

Abstract
The integrin α9β1 has been shown to be widely expressed on smooth muscle and epithelial cells, and to mediate adhesion to the extracellular matrix proteins osteopontin and tenascin-C. We have found that the peptide sequence this integrin recognizes in tenascin-C is highly homologous to the sequence recognized by the closely related integrin α4β1, in the inducible endothelial ligand, vascular cell adhesion mole-cule-1 (VCAM-1). We therefore sought to determine whether α9β1 also recognizes VCAM-1, and whether any such interaction would be biologically significant. In this report, we demonstrate that α9β1 mediates stable cell adhesion to recombinant VCAM-1 and to VCAM-1 induced on human umbilical vein endothelial cells by tumor necrosis factor-α. Furthermore, we show that α9β1 is highly and selectively expressed on neutrophils and is critical for neutrophil migration on VCAM-1 and tenascin-C. Finally, α9β1 and α4 integrins contribute to neutrophil chemotaxis across activated endothelial monolayers. These observations suggest a possible role for α9β1/VCAM-1 interactions in extravasation of neutrophils at sites of acute inflammation.
Keywords: integrin, α9β1, α4, neutrophil migration, vascular cell adhesion molecule-1
Integrins are heterodimeric receptors for extracellular matrix and cell surface counter-receptors which play important roles in embryonic development, inflammation, wound healing, and tumorigenesis (Hynes, 1987, 1992; Ruoslahti and Pierschbacher, 1987). Integrin ligand-binding specificity is determined by structural features of each subunit, but there is considerable ligand-binding overlap among integrin heterodimers. One clue to ligand-binding overlap has been the degree of sequence homology among integrin α subunits. For example, the integrin α subunits α5, αv, αIIβ, and α8 are all closely related, and integrin heterodimers containing these α subunits recognize ligands containing the peptide sequence arginine-glycine-aspartic acid (Hynes, 1992; Schnapp et al., 1995). Similarly, the αm, αL, and αx subunits are highly homologous to one another and recognize closely related immunoglobulin family members as ligands (Hynes, 1992). We previously cloned and sequenced the integrin α9 subunit, and have shown that it forms a single integrin heterodimer, α9β1 (Palmer et al., 1993). The α9 subunit cDNA sequence is 41% identical to the integrin α4 subunit sequence, but ≤27% identical to any other integrin subunit, identifying α9 and α4 as sole members of a subfamily of integrin α subunits.
In an effort to understand the structural basis of α9β1 ligand-binding in more detail, we recently mapped the α9β1 ligand-binding site in the extracellular matrix protein tenascin-C (Yokosaki et al., 1994). α9β1 binds to a single exposed loop in the third fibronectin type III repeat of tenascin-C (B–C loop) to a minimal sequence EIDGIEL (Schneider et al., 1998; Yokosaki et al., 1998). We noticed that a critical portion of this sequence (IDG) is homologous to the tripeptide sequence IDS present in the previously mapped ligand-binding site for the α4β1 ligand, vascular cell adhesion molecule-1 (VCAM-11; Clements et al., 1994; Yokosaki et al., 1998). Therefore, we undertook the current study to determine whether α9β1 recognizes VCAM-1 as a ligand and whether or not any such interaction is biologically significant.
Materials and Methods
Reagents
BSA, formyl-methionylleucylphenylalanine (FMLP), and dextran were purchased from Sigma Chemical Co. Recombinant human tumor necrosis factor (TNF)-α, recombinant human interferon (IFN)-γ (specific activity of 107 U/mg), and recombinant interleukin 8 (IL-8) were obtained from R&D Systems, Inc. Fluorescent reagent, 2′,7′-bis-(carboxyethyl)-5,6-carboxy-fluorescein acetoxymethyl ester (BCECF-AM) was purchased from Molecular Probes, Inc. A recombinant form of the third fibronectin type III repeat of chicken tenascin-C (Prieto et al., 1993) containing alanine substitution mutations within the RGD site (TNfn3RAA), was obtained from Anita Prieto and Kathryn Crossin (Scripps Research Institute, La Jolla, CA) and prepared in Escherichia coli. A recombinant VCAM-1/IgG chimera (Yednock et al., 1995) was produced in baculovirus as previously described. Recombinant intercellular adhesion molecule-1 (ICAM-1)-Cκ fusion protein was a gift from B. Imhof (Centre Medicale Universitaire, Geneva, Switzerland) to D. Erle (University of California, San Francisco, CA). Ficoll-hypaque plus for isolation of neutrophils from venous blood was purchased from Pharmacia Biotech, Inc. and used according to the manufacturer's specifications.
Antibodies, Cells, and Cell Culture
Mouse mAbs, Y9A2 against human α9β1 (Wang et al., 1996) and AN100226M (100226) against α4 (Kent et al., 1995), were prepared as previously described. Mouse mAbs, W6/32 against human MHC and IB4 against the integrin β2 subunit, were prepared from hybridomas obtained from American Type Tissue Collection. Mouse monoclonal antihuman VCAM-1 (CD106) was purchased from R&D Systems. FITC-labeled mouse monoclonal anti-CD16 antibody was purchased from Caltag. Human umbilical vein endothelial (HUVE) cells were purchased from Clonetics and grown in endothelial cell growth media (EGM) containing 2% FBS, 10 ng/ml human recombinant EGF, 50 μg/ml gentamycin, 50 ng/ ml amphotericin B, 12 μg/ml bovine brain extract, and 1 μg/ml hydrocortisone and were used between passage 3 and 10. α9- and mock-transfected SW480 and CHO cells were generated by transfection with the previously described full-length α9 expression plasmid pcDNAIneoα9 (Yokosaki et al., 1994) or the empty vector pcDNAIneo (Invitrogen Corp.) by calcium phosphate precipitation. Transfected cells were maintained in Dulbecco's minimal essential medium(DMEM) supplemented with 10% FCS and the neomycin analogue G-418 (1 mg/ml; Life Technologies, Inc.). Both cell lines continuously expressed high surface levels of α9β1 as determined by flow cytometry with Y9A2 (Yokosaki, 1996, 1998).
Flow Cytometry
Cultured cells were harvested by trypsinization and rinsed with PBS. Nonspecific binding was blocked with normal goat serum at 4°C for 10 min. Cells were then incubated with primary antibodies (unconjugated or conjugated with FITC) for 20 min at 4°C, followed by secondary antibodies conjugated with phycoerythrin (Chemicon International, Inc.). Between incubations, cells were washed twice with PBS. The stained cells were resuspended in 100 μl of PBS and fluorescence was quantified on 5,000 cells with a FACScan® (Becton Dickinson and Co.).
Immunoprecipitation and Western Blotting
Cells were lysed in immunoprecipitation buffer (100 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM CaCl2, 1% Triton X-100, 0.1% SDS, and 0.1% NP-40) supplemented with 10 μg/ml pepstatin (Sigma Chemical Co.), 10 μg/ml leupeptin, 5 μg/ml aprotinin (Calbiochem-Novabiochem Corp.), and 1 mM phenylmethylsulfonyl fluoride (Sigma Chemical Co.). Human neutrophils (107) were incubated with 1 mM diisopropyl flurophosphate (Sigma Chemical Co.) for 15 min before cell lysis. After preclearing with protein G–Sepharose, the supernatant was incubated with primary antibody for 2 h at 4°C and immune complexes were captured by protein G–Sepharose for 45 min at 4°C. The beads were washed five times, and boiled in 2.5× nonreducing Laemmli sample buffer, and samples were separated by SDS-PAGE on 7.5% gels under reducing conditions and transferred to Immobilon membranes. Membranes were blocked with 4% casein, incubated with affinity-purified anti-α9 cytoplasmic domain antiserum 1057 (Palmer et al., 1993), and developed with luminol.
Cell Adhesion Assays
Wells of nontissue culture treated polystyrene 96-well flat bottomed microtiter plates (Nunc Inc.) were coated by incubation with 100 μl VCAM-1/Ig or TNfn3RAA for 1 h at 37°C. After incubation, wells were washed with PBS, then blocked with 1% BSA in DMEM at 37°C for 30 min. Control wells were filled with 1% BSA in DMEM. SW480 or CHO cells were detached using trypsin/EDTA and resuspended in serum-free DMEM. For blocking experiments, cells were incubated with 10 μg/ml Y9A2 and/ or 100226, for 15 min at 4°C before plating. The plates were centrifuged (top side up) at 10 g for 5 min before incubation for 1 h at 37°C in humidified 5% CO2. Nonadherent cells were removed by centrifugation (top side down) at 48 g for 5 min. Attached cells were fixed with 1% formaldehyde and stained with 0.5% crystal violet, and the wells were washed with PBS. The relative number of cells in each well was evaluated after solubilization in 40 μl of 2% Triton X-100 by measuring the absorbance at 595 nm in a microplate reader (Bio-Rad Laboratories). All determinations were carried out in triplicate.
For adhesion assays on HUVE cells, confluent monolayers of HUVE cells were prepared in 96-well plates in 250 μl of EGM with 2% FBS. Plates were washed twice with serum-free DMEM, then stimulated for 24 h at 37°C with TNF-α (3 ng/ml) or IFN-γ (3 ng/ml) in serum-free DMEM. SW480 cells were detached using trypsin/EDTA and labeled with 2 μM BCECF-AM at room temperature for 30 min. Then cells were washed three times with serum-free DMEM and incubated with blocking antibody, Y9A2 (10 μg/ml), 100226 (10 μg/ml), or combinations of these antibodies for 15 min on ice. In some experiments, HUVE cells were incubated with CD106 (5 μg/ml) for 15 min at 37°C. 50,000 cells in 200 μl of serum-free DMEM were added to each well, and plates were centrifuged at 20 g for 5 min, and covered with aluminum foil to prevent photobleaching. Plates were then incubated for 60 min at 37°C in 5% CO2. After incubation, nonadherent cells were removed by washing twice with serum-free DMEM. Finally, 200 μl of the same medium was added to each well, and fluorescence was quantified with a fluorometer (Fluoroskan II; Labsystems) at excitation wavelength 485 nm and emission wavelength 538 nm. The adherent ratio (%) was calculated as follows: (fluorescence from experimental sample − fluorescence from negative control sample) ÷ total fluorescence added to chamber. All determinations were carried out in triplicate.
Neutrophil Migration Assays
Neutrophils were purified from human peripheral venous blood containing 20 U/ml of heparin. Neutrophils were isolated by ficoll-hypaque density gradient centrifugation, followed by 3% dextran sedimentation (Gresham et al., 1986). Erythrocytes were subjected to hypotonic lysis, remaining neutrophils were washed and resuspended in PBS. The isolated neutrophils were >95% pure and >95% viable as assessed by Wright-Giemsa staining and trypan blue exclusion, respectively. Cell migration was analyzed essentially as described by Marks et al. (1991). In brief, glass coverslips were placed in 35-mm culture dishes and incubated with 100 μl serum-free media containing 10 μg/ml VCAM-1/Ig, 10 μg/ml TNfn3RAA, and 5 μg/ml of ICAM-1 or 1% BSA for 60 min at 37°C, washed, and then incubated with 1% BSA for 30 min. Neutrophils were incubated with no antibody, Y9A2 (10 μg/ml), 100226 (10 μg/ml), IB4 (20 μg/ml), or combinations of antibodies for 15 min at 4°C, and were then incubated for 10 min at 37°C with or without 10 nM FMLP. 104 cells were plated onto the coverslip area of each well and allowed to attach at 37°C for 5 min. Dishes were then placed on a videomicroscope stage and individual fields (200×) were recorded for 3 min. Three different fields were examined in each chamber. To count the number of migrating cells in a given field, outlines were made of each cell. Cells were considered to have migrated when both the leading edge and tail of the cell moved ≥7 μm from their initial position. At least 40 neutrophils were analyzed per field and the ratio of migrating to total cells was calculated.
Neutrophil Transmigration Assays
Transendothelial neutrophil migration was assessed as described by Cooper et al. (1995). HUVE cells were plated onto polycarbonate inserts (Transwell, 6.5-mm diameter, 8-μm pore for 24-well plate; Costar Corp.) in 200 μl of serum-containing EGM, and allowed to grow to confluence over 72 h. 500 μl serum-free DMEM was added to the lower chamber of each well. 24 h before addition of neutrophils, upper chambers were washed twice with serum-free media and new medium with or without 3 ng/ml of TNF-α. Immediately before the addition of neutrophils, the upper chambers were washed twice with serum-free DMEM and medium in the lower chamber was replaced with 500 μl serum-free DMEM or serum-free DMEM with 10 nM FMLP or 50 ng/ml IL-8. In some experiments HUVE cells were incubated with CD106 (5 μg/ml) at 37°C for 15 min. Purified neutrophils were incubated with no antibody, Y9A2 (10 μg/ml), 100226 (10 μg/ml), IB4 (20 μg/ml), W6/32 (10 μg/ml), or combinations of antibodies for 15 min at 4°C, and 2 × 105 cells in 200 μl of media were added to each upper chamber. After 3 h at 37°C in 5% CO2, nonadherent cells in the upper chamber were removed. Medium, including migrated neutrophils from the lower chamber, was collected, the lower chamber was rinsed several times to collect all the neutrophils that had transmigrated, and the absence of additional adherent neutrophils was confirmed microscopically. The medium and all washes were pooled and resuspended, and cells were counted with a hemocytometer. All determinations were carried out in duplicate and repeated at least twice.
Results
α9β1 Mediates Static Adhesion of Resting α9-transfected SW480 Cells and CHO Cells to VCAM-1
To determine whether VCAM-1 could function as a ligand for α9β1, we performed cell adhesion assays with two different cell lines, SW480 and CHO, that had been stably transfected with either an α9-expression plasmid or empty vector. Both cell lines stably expressed α9β1 on the cell surface as demonstrated by flow cytometry with the anti-α9β1 antibody Y9A2 (Fig. 1, A and B). Adhesion assays were performed on plates coated with either the known α9β1 ligand, recombinant TNfn3RAA (Fig. 1, C and D), or recombinant VCAM-1/Ig (Fig. 1, E and F). For both cell lines, α9-transfectants adhered to both TNfn3 and to VCAM-1 in a concentration-dependent manner, whereas mock-transfectants did not adhere to either substrate. Adhesion of each α9-transfected cell line was completely inhibited by the anti-α9β1 antibody, Y9A2, demonstrating that this effect was mediated by α9β1.
Figure 1.

Adhesion of α9- and mock-transfected SW480 and CHO cells to TNfn3RAA or VCAM-1. Flow cytometric evaluation of cell surface expression of integrin α9 on CHO cells (A), or SW480 cells (B). Open peaks represent fluorescence of mock-transfected cells and shaded peaks represent fluorescence of α9-transfected cells stained with anti-α9β1 antibody, Y9A2. α9- or mock-transfected CHO (C and E) or SW480 cells (D and F) were added to 96-well plates coated with a range of concentrations of TNfn3RAA (C and D) or VCAM-1/Ig (E and F). Cells were allowed to attach for 1 h, nonadherent cells were removed by centrifugation, and adherent cells were stained with crystal violet and quantified by measurement of absorbance at 595 nm. Data for a typical experiment are shown and are expressed as the mean (+ SD) of triplicate measurements for untreated α9-transfected cells (squares), untreated mock-transfected cells (circles), α9-transfected cells treated with Y9A2 (10 μg/ml; diamonds), and mock-transfected cells treated with Y9A2 (10 μg/ml; triangles). Similar results were obtained in four separate experiments.
α9β1 Mediates Adhesion to TNF-α–activated, but not to IFN-γ–activated HUVE Cells, Via Interaction with Induced VCAM-1
To determine whether α9β1-mediated adhesion to VCAM-1 was biologically significant, we next examined the role of this integrin in adhesion of cells to resting HUVE cells, and to HUVE cells that had been activated by incubation with TNF-α (3 ng/ml), a well characterized inducer of VCAM-1 expression, or IFN-γ (3 ng/ml), a cytokine that does not induce VCAM-1 expression. The effects of each cytokine on VCAM-1 expression under the conditions used in these experiments were examined by flow cytometry with anti–VCAM-1 antibody CD106 (Fig. 2, B–D). As expected, resting HUVE cells (Fig. 2 B) and HUVE cells stimulated with IFN-γ (Fig. 2 D) did not express detectable levels of VCAM-1, but VCAM-1 was dramatically induced by TNF-α (Fig. 2 C). All cell lines examined demonstrated baseline adhesion to resting HUVE cells, and demonstrated a similar level of adhesion to HUVE activated by IFN-γ, and this baseline adhesion was unaffected by anti-α9β1 antibody (Fig. 2 A). However, only α9-transfected cells demonstrated enhanced adhesion to TNF-α–treated HUVE. This enhanced adhesion was returned completely to basal levels by antibody to either α9β1(Y9A2) or to VCAM-1 (CD106), demonstrating that it was due to an interaction between α9β1 and VCAM-1.
Figure 2.

Adhesion of α9- or mock-transfected SW480 cells to HUVE cells. (A) Confluent monolayers of HUVE cells were incubated for 24 h with medium alone (no activation), TNF-α (3 ng/ml), or IFN-γ (3 ng/ml). Fluorescently labeled α9- or mock-transfected SW480 cells were allowed to adhere to HUVE cell monolayers for 60 min in the presence or absence of the α9β1 blocking antibody Y9A2 (10 μg/ ml) or the VCAM-1 blocking antibody CD106 (5 μg/ml). Nonadherent cells were removed by gentle washing and the percent of adherent cells was calculated based on fluorescence. Data are presented as the mean (+ SD) of triplicate measurements. Similar results were obtained in two separate experiments. Flow cytometric evaluation of cell surface expression of VCAM-1 on HUVE cells treated with medium alone (B), TNF-α (3 ng/ml; C), or IFN-γ (3 ng/ml; D). Open peaks represent fluorescence of unstained HUVE cells and shaded peaks represent fluorescence of cells stained with the anti–VCAM-1 antibody, CD106.
α9β1 Is Expressed on Neutrophils
We have previously demonstrated that α9β1 is widely expressed on epithelial and smooth muscle cells (Palmer et al., 1993), but expression on leukocytes has not been reported. To determine whether α9β1 is expressed on cells likely to encounter activated endothelial cells, we performed flow cytometry on whole blood leukocytes with the α9β1 antibody Y9A2. We evaluated expression on neutrophils, monocytes, and lymphocytes by gating on each population separately, based on differential light scattering. From a separate atopic donor we evaluated expression on eosinophils, which were separated from other leukocytes based on light scattering and the absence of surface expression of CD16. In parallel, we examined expression of the structurally related integrin subunit, α4. α9β1 was not detected on lymphocytes or eosinophils and was expressed at low levels on monocytes (Fig. 3 A). In contrast, α9β1 was highly and uniformly expressed on human neutrophils. As expected, α4 was highly expressed on lymphocytes, monocytes, and eosinophils, but was also detected on neutrophils, albeit at considerably lower levels.
Figure 3.
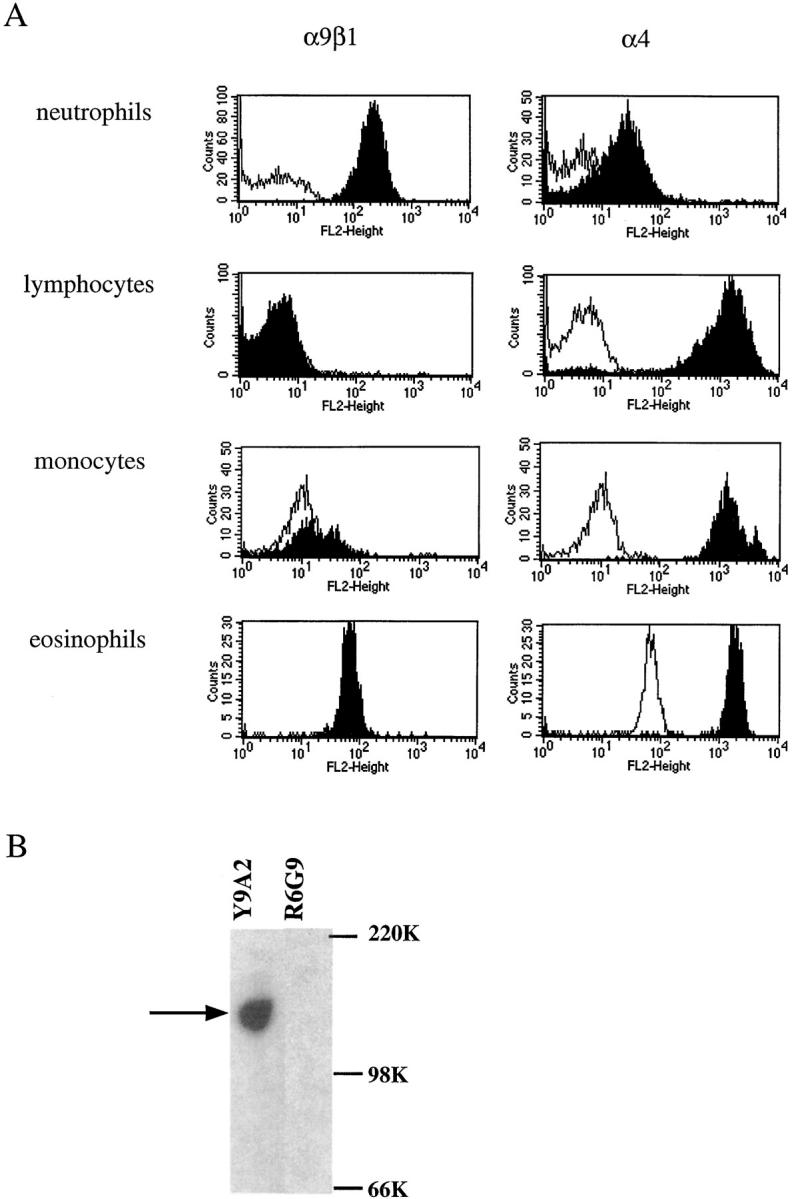
Expression of α4 or α9 integrins on leukocytes. (A) Whole blood leukocytes were stained with control antibody E7P6 that recognizes αvβ6, an integrin not expressed on leukocytes (unshaded peaks), Y9A2 against α9β1, or 100226 against α4 (shaded peaks). Fluorescence of lymphocytes, neutrophils, eosinophils, and monocytes were analyzed separately by gating on each population on the basis of a plot of forward versus side scattering of light. Fluorescence of eosinophils was analyzed from separate atopic donor by gating on eosinophils based on light scattering and the absence of expression CD16. (B) Western blot with anti-α9 antiserum 1057 of lysates of human neutrophils that had been immunoprecipitated with anti-α9β1 antibody Y9A2 or the control antibody R6G9 against the irrelevant integrin αvβ6 (lane 2). The expected molecular mass of the α9 subunit (160 kD) is shown by the lefthand arrow. The position of molecular size marker (kD) is shown to the right.
Expression of α9 on neutrophils was further confirmed by immunoprecipitation with Y9A2 followed by Western blotting with an affinity-purified antiserum raised against a unique portion of the α9 cytoplasmic domain. A band of 160 kD (appropriate molecular mass for α9) was detected in lysate of human neutrophils after immunoprecipitation with Y9A2, but not after immunoprecipitation with the control antibody R6G9 (Fig. 3 B).
α9β1 Mediates Migration of FMLP-activated Neutrophils on TNfn3 or VCAM-1
To determine whether α9β1 expression on neutrophils was biologically significant, we initially sought to examine static adhesion of neutrophils to dishes coated with either TNfn3RAA or VCAM-1. However, in the absence of antibodies against β2 integrins, neutrophils avidly adhered to all surfaces examined, and in the presence of β2 integrin blocking antibodies, neutrophils could not be induced to adhere to either VCAM-1 or TNfn3RAA by incubation with MnCl2, FMLP, phorbol esters, or the β1 activating antibody TS2/16 (data not shown). Therefore, we examined the possible role of α9β1 in another important neutrophil function, cell migration. Migration was examined by counting the numbers of individual neutrophils that migrated on chambers coated with either TNfn3RAA or VCAM-1 in the presence or absence of the activating agonist FMLP (10 nM). In the absence of FMLP, very few neutrophils migrated on either substrate (Fig. 4 A), and antibodies against α9β1, α4, or β2 integrins had no effect. In the presence of FMLP, neutrophil migration was significantly enhanced on TNfn3RAA, an effect that was abolished by antibody against α9β1. FMLP also enhanced neutrophil migration on VCAM-1, and this effect was partially inhibited by antibodies against α9β1 or α4, and completely inhibited by the combination of both antibodies. These data demonstrate a significant role for α9β1 in mediating neutrophil migration on both substrates. Antibody against β2 integrins had no effect on neutrophil migration on FMLP-induced neutrophil migration on TNfn3RAA or VCAM-1. However, as expected, antibody against β2 inhibited FMLP-induced migration on the β2 integrin ligand ICAM-1, whereas antibodies against α9β1 or α4 had no effect (Fig. 4 B).
Figure 4.

Neutrophil migration on VCAM-1 or TNfn3RAA. Neutrophils were allowed to migrate on glass coverslips coated with: (A) 1% BSA or TNfn3RAA (10 μg/ml); or (B) VCAM-1 (10 μg/ml) or ICAM-1 (5 μg/ml), for 3 min in the presence or absence of FMLP (10 nM), and in the presence of: no antibody; α9β1 antibody, Y9A2 (10 μg/ml); α4 antibody, 100226 (10 μg/ml); antibody IB4 (20 μg/ml); or the combination of these antibodies. The percentage of migrating cells was determined by analyzing ≥40 cells from each of three microscopic fields, and is expressed as the mean (+ SD) of triplicate values from two separate experiments.
α9β1 Mediates Migration of Neutrophils through Activated HUVE Cell Monolayers
We next sought to determine whether the effect of α9β1 and α4 integrin(s) described above was relevant to an in vitro model of neutrophil extravasation–migration across endothelial monolayers. HUVE cells were grown to confluence on the top side of permeable filter supports and incubated in the presence or absence of TNF-α (3 ng/ml). Purified neutrophils were added to the apical compartment in the presence or absence of FMLP added to the basal compartment. These studies were performed in the absence of blocking antibodies, or in the presence of antibodies against α9β1, α4, β2, VCAM-1, control antibody against MHC, or combinations of these antibodies. As expected, in the absence of blocking antibodies, FMLP greatly increased neutrophil migration into the bottom compartment, and this effect was augmented by pretreatment of HUVE cells with TNF-α (Fig. 5 A). No antibody affected basal migration across unstimulated HUVE cells or FMLP-induced migration across unstimulated HUVE cells (Fig. 5 B). However, antibody against either α9β1 or α4 inhibited the augmented migration induced by TNF-α. Antibody against VCAM-1 was equally effective in inhibiting migration across TNF-α–treated HUVE cells, suggesting that TNF-α augmented transmigration was mediated by an interaction between α9β1 and α4 integrins and VCAM-1. As previously reported, antibody against β2 integrins also partially inhibited transmigration in response to FMLP, but this effect was surprisingly small. Essentially identical results were obtained when IL-8 was used as a chemoattractant in place of FMLP (data not shown).
Figure 5.

Transmigration of neutrophils across activated HUVE cell monolayers. Purified human neutrophils that had been incubated with no antibody or antibody to α9β1 (Y9A2, 10 μg/ml), α4β1 (100226, 10 μg/ml), β2 (IB4, 20 μg/ml), a combination of these antibodies, or human MHC (W6/32, 10 μg/ml) were added to the top chambers above microporous chambers containing confluent monolayers of HUVE cells that had been incubated with (A) or without TNF-α (3 ng/ml; B) for 24 h. DMEM containing FMLP (10 nM) or DMEM alone was added to the bottom chamber. After 3 h at 37°C in 5% CO2, neutrophils that had migrated across the monolayer were collected from the bottom chamber and counted. In additional chambers, untreated neutrophils were added to HUVE cells that had been preincubated for 15 min with antibody to VCAM-1 (CD106, 5 μg/ml). Data are expressed as the mean (+ SD) of quadruplicate measurements from two separate experiments.
Discussion
The results of the current study demonstrate that the inducible endothelial cell immunoglobulin family member, VCAM-1, is an effective ligand for the integrin α9β1. This receptor–ligand interaction is sufficient to support adhesion of α9-transfected cell lines to VCAM-1 and to TNF-α–activated HUVE cells, an effect that is mediated by the binding of α9β1 to VCAM-1. Furthermore, α9β1 is uniformly and specifically expressed on normal resting human neutrophils, and mediates both neutrophil migration on a fragment of tenascin-C or VCAM-1 and transmigration of neutrophils across TNF-α–activated endothelial monolayers. Together, these data suggest a previously unsuspected role for α9β1 and VCAM-1 in extravasation of neutrophils at sites of acute inflammation.
In addition to α9β1, we found detectable, albeit low, levels of the structurally related integrin α4 subunit on resting human neutrophils. This finding is consistent with several previous reports of α4 expression on neutrophils from a variety of species (Issekutz et al., 1996; Gao and Issekutz, 1997; Davenpeck et al., 1998). Although the level of expression of α4 we detected on human neutrophils was one to two orders of magnitude lower than expression on eosinophils, monocytes, and lymphocytes, this low level expression appeared to be biologically significant, since antibody against α4 partially inhibited migration of neutrophils on VCAM-1 and migration across TNF-activated endothelial monolayers. Recently, α4β1 has been shown to mediate both neutrophil adhesion to VCAM-1 (Davenpeck et al., 1998) and neutrophil transmigration across fibroblast monolayers (Gao and Issekutz, 1997). As expected, α4 integrins did not contribute to migration on TNfn3RAA, since this fragment of tenascin is not a ligand for either α4 integrin.
Adhesion of activated neutrophils to endothelial cells at sites of inflammation is well known to require the participation of integrins sharing the β2 subunit (Arfors et al., 1987) which bind to two other members of the immunoglobulin family expressed on endothelial cells, ICAM-1 (Marlin and Springer, 1987; Diamond et al., 1990) and ICAM-2 (Staunton et al., 1989). ICAM-1 is constitutively expressed on many epithelia, but expression is dramatically induced by a variety of inflammatory stimuli, including TNF-α. Our data do not address the role of α9β1 or α4 integrins in stable adhesion of neutrophils, since we were not able to maintain adhesion of these cells to any substrate in the presence of β2 integrin blocking antibodies. This effect could be due to a critical role of these integrins in adhesion or to an inhibitory signaling pathway through which antibody-mediated ligation of β2 integrins inhibits the function of other integrins, such as α9β1. However, the mechanisms underlying the subsequent steps in neutrophil extravasation, including detachment from sites of initial adhesion and subsequent migration across the endothelial cell surface and components of the underlying extracellular matrix, are not as well understood. The data in this manuscript suggest, at least in the model system used, that α9β1 and α4 integrins are likely to play important roles. Both integrins could contribute to migration across VCAM-1 expressing endothelial cells and shared ligands such as osteopontin (Smith et al., 1996; Bayless et al., 1998), and α9β1 could be critical for migration across tenascin-C that is present outside the vasculature at sites of inflammation (Erickson, 1993).
A role for β2 integrin–independent processes in neutrophil extravasation in vivo has been suggested by several sets of observations, including studies of neutrophil extravasation into the liver in response to endotoxin (Essani et al., 1997) and neutrophil migration into the alveolar spaces of the lung in response to intratracheal instillation of live bacteria (Doerschuk et al., 1990). Recent studies demonstrating neutrophil extravasation into the lungs and peritoneal cavity in β2 integrin knockout mice also demonstrate the importance of mechanisms independent of β2 integrins (Mizgerd et al., 1997). The extent to which these events are mediated by α9β1 and/or α4 integrins needs to be determined from in vivo studies. We have recently succeeded in generating mice expressing a null mutation in the α9 subunit gene, but these mice die within 10 d of birth (unpublished observation). However, the development of bone marrow chimeras from this line should allow us to directly examine these questions.
In addition to the expression on neutrophils described in this report, α9β1 is widely expressed on muscle cells, surface epithelial cells, and hepatocytes (Palmer et al., 1993). It is unclear what role, if any, interactions with VCAM-1 might have at these sites. VCAM-1 has also been reported to be expressed on muscle cells under various conditions (Rosen et al., 1992; Sheppard et al., 1994), so it is conceivable that α9β1/VCAM-1 interactions may be biologically significant in muscle as well. Such an effect could explain the apparent contradiction between reports, based on antibody inhibition, that α4β1/VCAM-1 binding plays a critical role in myotube formation (Rosen et al., 1992) and the normal muscle development of α4 knockout cells in chimeric mice (Yang et al., 1996), if the α4 knockout led to a developmentally regulated increase in α9β1 expression.
In summary, we have identified VCAM-1 as a novel and biologically significant ligand for the integrin α9β1, have demonstrated that this integrin is expressed on neutrophils and mediates neutrophil migration on two relevant ligands and neutrophil transmigration across activated endothelial monolayers. These findings support a role for α9β1/ VCAM-1 interactions in extravasation of neutrophils at sites of inflammation.
Acknowledgments
We thank Amha Atakilit and David Erle for technical and intellectual assistance with flow cytometric analysis.
This work was supported by National Institutes of Health grants HLAI33259, HL47412, HL53949, and HL56385 to D. Sheppard.
Abbreviations used in this paper
- EGM
endothelial cell growth media
- FMLP
formyl-methionylleucylphenylalanine
- HUVE
human umbilical vein endothelial
- ICAM
intercellular adhesion molecule
- IFN
interferon
- TNF
tumor necrosis factor
- VCAM-1
vascular cell adhesion molecule-1
References
- Arfors K, Lundberg C, Lindblom L, Lundberg K, Beatty PG, Harlan JM. A monoclonal antibody to the membrane glycoprotein complex CD18 inhibits polymorphonuclear leukocyte accumulation and plasma leakage in vivo. Blood. 1987;69:338–340. [PubMed] [Google Scholar]
- Bayless KJ, Meininger GA, Scholtz JM, Davis GE. Osteopontin is a ligand for the alpha4beta1 integrin. J Cell Sci. 1998;111:1165–1174. doi: 10.1242/jcs.111.9.1165. [DOI] [PubMed] [Google Scholar]
- Clements JM, Newham P, Shepherd M, Gilbert R, Dudgeon TJ, Needham LA, Edwards RM, Berry L, Brass A, Humphries MJ. Identification of a key integrin-binding sequence in VCAM-1 homologous to the LDV active site in fibronectin. J Cell Sci. 1994;107:2127–2135. doi: 10.1242/jcs.107.8.2127. [DOI] [PubMed] [Google Scholar]
- Cooper D, Lindberg FP, Gamble JR, Brown EJ, Vadas MA. Transendothelial migration of neutrophils involves integrin-associated protein (CD47) Proc Natl Acad Sci USA. 1995;92:3978–3982. doi: 10.1073/pnas.92.9.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenpeck KL, Sterbinsky SA, Bochner BS. Rat neutrophils express alpha4 and beta1 integrins and bind to vascular cell adhesion molecule-1 (VCAM-1) and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) Blood. 1998;91:2341–2346. [PubMed] [Google Scholar]
- Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia AJ, Hibbs ML, Springer TA. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18) J Cell Biol. 1990;111:3129–3139. doi: 10.1083/jcb.111.6.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerschuk CM, Winn RK, Coxson HO, Harlan JM. CD18-dependent and -independent mechanisms of neutrophil adherence in the pulmonary and systemic microvasculature of rabbits. J Immunol. 1990;114:2327–2333. [PubMed] [Google Scholar]
- Erickson HP. Tenascin-C, tenascin-R, tenascin-X: a family of talented proteins in search of functions. Curr Opin Cell Biol. 1993;5:869–876. doi: 10.1016/0955-0674(93)90037-q. [DOI] [PubMed] [Google Scholar]
- Essani NA, Bajt ML, Farhood A, Vonderfecht SL, Jaeschke H. Transcriptional activation of vascular cell adhesion molecule-1 gene in vivo and its role in the pathophysiology of neutrophil-induced liver injury in murine endotoxin shock. J Immunol. 1997;158:5941–5948. [PubMed] [Google Scholar]
- Gao JX, Issekutz AC. The beta 1 integrin, very late activation antigen-4 on human neutrophils can contribute to neutrophil migration through connective tissue fibroblast barriers. Immunology. 1997;90:448–454. doi: 10.1111/j.1365-2567.1997.00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham HD, Clement LT, Lehmeyer JE, Griffin FM., Jr Simulation of human neutrophil Fc receptor-mediated phagocytosis by a low molecular weight cytokine. J Immunol. 1986;137:868–875. [PubMed] [Google Scholar]
- Hynes RO. Integrins: a family of cell surface receptors. Cell. 1987;48:549–554. doi: 10.1016/0092-8674(87)90233-9. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Issekutz TB, Miyasaka M, Issekutz AC. Rat neutrophils express very late activation antigen 4 and it mediates migration to arthritic joint and dermal inflammation. J Exp Med. 1996;183:2175–2184. doi: 10.1084/jem.183.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent SJ, Karlik SJ, Cannon C, Hines DK, Yednock TA, Fritz LC, Horner HC. A monoclonal antibody to alpha 4 integrin suppresses and reverses active experimental allergic encephalomyelitis. J Neuroimmunol. 1995;58:1–10. doi: 10.1016/0165-5728(94)00165-k. [DOI] [PubMed] [Google Scholar]
- Marks PW, Hendey B, Maxfield FR. Attachment to fibronectin or vitronectin makes human neutrophil migration sensitive to alterations in cytosolic free calcium concentration. J Cell Biol. 1991;112:149–158. doi: 10.1083/jcb.112.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1) Cell. 1987;51:813–819. doi: 10.1016/0092-8674(87)90104-8. [DOI] [PubMed] [Google Scholar]
- Mizgerd JP, Kubo H, Kutkoski GJ, Bhagwan SD, Scharffetter-Kochanek K, Beaudet AL, Doerschuk CM. Neutrophil emigration in the skin, lungs and peritoneum: differential requirements for CD11/CD18 revealed by CD18-deficient mice. J Exp Med. 1997;186:1357–1364. doi: 10.1084/jem.186.8.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer EL, Ruegg C, Ferrando R, Pytela R, Sheppard D. Sequence and tissue distribution of the integrin alpha 9 subunit, a novel partner of beta 1 that is widely distributed in epithelia and muscle. J Cell Biol. 1993;123:1289–1297. doi: 10.1083/jcb.123.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto AL, Edelman GM, Crossin KL. Multiple integrins mediate cell attachment to cytotactin/tenascin. Proc Natl Acad Sci USA. 1993;90:10154–10158. doi: 10.1073/pnas.90.21.10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen GD, Sanes JR, La Chance R, Cunningham JM, Roman J, Dean DC. Roles for the integrin VLA-4 and its counter receptor VCAM-1 in myogenesis. Cell. 1992;69:1107–1119. doi: 10.1016/0092-8674(92)90633-n. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- Schnapp LM, Hatch N, Ramos D, Kliminskaya IV, Sheppard D, Pytela R. The human integrin α8β1 functions as a receptor for tenascin, fibronectin, and vitronectin. J Biol Chem. 1995;270:23196–23202. doi: 10.1074/jbc.270.39.23196. [DOI] [PubMed] [Google Scholar]
- Schneider H, Harbottle RP, Yokosaki Y, Kunde J, Sheppard D, Coutelle C. A novel peptide, PLAEIDGIELTY, for the targeting of alpha9/beta1-integrins. FEBS Lett. 1998;429:269–273. doi: 10.1016/s0014-5793(98)00612-7. [DOI] [PubMed] [Google Scholar]
- Sheppard AM, Onken MD, Rosen GD, Noakes PG, Dean DC. Expanding roles for alpha 4 integrin and its ligands in development. Cell Adhes Commun. 1994;2:27–43. doi: 10.3109/15419069409014200. [DOI] [PubMed] [Google Scholar]
- Smith LL, Cheung H-K, Ling LE, Chen J, Sheppard D, Pytela R, Giachelli CM. Osteopontin N-terminal domain contains a cryptic adhesive sequence recognized by α9β1 integrin. J Biol Chem. 1996;271:28485–28491. [PubMed] [Google Scholar]
- Staunton DE, Dustin ML, Springer TA. Functional cloning of ICAM-2, a cell adhesion ligand for LFA-1 homologous to ICAM-1. Nature. 1989;339:61–64. doi: 10.1038/339061a0. [DOI] [PubMed] [Google Scholar]
- Wang A, Yokosaki Y, Ferrand R, Balmas J, Sheppard D. Differential regulation of airway epithelial integrins by growth factors. Am J Respir Cell Mol Biol. 1996;15:664–672. doi: 10.1165/ajrcmb.15.5.8918373. [DOI] [PubMed] [Google Scholar]
- Yang JT, Rando TA, Mohler WA, Rayburn H, Blau HM, Hynes RO. Genetic analysis of alpha 4 integrin functions in the development of mouse skeletal muscle. J Cell Biol. 1996;135:829–835. doi: 10.1083/jcb.135.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yednock TA, Cannon C, Vandevert C, Goldbach EG, Shaw G, Ellis DK, Liaw C, Fritz LC, Tanner LI. Alpha 4 beta 1 integrin-dependent cell adhesion is regulated by a low affinity receptor pool that is conformationally responsive to ligand. J Biol Chem. 1995;270:28740–28750. doi: 10.1074/jbc.270.48.28740. [DOI] [PubMed] [Google Scholar]
- Yokosaki Y, Palmer EL, Prieto AL, Crossin KL, Bourdon MA, Pytela R, Sheppard D. The integrin α9β1 mediates cell attachment to a non-RGD site in the third fibronectin type III repeat of tenascin. J Biol Chem. 1994;269:26691–26696. [PubMed] [Google Scholar]
- Yokosaki Y, Monis H, Chen J, Sheppard D. Differential effects of the integrins α9β1, αvβ3, and αvβ6 on cell proliferative responses to tenascin. Roles of the β subunit extracellular and cytoplasmic domains. J Biol Chem. 1996;271:24144–24150. doi: 10.1074/jbc.271.39.24144. [DOI] [PubMed] [Google Scholar]
- Yokosaki Y, Matsuura N, Higashiyama S, Murakami I, Obara M, Yamakido M, Shigeto N, Chen J, Sheppard D. Identification of the ligand binding site for the integrin alpha9/beta1 in the third fibronectin type III repeat of tenascin-C. J Biol Chem. 1998;273:11423–11428. doi: 10.1074/jbc.273.19.11423. [DOI] [PubMed] [Google Scholar]