

Abstract
The baculovirus fusogenic activity depends on the low pH conformation of virally-encoded trimeric glycoprotein, gp64. We used two experimental approaches to investigate whether monomers, trimers, and/or higher order oligomers are functionally involved in gp64 fusion machine. First, dithiothreitol (DTT)- based reduction of intersubunit disulfides was found to reversibly inhibit fusion, as assayed by fluorescent probe redistribution between gp64-expressing and target cells (i.e., erythrocytes or Sf9 cells). This inhibition correlates with disappearance of gp64 trimers and appearance of dimers and monomers in SDS-PAGE. Thus, stable (i.e., with intact intersubunit disulfides) gp64 trimers, rather than independent monomers, drive fusion. Second, we established that merger of membranes is preceded by formation of large (greater than 2 MDa), short-lived gp64 complexes. These complexes were stabilized by cell–surface cross-linking and characterized by glycerol density gradient ultracentrifugation. The basic structural unit of the complexes is stable gp64 trimer. Although DTT-destabilized trimers were still capable of assuming the low pH conformation, they failed to form multimeric complexes. The fact that formation of these complexes correlated with fusion in timing, and was dependent on (a) low pH application, (b) stable gp64 trimers, and (c) cell–cell contacts, suggests that such multimeric complexes represent a fusion machine.
Keywords: viral fusion, baculovirus gp64, fusion protein assembly, fusion inhibitors, thiol/disulfide exchange
Numerous viruses enter their host cell by fusion of the viral envelope with the cell membrane. The fusogenic activity depends on the virally encoded fusion proteins such as baculoviral gp64, influenza hemagglutinin (HA)1 or human immunodeficiency virus (HIV) gp120/gp41 which upon interaction with the specific trigger (e.g., acidic pH or specific receptor) assume fusion-competent conformations (Hernandez et al., 1996; Dimitrov, 1997). Recent studies revealed striking structural similarities between different viral fusion proteins as well as common functional motifs in other types of biological fusions such as exocytosis or protein trafficking (Zimmerberg et al., 1993; Hernandez et al., 1996; Monck and Fernandez, 1996; Hay and Scheller, 1997). However, there is still very little known about the molecular mechanisms of protein-mediated fusion, even for the best characterized viral fusion proteins.
Similar to many other fusion proteins, baculovirus gp64 and influenza virus HA are present on the viral membrane and on the membrane of the infected cells as homotrimers. Interaction with H+ in the acidified endosome milieu triggers fusion protein refolding which is a prerequisite for membrane fusion. It is not yet clear whether the initial oligomeric organization of viral fusion proteins is conserved upon their refolding into the fusion-competent conformation. For instance, trigger-dependent activation of the proteins can result in dissociation of existing HA trimers followed by monomer reassembly into new fusion-competent oligomers (Doms et al., 1986). Alternatively, rather than causing trimer dissociation, low pH triggering may in fact stabilize the initial oligomeric structure (Carr and Kim, 1993; Bullough et al., 1994).
Low pH-restructured fusion proteins may mediate fusion acting as individual monomers (Bundo-Gibson et al., 1986; Morita et al., 1987), trimers or complexes of higher order (Hernandez et al., 1996). A number of indirect lines of evidence suggest that multiple trigger-activated fusion proteins work together to mediate a merger of membrane bilayers. Functional studies, including the analysis of fusion rate dependence upon the surface density of HA, imply that fusion may be mediated by multiple HA molecules acting in a cooperative manner (Clague et al., 1991; Ellens et al., 1990; Danieli et al., 1996; Blumenthal et al., 1996). The importance of the HA mobility for fusion, along with data on the decrease in HA lateral diffusion in membrane after low pH application (Junankar and Cherry, 1986; Gutman et al., 1993) indicate that fusion may involve aggregation of several activated HA molecules. The restriction of lipid flow between membranes at early stages of HA-mediated fusion also suggests that the site of the actual merger of membranes is surrounded by a complex of activated HA molecules (Tse et al., 1993; Zimmerberg et al., 1994; Chernomordik et al., 1998). For gp64-mediated fusion, analysis of the electrophysiological data on the kinetics of the fusion pore formation and the size of the initial fusion pore indicate that this fusion involves formation of multiprotein complex comprised of ∼5–8 gp64 trimers (Plonsky and Zimmerberg, 1996).
To characterize the oligomeric state of viral fusion proteins in the functional fusion machine, in the present work we studied the dependence of degree of gp64 oligomerization on the extent of fusion. Baculovirus gp64-mediated fusion of viral envelope with endosome membrane upon acidification of the endosome can be reconstructed and studied in a simpler experimental system: low pH-induced cell–cell fusion. Gp64-expressing host cells (baculovirus-infected or transfected) fuse with contacting target cells that do, or do not express gp64 (Leikina et al., 1992; Blissard and Wenz, 1992).
Baculovirus gp64 is rather well characterized biochemically (Volkman and Goldsmith, 1984; Volkman et al., 1984; Roberts and Faulkner, 1989; Monsma and Blissard, 1995; Oomens et al., 1995). Monomeric forms of gp64 are not transported to the surface of the infected cell, and they are usually degraded within 30–45 min after synthesis (Oomens et al., 1995). Trimers of phosphorylated, acylated and N-glycosylated gp64 expressed in the membranes of infected cells and in the viral envelope are stabilized by intermolecular disulfide bonds formed posttranslationally (Volkman and Goldsmith, 1984). Interestingly, there are two isomeric gp64 trimers present on the membranes which were identified as distinct bands in nonreducing SDS-PAGE. It is not known whether these two trimeric gp64 forms, which apparently differ in the pattern of disulfide bonding (Oomens et al., 1995), differ in any way in their functional properties.
In the present study we found that the fusogenic activity of gp64 is dependent on the presence of stable trimers in the membrane. The term stable trimer is used here to denote gp64 trimer with intact intersubunit disulfides. DTT treatment of gp64-expressing cells reduced intersubunit disulfide bonds mostly in one of the two trimeric isoforms, and reversibly inhibited fusion. Although monomers in the reduced trimer underwent a low pH conformational change, restoration of disulfide bonds was essential for fusion. These results argue against the possibility that monomeric subunits act independently in fusion.
Our data also indicate that the functional fusion machine is composed of up to 10 stable, low pH-activated gp64 trimers assembled into a multiprotein complex. The appearance of gp64 complexes was limited to a narrow time span and depended on low pH triggering, stable trimers, and the presence of target membrane.
Materials and Methods
Materials
The current study focused on gp64 from two different strains of the baculovirus, i.e., Autographa californica (Ac) and Orgyia pseudotsugata (Op). Ac gp64 was expressed on the surface of infected Sf9 cells, whereas Op gp64 was expressed on the surface of Op1D cells (i.e., stably transfected Sf9 cells). The Ac strain of baculovirus was purchased from Invitrogen (San Diego, CA). Op1D cells and the tissue culture supernatant of monoclonal antibodies (mAb) OpE6A (specific for Op gp64), AcV5 (specific for both Op and Ac gp64), and AcV1 (specific for Ac gp64) were kindly provided by G. Blissard (Cornell University, Ithaca, NY). Goat anti– mouse IgG conjugated with horseradish peroxidase and alkaline phosphatase as well as paranitrophenyl phosphate were purchased from Pierce Chemical Co. (Rockford, IL). Cross-linking reagents cleavable N-hydroxysuccinimide ester (DTSSP), BS3, and sulfo NHS biotin were also obtained from Pierce Chemical Co. Protease inhibitor cocktail tablets were purchased from Boehringer Mannheim (Mannheim, Germany). Enhanced chemiluminescence (ECL) and enhanced chemifluorescence (ECF) reagents were obtained from Amersham (Buckinghamshire, UK). TNM-FH medium was obtained from Invitrogen (San Diego, CA). The lipid-soluble probe, PKH26, was purchased from Sigma Chemical Co. (St. Louis, MO) whereas, the water-soluble probe, 6-carboxyfluorescein (CF), was purchased from Molecular Probes (Eugene, OR). Immobilon-P filters were obtained from Millipore (Bedford, MA). Dithiothreitol (DTT) was purchased from ICN Biomedicals, Inc. (Irvine, CA); glutathione disulfide (GSSG), and sodium iodoacetate (NaIA) were purchased from Sigma Chemical Co. Lauroyl lysophosphatidylcholine (LPC) was purchased from Avanti Polar Lipids (Birmingham, AL).
Cells
Sf9 and Op1D cells were grown in 25-cm2 flasks at 27°C in complete TNM-FH medium (Summers and Smith, 1987) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS). In the experiments involving baculovirus-infected cells, confluent monolayers of Sf9 cells were infected with the budded form of the Ac strain of baculovirus at a multiplicity of infection (m.o.i.) of 1. After 1 h of viral adsorption period, the unbound virus was removed and the cells were incubated for 24 h at 27°C.
Human red blood cells (RBC) were freshly isolated from the whole blood obtained from the National Institutes of Health (Bethesda, MD) blood bank and labeled with the fluorescent dyes. RBC were loaded with both 5 mM of CF and 5 μM of PKH26 or, in most experiments, with only 7 μM of PKH26 as described previously (Chernomordik et al., 1998). Excess of dye was removed by three successive washes of RBC in phosphate buffered saline (PBS). In the experiments on Sf9-Op1D cell fusion, Sf9 cells were labeled with calcein AM by injection of 300 μl of the probe stock solution (1 mg/ml) into 5 ml of the culture medium/25-cm2 flask. The unincorporated dye was removed by two 5-ml washes with fresh medium.
Quantification of Cell–Cell Fusion and Binding
Fusion was assayed by fluorescent dye redistribution from labeled cells (RBC or noninfected Sf9 cells) to unlabeled gp64-expressing cells (i.e., Op1D or infected Sf9 cells). In a few experiments we used RBC labeled by membrane- and water-soluble dye in order to monitor both content (defined as CF redistribution) and lipid mixing (defined as PKH26 redistribution). Most fusion assays, however, were conducted with RBC labeled only with the lipid-soluble probe (i.e., PKH26). Labeled RBC were resuspended and mixed with unlabeled Op1D or infected Sf9 cells in 1:2 ratio. After incubation of RBC with Op1D or infected Sf9 cells and subsequent washing, 0–2 labeled RBC remained bound per gp64-expressing cell. In the experiments where Sf9–Op1D cell fusion was followed, calcein AM-labeled cells were resuspended and mixed with the unlabeled cells in a 1:3 ratio.
Fusion was triggered by a 1-min treatment with pH 5.1 medium. Low pH medium was prepared by titrating PBS with citric acid. Fusion extent was quantified using fluorescence microscopy as the ratio of redistributed dye bound RBC to the total number of the bound RBC. Fusion extents were assayed starting 1 min after the low pH pulse. Longer incubation at low pH (i.e., 30 min) did not increase the extent of fusion.
Cell–cell binding was quantified using a phase–contrast or fluorescence microscopy and expressed as a percentage of gp64-expressing cells with bound target cells normalized by the total number of cells in the field. An average of 5–10 fields were quantified.
SDS-PAGE and Western Blotting
Most of the fusion assays were complemented with sodium dodecyl sulfate-PAGE (SDS-PAGE) and Western blot analyses. Cells were prepared in the same manner as for the fusion assays and lysed in 200 μl of nonreducing SDS-PAGE lysis buffer (50 mM Tris-HCl, 20 mM EDTA, pH 7.2, 1.5% SDS, 37.5 mM iodoacetamide, 10% glycerol, 0.01% bromphenol blue). Immediately after lysis, samples were transferred to ice, heated to 100°C for 5 min, and stored at −25°C until use. If required, samples were reduced with 50 mM DTT postlysis to yield monomers. Prepared lysate was analyzed by 6% continuous, or 4–12% gradient SDS-PAGE at 4 μg protein/gel lane. Gels were typically run at constant voltage (i.e., 120 V) until the bromphenol blue front reached the end of the gel. For Western blot analysis, proteins were blotted to Immobilon-P filters, blots were then blocked with 6% BSA (wt/vol) in T-PBS (PBS supplemented with 0.05% vol/vol, Tween-20) and reacted with mouse mAb AcV5 at 1:100 dilution. Blots were subsequently washed and incubated in goat anti–mouse IgG (1:4,000) conjugated with horseradish peroxidase. To detect protein immunoreactivity, blots were incubated in the ECL reagent followed by exposure to film.
Quantitative Western blot analysis was performed on gp64 immunoreactive bands (i.e., monomers, dimer, and trimers) after Op1D cells were subjected to DTT or GSSG-treatments. Blots were incubated in AcV5 (1:100) followed by goat anti–mouse (1:11,000) IgG conjugated with alkaline phosphatase, and then proteins were detected by ECF. Dried blots were photographed and scanned on a STORM 860 Macintosh scanner (Molecular Dynamics, Sunnyvale, CA). Analysis of SDS-PAGE images and quantification of individual bands were carried out with ImageQuant software package (Molecular Dynamics). Relative band intensities are presented as a percentage of the total band intensity within the sample lane.
Cell Surface Enzyme-linked Immunosorbent Assay
To examine whether gp64 trimers lacking intersubunit disulfide bonds are capable of undergoing the low pH-induced refolding, cell surface enzyme-linked immunosorbent assay (CELISA) was performed with mAb OpE6A which recognizes the native, but not the low pH conformation of gp64. The relative levels of native (or low pH-triggered) gp64 was compared in 5 or 10 mM DTT-reduced versus unreduced (control) cells across different pH media (i.e., 4.9–7.4). The cells were rinsed in PBS and treated with 5 or 10 mM DTT in PBS or with only PBS (control) for 30 min. DTT was removed with two washes of PBS. Cells were then alkylated in NaIA for 15 min followed by two additional washes. Next, Op1D cells were treated with different pH media and fixed in 4% (wt/vol) paraformaldehyde followed by rinsing in Tris-buffered saline (TBS, 20 mM Tris-HCl, 0.5 M NaCl, pH 7.5). Cells were then blocked in 6% BSA (wt/vol) in TBS and the surface gp64 reacted with 1:100 dilution of mouse IgG (i.e., OpE6A) followed by 1:5,000 goat anti–mouse IgG conjugated with alkaline phosphatase. Paranitrophenyl phosphate (an alkaline phosphatase substrate) was used for detection at 1 mg/ml in 10% (vol/vol) diethanolamine buffer. The absorbance at 405 nm was measured on a microplate reader (THERMOmax; Molecular Devices, Sunnyvale, CA). The relative level of gp64 in the native conformation across different pH media is presented as a percentage of gp64, pH 7.4, which was taken as 100% (i.e., maximal extent of gp64 in the native conformation).
Immunoprecipitation
Biotinylated gp64-containing cell lysate was immunoprecipitated with AcV1 mAb. Immobilized antibodies were prepared by incubating 500 μl of AcV1 (1:50 in PBS) with 30 μl of protein G–agarose bead slurry (ImmunoPure Plus G Immobilized Protein G; Pierce Chemical Co.) overnight at 4°C. To the mAb protein G–agarose complex, 100 μl of the cell lysate was added and incubated at 4°C for 4 h. Beads were then pelleted and washed three times with 0.5 ml of immunoprecipitation buffer (150 mM NaCl, 1.5% Triton X-100, and 50 mM Tris, pH 8) and boiled in 1.5% SDS sample buffer. Beads were removed from the sample by filtration through 0.45-μm filters. Samples were then subjected to either reducing or nonreducing SDS-PAGE followed by Western blot analysis.
Detection of High Molecular Weight gp64-containing Aggregates
After low pH application (pH 4.9, 1–30 min), baculovirus-infected Sf9 or transfected Op1D cells were surface cross-linked to arrest the putative protein complexes. Cells treated with PBS, pH 7.4, were used as a control. Cell–surface cross-linking was performed with a water-soluble DTSSP which preferentially reacts with amino groups. Cells were incubated in 5 mM DTSSP in PBS for 20 min. Unreacted DTSSP was washed out and the cells were lysed in 0.5 ml of the lysis buffer (150 mM NaCl, 1.5% Triton X-100, 10% glycerol, 5 mM EDTA, 1 tablet of protease inhibitor cocktail and 50 mM Tris, pH 8) followed by a 30-min agitation at 4°C. Lysate was cleared of the detergent-insoluble material by 30 min centrifugation at 90,000 g in a TLA 100.1 rotor (Beckman, Fullerton, CA) at 4°C. 75 μl of the cleared supernatant was subsequently used for the glycerol density gradient ultracentrifugation. A glycerol gradient was initially prepared in steps by layering 16 0.3-ml cushions of 10–47.5% (wt/vol) glycerol in 2.5% increments. The gradient was allowed to become continuous during storage at 4°C for 1 h. Protein aggregates of different sizes were separated by centrifugation at 237,000 g in a SW 50 rotor (Beckman) for 5 h at 4°C using slow acceleration and deceleration. After centrifugation was completed, 31 equal fractions were collected by piercing the bottom of the centrifuge tube. An aliquot (i.e., 15 μl) of each fraction was denatured in 1.5% SDS sample buffer with 50 mM DTT to release the individual gp64 subunits from the cross-linked protein complex. Groups of corresponding fractions from each variant within the experiment were simultaneously analyzed in 10% SDS-PAGE followed by Western blotting and scanning the films with a Dicomed digital camera, v 2.5 Apple Macintosh (Dicomed, Burnsville, ME). Thus, our conclusions are based on the comparison of the gels run in parallel and processed simultaneously. All the patterns were reproducible upon reruns of the gels. External protein molecular weight standards (i.e., 66 kD albumin, 232 kD catalase, and 750 kD macroglobulin) and fluorescein-conjugated dextran (500 kD and 2 MD) were run in parallel with the test samples to estimate the molecular weights of the cross-linked protein complexes. Protein molecular weight standards were detected by Coomassie brilliant blue (Bio-Rad, Hercules, CA) staining. Fluorescein-conjugated dextran was detected in sample fractions by a fluorescence plate reader (CytoFluor™ 2350; Millipore) at excitation and emission wavelengths of 480 ± 20 and 530 ± 25 nm, respectively.
To study the effect of the cell–cell contacts on gp64 aggregation, baculovirus-infected cells were seeded at identical number into 25-cm2 (high cell density/high frequency of cell–cell contacts) and 75-cm2 (low cell density/low frequency of cell–cell contacts) culture flasks. In some experiments infected cells seeded in the 75-cm2 flask at the low density were supplemented by the noninfected cells added to increase the cell density to that in the high cell density culture. Addition of all reagents to the large flask was adjusted based on the area of the flask.
Reduction of gp64 Disulfides by DTT
Op1D or infected Sf9 cells were treated with 0.1–30-mM DTT in PBS for 30 min at 27°C, followed by a 15-min alkylation with 10 mM NaIA in PBS at 27°C. NaIA was added to block the free thiol groups liberated by DTT treatment. Reduced and alkylated cells were either lysed and used in SDS-PAGE, or they were combined with labeled RBC, acidified, and then used to test the fusion. Fusion of noninfected Sf9 cells, labeled with calcein AM, with Op1D cells was monitored in four different treatment combinations: (a) neither Sf9 nor Op1D cells were treated (control); (b) only Sf9 cells were treated with 10 mM DTT for 30 min followed by alkylation with 10 mM NaIA for 15 min; (c) only Op1D cells were treated with 10 mM DTT for 30 min followed by alkylation with 10 mM NaIA for 15 min; and (d) both Sf9 and Op1D cells were treated with 10 mM DTT for 30 min followed by 15 min of alkylation with 10 mM NaIA.
To investigate whether lack of intersubunit disulfides affects formation of gp64 aggregates, Op1D and infected Sf9 cells were treated with 20 mM DTT for 30 min at 27°C. Then cells were incubated in pH 4.9 medium for 5 min. After low pH application, cross-linking and glycerol density gradient ultracentrifugation were performed as described above.
Oxidation of Thiols by GSSG
GSSG in concentration of 1–30 mM in PBS was used to restore disulfides in gp64 trimers after DTT treatment. DTT was removed from cells by two 1-ml washes of PBS. After GSSG application, fusion of such newly oxidized Op1D cells with RBC was monitored by fluorescence probe (i.e., PKH26) redistribution.
Cross-linking of DTT-reduced gp64 Trimers
To assess whether removal of intersubunit disulfide linkages resulted in a complete dissociation of gp64 trimers, Op1D cells were treated with 10 or 30 mM DTT followed by cross-linking of the reduced surface proteins. Cross-linking was performed with water soluble, noncleavable N-hydroxysuccinimide ester, BS3, which reacts with the same groups as DTSSP. Cells were incubated in 5 mM BS3 in PBS for 20 min. Unreacted BS3 was washed out and the cells were lysed in reducing SDS-PAGE lysis buffer. Lysates were analyzed on 4–12% gradient SDS-PAGE gels and immunoreactive bands detected by AcV5 Mab.
Fusion Inhibition by LPC
LPC was used to reversibly arrest fusion downstream of a low pH-induced conformational change in gp64 (Chernomordik et al., 1995). A stock solution of LPC was freshly prepared as a 0.5% (wt/wt) aqueous dispersion and vortexed until clear. Within a single set of experiments all LPC-containing media had the same concentration of LPC.
In the first set of experiments involving LPC, Op1D cells with prebound RBC were incubated for 5 min in the presence of LPC supplemented-PBS before low pH activation. After pH 5.1 application in the presence of LPC, cells were incubated for 20 min in 20 mM DTT at neutral pH. Then, both LPC and DTT were removed by washing the cells with PBS, and fusion was assayed (see Fig. 5 C).
Figure 5.

Reduction of gp64 intersubunit disulfides does not affect low pH protein refolding. (A) Fraction of gp64 that underwent low pH-induced refolding was determined by CELISA for DTT-treated versus untreated surface gp64. Each data point represents the mean ± SE CELISA signal from four replicate wells normalized to the CELISA signal of gp64, pH 7.4 (100%). (B) Fusion of Op1D cells with RBC, both incubated in 120 μM LPC for 5 min followed by pH 5.1 application for 1 min and LPC removal (second bar) or not (third bar), was measured. (Fourth bar) In the experiment presented, after pH 5.1 application still in the presence of LPC, cells were incubated for 20 min in 20 mM DTT already at neutral pH. Then, both LPC and DTT were removed and fusion was assayed. This experimental protocol is illustrated by the diagram in C. (Fifth bar) The percentage of fusion of Op1D cells with RBC that were treated with 20 mM DTT followed by pH 5.1 application. Each bar represents mean fusion extent ± SE for 200 cells counted.
In the second set of experiments using LPC, Op1D cells with prebound RBC were first incubated in LPC-supplemented PBS for 5 min and then DTT was applied for 20 min still in the presence of LPC. After DTT application, cells were treated with pH 5.1 medium supplemented with LPC. LPC was washed out after returning to neutral pH and cells were incubated in 10 mM GSSG for 15 min. Then fusion was assayed (see Fig. 6 B).
Figure 6.

Fusion is recovered by GSSG-related restoration of intersubunit disulfides of gp64 trimers in their low pH conformation. (A) Fusion of Op1D cells with PKH26-labeled RBC, both incubated in 120 μM LPC for 5 min followed by pH 5.1 application for 1 min and LPC removal (second bar) or not (third bar), was measured. After incubation in 20 mM DTT for 20 min still in the presence of LPC, cells were activated by pH 5.1 medium for 1 min. Then LPC was removed and fusion was assayed (fourth bar). (Fifth bar) Cells (i.e., Op1D and RBC) that were treated with DTT and pH 5.1 medium for 1 min in presence of LPC, then after LPC and DTT removal were incubated in 10 mM GSSG for 15 min, and then their fusion was assayed. The protocol of this experiment is illustrated in B. Each bar represents mean fusion extent ± SE for 200 cells counted.
In the last set of experiments LPC was used to test its effect on formation of the gp64-containing complexes. Gp64-expressing cells were treated with LPC alone for 5 min then, in the presence of LPC, activated with pH 4.9 medium for 5 min and then cross-linked with DTSSP. After LPC removal, the cells were lysed and subjected to glycerol density gradient ultracentrifugation as described above.
Cell–Surface Biotinylation
Biotinylation was used in two experiments to label the surface proteins on the cell. Biotin labeling was performed with 0.05 and 0.005 mg/ml biotin, respectively, in PBS for 30 min at 4°C followed by one wash with ice-cold PBS. In the first experiment the goal was to determine whether low pH activation of gp64 increases the number of accessible cross-linkable sites. To test this we used sulfo NHS biotin which reacts with the same groups as DTSSP and BS3. After the low pH pulse, proteins were biotinylated and lysed in 1.5% Triton X-100. Gp64 was then immunoprecipitated from the lysate and analyzed by SDS-PAGE and Western blotting. The objective of the second experiment was to assess whether the gp64-containing protein complex includes any proteins other than gp64. Biotinylated cells were low pH-triggered and the surface proteins were cross-linked. Cells were then lysed and the proteins purified by glycerol density gradient centrifugation. The 10 heaviest fractions from glycerol density gradient were combined and gp64 was immunoprecipitated. Analysis of gp64 was done by SDS-PAGE and Western blotting. To detect the proteins of interest, blots were reacted with streptavidin (1:3,000) conjugated with horseradish peroxidase.
Results
Fusion of gp64-expressing Cells with RBC
It has previously been shown that gp64-expressing cells (either infected Sf9 or Op1D cells) fuse with each other or with non-infected Sf9 cells when exposed to a low pH medium (Leikina et al., 1992; Plonsky et al., 1998). In this study we tested whether gp64-expressing cells would fuse with RBC ghosts. RBC ghosts are widely used in studies involving viral fusion proteins (Hoekstra et al., 1984; Kaplan et al., 1991). They appear to be a better defined target membrane than Sf9 cells, and they can be conveniently labeled both by membrane and water-soluble dyes.
Binding of RBC to Op1D or infected Sf9 cells was independent of gp64. This is supported by the fact that RBC bind naive (i.e., non-infected) Sf9 cells to the same extent as they bind gp64-expressing cells (Leikina, E., unpublished data). Application of low pH medium to Op1D– RBC complex resulted in dye (e.g., PKH26 and CF) redistribution. However, neither lipid (i.e., PKH26 transfer) nor content (i.e., CF transfer) mixing were observed in the control experiments when a low pH pulse was not applied, or when it was applied to the noninfected Sf9–RBC complex. Similar to Op1D, infected Sf9 cells fused to the RBC in a low pH-dependent manner (data not shown).
Conversion of Intersubunit Disulfides of gp64 into Thiols Causes Inhibition of Fusion
Gp64-mediated fusion significantly depends on the presence of intersubunit disulfide bonds. Op1D–RBC fusion was inhibited by DTT treatment in a concentration-dependent manner (Fig. 1 A). For instance, 1 mM DTT caused 28% inhibition of fusion whereas 20 mM caused more than 60% inhibition. DTT similarly inhibited both lipid and content mixing (data not shown). Since no change in binding of Op1D or infected Sf9 cells to RBC was observed at fusion-inhibiting concentrations of DTT (data not shown), we can conclude that DTT affected fusion, rather than binding.
Figure 1.
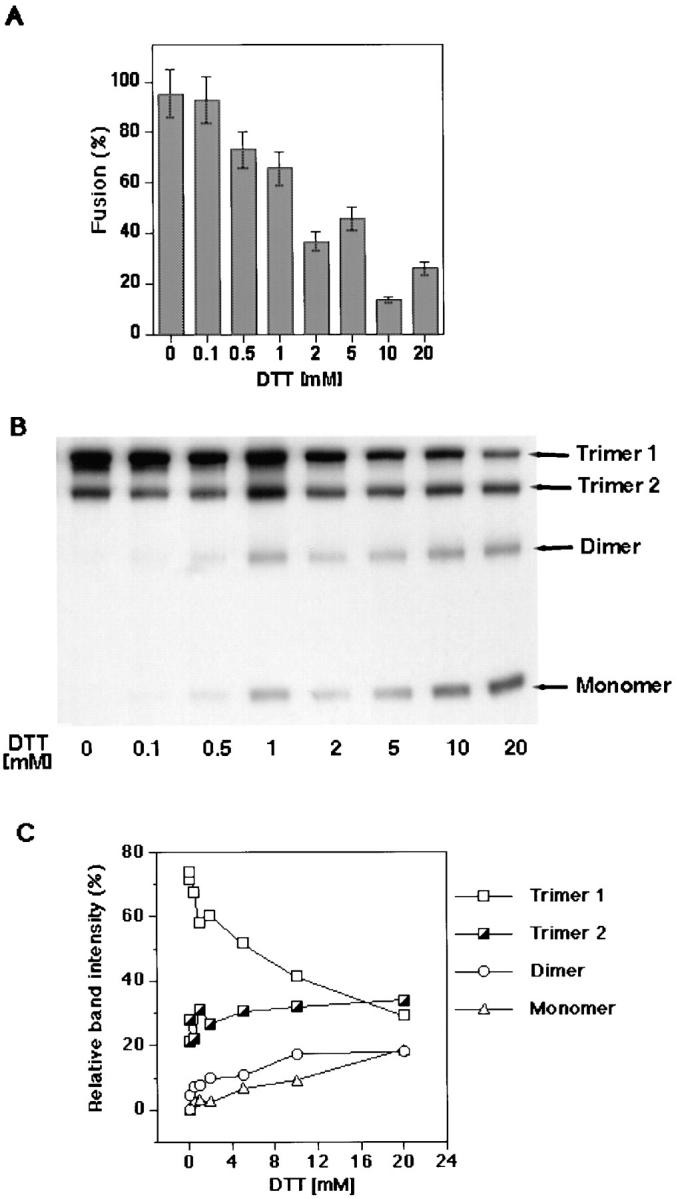
Effects of DTT-dependent disulfide-to-thiol exchange on fusion and gp64 trimerization. (A) Increasing concentrations of DTT significantly inhibited fusion between RBC and gp64-expressing Op1D cells. Op1D cells were treated with 0.1–20 mM DTT for 30 min followed by 15 min alkylation with 10 mM NaIA. PKH26-labeled RBC were then added to Op1D cells and the medium was acidified (i.e., pH 5.1) to trigger the fusion. Each bar represents mean fusion ± SE for 200 cells counted. (B) Western blot analysis of 0.1–20 mM DTT-treated and 10 mM NaIA-alkylated Op1D cells. Cells were lysed in nonreducing 1.5% SDS lysis buffer and proteins were separated by 6% SDS-PAGE. Increasing concentrations of DTT reduced gp64 trimers into monomers and dimers. (C) Quantitative Western blot analysis of DTT-reduced gp64 immunoreactive bands. Relative band intensities, measured by ECF, are presented as a percentage of the total band intensity within the sample lane. DTT caused reduction of mostly trimer 1.
Fusion inhibition correlated with Op gp64 reduction detected by SDS-PAGE and Western blot analysis. In the absence of DTT, gp64 exists as two distinct bands corresponding to two isoforms of the protein, i.e., trimer 1 and 2 (Oomens et al., 1995). Fusion-inhibiting concentrations of DTT (e.g., 1–20 mM) caused a significant decrease in the intensity of the trimer 1 band accompanied by appearance of additional bands corresponding to gp64 monomers and dimers (Fig. 1 B). Interestingly, trimer 2 appeared less affected by DTT treatment than trimer 1. Quantitative analysis of relative gp64 band intensities supported this finding (Fig. 1 C). Correlation of the disappearance of trimer 1 with a decrease in fusion argues in support of the hypothesis that trimer 1 is functionally involved in fusion.
The correlation between DTT concentration-dependent destabilization of trimer 1 and fusion inhibition (Fig. 1), suggests that such an inhibition was due to the loss of intersubunit disulfides from gp64. Still, we examined the possibility that DTT-based inhibition of fusion was caused by its effect on some proteins other than the gp64. To examine this possibility fusion was assayed by fluorescent dye (Calcein AM) redistribution from labeled, naive Sf9 into Op1D cells. The only known difference between these cell lines is the expression of gp64 on the Op1D cell surface. DTT treatment of naive Sf9 cells did not inhibit fusion. In contrast, DTT treatment of gp64-expressing cells (Op1D), or both Sf9 and Op1D cells, significantly reduced fusion (Fig. 2). This finding supports the notion that DTT inhibits fusion by acting on gp64.
Figure 2.

DTT inhibits fusion via its action on gp64. Fusion of Op1D with calcein AM–labeled naive Sf9 cells was assayed by fluorescent dye redistribution between fusing cells. As shown in the right portion of the diagram, four different treatment combinations were used, i.e., (1) neither Sf9 nor Op1D cells were treated; (2) only Sf9 cells were treated with DTT/NaIA (10 mM DTT for 30 min followed by alkylation with 10 mM NaIA for 15 min); (3) only Op1D cells were treated with DTT/NaIA; and (4) both Sf9 and Op1D cells were treated with DTT/NaIA. Each bar represents mean fusion extent ± SE for 200 cells counted.
To test whether DTT inhibits fusion mediated by another strain of gp64 we carried out similar experiments for Ac gp64. Similar to Op gp64, DTT treatment (1–20 mM) of Ac gp64 expressed in infected Sf9 cells significantly reduced the percentage of cell–cell fusion and SDS-PAGE– resolved trimeric band intensity (data not shown).
Conversion of DTT-released gp64 Thiols to Disulfides before Low pH Application Restores Fusion
Fusion competence of Op1D cells spontaneously recovered 3 h after DTT treatment to the extent observed in the nonreduced control. Blocking free thiols with an alkylating agent (i.e., NaIA) eliminated this fusion recovery. Control experiment have shown that application of only NaIA (1–10 mM) to Op1D cells does not affect their fusion with RBC (data not shown).
Oxidation of Op1D cells with 1–30 mM GSSG in the absence of alkylation resulted in thiol-to-disulfide exchange in DTT-reduced gp64 trimers. Restoration of the trimers by such treatment was evidenced by increase in Op1D– RBC fusion (Fig. 3 A) as well by increased trimeric band intensity in SDS-PAGE (Fig. 3 B). The dose-response curve of fusion restoration reached a plateau at 10 mM GSSG. Quantitative Western blot analysis of relative band intensities of restored trimers indicated that recovery of trimer 1 was greater than that of trimer 2 (Fig. 3 C).
Figure 3.
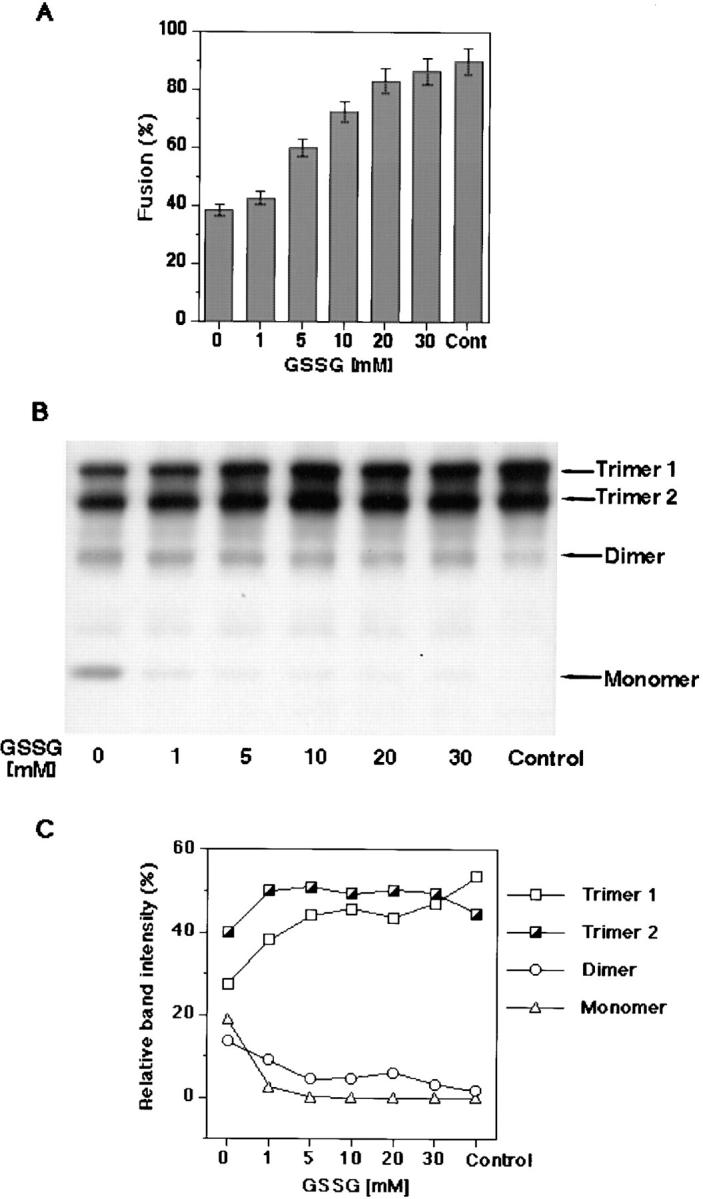
Effects of GSSG-dependent thiol-to-disulfide exchange on fusion and gp64 trimerization. (A) GSSG after DTT treatment, in absence of alkylation, fully recovered fusion. Op1D cells were first treated with 10 mM DTT for 30 min followed by a 15-min application of 1–30 mM GSSG. PKH26-labeled RBC were then added to Op1D cells and the medium was acidified (i.e., pH 5.1) to trigger the fusion. Each bar represents mean fusion extent ± SE for 200 cells counted. (B) Western blot analysis GSSG-treated Op1D cells. Cells were lysed in nonreducing 1.5% SDS lysis buffer and proteins were separated by 4–12% gradient SDS-PAGE. GSSG, after DTT, treatment restored gp64 trimers. (C) Quantitative Western blot analysis of GSSG-oxidized gp64 immunoreactive bands. Relative band intensities, measured by ECF, are presented as a percentage of the total band intensity within the sample lane. Restoration of trimer 1 was greater than that of trimer 2.
Gp64 Monomers with Reduced Intersubunit Disulfide Bridges Do Not Dissociate
To examine whether removal of intermonomeric disulfide linkages resulted in dissociation of gp64 subunits, a cell– surface cross-linking was performed with a water-soluble, homobifunctional cross-linker, BS3. Reducing SDS-PAGE of the noncross-linked control cells gave only a monomeric band (Fig. 4, lane 4). Cross-linking of Op1D cells resulted in the appearance of the distinct trimeric gp64 band in SDS-PAGE. Interestingly, after cross-linking and lysis in a DTT-containing buffer, the two trimers which formerly migrated as two distinct bands now migrated as a single band. Trimeric band intensities of samples derived from cells treated with 10 or 30 mM DTT (Fig. 4, lanes 1 and 2) and samples derived from untreated cells were approximately equal (Fig. 4, lane 3). This indicated that besides disulfide linkages, additional interactions keep monomers together in the membrane.
Figure 4.

Removal of intermonomeric disulfide bridges did not cause dissociation of gp64 subunits. Op1D cells were treated with 10 or 30 mM DTT (lanes 1 and 2), or no DTT (lane 3), followed by cross-linking with 5 mM BS3 for 20 min. Reducing SDS-PAGE in absence of cross-linking gave only a monomeric band (lane 4). Cells were lysed in reducing, 1.5% SDS lysis buffer, and analyzed on 4–12% gradient SDS-PAGE gels. The two trimers which formerly migrated as two distinct bands now migrated as a single band. A monomeric band detected in lanes 1, 2, and 3 suggests that cross-linking was not 100% efficient.
Low pH-induced Refolding of gp64 Does Not Require Intersubunit Disulfide Bonds
To determine the fraction of surface gp64 that underwent the low pH-induced conformational change, CELISA and OpE6A antibodies were used. These antibodies bind to gp64 in the neutral, but not in the low pH-induced conformation (Fig. 5 A). Thus, the binding of conformationally sensitive antibodies to a low pH-triggered gp64 was representative of the protein fraction which was changed from the initial to a low pH conformation. Treatment of Op1D cells with 5 and 10 mM DTT, or PBS caused no significant change in antibody binding to reduced versus control (i.e., DTT-untreated) gp64. Likewise, lowering the pH of the medium from 7.4 to 4.9 similarly decreased the antibody binding to both DTT-treated and untreated cells. Thus, the low pH gp64 refolding was apparently not affected by reduction of protein intersubunit disulfides.
The notion that DTT affects fusion downstream of gp64 refolding was further substantiated by experiments involving DTT application at the LPC-arrested stage of fusion. LPC is known to reversibly inhibit fusion after a low pH-triggered refolding of the protein but before membrane merger (Chernomordik et al., 1995). Threefold inhibition of fusion was observed when the low pH pulse was applied to the Op1D cells with prebound RBC in the presence of LPC (Fig. 5 B). Washing out the LPC led to significant recovery of the fusion. Treatment of the cells with 20 mM DTT in the time interval between the end of a low pH pulse and the removal of LPC blocked fusion. These DTT-treated cells failed to recover 81% of the fusion observed after LPC withdrawal from the DTT-untreated cells. Thus, reduction of gp64 intersubunit disulfides inhibited fusion downstream of the low pH-dependent conformational change.
Although DTT/NaIA-treated gp64 underwent low pH-triggered conformational change they were unable to mediate fusion. Surprisingly, using GSSG we found that restoration of disulfide bonds, at neutral pH, of DTT-reduced and low pH-activated proteins lead to recovery of the fusion. In this experiment we used LPC to reversibly block fusion and to minimize gp64 inactivation until GSSG-dependent restoration of disulfide bonds. Cells treated by DTT before application of LPC-containing low pH medium did not demonstrate fusion recovery upon removal of LPC (Fig. 6). However, when LPC removal was immediately followed by GSSG application, gp64 recovered most of its fusion activity. Thus, reduced and activated gp64 resumed the fusion competent-conformation upon recovery of the intersubunit disulfide bonds.
Analysis of the Cell–Surface gp64-containing Aggregates
Studying individual gp64 trimers provided no insight into the mechanism of DTT-based fusion inhibition. Thus, we decided to test the possibility that trimer destabilization affects assembly of gp64 into the functional multimeric complex. To stabilize gp64-containing aggregates, low pH-triggered cells were cell–surface cross-linked. Fractionated protein complexes separated by glycerol density gradient ultracentrifugation dissociated into monomers after reduction of the cross-linker (i.e., DTSSP) and gp64 disulfides in the presence of SDS. This allowed us to study the distribution of gp64 across different fractions of a glycerol density gradient using conventional SDS-PAGE. For infected Sf9 cells, untreated by low pH (but cross-linked), gp64 was distributed mainly across fractions 22–31 which corresponds to molecular weight (MW) of the trimers, dimers, and monomers (Fig. 7 A). In ∼50% of experiments, gp64 was present in the neutral pH control in fractions 10–18, which corresponds to an apparent MW higher than that of macroglobulin marker (i.e., 750 kD). The reason for the appearance of these higher MW gp64 aggregates in control experiments is not clear; however, it could be related to variation in the density of gp64 on the cell surface or in the density of cells in the culture.
Figure 7.

Gp64 HC were formed in response to low pH activation. (A) Sf9 cells infected with baculovirus at m.o.i. of 1 were treated with neutral (I) or pH 4.9 (II and III) medium for 5 min. After this treatment, cell–surface cross-linking with DTSSP was performed to arrest gp64 protein complexes. Cross-linked cells were lysed in 1.5% Triton X-100 (I and II) or SDS (III) lysis buffer and subjected to 10– 47.5% glycerol density gradient ultracentrifugation. 31 fractions were collected and a 75-μl aliquot of each fraction was reduced with 50 mM DTT and analyzed in 10% SDS-PAGE. Reduction of each fraction released monomers from the cross-linked protein complex. External protein molecular weight standards (i.e., 66 kD albumin, 232 kD catalase, and 750 kD macroglobulin) and fluorescein-conjugated dextran (500 kD and 2 MD) were run in parallel with the test samples to estimate the MW of the cross-linked protein complexes. Replacing Triton X-100 by 1.5% SDS in III resulted in disappearance of the gp64 HC. (B) To determine whether besides gp64 our protein complex contains other unrelated proteins, infected Sf9 cells were cell–surface biotinylated, treated with pH 7.4 medium (lane 1) or pH 4.9 (lane 2) for 5 min, and then cross-linked with 5 mM DTSSP. Lysis was performed in 1.5% Triton X-100 followed by glycerol density gradient ultracentrifugation. The 10 heaviest fractions from glycerol density gradient were pooled and AcV1 mAb-immunopurified gp64 was analyzed in 10% SDS-PAGE under reducing conditions. Biotinylated proteins were detected with streptavidin (1:3,000) conjugated to horseradish peroxidase. Lane 3 represents unimmunopurified monomer control detected by AcV5 Mab (1:100) ran in parallel with biotinylated samples.
The profile of gp64 distribution for low pH-triggered confluent monolayer of Sf9 cells revealed the appearance of gp64 bands in fractions 1–9 which roughly correspond to MW of 2 MD based on comparable migration of fluorescein-conjugated 2 MD dextran. Gp64 immunoreactive bands in fractions 1–9, further referred to as gp64-containing heavy complexes (gp64 HC), in contrast to fractions 10–18, were only observed after low pH treatment followed by cross-linking. Under these conditions, the gp64 HC were detected in 27 out of 32 experiments. High concentrations (i.e., 0.05 mg/ml) of sulfo NHS biotin which blocked the cross-linkable sites significantly reduced the amount of gp64 HC (data not shown). This indicates that cross-linking is necessary to stabilize the complexes which otherwise disintegrated during our handling procedure. Besides our standard cross-linking conditions (i.e., 20-min incubation at 27°C), gp64 HC were also detected after 5 min of incubation with cross-linker at 4°C. Gp64 HC were not observed when cross-linking was omitted. Similar to infected Sf9, Op1D cells showed gp64 immunoreactive bands in the fractions 1–9 of the glycerol density gradient only in response to low pH activation (see below). Extending the gradient from 10–47.5% to 10–60% did not reveal gp64 bands in the fractions corresponding to glycerol density above 47.5%, arguing against the presence of the gp64 aggregates several fold heavier than those resolved in our working conditions (data not shown).
Replacing Triton X-100, used for cell lysis, by 1.5% SDS resulted in disappearance of the gp64 HC (Fig. 7 A). This indicates that some of gp64 in HC remained uncross-linked. Similarly, no distinct bands of higher MW than trimers were detected in 1–2% SDS agarose gel electrophoresis suitable for analysis of proteins of 2 MD (Thornton et al., 1995) (data not shown).
To determine all the components involved in formation of gp64 HC, silver staining and biotin-streptavidin detection of the relevant complexes were performed. Silver staining of polyacrylamide gels containing cross-linked, immunopurified gp64 HC revealed, with the exception of IgG, only one protein band with the mobility corresponding to that of gp64 (data not shown). To exclude the possibility of selective protein staining by silver stain, cell surface proteins were biotinylated so as not to significantly affect the formation of the gp64 HC. Analysis of the biotinylated immunopurified gp64 HC indicated that gp64 is a sole protein component of such complexes (Fig. 7 B).
One may suggest that formation of the gp64 HC after low pH application can be explained merely by an increase in the accessibility of cross-linkable amino groups in the low pH-refolded gp64. However, we found no difference between trimeric band intensities of neutral versus low pH-treated protein using sulfo NHS biotin surface labeling. This indicates that low pH-dependent protein activation did not expose additional amino groups available for interaction with the cross-linking reagents. Control experiments confirmed that biotinylation does not inhibit fusion (data not shown).
Specific Factors Affecting Fusion May Also Influence Formation of gp64 HC
As previously shown, cell–cell fusion reaches final extent after 5 min of low pH activation (Chernomordik et al., 1995). To test the possible functional role of the gp64 HC, we compared the timing of the complex formation with that of the fusion reaction. We also studied how some of the factors known to affect fusion affect the complex formation. Increasing the acidic pH exposure time decreased the amount of gp64 HC (Fig. 8). For instance, a 10-min incubation at low pH reduced the amount of gp64 in fractions 1–9 compared with 1- and 5-min low pH treatment, whereas 20- and 30-min incubations resulted in complete loss of gp64 immunoreactive bands in corresponding fractions. This indicates that low pH-dependent gp64 complexes are short lived. The transient character of the gp64 HC was confirmed by another experiment. Nine bottom fractions were combined for each time point, applied on a single gel, and gp64 was quantified using two independent approaches (i.e., ECF and ECL). Both assays generated similar patterns in band distribution with the peak of gp64 HC at 1 and 5 min and their disappearance after longer (e.g., 30 min) low pH application (data not shown).
Figure 8.

Gp64 HC disappear with increased time exposure post-low pH triggering. Sf9 cells infected with baculovirus at m.o.i. of 1 were treated with neutral (I), or pH 4.9 medium for 1 (II), 5 (III), 10 (IV), 20 (V), or 30 min (VI). After the low pH activation, cell–surface cross-linking with DTSSP was performed to arrest gp64 protein complexes. Such low pH-triggered and cross-linked cells were lysed in 1.5% Triton X-100 lysis buffer and subjected to 10–47.5% glycerol density gradient ultracentrifugation.
Blocking the merger of membranes with LPC did not inhibit formation of the gp64 HC (Fig. 9 A). This supported the notion that gp64-specific assembly into gp64 HC preceded, rather than followed, merger of membranes.
Figure 9.

Specific factors affecting fusion may also influence formation of gp64 HC. (A) Sf9 cells infected with baculovirus at m.o.i. of 1 were treated with 120 μM LPC for 5 min then still in presence of LPC treated with neutral (I) or pH 4.9 (II) medium for 5 min and cell–surface cross-linked with 20 mM DTSSP. After LPC removal the cells were lysed in 1.5% Triton X-100 lysis buffer and subjected to 10– 47.5% glycerol density gradient ultracentrifugation. (B) Infected Sf9 cells were seeded at identical number into 25-cm2 (high cell density/high contact area, HCA) (I) and 75-cm2 (low cell density/ low contact area, LCA) culture flask (II and III). In II, infected cells were supplemented by the noninfected cells added to increase the cell density to that in the high cell density culture (I). Addition of all reagents to the large flask was adjusted based on the area of the flask. Cells were triggered to fuse with pH 4.9 medium for 5 min, surface cross-linked with 20 mM DTSSP, and then lysed in 1.5% Triton X-100. From there on the sample was treated in the same way as described in A. (C) Gp64-expressing Op1D cells were either treated with PBS (control) (I) or with 20 mM DTT (II) for 30 min before pH 4.9 treatment for 5 min. After DTT removal and low pH treatment, cross-linking with DTSSP and glycerol density gradient ultracentrifugation were performed as described above.
Cell–cell contact is an obvious prerequisite for fusion. The frequency of cell–cell contacts (assayed by phase–contrast microscopy as the average number of contacts per gp64 expressing cell) and the fusion efficiency (assayed by syncytium formation) were significantly reduced by a threefold decrease in the cell density. In baculovirus-infected cultures plated at high density (Materials and Methods), 91% cells were contacting other cells with 1.05 contact sites/cell resulting in 60% fusion triggered by low pH. In the low-density culture of infected cells, 32% cells were contacting other cells with 0.24 contact sites/cell resulting in 20% fusion.
Like fusion, low pH-induced assembly of gp64 requires cell–cell contacts (Fig. 9 B). Although infected cells plated at low density (Fig. 9 B, III) did not produce gp64 HC, adding noninfected cells to the low-density culture of the infected cells (to increase the total area of cell–cell contacts) (Fig. 9 B, II), yielded gp64 HC.
In contrast to LPC, DTT inhibited both the fusion and formation of the gp64 HC. As shown in Fig. 9 C, low pH treatment of DTT-reduced Op1D cells did not yield gp64 HC. DTT similarly inhibited formation of the gp64 HC for baculovirus-infected Sf9 cells treated with low pH medium (data not shown). These findings suggest that intersubunit gp64 disulfides are required for low pH-induced assembly of multimeric complexes of gp64 on the cell surface.
Discussion
The baculovirus homotrimeric enveloped protein (i.e., gp64) mediates low pH-triggered membrane fusion required for viral entry. To elucidate the molecular mechanism of the fusion reaction, we studied the role of oligomeric organization in fusion. This work presents the evidence that assembly of the low pH-activated gp64 trimers into a multimeric structure is an intermediate step in formation of the functional fusion machine. The appearance of these large, short-lived aggregates precedes merger of membranes. We also demonstrated that the basic structural unit of the multimeric aggregate is a stable gp64 trimer with the intact intersubunit disulfide bonds. Although reduction of intermolecular disulfide bonds destabilized trimers, it did not prevent them from undergoing conformational changes at low pH. Loss of the fusion activity under such conditions was associated with loss of the multimeric gp64 aggregates. These results pertain to both Ac and Op gp64 expressed in either infected or transfected cells. The structural similarity between these two strains has been reported earlier (Blissard and Rohrmann, 1989, 1991; Jarvis and Garcia, 1994; Hill and Faulkner, 1994; Oomens et al., 1995).
Membrane Merger Is Preceded by Formation of the Multimeric gp64 Complex
It has previously been suggested that viral glycoproteins are required to assemble into a multimeric complex in order to mediate fusion (Blumenthal et al., 1996; Danieli et al., 1996). Although these previous studies presented indirect experimental evidence, the current work provides the first direct evidence for the appearance of multimeric fusion protein complexes (i.e., gp64 HC). Like fusion, gp64 HC assembly requires low pH and cell–cell contacts. Dependence of complex formation on cell–cell contacts suggests that gp64 interaction with the lipid and/or protein receptor in the target membrane may be a prerequisite for protein assembly and the resulting fusion. Our finding that lack of cell contacts between baculovirus-infected cells can be compensated by contacts with noninfected cells (Fig. 9 B), indicates that gp64 aggregates are formed by the lateral assembly of gp64 in the same membrane rather than by interaction between gp64 proteins expressed in the apposing membranes.
The appearance of gp64 HC is coincident with fusion. Complex formation precedes, rather than follows membrane merger, as shown by the finding that LPC does not affect gp64 assembly. The time of the complex dissociation roughly corresponds to the time when fusion is completed.
Given that gp64 was the sole protein component in the gp64 HC detected by silver staining and biotin-streptavidin recognition, we conclude that such complexes are multimeric aggregates composed of only gp64. Present experimental approach, however, does not provide information whether gp64 HC are formed from a fixed or a variable number of trimers. The estimated size of the multimeric complexes may be rather approximate due to the potential difference in sedimentation characteristics between gp64 HC and MW markers. In addition, our experimental procedure may have led to either over- or underestimate the complex size. For instance, the actual fusion complexes on the cell membrane may be smaller than our detected 2 MD if they are formed from smaller cross-linked units that have a higher affinity towards each other after membrane solubilization with Triton X-100. Alternatively, such complexes may in reality be bigger than 2 MD but our detection may possibly be limited to the breakdown products resulting from the handling procedure. Although the estimate of the number of gp64 molecules in our complexes (2 MD corresponding to ∼10 trimers) may be approximate, this is comparable to what was reported by Plonsky and Zimmerberg (1996) based on electrophysiological analysis of the fusion pores.
The driving force for gp64 aggregation and the character of interactions between gp64 molecules in the complexes is poorly understood. These complexes were labile under our experimental conditions, and stabilization by cross-linking was required for their isolation and characterization. Dissociation of these partially cross-linked complexes by SDS indicates that on the membrane they are likely to be stabilized by noncovalent interactions among gp64 trimers. The relatively weak nature of trimer–trimer interactions in gp64 HC suggested by their transient existence may be important for expansion of the fusion pore. Since membrane contacts promote gp64 HC formation, decrease in the contact area upon fusion site expansion can explain the short lifetime of gp64 aggregates after fusion. Note that fusion machinery inactivates very slowly when fusion is blocked by LPC (Figs. 5 and 6).
Stable gp64 Trimers As Functional Units in Fusion
To determine whether the initial trimeric structure of gp64 is critical for fusion we attempted to destabilize the existing trimer. Reduction of the intersubunit gp64 disulfide bonds by DTT correlated with significant inhibition of fusion. The notion that this inhibition was caused by DTT's effect on gp64 was supported by the finding that fusion was not diminished by DTT treatment of the gp64-minus cells (Sf9) (Fig. 2).
Intramolecular disulfide bonds of membrane proteins are usually inaccessible to DTT (Tatu et al., 1993). Indeed, gp64 intramolecular disulfides were apparently not affected by DTT as suggested by the identical mobility of reduced versus unreduced trimers in SDS-PAGE and by their recognition by the same conformation-specific mAb. Thus, it appears that DTT in our experiments acted as a reversible and relatively specific reducing agent breaking inter-, but not intramonomeric, disulfide bonds of gp64 proteins on the cell surface. Future characterization of the specific disulfide bonds involved in oligomerization may be needed to confirm such assertion.
Inhibition of fusion via reduction of intersubunit disulfides indicates that oligomeric structure of gp64 is critical for fusion rather than just being a common structural motif of the viral envelope proteins regardless of their function. The importance of oligomerization for different viral proteins had been reviewed by Doms et al. (1993).
The specific role of the two trimers of gp64 in fusion is yet unknown. Correlation between disappearance of the only DTT-sensitive trimer (i.e., trimer 1) and fusion inhibition proves that trimer 1 is certainly involved in fusion. There are two possible explanations for the observed correlation. One may suggest that only trimer 1 mediates fusion. If so, than trimer 2 may either be a misfolded (i.e., inactive) form, or it may play a role at some other step of virus host–cell interaction. Alternatively, both types of gp64 trimers could be equally fusion-competent and fusion inhibition with DTT merely reflects the decrease in the total pool of stable (i.e., fusible) trimers.
What fusion stage was affected by trimer destabilization? Recent studies dissected membrane rearrangement into hemifusion (i.e., merger of the lipids but not the aqueous contents of the membranes) and fusion pore opening (Stamatatos and Duzgunes, 1993; Kemble et al., 1994; Melikyan et al., 1995, 1997; Chernomordik et al., 1998). In our system, DTT inhibited both lipid and content mixing, indicating that the affected stage precedes both hemifusion and fusion pore opening.
Trimer destabilization inhibited fusion downstream of the conformational change of fusion proteins. The pH dependence of gp64 refolding was not changed by reducing the trimers, indicating that a low pH sensor remained unaltered by trimer destabilization. The actual refolding of gp64 also appeared not to be affected by reduction of intersubunit disulfides. This conclusion is substantiated by the fact that DTT inhibited fusion at the LPC-arrested stage at which time gp64 has undergone the low pH refolding (Chernomordik et al., 1995). However, we can not exclude the possibility that the low pH form of DTT-treated gp64 is conformationally different from the untreated. If so, the former form after returning to the neutral pH has the full potential for refolding into the fusion-competent conformation upon restoration of the disulfide bonds (Fig. 6). This supports the notion that low pH refolded gp64 in the absence of intersubunit disulfides is a relatively long-lived form that is committed to fusion.
Although low pH refolding of individual trimers was not affected by their destabilization, this destabilization inhibited the subsequent stage, i.e., gp64 assembly into the multimeric complexes (Fig. 9 C). The fact that DTT inhibited fusion at the LPC-arrested stage when the gp64 HC were formed suggests that stable trimers are required not only for the formation, but also for the stabilization of the existing multimeric complexes.
Assembly of gp64 Trimers As a Pivotal Stage in Membrane Fusion
Our results documented assembly of a functional multiprotein fusion machine that follows trigger-dependent conformational change in viral fusion proteins, and precedes actual membrane rearrangements. We found that low pH-induced refolding of individual gp64 trimers is required but not sufficient for switching on the fusion machine of baculovirus. Although low pH triggering of a conformational change in gp64 did not require the presence of the contacting membrane (∼100% of gp64 at cell surface change their conformation at low pH, Fig. 5 A) (Chernomordik et al., 1995), assembly of gp64 into multimeric complexes was a contact-dependent process. For the membranes in the contact, fusion competence of activated gp64 trimers pretreated with DTT was restored by reestablishing the intersubunit disulfides and thus gp64 capability to form the multimeric complexes.
These fusion-competent gp64 complexes are apparently formed by a stepwise trimer aggregation mediated by noncovalent interactions among adjoining trimers. We hypothesize that the size and stability of the forming complex is determined by the energetics of these interactions and surface density of low pH-activated trimers. Destabilizing the trimers by breaking the disulfide bonds between the monomers may inhibit the growth of the multimeric fusion machine by permitting monomer dissociation from the trimer (Fig. 10). Alternatively, the low pH form of DTT-treated gp64 trimer could differ from the fusion-competent, untreated trimer in its ability to interact with neighboring trimers. For instance, protein sites required for such interactions may be less accessible in low pH-activated gp64 due to conformational modification in the absence of intersubunit disulfides.
Figure 10.

Proposed mechanism by which the absence of intersubunit disulfides affects aggregation of gp64 trimers. As illustrated, DTT-dependent destabilization of trimers may promote dissociation of monomers during trimer assembly and thus inhibit formation of the multimeric complexes.
For baculovirus, both an envelope protein responsible for virus binding and the receptor(s) remain to be identified (Wang et al., 1997). Op1D–RBC fusion system can lack these binding components which, in analogy to some other viruses (e.g., paramyxovirus [Bagai and Lamb, 1995] and HIV [for review see Dimitrov, 1997]), might modify fusion. However, as both DTT-based inhibition of fusion and the formation of gp64 HC were observed in infected Sf9 cells (expressing all viral envelope proteins including the hypothetical “binding” protein), we believe that main conclusions of our study pertain to biologically relevant gp64-mediated fusion. Similar to baculovirus, influenza HA fusion also appears to be independent of the specific binding mechanism (Schoen et al., 1996).
In summary, our results substantiate the hypothesis that fusion is mediated by short-lived multimeric complexes assembled in cell–cell contact regions by lateral association of low pH-activated, stable gp64 trimers. Further biochemical and morphological characterization of the multimeric complexes is needed to elucidate the character of the protein interactions, timing, location, and the specific role of the cell–cell contacts. The assembly of multiprotein fusion complexes investigated here for membrane fusion mediated by baculovirus gp64 can be a common motif for biological fusions.
Abbreviations used in this paper
- Ac
Autographa californica strain of baculovirus
- CELISA
cell surface enzyme-linked immunoabsorbent assay
- CF
6-carboxyfluorescein
- DTSSP
DTT-cleavable N-hydroxysuccinimide ester
- ECL
enhanced chemiluminescence
- ECF
enhanced chemifluorescence
- gp64 HC
gp64-containing heavy complexes
- GSSG
glutathione disulfide
- HA
influenza virus hemagglutinin
- LPC
lysophosphatidylcholine
- m.o.i.
multiplicity of infection
- MW
molecular weight
- NaIA
sodium iodoacetate
- Op
Orgyia pseudotsugata strain of baculovirus
- RBC
red blood cells
Footnotes
We greatly acknowledge E. Leikina (National Institute of Child Health and Human Development [NICHD], National Institutes of Health [NIH], Bethesda, MD) for valuable help and insightful discussions. We are grateful to J. Zimmerberg (NICHD, NIH) for continuous and enthusiastic support of the present work and creative discussions. We appreciate G.W. Blissard (Boyce Thompson Institute, Cornell University, Ithaca, NY) for his generosity in supplying us with Op1D cells and antibodies and for valuable discussion. We acknowledge P.S. Backlund, Jr. and A.L. Bailey (both from NICHD, NIH) for critical review of the manuscript and discussions. We thank A. Chrambach (NICHD, NIH) for his support and S.T. Case (University of Mississippi Medical Center, Jackson, MS) for providing us with high MW protein standard. We also thank other members in the Laboratory of Cellular and Molecular Biophysics for their input.
References
- Bagai S, Lamb R. Quantitative measurement of paramyxovirus fusion: differences in requirements of glycoproteins between simian virus 5 and human parainfluenza virus 3 or Newcastle disease virus. J Virol. 1995;69:6712–6719. doi: 10.1128/jvi.69.11.6712-6719.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blissard GW, Rohrmann GF. Location, sequence, transcriptional mapping, and temporal expression of the gp64 envelope glycoprotein gene of the Orgyia pseudotsugata multicapsid nuclear polyhedrosis virus. Virology. 1989;170:537–555. doi: 10.1016/0042-6822(89)90445-5. [DOI] [PubMed] [Google Scholar]
- Blissard GW, Rohrmann GF. Baculovirus gp64 gene expression: analysis of sequences modulating early transcription and transactivation by IE1. J Virol. 1991;65:5820–5827. doi: 10.1128/jvi.65.11.5820-5827.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blissard GW, Wenz JR. Baculovirus gp64 envelope glycoprotein is sufficient to mediate pH-dependent membrane fusion. J Virol. 1992;66:6829–6835. doi: 10.1128/jvi.66.11.6829-6835.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal R, Sarkar DP, Durell S, Howard DE, Morris SJ. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell-cell fusion events. J Cell Biol. 1996;135:63–71. doi: 10.1083/jcb.135.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza hemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- Bundo-Morita K, Gibson S, Lenard J. Estimation by radiation inactivation of the size of functional units governing Sendai and influenza virus fusion. Biochemistry. 1987;26:6223–6227. doi: 10.1021/bi00393a040. [DOI] [PubMed] [Google Scholar]
- Carr CM, Kim PS. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- Chernomordik L, Leikina E, Zimmerberg J. Control of baculovirus gp64-induced syncytia formation by membrane lipid composition. J Virol. 1995;69:3049–3058. doi: 10.1128/jvi.69.5.3049-3058.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik LV, Frolov VA, Leikina E, Bronk P, Zimmerberg J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol. 1998;140:1369–1382. doi: 10.1083/jcb.140.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague MJ, Schoch C, Blumenthal R. Delay time for influenza virus hemagglutinin-induced membrane fusion depends on hemagglutinin surface density. J Virol. 1991;65:2402–2407. doi: 10.1128/jvi.65.5.2402-2407.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danieli T, Pelletier SL, Henis YI, White JM. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol. 1996;133:559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov DS. How do viruses enter cells? The HIV coreceptors teach us a lesson of complexity. Cell. 1997;91:721–730. doi: 10.1016/s0092-8674(00)80460-2. [DOI] [PubMed] [Google Scholar]
- Doms RW, Gething MJ, Henneberry J, White J, Helenius A. Variant influenza virus hemagglutinin that induces fusion at elevated pH. J Virol. 1986;57:603–613. doi: 10.1128/jvi.57.2.603-613.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doms RW, Lamb RA, Rose JK, Helenius A. Folding and assembly of viral membrane proteins. Virology. 1993;193:545–562. doi: 10.1006/viro.1993.1164. [DOI] [PubMed] [Google Scholar]
- Ellens H, Bentz J, Mason D, Zhang F, White JM. Fusion of influenza hemagglutinin-expressing fibroblasts with glycophorin-bearing liposomes: role of hemagglutinin surface density. Biochemistry. 1990;29:9697–9707. doi: 10.1021/bi00493a027. [DOI] [PubMed] [Google Scholar]
- Gibson S, Jung CY, Takahashi M, Lenard J. Radiation inactivation analysis of influenza virus reveals different target sizes for fusion, leakage, and neuraminidase activities. Biochemistry. 1986;25:6264–6268. doi: 10.1021/bi00368a064. [DOI] [PubMed] [Google Scholar]
- Gutman O, Danieli T, White JM, Henis YI. Effects of exposure to low pH on the lateral mobility of influenza hemagglutinin expressed at the cell surface: correlation between mobility inhibition and inactivation. Biochemistry. 1993;32:101–106. doi: 10.1021/bi00052a014. [DOI] [PubMed] [Google Scholar]
- Hay JC, Scheller RH. SNAREs and NSF in targeted membrane fusion. Curr Opin Cell Biol. 1997;9:505–512. doi: 10.1016/s0955-0674(97)80026-9. [DOI] [PubMed] [Google Scholar]
- Hernandez LD, Hoffman LR, Wolfberg TG, White JM. Virus-cell and cell-cell fusion. Annu Rev Cell Dev Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- Hill JE, Faulkner P. Identification of the gp67 gene of a baculovirus pathogenic to the spruce budworm, Choristoneura fumiferanamulticapsid nuclear polyhedrosis virus. J Gen Virol. 1994;75:1811–1813. doi: 10.1099/0022-1317-75-7-1811. [DOI] [PubMed] [Google Scholar]
- Hoekstra D, de Boer T, Klappe K, Wilschut J. Fluorescence method for measuring the kinetics of fusion between biological membranes. Biochemistry. 1984;23:5675–5681. doi: 10.1021/bi00319a002. [DOI] [PubMed] [Google Scholar]
- Jarvis DL, Garcia AJ. Biosynthesis and processing of the Autographa californicanuclear polyhedrosis virus gp64 protein. Virology. 1994;205:300–313. doi: 10.1006/viro.1994.1646. [DOI] [PubMed] [Google Scholar]
- Junankar PR, Cherry RJ. Temperature and pH dependence of the haemolytic activity of influenza virus and of the rotational mobility of the spike glycoproteins. Biochim Biophys Acta. 1986;854:198–206. doi: 10.1016/0005-2736(86)90111-2. [DOI] [PubMed] [Google Scholar]
- Kaplan D, Zimmerberg J, Puri A, Sarkar DP, Blumenthal R. Single cell fusion events induced by influenza hemagglutinin: studies with rapid-flow, quantitative fluorescence microscopy. Exp Cell Res. 1991;195:137–144. doi: 10.1016/0014-4827(91)90509-s. [DOI] [PubMed] [Google Scholar]
- Kemble GW, Danieli T, White JM. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 1994;76:383–391. doi: 10.1016/0092-8674(94)90344-1. [DOI] [PubMed] [Google Scholar]
- Leikina E, Onaran HO, Zimmerberg J. Acidic pH induces fusion of cells infected with baculovirus to form syncytia. FEBS (Fed Eur Biochem Soc) Lett. 1992;304:221–224. doi: 10.1016/0014-5793(92)80623-O. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, White JM, Cohen FS. GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J Cell Biol. 1995;131:679–691. doi: 10.1083/jcb.131.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, Brener SA, Ok DC, Cohen FS. Inner but not outer membrane leaflets control the transition from glycosylphosphatidylinositol-anchored influenza hemagglutinin-induced hemifusion to full fusion. J Cell Biol. 1997;136:995–1005. doi: 10.1083/jcb.136.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monck JR, Fernandez JM. The fusion pore and mechanisms of biological membrane fusion . Curr Opin Cell Biol. 1996;8:524–533. doi: 10.1016/s0955-0674(96)80031-7. [DOI] [PubMed] [Google Scholar]
- Monsma SA, Blissard GW. Identification of a membrane fusion domain in the baculovirus gp64 envelope fusion protein. J Virol. 1995;69:2583–2595. doi: 10.1128/jvi.69.4.2583-2595.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oomens AG, Monsma SA, Blissard GW. The baculovirus GP64 envelope fusion protein: synthesis, oligomerization, and processing. Virology. 1995;209:592–603. doi: 10.1006/viro.1995.1291. [DOI] [PubMed] [Google Scholar]
- Plonsky I, Zimmerberg J. The initial fusion pore induced by baculovirus GP64 is large and forms quickly. J Cell Biol. 1996;135:1831–1839. doi: 10.1083/jcb.135.6.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plonsky, I., M.S. Cho, A. Oomens, G. Blissard, and J. Zimmerberg. 1998. An analysis of the role of the target membrane on the gp64-induced fusion pore. Virology. In press. [DOI] [PubMed]
- Roberts TE, Faulkner P. Fatty acid acylation of the 67K envelope glycoprotein of a baculovirus: Autographa californicanuclear polyhedrosis virus. Virology. 1989;172:377–381. doi: 10.1016/0042-6822(89)90145-1. [DOI] [PubMed] [Google Scholar]
- Schoen P, Leserman L, Wilschut J. Fusion of reconstituted influenza virus envelopes with liposomes mediated by streptavidin/biotin interactions. FEBS (Fed Eur Biochem Soc) Lett. 1996;390:315–318. doi: 10.1016/0014-5793(96)00682-5. [DOI] [PubMed] [Google Scholar]
- Stamatatos L, Duzgunes N. Simian immunodeficiency virus (SIVmac251) membrane lipid mixing with human CD4+ and CD4−cell lines in vitro does not necessarily result in internalization of the viral core proteins and productive infection. J Gen Virol. 1993;74:1043–1054. doi: 10.1099/0022-1317-74-6-1043. [DOI] [PubMed] [Google Scholar]
- Summers, M.D., and G.E. Smith. 1987. A Manual of Methods for Baculovirus Vectors and Insect Cell Culture Procedures. Tex. Agric. Exp. Stn. Bull. B1555:1–56.
- Tatu U, Braakman I, Helenius A. Membrane glycoprotein folding, oligomerization and intracellular transport: effects of dithiothreitol in living cells. EMBO (Eur Mol Biol Organ) J. 1993;12:2151–2157. doi: 10.1002/j.1460-2075.1993.tb05863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JR, Daum HAI, Case ST. Agarose gel electrophoresis of high molecular mass protein complexes. Biotechniques. 1995;18:324–327. [PubMed] [Google Scholar]
- Tse FW, Iwata A, Almers W. Membrane flux through the pore formed by a fusogenic viral envelope protein during cell fusion. J Cell Biol. 1993;121:543–552. doi: 10.1083/jcb.121.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman LE, Goldsmith PA. Budded Autographa californicaNPV 64 K protein: further biochemical analysis and effects of postimmunoprecipitation sample preparation conditions. Virology. 1984;139:295–302. doi: 10.1016/0042-6822(84)90375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman LE, Goldsmith PA, Hess RT, Faulkner P. Neutralization of budded Autographa californicaNPV by a monoclonal antibody: identification of the target antigen. Virology. 1984;133:354–362. doi: 10.1016/0042-6822(84)90401-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Hammer DA, Granados RR. Binding and fusion of Autographa californicanucleopolyhedrovirus to cultured insect cells. J Gen Virol. 1997;78:3081–3089. doi: 10.1099/0022-1317-78-12-3081. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Blumenthal R, Curran M, Sarkar D, Morris S. Restricted movement of lipid and aqueous dyes through pores formed by influenza hemagglutinin during cell fusion. J Cell Biol. 1994;127:1885–1894. doi: 10.1083/jcb.127.6.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerberg J, Vogel SS, Chernomordik LV. Mechanisms of membrane fusion. Annu Rev Biophys Biomol Struct. 1993;22:433–466. doi: 10.1146/annurev.bb.22.060193.002245. [DOI] [PubMed] [Google Scholar]