

Abstract
Autocrine EGF-receptor (EGFR) ligands are normally made as membrane-anchored precursors that are proteolytically processed to yield mature, soluble peptides. To explore the function of the membrane-anchoring domain of EGF, we expressed artificial EGF genes either with or without this structure in human mammary epithelial cells (HMEC). These cells require activation of the EGFR for cell proliferation. We found that HMEC expressing high levels of membrane- anchored EGF grew at a maximal rate that was not increased by exogenous EGF, but could be inhibited by anti–EGFR antibodies. In contrast, when cells expressed EGF lacking the membrane-anchoring domain (sEGF), their proliferation rate, growth at clonal densities, and receptor substrate phosphorylation were not affected by anti–EGFR antibodies. The sEGF was found to be colocalized with the EGFR within small cytoplasmic vesicles. It thus appears that removal of the membrane-anchoring domain converts autocrine to intracrine signaling. Significantly, sEGF inhibited the organization of HMEC on Matrigel, suggesting that spatial restriction of EGF access to its receptor is necessary for organization. Our results indicate that an important role of the membrane-anchoring domain of EGFR ligands is to restrict the cellular compartments in which the receptor is activated.
Keywords: epidermal growth factor, autocrine, intracrine, receptors, epithelium
The epidermal growth factor receptor system is perhaps the best characterized of all growth factor or cytokine systems. Isolated over 35 yr ago, EGF has been found to stimulate growth in a wide variety of epithelial cell types (Carpenter and Cohen, 1990). EGF was the first of what turned out to be a family of growth factors that bind to the EGF receptor (EGFR).1 These include TGFα (Derynck, 1992), amphiregulin (Shoyab et al., 1989), heparin binding EGF-like growth factor (HB-EGF; Higashiyama et al., 1991), betacellulin (Shing et al., 1993), and epiregulin (Toyoda et al., 1995). All these ligands are made as membrane-spanning prohormones that are processed and released through regulated proteolysis (Massagué and Pandiella, 1993). Similarly, the EGF receptor is also one member of a family that includes erbB-2, -3, and -4 (Lupu et al., 1995). It is thought that ligand binding to EGF receptors leads to homo- and heterodimerization of these family members, perhaps leading to diverse responses depending on the patterns of pairings (Earp et al., 1995; Alroy and Yarden, 1997). Whether different ligands promote different patterns of dimerization is not certain, although some data suggests that this may be the case (Beerli and Hynes, 1996). What is clear, however, is that a variety of different EGFR ligands are found throughout adult tissues such as the gut, the kidneys, and the skin (Fisher et al., 1989; Saeki et al., 1992; Downing et al., 1997). Whether the different EGFR ligands play different roles in normal tissue homeostasis is unknown.
The EGF receptor is known to play an important role during development. Knockout of the EGFR gene results in numerous developmental abnormalities in the brain, skin, and gut (Miettinen et al., 1995; Sibilia and Wagner, 1995; Threadgill et al., 1995). Interestingly, knockout of the TGFα gene results in only a mild phenotype, such as disoriented hair follicles, indicating that either other members of this ligand family may be more important, or there is redundancy of ligand function (Luetteke et al., 1993; Mann et al., 1993). It is possible that diverse functions of the EGFR depend on structurally different ligands. It has been shown that the postendocytic trafficking pattern of different EGFR ligands depends on their pH-sensitive dissociation from the EGFR (Ebner and Derynck, 1991; French et al., 1995). This may control persistence of receptor signaling, which, in turn, may alter the response of cells to EGFR activation (Traverse et al., 1994). Although there is evidence that TGFα is more effective than EGF in stimulating some cellular responses, such as migration and angiogenesis (Schreiber et al., 1986; Barrandon and Green, 1987), all EGFR ligands display very similar cellular and biochemical effects in vitro (Riese et al., 1996). This suggests that, in vivo, patterns of tissue distribution or methods of presentation may be more important than structure in dictating biological action of the EGFR ligands. Nevertheless, different ligand structures could potentially generate different biological responses depending on the context (Besner et al., 1992; Tzahar et al., 1997).
All the EGFR ligands consist of a conserved receptor-binding core domain flanked on the carboxy side by a membrane-spanning domain and on the amino side by a highly variable extracellular extension (Massagué and Pandiella, 1993). These extensions can be proteolytically removed before release of the ligand, such as the case with TGFα (Derynck, 1992). In other ligands, such as HB-EGF, most of the amino terminus is retained, which allows binding to extracellular glycosaminoglycans or to other cell surface molecules (Thompson et al., 1994). This extra-receptor binding can have profound effects on cell responsiveness in vitro and presumably in the intact animal (Cook et al., 1995). The proteolytic release of ligands, such as HB-EGF, can change their activity from juxtacrine to paracrine (Goishi et al., 1995). The transmembrane and cytoplasmic domains of the different ligands are also diverse, and may regulate cellular transport, localization, or proteolytic release (Dempsey et al., 1997). Although the membrane anchoring domain of EGFR ligands may regulate their cellular distribution, it remains to be demonstrated that altered cellular distribution has an impact on their biological activity.
Understanding the role of the membrane-anchoring domain of EGFR ligands is complicated by the fact that most cells making EGFR ligands also express the EGFR. Disruption of the EGFR gene in mice has shown that epithelial cells are most profoundly affected by receptor loss (Miettinen et al., 1995; Sibilia and Wagner, 1995; Threadgill et al., 1995). These cells, such as those found in the gut, the kidneys, and epidermis, have all been shown to express one or more EGFR ligands (Fisher et al., 1989; Barnard et al., 1994; Hashimoto et al., 1994; Sakurai et al., 1997). Although membrane-anchored growth factors have been shown to be biologically active in a juxtacrine fashion (Brachmann et al., 1989; Wong et al., 1989; Anklesaria et al., 1990; Higashiyama et al., 1995), these studies have used experimental systems in which the cell type expressing the ligand is distinct from the cell type expressing the receptor. In this situation, it is relatively simple to envision how spatially restricted juxtacrine signaling could play an important role in tissue organization. If a cell expresses both a receptor and a membrane-anchored growth factor, however, then juxtacrine signaling is unlikely to indicate cellular context. In addition, membrane-anchored EGFR ligands can be converted into soluble forms that are also biologically active (Derynck, 1992; Massagué and Pandiella, 1993). Thus, the function of the membrane-anchoring domain in autocrine signaling is unclear.
To determine the role that ligand structure and distribution play in the function of the EGFR system, we have employed human mammary epithelial cells (HMEC; Stampfer et al., 1997). These cells require activated EGFR for both cell division and motility (Matthay et al., 1993; Stampfer et al., 1993). They express a number of EGFR ligands and can thus grow in the absence of exogenous EGF (Li et al., 1992). Blocking the EGFR autocrine loop by the addition of antagonistic anti–EGFR antibodies, however, causes the cells to enter G0. Removal of the antibodies and addition of EGF causes the cells to synchronously reenter the cell cycle (Stampfer et al., 1993). Therefore, autocrine EGFR signaling in these cells is involved in a variety of different functions.
To explore the role of the membrane anchoring domain in EGFR ligand function, we constructed two derivatives of EGF: one lacking and one possessing the natural transmembrane domain. These artificial ligands were then expressed in HMEC cells to determine how they affected cell behavior. Surprisingly, we found that removal of the transmembrane domain resulted in a noninterruptible autocrine loop, apparently by an intracrine mechanism. Significantly, these cells could not organize into complex structures when grown on a reconstituted basement membrane. Our results suggest that an important function of the membrane-anchoring domain of EGF is to restrict the cellular location of receptor-ligand binding.
Materials and Methods
General
mAb 225 directed against the EGFR (Gill et al., 1984) was isolated from a hybridoma cell line obtained from the American Type Culture Collection (ATCC). Monoclonal antibody 13A9, which binds to both occupied and empty EGFR (Winkler et al., 1989) was obtained from Genentech Inc. (South San Francisco, CA). Monoclonal antibody HA directed against EGF was a kind gift from Katsuzo Nishikawa of the Kanazawa Medical University, Uchinoda, Ishikawa, Japan (Yoshitake and Nishikawa, 1988). Recombinant EGF (QCB, Inc., Hopkinton, MA) was conjugated to keyhole limpet hemocyanin (KLH) using sulfo-MBS (Pierce Chemical Co., Rockford, IL) after first introducing a sulfhydryl group using Traut's Reagent according to the manufacture's instruction. The KLH-EGF conjugate was used as an antigen to produce rabbit antisera. Polyclonal antibodies against the EGFR were raised in rabbits against affinity-purified EGFR (Gill and Weber, 1987). Vector pEGF-1 containing the mature sequence of human EGF was a gift from Salil Niyogi (Engler et al., 1988). LambdaEGF116 containing the entire coding sequence for human EGF was obtained from the ATCC. Human mammary epithelial cells 184 and line 184A1 (substrain L5) (Stampfer et al., 1993) were obtained from Dr. Martha Stampfer (Lawrence Berkeley Laboratories, Berkeley, CA) and cultured in either MCDB 170 (Hammond et al., 1984) or medium DFCI-1 as described (Band and Sager, 1989). Antibodies coupled to Protein A Sepharose beads were cross-linked to the beads using dimethyl pimelimidate and quenched using ethanolamine as described (Schneider et al., 1982). Beads were washed extensively and then directly added to the cell extracts.
Construction of sEGF and EGF-Ct
An artificial secreted form of human EGF (sEGF) was constructed using an artificial DNA sequence derived from the amino acid sequence of mature human EGF (Engler et al., 1988) fused to a 200-bp fragment of the 5′ untranslated region and adjacent signal sequence of the EGFR. The EGF DNA was removed from pEGF-1 by digesting with EagI, endfilling with Klenow, and digesting with EcoRI. The EGF DNA was ligated to pBluescript (Stratagene Inc., La Jolla, CA) that was digested with HindIII, endfilled with Klenow, and digested with EcoRI to create pBluescript-EGF. The 5′-untranslated region and signal sequence of the EGFR were isolated by PCR with primers to the SP6 promoter (5′-GTA TTC TAT AGT GTC ACC TA-3′) and the EGFR signal sequence (5′-GCC CGA CTC GCC GGG CAG AG-3′) using pLOLB (Opresko and Wiley, 1990) as the template. The PCR product was digested with XbaI to remove unwanted vector sequences, resulting in an insert with a 5′ XbaI end and a single 3′ A-overhang left by the Taq polymerase. This insert was ligated into pBluescript-EGF that was first digested with EcoRI and endfilled with Klenow, followed by addition of T overhangs with Taq polymerase and digestion with XbaI. EGF-Ct was made by inserting the entire membrane anchoring and cytoplasmic domain from lambdaEGF116 into pBluescript-sEGF. Both lambdaEGF116 and pBluescript-sEGF were digested with SphI and XhoI. After gel purification, the 760-bp fragment from pEGF was ligated into pBluescript-sEGF. All constructs were verified by sequencing.
For insertion into the MFG retrovirus vector (Eming et al., 1995), StyI and BglII sites were made at the 5′ and 3′ end of the sEGF construct using the primers 5′-CTT CGG GGA GCA GCC ATG GGA CCC TCC G-3′ and 5′-AGA TCT AAC GGA GCT CCC ACC ACT-3′. This set amplified the entire sEGF gene with the appropriate new restriction sites. The product was then ligated into pBluescript after digestion with SmaI and addition of T overhangs with Taq polymerase. The same protocol was followed for the EGF-Ct construct, except that a compatible NcoI site was used instead of the StyI site using the primer pair 5′-CCA TGG GAC CCT CCG GGA CG-3′ and 5′-AGA TCT ACT GAG TCA GCT CC-3′. The PCR reaction mixture included 100 pmol of each primer, 20 ng of temple, 200 μM of each dNTP, 25 mM MgCl2 and 2.5 U of Taq polymers. A DNA thermal cycler (Perkin Elmer Cetus Instruments, Emeryville, CA) was used for 25 cycles with an annealing temperature at 50°C. Final products were confirmed by DNA sequencing.
DNA fragments encoding sEGF or EGF-Ct were gel purified and ligated into the Nco1/BamH1 sites of the retrovirus vector MFG as previously described (Eming et al., 1995). The fidelity of the insert was verified by DNA sequencing. To generate cell lines producing recombinant retrovirus, plasmid DNAs encoding MFG-sEGF and MFG-EGF-Ct were transfected into the Ψ-CRIP packaging cell line as described (Danos and Mulligan, 1988). Clones of transfectants were isolated and screened for those producing the highest viral titer.
Cells were transfected with retrovirus stock using polybrene and grown for 2 d before plating at clonal density in medium lacking EGF. Individual colonies were isolated using cloning rings and then screened by immunofluorescence and by measuring the medium for the presence of EGF as described below. All experiments were done with several independently isolated colonies and all yielded the same results.
Organization of HMEC
Matrigel was brought to 4°C and 0.7 ml was placed in each well of a 12-well plate on ice. The matrix was carefully overlaid with 1 ml of ice-cold MCDB 170 to achieve a flat interface and the plates were transferred to a 37°C incubator for 1 h to solidify the Matrigel. The matrix was allowed to equilibrate overnight with 2 ml of appropriate growth medium before adding cells. The cells were removed from stock plates with trypsin, counted, and then 200,000 cells/well were added to the equilibrated Matrigel. After plating, the cells were examined daily and photographed.
Measurement of EGF and EGFR
A sandwich ELISA was developed to measure EGF levels in the medium. High binding ELISA plates (Corning Glass Works, Corning, NY) were coated with 50 μl of monoclonal antibody HA against EGF (5–10 μg/ml) diluted in phosphate-buffered saline, pH 7.4, with 0.02% sodium azide (PBSN). The plates were rinsed four times with wash buffer (0.05% Tween-20 in PBSN) before each new addition. The plates were then blocked using blocking buffer (10% horse sera in PBSN). Human recombinant EGF was diluted in blocking buffer for a standard curve ranging from 3 to 100 pg. A rabbit polyclonal serum directed against EGF was used as a secondary antibody diluted 1:100 in blocking buffer. Alkaline phosphatase-conjugated goat anti–rabbit antibody (Sigma Chemical Co., St. Louis, MO) was used as the tertiary antibody at a dilution of 1:6,000. The ELISA was developed by rinsing the plates twice with 10 mM diethanolamine, 0.5 mM MgCl2, pH 9.5, and then adding 50 μl of 1 mg/ml dinitrophenol (Sigma Chemical Co.) dissolved in the same buffer. The reaction was allowed to go for 4–10 min, and then quenched with 0.1 M EDTA. The ELISA plates were read at 405 nm using a SpectraMax microplate reader.
A sandwich ELISA was developed to measure total EGFR levels in cell extracts. The protocol is the same as for sEGF above, with the substitution of monoclonal antibody 13A9 against the EGFR (10 μg/ml) and polyclonal anti–EGFR antiserum #448 at a 1:250. Cells were extracted (250 μl per 10-cm dish) in 50 mM HEPES (pH 7.0), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 4 mM sodium iodoacetate, 1 mM EGTA, and 10 μg/ml each of aprotinin, leupeptin, chymostatin, and pepstatin. Cells were removed by scraping, transferred to 1.5-ml microfuge tubes, and incubated at 0°C for 10 min. Cell debris was removed by centrifugation at 10,000 g for 10 min. Protein concentrations were normalized between all samples before the assay using the BCA assay (Pierce Chemical Co.). A431 cell membranes were used as relative EGFR standards (Wiley, 1988). The addition of EGF to the A431 cell membranes confirmed that the EGFR ELISA did not discriminate between empty and occupied receptors.
Growth Rates
To determine the relative growth rates of cells expressing the different EGF constructs, confluent cultures were removed from their plates (6 cm) by trypsin and resuspended in 10 ml of DFCI-1 medium lacking EGF. Aliquots of cells were counted and 15,000 cells were seeded into each 3.5-cm dish. After allowing the cells to attach overnight, the medium was changed to DFCI-1 lacking EGF or that containing either 12.5 ng/ml of EGF of 10 μg/ml anti–EGFR mAb 225. Every 2 d, duplicate plates from each group were harvested and cell number was determined with a counter (Coulter Immunology, Hialeah, FL). Culture medium was changed every 2 d.
To measure clonal growth of cells, confluent cultures of cells were removed from their plates with trypsin, diluted 1:800 with DFCI-1 medium lacking EGF, and plated in 6-cm dishes. ∼18 h later, the medium was changed to DFCI-1 lacking EGF or that containing either 12.5 ng/ml of EGF of 10 μg/ml anti–EGFR mAb 225. Cultures were allowed to grow for 3 wk and the media were changed every 3 d. The cells were fixed in 50% methanol and stained with 0.4% Giemsa (Sigma Chemical Co.).
Shc Protein Phosphorylation
Cells from 100-mm plates were removed by scraping, pelleted, and extracted for 10 min on ice using 100 μl of 1% Triton X-100, 50 mM Tris, pH 7.2, 150 mM NaCl, 10% glycerol, 10 mM Na pyrophosphate, 1 mM EGTA, 10 mM iodoacetic acid, 1 mM sodium orthovanadate, 10 mM NaF, 10 μg/ml aprotinin, chymostatin, leupeptin, and pepstatin. After centrifugation to remove debris, protein concentrations of all samples were normalized. Anti–Shc antibodies (Transduction Laboratories, Lexington, KY) cross-linked to Protein A Sepharose (20–30 μl packed beads, ∼2 μg of antibody) were added to each sample, which was incubated at 4°C with rocking for 1.5 h. The resulting Shc-anti–Shc bead complex was washed twice in 1% Triton extraction buffer (see above), and then boiled in SDS-PAGE sample buffer before electrophoresis on 5–15% gradient gels. Samples were transferred to nitrocellulose and probed with RC20 antiphosphotyrosine antibody coupled to horseradish peroxidase (Transduction Laboratories). The blots were then developed with Western View ECL reagent (Transduction Laboratories).
Fluorescence Microscopy
Cells were plated on fibronectin-coated coverslips 48 h before the experiment. Cells were fixed for 10 min with freshly prepared 3.6% paraformaldehyde and 0.024% saponin in Ca2+, Mg2+-free phosphate buffered saline. Free aldehyde groups were quenched with 0.1% NaBH4 for 5 min. Cells were incubated simultaneously with anti–EGFR mAb 225 (10 μg/ ml) and anti–EGF rabbit polyclonal Z-12 (Santa Cruz Biotechnology, Santa Cruz, CA) in 0.012% saponin for 1 h followed by staining with FITC-labeled goat anti–mouse and Texas red-labeled goat anti–rabbit IgG antibodies (Molecular Probes, Inc., Eugene, OR) for 45 min. Alternately, anti–EGFR mAb 13A9 and anti–EGF mAb HA were directly labeled with Alexa 488 and Alexa 594 dyes (Molecular Probes, Inc.) and used at a concentration of 1 μg/ml each. Coverslips were mounted in ProLong antifade medium (Molecular Probes, Inc.) and viewed with a Nikon inverted fluorescence microscope with 60 or 100× oil immersion objectives. Images (12 bit, 656 × 517) were acquired using a Photometrics cooled CCD camera with a Macintosh workstation running Openlab 2.0 software (Improvision, Inc., Boston, MA). For digital confocal microscopy, image triplets were acquired 0.4-μm apart centered on the perinuclear endosomes at 520 and 615 nm (for Alexa 488 and Alexa 594, respectively). The image sets were deconvolved using nearest-neighbor subtraction (Agard et al., 1989). The deconvolved images of both EGF and EGFR distributions were then used to generate binary images using grayscale values between 400 and 4,095. A logical “AND” between these images was then used to determine the colocalization between the EGF and the EGFR. The deconvolution routines were calibrated using 15-μm FocalCheck beads (Molecular Probes, Inc.).
Results
Expression of Modified EGF Ligands in HMEC
The proteolytic processing of membrane-anchored EGFR ligands can be complex, giving rise to multiple forms of both soluble and membrane-anchored proteins (Derynck, 1992; Thorne and Plowman, 1994; Goishi et al., 1995). To simplify the interpretation of our experiments, we constructed the two artificial EGF genes diagramed in Fig. 1 A. Both lack the amino terminus extension that is frequently proteolytically removed. A signal sequence derived from the EGFR was substituted for the normal amino terminus extension. The sEGF construct terminates at amino acid 1023, which corresponds to the last amino acid in the mature EGF ligand. The EGF-Ct construct retains the entire transmembrane and cytoplasmic carboxy terminus of the EGF precursor. These artificial EGF genes were inserted into the retrovirus vector MFG, which was transfected into the CRIP packaging cell line (Danos and Mulligan, 1988). The resulting recombinant retroviruses were used to transduce the immortalized 184A1 HMEC line. Clonal cell lines were then randomly selected and screened for both EGF mRNA and protein expression.
Figure 1.


Expression of modified EGFR ligands in HMEC. (A) Maps of the constructs expressed in HMEC. Top map defines the domains found in native EGF. The core domain binds to the EGFR and is responsible for its biological activity. (B) Size of the EGF constructs expressed in HMEC. Conditioned medium from cells expressing sEGF was concentrated and applied to a Sephadex G-75 column together with molecular weight markers. Samples were collected and evaluated for EGF levels by ELISA. Elution position of the markers are indicated by arrows. (Inset) Western blot analysis of concentrated medium from cells expressing either EGF-Ct or sEGF. The antibody used was a polyclonal against human EGF. The standard (rhEGF) was commercially purified recombinant human EGF. (C) Rate of EGF production by either parental HMEC (WT) or several lines expressing either sEGF or EGF-Ct. Monolayers of cells were changed to medium lacking EGF and either with (solid bars) or without (hatched bars) 10 μg/ml 225 mAb. After 24 h, the conditioned medium was collected and evaluated for EGF levels by ELISA. Cell number was determined at both the initial and collection time points and the average was used to correct for the secretion rate. (D) Downregulation of the EGFR in either parental HMEC (WT) or cells expressing either sEGF or EGF-Ct. Cells were incubated in the presence (solid bars) or absence (hatched bars) of 33 nM EGF for 24 h. Cells were then extracted with detergent and the total cellular EGFR content was determined by ELISA. Data was standardized to the receptor content per microgram protein in the parental cells.
Conditioned medium from positive cell lines was collected, concentrated, and analyzed by gel filtration and Western blot analysis. As shown in Fig. 1 B, EGF activity from supernatants of cells producing sEGF ran as a single peak, corresponding to a molecular weight of ∼6.6 kD. This is slightly larger than the 6.2 kD predicted from protein sequence. Western blot analysis demonstrated that protein released from cells producing EGF-Ct ran as two bands, with the predominant lower band corresponding to authentic recombinant human EGF. The sEGF migrated primarily as the higher molecular weight product. Based on the molecular weight values obtained from gel filtration studies, the two bands likely correspond to alternate cleavage sites in the artificial signal sequence. We found that the biological activity of sEGF from conditioned medium was the same as commercially available recombinant EGF as determined by its ability to stimulate EGFR autophosphorylation and cell proliferation (data not shown).
Shown in Fig. 1 C are the rates of EGF release from several typical cell lines expressing either sEGF or EGF-Ct. The parental HMEC did not release any measurable amount of EGF into the medium, but clones expressing either sEGF or EGF-Ct released comparable amounts of soluble EGF at rates up to 40 ng/106 cells per d. Accumulation of EGF in the medium could be substantially increased by adding the receptor blocking antibody 225, indicating that the cells were capable of using a large fraction of the released EGF. Interestingly, if cells produced less than ∼10 ng EGF/106 cells per d, then no EGF was detected in the medium unless the endogenous EGFR were blocked (Fig. 1 C). This indicates either that HMEC are able to efficiently capture low levels of autocrine ligands or that the released ligand does not enter the bulk medium before receptor binding.
The clonal line secreting high levels of sEGF displayed a 75% reduction in EGFR levels, which was not reduced further by the addition of exogenous EGF (Fig. 1 D), indicating a maximal level of receptor downregulation. The clonal line expressing lower levels of sEGF displayed a corresponding lower degree of receptor downregulation. A similar situation was observed for lines expressing EGF-Ct (data not shown). If EGF was found in the medium in the absence of antagonistic anti–EGFR antibodies, EGFR downregulation was always complete (compare sEGF clone 1 in Fig. 1, C and D). This suggests that autocrine EGF escapes into the medium only when the EGFR are saturated and that at least some of the cell lines make more EGF than they can consume.
The expression of sEGF and EGF-Ct in HMEC was also evaluated by immunofluorescence. Shown in Fig. 2 are cells stained for both the EGFR and for EGF. The EGFR in parental HMEC were predominantly at the cell surface and EGF staining was not above background levels (Fig. 2, top). Cells expressing either sEGF or EGF-Ct displayed greatly reduced levels of EGFR, which were predominantly located in lysosomal structures (Fig. 2, middle and bottom), as shown by staining parallel groups of cells with an antibody to LAMP-2 (data not shown). As expected for a membrane-anchored protein, EGF-Ct was predominantly located at the cell surface (Fig. 2, bottom right). Some intracellular staining was also observed that colocalized with the EGFR.
Figure 2.

Distribution of EGFR and modified ligands in HMEC. Parental cells (WT) and cells expressing either sEGF (clone #1) or EGF-Ct (clone #2) were fixed, permeabilized, and simultaneously stained for the EGFR (left) or EGF (right). Exposure times for visualizing each antigen were identical using the WT cells as the standard for EGFR and the EGF-Ct cells as the standard for EGF.
The sEGF displayed a very weak staining pattern, consistent with its lack of a membrane-anchoring domain (Fig. 2, middle right). The pattern of sEGF staining, however, appeared to be coincident with the distribution of EGFR. To verify this colocalization, we directly labeled anti– EGFR and anti–EGF monoclonals with fluorescent dyes to avoid any possible cross-reactivity of secondary antibodies. Cells expressing sEGF were then fixed, permeabilized, and stained simultaneously with the anti–EGF and anti–EGFR antibodies. The distribution of sEGF and EGFR was then determined using digital confocal microscopy (Agard et al., 1989). As shown in Fig. 3, both the EGFR and the sEGF were found in small cytoplasmic vesicles. The distribution of sEGF (Fig. 3, red) was more restricted than the EGFR (green), most likely due to the loss of soluble sEGF from the permeabilized cells. Virtually all sEGF in the cell was found colocalized with the EGFR, as shown by yellow (Fig. 3, left) and by performing a logical AND of the EGFR and EGF images (Fig. 3, right). This is in contrast to the situation with EGF-Ct, where most of the ligand was found associated with the cell surface (Fig. 2, bottom right).
Figure 3.

Colocalization of sEGF and EGFR in HMEC. Cells expressing sEGF (clone #1) were fixed, permeabilized with saponin, and incubated with directly labeled anti–EGFR antibody 13A9 (green) or anti–EGF antibody HA (red). Image triplets were acquired with a 100× objective and deconvolved using a nearest-neighbor routine (left). The red image was converted to a binary image (middle), and logically AND'ed with a binary image of the green image to determine colocalization (right). The small panels beneath the main images are enlarged sections of the image. Bar, 5 μm.
The Transmembrane Domain of the EGF Ligand Allows Interruption of Autocrine Signaling
Some HMEC lines produce more EGF than can be consumed by the endogenous EGFR and display a maximal extent of receptor downregulation (Fig. 1, C and D). Because receptor downregulation is thought to reduce the sensitivity of cells to subsequent EGF addition (Wiley, 1985), we were interested in determining the growth rates of these chronically stimulated cells. The parental 184A1 cells grew in the absence of exogenous EGF, but grew to a higher density when EGF was added (Fig. 4 A). Blocking their EGFR with antagonistic mAb 225 strongly inhibited cell growth, as previously reported (Stampfer et al., 1993). Although the addition of EGF to cells producing sEGF had no effect on their growth rate, these cells grew at the same rate as parental cells treated with high concentrations of exogenous EGF (Fig. 4 A, middle). This indicates that downregulation of EGFR does not affect the steady state response of the cells. Surprisingly, addition of high concentrations of antagonistic 225 mAb had no effect on the growth rate of these cells either, indicating that the sEGF-EGFR autocrine loop could not be interrupted. In the case of cells expressing EGF-Ct, growth rates were again similar to those observed for parental cells treated with high concentrations of EGF and addition of exogenous EGF again had little effect (Fig. 4 A, right). In contrast to the situation with sEGF, however, the addition of 225 mAb effectively inhibited their growth. Thus, interruption of autocrine signaling in HMEC by antagonistic EGFR antibodies appeared to require the membrane anchoring domain of the ligand.
Figure 4.

Autocrine signaling by sEGF cannot be interrupted. (A) Proliferation of HMEC. Equal numbers of each cell type were plated into dishes without EGF (○) or either with 20 nM EGF (•) or 10 μg/ml of 225 mAb (□). Cell number was determined in duplicate at the indicated times. The medium was changed every other day. (B) Phosphorylation of the EGFR substrate Shc. Cells treated either with 20 nM EGF for 5 min or 10 μg/ml of 225 mAb for 18 h were extracted and total cellular Shc was immunoprecipitated. After electrophoresis and transfer to nitrocellulose, the blots were probed with antiphosphotyrosine antibodies. Arrows indicate the 66-kD Shc-related protein and the 53 and 46 kD forms of Shc.
Because cells expressing EGF-Ct produce at least as much ligand as those expressing sEGF (see Fig. 1 C), their relative sensitivity to 225 treatment could not be explained simply as an effect of mass action. An alternate explanation could be a clonal variation between cells with respect to their dependence on EGFR activation. We repeated our analysis on several independently isolated clones and observed the same effect; all clonal lines expressing sEGF were resistant to the effect of 225 mAb. To verify that EGFR activation itself was resistant to the effect of 225 mAb in cells expressing sEGF, we examined the phosphorylation of the EGFR substrate Shc in the different cell lines (Ruff-Jamison et al., 1993). As shown in Fig. 4 B, addition of EGF to the parental HMEC line resulted in high levels of Shc tyrosine phosphorylation, but little Shc phosphorylation was observed in either the absence of EGF or in the presence of 225 mAb. In the case of cells expressing either sEGF or EGF-Ct, Shc phosphorylation was significant in the absence or presence of exogenous EGF. Significantly, the addition of 225 mAb had little effect on Shc phosphorylation in cells expressing sEGF, but was strongly inhibitory in cells expressing EGF-Ct. These data demonstrate that sEGF can activate the EGFR even in the presence of antagonistic antibodies. The simplest explanation for this observation is that removal of the membrane-anchoring domain allows EGF to bind to its receptor before arrival at the cell surface and thus to operate in an “intracrine” fashion.
In the experiments shown in Fig. 4, A and B, cells were grown at a relatively high density (between 0.03 and 1 × 105/cm2). As an alternate to the intracrine hypothesis, sEGF could be trapped between cells and thus could bind to EGFR before the antagonistic antibody could diffuse to the cell surface. The more slowly released EGF-Ct would potentially not have such a kinetic advantage. If sEGF was operating in an intracrine manner, then growth of cells producing sEGF should be independent of cell density. If sEGF was simply being trapped between cells, then lowering the cell density should allow the 225 mAb to block receptor activation. To test this idea, cells were plated at clonal densities (<100 per cm2) and grown for several weeks in the presence or absence of anti–EGFR antibodies. As shown in Fig. 5, 225 mAb was unable to block the growth of cells producing sEGF, but were completely effective in preventing growth of either the parental cell line or cells producing EGF-Ct. These data demonstrate that even at the single cell level, autocrine signaling mediated by sEGF cannot be interrupted.
Figure 5.

Expression of sEGF allows clonal growth of HMEC in the presence of anti–EGFR antibodies. Parental cells (WT) and cells expressing either sEGF (clone #1) or EGF-Ct (clone #2) were seeded at a density of <100 per cm2 into 60-mm dishes and cultured for 3 wk in the presence of control medium lacking exogenous EGF, or with either 2 nM EGF or 10 μg/ml 225 mAb. The cells were then stained with crystal violet.
Spatial Organization of HMEC Is Disrupted by Secreted EGF
It has been shown previously that activation of the EGFR is important in proliferation and motility of HMEC (Matthay et al., 1993; Stampfer et al., 1993). We have found that EGFR signaling is also required for these cells to form three-dimensional structures on a reconstituted basement membrane. As shown in Fig. 6, when cells from the 184 line were plated on extracellular matrix (Matrigel) derived from Englebreth-Holme-Swarm fibrosarcoma (Schuetz et al., 1988), they rapidly assemble into epithelial cords and form complex three-dimensional structures. Colonies mature over 4 to 10 d to form a complex branched network with multiple endbuds that superficially resemble alveolar complexes in vivo. Occupancy of the EGFR was essential for HMEC organization on Matrigel because antagonistic anti–EGFR mAb 225 efficiently inhibited formation of complex structures (Fig. 6). Blocking EGFR occupancy after formation of initial aggregates also prevented the appearance of mature structures (data not shown).
Figure 6.
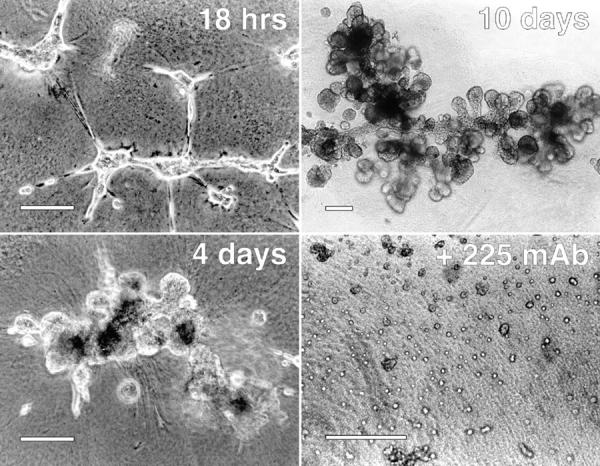
Organization of HMEC on Matrigel requires activation of EGFR. Cells (line 184) were plated on thick layers of Matrigel at a cell density of 2.5 × 104 cells/cm2 in the presence of 2 nM EGF for the indicated lengths of time. Cells incubated in 225 mAb (bottom right) were cultured for 2 d in the presence of 10 μg/ml of the antibody and in the absence of exogenous EGF. Photos were taken using phase optics. Bars, ∼200 μm.
The effect of mAb 225 on cell organization was not due to cell toxicity. Previously, it has been shown that mAb 225 causes a reversible entry into G0 of the cell cycle (Stampfer et al., 1993). We have found that high concentrations of EGF will readily reverse the effects of 225 mAb. In addition, no apoptosis was observed in 225-treated cells (data not shown).
Organization of 184A1 HMEC was dependent on both time and cell density. Increasing numbers of 184A1 cells were plated on Matrigel in either the presence or absence of exogenous EGF. As shown in Fig. 7, within 24 h the cells were able to form simple branching structures. Because the doubling time of these cells is 18–24 h (Stampfer and Yaswen, 1994), cell proliferation is unlikely to play a major role in formation of the initial structures. In the absence of exogenous EGF, a cell density of at least 105 cells/cm2 was required for organization. In the presence of EGF, a density of only 2 × 104 cells/cm2 was necessary. In the absence of exogenous EGF, the cells primarily formed branching structures that were stable over time (Fig. 7, left). The addition of exogenous EGF stimulated formation of endbud-like structures (Fig. 7, right), most likely by stimulating cell proliferation. The addition of anti–EGFR mAb 225 completely blocked organization of the cells at all time points (data not shown; also see below).
Figure 7.
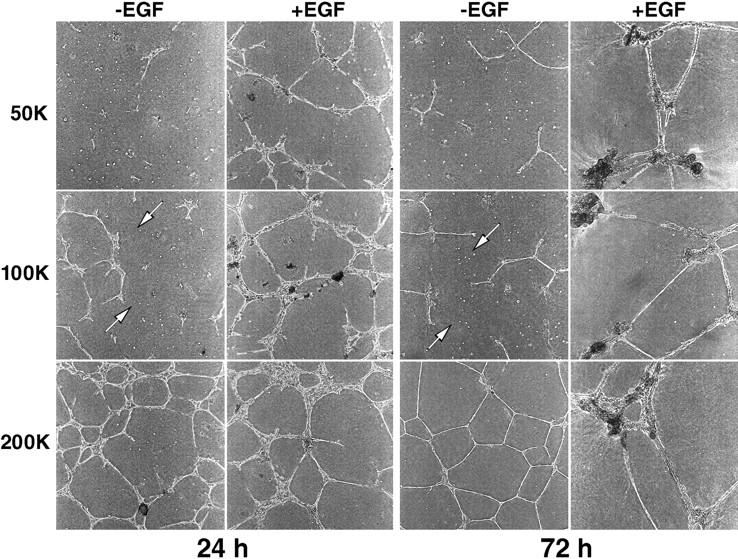
Organization of HMEC in the absence of exogenous EGF is dependent on cell density. The indicated number of cells (5 × 104, 105, and 2 × 105) were plated on thick layers of Matrigel cast in six well culture dishes either in the presence or absence of 2 nM EGF. Photographs were taken of the center of the wells (4× phase objectives) after either 24 (left) or 72 (right) h. Arrows indicate individual cells isolated from the main organizing structures.
There was also a time window of ∼12–24 h during which exogenous EGF facilitated organization of 184A1 cells. Visual observations suggested that this was related to invasion of the Matrigel by the cells. Thus, if EGF was provided to cells while they were still on the surface of the extracellular matrix, the cells could organize, but EGF had little effect once the cells entered the matrix. As indicated in Fig. 7 by the arrows, cells not included in a structure by 24 h remained as isolated colonies. The addition of EGF greatly increased the fraction of cells that joined organized structures, suggesting that cell migration and cell–cell contact were important aspects of EGF-stimulated organization.
We examined the effect of noninterruptible autocrine/ intracrine signaling on cell organization by observing the ability of HMEC expressing either sEGF or EGF-Ct to form complex structures. Cells were plated on Matrigel at a relatively low density (1.3 × 104/cm2) so that the effects of different EGF levels could be observed (see Fig. 7). After 6 d, their state of organization was evaluated. As shown in Fig. 8, parental cells formed small aggregates in the absence on exogenous EGF. As expected, the addition of EGF resulted in the formation of well defined complex structures. Again, addition of anti–EGFR antibody 225 blocked cell organization in the parental HMEC cells.
Figure 8.
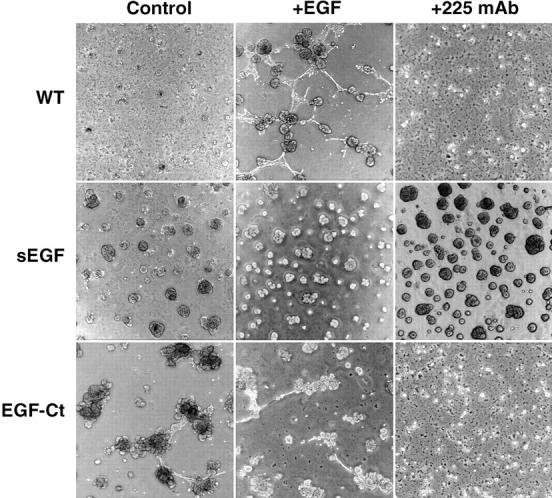
Expression of sEGF prevents HMEC from organizing into organotypic structures on Matrigel. Parental cells (WT) and cells expressing either sEGF (clone #1) or EGF-Ct (clone #2) were seeded at a density of 1.3 × 104/cm2 in the absence of EGF (Control), in the presence of 2 nM EGF (+EGF) or 10 μg/ml 225 anti–EGFR antibodies (+225). After 6 d, photographs were made using 4× phase objectives.
Cells expressing sEGF formed large aggregates that increased in size over time, but did not make organized structures (Fig. 8). In particular, the ability of these cells to form tubular or ductlike structures was severely affected. The addition of exogenous EGF or antagonistic 225 mAb had no effect on growth or organization of cells expressing sEGF.
In contrast to the effect of sEGF production, expression of EGF-Ct facilitated the formation of structures with clear lobular and ductal aspects (Fig. 8). The addition of exogenous EGF partially inhibited the formation of structures, whereas the addition of antagonistic 225 mAb completely blocked the process. If the cells were cultured for an additional 14 d, the structures formed by the parental HMEC were stable, but those formed by cells expressing EGF-Ct became less defined (data not shown). These observations indicate that both the concentration of EGF and its spatial presentation to receptors is important in its ability to facilitate the organization of HMEC in culture.
Discussion
The data presented here demonstrate that the membrane-anchoring domain of EGFR ligands may play an important role in regulating the cellular location of EGFR activation. Most studies on EGFR regulation have been focused at the receptor level, but ligand availability is generally rate-limiting in the overall activity of the EGFR pathway, as demonstrated by increased growth rate and density of autocrine cells after adding exogenous ligand (Atlas et al., 1992; Hashimoto et al., 1994). Release of EGFR ligands is itself a regulated process and appears to depend on the structure of the membrane-anchoring domain (Pandiella et al., 1992; Bosenberg et al., 1993; Massagué and Pandiella, 1993; Goishi et al., 1995; Baselga et al., 1996). Our investigation indicates that the membrane anchoring domain of the ligand also serves to restrict the site of ligand release. This spatial restriction appears to be important in normal cell function.
There is abundant evidence to show that EGFR function is critically important in both the formation and differentiated function of mammary glands (Snedeker et al., 1991; Fowler et al., 1995; Xie et al., 1997). Our HMEC experimental system appears to recapitulate some of the EGFR-dependent organization processes observed in vivo, although we did not observe differentiation of the cells into glandular epithelium (data not shown). Recent work suggests that differentiation requires the presence of multiple cell types (Gomm et al., 1997).
HMEC make a variety of different EGFR ligands, with TGFα and amphiregulin appearing the most important (Li et al., 1992). The simultaneous expression of a number of different EGFR ligands in a homogeneous cell population may indicate that different ligands have distinct functions. As a first step towards understanding structure–function relationships in EGFR ligands, we expressed EGF either with or without a membrane-anchoring domain. Removal of the membrane-anchoring domain of amphiregulin has been shown to alter the proteolytic processing of its amino-terminal extension, presumably by altering access to the processing enzymes (Thorne and Plowman, 1994). To simplify the analysis of our experiments, our EGF constructs lacked any amino-terminus extension.
We found that the most striking effect of removing the membrane anchoring domain of EGF was the loss of our ability to block EGFR signaling with antagonistic antireceptor antibodies. This was observed at all cell densities. Immunofluorescent imaging of cells expressing both sEGF and EGFR showed extensive colocalization in intracellular vesicles, indicating intracrine signaling. Because the ligand and antibody binding sites on the EGFR are overlapping and mutually exclusive (Gill et al., 1984), preformed EGF-receptor complexes arriving at the cell surface would not be affected by antagonistic antibodies. In contrast, we found that cells expressing membrane-anchored EGF-Ct were readily inhibited by antibodies. This indicates that membrane-anchored EGF and the EGFR are separate upon delivery to the cell surface. Thus, the formation of complexes between EGF-Ct and the EGFR either requires proteolytic release of EGF from the cell surface or any juxtacrine signaling must be restricted to the cell surface.
Juxtacrine activity has been described for membrane-anchored EGF and TGFα, as well as for HB-EGF (Brachmann et al., 1989; Mroczkowski et al., 1989; Wong et al., 1989; Higashiyama et al., 1995; Baselga et al., 1996). The relative contribution of membrane-anchored and soluble ligand forms to total ligand activity in vivo is unclear. If the membrane-anchored forms of the ligands do play a significant biological role, however, there must be specialized mechanisms to restrict receptor activation to the cell surface. CD9/DRAP 27 has been identified as an auxiliary molecule that facilitates juxtacrine signaling in the case of HB-EGF (Higashiyama et al., 1995). One of its roles could be to restrict juxtacrine signaling to the cell surface.
Restriction of EGFR signaling to the cell surface appears to be necessary for organization of HMEC on Matrigel. Cells incubated with exogenous EGF, or expressing membrane-anchored EGF, formed complex structures that superficially resembled ducts and endbuds. Expression of sEGF resulted in the disruption of the structures. This dominant, disorganizing effect of sEGF could indicate that intracrine signaling results in the phosphorylation of inappropriate intracellular substrates. However, we have not observed any significant differences between tyrosine-phosphorylated substrates in cells expressing sEGF versus EGF-Ct (data not shown). Alternately, the pattern of receptor occupancy at the cell surface could provide clues regarding the extracellular environment that could be important in cell organization. The uniform occupancy by sEGF of all EGFR before arrival at the cell surface would destroy any pattern of signaling imposed by the extracellular environment. The addition of exogenous EGF would not mimic the action of sEGF because its access to the cell surface is also spatially restricted by cell–cell and cell–matrix contacts. In either case, our results do show that the cellular location of EGFR signaling can have a significant effect on cell behavior.
All of the currently identified EGFR ligands, such as EGF, TGFα, amphiregulin, HB-EGF, and betacellulin, are made as membrane-associated precursors (Massagué and Pandiella, 1993). It is interesting to note that a number of virally encoded EGFR ligands have been identified, such as myxoma growth factor (Upton et al., 1987) and Shope fibroma growth factor (Chang et al., 1987). Unlike the normal EGFR ligands, these viral ligands lack a membrane anchoring domain. This may promote proliferation of infected cells by circumventing the normal mechanisms that regulate or interrupt autocrine signaling. Consistent with this hypothesis is the observation that Shope fibroma growth factor is a major virulence factor in malignant rabbit fibroma virus pathogenicity and is involved in promoting epithelial hyperplasia and squamous metaplasia (Opgenorth et al., 1992). Another pathological autocrine system is the v-sis/PDGF receptor system. Although PDGF is normally a paracrine growth factor (Battegay et al., 1994), inappropriate simultaneous expression of the ligand with the receptor results in uncontrolled cell proliferation (Chiu et al., 1984). An intracrine mechanism for v-sis signaling has been proposed as well (Bejcek et al., 1989), although the requirement of the ligand-receptor complex to reach the surface is controversial (Lee and Donoghue, 1992).
We found that unless cells made a greater amount of EGF than they could consume, little ligand was found in the extracellular medium unless the EGFR were blocked. A similar observation has been made in the case of MDCK cells expressing TGFα (Dempsey and Coffey, 1994). This suggests that either released ligand does not enter the bulk medium before binding to the EGFR, or that the cells are extremely efficient in binding low concentrations of ligands. It does show, however, that the lack of ligand in the extracellular medium does not indicate the absence of autocrine signaling.
A major theme emerging in the field of signal transduction is that alterations in the spatial distribution of signaling molecules is important for their activation (Carraway and Carraway, 1995; Leevers et al., 1994). This spatial distribution could drive morphogenic processes. It has already been shown that in Caenorhabditis elegans, the pattern of LET-23 receptor localization is important for lin-3–mediated vulval development (Simske et al., 1996). Because the LET-23/lin-3 pair is homologous to the mammalian EGFR system (Aroian et al., 1994), perhaps it is not surprising that disruption of autocrine ligand distribution should have a pronounced effect on cell organization. Because autocrine signaling is important in dictating tissue organization, it may play a more important role in development than previously suspected.
Acknowledgments
We thank Martha Stampfer for the 184 and 184A1 cells.
This work was supported by grant DAMD17-94-J-444 from the United States Army Breast Cancer Research Program (USABCRP). The authors also gratefully acknowledge financial support from a grant from the National Science Foundation Biotechnology Program, Division of Bioengineering and Environmental Systems. This work used the Flow Cytometry, Cell Imaging, and Oligonucleotide Synthesis core facilities of the Huntsman Cancer Institute, supported by National Cancer Institute Cancer Center Support Grant grant 5P30 CA 42014. P.M. Burke is a recipient of a predoctoral fellowship from the USABCRP.
Abbreviations used in this paper
- EGFR
EGF receptor
- HB-EGF
heparin binding EGF-like growth factor
- HMEC
human mammary epithelial cells
Footnotes
Address all correspondence to H. Steven Wiley, Department of Pathology, University of Utah Medical School, Salt Lake City, UT 84132. Tel.: (801) 581-5967. Fax: (801) 581-4517. E-mail: wiley@path.med.utah.edu
References
- Agard, D.A., Y. Hiraoka, P. Shaw, and J.W. Sedat. 1989. Fluorescence microscopy in three dimensions. In Fluorescence Microscopy of Living Cells in Culture. Vol. 30. D.L. Taylor and Y.L. Wang, editors. Academic Press, San Diego, CA. 353–377. [DOI] [PubMed]
- Alroy I, Yarden Y. The ErbB signaling network in embryogenesis and oncogenesis: signal diversification through combinatorial ligand–receptor interactions. FEBS Lett. 1997;410:83–86. doi: 10.1016/s0014-5793(97)00412-2. [DOI] [PubMed] [Google Scholar]
- Anklesaria P, Teixido J, Laiho M, Pierce JH, Greenberger JS, Massagué J. Cell–cell adhesion mediated by binding of membrane-anchored transforming growth factor alpha to epidermal growth factor receptors promotes cell proliferation. Proc Natl Acad Sci USA. 1990;87:3289–3293. doi: 10.1073/pnas.87.9.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroian RV, Lesa GM, Sternberg PW. Mutations in the Caenorhabditis eleganslet-23 EGFR-like gene define elements important for cell-type specificity and function. EMBO (Eur Mol Biol Organ) J. 1994;13:360–366. doi: 10.1002/j.1460-2075.1994.tb06269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas I, Mendelsohn J, Baselga J, Fair WR, Masui H, Kumar R. Growth regulation of human renal carcinoma cells: role of transforming growth factor alpha. Cancer Res. 1992;52:3335–3339. [PubMed] [Google Scholar]
- Band V, Sager R. Distinctive traits of normal and tumor-derived human mammary epithelial cells expressed in a medium that supports long-term growth of both cell types. Proc Natl Acad Sci USA. 1989;86:1249–1253. doi: 10.1073/pnas.86.4.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard JA, Graves-Deal R, Pittelkow MR, DuBois R, Cook P, Ramsey GW, Bishop PR, Damstrup L, Coffey RJ. Auto- and cross-induction within the mammalian epidermal growth factor-related peptide family. J Biol Chem. 1994;269:22817–22822. [PubMed] [Google Scholar]
- Barrandon Y, Green H. Cell migration is essential for sustained growth of keratinocyte colonies: the roles of transforming growth factor-alpha and epidermal growth factor. Cell. 1987;50:1131–1137. doi: 10.1016/0092-8674(87)90179-6. [DOI] [PubMed] [Google Scholar]
- Baselga J, Mendelsohn J, Kim YM, Pandiella A. Autocrine regulation of membrane transforming growth factor–alpha cleavage. J Biol Chem. 1996;271:3279–3284. doi: 10.1074/jbc.271.6.3279. [DOI] [PubMed] [Google Scholar]
- Battegay EJ, Rupp J, Iruela-Arispe L, Sage EH, Pech M. PDGF-BB modulates endothelial proliferation and angiogenesis in vitro via PDGF beta-receptors. J Cell Biol. 1994;125:917–928. doi: 10.1083/jcb.125.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli RR, Hynes NE. Epidermal growth factor–related peptides activate distinct subsets of ErbB receptors and differ in their biological activities. J Biol Chem. 1996;271:6071–6076. doi: 10.1074/jbc.271.11.6071. [DOI] [PubMed] [Google Scholar]
- Bejcek BE, Li DY, Deuel TF. Transformation by v-sis occurs by an internal autoactivation mechanism. Science. 1989;245:1496–1499. doi: 10.1126/science.2551043. [DOI] [PubMed] [Google Scholar]
- Besner GE, Whelton D, Crissman-Combs MA, Steffen CL, Kim GY, Brigstock DR. Interaction of heparin-binding EGF-like growth factor (HB-EGF) with the epidermal growth factor receptor: modulation by heparin, heparinase, or synthetic heparin-binding HB-EGF fragments. Growth Factors. 1992;7:289–296. doi: 10.3109/08977199209046411. [DOI] [PubMed] [Google Scholar]
- Bosenberg MW, Pandiella A, Massagué J. Activated release of membrane-anchored TGF-alpha in the absence of cytosol. J Cell Biol. 1993;122:95–101. doi: 10.1083/jcb.122.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann R, Lindquist PB, Nagashima M, Kohr W, Lipari T, Napier M, Derynck R. Transmembrane TGF-alpha precursors activate EGF/TGF-alpha receptors. Cell. 1989;56:691–700. doi: 10.1016/0092-8674(89)90591-6. [DOI] [PubMed] [Google Scholar]
- Carpenter G, Cohen S. Epidermal growth factor. J Biol Chem. 1990;265:7709–7712. [PubMed] [Google Scholar]
- Carraway KL, Carraway CA. Signaling, mitogenesis and the cytoskeleton: where the action is. Bioessays. 1995;17:171–175. doi: 10.1002/bies.950170212. [DOI] [PubMed] [Google Scholar]
- Chang W, Upton C, Hu SL, Purchio AF, McFadden G. The genome of Shope fibroma virus, a tumorigenic poxvirus, contains a growth factor gene with sequence similarity to those encoding epidermal growth factor and transforming growth factor alpha. Mol Cell Biol. 1987;7:535–540. doi: 10.1128/mcb.7.1.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Reddy EP, Givol D, Robbins KC, Tronick SR, Aaronson SA. Nucleotide sequence analysis identifies the human c-sis proto-oncogene as a structural gene for platelet-derived growth factor. Cell. 1984;37:123–129. doi: 10.1016/0092-8674(84)90307-6. [DOI] [PubMed] [Google Scholar]
- Cook PW, Ashton NM, Karkaria CE, Siess DC, Shipley GD. Differential effects of a heparin antagonist (hexadimethrine) or chlorate on amphiregulin, basic fibroblast growth factor, and heparin-binding EGF-like growth factor activity. J Cell Physiol. 1995;163:418–429. doi: 10.1002/jcp.1041630222. [DOI] [PubMed] [Google Scholar]
- Danos O, Mulligan RC. Safe and efficient generation of recombinant retroviruses with amphotropic and ecotropic host ranges. Proc Natl Acad Sci USA. 1988;85:6460–6464. doi: 10.1073/pnas.85.17.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey PJ, Coffey RJ. Basolateral targeting and efficient consumption of transforming growth factor-alpha when expressed in Madin-Darby canine kidney cells. J Biol Chem. 1994;269:16878–16889. [PubMed] [Google Scholar]
- Dempsey PJ, Meise KS, Yoshitake Y, Nishikawa K, Coffey RJ. Apical enrichment of human EGF precursor in Madin-Darby canine kidney cells involves preferential basolateral ectodomain cleavage sensitive to a metalloprotease inhibitor. J Cell Biol. 1997;138:747–758. doi: 10.1083/jcb.138.4.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R. The physiology of transforming growth factor-alpha. Adv Cancer Res. 1992;58:27–52. doi: 10.1016/s0065-230x(08)60289-4. [DOI] [PubMed] [Google Scholar]
- Downing MT, Brigstock DR, Luquette MH, Crissman-Combs M, Besner GE. Immunohistochemical localization of heparin-binding epidermal growth factor-like growth factor in normal skin and skin cancers. Histochem J. 1997;29:735–744. doi: 10.1023/a:1026417202351. [DOI] [PubMed] [Google Scholar]
- Earp HS, Dawson TL, Li X, Yu H. Heterodimerization and functional interaction between EGF receptor family members: a new signaling paradigm with implications for breast cancer research. Breast Cancer Res Treat. 1995;35:115–132. doi: 10.1007/BF00694752. [DOI] [PubMed] [Google Scholar]
- Ebner R, Derynck R. Epidermal growth factor and transforming growth factor-alpha: differential intracellular routing and processing of ligand-receptor complexes. Cell Regul. 1991;2:599–612. doi: 10.1091/mbc.2.8.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eming SA, Lee J, Snow RG, Tompkins RG, Yarmush ML, Morgan JR. Genetically modified human epidermis overexpressing PDGF-A directs the development of a cellular and vascular connective tissue stroma when transplanted to athymic mice—implications for the use of genetically modified keratinocytes to modulate dermal regeneration. J Invest Dermatol. 1995;105:756–763. doi: 10.1111/1523-1747.ep12325550. [DOI] [PubMed] [Google Scholar]
- Engler DA, Matsunami RK, Campion SR, Stringer CD, Stevens A, Niyogi SK. Cloning of authentic human epidermal growth factor as a bacterial secretory protein and its initial structure-function analysis by site-directed mutagenesis. J Biol Chem. 1988;263:12384–12390. [PubMed] [Google Scholar]
- Fisher DA, Salido EC, Barajas L. Epidermal growth factor and the kidney. Annu Rev Physiol. 1989;51:67–80. doi: 10.1146/annurev.ph.51.030189.000435. [DOI] [PubMed] [Google Scholar]
- Fowler KJ, Walker F, Alexander W, Hibbs ML, Nice EC, Bohmer RM, Mann GB, Thumwood C, Maglitto R, Danks JA, et al. A mutation in the epidermal growth factor receptor in waved-2 mice has a profound effect on receptor biochemistry that results in impaired lactation. Proc Natl Acad Sci USA. 1995;92:1465–1469. doi: 10.1073/pnas.92.5.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French AR, Tadaki DK, Niyogi SK, Lauffenburger DA. Intracellular trafficking of epidermal growth factor family ligands is directly influenced by the pH sensitivity of the receptor/ligand interaction. J Biol Chem. 1995;270:4334–4340. doi: 10.1074/jbc.270.9.4334. [DOI] [PubMed] [Google Scholar]
- Gill GN, Kawamoto T, Cochet C, Le A, Sato JD, Masui H, McLeod C, Mendelsohn J. Monoclonal anti-epidermal growth factor receptor antibodies which are inhibitors of epidermal growth factor binding and antagonists of epidermal growth factor-stimulated tyrosine protein kinase activity. J Biol Chem. 1984;259:7755–7760. [PubMed] [Google Scholar]
- Gill GN, Weber W. Purification of functionally active epidermal growth factor receptor protein using a competitive antagonistic monoclonal antibody and a competitive elution with epidermal growth factor. Methods Enzymol. 1987;146:82–88. doi: 10.1016/s0076-6879(87)46010-2. [DOI] [PubMed] [Google Scholar]
- Goishi K, Higashiyama S, Klagsbrun M, Nakano N, Umata T, Ishikawa M, Mekada E, Taniguchi N. Phorbol ester induces the rapid processing of cell surface heparin-binding EGF-like growth factor: conversion from juxtacrine to paracrine growth factor activity. Mol Biol Cell. 1995;6:967–980. doi: 10.1091/mbc.6.8.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomm JJ, Coope RC, Browne PJ, Coombes RC. Separated human breast epithelial and myoepithelial cells have different growth factor requirements in vitro but can reconstitute normal breast lobuloalveolar structure. J Cell Physiol. 1997;171:11–19. doi: 10.1002/(SICI)1097-4652(199704)171:1<11::AID-JCP2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Hammond SL, Ham RG, Stampfer MR. Serum-free growth of human mammary epithelial cells: rapid clonal growth in defined medium and extended serial passage with pituitary extract. Proc Natl Acad Sci USA. 1984;81:5435–5439. doi: 10.1073/pnas.81.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Higashiyama S, Asada H, Hashimura E, Kobayashi T, Sudo K, Nakagawa T, Damm D, Yoshikawa K, Taniguchi N. Heparin-binding epidermal growth factor-like growth factor is an autocrine growth factor for human keratinocytes. J Biol Chem. 1994;269:20060–20066. [PubMed] [Google Scholar]
- Higashiyama S, Abraham JA, Miller J, Fiddes JC, Klagsbrun M. A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science. 1991;251:936–939. doi: 10.1126/science.1840698. [DOI] [PubMed] [Google Scholar]
- Higashiyama S, Iwamoto R, Goishi K, Raab G, Taniguchi N, Klagsbrun M, Mekada E. The membrane protein CD9/DRAP 27 potentiates the juxtacrine growth factor activity of the membrane-anchored heparin-binding EGF-like growth factor. J Cell Biol. 1995;128:929–938. doi: 10.1083/jcb.128.5.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BA, Donoghue DJ. Intracellular retention of membrane-anchored v-sis protein abrogates autocrine signal transduction. J Cell Biol. 1992;118:1057–1070. doi: 10.1083/jcb.118.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- Li S, Plowman GD, Buckley SD, Shipley GD. Heparin inhibition of autonomous growth implicates amphiregulin as an autocrine growth factor for normal human mammary epithelial cells. J Cell Physiol. 1992;153:103–111. doi: 10.1002/jcp.1041530114. [DOI] [PubMed] [Google Scholar]
- Luetteke NC, Qiu TH, Peiffer RL, Oliver P, Smithies O, Lee DC. TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell. 1993;73:263–278. doi: 10.1016/0092-8674(93)90228-i. [DOI] [PubMed] [Google Scholar]
- Lupu R, Cardillo M, Harris L, Hijazi M, Rosenberg K. Interaction between erbB-receptors and heregulin in breast cancer tumor progression and drug resistance. Semin Cancer Biol. 1995;6:135–145. doi: 10.1006/scbi.1995.0016. [DOI] [PubMed] [Google Scholar]
- Mann GB, Fowler KJ, Gabriel A, Nice EC, Williams RL, Dunn AR. Mice with a null mutation of the TGF alpha gene have abnormal skin architecture, wavy hair, and curly whiskers and often develop corneal inflammation. Cell. 1993;73:249–261. doi: 10.1016/0092-8674(93)90227-h. [DOI] [PubMed] [Google Scholar]
- Massagué J, Pandiella A. Membrane-anchored growth factors. Annu Rev Biochem. 1993;62:515–541. doi: 10.1146/annurev.bi.62.070193.002503. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Thiery JP, Lafont F, Stampfer F, Boyer B. Transient effect of epidermal growth factor on the motility of an immortalized mammary epithelial cell line. J Cell Sci. 1993;106:869–878. doi: 10.1242/jcs.106.3.869. [DOI] [PubMed] [Google Scholar]
- Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376:337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- Mroczkowski B, Reich M, Chen K, Bell GI, Cohen S. Recombinant human epidermal growth factor precursor is a glycosylated membrane protein with biological activity. Mol Cell Biol. 1989;9:2771–2778. doi: 10.1128/mcb.9.7.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opgenorth A, Strayer D, Upton C, McFadden G. Deletion of the growth factor gene related to EGF and TGF alpha reduces virulence of malignant rabbit fibroma virus. Virology. 1992;186:175–191. doi: 10.1016/0042-6822(92)90072-w. [DOI] [PubMed] [Google Scholar]
- Opresko LK, Wiley HS. Functional reconstitutional of the human epidermal growth factor receptor system in Xenopusoocytes. J Cell Biol. 1990;111:1661–1671. doi: 10.1083/jcb.111.4.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandiella A, Bosenberg MW, Huang EJ, Besmer P, Massagué J. Cleavage of membrane-anchored growth factors involves distinct protease activities regulated through common mechanisms. J Biol Chem. 1992;267:24028–24033. [PubMed] [Google Scholar]
- Riese DJ, Kim ED, Elenius K, Buckley S, Klagsbrun M, Plowman GD, Stern DF. The epidermal growth factor receptor couples transforming growth factor-alpha, heparin-binding epidermal growth factor-like factor, and amphiregulin to Neu, ErbB-3, and ErbB-4. J Biol Chem. 1996;271:20047–20052. doi: 10.1074/jbc.271.33.20047. [DOI] [PubMed] [Google Scholar]
- Ruff-Jamison S, McGlade J, Pawson T, Chen K, Cohen S. Epidermal growth factor stimulates the tyrosine phosphorylation of SHC in the mouse. J Biol Chem. 1993;268:7610–7612. [PubMed] [Google Scholar]
- Saeki T, Stromberg K, Qi CF, Gullick WJ, Tahara E, Normanno N, Ciardiello F, Kenney N, Johnson GR, Salomon DS. Differential immunohistochemical detection of amphiregulin and cripto in human normal colon and colorectal tumors. Cancer Res. 1992;52:3467–3473. [PubMed] [Google Scholar]
- Sakurai H, Tsukamoto T, Kjelsberg CA, Cantley LG, Nigam SK. EGF receptor ligands are a large fraction of in vitro branching morphogens secreted by embryonic kidney. Am J Physiol. 1997;273:F463–F472. doi: 10.1152/ajprenal.1997.273.3.F463. [DOI] [PubMed] [Google Scholar]
- Schneider C, Newman RA, Sutherland DR, Asser U, Greaves MF. A one-step purification of membrane proteins using a high efficiency immunomatrix. J Biol Chem. 1982;257:10766–10769. [PubMed] [Google Scholar]
- Schreiber AB, Winkler ME, Derynck R. Transforming growth factor-alpha: a more potent angiogenic mediator than epidermal growth factor. Science. 1986;232:1250–1253. doi: 10.1126/science.2422759. [DOI] [PubMed] [Google Scholar]
- Schuetz EG, Li D, Omiecinski CJ, Muller-Eberhard U, Kleinman HK, Elswick B, Guzelian PS. Regulation of gene expression in adult rat hepatocytes cultured on a basement membrane matrix. J Cell Physiol. 1988;134:309–323. doi: 10.1002/jcp.1041340302. [DOI] [PubMed] [Google Scholar]
- Shing Y, Christofori G, Hanahan D, Ono Y, Sasada R, Igarashi K, Folkman J. Betacellulin: a mitogen from pancreatic beta cell tumors. Science. 1993;259:1604–1607. doi: 10.1126/science.8456283. [DOI] [PubMed] [Google Scholar]
- Shoyab M, Plowman GD, McDonald VL, Bradley JG, Todaro GJ. Structure and function of human amphiregulin: a member of the epidermal growth factor family. Science. 1989;243:1074–1076. doi: 10.1126/science.2466334. [DOI] [PubMed] [Google Scholar]
- Sibilia M, Wagner EF. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science. 1995;269:234–238. doi: 10.1126/science.7618085. [DOI] [PubMed] [Google Scholar]
- Simske JS, Kaech SM, Harp SA, Kim SK. LET-23 receptor localization by the cell junction protein LIN-7 during C. elegansvulval induction. Cell. 1996;85:195–204. doi: 10.1016/s0092-8674(00)81096-x. [DOI] [PubMed] [Google Scholar]
- Snedeker SM, Brown CF, DiAugustine RP. Expression and functional properties of transforming growth factor alpha and epidermal growth factor during mouse mammary gland ductal morphogenesis. Proc Natl Acad Sci USA. 1991;88:276–280. doi: 10.1073/pnas.88.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampfer MR, Bodnar A, Garbe J, Wong M, Pan A, Villeponteau B, Yaswen P. Gradual phenotypic conversion associated with immortalization of cultured human mammary epithelial cells. Mol Biol Cell. 1997;8:2391–2405. doi: 10.1091/mbc.8.12.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampfer MR, Pan CH, Hosoda J, Bartholomew J, Mendelsohn J, Yaswen P. Blockage of EGF receptor signal transduction causes reversible arrest of normal and immortal human mammary epithelial cells with synchronous reentry into the cell cycle. Exp Cell Res. 1993;208:175–188. doi: 10.1006/excr.1993.1236. [DOI] [PubMed] [Google Scholar]
- Stampfer MR, Yaswen P. Growth, differentiation, and transformation of human mammary epithelial cells in culture. Cancer Treat Res. 1994;71:29–48. doi: 10.1007/978-1-4615-2592-9_2. [DOI] [PubMed] [Google Scholar]
- Thompson SA, Higashiyama S, Wood K, Pollitt NS, Damm D, McEnroe G, Garrick B, Ashton N, Lau K, Hancock N, et al. Characterization of sequences within heparin-binding EGF-like growth factor that mediate interaction with heparin. J Biol Chem. 1994;269:2541–2549. [PubMed] [Google Scholar]
- Thorne BA, Plowman GD. The heparin-binding domain of amphiregulin necessitates the precursor pro-region for growth factor secretion. Mol Cell Biol. 1994;14:1635–1646. doi: 10.1128/mcb.14.3.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- Toyoda H, Komurasaki T, Ikeda Y, Yoshimoto M, Morimoto S. Molecular cloning of mouse epiregulin, a novel epidermal growth factor-related protein, expressed in the early stage of development. FEBS Lett. 1995;377:403–407. doi: 10.1016/0014-5793(95)01403-9. [DOI] [PubMed] [Google Scholar]
- Traverse S, Seedorf K, Paterson H, Marshall CJ, Cohen P, Ullrich A. EGF triggers neuronal differentiation of PC12 cells that overexpress the EGF receptor. Curr Biol. 1994;4:694–701. doi: 10.1016/s0960-9822(00)00154-8. [DOI] [PubMed] [Google Scholar]
- Tzahar E, Pinkas-Kramarski R, Moyer JD, Klapper LN, Alroy I, Levkowitz G, Shelly M, Henis S, Eisenstein M, Ratzkin BJ, et al. Bivalence of EGF-like ligands drives the ErbB signaling network. EMBO (Eur Mol Biol Organ) J. 1997;16:4938–4950. doi: 10.1093/emboj/16.16.4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton C, Macen JL, McFadden G. Mapping and sequencing of a gene from myxoma virus that is related to those encoding epidermal growth factor and transforming growth factor alpha. J Virol. 1987;61:1271–1275. doi: 10.1128/jvi.61.4.1271-1275.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley HS. Receptors as models for the mechanisms of membrane protein turnover and dynamics. Curr Top Membr Transp. 1985;24:369–412. [Google Scholar]
- Wiley HS. Anomalous binding of epidermal growth factor to A431 cells is due to the effect of high receptor densities and a saturable endocytic system. J Cell Biol. 1988;107:801–810. doi: 10.1083/jcb.107.2.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler ME, O'Connor L, Winget M, Fendly B. Epidermal growth factor and transforming growth factor alpha bind differently to the epidermal growth factor receptor. Biochemistry. 1989;28:6373–6378. doi: 10.1021/bi00441a033. [DOI] [PubMed] [Google Scholar]
- Wong ST, Winchell LF, McCune BK, Earp HS, Teixido J, Massagué J, Herman B, Lee DC. The TGF-alpha precursor expressed on the cell surface binds to the EGF receptor on adjacent cells, leading to signal transduction. Cell. 1989;56:495–506. doi: 10.1016/0092-8674(89)90252-3. [DOI] [PubMed] [Google Scholar]
- Xie W, Paterson AJ, Chin E, Nabell LM, Kudlow JE. Targeted expression of a dominant negative epidermal growth factor receptor in the mammary gland of transgenic mice inhibits pubertal mammary duct development. Mol Endocrinol. 1997;11:1766–1781. doi: 10.1210/mend.11.12.0019. [DOI] [PubMed] [Google Scholar]
- Yoshitake Y, Nishikawa K. Production of monoclonal antibodies with specificity for different epitopes on the human epidermal growth factor molecule. Arch Biochem Biophys. 1988;263:437–446. doi: 10.1016/0003-9861(88)90656-x. [DOI] [PubMed] [Google Scholar]