

Abstract
ERGIC-53, a homo-oligomeric recycling protein associated with the ER–Golgi intermediate compartment (ERGIC), has properties of a mannose-selective lectin in vitro, suggesting that it may function as a transport receptor for glycoproteins in the early secretory pathway. To investigate if ERGIC-53 is involved in glycoprotein secretion, a mutant form of this protein was generated that is incapable of leaving the ER. If expressed in HeLa cells in a tetracycline-inducible manner, this mutant accumulated in the ER and retained the endogenous ERGIC-53 in this compartment, thus preventing its recycling. Mistargeting of ERGIC-53 to the ER did not alter the gross morphology of the early secretory pathway, including the distribution of β′-COP. However, it impaired the secretion of one major glycoprotein, identified as the precursor of the lysosomal enzyme cathepsin C, while overexpression of wild-type ERGIC-53 had no effect on glycoprotein secretion. Transport of two other lysosomal enzymes and three post-Golgi membrane glycoproteins was unaffected by inactivating the recycling of ERGIC-53. The results suggest that the recycling of ERGIC-53 is required for efficient intracellular transport of a small subset of glycoproteins, but it does not appear to be essential for the majority of glycoproteins.
Keywords: cathepsin C, ER–Golgi intermediate compartment, glycoproteins, recycling, transport receptor
Secretory proteins are synthesized in the rough endoplasmic reticulum and packaged into COPII-coated transport vesicles budding from the transitional elements of the rough ER (Schekman and Orci, 1996). ER export involves transport signals (Kappeler et al., 1997; Nishimura and Balch, 1997), at least one of which directly binds COPII coats (Kappeler et al., 1997). Signal– coat interaction, either directly or indirectly, may lead to the known concentration of proteins during ER export (Mizuno and Singer, 1993; Balch et al., 1994). After losing their coat, COPII vesicles fuse with the ER–Golgi intermediate compartment (ERGIC)1 (according to the stable compartment/vesicle hypothesis) or fuse with one another to form the ERGIC (according to the maturation hypothesis) (for reviews see Bannykh and Balch, 1997; Farquhar and Hauri, 1997; Glick et al., 1997). The ERGIC is now best defined by the marker protein ERGIC-53 (Hauri and Schweizer, 1992, 1997) and in part also harbors the small GTPases rab1 (Griffiths et al., 1994; Saraste et al., 1995) and rab2 (Chavrier et al., 1990), the KDEL receptor (Griffiths et al., 1994), the SNARE syntaxin 5/Sed5 and rbet1 (Banfield et al., 1994; Zhang et al. 1997), and p23 (Rojo et al., 1997).
Protein transport from ERGIC to cis-Golgi is mediated by a COPI-dependent step, either reflecting a vesicular step or maturation (Bannykh and Balch, 1997; Presley et al., 1997; Scales et al., 1997). The ERGIC is the first site of segregation of anterograde and retrograde transported proteins (Aridor et al., 1995; Itin et al., 1995a ; Tang et al., 1995). Retrograde transport from ERGIC to ER as well as from cis-Golgi to ERGIC/ER is mediated by COPI-coated vesicles (Gaynor et al., 1994; Letourneur et al., 1994; Townsley and Pelham, 1994; Aridor et al., 1995). Some of the retrograde transported proteins are packaged into COPI-coated vesicles by direct interaction of their cytosolic dilysine ER retrieval signal with COPI components (Cosson and Letourneur, 1994; Kappeler et al., 1997; Tisdale et al., 1997).
It appears unlikely that all membrane proteins directly interact with COPII during ER exit, and secretory proteins, for reason of topology, are unable to bind cytosolic coats. Signal-mediated ER export of these proteins therefore requires transport receptors, the nature of which has remained elusive. Several features of the lectin ERGIC-53 suggest that this nonglycosylated membrane protein may operate as a transport receptor for glycoproteins in the early secretory pathway (Itin et al., 1996). ERGIC-53 is synthesized as a homo-oligomeric type I membrane protein (Schweizer et al., 1988; Schindler et al., 1993) and constitutively recycles between the ERGIC and ER (Lippincott-Schwartz et al., 1990; Saraste and Svensson, 1991; Aridor et al., 1995; Itin et al., 1995a ). The recycling of ERGIC-53 is controlled by a complex interplay of targeting signals, including a cytosolic dilysine ER retrieval signal interacting with COPI vesicle coat proteins (Schindler et al., 1993; Itin et al., 1995b ; Kappeler et al., 1997; Tisdale et al., 1997) and a cytosolic ER exit determinant binding to COPII vesicle coat proteins (Kappeler et al., 1997). The luminal domain of ERGIC-53 comprises a carbohydrate recognition domain (CRD) that exhibits homologies to the mammalian post-Golgi vesicle protein VIP36 and to leguminous lectins over a stretch of ∼200 amino acids (Fiedler and Simons, 1994). The CRD is more than 90% homologous in ERGIC-53 from Xenopus, rat, and man (Lahtinen et al., 1996). ERGIC-53 preferentially binds mannose in a calcium-dependent way, and mutating conserved amino acids in the CRD known to be essential for carbohydrate binding in leguminous lectins abolished mannose binding (Arar et al., 1995; Itin et al., 1996). These in vitro binding studies established ERGIC-53 as a mannose-selective lectin. The carbohydrate specificity in conjunction with the recycling properties led us to propose that ERGIC-53 may collect newly synthesized high-mannose glycoproteins in the ER and thereby facilitate their transport to the ERGIC (Itin et al., 1996).
In the present study, we asked if ERGIC-53 is required for glycoprotein secretion. To test this, we studied protein secretion in HeLa cells overexpressing a mutant of ERGIC-53 that is unable to leave the ER and compared it with that of HeLa cells overexpressing wild-type ERGIC-53. By using a tetracycline-driven inducible system for transient expression of stably transfected proteins, we observed that mistargeting of ERGIC-53 to the ER impairs the secretion of a major glycoprotein identified as procathepsin C.
Materials and Methods
Reagents
Antibodies used were: mAb G1/93 against human ERGIC-53 (Schweizer et al., 1988), mAb G1/296 against human p63 (Schweizer et al., 1993, 1995), mAb G1/139 against human Lamp1 (Schweizer et al., 1988), mAb G1/221 against human transferrin receptor (produced by the mouse hybridoma technique and confirmed by peptide sequencing of the purified antigen [Schweizer et al., 1988]), mAb HBB 3/153 against human small intestinal aminopeptidase N (Hauri et al., 1985), mAb A1/118 against monkey GPP130 (Linstedt et al., 1997), mAb G1/133 against human giantin (Linstedt and Hauri, 1993), mAb 9E10 against a c-myc epitope (American Type Culture Collection, Rockville, MD: ATCC CRL 1729), an mAb against human α-glucosidase (kindly provided by J. Fransen, University of Nijmegen, The Netherlands), an mAb against human cathepsin D (Calbiochem, La Jolla, CA), rabbit polyclonal antisera against human ERGIC-53 (Itin et al., 1995b ), and rat cathepsin C (kindly provided by E. Kominami, Juntendo University, Tokyo, Japan). The neoglycoprotein fluorescein- labeled (FITC) Man-BSA was kindly provided by A.C. Roche and M. Monsigny (University Orléans, France) (Itin et al., 1996). Concanavalin A (Con A)–Sepharose was from Pharmacia Biotech (Uppsala, Sweden). Protease inhibitors (used concentrations: 1 μg/ml pepstatin, 1 μg/ml aprotinin, 1 μg/ml benzamidin, 1 μg/ml antipain, and 0.2 mM phenylmethylsulfonyl fluoride) and tetracycline were from Sigma Chemical Co. (St. Louis, MO), and [35S]methionine was from New England Nuclear (Boston, MA).
Cells
HtTA-1 HeLa cells expressing the chimeric tetracycline (tet) regulatable transcription activator were kindly provided by H. Bujard (Zentrum für Molekulare Biologie, Heidelberg, Germany). They were cultured in DME supplemented with 10% heat-inactivated FCS, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 400 μg/ml geneticin (GIBCO BRL, Life Technologies, Basel, Switzerland). FAO cells were cultured as HeLa cells but without geneticin.
cDNA Constructs
Myc-tagged ERGIC-53 in pECE vector (p53cmyc; Itin et al., 1995a ) is termed KKFF in this study. An ERGIC-53 construct with its cytoplasmic tail mutated to R2A6KKAA in pECE (Itin et al.,1995b) was cut with AccI/XbaI, and the small fragment was subcloned into the AccI/XbaI site of KKFF. This construct is termed KKAA. Both KKFF and KKAA were cloned as SalI/XbaI fragments into the EcoRI site of the tetracycline- inducible expression plasmid pUHD10-3 (kindly provided by H. Bujard). This plasmid carries a human cytomegalovirus minimal promoter fused to seven repetitive tet operator sequences (tetO) upstream of the cloning site (Gossen and Bujard, 1992; Resnitzky et al., 1994).
Generation of Stably Transformed HtTa-1 Cell Lines
The procedure of Damke et al. (1995) was followed. Briefly, 1 × 106 cells were seeded into a 10-cm culture dish the day before transfection. After a 4-h preincubation with 2 μg/ml tet, the cells were transfected with the cDNA (20 μg) encoding either KKFF or KKAA under the control of the tTA promoter (pUHD10-3 plasmid) together with the plasmid pBSpac (1 μg) containing the puromycin resistance gene (de la Luna et al., 1988) by using the calcium phosphate precipitation method (Sambrook and Pollack, 1974). Clones were selected and expanded in the presence of 2 μg/ml tet, 500 ng/ml puromycin (Fluka AG, Buchs, Switzerland), and 400 μg/ml geneticin (GIBCO BRL). Puromycin-resistant clones were screened by immunofluorescence after incubation for 48 h in the absence of tet.
Immunofluorescence Microscopy
All experiments were carried out in eight-well multichamber glass slides (Miles Labs, Naperville, IL). The cells were plated and cultured for 48 h in the presence or absence (±) of tet and then fixed with paraformaldehyde, permeabilized with Triton X-100, and stained with antibodies using the indirect immunofluorescence procedure (Schweizer et al., 1988). For double immunofluorescence with FITC-Man-BSA and anti–ERGIC-53, the procedure of Itin et al. (1996) was used.
Metabolic Labeling, Immunoprecipitation, and Immunoblotting
Cells plated into 6-cm dishes were incubated ± tet (2 μg/ml) for 48 h and labeled with 100 μCi [35S]methionine for 30 min followed by a chase of varying length in the presence of 10 mM unlabeled methionine. ERGIC-53 was immunoprecipitated from Triton X-100–solubilized cells with mAb G1/93 against ERGIC-53 or mAb 9E10 against c-myc according to Schweizer et al. (1988). For the immunoprecipitation of cathepsin C, PBS-washed cells were solubilized in 10 mM phosphate, pH 7.4, 1% Triton X-100, 0.2% SDS, 0.2 mM PMSF (solubilization buffer) for 1 h on ice and centrifuged at 50,000 g for 1 h at 4°C. The resulting supernatant was incubated for 2 h with 2 μl of anti–rat cathepsin C antibody prebound to protein A–Sepharose CL4B beads, and the beads were washed three times with solubilization buffer. Cathepsin C was also immunoprecipitated from either methanol-precipitated culture medium or from a glycoprotein fraction prepared from the culture medium (see below). Immunoisolated proteins were subjected to SDS-PAGE and visualized by fluorography using Kodak X-omat AR films (Rochester, NY).
Digestion with Endoglycosidases
Immunoprecipitated proteins were boiled for 3 min in endoglycosidase H (endo H) buffer (1% SDS, 100 mM Na-citrate, pH 5.3) or N-glycosidase F (endo F) buffer (50 mM Na2HPO4, pH 7.2, 10 mM EDTA, 0.1% SDS, 1% Triton X-100). After cooling to room temperature, the samples were supplemented with additional buffer containing protease inhibitors, incubated with 3 μl of endo H or endo F for 24 h at 37°C, and analyzed by SDS-PAGE.
Isolation and Analysis of Secreted Glycoproteins
The cells were cultured in 6-cm dishes for 48 h ± tet (2 μg/ml) and subjected to the pulse-chase [35S]methionine labeling procedure. The chase medium was serum-free DME containing 10 mM methionine. Tetracycline was present or not during the preincubation, labeling, and chase periods, depending on whether noninducible or inducible conditions were studied. At the end of the chase, the medium was collected and centrifuged for 10 min at 10,000 rpm in a microfuge to remove cell debris. The medium was then incubated under constant agitation for 1 h at room temperature with 30 μl Con A beads in 1 ml Con A buffer (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 1 mM of CaCl2, MnCl2 and MgCl2). The beads were washed four times with 1 ml Con A buffer, and glycoproteins were eluted by incubating the beads for 30 min at room temperature with 100 μl of 0.5 M α-methyl-mannoside in Con A buffer under constant agitation. Glycoproteins were separated by one-dimensional SDS-PAGE and visualized by fluorography. Alternatively, secreted glycoproteins were analyzed by two-dimensional gel electrophoresis. The glycoproteins were isolated with Con A beads, precipitated with methanol, and separated in the first dimension for 15 h on a 110-mm pH 3–10 linear isoelectric focusing immobiline gradient gel DryStrip according to the instructions of the manufacturer (Pharmacia Biotech). A 10% SDS–polyacrylamide gel run with constant voltage was used in the second dimension.
Isolation and Microsequencing of the 57-kD Glycoprotein
Cells overexpressing the KKFF construct were plated into six 15-cm dishes. At 80% confluence, the cells were incubated with serum-free DME for 24 h. The medium was collected, centrifuged, and incubated with Con A beads as above. Eluted glycoproteins were separated on a 7–10% SDS–polyacrylamide gel and blotted onto a polyvinyl difluoride membrane (Millipore Corp., Bedford, MA) in 25 mM Tris, 190 mM glycine, 10% methanol for 3 h. Glycoproteins were stained with Coomassie blue R-250 for 1 h and destained in 50% methanol. The 57-kD area was excised and incubated with trypsin (Fernandez et al., 1992). Tryptic peptides were separated on a 1.0-mm Vydac218TP51 reverse-phase column in 0.1% TFA-acetonitrile and sequenced on a pulse–liquid phase sequencer (model 477A; PE Applied Biosystems, Foster City, CA) (Jenö et al., 1995).
Subcellular Fractionation
HeLa cells were fractionated by Nycodenz gradient centrifugation (Kappeler et al., 1997). Briefly, cells of four ∼80% confluent 15-cm plates were homogenized with a ball-bearing homogenizer and the postnuclear supernatant was loaded onto a 13–29% (wt/vol) linear Nycodenz gradient. After centrifugation at 25,500 rpm for 3 h (model TST-28.17 rotor; Kontron Elektronik GmbH, Eching, Germany), 14 fractions were collected, and organelle marker proteins were analyzed by SDS-PAGE followed by immunoblotting with specific antibodies (ECL procedure).
Results
Approach to Study the Involvement of ERGIC-53 in Protein Secretion
If ERGIC-53 operates as a transport receptor for glycoproteins in the early secretory pathway, mutations affecting its localization and recycling properties may lead to impaired glycoprotein secretion. In this study, we have tested this notion by mislocalizing ERGIC-53 to the ER. We have previously described a cytoplasmic tail mutant of ERGIC-53 (termed GMAA) that is unable to leave the ER. In this mutant, the two COOH-terminal phenylalanines were mutated to alanines, which inactivates the ER exit determinant of ERGIC-53 (Kappeler et al., 1997). The similar mutant KKAA used in the present study behaves identically to GMAA. It also accumulates in the ER (Kappeler, F., and H.-P. Hauri, unpublished). KKAA and wild-type ERGIC-53, termed KKFF, were tagged with a c-myc epitope at the NH2 terminus. It is important to note that the two constructs differed only in their cytosolic domain, whereas the luminal and transmembrane domains were identical (Fig. 1 A). Since ERGIC-53 is a homo- oligomeric protein, overexpression of KKAA in the ER can be expected to block the recycling of endogenous ERGIC-53 by mixed oligomer formation and thereby inactivate its (putative) transport receptor function.
Figure 1.



Expression of KKFF and KKAA in stably transformed HtTA-1 HeLa cells. (A) Schematic representation of the two ERGIC-53 constructs used in this study. Both carry a c-myc epitope after the signal sequence cleavage site (Itin et al., 1995b ). KKFF is the wild-type ERGIC-53. KKAA has the luminal and transmembrane domains of ERGIC-53 and a mutated cytoplasmic domain. The amino acid residues of the cytosolic tails are indicated in the single letter code. (B) Tet-dependent inducibility of the KKFF and KKAA constructs. Cells were incubated for 48 h ± tet, pulsed for 30 min with [35S]methionine, and immunoprecipitated with an mAb against ERGIC-53, and the proteins were separated by 4–10% SDS-PAGE followed by fluorography. (C) Coimmunoprecipitation of the KKAA construct with endogenous ERGIC-53. KKAA cells were cultured in the absence of tet in medium containing [35S]methionine for the indicated times. Immunoprecipitation with an antibody against the myc epitope (M, present only in the KKAA construct) coprecipitated endogenous ERGIC-53 (e) with the KKAA construct (m) (lanes 2 and 4). For reference, immunoprecipitation was also done with G1/93 (G), which recognizes an epitope present on both the KKAA construct and endogenous ERGIC-53 (lanes 1 and 3). The unequal signals are due to the known low immunoprecipitation efficiency of the anti-myc antibody. Proteins were visualized by fluorography after SDS-PAGE.
To minimize a possible upregulation of a compensatory mechanism for the lack of functional ERGIC-53, the mutants were expressed in the tet-controlled inducible system of Gossen and Bujard (1992). cDNAs encoding KKFF or KKAA were subcloned into the plasmid pUHD10-3 downstream of a human cytomegalovirus minimal promoter fused to seven repetitive tetO sequences (Damke et al., 1996). This vector allows inducible expression of cDNAs when transfected into HtTA-1 HeLa cells expressing constitutively a chimeric transactivator (tTA). In these cells and in the absence of tet, tTA binding to tetO efficiently activates transcription of the gene. In contrast, when tet is present, a conformational change in the tet repressor domain prevents tTA from binding to the tetO sequences, and the promoter is silent. Both constructs were transfected into HtTA-1 cells together with a puromycin-selectable marker (de la Luna et al., 1988). After selection and subcloning under noninducible conditions, positive clones were screened by immunofluorescence microscopy for the expression of recombinant protein 48 h after withdrawal of tet using an mAb against the c-myc epitope exclusively present on the transgenes.
Inducible Expression of KKAA in the ER
To analyze the inducibility of the ERGIC-53 constructs, stably transfected clones were incubated ± tet for 48 h and metabolically labeled with [35S]methionine for 30 min. ERGIC-53 was immunoprecipitated with an mAb and analyzed by SDS-PAGE (Fig. 1 B). Under inducible conditions (−tet), the rate of synthesis of both constructs was several-fold higher when compared with the endogenous ERGIC-53: up to fivefold for KKFF and up to 10-fold for KKAA. Similar values were obtained when total amounts of ERGIC-53 proteins were quantified by immunoblotting with a polyclonal antibody against ERGIC-53 (not shown). In the presence of tet, no (KKFF) or very little (KKAA) recombinant protein was detectable, demonstrating the tightness of the expression system under noninducible conditions.
We next analyzed by subcellular fractionation if the envisaged mislocalization of the KKAA construct to the ER was achieved. Postnuclear supernatants of KKFF or KKAA cells cultured for 48 h ± tet were separated by Nycodenz gradient centrifugation, and the positions of ERGIC-53, p63 (a marker for the ER; Schweizer et al., 1995), and GPP130 (a marker for cis/medial-Golgi; Linstedt et al., 1997) were determined by SDS-PAGE followed by immunoblotting. Fig. 2 A shows that the gradient, run with the KKFF (+tet) sample, separated ER from cis/medial-Golgi. Membranes containing endogenous ERGIC-53 cofractionated in part with ER but also appeared in a position overlapping with cis/medial-Golgi membranes, presumably representing the ERGIC. In the absence of tet, the KKFF protein was induced (recognizable by its slightly reduced electrophoretic mobility due to the additional presence of a myc epitope), but its fractionation was identical to that of endogenous ERGIC-53 (Fig. 2 B). Obviously, overexpressed KKFF together with the endogenous ERGIC-53 distributed normally among the organelles of the early secretory pathway. In noninduced KKAA cells (+tet), the endogenous ERGIC-53 also partitioned between ER and post-ER fractions (Fig. 2 C). The signal in the ER tended to be stronger than in post-ER fractions, presumably because of low level expression of the KKAA protein, which accumulated in ER fractions and may have retained some endogenous ERGIC-53 by mixed oligomer formation. Under inducible conditions (−tet), both the KKAA construct and endogenous ERGIC-53 cofractionated with the ER marker at the bottom of the gradient, and there was no indication for the presence of KKAA in post-ER fractions (Fig. 2 D). We conclude that both the KKAA protein and endogenous ERGIC-53 are retained in the ER under these conditions.
Figure 2.

Subcellular fractionation on Nycodenz gradients of KKFF (A and B) and KKAA (C and D) HeLa cells cultured in the presence (+tet) or absence (−tet, 48 h) of tetracycline. Fractions were collected from bottom (fraction 1) to top (fraction 13). The amount of marker protein in each fraction was assessed by immunoblotting. The blots were quantified by phosphorimaging, and total counts in the gradient were set to 100% with the exception of the KKFF and the KKAA constructs in B and D, which were drawn in relation to the endogenously expressed ERGIC-53. open circles, endogenous ERGIC-53; filled circles, transfected ERGIC-53 construct; triangles, cis/medial-Golgi marker GPP130; X, ER marker p63.
Immunofluorescence microscopy confirmed the strikingly different localization of the KKAA and KKFF constructs (Fig. 3 A). KKAA gave a typical ER pattern, while KKFF resulted in a pattern that is characteristic for the ERGIC with some ER staining because of the continuous recycling of this construct (Schweizer et al., 1988; Kappeler et al., 1997). KKAA was unable to recycle, as tested by immunofluorescence analysis of its localization in cells incubated for 3 h at 15°C and after rewarming from the 15°C block. The pattern of KKAA was unchanged by these temperature manipulations, very much in contrast to that of KKFF, which changed in a way that is characteristic for ERGIC-53's recycling (not shown; Lippincott-Schwartz et al., 1990; Itin et al. 1995b ; Kappeler et al., 1997).
Figure 3.


(A) Binding of fluorescein-labeled mannosylated bovine serum albumin (man-BSA). Transformed HeLa cell lines expressing KKFF and KKAA were incubated 48 h in the absence of tet, fixed/ permeabilized with paraformaldehyde/Triton X-100, and tested for binding of man-BSA (Itin et al., 1996). KKFF (a) and KKAA (c) were visualized with anti-myc followed by a rhodamine-labeled secondary antibody. Man-BSA binding was photographed in the fluorescein channel (b and d). (B) Localization of β′-COP and the KKAA construct (± tet) by double immunofluorescence microscopy. KKAA cells were fixed with paraformaldehyde, permeabilized with saponine, and subjected to immunolabeling. β′-COP (a and c) was detected with a specific rabbit antibody followed by a fluorescein-conjugated secondary antibody. The KKAA construct (b and d) was visualized with an mAb against the c-myc epitope followed by a rhodamine-conjugated secondary antibody. Note that the distribution of β′-COP is identical regardless of whether the KKAA construct is expressed (a and b) or not (c and d). Arrows indicate ERGIC clusters. Bars: (A) 15 μm; (B) 7 μm.
Coimmunoprecipitation experiments were performed to test if the KKAA construct can form disulfide-linked oligomers with the endogenous ERGIC-53. KKAA cells were cultured in tet-free medium containing [35S]methionine for 3 or 15 h followed by immunoprecipitation with the mAb against the myc epitope, which was present on the KKAA construct but not on the endogenous ERGIC-53. Although the immunoprecipitation with anti-myc was less efficient than that with the anti–ERGIC-53 mAb, the anti-myc antibody precipitated some endogenous ERGIC-53 together with the KKAA construct, suggesting that KKAA can form disulfide-linked mixed oligomers with endogenous ERGIC-53 (Fig. 1 C).
The restriction of the KKAA construct to the ER is not due to misfolding since the kinetics of oligomerization of KKAA and KKFF to dimers and hexamers were identical and comparable to that of endogenous ERGIC-53 (not shown, Schweizer et al., 1988). Additional evidence for correct folding was obtained by lectin binding experiments. Lectin binding was tested by a morphological binding assay using FITC-Man-BSA (Itin et al., 1996). 48 h after incubation with or without tet, the cells were simultaneously fixed and detergent-permeabilized and then double-stained with anti-myc and FITC-Man-BSA. Fig. 3 A shows colabeling for c-myc and FITC-Man-BSA binding sites predominantly at post-ER locations for KKFF and in the ER for KKAA. It should be noted that most of the immunofluorescence signal originated from the overexpressed constructs, since mock transfected cells exhibited almost no FITC-Man-BSA binding (not shown; Itin et al., 1996).
The KKAA construct has a functional dilysine ER retrieval signal (Jackson et al., 1990, 1993). Such dilysine signals are known to bind COPI vesicle coat proteins (Cosson and Letourneur, 1994). Based on the observation that an antibody to the ERGIC-53 cytoplasmic domain blocked the membrane recruitment of COPI in permeabilized cells and inhibited the transport of vesicular stomatitis virus G (VSV G) from ERGIC to Golgi, it was suggested that ERGIC-53 may be the major receptor for COPI in ERGIC and cis-Golgi and thereby controls ER to Golgi transport (Tisdale et al., 1997). In view of these findings, overexpression of the KKAA construct in the ER in conjunction with the retention of endogenous ERGIC-53 in this compartment may impair the organization of the early secretory pathway, since under these conditions the ERGIC would be depleted of ERGIC-53 because of its uninhibited retrograde transport. We tested this prediction by comparing the distribution of the coatomer subunit β′-COP in induced and noninduced KKAA cells by double immunofluorescence microscopy using a polyclonal antibody against β′-COP and an mAb against the c-myc epitope to visualize induction of the KKAA construct (Fig. 3 B). Surprisingly, the distribution of β′-COP was identical with the two conditions: Anti–β′-COP gave a strong signal in the Golgi area and labeled discrete dots in the cytoplasm (Fig. 3 B, a and c) known to represent ERGIC clusters (Oprins et al., 1993; Itin et al., 1995a ; Rowe et al., 1996). The cis/medial-Golgi was also unchanged as visualized with an mAb against giantin (not shown). Thus, the mistargeting of ERGIC-53 does not result in gross morphological changes of the early secretory pathway.
Glycoprotein Secretion in KKAA and KKFF Cells
Pulse-chase experiments were performed to test whether overexpression of the KKAA and KKFF constructs affects the secretion of glycoproteins. After a 48-h incubation ± tet, stably transfected cells were labeled with [35S]methionine for 30 min followed by a chase for up to 3 h. Secreted glycoproteins were affinity-isolated from the medium with Con A–Sepharose, eluted with 0.5 M α-methylmannoside, and analyzed by SDS-PAGE and fluorography. Fig. 4 A shows the pattern of secreted glycoproteins obtained for the KKAA and the KKFF cell lines. Endo F digestion ascertained that these proteins were indeed glycoproteins since all of them underwent a mobility shift (not shown). Within 1 h of chase, a major 57-kD glycoprotein showed reduced secretion in cells overexpressing KKAA (−tet), while the other major secreted glycoproteins were unaffected under these conditions. Similarly, at 2 and 3 h of chase, the 57-kD protein was the only major glycoprotein affected by overexpressing KKAA. Laser scanner densitometry of the fluorograms was used to quantify the difference in 57-kD secretion of induced versus noninduced cells. As indicated in Fig. 4 B, the secretion of this protein was reduced eightfold after 1 h, fourfold after 2 h, and twofold after 3 h of chase. To investigate if the reduced amounts of the 57-kD protein in the medium of KKAA cells is due to a delay of secretion or to reduced biosynthesis, pulse-chase experiments with extended chase times were performed. Fig. 4 C shows that the difference in secretion of the 57-kD protein gradually disappeared with time. It was still detectable after 6 h (2.5-fold reduction) and 12 h (1.7-fold reduction) but no longer after 24 h of chase, supporting the interpretation of a secretion delay. Cells overexpressing the KKFF construct exhibited normal glycoprotein secretion including the 57-kD protein (Fig. 4 A). Prolonged tet withdrawal for up to 9 d resulted in the same secretion phenotype (not shown). Thus, mistargeting of ERGIC-53 to the ER resulted in a selective secretion phenotype that did not affect the viability of HeLa cells.
Figure 4.


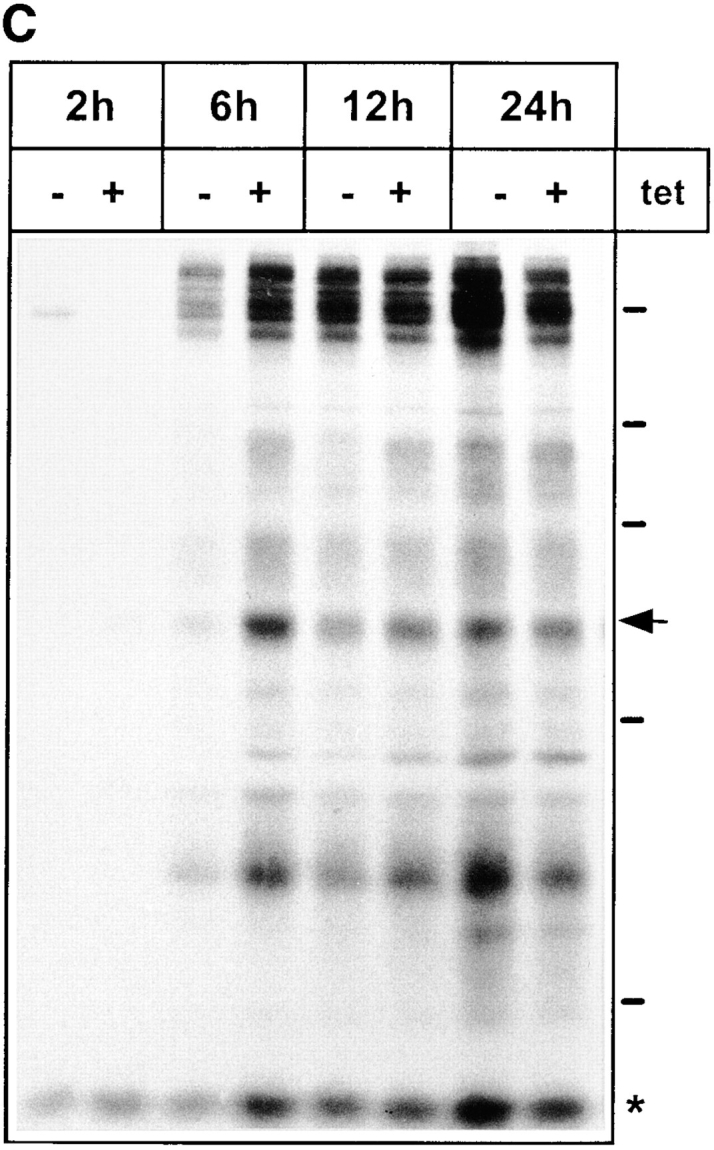
Secretion of glycoproteins in KKFF and KKAA HeLa cells. (A) Cells were incubated for 48 h ± tet, labeled for 30 min with [35S]methionine and chased for 1, 2, and 3 h. Glycoproteins were isolated with Con A beads and separated by 10% SDS-PAGE. 14C-labeled molecular mass markers are indicated at the right margin (200, 97.4, 66, 46, and 30 kD). The arrows indicate the position of the 57-kD protein. *, band used as a reference for quantification in B. The apparent absence of the 66-kD protein in the KKFF panel (3 h, −tet) is due to an artifactual inhomogeneity of the gel. (B) Quantification of the delay in secretion of the 57-kD glycoprotein. The amount of the 57-kD glycoprotein was determined by densitometry of fluorograms and normalized. Shown is the relative secretion as determined by dividing the value obtained for −tet by that for +tet. Values are means ± SEM of at least three independent experiments. white bars, KKFF; black bars, KKAA. (C) Tet- dependent difference in the secretion of the 57-kD protein disappears with increased chase times. KKAA cells were treated ± tet for 48 h, pulsed for 30 min with [35S]methionine, and chased for 2, 6, 12, and 24 h. Glycoproteins were isolated from the culture medium and analyzed by SDS-PAGE as in A.
Identification of the Inefficiently Secreted 57-kD Glycoprotein as Procathepsin C
To identify the 57-kD protein, it was purified from the culture medium of KKFF cells 24 h after plating the cells. Glycoproteins were isolated by Con A–Sepharose, separated by SDS-PAGE, and blotted onto a polyvinyl difluoride membrane. The 57-kD region was excised and subjected to tryptic digestion. Tryptic peptides were isolated by reverse-phase column chromatography and sequenced. The sequence of three peptides was established, all of which were found to belong to the sequence of procathepsin C, a precursor of the lysosomal enzyme cathepsin C, by database searches (Fig. 5).
Figure 5.

The sequence of three peptides isolated from the 57-kD protein can be accommodated within the amino acid sequence of the precursor of the human lysosomal peptidase procathepsin C (Paris et al., 1995). Shown is part of the deduced partial amino acid sequence of human procathepsin C and the sequence of three tryptic peptides determined by automatic sequencing (bold and underlined).
Because the 57-kD region may include several glycoproteins, it was important to establish if the inefficiently secreted glycoprotein was indeed procathepsin C. To test this, an antibody against rat cathepsin C was used (Muno et al., 1993), since no antibody against human cathepsin C was available. This antibody was found to cross-react with the human enzyme (Fig. 6 A). It precipitated a protein of ∼57-kD from metabolically labeled FAO rat hepatoma cells and human HeLa (KKAA) cells in reasonable agreement with the known molecular mass of procathepsin C (Muno et al., 1993). In the next step, we analyzed the culture media for secreted procathepsin C. KKAA cells were cultured ± tet for 48 h, labeled with [35S]methionine for 30 min, and chased for 2 h with unlabeled methionine in excess. Glycoproteins were isolated with Con A–Sepharose, immunoprecipitated with anti–procathepsin C, and analyzed by SDS-PAGE and fluorography. This experiment identified the inefficiently secreted 57-kD band as procathepsin C (Fig. 6 A, Cat C). Direct immunoprecipitation of the antigen from the medium omitting the Con A step gave identical results (Fig. 6 B, lanes 3 and 4) and showed that the majority of the glycoprotein in the 57-kD region of the gel is due to secreted procathepsin C. That the tet-dependent decrease of procathepsin C in the medium was not due to reduced synthesis was confirmed by metabolic labeling. After a 10-min pulse with [35S]methionine, identical amounts of metabolically labeled procathepsin C were immunoprecipitable from cells cultured ± tet (not shown). The inefficient secretion of procathepsin C was not due to clonal variation since three independent KKAA clones exhibited the same phenotype (Fig. 7).
Figure 6.


The inefficiently secreted 57-kD protein is procathepsin C. (A, left) Procathepsin C expression in FAO and HeLa cells. FAO and KKAA (± tet, 48 h) cells were labeled for 30 min with [35S]methionine and immunoprecipitated with an antibody against rat cathepsin C. Immunoprecipitated proteins were separated by 10% SDS-PAGE. (Right) Glycoproteins isolated by Con A beads and immunoprecipitation of procathepsin C. KKAA ± tet (48 h) cells were pulsed for 30 min and chased for 2 h. Secreted glycoproteins released from Con A beads (Con A) were lyophilized, immunoprecipitated with the cathepsin C antibody, and separated by 10% SDS-PAGE (Cat C). (B) Direct immunoprecipitation of procathepsin C from the culture medium and digestion with glycosidases. KKAA in the presence or absence of tet (48 h) cells were pulsed for 30 min and chased for 2 h. The medium (lanes 1–7) was subjected to methanol precipitation, and the precipitate was resuspended and immunoprecipitated with the cathepsin C antibody. Immunoprecipitates were digested with endo H under nonlimiting (lane 6) and limiting (lane 9, sample was from detergent-solubilized cells) conditions or with endo F as indicated and separated by SDS-PAGE.
Figure 7.

Inefficient secretion of procathepsin C is not due to clonal variation. Cells of clones KKAA 1, KKAA 15, KKAA 4 (used in this study), and KKFF 9 cells were cultured in the presence or absence of tet for 48 h followed by labeling for 30 min with [35S]methionine and a chase of 2 h. Proteins released into the culture medium were methanol precipitated. The precipitates were resuspended and immunoprecipitated with anti–cathepsin C antibody. The immunoprecipitates were separated by SDS-PAGE and radiolabeled proteins were visualized by fluorography.
The secreted glycoproteins were also analyzed by two-dimensional (2D) gel electrophoresis. To this end, cells cultured ± tet were labeled with [35S]methionine for 30 min and chased for 3 h with unlabeled methionine in excess. Secreted glycoproteins were isolated with Con A beads and separated by isoelectric focusing in the first dimension followed by SDS-PAGE in the second dimension. To identify procathepsin C, the protein was immunoprecipitated from a KKAA culture (+tet) labeled in parallel and also subjected to the 2D gel electrophoresis. Fig. 8 confirms that the major glycoprotein affected by tet withdrawal is procathepsin C. On 2D gels, procathepsin C appeared as a cohort of spots in the pI range 6 to 7, which is in reasonable agreement with the calculated pI 7.0 deduced from the primary sequence of human procathepsin C (Paris et al., 1995). The secretion of a few minor glycoproteins of unknown identity, e.g., in the 45-kD/pI 7 range, also appeared to be reduced in the absence of tet.
Figure 8.



2D gel analysis of secreted glycoproteins. KKAA cells were cultured in the absence (A) or presence (B) of tet for 48 h. Thereafter, the cells were labeled for 30 min with [35S]methionine and chased for 3 h. Secreted glycoproteins were isolated with Con A beads, methanol precipitated, and subjected to isoelectric focusing (15 h) in the first dimension using a 3–10 linear immobiline pH gradient. 10% SDS-PAGE was used for the second dimension. As a reference, cathepsin C was immunoprecipitated from the medium of KKAA cells (+tet) that had been labeled in parallel and subjected to the same 2D gel separation procedure (C). Proteins were visualized by fluorography.
To further confirm the identity of the 57-kD protein, the antigen was immunoprecipitated from the culture medium and digested with endo F and endo H. Endo F digestion reduced the apparent molecular mass to ∼5 kD (Fig. 6 B, lane 7). Endo H treatment resulted in several bands, suggesting considerable endo H sensitivity of the secreted form (Fig. 6 B, lane 6). Limited digestion with Endo H of the protein immunoprecipitated from cell extracts produced up to five bands (Fig. 6 B, lane 9). These results suggest that the 57-kD protein recognized by the anticathepsin antibody carries four N-linked glycans. Consistent with this, the cDNA of procathepsin C has four consensus sites for N-glycosylation (Paris et al., 1995).
Is delayed secretion associated with ERGIC-53's mislocalization a specific feature of lysosomal enzymes? This does not appear to be the case. Cathepsin D, known to carry two N-glycosidic side chains (Hasilik and Neufeld, 1980), was secreted with identical kinetics in KKAA cells ± tet (Fig. 9 B). α-Glucosidase, a lysosomal enzyme having seven potential N-glycosylation sites (Hermans et al., 1993) was not secreted into the culture medium. Previous studies suggested that this enzyme is membrane bound (Fransen et al., 1988; Wisselaar et al., 1993). Transport of α-glucosidase to lysosomes was unchanged by overexpressing KKAA in the ER, as indicated by the kinetics of appearance of the different maturation forms of the protein (Fig. 9 C). We next studied the transport of the endosomal marker transferrin receptor, a membrane glycoprotein with three N-linked glycans, one of which remains in a high mannose form (Enns et al., 1991). Transport from ER to medial-Golgi was found to be unaffected by overexpressing KKAA in the ER as monitored by the acquisition of endo H resistance (Fig. 9 A). Likewise, the transport of the plasma membrane protein aminopeptidase N (10 N-glycosylation sites; Olsen et al., 1988) and the lysosomal membrane glycoprotein Lamp 1 (18 N-glycans; Carlsson et al., 1988) was not affected by mislocalizing ERGIC-53 to the ER (not shown).
Figure 9.

Intracellular transport of identified glycoproteins in KKAA cells (treated in the presence or absence of tet for 48 h). (A) Transferrin receptor: Cells were pulsed for 10 min with [35S]methionine and chased as indicated. The receptor was immunoprecipitated from the Triton X-100–solubilized cells and digested (or not) with endo H. (B) Cathepsin D: Cells were metabolically labeled for 1 h followed by a chase for 3 or 6 h. The culture medium was precipitated with methanol. Cathepsin D was immunoprecipitated from the resuspended methanol precipitate. (C) α-Glucosidase: Cells were metabolically labeled for 1 h and chased as indicated, and the enzyme was immunoprecipitated after cell solubilization with Triton X-100. The immunoprecipitates were separated by SDS-PAGE and visualized by fluorography.
Overexpression of KKAA Delays ER Exit of Procathepsin C
Is the inefficient secretion of procathepsin C due to a delay in ER–Golgi transport? Because procathepsin C was found to remain endo H sensitive during its passage through the Golgi apparatus (not shown), we were unable to approach this question by monitoring the acquisition of complex glycans. Likewise, a morphological approach failed since the antibody was not useful for immunofluorescence microscopy. Therefore, we used subcellular fractionation. KKAA cells cultured for 48 h ± tet were pulsed for 15 min with [35S]methionine and chased for 30 and 60 min. Postnuclear supernatants were separated on Nycodenz gradients, the ER fractions (fractions 1–5) were pooled, and procathepsin C was immunoprecipitated and its radioactivity quantified by densitometric scanning after SDS-PAGE (Fig. 10). Under noninducible conditions, 25% of the initially synthesized procathepsin C disappeared from the pooled ER fractions after 30 min of chase and 59% after 60 min. Under inducible conditions, these values were 4% after 30 min and 35% after 60 min. These data suggest that overexpression of the KKAA construct delays transport of procathepsin C from the ER, although they do not indicate whether or not ER exit is the only rate-limiting transport step.
Figure 10.

Overexpression of the KKAA construct delays ER exit of procathepsin C. KKAA cells were cultured in presence or absence of tet for 48 h and pulse-labeled with [35S]methionine and chased for the indicated times. The cells were homogenized, and membranes were separated by Nycodenz gradient centrifugation (see Materials and Methods). Fractions 1–5 containing the ER marker p63 were pooled and Triton X-100 solubilized, and procathepsin C was immunoprecipitated. The immunoprecipitates were separated by SDS-PAGE, and procathepsin C was quantified from the fluorogram. The start signal after the pulse was set to 100%. All the lanes of this figure originate from the same fluorogram and were identically exposed. They had to be rearranged for reproduction purposes.
Discussion
In this study, we report that mistargeting of ERGIC-53 to the ER by overexpressing the KKAA construct led to a limited secretion phenotype resulting in the delayed secretion of procathepsin C from HeLa cells, while overexpression of KKFF, a myc-tagged wild-type ERGIC-53, had no effect on glycoprotein secretion.
The rationale of our approach was to mistarget an exogenous mutant ERGIC-53 to the ER and due to overexpression and mixed oligomer formation block the recycling of endogenous ERGIC-53 in this compartment. If ERGIC-53 is essential for glycoprotein transport, such a functional inactivation of ERGIC-53 should result in a secretion phenotype. To minimize the possibility of upregulation of a mechanism that may compensate for the functional inactivation of ERGIC-53, an inducible expression system was chosen. Our results suggest that in the KKAA clone the expected mistargeting of both KKAA and endogenous ERGIC-53 was indeed achieved under inducible conditions (−tet). Immunofluorescence microscopy and density gradient analysis suggest that both the KKAA and endogenous ERGIC-53 were apparently quantitatively retained in the ER. That KKAA constructs are unable to leave the ER has also been noted in other cell types (Kappeler et al., 1997).
The precise mechanism by which the KKAA construct retained endogenous ERGIC-53 in the ER remains to be elucidated. We assume that mixed oligomer formation is a major mechanism by which this is achieved. It is unlikely, however, that disulfide bond–mediated oligomerization entirely accounts for this effect. Only the newly synthesized but not the already existing endogenous ERGIC-53, which has a rather long half-life (Schweizer et al., 1988), can form disulfide bonds with the KKAA construct during induction. ERGIC-53 may form higher order oligomers that are not covalently linked and therefore do not survive coimmunoprecipitation as used in the present study. We also noted some downregulation of the endogenous ERGIC-53 by overexpressing the KKAA constructs (not shown).
ERGIC-53's cytosolic domain can bind COPI because of its KKFF dilysine ER retrieval signal (Schindler et al., 1993; Kappeler et al., 1997; Tisdale et al., 1997). An antibody against this domain was reported to block the recruitment of COPI to the ERGIC in permeabilized cells and to inhibit transport of VSV G glycoprotein from ERGIC to Golgi (Tisdale et al., 1997). The authors proposed that ERGIC-53 is required for selective transport through the early secretory pathway by serving as a major COPI-binding receptor in ERGIC and cis-Golgi. If ERGIC-53 would operate as the major receptor, its depletion from the ERGIC should lead to COPI dissociation from the membranes of the early secretory pathway. At variance with this notion, by immunofluorescence microscopy we find no change in the localization of COPI when ERGIC-53 is absent from post-ER sites by the mistargeting of KKAA to the ER. Indeed, our immunofluorescence experiments did not reveal any gross morphological changes of the early secretory pathway under these conditions. In particular, ERGIC clusters visualized by β′-COP were equally abundant in KKAA cells ± tet. The apparent discrepancy between Tisdale et al. (1997) and our data are not easy to explain but may be caused by experimental differences. While the study of Tisdale et al. (1997) was performed with semi-intact cells, our present study is based on intact cells and hence is closer to physiology. Clearly, cells not expressing any ERGIC-53 will be required to resolve the discrepancy.
Mistargeting of ERGIC-53 to the ER resulted in a rather limited secretion phenotype. It appears that in HeLa cells, the recycling of ERGIC-53 is essential for the efficient secretion of one major glycoprotein, procathepsin C, and a few minor glycoproteins, as indicated by our 2D gel analysis, whereas it is not essential for the majority of glycoproteins. Cathepsin D and α-glucosidase, two other lysosomal enzymes, were not affected by ERGIC-53's mistargeting. Likewise, locking ERGIC-53 to the ER did not impair the transport from ER to medial-Golgi of the plasma membrane protein aminopeptidase N, the endosomal membrane protein transferrin receptor, and the lysosomal membrane protein lamp1. That the transport of lamp1 was unaffected is particularly noteworthy since this protein carries 18 N-linked glycans (Carlsson et al., 1988).
How does the retention of ERGIC-53 in the ER affect the transport of procathepsin C? Consistent with its mannose-selective lectin activity in vitro, ERGIC-53 may be required for the efficient export of procathepsin C from the ER. This notion is supported by our finding that ER exit of procathepsin C is delayed by overexpressing the KKAA protein, although the data do not allow us to conclude that ER exit is the only delayed transport step. It is interesting to note that procathepsin C is a tetramer (Muno et al., 1993), in contrast to the nonaffected lysosomal enzymes cathepsin D and α-glucosidase, which are monomers (Hasilik and Neufeld, 1980). It is conceivable, therefore, that proteins carrying many N-linked glycans require ERGIC-53 for efficient transport. A tetramer of cathepsin C would amount to 16 glycans. However, the absolute number of oligosaccharide side chains may not be the only criterion since the transport of Lamp1 carrying 18 N-linked glycans was unaffected by the mistargeting of ERGIC-53. Our attempts to demonstrate direct interaction of procathepsin C and ERGIC-53 by coimmunoprecipitation have been unsuccessful, suggesting that this interaction, if existent, must be of low affinity. Alternatively, the effect on procathepsin C transport may be indirect. ER-retained ERGIC-53 may bind and disturb a glycoprotein cofactor required for the maturation or transport of cathepsin C. Whatever the precise mechanism, the result with cathepsin C demonstrates the first lectin effect of ERGIC-53 in vivo.
The role of carbohydrates in ER-to-Golgi traffic remains unclear. Even though ERGIC-53 does not appear to be essential for the transport of most glycoproteins in HeLa cells, the hypothesis of lectin-mediated transport (Fiedler and Simons, 1995; Itin et al., 1996) remains attractive. Carbohydrate-dependent transport may involve more than one lectin, so that the inactivation of one of them, as in the case of ERGIC-53, may be compensated largely by other lectins. If such lectins are not homologous to ERGIC-53 and VIP36, they may be difficult to detect. ERGIC-53 and VIP36 are so far the only members of this novel family of animal lectins. ERGIC-53 is conserved all the way from Caenorhabditis elegans to Xenopus, rat, and man. Of particular importance, this conservation includes the CRD in which the amino acids critical for sugar binding in leguminous lectins are invariant, and the cytosolic tail carrying ER export and ER retrieval signals (Kappeler et al., 1997). In a recent study, mutations in ERGIC-53 were found to lead to an autosomal recessive bleeding disorder in humans termed combined deficiency of coagulation factors V and VIII (Nichols et al., 1998). Although the mechanism by which ERGIC-53 is required for maintaining appropriate serum levels of the homologous coagulation factors V and VIII remains to be elucidated, this study is in agreement with our conclusion drawn from a cell culture system that ERGIC-53 is required for efficient secretion of a limited set of glycoproteins. ERGIC-53 may act as a traffic chaperone for these proteins. That ERGIC-53 is already expressed in C. elegans (our unpublished observations) strongly suggests that this protein has functions in addition to regulating blood coagulation.
The effect of inactivation of ERGIC-53 is reminiscent of a deletion of emp24 gene in yeast leading to a selective secretion phenotype involving the two proteins invertase and Gas1p (Schimmöller et al., 1995). This finding led to the hypothesis that Emp24p may act as a transport receptor for these proteins, but it is unknown if Emp24p directly binds these cargo proteins, and hence this suggestion remains to be proven.
In summary, while the precise function of ERGIC-53 remains to be established, the present study suggests that this protein is essential for the efficient transport of at least one glycoprotein in HeLa cells. Furthermore, mistargeting of ERGIC-53 to the ER does not alter the gross morphology of the early secretory pathway, indicating that this protein is not required for the maintenance of the ERGIC.
Acknowledgments
We thank Käthy Bucher for excellent technical assistance, Dieter Klopfenstein for help with the 2D gel electrophoresis, Paul Jenö for peptide sequencing, Hermann Bujard for the HtTA cells and the pUHD10-3 plasmid, Hanna Damke for the pBSpac plasmid and for discussions, Adrian Wiestner for the FAO cells, Eiki Kominami for the cathepsin C antibody, Jack Fransen for the α-glucosidase antibody, Thomas Kreis for the β′-COP antibody, and Annie-Claude Roche and Michel Monsigny for the FITC-Man-BSA. We are grateful to Graham Warren for many helpful comments and to Helena Andersson for reading the manuscript.
This study was supported by grants from the Kantons of Basel and the Swiss National Science Foundation.
Abbreviations used in this paper
- 2D
two-dimensional
- Con A
concanavalin A
- CRD
carbohydrate recognition domain
- endo H
endoglycosidase H
- ERGIC
ER–Golgi intermediate compartment
- tet
tetracycline
- tetO
tetracycline operator sequence
Footnotes
Address all correspondence to Hans-Peter Hauri, Department of Pharmacology, Biozentrum, University of Basel, Klingelbergstrasse 70, CH-4056 Basel, Switzerland. Tel.: +41-61 267 2222 (or 2229). Fax: +41-61 267 2208. E-mail: Hauri@ubaclu.unibas.ch
Christian Itin's present address is Department of Biochemistry, Stanford University, Stanford, CA 94305.
References
- Arar C, Carpentier V, Le Cear J-P, Monsigny M, Legrand A, Roche AC. ERGIC-53, a membrane protein of the endoplasmic reticulum-Golgi intermediate compartment, is identical to MR60, an intracellular mannose-specific lectin of myelomonocytic cells. J Biol Chem. 1995;270:3551–3553. doi: 10.1074/jbc.270.8.3551. [DOI] [PubMed] [Google Scholar]
- Aridor M, Bannykh SI, Rowe T, Balch WE. Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J Cell Biol. 1995;131:875–893. doi: 10.1083/jcb.131.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, McCaffery JM, Plutner H, Farquhar MG. Vesicular stomatitis virus glycoprotein is sorted and concentrated during export from the endoplasmic reticulum. Cell. 1994;76:841–852. doi: 10.1016/0092-8674(94)90359-x. [DOI] [PubMed] [Google Scholar]
- Banfiled DK, Louis C, Rabouille C, Warren G, Pelham HR. Localization of Sed5, a putative vesicle targeting molecule, to the cis-Golgi network involves both its transmembrane and cytoplasmic domains. J Cell Biol. 1994;127:357–371. doi: 10.1083/jcb.127.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannykh SI, Balch WE. Membrane dynamics at the endoplasmic reticulum–Golgi interface. J Cell Biol. 1997;138:1–4. doi: 10.1083/jcb.138.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson SR, Roth J, Piller F, Fukuda M. Isolation and characterization of human lysosomal membrane glycoproteins, h-lamp-1 and h-lamp-2. J Biol Chem. 1988;263:18911–18919. [PubMed] [Google Scholar]
- Chavrier P, Parton RG, Hauri H-P, Simons K, Zerial M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62:317–329. doi: 10.1016/0092-8674(90)90369-p. [DOI] [PubMed] [Google Scholar]
- Cosson P, Letourneur F. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science. 1994;263:1629–1631. doi: 10.1126/science.8128252. [DOI] [PubMed] [Google Scholar]
- Damke H, Baba T, Warnock DE, Schmid SL. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damke H, Gossen M, Bujard H, Schmid SL. Tightly regulated and inducible expression of dominant interfering dynamin mutant in stably transformed HeLa cells. Methods Enzymol. 1995;257:209–220. doi: 10.1016/s0076-6879(95)57026-8. [DOI] [PubMed] [Google Scholar]
- de la Luna S, Soria I, Pulido D, Ortín J, Jiménez A. Efficient transformation of mammalian cells with constructs containing a puromycin-resistance marker. Gene (Amst) 1988;62:121–126. doi: 10.1016/0378-1119(88)90585-9. [DOI] [PubMed] [Google Scholar]
- Enns CA, Clinton EM, Reckhow CL, Root BJ, Do S-I, Cook C. Acquisition of the functional properties of the transferrin receptor during its biosynthesis. J Biol Chem. 1991;266:13272–13277. [PubMed] [Google Scholar]
- Farquhar, M.G., and H.-P. Hauri. 1997. Protein sorting and vesicular traffic in the Golgi apparatus. In The Golgi Apparatus. E.G. Berger and J. Roth, editors. Birkhäuser Verlag, Basel, Switzerland. 63–129.
- Fernandez J, DeMott M, Atherton D, Mische SM. Internal protein sequence analysis: enzymatic digestion for less than 10 μg of protein bound to polyvinylidene difluoride or nitrocellulose membrane. Anal Biochem. 1992;201:255–264. doi: 10.1016/0003-2697(92)90336-6. [DOI] [PubMed] [Google Scholar]
- Fiedler K, Simons K. A putative novel class of animal lectins in the secretory pathway homologous to leguminous lectins. Cell. 1994;77:625–626. doi: 10.1016/0092-8674(94)90047-7. [DOI] [PubMed] [Google Scholar]
- Fiedler K, Simons K. The role of N-glycans in the secretory pathway. Cell. 1995;81:309–312. doi: 10.1016/0092-8674(95)90380-1. [DOI] [PubMed] [Google Scholar]
- Fransen JAM, Ginsel LA, Cambier PH, Klumperman J, Oude RPJ, Elferink, Tager JM. Immunocytochemical demonstration of the lysosomal enzyme α-glucosidase in the brush border of human intestinal epithelial cells. Eur J Cell Biol. 1988;47:72–80. [PubMed] [Google Scholar]
- Gaynor EC, te Heesen S, Graham TR, Aebi M, Emr SD. Signal-mediated retrieval of a membrane protein from the Golgi to the ER. J Cell Biol. 1994;127:653–665. doi: 10.1083/jcb.127.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick BS, Elston T, Oster G. A cisternal maturation mechanism can explain the asymmetry of the Golgi stack. FEBS Lett. 1997;414:177–181. doi: 10.1016/s0014-5793(97)00984-8. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Ericsson M, Krijnse-Locker J, Nilsson T, Goud B, Söling HD, Tang BL, Wong SH, Hong W. Localization of the Lys, Asp, Glu, Leu tetrapeptide receptor to the Golgi complex and the intermediate compartment in mammalian cells. J Cell Biol. 1994;127:1557–1574. doi: 10.1083/jcb.127.6.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasilik A, Neufeld EF. Biosynthesis of lysosomal enzymes in fibroblasts. J Biol Chem. 1980;255:4937–4945. [PubMed] [Google Scholar]
- Hauri H-P, Schweizer A. The endoplasmic reticulum-Golgi intermediate compartment. Curr Opin Cell Biol. 1992;4:600–608. doi: 10.1016/0955-0674(92)90078-Q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauri, H-P., and A. Schweizer. 1997. The ER-Golgi membrane system: compartmental organization and protein traffic. In Handbook of Physiology: Cell Physiology. J.F. Hoffman and J.D. Jamieson, editors. Oxford University Press. Sec. 14, 605–647.
- Hauri H-P, Sterchi EE, Bienz D, Fransen JAM, Marxer A. Expression and intracellular transport of microvillus membrane hydrolases in human intestinal epithelial cell. J Cell Biol. 1985;101:838–851. doi: 10.1083/jcb.101.3.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans MM, Wisselaar HA, Kroos MA, Oostra BA, Reuser AJ. Human lysosomal α-glucosidase: functional characterization of the glycosylation sites. Biochem J. 1993;289:681–686. doi: 10.1042/bj2890681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itin C, Foguet M, Kappeler F, Klumperman J, Hauri H-P. Recycling of the endoplasmic reticulum/Golgi intermediate compartment protein ERGIC-53 in the secretory pathway. Biochem Soc Trans. 1995a;3:541–544. doi: 10.1042/bst0230541. [DOI] [PubMed] [Google Scholar]
- Itin C, Schindler R, Hauri H-P. Targeting of protein ERGIC-53 to the ER/ERGIC/cis-Golgi recycling pathway. J Cell Biol. 1995b;131:57–67. doi: 10.1083/jcb.131.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itin C, Roche AC, Monsigny M, Hauri H-P. ERGIC-53 is a functional mannose-selective and calcium-dependant human homologue of leguminous lectin. Mol Biol Cell. 1996;7:483–493. doi: 10.1091/mbc.7.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MR, Nilsson T, Peterson PA. Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO (Eur Mol Biol Organ) J. 1990;9:3153–3162. doi: 10.1002/j.1460-2075.1990.tb07513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MR, Nilsson T, Peterson PA. Retrieval of transmembrane proteins to the endoplasmic reticulum. J Cell Biol. 1993;121:317–333. doi: 10.1083/jcb.121.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenö P, Mini T, Moes S, Hintermann E, Horst M. Internal sequences from proteins digested in polyacrylamide gels. Anal Biochem. 1995;224:75–82. doi: 10.1006/abio.1995.1010. [DOI] [PubMed] [Google Scholar]
- Kappeler F, Klopfenstein DRC, Foguet M, Paccaud J-P, Hauri H-P. The recycling of ERGIC-53 in the early secretory pathway. J Biol Chem. 1997;272:31801–31808. doi: 10.1074/jbc.272.50.31801. [DOI] [PubMed] [Google Scholar]
- Lahtinen U, Hellman U, Wernstedt C, Saraste J, Pettersson RF. Molecular cloning and expression of a 58-kDa cis-Golgi and intermediate compartment protein. J Biol Chem. 1996;271:4031–4037. doi: 10.1074/jbc.271.8.4031. [DOI] [PubMed] [Google Scholar]
- Letourneur F, Gaynor EC, Hennecke S, Demolliere C, Duden R, Emr SD. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell. 1994;79:1199–1207. doi: 10.1016/0092-8674(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Linstedt AD, Hauri H-P. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol Biol Cell. 1993;4:679–693. doi: 10.1091/mbc.4.7.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt AD, Mehta A, Suhan J, Reggio H, Hauri H-P. Sequence and overexpression of GPP130/GIMPc: evidence for saturable pH-sensitive targeting of a type II early Golgi membrane protein. Mol Biol Cell. 1997;8:1073–1087. doi: 10.1091/mbc.8.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Donaldson JG, Schweizer A, Berger EG, Hauri H-P, Yuan LC, Klausner RD. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell. 1990;60:821–836. doi: 10.1016/0092-8674(90)90096-w. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Singer SJ. A soluble secretory protein is first concentrated in the endoplasmic reticulum before transfer to the Golgi apparatus. Proc Natl Acad Sci USA. 1993;90:5732–5736. doi: 10.1073/pnas.90.12.5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muno D, Ishidoh K, Ueno T, Kominami E. Processing and transport of the precursor of cathepsin C during its transfer into lysosomes. Arch Biochem Biophys. 1993;306:103–110. doi: 10.1006/abbi.1993.1486. [DOI] [PubMed] [Google Scholar]
- Nichols WC, Seligson U, Zivelin A, Terry VH, Hertel CE, Wheatley MA, Moussalli MJ, Hauri H-P, Ciavarella N, Kaufman RJ, Ginsburg D. Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell. 1998;93:61–70. doi: 10.1016/s0092-8674(00)81146-0. [DOI] [PubMed] [Google Scholar]
- Nishimura N, Balch WE. A di-acidic signal required for selective export from the endoplasmic reticulum. Science. 1997;277:556–558. doi: 10.1126/science.277.5325.556. [DOI] [PubMed] [Google Scholar]
- Olsen J, Cowell GM, Konigshofer E, Danielsen EM, Moller J, Laustsen L, Hansen OC, Welinder KG, Engberg J, Hunziker W, et al. Complete amino acid sequence of human intestinal aminopeptidase N as deduced from cloned cDNA. FEBS Lett. 1988;238:307–314. doi: 10.1016/0014-5793(88)80502-7. [DOI] [PubMed] [Google Scholar]
- Oprins A, Duden R, Kreis TE, Geuze HJ, Slot JW. β-COP localizes mainly to the cis-Golgi side in exocrine pancreas. J Cell Biol. 1993;121:49–59. doi: 10.1083/jcb.121.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris A, Strukelj B, Pungercar J, Renko M, Dolenc I, Turk V. Molecular cloning and sequence analysis of human procathepsin C. FEBS Lett. 1995;369:326–330. doi: 10.1016/0014-5793(95)00777-7. [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol. 1994;14:1669–1679. doi: 10.1128/mcb.14.3.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo M, Pepperkok P, Emery G, Kellner R, Stang E, Parton RG, Gruenberg J. Involvement of the transmembrane protein p23 in biosynthetic protein transport. J Cell Biol. 1997;129:1119–1136. doi: 10.1083/jcb.139.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe T, Aridor M, McCaffery JM, Plutner H, Nuoffer C, Balch WE. COPII vesicles derived from mammalian endoplasmic reticulum microsomes recruit COPI. J Cell Biol. 1996;135:895–911. doi: 10.1083/jcb.135.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Pollack R. Basic methodology for cell culture-cell transformation. Methods Enzymol. 1974;32:583–592. doi: 10.1016/0076-6879(74)32058-7. [DOI] [PubMed] [Google Scholar]
- Saraste J, Svensson K. Distribution of the intermediate elements operating in ER to Golgi transport. J Cell Sci. 1991;100:415–430. doi: 10.1242/jcs.100.3.415. [DOI] [PubMed] [Google Scholar]
- Saraste J, Lahtinen U, Goud B. Localization of the small GTP-binding protein rab1p to early compartments of the secretory pathway. J Cell Sci. 1995;108:1541–1552. doi: 10.1242/jcs.108.4.1541. [DOI] [PubMed] [Google Scholar]
- Scales SJ, Pepperkok R, Kreis TE. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 1997;90:1137–1148. doi: 10.1016/s0092-8674(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Schimmöller F, Singer-Krüger B, Schröder S, Krüger U, Barlowe C, Riezman H. The absence of Emp24p, a component of ER-derived COPII-coated vesicles, causes a defect in transport of selected proteins to the Golgi. EMBO (Eur Mol Biol Organ) J. 1995;14:1329–1339. doi: 10.1002/j.1460-2075.1995.tb07119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler R, Itin C, Zerial M, Lottspeich F, Hauri H-P. ERGIC-53, a membrane protein of the ER-Golgi intermediate compartment, carries an ER retention motif. Eur J Cell Biol. 1993;61:1–9. [PubMed] [Google Scholar]
- Schweizer A, Fransen JAM, Bächi T, Ginsel L, Hauri H-P. Identification, by a monoclonal antibody, of a 53-kD protein associated with a tubular-vesicular compartment at the cis-side of the Golgi apparatus. J Cell Biol. 1988;107:1643–1653. doi: 10.1083/jcb.107.5.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer A, Ericsson M, Bächi T, Griffiths G, Hauri H-P. Characterization of a novel 63-kD membrane protein. Implications for the organization of the ER-to-Golgi pathway. J Cell Sci. 1993;104:671–683. doi: 10.1242/jcs.104.3.671. [DOI] [PubMed] [Google Scholar]
- Schweizer A, Rohrer J, Slot JW, Geuze HJ, Kornfeld S. Reassessment of the subcellular localization of p63. J Cell Sci. 1995;108:2477–2485. doi: 10.1242/jcs.108.6.2477. [DOI] [PubMed] [Google Scholar]
- Tang BL, Low SH, Hauri H-P, Hong W. Segregation of ERGIC53 and the mammalian KDEL receptor upon exit from the 15°C compartment. Eur J Cell Biol. 1995;68:398–410. [PubMed] [Google Scholar]
- Tisdale EJ, Pluttner H, Matteson J, Balch WE. p53/p58 binds COPI and is required for selective transport through the early secretory pathway. J Cell Biol. 1997;137:581–593. doi: 10.1083/jcb.137.3.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley FM, Pelham HR. The KKXX signal mediates retrieval of membrane proteins from the Golgi to the ER in yeast. Eur J Cell Biol. 1994;64:211–216. [PubMed] [Google Scholar]
- Wisselaar HA, Koos MA, Hermans MM, Beemen J, Reuser AJ. Structural and functional changes of lysosomal acid α-glucosidase during intracellular transport and maturation. J Biol Chem. 1993;268:2223–2231. [PubMed] [Google Scholar]
- Zhang T, Wong SH, Tang BL, Xu Y, Peter F, Subramaniam VN, Hong W. The mammalian protein (rbet1) homologous to yeast Bet1p is primarily associated with pre-Golgi intermediate compartment and is involved in vesicular transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol. 1997;139:1157–1168. doi: 10.1083/jcb.139.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]