

Abstract
The leading edge (∼1 μm) of lamellipodia in Xenopus laevis keratocytes and fibroblasts was shown to have an extensively branched organization of actin filaments, which we term the dendritic brush. Pointed ends of individual filaments were located at Y-junctions, where the Arp2/3 complex was also localized, suggesting a role of the Arp2/3 complex in branch formation. Differential depolymerization experiments suggested that the Arp2/3 complex also provided protection of pointed ends from depolymerization. Actin depolymerizing factor (ADF)/cofilin was excluded from the distal 0.4 μm of the lamellipodial network of keratocytes and in fibroblasts it was located within the depolymerization-resistant zone. These results suggest that ADF/cofilin, per se, is not sufficient for actin brush depolymerization and a regulatory step is required. Our evidence supports a dendritic nucleation model (Mullins, R.D., J.A. Heuser, and T.D. Pollard. 1998. Proc. Natl. Acad. Sci. USA. 95:6181–6186) for lamellipodial protrusion, which involves treadmilling of a branched actin array instead of treadmilling of individual filaments. In this model, Arp2/3 complex and ADF/cofilin have antagonistic activities. Arp2/3 complex is responsible for integration of nascent actin filaments into the actin network at the cell front and stabilizing pointed ends from depolymerization, while ADF/cofilin promotes filament disassembly at the rear of the brush, presumably by pointed end depolymerization after dissociation of the Arp2/3 complex.
Keywords: actin, Arp2/3 complex, actin depolymerizing factor/cofilin, locomotion, treadmilling
Actin dynamics play a central role in certain types of cell motility, primarily in protrusion of the leading edge of crawling cells (for review see Welch et al., 1997b; Carlier, 1998), but also in the rocketing motion of intracellular parasites (for review see Higley and Way, 1997). Polymerization at barbed ends of actin filaments has been proposed to provide the driving force for forward movement, while dissociation of actin subunits from free pointed ends allows for filament actin turnover.
Interpretation of polymerization-driven protrusion in terms of a molecular mechanism depends upon knowledge of the length distribution and organization of the actin filaments. Structural evidence pointing to the existence of long actin filaments in lamellipodia (Small, 1988) suggests a filament treadmilling model (Small, 1994, 1995). In this model, based on behavior of actin filaments demonstrated initially in vitro (Wegner, 1976; Pollard, 1986; Carlier and Pantaloni, 1997), subunits are continuously added to the forward-facing barbed ends and are lost from the rear-facing pointed ends.
In contrast to actin polymerization, which occurs preferentially at the leading edge, actin depolymerization occurs uniformly throughout the entire actin filament array (Theriot and Mitchison, 1991, 1992). According to the filament treadmilling model, depolymerization occurs only at free pointed ends. Therefore, actin filaments are proposed to have a graded length distribution, with their barbed ends concentrated at the leading edge and pointed ends distributed throughout the cytoplasm (Small, 1994, 1995).
We recently studied the supramolecular organization of actin filaments in a model motility system, namely, lamellipodia of fish keratocytes (Svitkina et al., 1997), and found a structural pattern differing significantly from the premise of the individual filament treadmilling model. Barbed ends were numerous near the leading edge, but we were unable to identify free pointed ends within the lamellipodium. Pointed ends, where they could be detected, were involved in structural association with the sides of other filaments, resulting in Y-junctions. We suggested that the Y-junctions arose as a result of tightly coupled nucleation and cross-linking of actin filaments at the leading edge (Svitkina et al., 1997). Such coupling would allow nascent filaments to push against the membrane immediately after formation and provide the structural basis for polymerization-driven protrusion. In general, our findings on the structural organization of the lamellipodial network suggest that actin dynamics at the leading edge should be discussed in terms of the behavior of an integrated actin filament array, rather than of a collection of individual filaments.
One recently characterized component, the Arp2/3 complex (for review see Machesky, 1997; Machesky and Way, 1998; Zigmond, 1998; Machesky and Gould, 1999), seemed to be a particularly good candidate for the role of Y-junction linker in the formation of an integrated actin filament array. The Arp2/3 complex, originally identified in Acanthamoeba castellanii (Machesky et al., 1994), consists of actin-related proteins 2 and 3, and five other proteins (Machesky et al., 1997; Mullins et al., 1997; Welch et al., 1997c). It localizes to the leading edge of crawling cells (Kelleher et al., 1995; Machesky et al., 1997; Mullins et al., 1997; Welch et al., 1997c) and is sufficient to induce actin polymerization at the surface of Listeria monocytogenes cells (Welch et al., 1997a). Biochemically, the Arp2/3 complex nucleates actin filaments (Mullins et al., 1998) and the nucleating activity is regulated by other proteins (Welch et al., 1998; Machesky et al., 1999; Rohatgi et al., 1999). The complex also binds to the sides of actin filaments (Mullins et al., 1997), caps pointed ends, and forms branched structures when mixed with actin filaments (Mullins et al., 1998).
Based on biochemical properties of the Arp2/3 complex, together with our previous structural data on the organization of the keratocyte lamellipodium (Svitkina et al., 1997), Mullins et al. (1998) proposed a dendritic nucleation model for the mechanism of actin assembly in cell protrusions (see also Machesky and Way, 1998). The model proposes that the Arp2/3 complex nucleates new actin filaments, caps their pointed ends, and anchors them to the sides of pre-existing filaments, which results in assembly of a branched actin network. In contrast to the filament treadmilling model, in which new growth occurs as elongation of pre-existing filaments (Small, 1994, 1995), the dendritic model proposes formation of new actin filaments by nucleation on pre-existing filaments. Previously, even in situations where massive formation of new sites of polymerization was expected, e.g., during chemotactic response, de novo nucleation was given less attention, as compared with two other mechanisms, uncapping or severing of pre-existing filaments. Capping of pointed ends is an additional feature of the dendritic model. The amount of Arp2/3 complex in Acanthamoeba has been estimated to be sufficient to cap all pointed ends in the lamellipodium (Mullins et al., 1998). Further, our structural investigation failed to reveal free pointed ends in the keratocyte lamellipodium (Svitkina et al., 1997). These findings challenge a critical assumption of the treadmilling model, namely, that pointed ends should be constitutively free to release actin subunits, thus allowing for actin turnover. Nevertheless, in any steady state mechanism, polymerization-driven protrusion at the leading edge must be balanced by depolymerization of actin filaments elsewhere. Consequently, a question directed to the dendritic model is how it may be adapted to explain the necessary depolymerization.
Actin depolymerizing factor (ADF)1/cofilin has been shown to play an important role in the depolymerization of actin filaments during actin-based motility (for recent reviews see Theriot, 1997; Maciver, 1998; Rosenblatt and Mitchison, 1998). Proteins of the ADF/cofilin family bind both G- and F-actin, but prefer ADP-bound forms (Maciver and Weeds, 1994). They enhance depolymerization by increasing the rate constant for actin dissociation from pointed ends (Carlier et al., 1997) and by severing actin filaments (Maciver et al., 1991, 1998; Maciver, 1998). The relative impact of these two pathways in vivo has not been established. In vivo, ADF/cofilin localizes to sites of intensive actin turnover (Theriot, 1997; Rosenblatt and Mitchison, 1998), including lamellipodia of crawling cells (Bamburg and Bray, 1987; Yonezawa et al., 1987; Meberg et al., 1998), and facilitates actin turnover in Listeria tails in cytoplasmic extracts (Carlier et al., 1997; Rosenblatt et al., 1997). ADF/cofilin may be regulated by multiple pathways, such as phosphorylation, phosphoinositides, and pH (for review see Moon and Drubin, 1995). A role for ADF/ cofilin is compatible with either the filament treadmilling model or the dendritic model, although the details of its structural organization and regulation are likely to differ.
The available evidence points to the idea that the mechanism of cell protrusion is driven by the polymerization of actin filaments organized in some sort of motile machinery. This study attempts to place likely key molecular regulators into the emerging picture of actin filament organization and to distinguish between the individual actin filament treadmilling model and the dendritic model. Specifically, we investigate the supramolecular organization and dynamics of the actin filament network in lamellipodia of two different cell types, keratocytes and fibroblasts, which use similar mechanisms for protrusion, but express remarkable differences in motile behavior. The smooth, persistent locomotion of stably shaped keratocytes contrasts with the jerky motion of ever changing fibroblasts. Comparison of these two cell types has the potential to reveal common functional elements, as well as variables in the mechanisms of actin-based motility. A key finding is that the Arp2/3 complex is located at Y-junctions of actin filaments at the leading edge and that this complex provides protection of pointed ends against depolymerization.
Materials and Methods
Cell Culture
Xenopus laevis keratocytes were isolated and cultured basically as described by Bereiter-Hahn and Vöth (1988). In brief, tails were amputated from anesthetized tadpoles at stages 48–55 (Nieuwkoop and Faber, 1956), cut into pieces with a razor blade, incubated in digestion solution (0.2% trypsin and 0.2% EDTA in PBS) for 5 min, and rigorously pipetted. Large tissue pieces were allowed to settle and the supernatant cell suspension was collected. Cells were centrifuged to remove digestion solution, resuspended in L-15 medium (Sigma Chemical Co.), diluted to 70% with distilled water, and supplemented with 20% FBS (HyClone Labs), 0.29 g/liter glutamine, and antibiotics plated onto glass coverslips and cultured at 27°C. After ∼1 h, cultures were washed free from unattached cells and debris, and used for experiments.
A spontaneously immortalized cell line of Xenopus embryo fibroblasts obtained as in Daniolos et al. (1990) was provided by Dr. V.I. Rodionov (University of Wisconsin, Madison, WI) and cultured using the same medium and temperature as for Xenopus keratocytes. Human 356 fibroblasts, rat REF-52, mouse MFT-6, and Swiss 3T3 cell lines were cultured as described in Verkhovsky et al. (1995) and Svitkina et al. (1996).
Cytochalasin D (CD; Sigma Chemical Co.), latrunculin A (LA; Molecular Probes), or staurosporine (Sigma Chemical Co.) were added to culture medium from concentrated stock solutions in DMSO. For recovery from serum starvation experiments, cells were kept in serum-free medium overnight and then returned to serum-containing medium. For energy starvation, cells were incubated for 30 min at 37°C in PBS supplemented with 1 mM MgCl2, 0.5 mM CaCl2, 10 mM NaN3, and 50 mM deoxyglucose (Sigma Chemical Co.).
Antibody Reagents
The following antibodies were used for immunofluorescence and immuno-EM. Arp2/3 complex: affinity-purified rabbit polyclonal antibodies prepared against mammalian Arp3, p34-Arc, and p21-Arc (Welch et al., 1997c) were kindly provided by Dr. M.D. Welch (University of California, Berkeley, CA); antibodies against mammalian p33-Arc (Machesky et al., 1997) were provided by Dr. L.M. Machesky (University of Birmingham, Edgbaston, UK). Each of these antibodies was tested against lysates of Xenopus fibroblasts by immunoblot analysis and gave a single band of the predicted mobility (results not shown). ADF/cofilin: affinity-purified rabbit polyclonal antibodies to Xenopus ADF/cofilin (XAC; Abe et al., 1996; Rosenblatt et al., 1997) were kindly provided by Dr. J. Rosenblatt (University of California, San Francisco, CA) and by Dr. J.R. Bamburg (Colorado State University, Fort Collins, CO). We confirmed that these antibodies gave a single band by immunoblot analysis. ABP 280: mouse mAbs to human ABP-280 (Gorlin et al., 1990) were generously provided by Dr. J. Hartwig and Dr. T. Stossel (Harvard Medical School, Boston, MA). This antibody did not cross-react with Xenopus and was used only with human fibroblasts. α-Actinin: mouse mAb to α-actinin was purchased from Sigma Chemical Co. Immunoblot analysis gave a single band with Xenopus lysates. Secondary antibodies: secondary TRITC-, FITC-, and 10-nm gold-conjugated antibodies were purchased from Sigma Chemical Co. 18-nm gold-conjugated anti–mouse IgM antibody was obtained from Jackson ImmunoResearch Laboratories.
Microscopy
Procedures for detergent extraction, immunostaining, S1 decoration, light, and EM were described previously (Svitkina et al., 1995, 1996, 1997; Verkhovsky et al., 1995; Svitkina and Borisy, 1998). In brief, cells were washed in PBS and extracted for 3 min at room temperature with 1% Triton X-100 in PEM buffer (100 mM Pipes, pH 6.9, 1 mM MgCl2, and 1 mM EGTA) containing 4% polyethylene glycol (PEG), mol wt 40,000 (Serva), and either 0.5 μM TRITC-phalloidin (for light microscopy) or 2 μM phalloidin (for EM; Sigma Chemical Co.). Extracted cells were briefly washed with the phalloidin-containing PEM buffer and fixed with 2% glutaraldehyde.
For some experiments, PEG and/or phalloidin were omitted from the extraction procedure. PEG without phalloidin preserved lamellipodial network similar to the regular extraction. Decrease in PEG concentration from 4 to 1% or less in the absence of phalloidin, produced gradual loss of actin filaments from lamellipodial rear (see Results). Omitting PEG in the presence of phalloidin resulted in loss of granular material from lamellipodia, which is usually present in minor amounts, yet actin filaments seemed to be completely preserved. This granular material became abundant after LA treatment, and to a lesser extent, after CD treatment. Presumably, it represented insoluble forms of G-actin and might be similar to G-actin structures revealed after introducing fluorescently labeled actin into living cells (Cao et al., 1993; Ballestrem et al., 1998). To remove granular material obscuring F-actin distribution, we omitted PEG from the extraction solution for LA- and CD-treated cells.
For some antibodies, we used modifications of the regular immunostaining procedure. The ABP-280 antibody, which only worked with unfixed antigen, was applied in the PEM buffer containing phalloidin to extracted cells before glutaraldehyde fixation. For immunolocalization of XAC, we excluded phalloidin from the extraction solution to prevent competitive displacement of XAC from actin filaments. The presence of PEG, as we mentioned earlier, provided as good a quality of cytoskeleton preservation as phalloidin. For immunofluorescence microscopy with antibodies to Arp2/3 complex components, which worked only with denatured proteins, glutaraldehyde-fixed cytoskeletons were treated with 100% methanol for 20 min at 37°C and rinsed with PBS before application of antibodies. This procedure provided the brightest staining, compared with lower concentrations of methanol, but at the electron microscopic level, it completely ruined filament structure. Therefore, for immuno-EM we used aqueous 33% methanol instead of pure methanol, which provided acceptable morphology and detectable immunoreactivity of samples. Comparable results were obtained when glutaraldehyde-fixed cells were treated with 0.5% SDS in Tris-buffered saline for 20 min at 37°C and rinsed with Tris-buffered saline before application of antibodies. All of the four Arp2/3 complex antibodies tested gave essentially identical immunofluorescence patterns of staining. The immuno-EM reaction was of varying intensity, with the p21-Arc antibody giving the strongest signal. In all cases, at both the light and electron microscopic levels, staining by secondary antibody reagents alone was negligible (not shown).
Results
Y-Junctions in the Actin Lamellipodial Network
Results of our previous study on fish keratocytes (Svitkina et al., 1997) suggested that actin filaments were organized into a branched network similar to that proposed by the dendritic model (Mullins et al., 1998). However, few antibodies are available which cross-react with components in fish cells. Consequently, to test the presence and determine the distribution of predicted molecular constituents, we focused on the lamellipodial network in Xenopus keratocytes and fibroblasts, systems better suited for immunolocalization studies.
Xenopus keratocytes had similar cytoskeletal organization to fish keratocytes. Actin filaments in lamellipodia (Fig. 1 a) formed an extensive network, with the highest filament density at the leading edge which gradually decreased with distance from the edge. Assaying for actin filament polarity using myosin S1 decoration demonstrated that barbed ends faced forward (not shown) as in fish keratocytes (Svitkina et al., 1997). Although ultrastructural observation, per se, does not permit one to say that actin filaments do not terminate in capping protein, for our operational purposes, their ends are designated as free if the filament did not terminate at another filament or any other visible structure. With this criterion, the most peripheral zone (up to ∼1 μm from the leading edge) was highly enriched in apparently free barbed ends, but some barbed ends were also found deeper in the lamellipodium. Many Y-junctions between filaments (Fig. 1, b–g), but no free pointed ends, were visualized within the lamellipodial network. As determined by quantification of visible Y-junctions per unit area on electron micrographs, Y-junctions were ∼2–3 times more abundant in the peripheral ∼1 μm zone, as compared with the zone immediately behind this region, namely, 1–2 μm from the leading edge. The real difference in Y-junction frequency between these zones may be even greater, since the number of Y-junctions near the edge was likely underestimated because of very high filament density in this region. Fibroblast lamellipodia (Fig. 1, h–o) had the same basic features of actin network organization, but had qualitatively lower network density and fewer free barbed ends at the leading edge, as compared with keratocytes. Thus, both Xenopus keratocytes and vertebrate fibroblasts confirmed the existence of abundant Y-junctions near the leading edge of motile cells.
Figure 1.

Multiple branching of actin filaments in lamellipodia. EM of lamellipodia of Xenopus keratocytes (a–g) and fibroblasts (h–o) showing overviews of the leading edge (a and h) and enlargements of the boxed regions (b–g and i–o). Many examples of filaments with tightly spaced multiple branches (cyan) can be visualized in lamellipodia despite the high overall density of the actin network. Bar, 0.5 μm.
The high filament density in lamellipodia hindered observation of individual filaments for a significant distance and determination of the frequency of Y-junctions per unit length of filament. Occasionally, we could see several Y-junctions belonging to the same tree of actin filaments and they were usually closely spaced, suggesting frequent branching of individual filaments (Fig. 1). To facilitate the visibility of branched filaments, we attempted to generate a sparser lamellipodial network by treatment with CD, which acts preferentially to cap barbed ends (Brown and Spudich, 1981; Goddette and Frieden, 1986; Sampath and Pollard, 1991). At low concentrations (0.2–0.5 μM), CD gradually suppressed lamellipodial protrusion in keratocyte. TRITC-phalloidin staining of lamellipodia of CD-treated keratocytes revealed low and uniform actin density from front to rear (not shown) in contrast to control cells displaying a pronounced gradient of actin staining (Small et al., 1995; Svitkina et al., 1997; see also Figs. 3 and 8).
Figure 3.
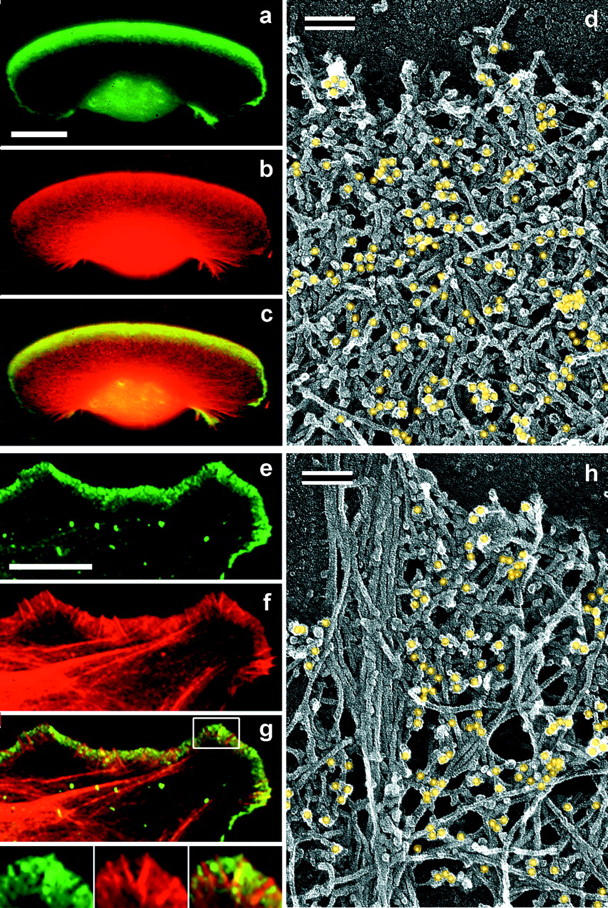
Localization of Arp2/3 complex in lamellipodia. (a–c and e–g) Fluorescence microscopy of Xenopus keratocyte (a–c) or fibroblast (e–g). Staining with p21 antibody (green) and TRITC-phalloidin (red) shows ARP2/3 complex highly enriched in lamellipodia. Boxed region in g is enlarged in insets; it shows several filopodia lacking and only one filopodium containing Arp2/3 complex. (d and h) Immuno-EM of lamellipodia of Xenopus keratocyte (d) or fibroblast (h) stained with p21 primary antibody and 10-nm gold-conjugated secondary antibody after glutaraldehyde fixation and SDS treatment of detergent-extracted cells. Gold particles are highlighted in yellow. Bars: (a and e) 10 μm; (d and h) 0.1 μm.
Figure 8.

Localization of XAC in Xenopus keratocytes. (a–e) Fluorescence microscopy of a whole cell (a–c) and the intensity profile (d) of the enlarged lamellipodium (e) from the boxed region in c, double stained with XAC antibody (green) and TRITC-phalloidin (red). XAC in lamellipodium is excluded from the narrow zone at the extreme leading edge. (f) Phase-contrast sequence of a locomoting cell; time is shown in min. (g–i) Immuno-EM with XAC antibody of the cell shown in f, which was lysed and processed at 8 min time point. (g) Cell overview. (h) Intermediate magnification of the boxed region from g showing distribution of gold particles (yellow) in lamellipodia. (i) High magnification of the boxed region from d showing leading edge. XAC is excluded from the extreme front of the lamellipodium (h and i). Bars: (a and f) 10 μm; (h) 10 μm.
EM demonstrated a significantly sparser actin network in lamellipodia of CD-treated cells, permitting observation of individual filaments for a significant length and improved visualization of Y-junctions. Depending upon the concentration of CD and time of treatment, as well as on the response of individual cells, it was possible to find the entire range of variation from an almost normal lamellipodial network to a sparse collection of branched filaments (Fig. 2, a and b). The average angle subtended by a Y-junction was 67 ± 12° (n = 212), similar to that reported for Arp2/3-nucleated branches in vitro (70 ± 7°; Mullins et al., 1998). The spacing between adjacent Y-junctions was variable. However, many Y-junctions occurred within 20–50 nm of each other, indicating a high probability for branching near the leading edge. Some filaments appeared to have axial functions: they held numerous secondary filaments alongside. Assay for filament polarity (not shown) demonstrated that free ends were barbed, and pointed ends were involved in junction formation, as in untreated cells (Svitkina et al., 1997). As in control preparations, no free pointed ends were identified in CD-treated lamellipodia. Since the actin network in CD-treated cells was sufficiently sparse to visualize free pointed ends if they were abundant, this observation suggests that free pointed ends were virtually absent or very transient.
Figure 2.

Improved visualization of actin filament branching in lamellipodia. EM of keratocyte or fibroblast lamellipodial actin network after CD treatment (a and b, 0.2 μM for 30 min or 0.5 μM for 10 min), 1 min recovery from serum starvation of a mouse fibroblast (c), LA treatment (0.2 μM for 10 min), or unprotected extraction (d). All examples demonstrate frequent branching of actin filaments. Bars, 0.1 μm; b and d are shown at the same magnification as c.
The high frequency of branching seemed remarkable. To test the possibility that frequent branching was artifactually induced by CD, we examined lamellipodia in other situations, which also allowed for better visualization of branched filaments. They included: short term release from serum or energy starvation, which led to formation of nascent comparatively loose lamellipodia; treatment with the actin monomer sequestering agent, LA (for review see Ayscough, 1998), which caused depolymerization of actin filaments in cells; and cell lysis under conditions allowing for actin filament depolymerization. In all three experimental conditions, we were able to visualize filaments with multiple branches (Fig. 2, c and d) and the spacing between branches was similar to that observed in CD-treated cells. Again, free pointed ends were not observed.
Thus, actin filaments in lamellipodia displayed frequent branches, which engaged virtually all detectable pointed ends into Y-junctions and left numerous barbed ends apparently free. Extensive filament branching was a general feature of lamellipodia in different situations, including expanding lamellipodia in starvation-release experiments, steady state conditions in untreated keratocyte lamellipodia, or declining protrusions after drug treatment.
Identification of Y-Junction Linker
Our next goal was to identify a molecule that localized to actin filament branch points. Arp2/3 complex seemed to be a primary candidate, but other cross-linking proteins which localize to lamellipodia, such as α-actinin or ABP-280/filamin, were also possibilities. We performed immunolocalization of these proteins in keratocytes and fibroblasts.
Antibodies to various components of the mammalian Arp2/3 complex have been shown to stain lamellipodia in cultured cells of mammalian origin (Machesky et al., 1997; Welch et al., 1997c). We found that they also stained lamellipodia in Xenopus cells (Fig. 3), but in fibroblasts, Arp2/3 complex was excluded from most filopodia (Fig. 3 g, insets).
The overall pattern of Arp2/3 staining at the electron microscopic level in keratocytes and fibroblasts (Fig. 3, d and h) correlated with the results obtained by light microscopy. The gold label was distributed all over the dense lamellipodial network and gradually declined toward the lamellipodial rear. The high density of the actin network, however, did not allow us to routinely attribute gold particles to any specific filaments or branches. As before, we used CD treatment to visualize individual Y-junctions and were able to stain branch points with antibody to p21-Arc as a result (Fig. 4). Distinctly labeled branches were seen clearly in regions with very sparse filament distribution. More commonly, clusters of gold particles with short filaments sticking out were observed (not shown). Such distribution of label would be predicted for multiple branches in close proximity to each other, as was observed (see above). Not all visible branches in the specimen were labeled, but incomplete labeling could be the result of a variety of factors, including suboptimal immunoreactivity because of the procedure used for structural preservation (Materials and Methods).
Figure 4.

Localization of Arp2/3 complex at actin filament branching points. Xenopus keratocytes and fibroblasts were treated with CD (0.2 μM for 30 min or 0.5 μM for 10 min), extracted in the presence of phalloidin, fixed with glutaraldehyde, treated with 33% methanol, and immunostained with p21 antibody followed by 10-nm gold-conjugated secondary antibody. Gold particles are highlighted in yellow. Bar, 50 nm.
To test whether a fraction of branches was mediated by other proteins, we performed immunolocalization of α-actinin and ABP-280, proteins which make side-to-side cross-links (for review see Matsudaira, 1994). Immunofluorescence of Xenopus fibroblasts and keratocytes with antibody to α-actinin, and of human fibroblasts with antibody to ABP-280 (Fig. 5, a–c), revealed some lamellipodial staining, but it was not as prominent as staining of internally located actin structures. Compared with Arp2/3 complex, α-actinin and ABP-280 in lamellipodia were much less abundant with respect to actin content (Fig. 5, a′–c′). A similar relationship between α-actinin and Arp2/3 complex distribution was found in Acanthamoeba (Mullins et al., 1998). Immuno-EM of CD-treated cells with antibody to α-actinin and ABP-280 demonstrated negligible staining of branched filaments near the leading edge (Fig. 5, e and f), but significant staining of internal actin networks, predominantly at points of filament crossovers or juxtapositions (Fig. 5, h and i), which contrasted with the opposite pattern of Arp2/3 staining (Fig. 5, d and g).
Figure 5.
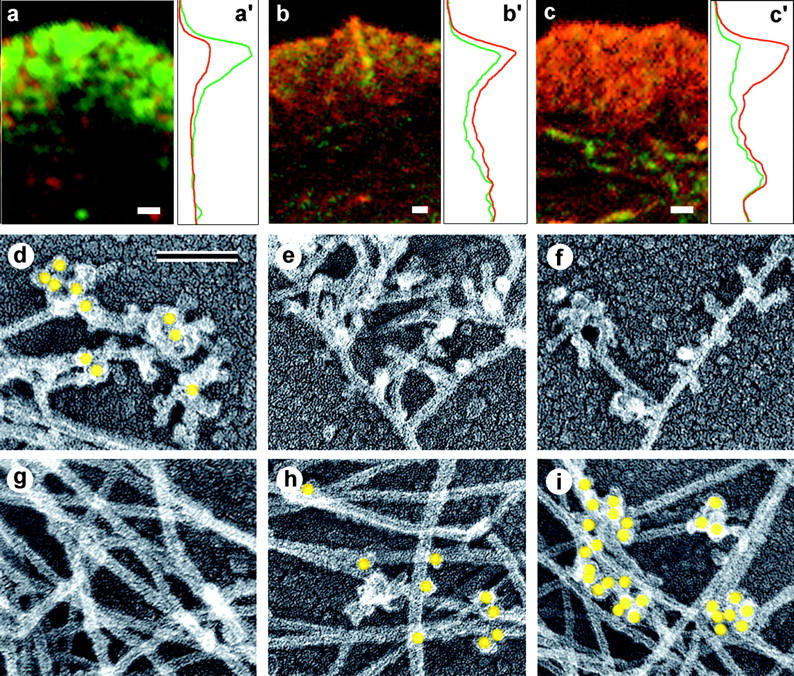
Localization of cross-linking proteins in fibroblast cytoskeleton. (a–c) Fluorescence microscopy and corresponding intensity profiles (a′–c′) of Xenopus (a and c) or human 356 (b) fibroblast lamellipodia double stained with TRITC-phalloidin (red) and either p21 (a and a′), ABP-280 (b and b′), or α-actinin (c and c′) antibodies (green). The protein/actin ratio at the leading edge of the lamellipodium is high for Arp2/3 complex (a and a′), medium for ABP-280 (b and b′), and low for α-actinin (c and c′) compared with internal actin structures. (d–i) Immuno-EM of the cell edge (d–f) or interior (g–i) of CD-treated Xenopus (d, f, g, and i) or human 356 (e and h) fibroblasts stained with p21 (d and g), ABP-280 (e and h), or α-actinin (f and i) primary antibody and 10-nm (d, e, g, and h) or 18-nm (f and i) gold-conjugated secondary antibody. Gold particles (yellow) reveal Arp2/3 complex at Y-junctions at cell edge and ABP-280 and α-actinin at filament crossovers in the cell interior. Bars: (a–c) 1 μm; (d–i) 0.1 μm.
Thus, our data demonstrate that Arp2/3 complex is located at Y-junctions, implying that it plays a role in their formation or maintenance. The structural evidence for ABP-280 and α-actinin suggests that they are unlikely to play a significant role in branching near the leading edge. Their impact on filament cross-linking is likely to be expressed more deeply in the cytoplasm.
Protection of Pointed Ends from Depolymerization
Involvement of pointed ends in Y-junction formation and localization of Arp2/3 complex at the same positions suggested that pointed end depolymerization in lamellipodia may be significantly blocked due to pointed end capping by the Arp2/3 complex (Mullins et al., 1998). We tested depolymerization properties of the lamellipodial network using two approaches, cytoskeletal preparations and living cells.
To allow for actin depolymerization in cytoskeletons, we omitted certain precautions in the process of detergent extraction, which are usually necessary to preserve the actin network in the lamellipodium. Our regular extracting solution (Svitkina et al., 1995; Svitkina and Borisy, 1998) contains two protective agents to stabilize the actin network: a nonspecific stabilizer, PEG; and a specific F-actin stabilizing drug, phalloidin. In the absence of these chemicals, significant loss of actin filaments was observed in the rear of lamellipodia, whereas the peripheral zone (∼1 μm) at the leading edge remained almost as dense as in control cytoskeletons (Fig. 6). This zone frequently looked barely connected to the rest of the cytoskeleton and usually contained numerous free barbed ends rendering it a brush-like appearance. Only a minor proportion of actin filaments seemed to be lost from this actin brush. CD (1 μM), when added to the extraction solution to suppress depolymerization from the barbed ends, did not prevent loss of actin filaments in the rear network (not shown), although the density of the actin brush at the front was similar to that of untreated cells. These results demonstrated that loss of actin filaments at the lamellipodial rear occurred mostly via pointed end depolymerization, whereas loss from barbed ends had only minor impact.
Figure 6.

Structural differentiation of actin network in lamellipodium. EM of Xenopus keratocytes (a–c) or Xenopus fibroblasts (d–f) after regular extraction in the presence of PEG and phalloidin (a and d) or after unprotected extraction without PEG and phalloidin (b, c, e, and f). Boxed areas from b and e are enlarged in c and f, respectively. Actin network at lamellipodial rear disassembled in the course of unprotected extraction, whereas front zone remained as dense as in control cells. Bar, 1 μm.
In living cells, we used LA under conditions which shifted the actin steady state toward depolymerization, but did not completely block motility. Observations on living keratocytes by phase-contrast microscopy demonstrated that at concentrations of 0.3 μM or higher, LA induced fast cessation of motility and collapse of lamellipodia (not shown). At lower concentrations (0.1–0.2 μM), protrusion of the keratocyte leading edge continued with a progressively slower rate (Fig. 7, A and B) and eventually ceased, after which the cell body continued to migrate forward for a short time approaching the leading edge, similar to what has been reported for CD-treated keratocytes (Anderson et al., 1996). Pseudopodial activity at cell edges persisted for up to 40 min, but became progressively irregular and frequently ended up with the formation of phase dense beads along the cell periphery. Thus, low level LA treatment permitted analysis of the pathway whereby sequestration of actin subunits leads to cessation of leading edge protrusion.
Figure 7.

Differential response of lamellipodial actin network to LA. (A) Phase-contrast sequence of a locomoting Xenopus keratocyte. After addition of 0.1 μm LA at 9 min time point, the cell continued to translocate, retaining the crescent-like shape. (B) Plot showing rate of front edge protrusion versus time of the cell shown in A. LA addition (arrow) decreased rate of protrusion from ∼4 μm/min before LA application to 1 μm/min at the end of the sequence. (C) Fluorescence microscopy of the boxed region of the cell shown in A, which was lysed at 34 min time point, fixed, and stained with TRITC-phalloidin. Actin-staining reveals narrow bright lamellipodium at the leading edge, separated by the wide actin-depleted zone from the internal actin structures. (D and E) EM of a Xenopus keratocyte (D) or Xenopus fibroblast (E) lamellipodium treated with LA (D, 0.1 μM for 30 min; E, 0.25 μM for 10 min) reveals actin depletion from the lamellipodial rear. Bars: (A) 10 μm; (D and E) 1 μm.
We studied actin filament organization in LA-treated keratocytes by TRITC-phalloidin staining and by EM. At the light microscopic level in most cells, which were still motile after LA treatment, the lamellipodium looked like a narrow band of phalloidin-stained actin at the cell edge, which was separated by a wide dark zone from internally located actin bundles, as if the lamellipodium ran away from the cell body (Fig. 7 C). As the motility of the lamellipodium decreased and the cell body caught up, the distance between the lamellipodium and the rest of the cytoskeleton decreased. At late stages of LA treatment, actin was found only in bright spots along the cell periphery (not shown).
At the electron microscopic level, the runaway lamellipodium of an LA-treated keratocyte was very similar to the actin brush revealed by unprotected extraction. It was a dense, narrow (0.5–1 μm) band of actin network containing numerous Y-junctions and free ends (Fig. 7 D). In cells treated for a longer time and/or with higher concentrations of LA, discontinuous foci of the actin brush were found instead of a continuous brush. In fibroblasts, LA treatment gave basically the same results as in keratocytes. After ∼10 min in 0.1 μM LA, numerous runaway lamellipodia composed of dense actin network were formed (Fig. 7 E). A possible mechanism for the formation of runaway lamellipodia is continued protrusion under nonsteady state conditions. Although the rate of actin polymerization is predicted to be decreased by LA, the rate of depolymerization would be unaffected, leading to an erosion of the brush from the rear. If actin filaments were free to depolymerize throughout the lamellipodium, such imbalance would result in fast and complete disassembly of the entire lamellipodial network.
Thus, our results on actin depolymerization in cytoskeletons and in live cells demonstrated that actin filaments are protected from depolymerization within a narrow zone at the leading edge, but were susceptible to depolymerization farther away from the edge. The most likely mechanism of protection is capping of filament pointed ends by the Arp2/3 complex. In addition, our results with LA treatment revealed a possible minimal system for actin turnover. Under conditions of G-actin deficiency, cells progressively depolymerized actin from the rear of the lamellipodium retaining just a narrow, runaway actin brush. Progressive locomotion of such cells suggested that actin turnover can occur within this narrow zone.
Localization of ADF/Cofilin in Lamellipodia
Differential depolymerization of the lamellipodial actin network suggested a spatially regulated mechanism for exposing the pointed ends, which would allow for actin depolymerization at the lamellipodial rear. Exposure of the pointed ends may occur by dissociation of the Arp2/3 complex or by filament severing. The mechanism of actin filament depolymerization in lamellipodia is likely to involve activity of ADF/cofilin. The exact mode of action of ADF/ cofilin in cells (pointed end depolymerization, severing, or both) is not clear. If we assume that ADF/cofilin severs actin filaments to expose pointed ends, then to explain local protection of the front actin brush from depolymerization we should expect ADF/cofilin to be excluded from the protected zone. Alternatively, if ADF/cofilin depolymerizes actin filaments only from pointed ends, then binding of ADF/cofilin to actin filaments within the protected area would not result in actin depolymerization until pointed ends were released. In this case, spatially regulated pointed end uncapping could be the rate-limiting mechanism for actin network disassembly. We performed immunolocalization of ADF/cofilin in keratocytes and fibroblasts to get insight into this problem. Since we found differences between the two cell types, we present results for keratocytes and fibroblasts separately.
Keratocytes.
Immunofluorescence staining of keratocytes with antibody to XAC revealed XAC in lamellipodia (Fig. 8 a), similar to what has been shown for other cells (Bamburg and Bray, 1987; Yonezawa et al., 1987; Meberg et al., 1998). However, upon double-staining with TRITC-phalloidin, we found a new feature of XAC distribution in keratocyte lamellipodia: exclusion of XAC from a narrow marginal zone. Immuno-EM demonstrated that XAC was absent from the most peripheral 0.3–0.7 μm of the lamellipodial actin network (Fig. 8, f–i). The intensity of XAC staining gradually declined toward the cell center and was at a minimum at the rear of the lamellipodium.
The absence of XAC from actin filaments at the leading edge is consistent with in vitro data which indicate that ADF/cofilin does not bind to actin filaments with bound ATP or ADP and Pi (Maciver et al., 1991; Carlier et al., 1997). If this were the explanation, the length of the XAC-free zone would be a measure of the polymerization velocity of actin filaments and hence, of the protrusive speed of the cell. To test for a possible relationship between the width of the XAC-free zone and the rate of keratocyte locomotion, we performed correlative light and immuno-EM of individual cells crawling at naturally varying speeds. To broaden the range of speed variations, we slowed cell locomotion by a protein kinase inhibitor, staurosporine (50 nM), or by decreasing serum concentration in the medium, and carried out a statistical analysis of the covariation of cell speed and width of XAC-free zone. Surprisingly, no correlation between these two parameters was evident (data not shown). All keratocytes in culture had XAC-free front zones, including cells locomoting at normal rate (Fig. 8, f–i) and round stationary cells, which were occasionally found in the culture (not shown). The lack of correlation of the XAC-free zone with cell speed suggests that additional or alternative factors influence the binding of XAC to actin filaments.
Unprotected extraction and LA treatment had the effect of depolymerizing the bulk of cellular actin, although a lamellipodial brush survived. If ADF/cofilin binding was sufficient for actin depolymerization, surviving filaments would be predicted to be devoid of XAC. However, immunostaining with XAC antibody showed that the depolymerization-resistant brush obtained during unprotected extraction contained XAC at its rear (Fig. 9 a). In LA-treated cells, XAC also localized to the posterior portion of the runaway lamellipodium leaving a narrow XAC-free zone at the front (Fig. 9, b and c). Since in LA-treated cells, the brush continued to move, these data suggest that the brush effectively treadmills with assembly of XAC-free actin filaments at the front and disassembly of XAC-containing filaments at the rear.
Figure 9.
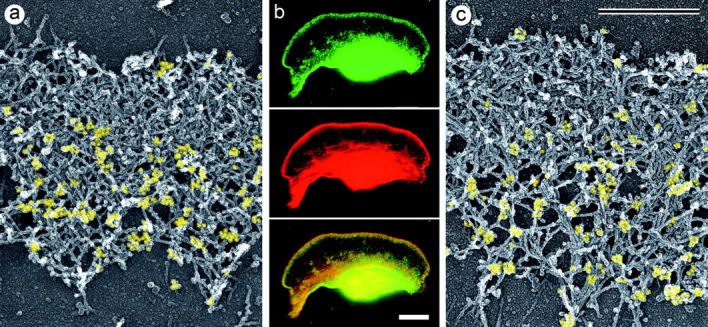
Localization of XAC to posterior regions of depolymerization-resistant actin brush. Electron (a and c) and fluorescence (b) microscopy of lamellipodia of Xenopus keratocytes after unprotected extraction (a) or LA treatment (b, 0.15 μM for 30 min; c, 0.25 μM for 10 min) and subsequent staining with XAC antibody (a and c) or double staining with TRITC-phalloidin and XAC antibody (b).
Fibroblasts.
Immunofluorescence also revealed XAC localizing to lamellipodia of Xenopus fibroblasts (Fig. 10 a). In contrast to keratocytes, no distinct XAC-free zone at fibroblast leading edges was revealed in double staining with TRITC-phalloidin: both proteins were found all the way to the cell edge (Fig. 10, a–e). Only in rare cases was actin staining slightly extended beyond the XAC staining. Most filopodia were not stained with XAC antibody, but some of them were stained. Immuno-EM confirmed that XAC in fibroblast lamellipodia was distributed all the way to the periphery (Fig. 10 i). The concentration of XAC was highest at the extreme outer margins of lamellipodia and gradually disappeared toward the rear.
Figure 10.
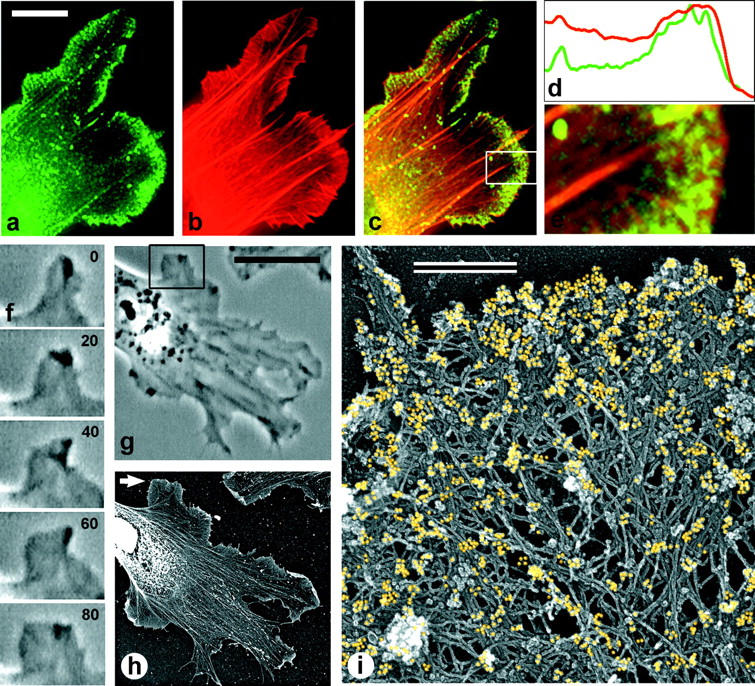
Localization of XAC in Xenopus fibroblasts. (a–e) Fluorescence microscopy of a cell fragment (a–c) and the intensity profile (d) of the enlarged lamellipodium (e) from the boxed region in c, double stained with XAC antibody (green) and TRITC-phalloidin (red). XAC is distributed throughout the entire lamellipodium. (f and g) Phase-contrast sequence showing protruding lamellipodium of a motile cell (f) and the overview of the same cell (g) at 80 s time point. (h and i) Immuno-EM with XAC antibody of the cell shown in g. (h) Cell overview. Nucleus and surrounding regions look bright because of high content of cellular proteins, which creates high electron density (brightness in inverted contrast). Immunogold-labeling in these central regions is very low. (i) High magnification of the protruding region pointed to by arrow in h. Gold particles (yellow) revealing XAC localization are found at the extreme leading edge, as well as throughout the lamellipodia.
Since protrusion of lamellipodia in fibroblasts is not as persistent as in keratocytes and may frequently alternate with withdrawals, we performed correlative analysis of locomotory behavior and XAC staining of fibroblast lamellipodia. In 13 examined cells, the vast majority of protruding (Fig. 10, f–i) or stationary lamellipodia, as well as ruffling lamellipodia, had XAC distributed all the way to the edge or very close (within 0.1 μm of the edge). Thus, XAC in fibroblast lamellipodia was found essentially throughout the depolymerization-resistant actin brush, as well as in the more labile rear parts of the lamellipodium. The difference between keratocytes and fibroblasts cannot be attributed simply to speed of protrusion. Although the net speed of a fibroblast cell is slow compared with that of a keratocyte, the speed of many fibroblast protrusions is comparable, ∼5–6 μm/min. Again, the lack of evident correlation between protrusive speed and distribution of XAC suggests that factors in addition to ATP or ADP-Pi regulate the binding of ADF/cofilin to actin filaments.
Discussion
Properties of the Dendritic Actin Brush
Our results indicate that the actin network at the leading edge of crawling cells, the dendritic brush, is distinctive in its structural organization, dynamics, and biochemical composition. Structurally, the brush is characterized by an extensively branched organization of actin filaments, with barbed ends facing approximately forward and pointed ends essentially all involved in Y-junctions. That barbed ends are enriched in the brush as compared with the rest of the lamellipodium is consistent with the idea that actin assembly occurs primarily within the dendritic brush. Further, the depth of the actin brush matches well with the depth of the zone, which has been demonstrated to incorporate actin (Symons and Mitchison, 1991; Chan et al., 1998). Pointed ends are more evenly distributed throughout the lamellipodium as compared with barbed ends, but they are more abundant in the actin brush. Remarkably, individual Y-junctions in the brush are frequently spaced by as little as several tens of nanometers. As a result, the dendritic brush contains numerous short filaments incorporated into the actin array by Y-junctions, as well as a proportion of longer filaments which continue into the more internal lamellipodial regions.
Dynamically, differential depolymerization experiments indicate that the dendritic brush is highly protected from disassembly. The fact that capping of barbed ends by CD during extraction did not significantly affect the differential depolymerization of the lamellipodial network implies that depolymerization indeed occurs from pointed ends and that the status of pointed ends (capped or uncapped) is responsible for the differential behavior of the actin brush and the internal actin network. Remarkably, protection of the actin brush from depolymerization does not interfere with its dynamic behavior. Indeed, cells which retained just the actin brush (runaway lamellipodium) and lost almost all the internal actin network were still able to locomote and therefore, to maintain continuous actin turnover within the actin brush. Thus, pointed end capping as a putative mechanism for protection of the actin brush from depolymerization is dynamic and probably regulated. Protection of newly formed barbed ends from capping, recently demonstrated in neutrophil extracts stimulated by Cdc42 (Zigmond et al., 1998), may also have an impact for dynamic persistence of the actin brush in addition to protection of pointed ends from depolymerization.
Biochemically, the dendritic actin brush contains significant amounts of the Arp2/3 complex, which localizes specifically to Y-junctions, whereas other possible cross-linking proteins, α-actinin and ABP-280, have their predominant association with X-junctions deeper in the cytoplasm. These results support the idea that the Arp2/3 complex is the major cross-linker in the actin brush and that it plays a role in stabilization of the actin brush by capping pointed ends (Mullins et al., 1998). Another lamellipodium-specific protein, ADF/cofilin, which plays a role in actin depolymerization and thus has an antagonistic activity to the Arp2/3 complex, is not a uniform component of the actin brush. Depending on cell type, it may associate with actin filaments throughout the brush, but it may also localize just to the brush's posterior portion. We propose that ADF/cofilin is functionally regulated in the actin brush and performs actin depolymerization after dissociation of the Arp2/3 complex, predominantly in the lamellipodial network behind the brush region.
Formation of the Dendritic Actin Brush
The central problem of dendritic brush formation is the origin of side branches: actin filaments which have pointed ends associated with Arp2/3 molecules at sides of other filaments. Two different, although nonexclusive, possibilities may be considered for the location of the Arp2/3 complex at Y-junctions: Arp2/3 complex nucleates filaments de novo or Arp2/3 complex captures pointed ends of pre-existing filaments nucleated elsewhere. Filament nucleation mediated by the Arp2/3 complex residing on pre-existing filaments is proposed by the dendritic nucleation model (Mullins et al., 1998) and is supported by biochemical data indicating that binding of the Arp2/3 complex to preformed filaments significantly increases its nucleating activity (Machesky et al., 1999). However, filament nucleation by the Arp2/3 complex near the leading edge, followed by docking via the Arp2/3 complex onto pre-existing filaments have not been formally excluded. In the second mechanism, the Arp2/3 complex residing on pre-existing filaments captures the pointed ends of filaments formed elsewhere, either new filaments nucleated near the leading edge by an Arp2/3-independent mechanism or older filaments in the process of depolymerization.
Constitutive locomotion similar to that expressed by keratocytes or Listeria theoretically could be maintained primarily by continuous elongation of pre-existing filaments. However, in many other systems actin-based motility is characterized by frequent protrusion–withdrawal cycles, like in locomoting fibroblasts, or by explosive actin polymerization in response to external stimuli, e.g., during chemotaxis. In such cases, generation of new sites for actin polymerization is unavoidable. In addition to nucleation, barbed end uncapping and filament severing (Condeelis, 1993; Zigmond, 1996) have been proposed as mechanisms. Our data, together with a growing mass of evidence indicating a role for the Arp2/3 complex in filament nucleation (Mullins et al., 1998; Machesky et al., 1999), are most consistent with the de novo nucleation mechanism mediated by Arp2/3 complex resulting in the formation of the dendritic brush. However, uncapping and severing mechanisms may also work in other systems or along with de novo nucleation (Hartwig, 1992; Hartwig et al., 1995; Schafer et al., 1996; Eddy et al., 1997). Dendritic nucleation of actin filaments may require a mechanism to ensure that most face forward. Preferential growth of barbed ends, perhaps by involvement of plasma membrane-associated factors, is one possibility. Recent data showing a role for the Ena/VASP protein family in directional motility of Listeria, supposedly by keeping growing barbed ends in a correct position (Laurent et al., 1999), is an example of such function. Additional possibilities include membrane-dependent regulation of nucleation or capping.
The high frequency of branches in the dendritic brush carries implications for its dynamics and regulation. If each Y-junction in the brush indeed represents an individual nucleation event, frequent branching is predicted to result in rapid, exponential growth of filament number, which may occur in expanding protrusions. However, extensively branched filaments also were observed in apparently steady state protrusions, such as keratocyte lamellipodia, suggesting that continuous de novo nucleation may be a constitutive mechanism for generating protrusions. One way to maintain steady state would be for most of the nucleated filaments to be capped soon after nucleation and only a small proportion of them continue to elongate and branch. This assumption is consistent with data showing that capping protein is localized at the leading edge (Schafer at al., 1998), the vast majority of free barbed ends are capped (Eddy et al., 1997), and stimulation of actin polymerization leads to association of capping protein with the cytoskeleton (Barkalow et al., 1996; Eddy et al., 1997). Although we have referred to free barbed ends in the dendritic brush, our structural observations do not distinguish between ends free to grow and ends terminated by capping protein. Frequent nucleation followed by capping should generate a significant number of short filaments, which is indeed what we observed in the actin brush. Increase in filament number, but not in their length during stimulated actin polymerization (Eddy et al., 1997; Zigmond et al., 1998), is also consistent with frequent nucleation as a pathway for actin polymerization. Estimates of a low average length of filaments in vivo (<1 μm; Podolski and Steck, 1990; Cano et al., 1991) are hard to explain otherwise. A nucleation-capping mechanism is attractive because it offers considerable flexibility in terms of organization and level of actin polymerization, which could be controlled via activity of the Arp2/3 complex and/or capping protein.
Disassembly of the Dendritic Actin Brush
Our working hypothesis is that the Arp2/3 complex protects actin filaments in the dendritic brush from depolymerization, whereas ADF/cofilin acts to promote disassembly of the actin network. Two mechanisms of ADF/ cofilin action in actin disassembly have been described in vitro, facilitated release of actin subunits from the pointed end and severing (Carlier et al., 1997; Maciver, 1998; Maciver et al., 1998). Which mechanism ADF/cofilin uses in vivo and how antagonistic activities of ADF/cofilin and Arp2/3 complex are coordinated in cells are important questions for understanding actin turnover in lamellipodia.
The extreme distal zone of keratocyte lamellipodia remains free of XAC, whereas the actin network toward the rear of lamellipodia is intensively stained by XAC antibody. The mechanism for XAC exclusion from the keratocyte leading edge is not clear. One possibility is preferential association of ADF/cofilin to ADP-bound forms of actin (Maciver and Weeds, 1994; Carlier et al., 1997). Because of the lag of ATP hydrolysis and Pi release following actin filament elongation, ADP-actin filament domains would not be present or would be less frequent at the extreme front. Such a mechanism implies almost synchronous growth of actin filaments at the keratocyte leading edge to account for the observed pattern of XAC staining. A different possibility suggests that some regulatory factors affecting XAC may inhibit its binding to actin filaments at the cell front. Spatially confined phosphorylation of XAC, production of inhibitory phosphoinositides at the leading edge, and/or a pH gradient may restrain XAC from binding to actin filaments. Finally, we cannot exclude the possibility that XAC was lost from the leading edge of keratocytes during permeabilization. In contrast to keratocytes, XAC in fibroblast lamellipodia was found even at the extreme leading edge. The different pattern of XAC distribution in keratocytes and fibroblasts seems not to be related to the difference in the instantaneous rate of protrusion, as we showed by correlating rate of protrusion and localization of XAC for individual lamellipodia. Asynchronous growth of actin filaments at the fibroblast leading edge or a different mode of regulation may permit XAC binding at the fibroblast leading edge.
Despite apparently different mechanisms for determining localization of XAC in two cell types, the presence of XAC within the actin brush in fibroblasts suggests that XAC binding to actin filaments is not sufficient for filament disassembly. The same conclusion can be drawn from XAC staining of actin brush in keratocytes after unprotected extraction or after LA treatment. In these preparations, XAC binding to posterior parts of the depolymerization-resistant actin brush demonstrates that the XAC-containing network and the network susceptible to depolymerization do not completely coincide. An XAC-independent step is consistent with the results of Rosenblatt et al. (1997) who showed that excess of XAC in cytoplasmic extracts was unable to shorten Listeria tails below a certain limit. These findings are hard to reconcile with a pure severing activity of XAC because rapid depolymerization would be expected to follow severing. The results more readily fit with the idea that severing activity of XAC is not significantly expressed in the actin brush in vivo. We suggest that XAC binds actin filaments within the actin brush and waits for the release of Arp2/3 complex to get a chance to facilitate subunit dissociation from pointed ends. In this framework, the XAC-independent step would be Arp2/3 dissociation from pointed ends, the signals for which remain to be determined.
Actin Array Treadmilling Model
Our data on the structural organization of the actin network at the leading edge and localization of the Arp2/3 complex at Y-junctions in vivo are fully consistent with the dendritic nucleation model (Mullins et al., 1998; Machesky et al., 1999) concerning cross-linking and pointed end capping activity of the Arp2/3 complex in vitro. Consequences of the dendritic model suggest a novel concept of actin turnover in lamellipodia in which the actin array as a whole treadmills, but not individual actin filaments, per se (Fig. 11).
Figure 11.

Two treadmilling models for actin turnover in lamellipodia. Left, treadmilling of individual filaments suggests that each actin filament in the actin network simultaneously assembles subunits at its barbed end and releases subunits from the pointed end, thus continuously reproducing itself by treadmilling mechanism. Treadmilling of individual filaments collectively results in the treadmilling of the lamellipodial network. Right, treadmilling of the dendritic array suggests frequent formation of new filaments by de novo nucleation, which occurs within the narrow zone at the leading edge, the actin brush. Newborn filaments become immediately incorporated into the actin array as branches of pre-existing filaments. Within the actin brush, filaments are protected from depolymerization at pointed ends. Nucleation, cross-linking, and pointed end capping are proposed to be mediated by the Arp2/3 complex. Many barbed ends are predicted to be capped to prevent exponential increase in filament mass. Release of the Arp2/3 complex from Y-junctions behind the actin brush, followed by ADF/cofilin-mediated dissociation of actin subunits from pointed ends, may be a major pathway for actin array disassembly. By this model, an individual filament in the dendritic array does not treadmill, but rather first grows at the barbed end and later shrinks at the pointed end. However, the actin filament array as a whole treadmills, reproducing itself at the cell front and dismantling itself at the lamellipodial rear. Growing barbed ends are shaded in gray.
All polymer turnover mechanisms require a balance in the steady state of formation and disassembly. In the treadmilling filament model, each actin filament in the actin network simultaneously adds subunits at its barbed end and releases subunits from its pointed end, thus continuously reproducing itself by balanced growth and disassembly. Treadmilling of individual filaments collectively results in the treadmilling of the lamellipodial network. In contrast, in the array treadmilling model, an individual filament does not treadmill, but rather first grows at the barbed end and later shrinks at the pointed end. However, the actin filament array as a whole treadmills, reproducing itself at the cell front and dismantling itself at the lamellipodial rear. Formation of new filaments occurs by Arp2/3-mediated nucleation, within a narrow zone at the leading edge, the dendritic actin brush. Newborn filaments become incorporated immediately into the actin array as branches of pre-existing filaments. Some of these nascent filaments continue to grow and branch, whereas others are predicted to be capped after a short period of elongation to prevent exponential increase in filament mass. Thus, in contrast to the individual filament treadmilling model which is characterized by low nucleation frequency and extensive filament growth, the array treadmilling model is characterized by high frequency of nucleation and limited filament growth. In the array treadmilling model, a debranching reaction must exist to balance the branching reaction. The dendritic brush assembled at the leading edge is protected from depolymerization due to pointed-end capping activity of the Arp2/3 complex. Disassembly of actin filaments is favored farther away from the leading edge through abrogation of the protection mechanism. Debranching of filaments would result from release of Arp2/3 complex from Y-junctions. Subsequently, depolymerization would result from ADF/cofilin-mediated dissociation of actin subunits from their pointed ends. Thus, the life cycle of an actin filament would consist of steps of nucleation with pointed-end capping, elongation, barbed-end capping, pointed-end uncapping, and disassembly. Depending on the rate constants and probabilities of the individual steps, an actin filament could undergo repeated reactions of capping and uncapping, growth and shortening, branching and debranching. Alternatively, it could arise as a new branch and undergo a single episode of growth balanced by a single episode of shortening at a later time. In contrast to the stochastic life cycle of an individual filament, the array, consisting of a large number of filaments, on average would add polymer continuously at its leading edge and disassemble polymer continuously toward the rear, resulting in a uniform treadmilling of the array as a whole.
From the functional point of view, a dendritic brush of actin filaments at the leading edge of locomoting cells seems well designed for lamellipodial protrusion. First, the brush naturally can support massive actin polymerization because of the presence of numerous nucleating sites and the level of polymerization, in principle, can be readily controlled by regulation of the activity of the Arp2/3 complex and/or capping protein. Second, the brush is mechanically organized for efficient polymerization-driven force generation because of its high filament density, extensive cross-linking of actin filaments, and the angular orientation of actin filaments (Mogilner and Oster, 1996). Finally, the brush is dynamically regulated so as to generate polymerization-driven protrusion by an array treadmilling mechanism which may play a role in the persistence of lamellipodial protrusion and in the ability to adapt to change in direction.
Acknowledgments
We thank Dr. Vladimir Rodionov for providing the Xenopus fibroblast cell line, Drs. Matt Welch, Laura Machesky, Jody Rosenblatt, James Bamburg, John Hartwig, and Tom Stossel for generous gifts of antibodies, John Peloquin for help with immunoblot analysis, Dr. Alexander Verkhovsky for constructive discussions and critical reading of this manuscript, Steve Limbach for excellent light and electron microscope support, and Zoya Svitkina for assistance in analyzing live sequences.
This work was supported by the American Cancer Society grant CB-95 and the National Institutes of Health grant GM 25062.
Abbreviations used in this paper
- ADF
actin depolymerizing factor
- CD
cytochalasin D
- LA
latrunculin A
- PEG
polyethylene glycol
- XAC
Xenopus ADF/cofilin
References
- Abe H, Obinata T, Minamide LS, Bamburg JR. Xenopus laevisactin-depolymerizing factor/cofilin: a phosphorylation-regulated protein essential for development. J Cell Biol. 1996;132:871–885. doi: 10.1083/jcb.132.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KI, Wang Y-L, Small JV. Coordination of protrusion and translocation of the keratocyte involves rolling of the cell body. J Cell Biol. 1996;134:1209–1218. doi: 10.1083/jcb.134.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayscough K. Use of latrunculin-A, an actin monomer-binding drug. Methods Enzymol. 1998;298:18–25. doi: 10.1016/s0076-6879(98)98004-1. [DOI] [PubMed] [Google Scholar]
- Ballestrem C, Wehrle-Haller B, Imhof BA. Actin dynamics in living mammalian cells. J Cell Sci. 1998;111:1649–1658. doi: 10.1242/jcs.111.12.1649. [DOI] [PubMed] [Google Scholar]
- Bamburg JR, Bray D. Distribution and cellular localization of actin depolymerizing factor. J Cell Biol. 1987;105:2817–2825. doi: 10.1083/jcb.105.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkalow K, Witke W, Kwiatkowski DJ, Hartwig JH. Coordinated regulation of platelet actin filament barbed ends by gelsolin and capping protein. J Cell Biol. 1996;134:389–399. doi: 10.1083/jcb.134.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereiter-Hahn J, Vöth M. Ionic control of locomotion and shape of epithelial cells. II. Role of monovalent cations. Cell Motil Cytoskel. 1988;10:528–536. doi: 10.1002/cm.970100409. [DOI] [PubMed] [Google Scholar]
- Brown SS, Spudich JA. Mechanism of action of cytochalasin: evidence that it binds to actin filament ends. J Cell Biol. 1981;88:487–491. doi: 10.1083/jcb.88.3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano ML, Lauffenburger DA, Zigmond SH. Kinetic analysis of F-actin depolymerization in polymorphonuclear leukocyte lysates indicates that chemoattractant stimulation increases actin filament number without altering the filament length distribution. J Cell Biol. 1991;115:677–687. doi: 10.1083/jcb.115.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao LG, Fishkind DJ, Wang YL. Localization and dynamics of nonfilamentous actin in cultured cells. J Cell Biol. 1993;123:173–181. doi: 10.1083/jcb.123.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier M-F. Control of actin dynamics. Curr Opin Cell Biol. 1998;10:45–51. doi: 10.1016/s0955-0674(98)80085-9. [DOI] [PubMed] [Google Scholar]
- Carlier M-F, Pantaloni D. Control of actin dynamics in cell motility. J Mol Biol. 1997;269:459–467. doi: 10.1006/jmbi.1997.1062. [DOI] [PubMed] [Google Scholar]
- Carlier M-F, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AY, Raft S, Bailly M, Wyckoff JB, Segall JE, Condeelis JS. EGF stimulates an increase in actin nucleation and filament number at the leading edge of the lamellipod in mammary adenocarcinoma cells. J Cell Sci. 1998;111:199–211. doi: 10.1242/jcs.111.2.199. [DOI] [PubMed] [Google Scholar]
- Condeelis J. Life at the leading edge: the formation of cell protrusions. Annu Rev Cell Biol. 1993;9:411–444. doi: 10.1146/annurev.cb.09.110193.002211. [DOI] [PubMed] [Google Scholar]
- Daniolos A, Lerner AB, Lerner MR. Action of light on frog pigment cells in culture. Pigm Cell Res. 1990;3:38–43. doi: 10.1111/j.1600-0749.1990.tb00260.x. [DOI] [PubMed] [Google Scholar]
- Eddy RJ, Han J, Condeelis JS. Capping protein terminates but does not initiate chemoattractant-induced actin assembly in Dictyostelium. . J Cell Biol. 1997;139:1243–1253. doi: 10.1083/jcb.139.5.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddette DW, Frieden C. Actin polymerization. The mechanism of action of cytochalasin D. J Biol Chem. 1986;261:15974–15980. [PubMed] [Google Scholar]
- Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ, Hartwig JH. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J Cell Biol. 1990;111:1089–1105. doi: 10.1083/jcb.111.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig JH. Mechanisms of actin rearrangements mediating platelet activation. J Cell Biol. 1992;118:1421–1442. doi: 10.1083/jcb.118.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig JH, Bokoch GM, Carpenter CL, Janmey PA, Taylor LA, Toker A, Stossel TP. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell. 1995;82:643–653. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- Higley S, Way M. Actin and cell pathogenesis. Curr Opin Cell Biol. 1997;9:62–69. doi: 10.1016/s0955-0674(97)80153-6. [DOI] [PubMed] [Google Scholar]
- Kelleher JF, Atkinson SJ, Pollard TD. Sequences, structural models, and cellular localization of the actin-related proteins Arp2 and Arp3 from Acanthamoeba. . J Cell Biol. 1995;131:385–397. doi: 10.1083/jcb.131.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent V, Loisel TP, Harbeck B, Wehman A, Grobe L, Jockusch BM, Wehland J, Gertler FB, Carlier M-F. Role of proteins of the Ena/ VASP family in actin-based motility of Listeria monocytogenes. . J Cell Biol. 1999;144:1245–1258. doi: 10.1083/jcb.144.6.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM. Cell motility: complex dynamics at the leading edge. Curr Biol. 1997;7:R164–R167. doi: 10.1016/s0960-9822(97)70079-4. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Way M. Actin branches out. Nature. 1998;394:125–126. doi: 10.1038/28039. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Gould KL. The Arp2/3 complex: a multifunctional actin organizer. Curr Opin Cell Biol. 1999;11:117–121. doi: 10.1016/s0955-0674(99)80014-3. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Atkinson SJ, Ampe C, Vandekerckhove J, Pollard TD. Purification of a cortical complex containing two unconventional actins from Acanthamoebaby affinity chromatography on profilin-agarose. J Cell Biol. 1994;127:107–115. doi: 10.1083/jcb.127.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM, Reeves E, Wientjes F, Mattheyse FJ, Grogan A, Totty NF, Burlingame AL, Hsuan JJ, Segal AW. Mammalian actin-related protein 2/3 complex localizes to regions of lamellipodial protrusion and is composed of evolutionarily conserved proteins. Biochem J. 1997;328:105–112. doi: 10.1042/bj3280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM, Mullins RD, Higgs HN, Kaiser DA, Blanchoin L, May RC, Hall ME, Pollard TD. Scar, a WASP-related protein, activates dendritic nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci USA. 1999;96:3739–3744. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciver SK. How ADF/cofilin depolymerizes actin filaments. Curr Opin Cell Biol. 1998;10:140–144. doi: 10.1016/s0955-0674(98)80097-5. [DOI] [PubMed] [Google Scholar]
- Maciver SK, Weeds AG. Actophorin preferentially binds monomeric ADP-actin over ATP-bound actin: consequences for cell locomotion. FEBS Lett. 1994;347:251–256. doi: 10.1016/0014-5793(94)00552-4. [DOI] [PubMed] [Google Scholar]
- Maciver SK, Zot HG, Pollard TD. Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. . J Cell Biol. 1991;115:1611–1620. doi: 10.1083/jcb.115.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciver SK, Pope BJ, Whytock S, Weeds AG. The effect of two actin depolymerizing factors (ADF/cofilins) on actin filament turnover: pH sensitivity of F-actin binding by human ADF, but not of Acanthamoebaactophorin. Eur J Biochem. 1998;256:388–397. doi: 10.1046/j.1432-1327.1998.2560388.x. [DOI] [PubMed] [Google Scholar]
- Matsudaira P. Actin cross-linking proteins at the leading edge. Semin Cell Biol. 1994;5:165–174. doi: 10.1006/scel.1994.1021. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Ono S, Minamide LS, Takahashi M, Bamburg JR. Actin depolymerizing factor and cofilin phosphorylation dynamics: response to signals that regulate neurite extension. Cell Motil Cytoskel. 1998;39:172–190. doi: 10.1002/(SICI)1097-0169(1998)39:2<172::AID-CM8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Mogilner A, Oster G. Cell motility driven by actin polymerization. Biophys J. 1996;71:3030–3045. doi: 10.1016/S0006-3495(96)79496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon A, Drubin DG. The ADF/cofilin proteins: stimulus-responsive modulators of actin dynamics. Mol Biol Cell. 1995;6:1423–1431. doi: 10.1091/mbc.6.11.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RD, Stafford WF, Pollard TD. Structure, subunit topology, and actin-binding activity of the Arp2/3 complex from Acanthamoeba. . J Cell Biol. 1997;136:331–343. doi: 10.1083/jcb.136.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop, P.D., and J. Faber. 1956. Normal Tables of Xenopus laevis (Daudin). A Systematical and Chronological Survey of the Development from Fertilized Egg Till the End of Metamorphosis. North Holland Publishing Co., Amsterdam.
- Podolski JL, Steck TL. Length distribution of F-actin in Dictyostelium discoideum. . J Biol Chem. 1990;265:1312–1318. [PubMed] [Google Scholar]
- Pollard TD. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J Cell Biol. 1986;103:2747–2754. doi: 10.1083/jcb.103.6.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Mitchison TJ. Actin, cofilin and cognition. Nature. 1998;393:739–740. doi: 10.1038/31597. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Agnew BJ, Abe H, Bamburg JR, Mitchison TJ. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the turnover of actin filaments in Listeria monocytogenestails. J Cell Biol. 1997;136:1323–1332. doi: 10.1083/jcb.136.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath P, Pollard TD. Effects of cytochalasin, phalloidin, and pH on the elongation of actin filaments. Biochemistry. 1991;30:1973–1980. doi: 10.1021/bi00221a034. [DOI] [PubMed] [Google Scholar]
- Schafer DA, Jennings PB, Cooper JA. Dynamics of capping protein and actin assembly in vitro: uncapping barbed ends by polyphosphoinositides. J Cell Biol. 1996;135:169–179. doi: 10.1083/jcb.135.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DA, Welch MD, Machesky LM, Bridgman PC, Meyer SM, Cooper JA. Visualization and molecular analysis of actin assembly in living cells. J Cell Biol. 1998;143:1919–1930. doi: 10.1083/jcb.143.7.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small JV. The actin cytoskeleton. Electron Microsc Rev. 1988;1:155–174. doi: 10.1016/s0892-0354(98)90010-7. [DOI] [PubMed] [Google Scholar]
- Small JV. Lamellipodia architecture: actin filament turnover and the lateral flow of actin filaments during motility. Semin Cell Biol. 1994;5:157–163. doi: 10.1006/scel.1994.1020. [DOI] [PubMed] [Google Scholar]
- Small JV. Getting the actin filaments straight: nucleation-release or treadmilling? . Trends Cell Biol. 1995;5:52–55. doi: 10.1016/s0962-8924(00)88939-4. [DOI] [PubMed] [Google Scholar]
- Small JV, Herzog M, Anderson K. Actin filament organization in the fish keratocyte lamellipodium. J Cell Biol. 1995;129:1275–1286. doi: 10.1083/jcb.129.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods Enzymol. 1998;298:570–592. doi: 10.1016/s0076-6879(98)98045-4. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, Borisy GG. Improved procedures for electron microscopic visualization of the cytoskeleton of cultured cells. J Struct Biol. 1995;115:290–303. doi: 10.1006/jsbi.1995.1054. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, Borisy GG. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol. 1996;135:991–1007. doi: 10.1083/jcb.135.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, McQuade KM, Borisy GG. Analysis of the actin-myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J Cell Biol. 1997;139:397–415. doi: 10.1083/jcb.139.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons MH, Mitchison TJ. Control of actin polymerization in live and permeabilized fibroblasts. J Cell Biol. 1991;114:503–513. doi: 10.1083/jcb.114.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot JA. Accelerating on a treadmill: ADF/cofilin promotes rapid actin filament turnover in the dynamic cytoskeleton. J Cell Biol. 1997;136:1165–1168. doi: 10.1083/jcb.136.6.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ. Actin microfilament dynamics in locomoting cells. Nature. 1991;352:126–131. doi: 10.1038/352126a0. [DOI] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ. Comparison of actin and cell surface dynamics in motile fibroblasts. J Cell Biol. 1992;119:367–377. doi: 10.1083/jcb.119.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovsky AB, Svitkina TM, Borisy GG. Myosin II filament assemblies in the active lamella of fibroblasts: their morphogenesis and role in the formation of actin filament bundles. J Cell Biol. 1995;131:989–1002. doi: 10.1083/jcb.131.4.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner A. A head to tail polymerization of actin. J Mol Biol. 1976;108:139–150. doi: 10.1016/s0022-2836(76)80100-3. [DOI] [PubMed] [Google Scholar]
- Welch MD, Iwamatsu A, Mitchison TJ. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. . Nature. 1997a;385:265–269. doi: 10.1038/385265a0. [DOI] [PubMed] [Google Scholar]
- Welch MD, Mallavarapu A, Rosenblatt J, Mitchison TJ. Actin dynamics in vivo. Curr Opin Cell Biol. 1997b;9:54–61. doi: 10.1016/s0955-0674(97)80152-4. [DOI] [PubMed] [Google Scholar]
- Welch MD, DePace AH, Verma S, Iwamatsu A, Mitchison TJ. The human Arp2/3 complex is composed of evolutionarily conserved subunits and is localized to cellular regions of dynamic actin filament assembly. J Cell Biol. 1997c;138:375–384. doi: 10.1083/jcb.138.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch MD, Rosenblatt J, Skoble J, Portnoy DA, Mitchison TJ. Interaction of human Arp2/3 complex and the Listeria monocytogenesActA protein in actin filament nucleation. Science. 1998;281:105–108. doi: 10.1126/science.281.5373.105. [DOI] [PubMed] [Google Scholar]
- Yonezawa N, Nishida E, Koyasu S, Maekawa S, Ohta Y, Yahara I, Sakai H. Distribution among tissues and intracellular localization of cofilin, a 21 kDa actin-binding protein. Cell Struct Funct. 1987;12:443–452. doi: 10.1247/csf.12.443. [DOI] [PubMed] [Google Scholar]
- Zigmond SH. Signal transduction and actin filament organization. Curr Opin Cell Biol. 1996;8:66–73. doi: 10.1016/s0955-0674(96)80050-0. [DOI] [PubMed] [Google Scholar]
- Zigmond SH. Actin cytoskeleton: the Arp2/3 complex gets to the point. Curr Biol. 1998;8:R654–R657. doi: 10.1016/s0960-9822(07)00415-0. [DOI] [PubMed] [Google Scholar]
- Zigmond SH, Joyce M, Yang C, Brown K, Huang M, Pring M. Mechanism of Cdc42-induced actin polymerization in neutrophil extracts. J Cell Biol. 1998;142:1001–1012. doi: 10.1083/jcb.142.4.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]