

Abstract
Deletion of the synapsin I genes, encoding one of the major groups of proteins on synaptic vesicles, in mice causes late onset epileptic seizures and enhanced experimental temporal lobe epilepsy. However, mice lacking synapsin I maintain normal excitatory synaptic transmission and modulation but for an enhancement of paired-pulse facilitation. To elucidate the cellular basis for epilepsy in mutants, we examined whether the inhibitory synapses in the hippocampus from mutant mice are intact by electrophysiological and morphological means. In the cultured hippocampal synapses from mutant mice, repeated application of a hypertonic solution significantly suppressed the subsequent transmitter release, associated with an accelerated vesicle replenishing time at the inhibitory synapses, compared with the excitatory synapses. In the mutants, morphologically identifiable synaptic vesicles failed to accumulate after application of a hypertonic solution at the inhibitory preterminals but not at the excitatory preterminals. In the CA3 pyramidal cells in hippocampal slices from mutant mice, inhibitory postsynaptic currents evoked by direct electrical stimulation of the interneuron in the striatum oriens were characterized by reduced quantal content compared with those in wild type. We conclude that synapsin I contributes to the anchoring of synaptic vesicles, thereby minimizing transmitter depletion at the inhibitory synapses. This may explain, at least in part, the epileptic seizures occurring in the synapsin I mutant mice.
Keywords: inhibitory, synapse, transmission, synapsin I, epilepsy
Synapsins are the alternatively spliced products of three genes (synapsin I, II, and III), and comprise >10% of synaptic vesicle proteins in mammals (Südhof et al., 1989; Hosaka and Südhof, 1998; Kao et al., 1998). They have been identified in both vertebrates (Südhof et al., 1989; Kao et al., 1998) and invertebrates (Klagges et al., 1996; Hilfiker et al., 1998), and are regarded as the anchor proteins that cross-link synaptic vesicles to each other and to the cytoskeletal meshwork at the presynaptic nerve terminals (Llinas et al., 1985, 1991; Hirokawa et al., 1989; Harada et al., 1990; Benfenati et al., 1992; Torri Tarelli et al., 1992; Hayashi et al., 1994; Ceccaldi et al., 1995; Pieribone et al., 1995). As these interactions are known to be regulated by calcium-calmodulin kinase II in the case of synapsin I, this protein has been believed to play an important role in presynaptic vesicle turnover during synaptic transmission (Llinas et al., 1985, 1991; Hirokawa et al., 1989; Harada et al., 1990; Benfenati et al., 1992; Torri Tarelli et al., 1992; Hayashi et al., 1994; Ceccaldi et al., 1995; Pieribone et al., 1995).
Contrary to our expectations, several previous studies showed that synapsin mutant mice survive without any appreciable defect in the macroscopic morphology of the brain or general behavior (Rosahl et al., 1993, 1995; Li et al., 1995; Takei et al., 1995). Although several perturbations in electrophysiological responses that are difficult to interpret occur (e.g., synapsin II mutant homozygotes and mice lacking both synapsin I and II show normal paired-pulse facilitation but lower posttetanic potentiation; in contrast, synapsin I mutant hemizygotes show an enhancement of paired-pulse facilitation but normal posttetanic potentiation), all these mutant mice show apparently normal long term potentiation (Rosahl et al., 1993, 1995; Li et al., 1995; Spillane et al., 1995; Takei et al., 1995). In the case of synapsin I mutant mice, behavioral analyses did not reveal any learning deficits (Silva et al., 1996). The only apparent abnormality was late onset epileptic seizures and enhanced stimulation-evoked epileptic seizures (experimental temporal lobe epilepsy) that were noted as the mice grew (Rosahl et al., 1993, 1995; Li et al., 1995; Takei et al., 1995). Thus, the mechanism of action of synapsin I in vivo still remains under debate.
To clarify a possible link between synapsin I deficiency and the epileptic phenotype, we examined whether the inhibitory synapses in the hippocampus in mutant mice are intact by electrophysiological and morphological analyses. Repeated application of a hypertonic solution to cultured hippocampal synapses in mutant mice significantly suppressed the subsequent transmitter release, associated with an accelerated vesicle replenishing time (Stevens and Tsujimoto, 1995) at the inhibitory synapses, but not at the excitatory synapses. Synaptic vesicles failed to accumulate after application of a hypertonic solution at the inhibitory preterminals, whereas their accumulation at the excitatory preterminals was unaffected. In the CA3 pyramidal cells in hippocampal slices (Edwards et al., 1989) from the mutant mice, inhibitory postsynaptic currents (IPSCs)1 evoked by the stimulation of an interneuron in the striatum oriens were characterized by a reduced quantal content compared with those in wild-type mice. Our results indicate that synapsin I at the inhibitory synapses plays an important role in minimizing transmitter depletion by anchoring synaptic vesicles. Impairment of inhibitory synaptic transmission may explain, at least in part, the epileptic phenotype in the synapsin I mutant mice.
Materials and Methods
Animals
Male hemizygotes and wild-type littermates derived from the crossing (>3 generations) of heterozygous synapsin I 129/Sv/C57Bl/6 mice were used for this study. The animals were housed under a 12-h light/12-h dark cycle with free access to water and food. All animal procedures were approved by the Institutional Animal Care and Use Committee.
Electrophysiological Procedures
The electrophysiological and culture techniques used have been described earlier (Edwards et al., 1989; Goslin and Banker, 1991; Stevens and Tsujimoto, 1995). Data acquisition and analyses were performed without knowledge of the genotype of the mouse under study. In brief, low density primary cultures of hippocampal neurons were prepared from 16.5-d-old embryonic mice and used for the electrophysiological experiments at 8 d in vitro (Goslin and Banker, 1991). All the electrophysiological experiments were performed at room temperature (24–28°C). Whole-cell currents were recorded from pyramidal cells at the holding potential of 0 mV for IPSCs, or −70 mV for excitatory postsynaptic currents (EPSCs). The bathing solution contained 137 mM NaCl, 3.5 mM KCl, 10 mM Hepes, 10 mM glucose, 0.7 mM CaCl2, 2 mM MgCl2, 10−4 mM tetrodotoxin (TTX), and 0.5 × 10−3 mM strychnine, pH 7.2. 2 mM kynurenic acid was added for recording IPSCs and 0.1 mM picrotoxin for EPSCs. A small region of the dendritic tree was superfused with a hypertonic solution (800 mosM with sucrose) of the same aforementioned ionic composition except for the presence of TTX at the concentration of 10−3 mM. The pipette solution contained the following: 122.5 cesium gluconate, 17.5 mM CsCl, 8 mM NaCl, 10 mM Hepes, 5 mM EGTA, 2 mM Mg ATP, and 0.3 mM Na3GTP, pH 7.2, for IPSC recordings, and 120 mM potassium gluconate, 12 mM KCl, 5 mM NaCl, 2 mM MgCl2, 1 mM CaCl2, 10 mM Hepes, 5 mM EGTA, and 2 mM Na2ATP, pH 7.2, for EPSC recordings.
We determined the parameters of vesicle turnover according to a previously described model (Stevens and Tsujimoto, 1995). The model assumes two pools of synaptic vesicles: the readily releasable pool and the infinite reservoir pool. The vesicle turnover rate of the readily releasable pool at time t can be described as,
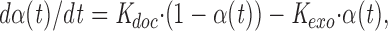 |
1 |
where α(t) designates the probability of the synaptic vesicle existence at a docking site in the readily releasable pool at time t (0 < α(t) < 1, α(0) ∼1), K doc, the rate of replenishing one vesicle from the infinite reservoir pool to a docking site, and K exo, the rate of releasing one vesicle for transmitter release from a docking site.
Hence,
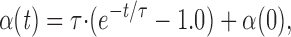 |
2 |
where
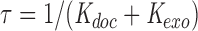 |
and
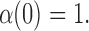 |
The cumulative number of events of miniature EPSCs or IPSCs (mEPSCs of mIPSCs) at time t (C(t)) can be described as,
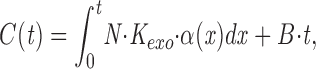 |
3 |
where B represents the background frequency of events derived from unperfused synapses, N is the total number of docking sites at synapses in a small dendritic region superfused with the hypertonic solution (800 mosM). K doc, K exo, and N can be determined by fitting the time course (0– 2.5 s) of cumulative mIPSCs or mEPSCs during depletion with equation 3.
Transverse hippocampal slices (300-μm-thick) were prepared from mice (10–14-d-old) after decapitation under halothane anesthesia (Edwards et al., 1989). Slices were maintained in an incubation chamber for 1 h at 37°C and at room temperature thereafter. The external solution contained the following: 119 mM NaCl, 2.5 mM KCl, 1.0 mM NaH2PO4, 26.2 mM NaHCO3, 11 mM glucose, 2.0 mM CaCl2, 1.0 mM MgCl2, 0.5 × 10−3 mM strychnine, 2 mM kynurenic acid, pH 7.2, equilibrated with 95% O2: 5% CO2. A stimulating electrode filled with the external solution was placed close to an interneuron in the striatum oriens of CA3. IPSCs evoked by short (100 μs) current pulses (3–10 V) at 0.05 Hz were recorded from a CA3 pyramidal neuron by whole-cell recording at a holding potential of 0 mV. The IPSCs were blocked reversibly by bicuculline (10 μM), indicating that they were mediated by γ-aminobutyric acid (GABA)- gated Cl− channels. Whole-cell currents were recorded by a patch-clamp amplifier (EPC-7; List), filtered at 5 kHz, and digitized at 10 kHz for further analysis.
Immunoelectron Microscopy and Morphometric Analysis
Cultured neurons were incubated in medium containing 10−3 mM TTX for 5 min, stimulated with the above mentioned 800-mosM hypertonic solution for 10 s, washed out with medium for 1 min, and fixed with 0.1 M cacodylate buffer, pH 7.2, containing freshly prepared 4% paraformaldehyde and 0.1% glutaraldehyde for 15 min. The samples were quenched with 100 mM glycine for 5 min, permeabilized, and blocked in 0.5% BSA, 0.1% gelatin, 0.05% Tween 20, 20 μM digitonin, 1% skim milk, and 500 mM NaCl for 30 min; both solutions were prepared in PBS, pH 7.2. Subsequently, they were incubated for 1 h with a polyclonal anti-GABA antibody (diluted 1:450 with the above mentioned blocking solution) and blocked again for 30 min.
Up to here all procedures were carried out at 37°C, thereafter the samples were incubated at room temperature for 10 min with 10 μg/ml biotin-labeled goat anti–rabbit IgG antibody (Nichirei), treated with 100 μg/ml HRP-labeled streptavidin (Nichirei) for 5 min, and processed for DAB staining. Subsequently, they were postfixed by dipping in 1% osmium tetroxide in 0.1 M cacodylate buffer, pH 7.2, for 10 min on ice, stained with 1% uranyl acetate for 1 h at room temperature, dehydrated in a graded series of ethanol concentrations, and embedded in Quetol-812 (Nisshin EM). Ultrathin sections were cut on a conventional ultramicrotome (Ultratome Nova; LKB Bromma), stained with uranyl acetate and lead citrate, and examined and photographed under a transmission electron microscope (2000EX; JEOL) at an accelerating voltage of 80 kV. The number of synaptic vesicles in presynaptic nerve terminals was determined by direct counting from the micrographs. These numbers were converted into the number of vesicles per unit area (μm2) by dividing the vesicle count by the presynaptic nerve terminal area as described before (Takei et al., 1995).
Results
Repeated Application of a Hypertonic Solution Suppressed the Subsequent Transmitter Release at the Cultured Inhibitory Synapses of Mutants
We tested the possibility of the inhibitory synapses becoming easily fatigued and recovering slowly in synapsin mutants. Spontaneous mIPSCs were recorded from cultured hippocampal neurons in the presence of 1 μM TTX, and a hypertonic sucrose solution (800 mosM) was applied locally to a neurite for 5 s, followed by application for 2 s every 5 min to monitor the size of the readily releasable pool of synaptic vesicles. Most of the readily releasable vesicles were released during the first 2 s of application, and the size of released quanta measured by second 2 s applications recovered in parallel with the amplitude of the evoked EPSCs (Rosenmund and Stevens, 1996). Assuming that the replenishing time constant of the readily releasable pool is ∼ 10 s, the 5-min intervals would be expected to minimize the effect of replenishment. When the hypertonic solution was applied for 5 s, the frequency of mIPSCs declined exponentially (Figs. 1 a and 2 a), presumably because of a depletion of available transmitter quanta or synaptic vesicles (Stevens and Tsujimoto, 1995). After a 5-min interval, the second application of hypertonic solution (2 s) caused a similar magnitude of increase in the frequency of mIPSCs in cells from the wild-type mice, whereas a much smaller increase was noted in the cells from synapsin I mutant mice (Fig. 1 a). The total amount of quanta released during the first 2 s of hypertonic application in the second sucrose application was 43 ± 14% (n = 6) in the case of mutant mice, whereas it remained at 97 ± 5% (n = 5) in the case of wild-type mice (Fig. 1 b). Subsequent applications of the hypertonic solution at 5-min intervals did not further reduce the amount of quantal release, either in the mutant or in wild-type mice. In contrast to the result at inhibitory synapses, no such depression was observed at the excitatory synapses in the mutant mice (95 ± 8% at the second application, n = 6) (Fig. 1 c).
Figure 1.

Impaired recovery of mIPSCs in cultured inhibitory synapses lacking synapsin I. (a) Sample traces of mIPSCs (middle) and their frequency (bottom). Hypertonic solution was applied for 5 s and thereafter for 2 s at intervals of 5 min, as shown by boxes (top). The left frame shows wild-type mice and the right one shows mutants. (b) The number of mIPSCs during 2 s relative to the first 2 of 5 s application of hypertonic solution in wild-type mice (open circles, n = 3) and mutant (filled circles, n = 6) mice. Bars represent SEM in this and the following figures. (c) Sample traces of mEPSCs and their frequency in the case of mutants after repeated applications of hypertonic solution.
Accelerated Vesicle Replenishing Time in Cultured Inhibitory Synapses from Mutants
We analyzed the changes in the kinetics of synaptic vesicle turnover. Paired-pulse application of the hypertonic solution at various intervals commonly is used to assess the recovery time following depletion. But in our case, repeated stimulation with the hypertonic solution suppressed the subsequent mIPSCs in mutants (Fig. 1) that suggested the possibility that either the replenishing time might be altered on the order of tens of minutes or that the vesicular recycling process, including endocytosis, might be severely retarded. If the latter were the case, it would not be appropriate to apply the paired superfusion method to inhibitory synapses in mutants. We applied the model described by Stevens and Tsujimoto (1995) to estimate the replenishing time constant after a single 5-s hypertonic application. Assuming an infinite reservoir pool and a readily releasable pool of synaptic vesicles, the vesicle turnover rate in the readily releasable pool can be described by a simple model equation (Stevens and Tsujimoto, 1995) (see Materials and Methods.). As shown in Fig. 2 a, because the mIPSCs in a mutant fail to respond to hypertonic stimulation after 3 s from the beginning of superfusion, we calculated the parameters for the synaptic vesicle turnover by equation 3 to the cumulative events of mIPSCs during 0–2.5 s of hypertonic application. (For these examples, the calculated replenishing time [1/K doc] was 10.1 s [wild type], 5.7 s [mutant]. The rate of releasing one vesicle from a docking site was 0.56 s−1 [wild type], 1.83 s−1 [mutant].) At 11 inhibitory synapses from 4 synapsin I–deficient mice and at 5 inhibitory synapses from 3 wild-type mice, the mean time to replenish vacant release sites (1/K doc) was 5.5 ± 3.7 s and 10.7 ± 3.7 s, respectively (Fig. 2 b). Thus, the replenishing time of synaptic vesicles from a reservoir pool to a readily releasable site was reduced in synapsin I mutants (*P < 0.05, t test; the asterisk indicates that the related probability demonstrates statistical significance). In contrast, at the excitatory synapses, no significant difference was observed in the replenishing time between synapsin I–deficient mice (8.3 ± 2.3 s, n = 12) and wild-type mice (10.4 ± 2.6 s, n = 12, P > 0.5) (Fig. 2 b). The releasing rate from a readily releasable pool (K exo) of mIPSCs was higher by a mean of 36% for the mutants (1.5 ± 0.8 s−1, n = 11) compared with the wild-type mice (1.1 ± 0.5 s−1, n = 5). However, because of a large variance, the difference was not statistically significant (P = 0.18). The releasing rate from a readily releasable pool of mEPSCs was unaltered between mutants (1.2 ± 0.4 s−1, n = 12) and wild-type mice (0.9 ± 0.2 s−1, n = 12, P > 0.5) (Fig. 2 c). It is suggested that the accelerated replenishment from the reservoir pool to the readily releasable pool in mutant inhibitory presynaptic terminals accelerates the vesicular depletion of the reservoir pool, especially after rigorous hypertonic stimulation, at mutant inhibitory synapses.
Figure 2.

Accelerated replenishing rate of the readily releasable pool at the inhibitory presynaptic terminals in the case of mutant mice. (a) Sample traces (upper) and cumulative number (lower) of mIPSCs. Abscissa indicates the time before, during, and after local application of hypertonic solution as indicated by boxes (top) in wild-type (left) and synapsin I mutant (right) cells. Fitting curves were derived from Eq. 3. (b) The replenishing time (1/K doc, s) was significantly reduced in the mutant inhibitory synapses (*P < 0.05). No significant difference was observed at the excitatory synapses. (c) The rates of releasing one vesicle from a docking site (K exo, s−1) at both inhibitory and excitatory presynaptic terminals were unaltered.
Synaptic Vesicles Failed to Accumulate after Hypertonic Application at Inhibitory Preterminals in Mutants
To obtain morphological correlates, we performed electron microscopic analysis of the presynaptic terminals of hippocampal neurons in culture. From a morphological perspective, at the 8-d stage in vitro, the synapses of cultured hippocampal neurons were still not fully mature, although functionally, their mIPSCs or mEPSCs corresponded to that of fully mature synapses. The presynaptic terminals were smaller, the synapses often lacked postsynaptic densities, and synaptic vesicles seemed less apparent. However on immunoelectron microscopy using the anti-GABA antibody, we could visualize inhibitory presynaptic terminals because they were filled with fuzzy DAB staining, especially intense just around the synaptic vesicles (Fig. 3, a–d). Without hypertonic stimulation, there was no difference between the two genotypes with respect to synaptic vesicle densities at the inhibitory presynaptic terminals as identified by positive staining with anti-GABA antibody (Fig. 3, a and b). However, after rigorous hypertonic stimulation, a clear loss of synaptic vesicles was observed at the inhibitory presynaptic terminals in synapsin I mutant mice (Fig. 3 d) but not in the wild-type mice (Fig. 3 c). No such morphological difference was observed between the mutant and wild-type mice at the excitatory presynaptic terminals as identified by the absence of anti-GABA staining (Fig. 3, e and f).
Figure 3.
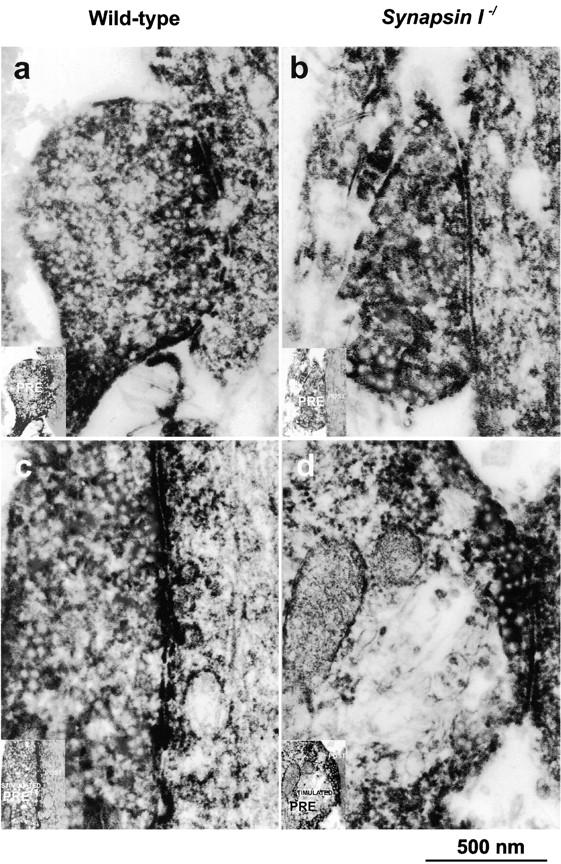
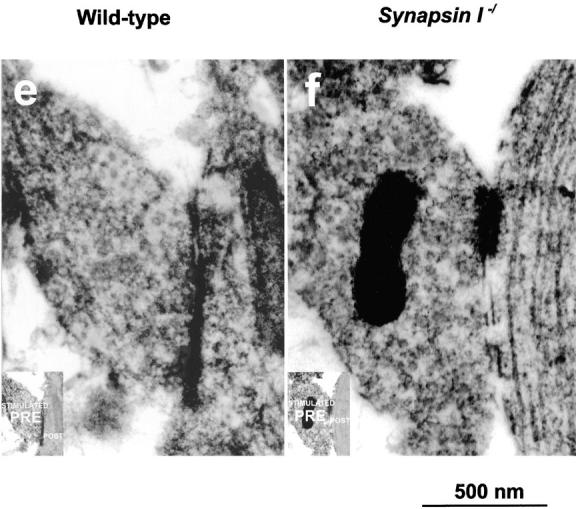
Immunoelectron microscopic analysis of presynaptic terminals of cultured hippocampal neurons. Frames a, c, and e are preparations from wild-type mice, whereas b, d, and f are from synapsin I–ablated mutants. Frames a and b show inhibitory presynaptic terminals before hypertonic application and c and d show inhibitory presynaptic terminals after hypertonic application. Frames e and f show excitatory presynaptic terminals after hypertonic application. Presynaptic terminals (a–f) are stained with anti-GABA antibody (see Materials and Methods). Note that retarded accumulation of synaptic vesicles after stimulation was noted only in inhibitory presynaptic terminals from mutant mice (d). Each inset shows the schematic representation of pre- and postsynaptic terminals. Bar, 500 nm.
We performed morphometrical analyses on the synaptic vesicle densities at the presynaptic terminals of each genotype and, as shown in Table I, the density was significantly reduced only in the stimulated inhibitory synapses from mutants (*P< 0.05, t test). To determine whether this reduction affects the distribution of synaptic vesicles, we further counted the number of synaptic vesicles within a distance of 200 nm from the synaptic clefts. This revealed that the vesicle density near the synaptic clefts was not statistically different between wild-type and mutant mice. Thus, the physiological and morphological results consistently suggest that the reservoir pool of inhibitory presynaptic terminals could be depleted after hypertonic stimulation in synapsin I–deficient mice.
Table I.
Morphometric Analysis of Presynaptic Terminals of Cultured Hippocampal Neurons before and after Hypertonic Stimulation
| Numbers of Synaptic Vesicles per Unit Area in Presynaptic Terminals | ||||||||
|---|---|---|---|---|---|---|---|---|
| Wild-type | Synapsin I−/ | |||||||
| Excitatory | Inhibitory | Excitory | Inhibitory | |||||
| Control | 233 ± 15 (n = 10) | 209 ± 60 (n = 14) | 225 ± 31 (n = 12) | 245 ± 47 (n = 10) | ||||
| Stimulated | 202 ± 29 (n = 12) | 176 ± 27 (n = 12) | 209 ± 20 (n = 8) | 139 ± 25* (n = 10) | ||||
| Numbers of Synaptic Vesicles per Unit Area within 200 nm from the Synaptic Cleft | ||||||||
| Wild-type | Synapsin I−/ | |||||||
| Excitatory | Inhibitory | Excitatory | Inhibitory | |||||
| Control | 341 ± 66 (n = 10) | 270 ± 76 (n = 14) | 310 ± 46 (n = 12) | 335 ± 28 (n = 10) | ||||
| Stimulated | 283 ± 45 (n = 12) | 259 ± 30 (n = 12) | 269 ± 50 (n = 8) | 304 ± 72 (n = 10) | ||||
Different from the value for wild-type at P < 0.05. The mean ± SD of the number of synaptic vesicles/μm2 (n represents the number of synapses examined) is shown. t test was used to determine the significance of the changes.
Inhibitory Postsynaptic Currents Characterized by Reduced Quantal Contents in CA3 Pyramidal Cells from Mutants
To assess the physiological role of synapsin I in synaptic transmission, we evoked IPSCs in pyramidal cells in the CA3 region of mouse hippocampal slices (Edwards et al., 1989) (Fig. 4). IPSCs in mutant mice showed a wider scatter in amplitude than those of wild-type mice (Fig. 4 a). Also, the mean amplitude of the evoked IPSCs was smaller in the case of mutants (159.0 ± 21.3 pA, n = 10) than in wild-type littermates (219.6 ± 31.1 pA, n = 9) (Fig. 4 b, *P < 0.05). The coefficient of variation estimated from the regression line for the data points in the relationship between mean amplitude and SD of IPSCs was larger in mutants (0.18 ± 0.02, n = 10) than in the wild-type mice (0.12 ± 0.02, n = 9) (Fig. 4 c, *P < 0.05). There was no significant difference between mutant and wild-type mice with respect to the rise time (10–90%) (wild-type, n = 9, 2.6 ± 0.3 ms; mutant, n = 10, 2.8 ± 0.3 ms) and biexponential decay time constants (wild-type, n = 9, 21.1 ± 0.9 ms and 39.9 ± 6.3 ms; mutant, n = 10, 22.5 ± 1.6 ms and 31.6 ± 3.3 ms) of evoked IPSCs.
Figure 4.
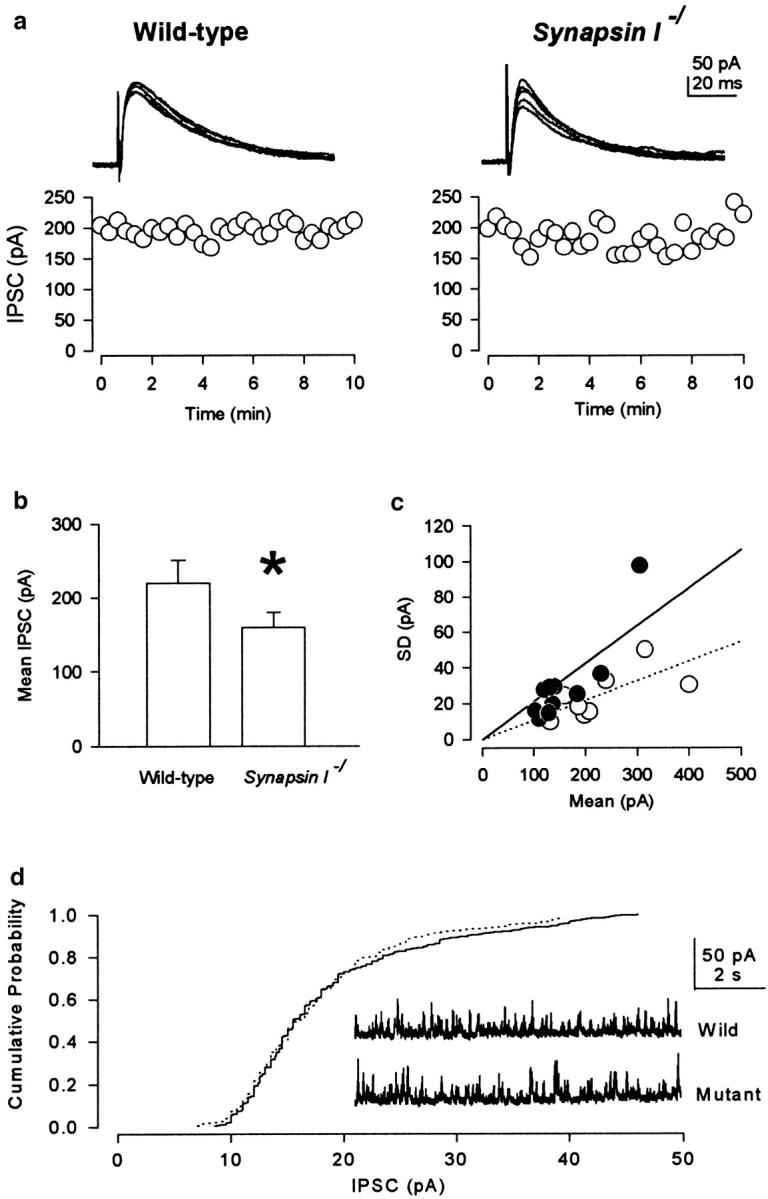
Evoked IPSCs from pyramidal cells in the hippocampal CA3 region. (a) Superimposed traces of evoked IPSCs at a holding potential of 0 mV (top). There was no significant difference between mutant and wild-type littermates with respect to the rise time (10– 90%; ms) and biexponential decay time constants (ms) of evoked IPSCs. Fluctuation in amplitude of the evoked IPSCs (bottom). (b) Smaller mean amplitude (pA) of evoked IPSCs in the case of mutants (*P < 0.05). (c) There is a larger coefficient of variation (SD/ Mean) in the case of mutants (filled circles and solid line), compared with the wild-type mice (open circles and dotted line) (*P < 0.05). Exclusion of extreme points from both mutant and wild-type cases gave a similar result (*P < 0.05). (d) Comparison of the cumulative probability distributions of the amplitude of mIPSCs from a wild-type cell (dotted line) and mutant cell (solid line). No significant difference in mIPSC amplitude (pA) was observed. The inset shows traces of mIPSCs from wild-type and mutant cells. The mIPSC amplitude distributions of the wild-type and mutant cells are not significantly different (Kolmogorov-Smirnov test; P > 0.1).
To examine whether the quantal size may be altered in synapsin I mutant mice, we recorded the spontaneous mIPSCs from hippocampal CA3 neurons in the presence of TTX (Fig. 4 d). No significant difference was observed in the amplitude of mIPSCs between the case of mutant (18.6 ± 3.7, n = 6) and wild-type mice (17.6 ± 4.0, n = 4). The mIPSC amplitude distributions of the wild-type and mutant cells are not significantly different (Kolmogorov-Smirnov test; P > 0.1). These results suggest that synapsin I deficiency reduces the inhibitory synaptic efficacy by reducing the quantal content (i.e., the number of synaptic vesicles released by a single presynaptic action potential).
Discussion
Synaptic vesicles normally are anchored to actin filaments by synapsins (Llinas et al., 1985, 1991; Hirokawa et al., 1989; Harada et al., 1990; Benfenati et al., 1992; Torri Tarelli et al., 1992; Hayashi et al., 1994; Ceccaldi et al., 1995; Pieribone et al., 1995). As the association and dissociation of synapsins to synaptic vesicles are regulated by protein kinases, it is believed that synapsins may play an important role in the regulation of synaptic transmission by the following mechanisms. First, synapsins may modulate the vesicular traffic from the reservoir pool to the readily releasable pool (predocking mechanism) (Llinas et al., 1985, 1991; Hirokawa et al., 1989; Harada et al., 1990; Benfenati et al., 1992; Torri Tarelli et al., 1992; Hayashi et al., 1994; Ceccaldi et al., 1995; Li et al., 1995; Pieribone et al., 1995; Takei et al., 1995). The fluorescence resonance energy transfer experiment revealed that the association and dissociation kinetics of synapsin I and synaptic vesicles is the same order of magnitude as the kinetics of synaptic vesicle recycling (Stefani et al., 1997). It has been reported that domain E of synapsins is responsible for maintaining the reservoir pool of synaptic vesicles (Pieribone et al., 1995; Hilfiker et al., 1998). In cultured synapses, the number of vesicles exocytosed during action potential trains and the total recycling vesicle pools are reduced in synapsin I knockout mice (Ryan et al., 1996). Also, synapsins have a binding activity to ATP and are predicted to transfer phosphate to an unidentified substrate that may suggest the possibility that synapsin I may be involved in the priming process (Esser et al., 1998; Hosaka and Südhof, 1998). Second, synapsins may inhibit synaptic vesicle fusion for exocytosis (Rosahl et al., 1995). Presynaptic injection of the synapsin domain E peptide reduced the size of EPSCs, accompanied by retarded kinetics of exocytosis (Hilfiker et al., 1998).
Recently, it has been postulated that the efficacy of neurotransmitter release may be regulated by the size of the readily releasable pool as supported by the results of the following experiments. The time courses of paired-pulse inhibition of action potentials and hypertonic solution– evoked release are correlated with each other at individual interpulse intervals when the exocytosis is evoked by action potentials followed by hypertonic solution application (Rosenmund and Stevens, 1996). The release probability as measured by the minimal stimulation technique is related directly to the size of the readily releasable pool as measured by repetitive nerve fiber stimulation (Dobrunz and Stevens, 1997). The replenishment of the readily releasable pool of giant presynaptic terminals in brainstem slices was accelerated by preceding high frequency action potentials in a calcium-dependent manner (Wang and Kaczmarek, 1998). The replenishing time of the readily releasable pool in our case was 10.4 ± 2.6 s (wild-type EPSC) and 10.7 ± 3.7 s (wild-type IPSC), comparable with values reported previously (12 s, Stevens and Tsujmoto, 1995; 8 s, von Gersdorff and Matthews, 1997). This variation may arise from variations in the intraterminal environment (e.g., the concentration of Ca2+ and protein kinases). Indeed, it is suggested that the replenishment process is accelerated by an elevation in the concentration of intracellular Ca2+ in hippocampal synapses in culture (Stevens and Wesseling, 1998), retinal bipolar cells (von Gersdorff and Matthews, 1997), and brainstem giant synapses (Wang and Kaczmarek, 1998). Activation of protein kinase C by phorbol ester also has been reported to reduce the replenishing time of the readily releasable pool (Stevens and Sullivan, 1998).
Our results in synapsin I mutants suggested a significant reduction in the replenishing time of inhibitory synaptic vesicles from the reservoir pool to a readily releasable site in synapsin I mutants. Synapsin I deficiency had no statistically significant (P = 0.18) effect on the rate of release from a readily releasable pool (K exo). Presumably, this was due to a large variance between the two populations, although the mean value was 36% higher in the case of mutants compared with the wild-type mice. Thus, our results are consistent with the proposed predocking mechanism of synapsin I in the inhibitory presynaptic terminals. However, we cannot exclude the possibility that synapsin I gene knockout may accelerate the release rate of synaptic vesicles during hypertonic superfusion.
Both synapsin I and II are contained in the excitatory mossy fiber terminals in the hippocampus, whereas the inhibitory terminals of cerebellar Purkinje cells lack synapsin IIa and express only a low level of synapsin IIb (Südhof et al., 1989). Also in the rat retina, glutamic acid decarboxylase-positive terminals lack synapsin II (Mandell et al., 1992). Although there is no direct evidence that hippocampal inhibitory synapses lack synapsin II, our results suggest at least that synapsin I plays a very specific role that cannot be compensated for by the presence of synapsin II. In addition, previous studies on synapsin mutants have shown that synapsin II knockouts, but not synapsin I knockouts, exhibit decreased posttetanic potentiation and severe synaptic depression on repetitive stimulation at the excitatory synapses (Rosahl et al., 1995). Considering together, we suppose that synapsin II may compensate for the absence of synapsin I (Ryan et al., 1996) at the excitatory synapses, whereas the deficiency of synapsin I exerted a more serious effect at the inhibitory synapses, presumably because of the poorer compensation.
During the baseline synaptic transmission, the apparent velocity to replenish the readily releasable pool is determined by the true replenishment velocity (the speed of transferring vesicles from the reservoir pool to the readily releasable pool), if the size of the reservoir pool is large enough. Alternatively, when the reservoir pool size reduces small enough because of the prolonged consumption of synaptic vesicles by repetitive stimulation, the apparent replenishing velocity will be limited by the velocity of refilling the reservoir pool. Indeed, our results indicated that the mutant inhibitory synapses in culture could not maintain the capability of subsequent transmitter release because of the exhaustion of reservoir pool after the first massive transmitter release. We consider that the size of reservoir vesicular pool is progressively reduced in the mutants in an activity-dependent manner, maybe by the accelerated exocytosis/replenishment, the impaired endocytosis, retarded incorporation of endocytosed vesicles into the reservoir pool, or slow uptake of neurotransmitter into the vesicles. At the equilibrated state, the exhausted reservoir pool can no longer support full replenishment of the readily releasable pool, causing reduction in amplitude of evoked IPSCs. If this is also the case in vivo, one could picture use-dependent changes in the reliability of synaptic transmission as follows. Briefly, in the mutant inhibitory synapse after a long enough quiescent period, synaptic vesicles are fully loaded to the reservoir pool and to the readily releasable pool. When bursts of action potentials reach the nerve terminal at this state, a larger amount of neurotransmitter (GABA) is released because of the accelerated replenishment of the readily releasable pool. This causes rapid exhaustion of the reservoir pool and the nerve terminal can no longer release GABA although the action potentials reach the terminal. This would be one of the underlying mechanisms for epilepsy observed in the mutant mice.
Primary epilepsy in humans is largely known to be unassociated with mental retardation (Murphy et al., 1995). Similarly, synapsin I mutants exhibiting epileptic seizures are normal in terms of conditioning and spatial learning (Silva et al., 1996), whereas most rodents with epileptic phenotypes are characterized by learning disabilities. In this respect, the synapsin I mutants may well be a model of epilepsy in humans. The cellular mechanisms underlying epilepsy are as follows: (1) an increased electrical excitability of neurons capable of firing at a high frequency (Kandel and Spencer, 1961; Wong and Prince, 1981); (2) frequency potentiation of excitatory synaptic transmission (Mallart and Martin, 1967); and (3) use-dependent depression of inhibitory synaptic transmission (Ben-Ari et al., 1979; McCarren and Alger, 1985) resulting in the removal of GABAergic inhibition. In an animal model of temporal lobe epilepsy, GABAergic inhibitory synaptic transmission from basket cells to CA1 pyramidal cells is known to be suppressed because of the reduced synaptic input to the inhibitory cells (Bekenstein and Lothman, 1993). Our results suggest that synaptic vesicle turnover at the inhibitory synaptic terminals can be altered by synapsin I deficiency. A subtle change in the turnover rate at inhibitory synaptic vesicles may reduce the inhibitory synaptic efficacy, thereby possibly causing epilepsy in synapsin I mutant mice. Any defect at the inhibitory synapses potentially can produce disintegration of the central nervous system such as epilepsy and in this respect, much attention must be paid to inhibitory synaptic transmission in various mutants with their molecules genetically ablated.
Acknowledgments
We thank H. Sato for her technical assistance in primary cell culture and H. Fukuda and M. Sugaya for their secretarial assistance.
This work was supported by a Grant-in-Aid for Center of Excellence Research from the Japan Ministry of Education, Science, Sports, and Culture to N. Hirokawa.
Abbreviations used in this paper
- EPSC
excitatory postsynaptic current;GABA, γ-aminobutyric acid
- IPSC
inhibitory postsynaptic current
- mEPSC
miniature EPSC
- mIPSC
miniature IPSC
- TTX
tetrodotoxin
Footnotes
S. Terada and T. Tsujimoto contributed equally to this work.
References
- Bekenstein JW, Lothman EW. Dormancy of inhibitory interneurons in a model of temporal lobe epilepsy. Science. 1993;259:97–100. doi: 10.1126/science.8093417. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Krnjevic K, Reinhardt W. Hippocampal seizures and failure of inhibition. Can J Physiol Pharmacol. 1979;57:1462–1466. [Google Scholar]
- Benfenati F, Valtorta F, Rubenstein JL, Gorelick FS, Greengard P, Czernik AJ. Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature. 1992;359:417–420. doi: 10.1038/359417a0. [DOI] [PubMed] [Google Scholar]
- Ceccaldi PE, Grohovaz F, Benfenati F, Chieregatti E, Greengard P, Valtorta F. Dephosphorylated synapsin I anchors synaptic vesicles to actin cytoskeleton: an analysis by videomicroscopy. J Cell Biol. 1995;128:905–912. doi: 10.1083/jcb.128.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recordings from neurones of the mammalian central nervous system. Pflügers Archiv. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- Esser L, Wang CR, Hosaka M, Smagula CS, Südhof TC, Deisenhofer J. Synapsin I is structurally similar to ATP-utilizing enzymes. EMBO (Eur Mol Biol Organ) J. 1998;17:977–984. doi: 10.1093/emboj/17.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin, K., and G. Banker. 1991. Rat hippocampal neurons in low-density culture. In Culturing Nerve Cells. G. Banker and K. Goslin, editors. MIT Press, Cambridge, MA. 251–281.
- Harada A, Sobue K, Hirokawa N. Developmental changes of synapsin I subcellular localization in rat cerebellar neurons. Cell Struct Funct. 1990;15:329–342. doi: 10.1247/csf.15.329. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Soulie F, Nakata T, Hirokawa N. Redistribution of synapsin I and synaptophysin in response to electrical stimulation in the rat neurohypophysial nerve endings. Cell Struct Funct. 1994;19:253–262. doi: 10.1247/csf.19.253. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Schweizer FE, Kao H-T, Czernik AJ, Greengard P, Augustine GJ. Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci. 1998;1:29–35. doi: 10.1038/229. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Sobue K, Kanda K, Harada A, Yorifuji H. The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin I. J Cell Biol. 1989;108:111–126. doi: 10.1083/jcb.108.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka M, Südhof TC. Synapsin I and II are ATP-binding proteins with differential Ca2+regulation. J Biol Chem. 1998;273:1425–1429. doi: 10.1074/jbc.273.3.1425. [DOI] [PubMed] [Google Scholar]
- Kandel E, Spencer WA. Electrophysiology of hippocampal neurons. II. After-potentials and repetitive firing. J Neurophysiol. 1961;24:243–259. doi: 10.1152/jn.1961.24.3.243. [DOI] [PubMed] [Google Scholar]
- Kao H-T, Porton B, Czernik AJ, Feng J, Yiu G, Haring M, Benfenati F, Greengard P. A third member of the synapsin gene family. Proc Natl Acad Sci USA. 1998;95:4667–4672. doi: 10.1073/pnas.95.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klagges BR, Heimbeck G, Godenschwege TA, Hofbauer A, Pflugfelder GO, Reifegerste R, Reisch D, Schaupp M, Buchner S, Buchner E. Invertebrate synapsins: a single gene codes for several isoforms in Drosophila. . J Neurosci. 1996;16:3154–3165. doi: 10.1523/JNEUROSCI.16-10-03154.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chin L-S, Shupliakov O, Brodin L, Sihra TS, Hvalby O, Jensen V, Zheng D, McNamara JO, Greengard P, Andersen P. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci USA. 1995;92:9235–9239. doi: 10.1073/pnas.92.20.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, McGuinness TL, Leonard CS, Sugimori M, Greengard P. Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc Natl Acad Sci USA. 1985;82:3035–3039. doi: 10.1073/pnas.82.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Gruner JA, Sugimori M, McGuinness TL, Greengard P. Regulation by synapsin I and Ca2+-calmodulin-dependent protein kinase II of the transmitter release in squid giant synapse. J Physiol. 1991;436:257–282. doi: 10.1113/jphysiol.1991.sp018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallart A, Martin AR. An analysis of facilitation of transmitter release at the neuromuscular junction of the frog. J Physiol. 1967;193:679–694. doi: 10.1113/jphysiol.1967.sp008388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell JW, Czernik AJ, De Camilli P, Greengard P, Townes-Anderson E. Differential expression of synapsins I and II among rat retinal synapses. J Neurosci. 1992;12:1736–1749. doi: 10.1523/JNEUROSCI.12-05-01736.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarren M, Alger BE. Use-dependent depression of IPSPs in rat hippocampal pyramidal cells in vitro. J Neurophysiol. 1985;53:557–571. doi: 10.1152/jn.1985.53.2.557. [DOI] [PubMed] [Google Scholar]
- Murphy CC, Trevathan E, Yeargin-Allsopp M. Prevalence of epilepsy and epileptic seizures in 10-year-old children: results from the Metropolitan Atlanta Developmental Disabilities Study. Epilepsia. 1995;36:866–872. doi: 10.1111/j.1528-1157.1995.tb01629.x. [DOI] [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC, Südhof TC. Short-term synaptic plasticity is altered in mice lacking synapsin I. Cell. 1993;75:661–670. doi: 10.1016/0092-8674(93)90487-b. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Li L, Chin LS, Greengard P, Smith SJ. Synaptic vesicle recycling in synapsin I knockout mice. J Cell Biol. 1996;134:1219–1227. doi: 10.1083/jcb.134.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Rosahl TW, Chapman PF, Marowitz Z, Friedman E, Frankland PW, Cestari V, Cioffi D, Südhof TC, Bourtchuladze R. Impaired learning in mice with abnormal short-lived plasticity. Curr Biol. 1996;6:1509–1518. doi: 10.1016/s0960-9822(96)00756-7. [DOI] [PubMed] [Google Scholar]
- Spillane DM, Rosahl TW, Südhof TC, Malenka RC. Long-term potentiation in mice lacking synapsins. Neuropharmacology. 1995;34:1573–1579. doi: 10.1016/0028-3908(95)00107-h. [DOI] [PubMed] [Google Scholar]
- Stefani G, Onofri F, Valtorta F, Vaccaro P, Greengard P, Benfenati F. Kinetic analysis of the phosphorylation-dependent interactions of synapsin I with rat brain synaptic vesicles. J Physiol. 1997;504:501–515. doi: 10.1111/j.1469-7793.1997.501bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci USA. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Czernik AJ, Kao H-T, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, De Camilli P, Greengard P. Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science. 1989;245:1474–1480. doi: 10.1126/science.2506642. [DOI] [PubMed] [Google Scholar]
- Takei Y, Harada A, Takeda S, Kobayashi K, Terada S, Noda T, Takahashi T, Hirokawa N. Synapsin I deficiency results in the structural change in the presynaptic terminals in the murine nervous system. J Cell Biol. 1995;131:1789–1800. doi: 10.1083/jcb.131.6.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torri Tarelli, F., M. Bossi, R. Fesce, P. Greengard, and F. Valtorta. Synapsin I partially dissociates from synaptic vesicles during exocytosis induced by electrical stimulation. Neuron. 1992;9:1143–1153. doi: 10.1016/0896-6273(92)90072-l. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Depletion and replenishment of vesicle pools at a ribbon-type synaptic terminal. J Neurosci. 1997;17:1919–1927. doi: 10.1523/JNEUROSCI.17-06-01919.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L-Y, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wong RK, Prince DA. Afterpotential generation in hippocampal pyramidal cells. J Neurophysiol. 1981;45:86–97. doi: 10.1152/jn.1981.45.1.86. [DOI] [PubMed] [Google Scholar]