

Abstract
The NH2-terminal domain (N-tail) of histone H3 has been implicated in chromatin compaction and its phosphorylation at Ser10 is tightly correlated with mitotic chromosome condensation. We have developed one mAb that specifically recognizes histone H3 N-tails phosphorylated at Ser10 (H3P Ab) and another that recognizes phosphorylated and unphosphorylated H3 N-tails equally well (H3 Ab). Immunocytochemistry with the H3P Ab shows that Ser10 phosphorylation begins in early prophase, peaks before metaphase, and decreases during anaphase and telophase. Unexpectedly, the H3 Ab shows stronger immunofluorescence in mitosis than interphase, indicating that the H3 N-tail is more accessible in condensed mitotic chromatin than in decondensed interphase chromatin. In vivo ultraviolet laser cross-linking indicates that the H3 N-tail is bound to DNA in interphase cells and that binding is reduced in mitotic cells. Treatment of mitotic cells with the protein kinase inhibitor staurosporine causes histone H3 dephosphorylation and chromosome decondensation. It also decreases the accessibility of the H3 N-tail to H3 Ab and increases the binding of the N-tail to DNA. These results indicate that a phosphorylation-dependent weakening of the association between the H3 N-tail and DNA plays a role in mitotic chromosome condensation.
Keywords: chromatin, polyamines, staurosporine, ultraviolet laser cross-linking
In human cells, 2 m of DNA must be organized in a nucleus 5 μm in diameter to allow regulated compaction and decompaction during the cell cycle. The basic packaging unit of chromatin is the nucleosome, an assembly of two molecules each of the four core histones, H2A, H2B, H3, and H4 around which DNA is wrapped, and of one linker histone, H1. Arrays of nucleosomes are further folded into a chromatin fiber (van Holde, 1989; Wolffe, 1995; Koshland and Strunnikov, 1996). The degree of compaction of the chromatin fiber varies locally according to the need for access: it is least compact in regions undergoing transcription and replication, and most compact in heterochromatin and during mitosis, when dense packing is required for accurate segregation of DNA to daughter cells. Little is known about the structure of the chromatin fiber and how its compaction is regulated.
Core histones are small basic proteins whose amino acid sequences have been remarkably conserved throughout evolution (van Holde, 1989). They contain a common structural motif, called the histone fold, through which the histone–histone and histone–DNA interactions that constitute the nucleosome take place. Each core histone also contains an NH2-terminal domain 15–45 amino acid residues in length, here termed the N-tail. N-tails of the core histones protrude on either side of the DNA gyres at the surface of the nucleosome and extend outwards (Luger et al., 1997). The N-tails are not required for the conformation or stability of the nucleosome itself (Rill and Oosterhof, 1982; Ausio et al., 1989; Hacques et al., 1990; Krajewski and Ausio, 1996).
An essential constraint on chromatin fiber formation is generally agreed to be the negative charge of DNA, particularly in the linker region (Clark and Kimura, 1990). Packing nucleosomes close together is expected to require neutralization of some of this charge to reduce repulsion between DNA segments. The N-tails contain a high density of positively charged residues and are sufficiently long enough to interact with negatively charged linker DNA between nucleosomes or the DNA and histones of adjacent nucleosomes. The N-tails also contain sites of posttranslational modifications such as acetylation, methylation, phosphorylation, and ubiquitination that affect their charge and function (for review see van Holde, 1989; Wolffe, 1995; Grunstein, 1997). Therefore, the N-tails are prime candidates for regulating the conformation of the chromatin fiber by mediating reversible interactions with linker DNA or adjacent nucleosomes.
The N-tail of histone H3 may play a particularly important role in chromatin conformation changes. The H3 N-tail emerges from the nucleosome at the entry and exit points of DNA (Luger et al., 1997) and was shown to bind linker DNA in vitro (Rill and Oosterhof, 1982; Hill and Thomas, 1990; Pruss and Wolffe, 1993). Acetylation, which is associated with gene activity, neutralizes positively charged lysine residues in the N-tails. It may increase access of DNA to transcription factors by promoting release of N-tails from DNA, or by altering higher order folding of the chromatin (Lee et al., 1993; Grunstein, 1997; Tse et al., 1998). Removal of the tails of H3 or H3 and H4 prevents compaction in vitro (Marion et al., 1983a,b; Krajewski and Ausio, 1996; Moore and Ausio, 1997; Tse and Hansen, 1997; Zlatanova et al., 1998). In yeast, the N-tails of H3 and H4 can also interact with nonhistone proteins that are required for the formation of heterochromatin and for transcriptional silencing (Grunstein, 1997).
Biochemical studies have shown that few H3 molecules are phosphorylated in interphase, whereas all molecules become phosphorylated at the Ser10 residue in the N-tail during mitosis (Gurley et al., 1978). There is a strong correlation between Ser10 phosphorylation and chromosome condensation under normal and experimental conditions (Roberge et al., 1990; Th'ng et al., 1994; Guo et al., 1995; Ajiro et al., 1996). However, it is not clear how the phosphorylation of Ser10, which decreases the overall positive charge of the H3 N-tail from +14 to +12, could increase its binding to DNA to shield charge and promote compaction. The H3 N-tail is unstructured in its free form, but adopts an α-helical conformation upon binding DNA (Banéres et al., 1997). It is possible that phosphorylation of Ser10 increases the α-helix potential of the H3 N-tail by neutralizing some of its positive charge. This leads to increased affinity of the tail for DNA and to chromatin fiber compaction. It is also conceivable that Ser10 phosphorylation could decrease the α-helix potential of the H3 N-tail and weaken binding of the tail to DNA to either permit the association of other factors with the vacated DNA or promote the association of the H3 N-tail with proteins required for chromatin compaction (Roth and Allis, 1992; Koshland and Strunnikov, 1996; Hendzel et al., 1997). Alternatively, more subtle changes in the interaction between the H3 N-tail and DNA could change the angle at which linker DNA enters and exits the nucleosome and promote closer packing of nucleosomes (van Holde and Zlanatova, 1996).
The role of the H3 N-tail in chromatin structure clearly warrants further study. However, results obtained using isolated chromatin are of limited value since significant changes in chromatin morphology take place during isolation (Woodcock and Horowitz, 1995). Studies with chromatin reconstituted from purified components are also of limited significance because they clearly do not reflect the complexity of the in vivo state (Bednar et al., 1995; van Holde and Zlatanova, 1995; Woodcock and Horowitz, 1995). We have used in situ and in vivo approaches that do not disrupt chromatin structure and demonstrate that a major rearrangement of the H3 N-tail takes place during chromosome condensation.
Materials and Methods
Antibody Production
mAbs to the N-tail of H3 were prepared using, as an immunogen, a synthetic peptide of residues 1–17 in which Ser10 was phosphorylated and coupled to a keyhole limpet hemocyanin. The antibodies were purified from mouse ascites fluid by cycling over a protein A column. The mAb recognizing histone H3 N-tails phosphorylated at Ser10 (H3P Ab) and the mAb recognizing both unphosphorylated and phosphorylated histone H3 N-tails (H3 Ab) were of the IgG2aκ and IgG1κ isotypes, respectively. Antibodies were stored at 3.7 mg/ml in 200 mM NaCl, 100 mM Hepes, pH 7.4, 7.7 mM NaN3, and 5 mg/ml BSA at 4°C.
Cell Culture and Drug Treatment
MCF-7 human breast carcinoma cells were cultured as monolayers (Anderson et al., 1997). To obtain mitotic populations, 100 ng/ml nocodazole (Sigma Chemical Co.), from a 10,000-fold stock in DMSO, was added to growth medium for 18 h. The loosely attached mitotic cells were released into the medium, by tapping the culture flask, and collected. Mitotic populations contained between 60 and 90% mitotic cells, as determined by microscopy. Treatment with staurosporine (Sigma Chemical Co.) lasted for 15–30 min at 100 ng/ml, from a 1,000-fold stock in DMSO.
ELISA and Western Blots
Extracts from samples containing either 3 or 60% mitotic cells were prepared in 1 mM EGTA, 1 mM PMSF, and 4 μg/ml leupeptin. Protein content was determined by protein assay (Bio-Rad Laboratories). For ELISA, cell extracts containing 1–10 μg protein, or samples in PBS containing 0–0.5 μg BSA conjugated H3 peptide (residues 1–17), either phosphorylated or not at Ser10, were applied to 96-well plates (Nunc-Immuno) and blocked for 1 h in KB (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% Triton X-100, 1% BSA). The plates were incubated with 100 μl of H3 or H3P Ab, serially diluted in KB and HRP-conjugated goat anti–mouse IgG secondary antibody (Pierce) diluted 1:7,500 in KB for 1 h at ambient temperature. The plates were washed five times with 10 mM Tris-HCl, pH 7.4, 0.02% Tween 20. Antigen–antibody complexes were visualized by incubating with 120 mM Na2HPO4, 100 mM citric acid, pH 4.0, 0.5 mg/ml 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid), 0.01% hydrogen peroxide. Absorbance was measured at 405 nm with a Biotek plate reader.
For Western blots, equal amounts of protein from cell extracts were separated using 15% SDS-PAGE. Proteins were transferred to nitrocellulose membranes by electroblot technique and membranes were blocked in PBS-T (PBS, 0.05% Tween 20) containing 5% nonfat dried milk, for 1 h at ambient temperature. The membranes were incubated for 1 h by shaking with antibodies diluted 1:106 in PBS-T and washed three times for 20 min each. The membranes were further incubated for 1 h with HRP-conjugated goat anti–mouse IgG secondary antibody (Pierce Chemical Co.) diluted 1:10,000 in PBS-T, washed three times for 10 min each, and washed once with PBS-T containing 1 M NaCl. Antibody binding was detected by enhanced chemiluminescence (Pierce Chemical Co.).
Immunofluorescence Microscopy
Indirect immunofluorescence was performed as described previously (Anderson et al., 1997), using the H3 Ab, H3P Ab, or H11-4 clone (Boehringer Mannheim) at a dilution of 1:400 in KB for 1 h at ambient temperature. Indirect immunofluorescence using mAb SPM8-2 was performed as in Delcros et al. (1997). Cells were photographed on Kodak Tmax 400 film using a Zeiss axiophot microscope. Immunofluorescence quantitation was performed by scanning the negatives with a scanner (Kodak professional plus RFS 2035 and measuring fluorescence intensity using image processing software (IPLab Gel 1.5e; Signal Analytics). To compensate for differences in photographic exposure between frames, the fluorescence intensity of mitotic cells was measured relative to that of adjacent interphase cells in the same frame.
UV Laser Irradiation
UV laser irradiation was performed using a Quanta-Ray pulsed Nd:YAG laser (model GCR14S; Spectra Physics) equipped with an HG-2 harmonic generator (Spectra Physics) and dichroic mirrors (DHS-2 Quanta-Ray dichroic harmonic separator) to give monochromatic 266-nm light with a beam diameter of 6.4 mm. Open 1.5-ml microfuge tubes, containing cell suspensions ready for irradiation, were placed horizontally in a 10-mm-diam hole drilled in a small Plexiglas™ sheet held in a Brinkmann micromanipulator. All experiments were performed using a single 5-ns pulse with an energy of 50 mJ measured with a power and energy meter (model AA30; Astral) equipped with a UV sensor (model AC25; Scientech) (Ho et al., 1994).
Immunoprecipitation and End-labeling of Cross-linked Histone H3–DNA Complexes
Cycling and mitotic cells were collected and samples removed to determine the percentage of cells in mitosis. The cells were counted with a hemacytometer, washed twice with ice-cold PBS, and diluted in PBS in 1.5-ml microfuge tubes to give 5 × 106 cells an optical density of 5 OD266/ml. Equal cell samples were irradiated with a single 5-ns, 50-mJ pulse of 266 nm light.
All of the following procedures were performed at 4°C unless stated otherwise. After irradiation, cells were lysed in RIPA buffer (PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS), DNA was sheared by passing the lysate through a 21-gauge needle, and lysates were precleared for 30 min with 20 μl of a 50% slurry of protein A–agarose beads (GIBCO BRL). Samples of precleared lysates were separated by SDS-PAGE and stained with Coomassie blue to ensure that they contained the same amount of protein. Free H3 and cross-linked H3–DNA complexes were immunoprecipitated from precleared lysates by incubation with 3 μl of H3 Ab for 1 h, followed by incubation with 20 μl protein A–agarose beads for 1–2 h. Samples were centrifuged at 200 g and beads were washed once with RIPA, twice with RIPA containing 1 M NaCl, once with RIPA containing 0.25 M LiCl, and once with PBS. Immunoprecipitated H3– DNA complexes were digested by incubating the beads with 15 U micrococcal nuclease (Pharmacia) for 30 min at 37°C. The beads were washed twice with PBS, and DNA was end-labeled with [γ32P]ATP and 10 U T4 polynucleotide kinase (Pharmacia) for 30 min at 37°C. The beads were washed twice with RIPA and were incubated overnight with RIPA containing 1 M NaCl. Immunoprecipitated free H3 and phosphorus-32– labeled cross-linked H3–DNA complexes were eluted from the beads by boiling in SDS sample buffer containing 1 M urea (Ho et al., 1994). In some experiments, one-half of the sample was digested with SV8 protease (Boehringer-Mannheim) in 20 mM ammonium bicarbonate, pH 8.0, and two times the sample buffer for 4–12 h at ambient temperature. All samples were analyzed by 15% SDS-PAGE. Phosphorus-32 was quantified using a PhosphorImager (Molecular Dynamics, Inc.).
Cellular Compartmentalization of [3H]Spermidine
Cycling or mitotic cells were collected, washed twice with PBS, suspended in serum-free DME (GIBCO BRL) containing 20 μCi [3H]spermidine (NEN), and incubated with or without 100 ng/ml staurosporine for 30 min at 37°C in humidified 5%CO2. The cells were collected by centrifugation and washed three times with PBS. Compartmentalization of [3H]spermidine was determined essentially as described in Mach et al. (1982). Cells were lysed by pipetting for exactly 20 s in 500 μl 30 mM Hepes, 200 mM sucrose, 40 mM NaCl, 5 mM MgCl2, pH 7.6, and centrifuged for 20 s at 200 g. The cytoplasmic component contained in the supernatant was quickly removed and the procedure was repeated. The supernatant, designated the wash, was removed and the pellet, representing the nuclear compartment, was resuspended in 500 μl of the same buffer and vortexed vigorously. 3H was detected by liquid scintillation counting.
Results
mAbs to the Histone H3 N-tail
To study the involvement of the H3 N-tail in chromosome condensation, we first studied its phosphorylation at Ser10. We generated mAbs to a synthetic peptide (ARTKQTARKSTGGKAPR) representing residues 1–17 of H3 in which the Ser10 residue (underlined) was phosphorylated. In ELISA, an mAb designated H3P Ab recognized the peptide phosphorylated at Ser10 but not the unphosphorylated peptide (Fig. 1 A), even when the antibody was used at high concentration. In Western blots, H3P Ab recognized a major band comigrating with H3 in mitotic cell lysates, where H3 was phosphorylated, and a very minor band at the same position in interphase cell lysates, where H3 was largely unphosphorylated (Fig. 1 B). A minor band at 42 kD was also observed, but only in the mitotic cell lysates. The 42-kD band may represent H3 dimers arising from oxidation of H3 cysteine residues forming a disulfide bond, or it may be an unrelated protein with a similar phosphoepitope. A second mAb designated H3 Ab reacted equally well in ELISA with phosphorylated and unphosphorylated H3 peptide and with extracts from interphase and mitotic cells (Fig. 1 C). In Western blots, it recognized H3 specifically and equally well in lysates from interphase or mitotic cells (Fig. 1 D).
Figure 1.

Specificity of H3P and H3 antibodies. (A) ELISA of H3P Ab binding to a peptide corresponding to residues 1–17 of H3 unphosphorylated (open square) or phosphorylated at Ser10 (closed square). (B) Western blot analysis of H3P Ab binding to total cellular proteins obtained from cycling cells (−N, containing 3% mitotic cells) or cells treated with nocodazole (+N, 60% mitotic cells). The unfilled arrow indicates the minor cross-reactive band. Molecular mass standards are shown on the left. (C) ELISA of H3 Ab binding to the unphosphorylated H3 peptide (open square), phosphorylated H3 peptide (closed square), total cellular proteins from cycling cells (open circle) or from cells treated with nocodazole (closed circle). (D) Western blot analysis of H3 Ab binding to cellular proteins performed as in B.
Phosphorylation of Ser10 at Different Stages of the Cell Cycle
To examine the phosphorylation of H3 at Ser10 during the different phases of the cell cycle we used H3P Ab in immunofluorescence microscopy in combination with the fluorescent DNA dye, bisbenzimide. Fig. 2 shows paired photographs of cycling MCF-7 cells with DNA fluorescence on the left (to define their cell cycle stage) and H3P Ab immunofluorescence on the right. In interphase cells (arrowheads), speckles of H3P Ab immunofluorescence were present in the nucleus (Fig. 2, B, D, and F) that corresponded to regions of less intense DNA staining (Fig. 2, A, C, and E). Early in prophase, before chromosomes were clearly delineated (Fig. 2 A), intense H3P Ab immunofluorescence appeared in the nucleus, associated with the condensing chromatin (Fig. 2 B). H3P Ab immunofluorescence was also intense in metaphase (Fig. 2, D and H), weaker in anaphase (Fig. 2 F), but remained strongly associated with the chromosomes (Fig. 2, C, E, and G). In telophase cells, H3P Ab immunofluorescence was considerably reduced. In many cases it was absent entirely (Fig. 2 H) although the chromatin still appeared condensed (Fig. 2 G), whereas in other cases, the periphery of each mass of chromatin showed no H3P Ab immunofluorescence but the center retained a patch of immunofluorescence (not shown). Throughout metaphase, anaphase, and telophase weak cytoplasmic immunofluorescence was also observed with the H3P Ab (Fig. 2, D, F, and H).
Figure 2.
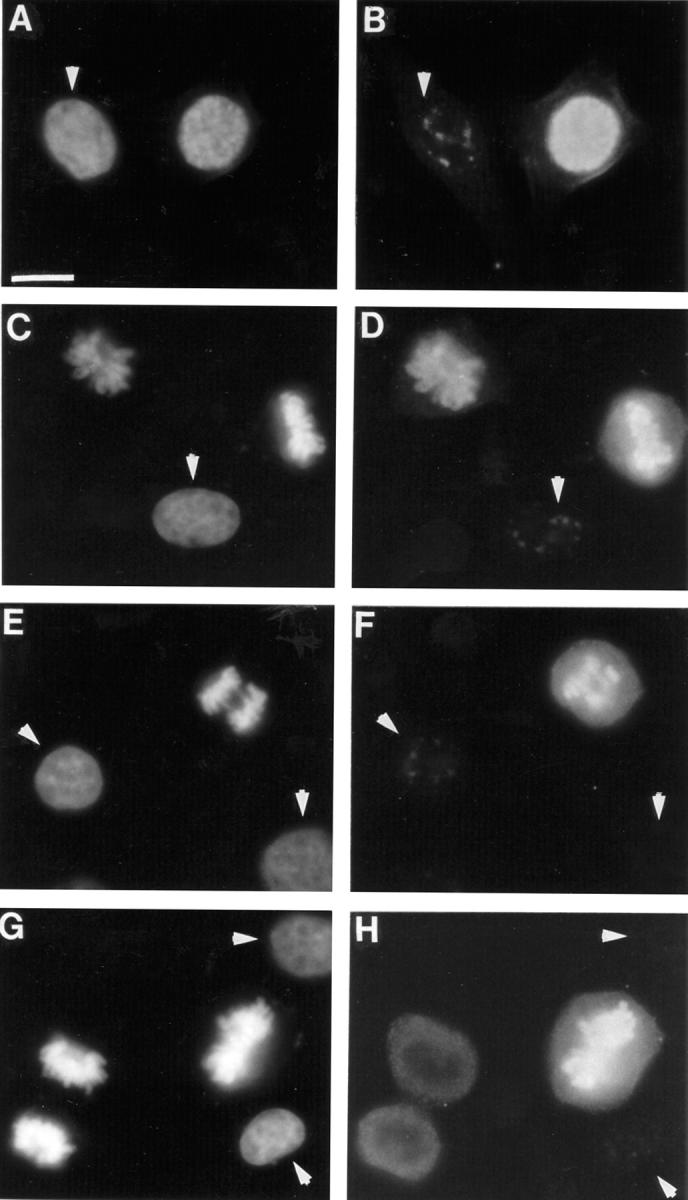
Phosphorylation of histone H3 Ser10 at mitosis. Paired photographs of cells showing bisbenzimide fluorescence of DNA (left) or H3P Ab immunofluorescence (right). Cells in interphase (indicated by arrowheads), prophase (A and B), metaphase (C, D, G, and H), anaphase (E and F), and telophase (G and H) are shown. Bar, 20 μm.
These results indicate that in interphase a few discrete regions of nucleus that are low in DNA content, probably representing regions with less condensed chromatin, contain H3 phosphorylated at Ser10. A burst of phosphorylation on Ser10 takes place early in prophase at the onset of chromosome condensation, the phosphorylation is maintained at metaphase, reduced in anaphase, and disappears in telophase before or at the onset of chromosome decondensation.
Increased Accessibility of the Histone H3 N-tail during Chromosome Condensation
We performed a parallel immunofluorescence study using H3 Ab that recognizes both the phosphorylated and unphosphorylated H3 N-tail. As expected, cells in all stages of the cell cycle showed H3 Ab immunofluorescence in their nuclei (Fig. 3, A–F). In interphase cells the pattern of H3 Ab immunofluorescence appeared the same as that of the DNA visualized with bisbenzimide (Fig. 3, A–F). Unexpectedly, the intensity of H3 Ab immunofluorescence was distinctly higher in mitotic cells than in interphase cells (Fig. 3, B, D, and F). Early in prophase, increased nuclear H3 Ab immunofluorescence was observed (Fig. 3 B) that was associated with the condensing chromatin (Fig. 3 A). H3 Ab immunofluorescence was intense in metaphase (Fig. 3, D and F) and somewhat weaker in anaphase (Fig. 3 D), but remained associated with the chromosomes. In telophase cells, H3 Ab immunofluorescence was reduced further to levels approaching those observed in interphase nuclei (Fig. 3 F). No cytoplasmic immunofluorescence was observed. The H3 Ab immunofluorescence from individual cells was measured by scanning densitometry. Immunofluorescence was significantly higher in prophase (2.4-fold), metaphase (3.3-fold), and anaphase (2.4-fold) cells than in interphase cells (Fig. 4 A). Quantitation of the H3 Ab immunofluorescence signal using flow cytometry also showed a two- to threefold increase in mitotic cells over G2 cells (not shown).
Figure 3.

Accessibility of the H3 N-tail during mitosis. Paired photographs of cells showing DNA fluorescence (left), and immunofluorescence (right) using the H3 Ab (B, D, and F) or mAb to all histones (H). The panels show cells in interphase as well as prophase (A and B, arrows), metaphase (C–H, arrows), anaphase (C and D, double arrows), and telophase (E and F, right angled arrow). Bar, 20 μm.
Figure 4.

Quantitation of H3 Ab immunofluorescence associated with interphase and mitotic cells. (A) The H3 Ab immunofluorescence associated with interphase cells (n = 60), prophase cells (n = 4), metaphase cells (n = 14), anaphase cells (n = 5), and telophase (n = 1) measured by scanning densitometry as described in Materials and Methods and expressed relative to that of interphase cells. (B) The total histone immunofluorescence associated with interphase cells (n = 6) or mitotic cells (n = 7) was measured and expressed as in A. Bars and error bars show mean and standard deviation.
As an explanation for the increased H3 Ab immunofluorescence in mitosis, we considered the possibility that breakdown of the nuclear envelope in mitosis might generally increase the accessibility of nuclear targets to antibodies. However, a commercially available mAb (clone H11-4; Boehringer-Mannheim) that reacts with most histones (H1 > H3 = H2A = H2B ≫ H4) showed equal staining of interphase and mitotic cells under identical experimental conditions (Fig. 3, G and H) and quantitation of the fluorescence signal by scanning densitometry showed no significant difference between interphase and mitotic cells (Fig. 4 B). This indicates that antibodies can reach nuclear targets as readily in interphase as in mitotic cells. The increased binding of the H3 Ab to mitotic chromosomes could not be due to preferential binding to phosphorylated H3 (Fig. 1) or binding to additional chromosomal proteins at mitosis since Western blots showed that the H3 Ab recognized only a single band with the molecular mass of H3 in whole cell lysates from interphase or mitotic cells (Fig. 1 D). Nor could the weaker binding of the H3 Ab in interphase nuclei be due to masking of the H3 Ab epitope by a posttranslational modification of the H3 tail, such as methylation or acetylation, because the H3 Ab reacted equally well with H3 from interphase or mitotic cells in Western blots and ELISA (Fig. 1, C and D). These results show that the accessibility of the H3 N-tail to the H3 Ab increases when cells enter mitosis and is temporally correlated with the phosphorylation of H3 at Ser10.
Decreased Binding of the Histone H3 N-tail to DNA during Chromosome Condensation
We considered the possibility that increased accessibility of the H3 N-tail to H3 Ab as chromatin changes from its decondensed interphase state to its condensed mitotic state, might occur because of a decrease in binding of the H3 N-tail to DNA. To examine the state of binding of the H3 N-tail to DNA in interphase and mitotic chromatin, we performed in vivo UV laser cross-linking using a single short (5 ns) pulse of irradiation. We developed this approach because of concerns that experiments carried out in vitro with isolated interphase nuclei and metaphase chromosomes do not reflect the in vivo situation, and because current procedures for cross-linking or footprinting in living cells require modification of DNA structure or integrity and long treatments that might disrupt interactions between the H3 N-tail and DNA.
Stefanovsky et al. (1989a,b) showed that irradiation of isolated nuclei or chromatin using short pulses of UV laser irradiation causes covalent cross-links between histones and DNA, via the histone tails exclusively. Irradiation with nano- or pico-second lasers causes the cross-links to form in <1 μs (Hockensmith et al., 1991), well below the 100-μs timescale for conformational transitions of macromolecules (Careri et al., 1975). Therefore, we used a laser to irradiate living cells with a single 50-mJ, 5-ns pulse of 266 nm UV light. We lysed the cells, immunoprecipitated H3 under stringent conditions, enzymatically trimmed the DNA cross-linked to H3, end-labeled it with phosphorus-32, released the complexes from the beads used in immunoprecipitation, and analyzed them using SDS-PAGE and quantitation with a PhosphorImager. The radiolabeled band seen in SDS-PAGE represents H3 cross-linked to DNA. As outlined in Materials and Methods, irradiation was performed on samples containing the same number of cells at the same absorbance at 266 nm and immunoprecipitations were performed on samples containing the same amount of protein.
H3 Ab immunoprecipitates from irradiated cycling cells contained a major radiolabeled band corresponding to H3 (Fig. 5 A, second lane) that was not observed in unirradiated controls (Fig. 5 A, first lane). Microscopy of a sample of the original cell population showed it to contain 3% mitotic cells. The H3 Ab immunoprecipitates from an irradiated mitotic population (85% mitotic cells) also contained a radiolabeled band corresponding to H3 (Fig. 5 A, third lane) but its intensity was reduced to 56% of the interphase band. This result was observed in five experiments, with reductions ranging from 46% (Fig. 5 A) to ∼100% (not shown). Therefore, H3 is cross-linked to DNA to a larger extent in interphase cells than in mitotic cells. These results indicate that when cells enter mitosis a phosphorylation-dependent mechanism weakens the association of the H3 N-tail with DNA and that this weakening is important for chromosome condensation.
Figure 5.

Association of the H3 N-tail with DNA in vivo. (A) Cells were treated or not with nocodazole (N), staurosporine (Stsp), and irradiation using a single 5-ns 50-mJ pulse of UV light as indicated. H3 was immunoprecipitated and cross-linked DNA was end-labeled with phosphorus-32. Cross-linked H3–DNA complexes were separated by SDS-PAGE and detected using a PhosphorImager. The extent of cross-linking expressed as a percentage of that observed in interphase cells is shown at the bottom for one representative experiment. (B) Phosphorus-32– labeled cross-linked H3–DNA complexes obtained from interphase cells were treated or not with SV8 protease to cleave the N-tail and were analyzed as in A. The positions of H3 and of the H3 N-tail are indicated.
To confirm cross-linking had occurred via the H3 N-tail and not via the histone fold domain, cross-linked H3– DNA complexes immunoprecipitated from interphase cells and labeled with phosphorus-32 were digested with SV8 protease. SV8 protease cleaves H3 into one 6-kD fragment corresponding to the N-tail (residues 1–50), and several fragments too small to visualize on 15% SDS-PAGE. The band corresponding to intact H3 (Fig. 5 B, first lane) disappeared after SV8 protease digestion and a band of similar intensity appeared at a position corresponding to the 6-kD cleavage fragment (Fig. 5 B, second lane). This result demonstrates that cross-linking of H3 to DNA occurred via the N-tail. A radioactive band below the H3 N-tail band in the SV8-digested sample was also observed in unirradiated samples and in samples immunoprecipitated with unrelated antibodies. Therefore, it does not represent DNA cross-linked to a histone H3 proteolytic fragment, but rather it is a proteolytic fragment of immunoglobulins that became phosphorylated during incubation with [γ-32P]ATP.
Accessibility of the Histone H3 N-tail to Antibodies and Its Association with DNA Are Controlled by a Staurosporine-sensitive Kinase
To examine whether the rearrangement of the H3 N-tail with DNA at mitosis resulted from Ser10 phosphorylation, we treated mitotic cells with the protein kinase inhibitor, staurosporine, which causes rapid dephosphorylation of histone H1 and of H3 Ser10 (Th'ng et al., 1994), and performed immunofluorescence and cross-linking experiments.
Cells arrested in mitosis with nocodazole were treated with or without 100 ng/ml staurosporine for 20 min and prepared for fluorescence microscopy using H3 or H3P Abs and bisbenzimide. Some cells were prepared as chromosome spreads so that the state of condensation could be evaluated more clearly. In all cells, staurosporine caused rapid chromosome decondensation, but the extent varied between individual cells. This is best shown in the chromosome spreads (Fig. 6, A and B). In most cases staurosporine caused the discrete highly condensed mitotic chromosomes (Fig. 6 A) to decondense into a mass of chromatin (Fig. 6 B, lower cell). In other cases chromosomes, although decondensed, could still be discerned (Fig. 6 B, upper cell).
Figure 6.

Effect of staurosporine on chromosome condensation and on the accessibility of the H3 N-tail at mitosis. (A and B) Mitotic spreads of cells arrested in mitosis with nocodazole and treated with (B) or without (A) staurosporine for 15 min and stained with bisbenzimide are shown. (C–H) Paired photographs of cells arrested in mitosis with nocodazole and treated without (C, D) or with (E–H) staurosporine, showing DNA fluorescence (left column) or H3 Ab immunofluorescence (right). (I and J) Paired photographs of cells arrested in mitosis with nocodazole and treated with staurosporine, showing DNA fluorescence (left) or H3P Ab immunofluorescence (right). The arrows indicate mitotic cells with decondensed chromosomes as a result of staurosporine treatment. Bar, 20 μm.
Immunofluorescence microscopy showed that after treatment with staurosporine, not only were mitotic chromosomes decondensed into fuzzy masses of chromatin (Fig. 6, E and G, arrows), but the intense H3 Ab immunofluorescence associated with mitotic chromosomes of untreated cells (Fig. 6 D) was reduced significantly (Fig. 6, F and H). Each frame in Fig. 6 was exposed differently to optimize detail, but in each case the intensity of H3 Ab immunofluorescence in mitotic cells can be compared with an interphase cell in the same preparation. In untreated cells (Fig. 6 D), the H3 Ab immunofluorescence intensity of the two mitotic cells is sufficiently high that at an appropriate exposure no immunofluorescence was seen in the interphase cell. After treatment with staurosporine, the partially decondensed chromosomes seen in the cell in Fig. 6 E (arrow) show much less H3 Ab immunofluorescence (Fig. 6 F) than untreated cells (Fig. 6 D), even though a longer exposure was used so that H3 Ab immunofluorescence may be detected in the adjacent interphase cells (Fig. 6 F). In some cells, staurosporine treatment resulted in more extensive decondensation of chromosomes (Fig. 6 G, arrow) and the H3 Ab immunofluorescence was correspondingly weaker (Fig. 5 H, arrow), approaching that of adjacent interphase cells. Quantitation using scanning densitometry showed that the fluorescence signal decreased from 3.26 ± 1.35 (n = 6) to 1.15 ± 0.36 (n = 6) during a 20-min staurosporine treatment.
Similar experiments performed using the H3P Ab confirmed that staurosporine caused H3 dephosphorylation; a cell with partially decondensed chromosomes (Fig. 6 I, arrow) no longer showed chromosomal H3P Ab immunofluorescence (Fig. 6 J, arrow), but showed cytoplasmic staining at an exposure long enough to show the speckled H3P Ab immunofluorescence of interphase nuclei (Fig. 6 J).
We also examined the effects of staurosporine treatment on the association of the H3 N-tail with DNA. Samples containing equal numbers of cells arrested in mitosis with nocodazole were either treated or not treated with staurosporine for 20 min and irradiated. Staurosporine treatment led to an increase in the amount of radiolabeled H3 from 56% to 84% (Fig. 5 A), indicating that binding of the H3 N-tail to DNA increased during staurosporine-induced chromosome decondensation.
These results show that experimental induction of chromosome decondensation and H3 dephosphorylation leads to a decrease in the accessibility of the H3 N-tail to antibodies and an increase in its association with DNA. The results strengthen our earlier correlation between the conformation of H3 N-tail and chromosome condensation and Ser10 phosphorylation. The results also show that a staurosporine-sensitive kinase regulates the accessibility of the H3 N-tail to antibodies and its binding to DNA.
Association of Spermidine and Spermine with Chromosomes Is Controlled by a Staurosporine-sensitive Kinase
Close packing of nucleosomes is expected to require neutralization of some DNA negative charge to reduce repulsion between DNA segments (Clark and Kimura, 1990). It was presumed that this is accomplished in part by the positively charged H3 N-tail associating with linker DNA (Moore and Ausio, 1997; Tse and Hansen, 1997). According to this view, the dissociation of the N-tail from DNA during chromosome condensation that we observe would be expected to hinder chromatin compaction, unless it served to facilitate the binding of other positively charged molecules or is a consequence of binding of other positively charged molecules to DNA. The most abundant cellular polycationic molecules are the polyamines, which can bind to DNA and associate with chromosomes at mitosis (for review see Hougaard, 1992).
To determine the dependence of this association on protein phosphorylation, interphase cells, mitotic cells, and mitotic cells treated with staurosporine were incubated with [3H]spermidine, which accumulates rapidly in cells. After brief fixation, cytoplasmic and nuclear compartments were separated quickly (Mach et al., 1982) and the association of [3H]spermidine with these two compartments was determined (Fig. 7). In all samples, most [3H]spermidine was in the cytoplasmic compartment. However, a larger proportion was in the cytoplasmic fraction of interphase cells than of mitotic cells. Conversely, the nuclear compartment of mitotic cells contained more [3H]spermidine (33%) than that of interphase cells (15%). Staurosporine treatment of mitotic cells caused a decrease in [3H]spermidine in the nuclear compartment to 21% accompanied by an increase in the cytoplasmic compartment to 65%. Therefore, in mitotic cells more spermidine is associated with the nuclear compartment than in interphase cells but treatment with staurosporine reduced this association. We were unable to use the UV laser cross-linking procedure to examine polyamine binding to DNA in vivo because polyamines do not form covalent cross-links at 266 nm (Sauvé, D.M., unpublished results).
Figure 7.

Intracellular distribution of [3H]spermidine in interphase (−N), mitotic (+N) and mitotic cells treated with staurosporine (+N + Stsp).
Further evidence for the sensitivity of chromosomal association of polyamines to staurosporine was obtained by indirect immunofluorescence with mAb SPM8-2 that recognizes spermine and spermidine (Delcros et al., 1997). Polyamines are small and present in compartments unreachable by antibodies. To allow antibody access, cells rapidly fixed in formaldehyde were digested with DNaseI and RNaseA before incubation with antibody (Delcros et al., 1997), although this treatment considerably reduces the quality of the preparations. In mitotic cells, polyamine immunofluorescence was more intense in the chromosomes than in the cytoplasm (Fig. 8 A). After treatment with staurosporine for 15 min this difference was lost, leading to uniform immunofluorescence throughout the cell (Fig. 8 B), indicating a redistribution of polyamines from the chromosomes to the cytoplasm and supporting the results obtained with [3H]spermidine.
Figure 8.

Effect of staurosporine on the distribution of polyamines in mitotic cells. SPM8-2 antibody immunofluorescence shows that the predominantly nuclear localization of spermine and spermidine in cells arrested in mitosis with nocodazole (A) is lost after 15 min of staurosporine treatment (B). Bar, 20 μm.
Discussion
Histone H3 Phosphorylation and Chromosome Condensation
Our H3P Ab immunofluorescence studies showed that Ser10 phosphorylation starts early in prophase, at the onset of chromosome condensation. The seemingly all or none appearance of Ser10 phosphorylation indicates that it occurs over a very short time period, attaining a maximum before the chromosomes become fully condensed at metaphase. In contrast to phosphorylation, dephosphorylation of Ser10 takes place over a longer time period because anaphase and telophase cells with intermediate levels of phosphorylation were often observed. Dephosphorylation precedes chromosome decondensation since telophase cells with condensed chromosomes showing little or no H3P Ab immunofluorescence were observed. The pattern of loss of H3P Ab immunofluorescence in telophase cells indicated that Ser10 dephosphorylation initiates at the periphery of the chromosome mass, suggesting that the dephosphorylation signal originates in the cytoplasm rather than within the chromosomes.
Weak speckles of H3P Ab immunofluorescence were also observed in the nuclei of all interphase cells. In contrast with H3P Ab immunofluorescence in mitotic cells that is associated with all chromatin, these speckles were restricted to regions of less condensed chromatin. Western blots of interphase cell extracts showed a single minor band at the position of H3, implying that a small proportion of H3 is phosphorylated at Ser10 in interphase. This result is compatible with the observation that mitogen stimulation of quiescent interphase cells causes the rapid phosphorylation of a small fraction of H3 (Mahadevan et al., 1991). This H3 fraction is very sensitive to acetylation (Barratt et al., 1994), implying that it is associated with new gene expression that requires the opening up of chromatin.
Our observations generally support the pioneering biochemical studies that first established a temporal link between H3 phosphorylation and chromosome condensation in mammalian cells (Gurley et al., 1978) and recent studies using a rabbit antiserum that recognizes phosphorylated H3 (Hendzel et al., 1997; Van Hooser et al., 1998). Gurley et al. (1978) suggested that Ser10 phosphorylation began at the time of chromosome formation in prophase, reached a maximum in metaphase, diminished in anaphase, and was absent in telophase. However, we place the onset of Ser10 phosphorylation earlier, at the very beginning of prophase, when chromatin is beginning to condense but chromosomes are not clearly delineated. Hendzel et al. (1997) and Van Hooser et al. (1998) reported Ser10 phosphorylation to begin even earlier, in G2 and to initiate in pericentromeric heterochromatin. In our study, all cells showing increased H3P Ab immunofluorescence also showed morphological evidence of being in mitosis, suggesting that H3 phosphorylation takes place at the onset of mitosis and not in G2. We did not attempt to delineate precisely whether H3 phosphorylation initiates at specific positions within chromosomes.
Our results are compatible with Ser10 phosphorylation playing a causative role in the initial stages of chromosome condensation, which involve compaction of the chromatin fiber, since phosphorylation is strong in early prophase, but not in the later stages of condensation, which involve coiling of the chromatids (Anderson and Roberge, 1996; Koshland and Strunnikov, 1996), since H3 phosphorylation peaks before maximal chromosome condensation and is absent in the condensed chromosomes of telophase cells. Similarly, Van Hooser et al. (1998) observed that hypotonic treatment of mitotic cells can cause H3 dephosphorylation without accompanying chromosome decondensation and suggested that H3 phosphorylation is not required for maintaining high levels of chromosome condensation.
Decreased Binding of the Histone H3 N-tail to DNA during Chromosome Condensation
Whereas the H3P Ab recognized only a small subset of interphase H3 molecules, the H3 Ab showed staining of all interphase chromatin in a pattern indistinguishable from that obtained with the DNA dye, bisbenzimide. Unexpectedly, the accessibility of the H3 N-tail to the H3 Ab changed during mitosis: it increased sharply at prophase, was high at metaphase, decreased at anaphase, and decreased further to approach interphase levels at telophase. This pattern closely resembled that seen with the H3P Ab, suggesting that Ser10 phosphorylation and increased accessibility of the H3 N-tail are closely associated during mitosis.
Our UV laser cross-linking experiments provide evidence that the H3 N-tails are indeed bound to DNA in vivo. They showed that the H3 N-tail is associated with DNA during interphase and that binding of the N-tail to DNA is decreased in cells arrested in metaphase by nocodazole. The most straightforward interpretation of our observation that the H3 N-tail becomes more accessible to the H3 Ab during mitosis, is that this occurs as a result of its dissociation from DNA. The results have several important implications. They are incompatible with compaction being driven by increased association of the H3 N-tail with DNA, but rather they provide evidence that compaction may be driven by dissociation of the tails from DNA. Dissociation could free the tails to bind proteins required for chromosome condensation, such as topoisomerase II or the SMC class (Roth and Allis, 1992; Anderson and Roberge, 1996; Hendzel et al., 1997), or allow access of the vacated DNA to factors required for condensation, as discussed below.
Our observations also bear importantly on our understanding of the structure of the compact chromatin fiber. According to a widely accepted model (Finch and Klug, 1976), the compact fiber is a solenoid with about six nucleosomes per helical turn in which the nucleosomes are stacked with their long axis parallel to the fiber, with the DNA entry and exit sites and linker DNA inside the fiber. In this model, the H3 N-tails extend into the interior of the fiber, in a central hole too small to allow access to large antibody–secondary antibody complexes, an arrangement incompatible with our immunofluorescence observations. Previous biochemical observations asserting that linker DNA is more accessible to nuclease than intranucleosomal sites in condensed chromatin, and that histone H1 is the first histone degraded by trypsin (Marion et al., 1983a; van Holde, 1989) are also incompatible with this model.
Our results are more compatible with models where the H3 N-tail is not expected to preferentially face the inside of the fiber. These include the coiled linker model, in which linker DNA is on the outside of the fiber (Felsenfeld and McGee, 1986), different zigzag ribbon models containing two parallel rows of nucleosomes (Woodcock et al., 1984), and results from scanning force microscopy and cryoelectron microscopy showing an irregular pattern of folding, possibly a three-dimensional zigzag without face to face aggregation of the nucleosomes (van Holde and Zlatanova, 1995). Our results are also compatible with recent data obtained from X-irradiation of living cells that suggested a zigzag model of the chromatin fiber (Rydberg et al., 1998).
Involvement of Polyamines in Chromosome Condensation
Regardless of the organization of DNA and histones within the chromatin fiber, an essential constraint on the formation of compact chromatin is probably the neutralization of some of the negative charge of the linker DNA, to reduce repulsion between linker DNA segments (Clark and Kimura, 1990; Zlatanova et al., 1998). Dissociation of the H3 N-tail from DNA would achieve the opposite effect. Therefore, DNA charge neutralization during chromosome condensation must be accomplished by a different mechanism.
Polyamines can displace histone N-tails in nucleosome core particles at concentrations >0.7 spermidine molecules per DNA turn (Dumuis-Kervabon et al., 1986) and can cause chromatin compaction in vitro, with maximal compaction at about 1.5–2 polyamines per DNA turn (Encontre and Parello, 1988; Smirnov et al., 1988). Human cells in culture typically contain 0.6–3 × 109 molecules of spermidine and spermine (Sunkara et al., 1983; Snyder, 1989) and 5–10 pg DNA, corresponding to 0.6–1.2 × 109 DNA turns. Therefore, the cellular concentration of polyamines appears sufficient to play a role in chromatin compaction in vivo.
We examined whether polyamines play a role in mitotic chromosome condensation in vivo by labeling cells with [3H]spermidine, rapidly separating the nuclear and cytoplasmic compartments, and performing immunofluorescence with an mAb to spermine and spermidine. We found that there was an increased association of [3H]spermidine with the nuclear fraction in mitotic cells compared with interphase cells. In addition, polyamine immunofluorescence was found to associate preferentially with chromosomes. These findings agree with early fractionation studies showing high spermidine and spermine concentrations in mitotic chromosomes (Goyns, 1979), and with immunocytochemical studies using a polyclonal antiserum to spermine and spermidine showing that polyamines are almost entirely cytoplasmic during interphase but associate with chromosomes in mitosis (Hougaard et al., 1987; Hougaard, 1992). Interestingly, Hougaard et al. (1987) observed that the association of spermine and spermidine with chromosomes occurred only in prophase, metaphase, and early anaphase, when we find that H3 is phosphorylated and more accessible to H3 Ab binding, and not in telophase, when H3 is dephosphorylated and becomes less accessible to H3 Ab binding.
Control of Chromosome Condensation by Protein Phosphorylation
Treatment of mitotic cells with staurosporine concurrently caused the following: dephosphorylation of H3 Ser10, decondensation of chromosomes, decreased availability of the H3 N-tail to antibody binding, increased binding of H3 N-tail to DNA, and decreased association of polyamines with DNA. These events are interconnected and controlled by a staurosporine-sensitive protein kinase.
We do not know the nature of the protein kinase(s) or its relevant substrates but it is tempting to speculate that the substrate may be H3 Ser10 itself. This is suggested by the temporal link between Ser10 phosphorylation and the onset of chromosome condensation. Phosphorylation of H3 Ser10 at prophase could weaken the interaction of the N-tail with DNA to allow binding of polyamines to DNA to drive chromosome compaction. Alternatively, the staurosporine-sensitive kinase(s) may control phosphorylation of other proteins, including histone H1, that also participate in the control of chromosome condensation. Currently, we are seeking this kinase. The observation that staurosporine treatment had no effect on the interphase speckles of H3P Ab immunofluorescence suggests that the kinase responsible for interphase H3 phosphorylation is not the mitotic H3 kinase.
Model for the Role of Histone H3 Ser10 Phosphorylation in Chromosome Condensation
The results presented in this study show that a structural rearrangement of the H3 N-tail accompanies Ser10 phosphorylation at mitosis and are consistent with the following model. During interphase, the H3 N-tail is bound to DNA and participates in chromatin compaction, in concert with other histone tails and factors. The tails of H3 and other histones can be modified extensively during interphase by acetylation, methylation, ubiquitination, and phosphorylation. These modifications may serve to fine tune the degree of chromatin compaction to allow the regulated access to DNA that is required for diverse transactions such as transcription, replication, and repair. In calf thymus chromatin, 50% of the DNA charge is neutralized by histones (Smirnov et al., 1988; Clark and Kimura, 1990). This may be sufficient for regulated chromatin compaction during interphase, but not for the extensive compaction required in mitosis. This function may be carried out by a distinct set of polycationic molecules more suited for bulk compaction, such as polyamines. Phosphorylation of the H3 N-tail at mitosis weakens N-tail–DNA binding and favors DNA–polyamine binding. Polyamines neutralize the negative charge of DNA more extensively, thus minimizing repulsion between nucleosomes and allowing formation of the highly compacted mitotic chromosome. Displacement of the H3 N-tail from DNA may also promote its association with other histones in adjacent nucleosomes or with nonhistone proteins to further stabilize condensed chromatin. Dephosphorylation of H3 Ser10 in anaphase and telophase would increase the affinity of the H3 N-tail to DNA and favor displacement of the polyamines, promoting chromatin decondensation.
Finally, we saw no evidence of increased accessibility of the H3 N-tail in condensed heterochromatin compared with decondensed euchromatin in interphase nuclei. This implies that condensed mitotic chromosomes may be a form of compaction distinct from that of interphase heterochromatin in both structure and regulation.
Acknowledgments
We thank Jacques Moulinoux for his gift of SPM8-2 antibody, Allison Buchan for use of her microscope, and Stephen Friend for helpful discussions.
This work was supported by the Canadian Breast Cancer Research Initiative and the Medical Research Council of Canada (MT-11375).
References
- Ajiro K, Yoda K, Utsumi K, Nishikawa Y. Alteration of cell cycle– dependent histone phosphorylations by okadaic acid. Induction of mitosis-specific H3 phosphorylation and chromatin condensation in mammalian interphase cells. J Biol Chem. 1996;271:13197–13201. doi: 10.1074/jbc.271.22.13197. [DOI] [PubMed] [Google Scholar]
- Anderson H, Roberge M. Topoisomerase II inhibitors affect entry into mitosis and chromosome condensation in BHK cells. Cell Growth Diff. 1996;7:83–90. [PubMed] [Google Scholar]
- Anderson HJ, Coleman JE, Andersen RJ, Roberge M. Cytotoxic peptides hemiasterlin, hemiasterlin A, and hemiasterlin B induce mitotic arrest and abnormal spindle formation. Cancer Chemother Pharmacol. 1997;39:223–226. doi: 10.1007/s002800050564. [DOI] [PubMed] [Google Scholar]
- Ausio J, Dong F, van Holde KE. Use of selectively trypsinized nucleosome core particles to analyze the role of the histone tails in the stabilization of the nucleosome. J Mol Biol. 1989;206:451–463. doi: 10.1016/0022-2836(89)90493-2. [DOI] [PubMed] [Google Scholar]
- Banéres J-L, Martin A, Parello J. The N tails of histones H3 and H4 adopt a highly structured conformation in the nucleosome. J Mol Biol. 1997;273:503–508. doi: 10.1006/jmbi.1997.1297. [DOI] [PubMed] [Google Scholar]
- Barratt MJ, Hazzalin CA, Cano E, Mahadevan LC. Mitogen-stimulated phosphorylation of histone H3 is targeted to a small hyperacetylation-sensitive fraction. Proc Natl Acad Sci USA. 1994;91:4781–4785. doi: 10.1073/pnas.91.11.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednar J, Horowitz RA, Dubochet J, Woodcock CL. Chromatin conformation and salt-induced compaction: three-dimensional structural information from cryoelectron microscopy. J Cell Biol. 1995;131:1365–1376. doi: 10.1083/jcb.131.6.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careri G, Fasella P, Gratton E. Statistical time events in enzymes: a physical assessment. CRC Crit Rev Biochem. 1975;3:141–164. doi: 10.3109/10409237509102555. [DOI] [PubMed] [Google Scholar]
- Clark DJ, Kimura T. Electrostatic mechanism of chromatin folding. J Mol Biol. 1990;211:883–896. doi: 10.1016/0022-2836(90)90081-V. [DOI] [PubMed] [Google Scholar]
- Delcros J-G, Loeuillet L, Chamaillard L, Royou A, Bouille N, Seiler N, Moulinoux J-P. Flow cytometric analysis of in vivo polyamine deprivation in Lewis lung carcinoma (3LL) cells using the monoclonal antibody SPM8-2. Cytometry. 1997;27:255–261. [PubMed] [Google Scholar]
- Dumuis-Kervabon A, Encontre I, Etienne G, Jauregui-Adell J, Méry J, Mesnier D, Parello J. A chromatin core particle obtained by selective cleavage of histones by clostripain. EMBO (Eur Mol Biol Organ) J. 1986;5:1735–1742. doi: 10.1002/j.1460-2075.1986.tb04418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encontre I, Parello J. Chromatin core particle obtained by selective cleavage of histones H3 and H4 by clostripain. J Mol Biol. 1988;202:673–676. doi: 10.1016/0022-2836(88)90296-3. [DOI] [PubMed] [Google Scholar]
- Felsenfeld G, McGee JD. Structure of the 30 nm chromatin fiber. Cell. 1986;44:375–377. doi: 10.1016/0092-8674(86)90456-3. [DOI] [PubMed] [Google Scholar]
- Finch JT, Klug A. Solenoidal model for superstructure in chromatin. Proc Natl Acad Sci USA. 1976;73:1897–1901. doi: 10.1073/pnas.73.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyns MH. Polyamine content of a non-aqueously isolated chromosome preparation. Exp Cell Res. 1979;122:377–380. doi: 10.1016/0014-4827(79)90313-6. [DOI] [PubMed] [Google Scholar]
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- Guo XW, Th'ng JP, Swank RA, Anderson HJ, Tudan C, Bradbury EM, Roberge M. Chromosome condensation induced by fostriecin does not require p34cdc2kinase activity and histone H1 hyperphosphorylation, but is associated with enhanced histone H2A and H3 phosphorylation. EMBO (Eur Mol Biol Organ) J. 1995;14:976–985. doi: 10.1002/j.1460-2075.1995.tb07078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley LR, D'Anna JA, Barham SS, Deaven LL, Tobey RA. Histone phosphorylation and chromatin structure during mitosis in chinese hamster cells. Eur J Biochem. 1978;84:1–15. doi: 10.1111/j.1432-1033.1978.tb12135.x. [DOI] [PubMed] [Google Scholar]
- Hacques M-F, Muller S, De Murcia G, Van Regenmortel MHV, Marion C. Use of an immobilized enzyme and specific antibodies to analyze the accessibility and role of histone tails in chromatin structure. Biochem Biophys Res Commun. 1990;168:637–643. doi: 10.1016/0006-291x(90)92368-a. [DOI] [PubMed] [Google Scholar]
- Hendzel MJ, Wei Y, Mancini MA, van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- Hill CS, Thomas JO. Core histone–DNA interactions in sea urchin sperm chromatin. The N-terminal tail of H2B interacts with linker DNA. Eur J Biochem. 1990;187:145–153. doi: 10.1111/j.1432-1033.1990.tb15288.x. [DOI] [PubMed] [Google Scholar]
- Ho DT, Sauvé DM, Roberge M. Detection and isolation of DNA-binding proteins using single-pulse ultraviolet laser crosslinking. Anal Biochem. 1994;218:248–254. doi: 10.1006/abio.1994.1174. [DOI] [PubMed] [Google Scholar]
- Hockensmith JW, Kubasek WL, Vorachek WR, Evertsz EM, von Hippel PH. Laser cross-linking of protein-nucleic acid complexes. Methods Enzymol. 1991;208:211–235. doi: 10.1016/0076-6879(91)08015-a. [DOI] [PubMed] [Google Scholar]
- Hougaard DM. Polyamine cytochemistry: localization and possible functions of polyamines. Intl Rev Cytol. 1992;138:51–88. doi: 10.1016/s0074-7696(08)61587-9. [DOI] [PubMed] [Google Scholar]
- Hougaard DM, Bolund L, Fujiwara K, Larsson L-I. Endogenous polyamines are intimately associated with highly condensed chromatin in vivo. A fluorescence cytochemical and immunocytochemical study of spermine and spermidine during the cell cycle and in reactivated nuclei. Eur J Cell Biol. 1987;44:151–155. [PubMed] [Google Scholar]
- Koshland D, Strunnikov A. Mitotic chromosome condensation. Annu Rev Cell Dev Biol. 1996;12:305–313. doi: 10.1146/annurev.cellbio.12.1.305. [DOI] [PubMed] [Google Scholar]
- Krajewski WA, Ausio J. Modulation of the higher-order folding of chromatin by deletion of histone H3 and H4 terminal domains. Biochem J. 1996;316:395–400. doi: 10.1042/bj3160395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Hayes JJ, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Mach M, Ebert P, Popp R, Ogilvie A. Compartmentalization of polyamines in mammalian cells. Biochem Biophys Res Commun. 1982;104:1327–1334. doi: 10.1016/0006-291x(82)91395-x. [DOI] [PubMed] [Google Scholar]
- Mahadevan LC, Willis AC, Barratt MJ. Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid and protein synthesis inhibitors. Cell. 1991;65:775–783. doi: 10.1016/0092-8674(91)90385-c. [DOI] [PubMed] [Google Scholar]
- Marion C, Roux B, Pallotta L, Coulet PR. Study of chromatin organization with trypsin immobilized on collagen membranes. Biochem Biophys Res Commun. 1983a;114:1169–1175. doi: 10.1016/0006-291x(83)90685-x. [DOI] [PubMed] [Google Scholar]
- Marion C, Roux B, Coulet PR. Role of histones H1 and H3 in the maintenance of chromatin in a compact form. Study with an immobilized enzyme. FEBS (Fed Eur Biochem Soc) Lett. 1983b;157:317–321. doi: 10.1016/0014-5793(83)80568-7. [DOI] [PubMed] [Google Scholar]
- Moore SC, Ausio J. Major role of the histones H3-H3 in the folding of the chromatin fiber. Biochem Biophys Res Commun. 1997;230:136–139. doi: 10.1006/bbrc.1996.5903. [DOI] [PubMed] [Google Scholar]
- Pruss D, Wolffe AP. Histone–DNA contacts in a nucleosome core containing a Xenopus5S rRNA gene. Biochemistry. 1993;32:6810–6814. doi: 10.1021/bi00078a002. [DOI] [PubMed] [Google Scholar]
- Rill RL, Oosterhof DK. The accessibilities of histones in nucleosome cores to an arginine-specific protease. J Biol Chem. 1982;257:14875–14880. [PubMed] [Google Scholar]
- Roberge M, Th'ng J, Hamaguchi J, Bradbury EM. The topoisomerase II inhibitor VM-26 induces marked changes in histone H1 kinase activity, histones H1 and H3 phosphorylation, and chromosome condensation in G2 phase and mitotic BHK cells. J Cell Biol. 1990;111:1753–1762. doi: 10.1083/jcb.111.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth SY, Allis CD. Chromatin condensation: does histone H1 dephosphorylation play a role? . Trends Biochem Sci. 1992;17:93–98. doi: 10.1016/0968-0004(92)90243-3. [DOI] [PubMed] [Google Scholar]
- Rydberg B, Holley WR, Mian S, Chatterjee A. Chromatin conformation in living cells: support for a zig-zag model of the 30 nm chromatin fiber. J Mol Biol. 1998;284:71–84. doi: 10.1006/jmbi.1998.2150. [DOI] [PubMed] [Google Scholar]
- Smirnov IV, Dimitrov SI, Makarov VL. Polyamine-DNA interactions. Condensation of chromatin and naked DNA. J Biomol Struct Dyn. 1988;5:1149–1161. doi: 10.1080/07391102.1988.10506455. [DOI] [PubMed] [Google Scholar]
- Snyder RD. Polyamine depletion is associated with altered chromatin structure in HeLa cells. Biochem J. 1989;260:697–704. doi: 10.1042/bj2600697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovsky VY, Dimitrov SI, Angelov D, Pashev IG. Interactions of acetylated histones with DNA as revealed by UV laser induced histone-DNA crosslinking. Biochem Biophys Res Commun. 1989a;164:304–310. doi: 10.1016/0006-291x(89)91718-x. [DOI] [PubMed] [Google Scholar]
- Stefanovsky VY, Dimitrov SI, Russanova VR, Angelov D, Pashev IG. Laser-induced crosslinking of histones to DNA in chromatin and core particles: implications in studying histone-DNA interactions. Nucleic Acids Res. 1989b;17:10069–10081. doi: 10.1093/nar/17.23.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkara PS, Chang CC, Prakash NJ. Role of polyamines during chromosome condensation of mammalian cells. Cell Biol Int Rep. 1983;7:455–463. doi: 10.1016/0309-1651(83)90135-2. [DOI] [PubMed] [Google Scholar]
- Th'ng JPH, Guo X-W, Swank RA, Crissman HA, Bradbury EM. Inhibition of histone phosphorylation by staurosporine leads to chromosome decondensation. J Biol Chem. 1994;269:9568–9573. [PubMed] [Google Scholar]
- Tse C, Hansen JC. Hybrid trypsinized nucleosomal arrays: identification of multiple functional roles of the H2A/H2B and H3/H4 N-termini in chromatin fiber compaction. Biochemistry. 1997;36:11381–11388. doi: 10.1021/bi970801n. [DOI] [PubMed] [Google Scholar]
- Tse C, Sera T, Wolffe AP, Hansen JC. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol Cell Biol. 1998;18:4629–4638. doi: 10.1128/mcb.18.8.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Holde K, Zlatanova J. Chromatin higher order structure: chasing a mirage? . J Biol Chem. 1995;270:8373–8376. doi: 10.1074/jbc.270.15.8373. [DOI] [PubMed] [Google Scholar]
- van Holde, K.E. 1989. Chromatin. Springer-Verlag New York Inc., New York. 497 pp.
- van Holde KE, Zlanatova J. What determines the folding of the chromatin fiber? . Proc Natl Acad Sci USA. 1996;93:10548–10555. doi: 10.1073/pnas.93.20.10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hooser A, Goodrich DW, Allis CD, Brinkley BR, Mancini MA. Histone H3 phosphorylation is required for the initiation, but not maintenance, of mammalian chromosome condensation. J Cell Sci. 1998;111:3497–3506. doi: 10.1242/jcs.111.23.3497. [DOI] [PubMed] [Google Scholar]
- Wolffe, A.P. 1995. Chromatin Structure and Function. Academic Press, London. 299 pp.
- Woodcock CL, Horowitz RA. Chromatin organization re-viewed. Trends Cell Biol. 1995;5:272–277. doi: 10.1016/s0962-8924(00)89038-8. [DOI] [PubMed] [Google Scholar]
- Woodcock CLF, Frado LL, Rattner JB. The higher-order structure of chromatin: evidence for a helical ribbon arrangement. J Cell Biol. 1984;99:42–52. doi: 10.1083/jcb.99.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatanova J, Leuba SH, van Holde K. Chromatin fiber structure: morphology, molecular determinants, structural transitions. Biophys J. 1998;74:2554–2566. doi: 10.1016/S0006-3495(98)77963-9. [DOI] [PMC free article] [PubMed] [Google Scholar]