMed] [Google Scholar]
- Ho MN, Hill KJ, Lindorfer MA, Stevens TH. Isolation of vacuolar membrane H+-ATPase-deficient yeast mutants; the VMA5 and VMA4 genes are essential for assembly and activity of the vacuolar H+-ATPase. J Biol Chem. 1993a;268:221–227. [PubMed] [Google Scholar]
- Ho MN, Hirata R, Umemoto N, Ohya Y, Takatsuki A, Stevens TH, Anraku Y. b. VMA13 encodes a 54-kD vacuolar H+-ATPase subunit required for the activity but not the assembly of the enzyme complex in Saccharomyces cerevisiae. . J Biol Chem. 1993;268:18286–18292. [PubMed] [Google Scholar]
- Horazdovsky BF, Emr SD. The VPS16gene product associates with a sedimentable protein complex and is essential for vacuolar protein sorting in yeast. J Biol Chem. 1993;268:4953–4962. [PubMed] [Google Scholar]
- Jackson DD, Stevens TH. VMA12 encodes a yeast endoplasmic reticulum protein required for vacuolar H+-ATPase assembly. J Biol Chem. 1997;272:25928–25934. doi: 10.1074/jbc.272.41.25928. [DOI] [PubMed] [Google Scholar]
- Kane PM, Kuehn MC, Howald-Stevenson I, Stevens TH. Assembly and targeting of peripheral and integral membrane subunits of the yeast vacuolar H+-ATPase. J Biol Chem. 1992;267:447–454. [PubMed] [Google Scholar]
- Klionsky D, Emr S. Membrane protein sorting: biosynthesis, transport and processing of yeast vacuolar alkaline phosphatase. EMBO (Eur Mol Biol Organ) J. 1989;8:2241–2250. doi: 10.1002/j.1460-2075.1989.tb08348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn MJ, Schekman R, Ljungdahl PO. Amino acid permease require COPII components and the ER resident membrane protein Shr3p for packaging into transport vesicles in vitro. . J Cell Biol. 1996;135:585–595. doi: 10.1083/jcb.135.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungdahl PO, Gimeno CJ, Styles CA, Fink GR. SHR3: a novel component of the secretory pathway specifically required for localization of amino acid permeases in yeast. Cell. 1992;71:463–478. doi: 10.1016/0092-8674(92)90515-e. [DOI] [PubMed] [Google Scholar]
- Manolson MF, Proteau D, Preston RA, Stenbit A, Roberts BT, Hoyt MA, Preuss D, Mulholland J, Botstein D, Jones EW. The VPH1 gene encodes a 95-kD integral membrane polypeptide required for in vivoassembly and activity of the yeast vacuolar H(+)-ATPase. J Biol Chem. 1992;267:14294–14303. [PubMed] [Google Scholar]
- Markwell MA, Haas SM, Bieber LL, Tolbert NE. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal Biochem. 1978;87:206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- Nakano A, Brada D, Schekman R. A membrane glycoprotein, Sec12p, required for protein transport from the endoplasmic reticulum to the Golgi apparatus in yeast. J Cell Biol. 1988;107:851–863. doi: 10.1083/jcb.107.3.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothwehr SF, Conibear E, Stevens TH. Golgi and vacuolar membrane proteins reach the vacuole in vps1mutant cells via the plasma membrane. J Cell Biol. 1995;129:35–46. doi: 10.1083/jcb.129.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper RC, Bryant NJ, Stevens TH. The membrane protein alkaline phosphatase is delivered to the vacuole by a route that is distinct from the VPS-dependent pathway. J Cell Biol. 1997;138:531–545. doi: 10.1083/jcb.138.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JH, Stevens TH. Protein sorting in yeast: mutants defective in vacuole biogenesis mislocalize vacuolar proteins into the late secretory pathway. Cell. 1986;47:1041–1051. doi: 10.1016/0092-8674(86)90819-6. [DOI] [PubMed] [Google Scholar]
- Stack JH, Herman PK, Schu PV, Emr SD. A membrane-associated complex containing the Vps15 protein kinase and the Vps34 PI 3-kinase is essential for protein sorting to the yeast lysosome-like vacuole. EMBO (Eur Mol Biol Organ) J. 1993;12:2195–2204. doi: 10.1002/j.1460-2075.1993.tb05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens TH, Forgac M. Structure, function, and regulation of the vacuolar (H+)-ATPase. Annu Rev Cell Dev Biol. 1997;13:779–808. doi: 10.1146/annurev.cellbio.13.1.779. [DOI] [PubMed] [Google Scholar]
- Tomashek JJ, Sonnenburg JL, Artimovich JM, Klionsky DJ. Resolution of subunit interactions and cytoplasmic subcomplexes of the yeast vacuolar proton-translocating ATPase. J Biol Chem. 1996;271:10397–10404. doi: 10.1074/jbc.271.17.10397. [DOI] [PubMed] [Google Scholar]
- Yamashiro CT, Kane PM, Wolczyk DF, Preston RA, Stevens TH. Role of vacuolar acidification in protein sorting and zymogen activation: a genetic analysis of the yeast vacuolar proton-translocating ATPase. Mol Cell Biol. 1990;10:3737–3749. doi: 10.1128/mcb.10.7.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]