

Abstract
Activation of integrins upon binding to extracellular matrix proteins is believed to be a crucial step for the regulation of cell survival and proliferation. We have used integrin α1-null mice to investigate the role of this collagen receptor in the regulation of cell growth and survival in vivo. α1-deficient animals, which are viable and fertile, have a hypocellular dermis and a deficiency in dermal fibroblast proliferation as embryos. In vitro analysis of α1-null embryonic fibroblasts has revealed that their proliferation rate is markedly reduced when plated on collagenous substrata, despite normal attachment and spreading. Moreover, on the same collagenous matrices, α1-null fibroblasts fail to recruit and activate the adaptor protein Shc. The failure to activate Shc is accompanied by a downstream deficiency in recruitment of Grb2 and subsequent mitogen-activated protein kinase activation. Taken together with the growth deficiency observed on collagens, this finding indicates that the α1β1 is the sole collagen receptor which can activate the Shc mediated growth pathway. Thus, integrin α1 has a unique role among the collagen receptors in regulating both in vivo and in vitro cell proliferation in collagenous matrices.
Keywords: α1β1 integrin, collagen, proliferation, signal transduction, dermis
Integrins are transmembrane heterodimeric receptors for extracellular matrix (ECM)1 molecules (Hynes, 1992). They are composed of α and β subunits that heterodimerize to produce >20 different receptors. One family of integrins, which all have the β1 subunit in common, include high affinity receptors for many ECM proteins including fibronectin, laminin, and the collagens. Three members of this family are known to be the major receptors for collagens, namely α1β1, α2β1, and α3β1 (Yamamoto and Yamamoto, 1994; Gardner et al., 1996). All three of these receptors are expressed by fibroblasts, and they seem to regulate different functions. Integrin α1β1, for example, appears to regulate collagen synthesis, whereas α2β1 mediates collagen gel contraction (Schiro et al., 1991; Langholz et al., 1995; Broberg and Heino, 1996).
Integrin α1β1 is widely expressed in the adult animal, being found in visceral and some vascular smooth muscle, liver, microvascular endothelium, activated lymphocytes, and dermis (see references cited in Gardner et al., 1996). During embryonic development, α1 is first expressed in trophoblast immediately after implantation (Sutherland et al., 1993), suggesting an essential role of this molecule during early stages of development. To analyze the role of α1 integrin during mouse formation, we have generated mice deficient in integrin α1 by gene targeting (Gardner et al., 1996). This disruption prevents the formation of the α1β1 receptor without altering other β1 integrin heterodimers. Animals homozygous for the disruption develop normally and are fertile, indicating that despite its prominent expression during development, α1 integrin is not essential for formation of the adult mouse. Using embryonic fibrobasts derived from mutant animals, we have shown that α1 uniquely confers on fibroblasts the ability to adhere and migrate on collagen IV, while being only one of three integrin collagen type I receptors present on fibroblasts, together with α2β1 and α3β1. Recent studies have also implicated DDR1 and 2, “orphan” tyrosine kinase receptors, as receptors for collagens (Shrivastava et al., 1997; Vogel et al., 1997).
The interaction between integrins and their ligands, besides mediating cell adhesion, plays a role in a number of cellular processes, including cell differentiation (Adams and Watt, 1993), cell cycle progression (Giancotti, 1997), and cell survival (Frisch and Ruoslahti, 1997). The regulation of these different events requires the activation of specific cellular targets, including protein kinase C (Clark and Brugge, 1995), PI3-OH kinase (Khwaja et al., 1997), and the small GTPase Rho (Schwartz et al., 1996). Moreover, there is evidence that integrins activate shared as well as subgroup-specific signaling pathways that contribute to organization of the cytoskeleton and activation of the mitogen-activated protein kinase (MAPK) cascades, thereby influencing immediate early gene expression (Clark and Hynes, 1997; Giancotti, 1997). Recent studies have indicated that a class of integrins that includes the α1β1, α5β1, α6β4, and αvβ3 receptors, is coupled to the Ras–MAPK signaling pathway by the adaptor protein Shc (Maniero et al., 1995; Wary et al., 1996; Maniero et al., 1997). Adhesion mediated by Shc-linked integrins promotes cell survival and progression through the G1 phase of the cell cycle in response to mitogenic growth factors, whereas adhesion mediated by other integrins results in exit from the cell cycle and, in certain cases, cell death (Wary et al., 1996; Maniero et al., 1997). These in vitro studies suggest that integrin α1β1 may regulate cell survival and growth in response to ECM components. We have used integrin α1–null mice to investigate the role of this collagen receptor in the regulation of cell survival and proliferation in vivo. The observation that α1-null dermis is hypocellular, prompted us to analyze fibroblasts from control and integrin α1–deficient mice, and to compare their growth and survival in response to ECM. α1-null fibroblasts proliferate less than their normal counterparts both in vivo and in vitro, suggesting that survival and proliferation on collagenous substrata are events mediated specifically and uniquely by activation of integrin α1β1. These results suggest that α1β1 has, among many integrin and non-integrin collagen receptors, a unique signaling function in vivo.
Materials and Methods
Cell Culture and Matrix Components
Mouse embryonic fibroblasts (EFs) were isolated from wild-type and α1-null E 14.5 embryos by trypsinization (Li et al., 1992). All cells were used for in vitro studies at matched passage numbers 3 or 4. Cells were cultured in DME supplemented with 2% or 10% FCS (GIBCO BRL, Gaithersburg, MD). Matrix components used in the study were bovine skin collagen I (Vitrogen 100; Collagen Corp., Palo Alto, CA), human placental collagen IV (Sigma Chemical Co., St. Louis, MO), human fibrinogen (gift of Z. Ruggeri, The Scripps Research Institute, La Jolla, CA), and bovine plasma fibronectin (GIBCO BRL).
Evaluation of Fibroblast Density in the Dermis
129Sv/ter wild-type and integrin α1-null mice (6-, 12-, and 24-wk old) were killed by cervical dislocation. The dorsal skin was shaved, removed, fixed in formalin, and then embedded in paraffin. Sections were stained with hematoxylin and eosin, and the number of dermal nuclei was determined by random evaluation of six high power field views of normal, as well as α1-null dermis.
Measurement of In Vivo Proliferative Index by PCNA Labeling
Integrin α1–null and –positive control mouse embryos at day 16.5 of gestation, were obtained from timed matings. Embryos were fixed in formalin and paraffin embedded. For proliferating cell nuclear antigen (PCNA) staining, transverse midsections of three wild-type, as well as three α1- deficient embryos, were stained by using the mouse monoclonal IgG2a antibody PC10 (Zymed Laboratories, Inc., South San Francisco, CA) at a dilution of 1:200. Biotinylated secondary anti–mouse antibody and avidin/ biotin-HRP–conjugated complex were used to detect bound antibody. Sections were then counterstained with hematoxylin and the dermal proliferative index (PCNA-positive cells/total number of counted cells × 100) was determined by random evaluation of 12 40× field views of dermis of each embryo. At least 900 cells around the entire circumference of the dermis of each embryo were thus counted.
For wound healing experiments, 1-cm dorsal full thickness, cutaneous incisions were made and immediately closed with interrupted 6/0 prolene sutures in four wild-type and four age-matched α1-null animals. 12 d after the incision, animals were killed and the dorsal skin was shaved, removed, fixed in formalin, and then paraffin embedded. Sections were then subjected to PCNA staining as described above. Fibroblasts in three 40× fields encompassing the wound were counted, and the proliferation index expressed as number of PCNA-positive nuclei/total fibroblast number. At least 300 cells in each wound were counted.
Cell Proliferation Assay
Control and integrin α1-null EFs were obtained from E13.5 embryos, according to the method of Li et al. (1992). Cells were detached from culture dishes by trypsin, washed, resuspended in DME containing 2% or 10% FCS, and then plated at the density of 10 × 104 cells on 24-well plates uncoated, or coated with fibrinogen (10 μg/ml), collagen I (100 μg/ml), or a mixture of collagen I (100 μg/ml) and IV (30 μg/ml). Fibrinogen solution was prepared in PBS, while the solutions containing collagen I were prepared in 0.1 M acetic acid, as described by Koyama et al. (1996). Cells were trypsinized and counted every 24 h, for a total of 72 h.
Measurement of In Vitro Proliferative Index by BrdU Labeling and FACS®
To evaluate the proliferative index, control and integrin α1–deficient EFs were plated in presence of 2% FCS, at a density of 5 × 104 cells on 16-well tissue culture chamber slides (Nunc, Inc., Naperville, IL) that were uncoated or coated with fibrinogen (10 μg/ml), collagen I (100 μg/ml), or a mixture of collagens I (100 μg/ml) and IV (30 μg/ml). After 24 h, cells were labeled with 10 μM 5′-bromo-2′-deoxy-uridine (BrdU; Sigma Chemical Co.), and incubated for a further 24 h. Cells were then washed, fixed, and stained with anti-BrdU mAbs (Sigma Chemical Co.), followed by biotinylated secondary anti–mouse antibody and avidin/biotin-HRP–conjugated complex (Vector Laboratories, Burlingame, CA). The BrdU labeling index (positive cells/total number of counted cells × 100) was determined by random evaluation of 10 40× fields, counting a minimum of 200 cells. For FACS® analysis, 3 × 105 serum-starved cells were plated on 6-well plates coated with fibronectin (10 μg/ml), fibrinogen (10 μg/ml) or collagen I (100 μg/ml) in 2% FCS. After 12 h, they were trypsinized and fixed in 70% ethanol. Cells were stained with PBS/0.1% Triton X-100/0.2 mg/ml RNase A/20 μg/ml propidium iodide, and analyzed on a Becton Dickinson FACScan® machine. S phase measurements were obtained using the Mod Fit LT v.2 program (Verity Software, Topsham, ME).
Measurement of In Vitro Apoptotic Index by DAPI Labeling
To evaluate the effect of ECM proteins on cell survival, control and integrin α1–deficient EFs were either kept in suspension, or plated in absence of serum, at the density of 7 × 104 cells on coverslips coated with fibrinogen (10 μg/ml), collagen I (20 μg/ml), or a mixture of collagen I (20 μg/ml) and IV (10 μg/ml). After 16 h, coverslips were washed with PBS to remove unattached cells, and then fixed and stained with 0.5 μg/ml DAPI (Sigma Chemical Co.)/PBS/0.1% Triton X-100. Chromatin condensation was used as morphological index for apoptotic cells. The apoptotic labeling index (cells with condensed chromatin/total number of counted cells × 100) was determined by random evaluation of 5–10 40× fields, counting a minimum of 300 cells.
In Vitro Immunoprecipitation Assay
Cell lysates were prepared according to the procedure described by Wary et al., 1996. Briefly, control and integrin α1–null EFs were growth factor starved for 8 h, and subsequently trypsinized. Cells were then resuspended in DME containing 0.1% BSA and kept at room temperature for 30 min. They were then either left in suspension or plated on dishes coated with fibronectin (10 μg/ml), collagen I (20 μg/ml), or a mixture of collagen I (20 μg/ml) and IV (10 μg/ml) all diluted in PBS. After 1 h at 37°C, the cells were washed with PBS, scraped with a rubber spatula in 50 mM Hepes, pH 7.5, 150 mM NaCl, 1% Triton X-100, and then centrifuged at 14,000 rpm at 4°C for 10 min. Protein concentration was determinded using the micro BCA assay (Pierce Chemical Co., Rockford, IL) with BSA as the standard. Immunoprecipitation and immunoblotting were performed as described by Maniero et al. (1995). Briefly, lysates containing equal amount of proteins were immunoprecipitated with affinity-purified anti-Shc polyclonal antibody (Maniero et al., 1995), and subjected to immunoblotting with anti-phosphotyrosine mAb RC-20H (Transduction Laboratories, Lexington, KY), anti-Grb2 polyclonal antibody (C-23; Santa Cruz Biotechnology, Santa Cruz, CA), and anti-Shc polyclonal antibody. Nitrocellulose-bound antibodies were detected by chemiluminescence with enhanced chemiluminescence (ECL; Amersham, Life Sciences, Little Chalfont, UK). To examine Erk activity, cell lysates containing equal amount of proteins were immunoprecipitated with anti-Erk2 polyclonal antibody C-14 (Santa Cruz Biotechnology) and subjected to in vitro kinase assay in 50 mM Tris, pH 7.5, 10 mM MgCl2 containing 5 μCi of [γ32P]ATP (4,500 Ci/mmol; ICN Biomedicals, Inc., Costa Mesa, CA) and 2.5 μg of myelin basic protein (Sigma Chemical Co.).
Results
α1-deficient Dermis Is Hypocellular
Histological analysis of skin sections from wild-type and α1-deficient mice did not reveal any substantial differences in dermal morphology and thickness (Fig. 1 A). However, we observed that the number of dermal nuclei was reduced in the α1-null skin when compared with the wild-type group (Fig. 1 A). To quantify this difference, pairs of wild-type and knockout skins obtained from 6-, 12-, and 24-wk-old 129Sv/ter mice were stained with hematoxylin and eosin, and the number of dermal nuclei was determined by random evaluation of six high power field views of dermis. The number of dermal nuclei in α1-null dermis was reduced at all stages examined as compared with the wild-type dermis (Fig. 1 B). To assess whether the difference in cell number was due to dermal fibroblasts, rather than dermal lymphocytes, the dermis of both control and α1-deficient animals were stained with CD5, a specific T-lymphocyte marker. No CD5 dermal staining was observed in the dermis of wild-type or α1-null animals (data not shown), suggesting that dermal fibroblasts rather than lymphocytes account for the difference in cell number observed in the two groups.
Figure 1.


Fibroblast density in dermis of wild-type and α1-deficient animals. (A) Representative sections of wild-type (WT) and α1-deficient (KO) 6-wk-old skin stained with hematoxylin and eosin. The α1-deficient skin has apparently normal architecture but reduced numbers of nuclei in the dermis. (B) Counts of dermal nuclei in pairs of wild-type and knockout skin obtained from 6-, 12-, and 24-wk-old 129 Sv/ter mice. Each pair of bars represents one pair of apposed sections of back skin sectioned at 5 μm. Note the lower number of dermal nuclei in α1-null dermis compared with that observed in wild-type samples. Bars and errors show mean and standard deviation of counts of dermal nuclei in six high power field views of dermis. Differences between wild-type and α1-null groups were significant with P < 0.05 by Student's t test in each case. Bar, 100 μm.
The observation that there are fewer fibroblasts in the dermis of α1-null animals than in the control group suggests the possibility of a deficiency in survival or proliferation of α1-null fibroblasts in the dermis.
α1-null Embryonic Fibroblasts Show Marked Reduction in Proliferation Compared with Their Wild-Type Counterparts
To test the hypothesis that α1β1 integrin delivers a growth signal, the proliferative index of wild-type and α1-deficient dermal fibroblasts was assessed by staining both embryonic and adult skin with antibodies to PCNA. PCNA is a DNA polymerase ε– and γ–associated protein, which is bound tightly to DNA only during S phase (Toschi and Bravo, 1988); in formalin-fixed, paraffin-embedded tissues PCNA is preferentially retained in S phase cells. Whereas results from PCNA staining are not numerically comparable to other methods of estimating S phase fraction, they do provide robust comparisons between similarly processed samples (Holt et al., 1997). As expected, there was no appreciable proliferation in sections of adult dermis of either genotype (data not shown). Analysis of 16.5-d-old embryos, however, (Fig. 2 A) revealed a marked reduction in the proliferative index of α1-null dermis as compared with the wild-type (Fig. 2 B).
Figure 2.


Proliferative index of wild-type and α1-null embryonic dermal fibroblasts. (A) Mid cross-section of 16.5 embryos were stained with anti-PCNA antibodies, and subsequently counterstained with hematoxylin. Note the higher number of dermal PCNA-positive cells in the wild-type (WT) dermis, as compared with their α1-null (KO) counterparts. (B) The dermal proliferative index (number of PCNA positive cells/total number of cells × 100) of three wild-type and three α1-null 16.5 embryos was determined by random evaluation of 12 different 40× microscopic fields for each sample, counting a minimum of 900 cells around the entire circumference of embryonal dermis. Bars and errors indicate the mean and standard deviation. Differences between wild-type and α1-null groups were significant with P < 0.05 by Student's t test. Bar, 30 μm.
To induce fibroblast proliferation in adult dermis 1-cm full-thickness, dorsal cutaneous incisions were generated in age-matched wild-type and α1-null animals, and allowed to heal by primary intention. Sections of 12-d-old wounds were stained with antibodies to PCNA (Fig. 3 A). No differences in the proliferative index of dermal fibroblasts of either genotype were evident in the wounded areas (Fig. 3 B). We also investigated the proliferative index of different embryonic, as well as adult tissues known to express α1β1 integrin, such as liver hepatocytes (Gullberg et al., 1990) and smooth muscle. No appreciable differences were observed between wild-type and α1-null embryos or adults in these tissues.
Figure 3.
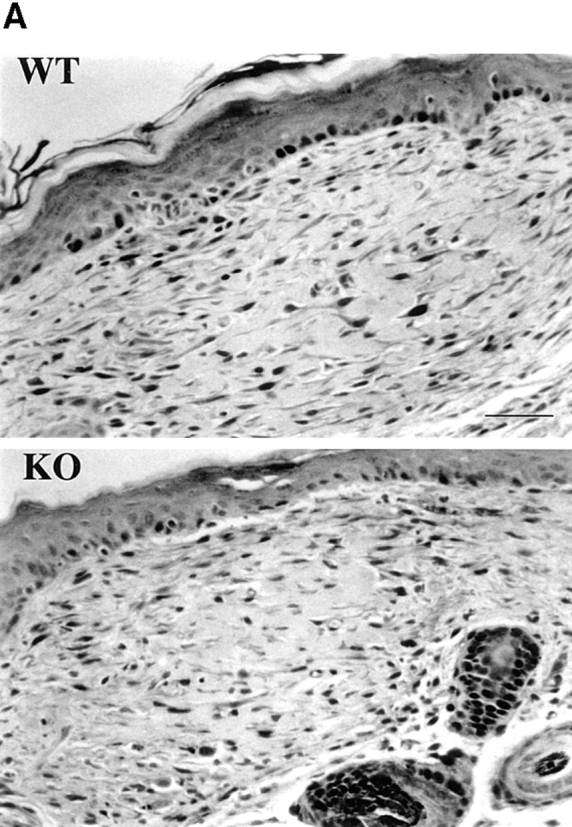
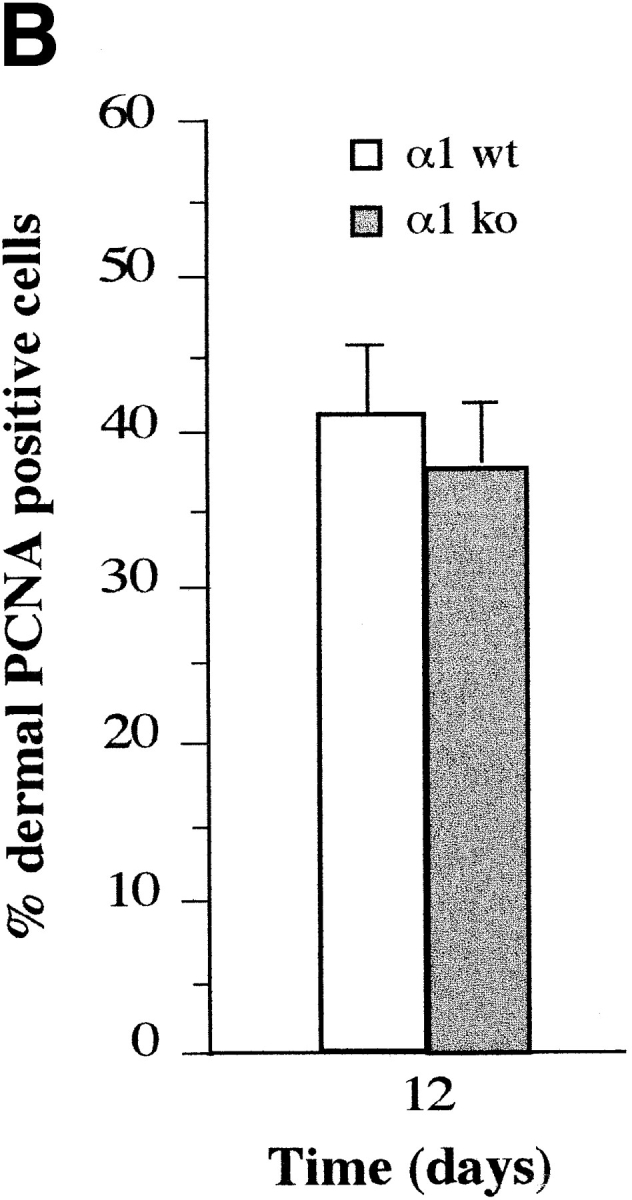
Proliferative index of wild-type and α1-null adult fibroblasts in wounded dermis. (A) Sections of wild-type (WT) and α1-deficient (KO) wounded skin, 12 d after incision and primary closure, stained with anti-PCNA antibodies, and subsequently counterstained with hematoxylin. The proportion of PCNA-positive cells in the dermal wound (seen as dark nuclei in the micrograph) was similar in wild-type and α1-null animals. (B) The dermal proliferative index (number of PCNA positive cells/total number of cells × 100) was determined by random evaluation of 3 different 40× microscopic fields for each sample, counting a minimum of 300 cells. Bars and errors indicate the mean and standard deviation. Bar, 100 μm.
The finding on embryonic dermis prompted us to determine whether the differences in cell survival and proliferation observed in vivo were due to the interaction of specific ECM components with the α1β1 receptor. Therefore, we analyzed the growth profile of fibroblasts in vitro, by using embryonic fibroblasts, derived from control and α1-null animals, plated in the presence of low or high serum concentration on different substrata (Fig. 4, A and B). Whereas both wild-type and α1-null EFs showed similar growth profiles on tissue culture plastic, fibrinogen, (Fig. 4, A and B) or fibronectin (not shown), on collagen substrata α1-deficient EFs showed a marked reduction in proliferation in comparison to wild-type cells. Similar deficiency in α1-null cell growth was seen when cells were plated on collagen IV alone (not shown). As expected, the differences in cell growth on collagen substrata were most pronounced in growth media containing 2% serum, and were less pronounced in the presence of 10% serum (Fig. 4 B), where both growth factors and ligands for αv and α5 integrins, vitronectin and fibronectin, were more abundant. Attempts to grow EFs in defined media in the absence of serum but containing bFGF and EGF (Wary et al., 1996) were unsuccessful.
Figure 4.

Growth of wild-type and α1-deficient embryonic fibroblasts on different substrata. 10 × 104 log phase wild-type (□) and α1-null (•) EFs were plated in presence of 2% FCS (A) or 10% FCS (B) on 24-well plates uncoated, or coated with fibrinogen (10 μg/ml), collagen I (100 μg/ml), or a mixture of collagen I (100 μg/ml) and IV (30 μg/ml). Two wells per group for each treatment were counted at the time point indicated. This experiment is representative of three repeated experiments. Note the reduced growth of α1-null EFs, on collagen-coated dishes, as compared with the wild-type cells. Bars and errors indicate the mean and the higher number from two dishes per each group.
The differences in fibroblast proliferation on collagens was also confirmed by BrdU incorporation and anti-BrdU staining. After 48 h of culture in 2% serum, a similar proportion of wild-type and α1-deficient EFs was BrdU positive when plated on tissue culture plastic, or fibrinogen (Fig. 5 A). In contrast, a marked reduction in BrdU incorporation was observed in α1-null EFs plated on collagen I (Fig. 5 A), or a mixture of collagen I and IV (Fig. 5 A). Similar results were observed when serum-starved cells were plated on collagenous or non-collagenous substrata in presence of 2% FCS, and then analysed by FACS® after 12 hours (Fig. 5 B).
Figure 5.

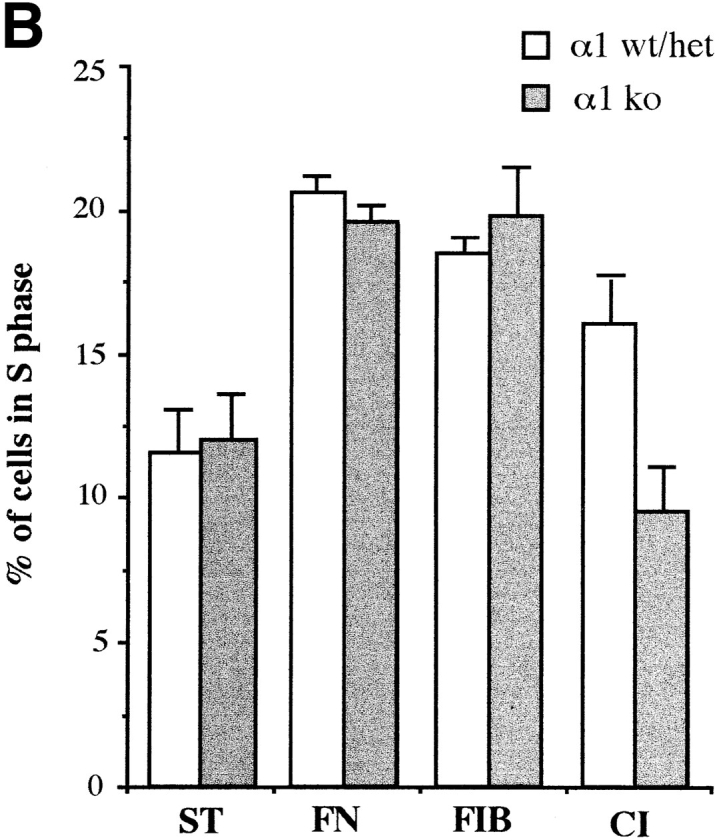
Profilerative index of wild-type and α1-null embryonic fibroblasts in vitro. (A) EFs were plated in presence of 2% FCS onto dishes uncoated or coated with fibrinogen (10 μg/ml), collagen I (100 μg/ml), or a mixture of collagen I (100 μg/ml) and collagen IV (30 μg/ml). 24 h after plating, cells were labeled with 10 μM BrdU (Sigma Chemical Co.), and incubated for a further 24 h. After staining with anti-BrdU mAbs, the Brdu labeling index (positive cells/total number of counted cells × 100) was determined by random evaluation of five different 40× microscopic fields for each duplicate sample, counting a minimum of 200 cells. Bars and errors indicate the mean and standard deviation. The differences seen between wild-type and α1-null groups on collagens are significant with P < 0.05 using a Student's t-test on the 10 microscopic fields counted for each sample. (B) S phase entry of wild-type and α1-null embryonic fibroblasts after serum starvation. Passage 4 EFs were starved for 24 h, before plating on various substrata in the presence of 2% FCS. After 12 h cells were recovered by trypsinization, and subjected to FACS® analysis as described in Materials and Methods. Bars indicate the mean and error bars the distance to the higher value for each pair of samples analyzed. TC, tissue culture; FN, fibronectin; FIB, fibrinogen; CI, collagen I; CI + IV, collagen I + IV; ST, serum starved.
While it is clear that apoptosis occurs via distinct cellular pathways, it is a frequent observation that circumstances preventing cell proliferation are associated with an increase in cell death. To determine whether α1-null EFs were more susceptible to apoptosis than their wild-type counterparts, cells were plated on different substrata, or kept in suspension, in the absence of serum, and the apoptotic index was determined by DAPI staining after 16 h. As expected, 60–70% of cells of either genotype became apoptotic in suspension (Fig. 6 B), while only 2% of either genotype plated on fibrinogen became apoptotic (Fig. 6, A and B). On collagenous matrices however (Fig. 6, A and B), α1-null EFs showed a sevenfold increase in apoptosis over wild-type EFs.
Figure 6.


Apoptotic index of wild-type and α1-null EFs in vitro. (A) EFs were plated in absence of serum on coverslips coated with collagen I (20 μg/ml) or fibrinogen (10 μg/ ml). 16 h after plating, coverslips were washed to remove unattached cells, fixed, and then stained with 0.5 μg/ml DAPI. Chromatin condensation was used to score positive apoptotic cells. Note the higher number of apoptotic-positive cells in α1-null EFs as compared with wild-type cells when plated on collagen matrix. (B) The apoptotic index (cells with condensed chromatin/total number of counted cells × 100) of EFs, kept in suspension or plated in dishes coated as indicated, was determined by random evaluation of five different 40× microscopic fields for each duplicate sample, counting a minimum of 300 cells. Bars and errors indicate the mean and standard deviation. Significant differences were seen between wild-type and α1-null groups on collagens with P < 0.05 using a Student's t-test on the 10 microscopic fields counted for each sample. SUSP, suspension; FIB, fibrinogen; CI, collagen I; C I + IV, collagen I + IV. Bar, 40 μm.
In conclusion, the in vitro results are in agreement with the in vivo observation, and suggest that the reduction in dermal fibroblast number observed in α1-null mice is caused by a combination of reduced survival and reduced proliferation.
α1-null Embryonic Fibroblasts Fail to Activate the Adaptor Protein Shc When Plated on Collagens
The differences in proliferation between α1-null and wild-type EFs on collagen substrata suggests that the interaction between integrin α1β1 and collagens is required for the activation of a specific survival/proliferation pathway. It has been shown (Wary et al., 1996) that activation of α1β1 causes recruitment and tyrosine phosphorylation of the adaptor protein Shc. This protein, via recruitment of Grb2 and activation of the the Ras-MAP kinase pathway, cooperates with mitogens to mediate cell survival and proliferation. Based on this observation, we investigated the activation of Shc by wild-type and α1-deficient EFs upon adhesion on different substrata. We have previously shown that α1-deficient fibroblasts fail to adhere to collagen IV (Gardner et al., 1996). Thus, to analyze the contribution of this molecule in the activation of Shc via α1 integrin, α1-deficient cells were plated on a mixture of collagen I and IV, or on collagen I alone, and the level of Shc activation compared with that observed in their normal counterparts. With these substrata, the extent of adhesion of wild-type and α1-null cells is indistinguishable. Cells kept in suspension, or plated on fibronectin were used, respectively, as negative and positive controls for activation of the Shc pathway. Cell lysates were immunoprecipitated with anti-Shc antibody and subjected to immunoblotting with anti-phosphotyrosine antibody to determine Shc activation, with anti-Grb2 antibody to examine recruitment of Grb2, and with anti-Shc antibody to check for equal loading. Cell lysates were also immunoprecipitated with anti-Erk2 antibody and subjected to in vitro kinase assay to determine MAPK activation. As expected, EFs of both genotype failed to cause Shc phosphorylation when kept in suspension (Fig. 7 B), despite containing normal levels of protein (Fig. 7 A). Consequently, no recruitment of Grb 2 (Fig. 7 C), or activation of MAP kinase (Fig. 7 D) was observed. In contrast, both cell types activated Shc protein and its related downstream cascade, when plated on fibronectin (Fig. 7, A–D). Striking differences in the activation of Shc, and its related downstream events, however, were observed between wild-type and α1-null EFs after adhesion on collagen I or a mixture of collagen I and IV. α1-null EFs entirely failed to activate Shc when plated for 1 h on substrata of collagen I or a mixture of collagen I and IV in contrast to wild-type cells (Fig. 7 B). The failure to activate Shc by α1-deficient cells was accompanied by a downstream deficiency in recruitment of Grb 2 (Fig. 7 C) and subsequent MAPK activation (Fig. 7 D). Taken together with the growth deficiency we have observed in α1-null EFs on collagen substrata (Fig. 4), this finding demonstrates that α1 collagen receptor has a unique and pivotal role in regulating cell survival and proliferation on collagenous matrices.
Figure 7.

α1-null EFs fail to recruit and activate Shc and subsequent downstream events when plated on collagens. EFs were serum starved for 8 h and then trypsinized. Cells were then resuspended in DME/0.1% BSA and kept at room temperature for 30 min. They were then either left in suspension or plated on dishes coated with fibronectin (10 μg/ml), collagen I (20 μg/ml), or a mixture of collagen I (20 μg/ml) and IV (10 μg/ml). After 1 h at 37°C, cells were lysed and an equal amount of proteins were immunoprecipitated with anti-Shc polyclonal antibody (A–C), and subjected to immunoblotting with anti-Shc polyclonal antibody (A), anti–P-Tyr mAb (B), or anti–Grb-2 mAb (C). In D, equal amount of proteins were immunoprecipitated with anti-ERK2 polyclonal antibody and subjected to in vitro kinase assay with myelin basic protein as substrate. Note that when plated on collagens, α1-null EFs fail to phosphorylate Shc (B) or to activate events downstream of Shc (C and D), despite containing normal level of Shc protein (A). SUSP, suspension; FN, fibronectin; CI, collagen I; C I + IV, collagen I + IV.
Discussion
In this study we have used integrin α1–deficient mice to investigate the role of α1β1 receptor in the regulation of dermal fibroblast growth. We provide evidence that integrin α1β1, the major collagen-binding receptor, controls the proliferation of dermal fibroblasts both in vivo and in vitro, and that cell survival and proliferation on collagen substrata are mediated specifically and uniquely by integrin α1β1.
In fibroblasts integrin α1β1 regulates different functions upon binding to collagen matrices. One of these is the regulation of collagen synthesis. We have found that the α1-null animals lack negative feedback regulation of collagen synthesis and synthesize excess dermal collagen in vivo. This is not apparent morphologically, as the animals also show increased synthesis of collagenases, and thus compensate excess synthesis with increased breakdown (Gardner, H., A. Broberg, A. Pozzi, J. Heino, submitted manuscript). This dysregulation appears to cause a small reduction in tensile strength of intact skin (Davidson, J., unpublished results).
Another function of integrin α1β1, analyzed in the present paper, is the control of cell growth and proliferation. We have shown that fibroblast growth, proliferation, and survival are events mediated by integrin α1 activation of the Ras-MAPK pathway, as previously suggested by the in vitro studies of Wary et al. (1996). We have demonstrated that when plated on collagen matrices, α1-deficient fibroblasts fail to activate the adaptor protein Shc, showing a reduced cell proliferation compared with their wild-type counterparts. This observation is complemented by our observation of dermal fibroblasts in vivo. Analysis of embryonic skin indicates that wild-type dermal fibroblasts proliferate more than their α1-null counterparts, supporting the hypothesis that α1 integrin plays a role in controlling cell growth. This difference in cell growth was not evident in adult skin, probably because the overall rate of proliferation in the adult dermis is negligible. Thus, our observation that adult α1-null dermis is hypocellular as compared with wild-type can be interpreted as the result of a defective proliferation of α1-deficient fibroblasts during embryogenesis, although a very slowly accumulated deficiency in proliferation in the adult α1-null dermis cannot be ruled out by our studies.
To induce fibroblast proliferation in adult dermis, we have performed dermal wound healing experiments on wild-type and α1-deficient animals. 12 d after the incision, no differences in the proliferative index of dermal fibroblasts of either genotype were evident in the wounded area. This may be explained by the fact that, during the first stages of wound healing fibrinogen, rather than collagen, is the major ECM component to be deposited in the wound, accompanied by local synthesis of fibronectin. These molecules could regulate, via αvβ3 and α5β1 integrins, respectively, cell growth of both wild-type and α1-null fibroblasts, reducing or eliminating the differences in cell proliferation visible on collagen substrata. In this context, we have observed that when plated on fibronectin both wild-type and α1-deficient embryonic fibroblasts activate the Shc pathway, and show a similar growth profile. Fibroblasts express α5β1 (Dalton et al., 1992) and it has been shown that activation of α5β1 leads to recruitment of Shc (Wary et al., 1996). Similarly, our results on the proliferative index of wild-type and α1-deficient hepatocytes suggest the possibility that different ECM–integrin interactions could take place in these cells, and be dominant over the lack of α1 integrin in the control of cell survival and proliferation. In fact, given the variety of possible ligand integrin interactions, which do promote proliferation, it is striking that embryonic dermal fibroblasts have a perceptible dependence on the α1–collagen interaction for proliferation in vivo.
Our in vitro observation that activation of Shc on collagenous matrices occurs only in wild-type cells indicates that, although fibroblasts express three collagen receptors α1β1, α2β1, and α3β1 (Zutter and Santoro, 1990; Gardner et al., 1996), α1β1 integrin is the only one of these able to deliver a specific collagen-induced signal to the Ras-MAPK pathway, via Shc activation. Recently, it has been observed that the Shc phosphotyrosine binding domain can also bind to the DDR tyrosine kinase receptor after collagen stimulation and receptor autophosphorylation (Vogel et al., 1997), suggesting the possibility that DDR and integrin receptors may cooperate in the regulation of cell survival and proliferation. However, the observation that the MAPK pathway is not activated upon binding of DDR to collagen (Vogel et al., 1997), strongly supports the hypothesis that collagen- dependent proliferation is mainly mediated via activation of integrin α1β1.
Integrin α1β1 is only one of a larger family of integrins able to activate Shc. This subset of receptors includes integrin α5β1, the receptor involved in the control of cell cycle progression and proliferation in human umbilical vein endothelial cells (HUVECs) upon binding to fibronectin (Wary et al., 1996) and α6β4, which plays a role in regulating keratinocyte proliferation, via activation of the same Ras-MAPK pathway (Maniero et al., 1997). Thus, the same adaptor molecule, activated by different integrin–ligand interaction (α1β1–collagen, α5β1–fibronectin, α6β4–laminin, and αvβ3–fibrinogen) is able to regulate cell growth of different cell types. It is interesting to note in this context that integrin α5–null embryos, which die in mid-gestation, show a failure of proliferation of neural crest derivatives (Goh et al., 1997), suggesting that these may depend upon the α5β1–fibronectin interaction for proliferation in a manner analagous to the α1β1–collagen interaction in the dermis.
Whereas activation of Shc leads to cell growth and proliferation, adhesion mediated by integrins not linked to Shc appears unable to prevent apoptotic death (Wary et al., 1996). We have observed that α1-null cells, although able to adhere on collagen I or a mixture of collagen I and IV indistinguishably from their wild-type counterparts, are more susceptible to apoptosis, indicating that α1 integrin effects both fibroblast proliferation and survival. Shc signaling may contribute to protection from apoptosis by activating Ras and thereby PI-3 kinase, as activated forms of Ras and PI-3 kinase protect from suspension induced apoptosis (Khwaja et al., 1997). FAK activation by various integrins has also been implicated in cell survival (Frisch and Ruoslahti, 1997; Hungerford et al., 1996), and may too act via PI-3 kinase (Chen and Guan, 1994).
It is striking that the adhesive function of integrin α1β1 is apparently dispensable, permitting normal morphogenesis in the null, it is its role in regulating cell proliferation which, in its absence, gives rise to a perceptible phenotype in vivo. The observation that only one of the three dermal fibroblast collagen receptors specifically controls proliferation suggests that different integrin receptors for a single ligand accomplish different and unique functions within a single cell. Such functions may be quite distinct from, and in some instances more important than, their adhesive functions.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant R29-AR4415 and the Dr. Mark Flapan Memorial Grant Award from the Scleroderma Federation/United Scleroderma Foundation to H. Gardner, and by NIH grant CA78901 to F.G. Giancotti.
Abbreviations used in this paper
- BrdU
5′-bromo-2′-deoxy-uridine
- ECM
extracellular matrix
- EF
embryonic fibroblasts
- MAPK
mitogen-activated protein kinase
- PCNA
proliferating cell nuclear antigen
Note Added in Proof
A recent study provides further evidence that integrin-mediated Shc signaling regulates cell proliferation in vivo (Murgia, C., P. Blaikie, N. Kim, M. Dans, H.T. Petrie, and F.G. Giancotti. 1998. EMBO (Eur. Mol. Biol. Organ.) J. In press).
Footnotes
H. Gardner thanks J. Trotter for training in flow cytometry, and J. Leopard for histotechnology.
Address all correspondence to Humphrey A. Gardner, Department of Cell Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037. Tel.: (619) 784-9821. Fax: (619) 784-9927. E-mail: humphrey@scripps.edu
References
- Adams JC, Watt FM. Regulation of development and differentiation by the extracellular matrix. Development (Camb) 1993;117:1183–1198. doi: 10.1242/dev.117.4.1183. [DOI] [PubMed] [Google Scholar]
- Broberg A, Heino J. Integrin α2β1-dependent contraction of floating collagen gels and induction of collagenase are inhibited by tyrosine kinase inhibitors. Exp Cell Res. 1996;228:29–35. doi: 10.1006/excr.1996.0295. [DOI] [PubMed] [Google Scholar]
- Chen HC, Guan JL. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1994;91:10148–10152. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Clark EA, Hynes RO. 1997 Keystone symposium on signal transduction by cell adhesion receptors. Biochim Biophys Acta. 1997;1333:9–16. doi: 10.1016/s0304-419x(97)00028-0. [DOI] [PubMed] [Google Scholar]
- Dalton SL, Marcantonio EE, Assoian RK. Cell attachment controls fibronectin and α5β1 integrin level in fibroblasts. Implication for anchorage-dependent and -independent growth. J Biol Chem. 1992;276:8186–8191. [PubMed] [Google Scholar]
- Frisch SM, Ruoslahti R. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- Gardner HAR, Kreidberg JA, Koteliansky VE, Jaenisch R. Deletion of integrin α1 by homologous recombination permits normal murine development but gives rise to a specific deficit in cell adhesion. Dev Biol. 1996;175:301–313. doi: 10.1006/dbio.1996.0116. [DOI] [PubMed] [Google Scholar]
- Giancotti FG. Integrin signaling: specificity and control of cell survival and cell progression. Curr Opin Cell Biol. 1997;9:691–700. doi: 10.1016/s0955-0674(97)80123-8. [DOI] [PubMed] [Google Scholar]
- Goh KL, Yang JT, Hynes RO. Mesodermal defects and cranial neural crest apoptosis in alpha5 integrin-null embryos. Development (Camb) 1997;124:4309–4319. doi: 10.1242/dev.124.21.4309. [DOI] [PubMed] [Google Scholar]
- Gullberg D, Turner DC, Borg TL, Terracio L, Rubin K. Different β1-integrin collagen receptors on rat hepatocytes and cardiac fibroblasts. Exp Cell Res. 1990;190:254–264. doi: 10.1016/0014-4827(90)90194-f. [DOI] [PubMed] [Google Scholar]
- Holt PR, Moss SF, Kapetanakis AM, Petrotos A, Wang S. Is Ki-67 a better proliferative marker in the colon than proliferating cell nuclear antigen? . Cancer Epidemiol Biomarkers Prev. 1997;6:131–135. [PubMed] [Google Scholar]
- Hungerford JE, Compton MT, Matter ML, Hoffstrom BG, Otey CA. Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO (Eur Mol Biol Organ) J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama H, Raines EW, Bornfeldt KE, Roberts JM, Ross R. Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of Cdk2 inhibitors. Cell. 1996;87:1069–1078. doi: 10.1016/s0092-8674(00)81801-2. [DOI] [PubMed] [Google Scholar]
- Langholz O, Rockel D, Mauch C, Kozlowska E, Bank I, Krieg T, Eckes B. Collagen and collagenase gene expression in three dimensional collagen lattices are differentially regulated by α1β1 and α2β1 integrins. J Cell Biol. 1995;131:1903–1915. doi: 10.1083/jcb.131.6.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutaion of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Maniero F, Pepe A, Wary KK, Spinardi L, Mohammadi M, Schlessinger J, Giancotti FG. Signal transduction by the α6β4 integrin: distinct β4 subunit sites mediate recruitment of Shc/Grb2 and association with the cytoskeleton of hemidesmosomes. EMBO (Eur Mol Biol Organ) J. 1995;14:4470–4481. doi: 10.1002/j.1460-2075.1995.tb00126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniero F, Murgia C, Wary KK, Curatola AM, Pepe A, Blumemberg M, Westiwick JK, Der CJ, Giancotti F. The coupling of α4β4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. EMBO (Eur Mol Biol Organ) J. 1997;16:2365–2375. doi: 10.1093/emboj/16.9.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiro JA, Chan BM, Roswit WT, Kassner PD, Pentland AP, Hemler ME, Eisen AZ, Kupper TS. Integrin α2 β1 (VLA-2) mediates reorganization and contraction of collagen matrices by human cells. Cell. 1991;67:403–410. doi: 10.1016/0092-8674(91)90191-z. [DOI] [PubMed] [Google Scholar]
- Schwartz MA, Toksoz D, Khosravi-Far R. Transformation by Rho exchange factor oncogene is mediated by activation of an integrin-dependent pathway. EMBO (Eur Mol Biol Organ) J. 1996;15:6525–6530. [PMC free article] [PubMed] [Google Scholar]
- Shrivastava A, Radziejeweski C, Campbell E, Kovac L, McGlynn M, Ryan TE, Davis S, Goldfarb MP, Glass DJ, Lemke G, Yancopoulos GD. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell. 1997;1:25–34. doi: 10.1016/s1097-2765(00)80004-0. [DOI] [PubMed] [Google Scholar]
- Sutherland AE, Calarco PG, Damsky CH. Developmental regulation of integrin expression at the time of implantation in the mouse embryo. Development (Camb) 1993;119:1175–1186. doi: 10.1242/dev.119.4.1175. [DOI] [PubMed] [Google Scholar]
- Toschi L, Bravo R. Changes in cyclin/proliferating cell nuclear antigen distribution during DNA repair synthesis. J Cell Biol. 1988;107:1623–1628. doi: 10.1083/jcb.107.5.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1998;1:13–23. doi: 10.1016/s1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- Wary KK, Maniero F, Isakoff SJ, Marcantonio EE, Giancotti FG. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Yamamoto M. Cell adhesion receptors for native and denatured type I collagens and fibronectin in rabbit arterial smooth muscle cells in culture. Exp Cell Res. 1994;214:258–263. doi: 10.1006/excr.1994.1256. [DOI] [PubMed] [Google Scholar]
- Zutter MM, Santoro SA. Widespread histologic distribution of the α2β1 integrin cell-surface collagen receptor. Am J Pathol. 1990;137:113–120. [PMC free article] [PubMed] [Google Scholar]