

Abstract
Kinetochores are smaller and more accessible experimentally in budding yeast than in any other eukaryote. Believing that simple and complex kinetochores have important structural and functional properties in common, we characterized the structure of CBF3, the essential centromere-binding complex that initiates kinetochore formation in Saccharomyces cerevisiae. We find that the four subunits of CBF3 are multimeric in solution: p23Skp1 and p58Ctf13 form a heterodimer, and p64Cep3 and p110Ndc10 form homodimers. Subcomplexes involving p58 and each of the other CBF3 subunits can assemble in the absence of centromeric DNA. In these subcomplexes, p58 appears to function as a structural core mediating stable interactions among other CBF3 proteins. p58 has a short half-life in yeast, being subject to ubiquitin-dependent proteolysis, but we find that it is much more stable following association with p64. We propose that p23Skp1-p58-p64 complexes constitute the primary pool of active p58 in yeast cells. These complexes can either dissociate, reexposing p58 to the degradation pathway, or can bind to p110 and centromeric DNA, forming a functional CBF3 complex in which p58 is fully protected from degradation. This pathway may constitute an editing mechanism preventing the formation of ectopic kinetochores and ensuring the fidelity of chromosome segregation.
Keywords: kinetochore, CBF3, centromere, chromosome segregation, Saccharomyces cerevisiae
During mitosis, chromosomes attach to the microtubules of the mitotic spindle via kinetochores, specialized DNA-protein structures that form on centromeric DNA. In budding yeast, as in most other eukaryotes, the failure to form an active kinetochore or the formation of two or more kinetochores per chromatid prevents sister chromatids from disjoining correctly at anaphase. Because each of the 16 chromosomes in Saccharomyces cerevisiae is lost in fewer than 1 in 105 cell divisions, the accuracy of kinetochore assembly must be very high (Hieter et al., 1985a). In this paper, we analyze in detail the assembly and activation of CBF3, a complex that appears to constitute the centromere-bound scaffold on which the microtubule binding components of yeast kinetochores assemble (Lechner and Carbon, 1991; Kingsbury and Koshland, 1993; Sorger et al., 1994).
S. cerevisiae centromeres contain three conserved sequence elements, CDEI, CDEII, and CDEIII (Fitzgerald-Hayes et al., 1982; Hieter et al., 1985b). CDEIII is unique among the elements in that single point mutations in CDEIII abolish centromere function. CDEIII is bound by a four-protein complex, CBF3, that is composed of p110, p64, p58, and p23Skp1 subunits (for review see Clarke, 1998). In cells, all four of the CBF3-encoding genes are essential for growth (Doheny et al., 1993; Goh and Kilmartin, 1993; Jiang et al., 1993; Lechner, 1994; Strunnikov et al., 1995; Connelly and Hieter, 1996; Stemmann and Lechner, 1996) and in vitro, the four proteins are necessary and sufficient to form a complex that binds specifically to centromeric DNA (Stemmann and Lechner, 1996; Espelin et al., 1997; Kaplan et al., 1997). The shortest piece of CDEIII DNA to which a core CBF3 complex can bind in vitro is 56 bp. However, in the presence of an 88-bp centromeric fragment containing the minimal 56-bp CDEIII sequence and a further 32 bp of DNA, a CBF3 complex containing two additional 110 subunits forms (Espelin et al., 1997). In both this extended complex and in the core complex, p58, p64, and p110 are in direct contact with DNA. However, only p64 contains a recognizable DNA binding motif. The amino terminus of p64 is highly homologous to Zn2Cys6 zinc cluster domains found in Gal4p and in other fungal transcription factors (Schjerling and Holmberg, 1996). Like the well-characterized zinc cluster proteins Gal4p and Hap1p (Marmorstein et al., 1992; Marmorstein and Harrison, 1994; Zhang and Guarente, 1994), p64 binds to a CCG half-site, but unlike other zinc cluster proteins, p64 cannot form a stable complex with DNA in the absence of additional DNA-binding proteins.
p23Skp1 is a protein that appears to have at least two distinct functions in the cell. First, it mediates the phosphorylation-dependent activation of p58 by an as yet unidentified kinase (Kaplan et al., 1997). Second, it is required for the activity of SCFs, E3 complexes that transfer ubiquitin to critical cell cycle regulators and thereby target them for proteosome-dependent degradation (Feldman et al., 1997; Skowyra et al., 1997; Verma et al., 1997). In addition to p23Skp1, SCFcdc4 contains the cell division control protein Cdc4p, the cullin Cdc53p, and the E2 enzyme Cdc34p. Mutations in these cdc gene products cause cells to arrest at the G1-S transition, apparently because they prevent the degradation of the p34Cdc28 inhibitor Sic1p (Feldman et al., 1997; Skowyra et al., 1997). Among the mutations that have been isolated in p23Skp1 are ones that arrest cells at G1-S and others that cause arrest at G2-M (Bai et al., 1996; Connelly and Hieter, 1996). An appealing but as yet unproven possibility is that a G1-S arrest arises from the failure of some p23Skp1 mutants to assemble functional SCF, and a G2-M arrest arises from a failure of other p23Skp1 mutants to activate CBF3.
The analysis of two p23Skp1-binding proteins, Cdc4p from yeast and Cyclin F from humans, has shown that a conserved sequence, termed the F-box, is required for association with p23Skp1 (Bai et al., 1996). Homologies to the F-box have been found in several dozen proteins from various species. These proteins are not involved in a common physiological pathway and the common function of p23Skp1 binding is not known. In SCF, the function of p23Skp1 appears to be to stabilize the association of Cdc4p with Cdc53p, implicating p23Skp1 in SCF assembly (Feldman et al., 1997; Skowyra et al., 1997). A similar role for p23Skp1 has been proposed for the variant SCF complex SCFGrr1, which contains the F-box protein Grr1p in place of Cdc4p (Li and Johnston, 1997; Kishi et al., 1998).
p58 is subject not only to positive regulation by phosphorylation in a Skp1-dependent manner, but also to negative regulation by ubiquitin-dependent degradation (Kaplan et al., 1997). Whereas p23, p64, and p110 are stable proteins, p58 has a very short half-life (15 min). We have shown recently that p58 instability requires active SCFcdc4 (Kaplan et al., 1997). Thus, p58 is activated in a step that requires p23Skp1 and degraded in a step that requires SCFcdc4, a p23Skp1-containing complex. To explain these findings we have postulated the existence of an editing process in which coupled cycles of p58 activation and destruction serve to tightly limit the amount of active CBF3. This may prevent the formation of ectopic kinetochores and help to maintain the fidelity of chromosome segregation.
Our working model for kinetochore formation in budding yeast is that CBF3 forms a DNA-bound scaffold onto which microtubule binding proteins assemble (Sorger et al., 1994). If this hypothesis is correct, then CBF3 plays a critical role in determining the specificity of kinetochore formation and in anchoring the kinetochore on centromeric DNA in the face of the strong microtubule-dependent forces that move chromosomes during mitosis. To understand how CBF3 functions, we need to determine how it becomes activated and assembled into a functional DNA-binding complex. In this paper, we examine the organization of CBF3 complexes in solution and when bound to DNA, focusing on the tightly regulated subunit p58. We show that p58 can form binary complexes with either p23Skp1, p64, or p110 in solution. Using p58 mutations that disrupt p23Skp1-p58 interaction, we show that p23Skp1-p58 association is essential for p58 activation but is not required for p58 degradation. The half-life of p58 is regulated primarily by its association with p64. Whereas free p58 has a half-life of <15 min, p58 assembled into a p58/ p64 complex has a half-life of many hours. Thus p58 is an example of a protein whose stability is regulated by assembly into a multiprotein complex. The purpose of this regulation may be to ensure high accuracy in kinetochore formation.
Materials and Methods
Yeast Strains, Plasmids, and Extracts
The strain YK113 (MAT a, ura3-52, lys2-801, ade2-101, his3Δ200, trp1Δ1, leu2Δ1, Δctf13::HIS3, pCTF13(URA3), C.F.TRP, Rad2 distal; Doheny et al., 1993) was used to assay the function of ctf13 F-box mutations. Mutant ctf13 alleles were integrated at the leu2 locus and resulting LEU+ transformants were tested for growth in the presence of 5-fluororotic acid (5-FOA). Protein half-lives were determined by promoter shut-off experiments in the protease deficient strain JB811 (Mat a, leu2, trp1, ura3-52, prb1-1122, pep4-3, prc1-407, GAL). Extracts were prepared as described (Kaplan and Sorger, 1997). F-box point mutations in p58 and Cdc4p were created by site-directed mutagenesis of plasmids pIR112 (pRS305 containing the 2.2-kb CTF13 fragment from pKF11; Doheny et al., 1993) and pIR329 (pRS316::PY2-HA3-CDC4), respectively, essentially as described (Kunkel et al., 1991). Mutant sequences were confirmed by sequencing through the region homologous to the mutagenic oligonucleotide. For targeted integration at leu2, pIR112 was linearized using HpaI. Yeast manipulations were performed as described (Guthrie and Fink, 1991).
Expression of Recombinant CBF3 in Insect Cells
CBF3 genes were subcloned into the pFastBac plasmid (GIBCO BRL) and baculovirus DNA was prepared as recommended. p23Skp1, p64, and p110 constructs were created containing the sequence MRGSH6 fused to the initiator methionine. p110 has methionines at residues 1 and 12 of the published sequence, and we have found that p110 fused at Met-12 is more active in DNA binding than p110 fused at Met-1. Thus, p110 fused at Met-12 was used for the work described here. As in yeast, p58 produced in insect cells has a short half-life and is expressed at lower levels than the other CBF3 subunits. The relatively low levels of p58 in insect cell lysates made it difficult to purify and, in this paper, we primarily use recombinant p58 in unfractionated extracts. Nevertheless, in several cases we show that the properties of unpurified and purified CBF3 proteins are indistinguishable.
Proteins were expressed in High Five insect cells (Invitrogen Corp.) by infecting cells with one or more baculoviruses and harvesting after 48–72 h. To minimize protein degradation, all steps of the extraction were performed on ice. Cytoplasmic extracts were prepared by swelling cells in hypotonic lysis buffer (10 mM Tris-HCl, pH 8.0, 10 mM KCl, 1.5 mM MgCl2, 10 mM β-mercaptoethanol, 10 μg/ml each of leupeptin, pepstatin, and chymostatin, 50 mM TPCK, 1 mM PMSF), breaking the cells with a Dounce tissue grinder (Wheaton), and centrifuging the lysate to pellet the nuclei. Supernatants were adjusted to 10% glycerol, 150 mM KCl, and 50 mM β-glycerophosphate before freezing for storage. The nuclear pellet was extracted for 30 min in nuclear extraction buffer (10 mM Hepes, pH 8.0, 50 mM β-glycerophosphate, 0.1 mM EDTA, 0.5 M KCl, 5 mM MgCl2, 10% glycerol, 50 mM NaF, 10 mM β-mercaptoethanol, 10 μg/ml each of leupeptin, pepstatin, and chymostatin, 50 mM TPCK, 1 mM PMSF) and centrifuged to remove DNA and other insoluble material. Under these extraction conditions, p64, p58, and p110 were found primarily in nuclear fractions while p23Skp1 partitioned mostly to the cytoplasmic fraction. Nuclear extracts used for purification were prepared in the absence of EDTA so that material could be incubated directly with metal chelate resin (see below). 1% Triton X-100 was included in nuclear extraction buffer when p110 was prepared for purification to improve solubility.
His6-tagged proteins were purified using Ni-NTA Superflow resin (Qiagen, Inc.). Extracts were adjusted to 3 mM imidazole, added to resin, and bound in batch overnight at 4°C. For p64 and p23Skp1, resin was washed in Ni buffer (20 mM Hepes, pH 8.0, 500 mM NaCl, 10% glycerol, 10 mM β-mercaptoethanol, 50 mM imidazole) before elution in the same buffer but containing 500 mM imidazole. For p110, resin was washed in Ni buffer (3 mM imidazole), eluted in batch with Ni buffer (250 mM imidazole), and diluted to 150 mM NaCl before loading onto a column containing a small volume of Poros 20 HQ resin (PerSeptive Biosystems) to concentrate the protein. p110 was then eluted in Ni buffer containing 800 mM NaCl and no imidazole.
Gel Filtration Chromatography
Gel filtration chromatography was performed on a SMART System (Pharmacia Biotech) using Superose 6 and Superose 12 columns. To determine diffusion coefficients, standard curves were generated by plotting elution volume versus 1/D, where D is the diffusion constant, using the following protein standards (Bio-Rad): thyroglobulin (molecular mass = 670 kD, D = 2.63 × 10−7 cm2s−1), bovine gamma globulin (molecular mass = 158 kD, D = 4.1), chicken ovalbumin (molecular mass = 44 kD, D = 7.8), and equine myoglobulin (molecular mass = 17 kD, D = 11.3). Diffusion coefficients for protein standards were obtained from Sober (1970) and represent values determined in water at 20°C (D20,w). Elution from a gel filtration column correlates with Stokes radius (Siegel and Monty, 1966). All columns were run at 4°C with column buffer (10 mM Hepes, pH 8.0, 6 mM MgCl2, 10% glycerol, 150 or 300 mM NaCl, 10 mM β-mercaptoethanol). Analyses were repeated at least two times and values for 1/D varied by no more than 4% between experiments.
Chromatography of Purified Proteins.
Purified p23Skp1 and p110 were diluted to 150 mM NaCl with column buffer lacking NaCl. Because p64 was only partially soluble in 150 mM NaCl, it was diluted to 300 mM NaCl and separated using column buffer containing 300 mM NaCl. Diluted proteins were centrifuged to remove insoluble material before loading onto the column. Elution was monitored by absorbance at 280 nm and fractions were collected and analyzed by immunoblotting and by bandshift assays.
Chromatography of Proteins in Insect Cell Extracts.
Because it was difficult to express and purify sufficient quantities of p58, all analyses of p58-containing mixtures were performed using insect cell extracts. Samples were prepared by dialyzing nuclear extracts into column buffer using a microdialysis system (GIBCO BRL) and then centrifuging to remove insoluble material. To compare directly the elution profiles of different sets of coexpressed proteins, the fraction size, the number of fractions, and the starting point for fraction collection were kept constant.
Glycerol Gradient Sedimentation
Gradients were prepared in 2-ml volumes by layering successive 0.4-ml aliquots of column buffer containing decreasing concentrations of glycerol and incubating the gradient at 4°C for 1 h to equilibrate. Purified p64 was analyzed in 300 mM NaCl and all other proteins were analyzed in 150 mM NaCl. p110 and p64 were sedimented in 15–35% gradients while p23Skp1 and p23Skp1/p58 were sedimented in 5–25% gradients. Purified p23Skp1, p64, and p110 were prepared by diluting to the same salt concentration as the gradient using column buffer that lacked glycerol and NaCl. Insect cell extract containing p58 and p23Skp1 was dialyzed into column buffer lacking glycerol but containing 150 mM NaCl. For p110 and p64, gradient standards were added directly to the protein samples; for p23Skp1 and p58, the standards were run separately. Gradients were centrifuged at 50,000 rpm 10–16 h at 4°C in a TL-S55 swinging bucket rotor (Beckman Instruments) and fractionated by removing 100-μl aliquots from the top of the gradient. Fractions were then analyzed either by bandshifts or by TCA precipitation followed by immunoblotting. Protein standards (Boehringer Mannheim) cytochrome c (molecular mass = 12.5 kD, s = 1.9 S), chymotrypsinogen A (molecular mass = 25 kD, s = 2.58), hen egg albumin (molecular mass = 45 kD, s = 3.55), bovine serum albumin (molecular mass = 68 kD, s = 4.22), aldolase (molecular mass = 158 kD, s = 7.4), and catalase (molecular mass = 240 kD, s = 11.3) were separated on SDS-polyacrylamide gels, stained with Coomassie blue and quantified using IPLab Gel image processing software (Signal Analytics). All samples were analyzed at least two times, and sedimentation coefficients varied by no more than 15% between experiments.
Calculations
Native molecular mass was calculated using the expression molecular mass = RTs/D(1 − νρ), where R is the ideal gas constant; T, absolute temperature; s, sedimentation coefficient; D, diffusion coefficient; ν, partial specific volume; and ρ, density of water at 20°C. Partial specific volumes (0.715 cm3g−1 for p110, 0.724 for p64, 0.697 for p23Skp1, and 0.719 for a p23Skp1/p58 heterodimer) were estimated from amino acid content. Stokes radii were calculated using the equation a = kT/6πηD, where a is Stokes radius and η is viscosity of water at 20°C. f/f0 was calculated using the relationship f/f 0 = a/(3νM/4πN)1/3, where M is molecular mass and N is Avogadro's number.
Bandshift and Reticulocyte Lysate Binding Assays
Bandshift assays were performed as described (Kaplan and Sorger, 1997). Bacterial GST-fusion proteins were expressed in the strain BL21ΔE3 using the plasmid pGEX-4T-2 as recommended (Pharmacia Biotech). Cells were lysed by sonication in breakage buffer (20 mM Hepes, pH 8.0, 150 mM NaCl, 2 mM EDTA, 10% glycerol, 1 mM DTT, and 1 mM PMSF), the cell extracts were cleared by centrifugation and the concentration of GST-fusion was estimated by comparing Coomassie-stained bands on a 10% SDS-PAGE gel to a protein standard. Extracts containing GST-fusion proteins were adjusted to 1% Triton X-100 and bound to glutathione-Sepharose beads for 1 h at 20°C as recommended (Pharmacia Biotech). Beads were spun down and washed three times with IP buffer (1% Triton X-100, 50 mM Hepes, pH 8.0, 150 mM NaCl, 50 mM NaF, 50 mM β-glycerophosphate, and 1% glycerol). 40 μl of a 50% bead slurry containing 2.5 μg of immobilized GST-fusion protein was used for each binding reaction.
p58 mutants and fragments were amplified from plasmids by PCR using 5′ primers designed to include a T7 promoter and Kozak consensus start ATG codon (5′-tgtaatacgactcactatagggccaccatg-3′). To generate 35S-labeled protein in vitro, mRNA was synthesized from the PCR products using T7 RNA polymerase as recommended (Promega). Approximately 500 ng of mRNA was incubated in a 100-μl rabbit reticulocyte lysate in vitro translation reaction for 2 h at 30°C.
200-μl binding reactions containing 20 μl of 35S-labeled protein, 40 μl of 50% bead slurry, 2 μl of PMSF, and 138 μl of IP buffer were incubated for 4 h at 4°C with end-over-end tube rotation. The beads were then recovered by centrifugation, washed three times with 1 ml IP buffer, and bead associated 35S-labeled protein analyzed by SDS-PAGE and autoradiography.
p58 Stability Experiments
Yeast strain JB811 containing plasmid borne GAL1 driven GST-CTF13 or GST-ctf13 truncations were grown to late-log phase in media containing 2% galactose and 2% raffinose at 30°C for ∼14 h. A sample was chilled at time 0 and the remaining cells were washed into media containing 2% glucose, grown at 30°C, and aliquots placed on ice at the indicated times after addition of glucose. Extracts were prepared and duplicate samples of GST-fused proteins were purified from 2 mg of total yeast protein by binding to glutathione-Sepharose (Pharmacia Biotech). Beads were washed three times in IP buffer (see above) and bead-bound protein was loaded on 12.5% SDS-PAGE gels. Protein was transferred to nitrocellulose membranes and the GST moiety was detected by immunoblotting with an anti-GST antibody. Antibody binding was quantified using 125I-labeled protein A (DuPont/NEN) and a PhosphorImager (Molecular Dynamics).
Partial Proteolysis
p64 was treated with dilute proteolytic enzymes in 50 mM Hepes, pH 8.0, 300 mM NaCl, 10% glycerol, 1 mM EDTA at 30°C. Aliquots were removed at different time points and proteolysis stopped by boiling the sample in SDS-PAGE loading buffer containing 5 mM PMSF. Microsequencing of proteolytic fragments was performed at the MIT Biopolymers Lab from PVDF blots of SDS-PAGE gels.
Results
To understand the regulation and function of CBF3, we need to determine its mode of assembly and its structure when bound to DNA. In this paper, we use a combination of biochemical and molecular techniques to study CBF3 complexes in solution and in association with CDEIII. Hydrodynamic measurements have proven particularly valuable for determining the shape and composition of various CBF3 complexes. Protein hydrodynamics generates values for the native mass and size (the radius of gyration or Stokes radius) of a complex. By systematically determining the native masses of complexes produced by mixtures of two or three CBF3 subunits, we have deduced which subunits touch each other. To extend this interaction map and determine the consequences of disrupting various subunit-subunit interactions, we have used molecular techniques to map binding domains. The method is quite laborious but the net result is simple: an experimentally derived model for the organization of CBF3.
To obtain CBF3 in reasonable yield and free of contaminating yeast proteins, recombinant CBF3 proteins were produced in insect cells. We have shown previously that recombinant CBF3 is indistinguishable in its DNA-binding properties from CBF3 isolated from yeast (Kaplan et al., 1997). The native molecular masses and radii of gyration of proteins were calculated using hydrodynamic data obtained from gel filtration columns and glycerol velocity gradients. Fractions from columns or gradients were collected and the levels of a particular CBF3 subunit determined either by Western blotting, absorbance measurements, or “bandshift complementation” assays. In the complementation assay, the amount of an active CBF3 subunit was measured by adding an excess of the other CBF3 proteins and radiolabeled CEN DNA, and then quantifying CBF3-DNA complexes on nondenaturing bandshift gels. We have determined previously that CBF3 subunits can exchange in and out of various complexes before binding to centromeric DNA. Thus, the complementation approach permits a selective analysis of a single active protein in a multicomponent mixture.
p110 and p64 Are Homodimers in Solution
The p110 subunit of CBF3 eluted as a single symmetrical peak from gel filtration columns and glycerol velocity gradients, indicating that it was present as a relatively homogenous species (Fig. 1). Preparations of recombinant p110 often contained a major p110 breakdown product, but its presence did not interfere significantly with molecular mass determinations: on gel filtration columns and gradients it fractionated away from full-length p110. On sizing columns, the apparent mass of p110 was nearly 670 kD but on velocity gradients it sedimented near the 158-kD aldolase standard. Such differences in apparent mass are expected for proteins that are not globular, but a simple formula can be used to calculate a native molecular mass independent of protein shape (see Materials and Methods). For these calculations, a diffusion constant was obtained from the elution volume of p110 on Superose sizing columns relative to the elution volumes of protein standards (Fig. 1 c), a sedimentation coefficient was obtained from the migration of p110 on glycerol velocity gradients relative to standards (Fig. 1 d), and a partial specific volume was estimated by extrapolation from the amino acid composition. Based on these data, we obtained a native molecular mass for p110 of 197 kD ± 10%, leading us to conclude that p110, whose calculated monomer molecular mass is 112 kD, forms a homodimer in solution (Table I). We have shown previously that p110 is in close contact with bases in the major groove of CDEIII DNA (Espelin et al., 1997) and the hydrodynamic data reveal that the protein is indeed elongated and has a long dimension about the same as 4.5 helical turns of B-form DNA. To obtain further evidence that p110 can self-associate, we expressed native and GST-tagged p110 either individually or in combination, and then isolated GST p110 using glutathione-Sepharose. We observed that native p110 bound GST-tagged p110 (Fig. 1 e, lane 2) but not, as a negative control, GST alone (lane 3). The ability of GST p110 to pull down native p110 is good evidence that p110 indeed forms multimers. From these findings, we conclude that p110 exists in solution as a dimer, and possibly also as higher-order oligomers.
Figure 1.

Hydrodynamic analysis of p110. (a) Purified p110 was applied to Superose 6 gel filtration columns and the eluate analyzed by absorption at 280 nm (dotted line), by bandshift complementation (solid line), and by Western blotting (gel). Arrows indicate the peak elution volumes for the molecular mass standards. For the complementation assay, excess p23Skp1, p58, p64, and radiolabeled CDEIII-containing probe were added and DNA-protein complexes were detected on bandshift gels. For the Western blot, fractions were TCA-precipitated, separated by SDS-PAGE, and immunoblotted using polyclonal rabbit anti-p110 antibodies and 125I-labeled protein A. A Coomassie-stained gel of recombinant p110 is shown to the right of the graph. The upper band is full-length p110 and the lower band is a p110 breakdown product, as determined by protein sequencing. (b) Western blot of p110 analyzed on a 15–35% glycerol gradient. The positions on the gradient of the protein standards are indicated above the gel panel. (c) Data and standard curves for three independent chromatographic separations or (d) glycerol gradient analyses. The data points show the positions of protein standards (see Materials and Methods) and the arrow shows the elution volume of p110. (e) Pull-down experiment demonstrating that p110 self-associates. GST-p110 or GST was expressed in insect cells alone or in combination with native p110, protein lysates were prepared, and GST protein complexes were isolated on glutathione-Sepharose beads. Bead-bound proteins were separated on 10% SDS-PAGE and immunoblotted with anti-p110 antibodies.
Table I.
Hydrodynamic Parameters for CBF3 Proteins
| Subunit | Diffusion Coefficient-D (× 10−7 cm) | Sedimentation Coefficient-s (Svedbergs) | Calculated molecular mass (kD) | Monomer molecular mass (kD) | Stoichiometry‡ | Stokes radius (Å) | f/f0 § | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p110 | 2.66 | 6.14 | 197.2 | 112.2 | 1.76 | 80.1 | 2.09 | |||||||
| (2.64–2.68) | (5.70–6.58) | (184.4–209.7) | (1.64–1.87) | (79.5–80.7) | (2.03–2.15) | |||||||||
| p64 | 4.75 | 7.09 | 131.7 | 73.9 | 1.78 | 44.8 | 1.34 | |||||||
| (4.69–4.80) | (6.70–7.49) | (126.0–137.6) | (1.71–1.86) | (44.4–45.4) | (1.30–1.37) | |||||||||
| p58/p23Skp1 | 6.00 | 5.26 | 76.0 | 56.3 [86.6]‖ | 0.88‖ | 35.5 | 1.27 | |||||||
| (5.70–6.34) | (4.95–5.58) | (75.2–76.3) | (0.87–0.88)‖ | (33.6–37.4) | (1.20–1.34) | |||||||||
| p23Skp1 | 7.02 | 2.65 | 30.3 | 23.9 | 30.3 | 1.48 | ||||||||
| (6.90–7.15) | (2.49–2.81) | (29.0–31.6) | — | (29.8–30.9) | (1.43–1.52) | |||||||||
| p23Skp1 (bacterial) | 7.01 | 2.18 | 25.0 | 23.9 | 1.05 | 30.4 | 1.58 | |||||||
| (6.89–7.14) | (2.02–2.33) | (23.6–26.2) | (0.99–1.10) | (29.8–30.9) | (1.52–1.63) |
These data are the average of 2–4 independent determinations. The numbers in parentheses indicate the range for each value as dictated by the size of gel filtration or glycerol gradient fractions.
Calculated from the predicted amino acid sequence of protein plus the hexa-histidine tag, if used.
Determined by dividing calculated molecular mass by monomer molecular mass.
Proteins with f/f0 ratios <1.5 are considered “globular,” while proteins with ratios >1.5 are considered “elongated.”
Indicates values that include the monomer molecular mass of p58 plus the calculated molecular mass of p23Skp1.
Next, we analyzed p64 using similar methods (Fig. 2, a and b). The calculated native molecular mass of p64 was 132 kD, suggesting that p64 is a homodimer in solution (the p64 homodimer seems to be roughly spherical; Table I). In addition to the dimer peak, complementation assays revealed a second, later eluting peak of p64 on sizing columns (Fig. 2 a). We have not been able to determine a sedimentation coefficient for this material, but its migration on gel filtration columns suggests that it represents monomeric p64. To confirm that p64 can self-associate, we asked whether GST p64 and His6-tagged p64 would interact when coexpressed. Pull-downs using glutathione-Sepharose beads showed that p64 binds to GST p64 but not (as a control) to GST alone (Fig. 2 c). We conclude that p64 is primarily a dimer in solution but that it is in equilibrium with a small pool of monomeric protein.
Figure 2.


Hydrodynamic analysis and domain mapping of p64. (a) Purified p64 was applied to a Superose 12 gel filtration column and the eluate analyzed by absorption at 280 nm (dotted line), by bandshift complementation (solid line), and by Western blotting (gel; see Fig. 1 for details). To the right of the graph, a Coomassie-stained gel of recombinant p64 shows that it is present as a single pure species. (b) Western blot of p64 analyzed on a 15– 35% glycerol gradient. Hydrodynamic data were obtained from three independent experiments, as described in Fig. 1. (c) Pull-down experiment demonstrating that p64 self-associates. GSTp64 or GST were expressed in insect cells either alone or in combination with tagged p64 (carrying an amino-terminal MRGSHis6 tag; Qiagen), protein lysates were prepared, and GST protein complexes were isolated using glutathione-Sepharose. Bead-bound proteins were separated on 10% SDS-PAGE and immunoblotted with anti-MRGSHis6 tag antibodies. (d) Coomassie-stained gel of purified bacterially expressed p64 treated with limiting amounts of proteolytic enzymes to identify hypersensitive sites and delineate physical domains. The bacterially expressed p64 used in this experiment and the insect cell–expressed p64 analyzed in a and b, have identical specific activities for DNA binding (data not shown). 0.2 mg/ml p64 (lane 1) was treated at 30°C with 5 μg/ml trypsin for 10 min (lane 2), 20 min (lane 3), 40 min (lane 4), or 80 min (lane 5) or with 1 μg/ml chymotrypsin (lane 6) or 2 μg/ml V8 protease (lane 7) for 80 min. Digestions were stopped by boiling samples in SDS-PAGE loading buffer that contained PMSF. (e) The amino-terminal 100 residues of p64 are shown and the sites of tryptic cleavage for the fragments marked with arrows in c are indicated. The tryptic cleavage sites were identified by NH2-terminal microsequencing. The Zn2Cys6 zinc cluster domain of p64 (boxed) contains multiple potential trypsin recognition sequences (basic amino acids, shaded). No major fragments with cleavage sites within the zinc cluster were recovered, suggesting that the zinc cluster is indeed a compact folded structure. (f) Competition analysis on bandshift gels of wild-type (WT) and mutant 88-bp CDEIII DNA sequences. CBF3 and radiolabeled probe were mixed with a 4–200-fold excess of unlabeled centromeric competitor DNAs (as indicated) and analyzed on bandshift gels. Values are expressed as a percentage of the binding observed in the absence of competitor. Error bars show the mean and range of independent determinations. (g) Sequences of the various competitor DNAs showing only a relevant 21-bp segment of the 88-bp probes. Multiple point mutations in mutants 1 and 2 were introduced using PCR, and the 3-bp deletion in mutant 3 introduced as previously described (Espelin et al., 1997).
p64 is the only subunit of CBF3 that is detectably homologous to proteins known to bind to DNA. The amino terminus of p64 contains a Zn2Cys6 “zinc cluster” DNA-binding module similar in sequence to the zinc clusters found in other fungal DNA binding proteins (Schjerling and Holmberg, 1996). The Gal4p zinc cluster, whose structure has been determined by x-ray crystallography, forms a compact domain that is attached to the rest of the protein by a flexible linker (Marmorstein et al., 1992). To determine whether p64 has a similar domain organization, we treated purified protein with limiting amounts of protease for varying times and looked for the accumulation of discrete protein fragments (Fig. 2 c). In multidomain proteins, proteolytic mapping can be used to identify domain boundaries because exposed loops between domains are often significantly more susceptible to digestion than the domains themselves. After cleaving p64 with limiting amounts of trypsin for 10 min, essentially all of the intact protein was converted into a 58-kD form (Fig. 2 d, lane 2, solid arrowhead); more extensive proteolysis led to the accumulation of 51- and 48.5-kD species (Fig. 2 d, lanes 3–5, open arrowhead). The amino terminus of the 58-kD fragment was residue 48 (as determined by NH2-terminal sequencing), indicating that cleavage had occurred just downstream of the zinc cluster domain (Fig. 2 e). Similarly sized cleavage products were generated, albeit less efficiently, by digestion with chymotrypsin (Fig. 2 d, lane 6) or V8 protease (Fig. 2 d, lane 7). From these data, we conclude that the zinc cluster of p64 is attached to the remainder of the protein via a proteolytically accessible and potentially flexible arm.
Well characterized zinc cluster proteins such as Gal4p and Hap1p bind, in the absence of other proteins, to direct or inverted repeats of the sequence CCG (Marmorstein et al., 1992; Zhang and Guarente, 1994). CDEIII contains only one CCG half-site, but DNA-protein cross-linking shows that p64 contacts this CCG and a second sequence, TGT (in CEN3), 12 bp to the left (Espelin et al., 1997). We wondered whether this TGT might represent a degenerate CCG half-site and thus whether p64 might be able to bind on its own to a CDEIII sequence in which a second complete CCG half-site had been introduced. Two CDEIII variants were synthesized so as to create a second CCG half-site in one of three different positions while retaining the highly conserved 8G residue that contacts p64 (see Fig. 2 g for an explanation of these sequences; Espelin et al., 1997). Competition experiments showed that neither variant CDEIII was able to bind efficiently to CBF3 and in no case could we obtain evidence from bandshift gels that p64 could bind to the variant CDEIIIs (Fig. 2, f and g, and data not shown). Moreover, it appeared that mutations in the bases surrounding 8G were nearly as deleterious for CBF3 binding as mutations in the absolutely conserved CCG centered on 20C (compare mutants 2 and 3). We therefore conclude that the inability of p64 to bind to DNA on its own is unlikely to be a simple consequence of the absence of a second CCG half-site.
Probing the p23Skp1-p58 Heterodimer in Solution
We have shown previously that p58 is active for DNA binding only if phosphorylated in a p23Skp1-dependent manner. p58 has proven quite difficult to purify in large quantities, and we therefore analyzed nuclear extracts from insect cells coexpressing p23Skp1 and p58. Only some of the p58 expressed in insect cells is active (this is also true in yeast; data not shown and Kaplan et al., 1997), and we therefore used a complementation assay to detect specifically the active pool of p58. The calculated native molecular mass of p58 derived from insect cells coexpressing p23Skp1 and p58 was 76 kD (Fig. 3, a–c, and Table I). p58 has been shown previously to associate with p23Skp1 in yeast and in insect cells (Stemmann and Lechner, 1996; Kaplan et al., 1997), but following p23Skp1-mediated phosphorylation, p58 can dissociate from p23Skp1 and remain active (Kaplan et al., 1997). Thus, we needed to determine whether our active p58 was actually associated with p23Skp1. We reasoned that by generating a fusion between p23Skp1 and GST we would alter the mobility of p23Skp1 sufficiently that free and p23Skp1-bound p58 could be distinguished. As shown in Fig. 3 b, the coexpression of p58 with GST p23Skp1 shifted all of the p58 to a higher molecular mass, demonstrating that p58 forms a stable complex with GST p23Skp1 and, thus, probably also with untagged p23Skp1. We conclude that the 76-kD native molecular mass of p58 reflects the formation of a stable heterodimer between one p58 polypeptide and one p23Skp1.
Figure 3.
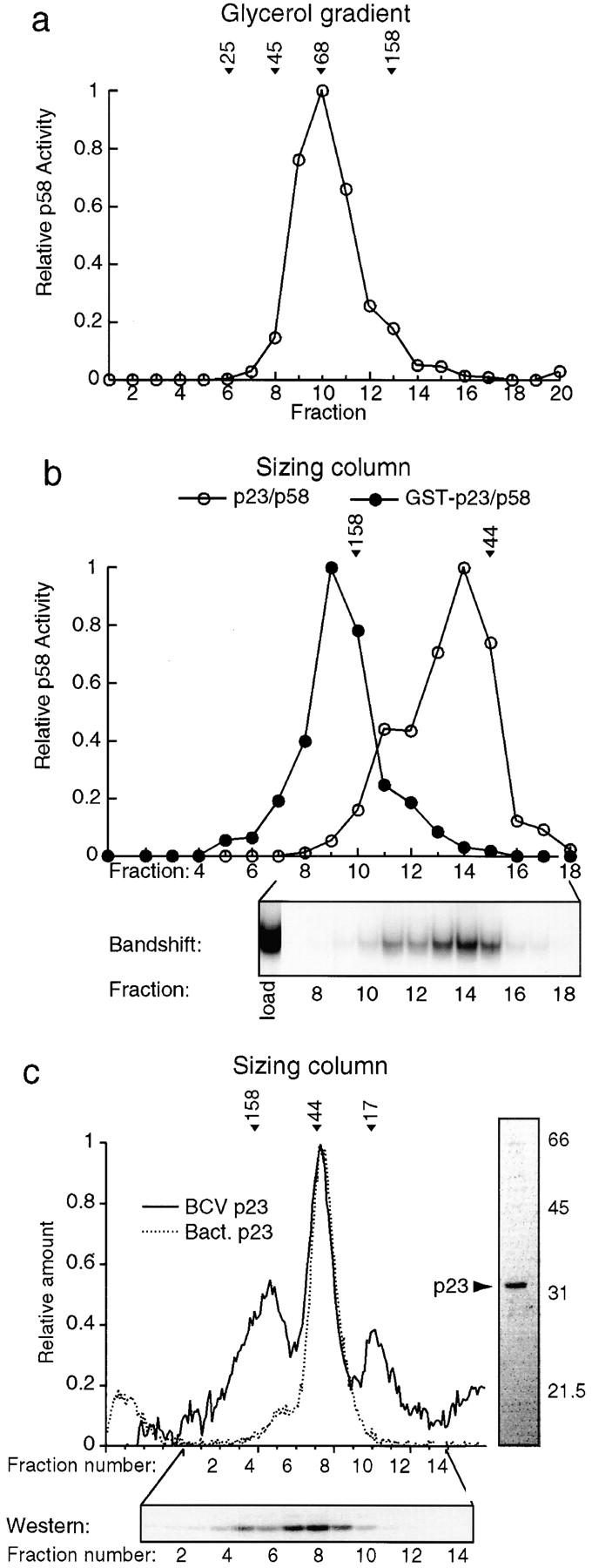
Probing the binding of p23Skp1 to p58. (a) 5–25% glycerol gradient of extract from cells coexpressing p58 and p23Skp1 analyzed using a bandshift complementation assay for p58. (b) Extracts from baculovirus-infected insect cells coexpressing p58 and p23Skp1 (open circles) or p58 and GST p23Skp1 (solid circles) were applied to a Superose 12 gel filtration column and the eluate analyzed using a bandshift complementation assay. The resulting bandshift complementation gel of the p58/p23Skp1 extract is shown below. (c) Purified p23Skp1 expressed in either insect cells or bacteria was applied to a Superose 12 gel filtration column and the eluate analyzed by absorption at 280 nm (dotted line) and by Western blotting (solid line; see Fig. 1 for details). To the right of the graph, a Coomassie-stained gel of recombinant p23 shows that it is present as a single pure species. Because activity assays for p23Skp1 are nonquantitative, no activity plot is shown.
We are somewhat uncertain about the oligomeric state and mass of p23Skp1. Data for bacterially expressed p23Skp1 are clear: at a concentration of ∼100 μM, p23Skp1 is monomeric and has the mass predicted from its amino acid sequence. However, the data for insect cell–expressed p23Skp1 are more ambiguous. The majority of p23Skp1 is monomeric, but ∼30% of the protein migrates as a larger species that may represent an oligomer. Unfortunately, in the absence of a reliable assay for functional p23Skp1, it is not possible to determine whether the activity of the potentially oligomeric forms is different from that of monomer.
p58 Can Bind to p64 and to p110
A great advantage of hydrodynamic measurements is that they not only allow protein-protein interactions among CBF3 subunits to be probed, but the stoichiometry of the hetero-oligomeric complexes to be determined. We used mobility shifts on gel filtration columns as an assay for the interaction of p58 with p64 and p110. First, we looked for an association between p58 and p64 by coexpressed p58 either with p23Skp1 alone or with both p23Skp1 and p64, and then determined the elution volume of p58 on gel filtration columns using bandshift complementation assays (Fig. 4 and data not shown). Active p58 eluted earlier from columns in the presence of p64 (Fig. 4 a, fraction 10) than in its absence (Fig. 4 a, fraction 14), indicating that interaction with p64 had shifted p58 into a larger complex. To confirm this interpretation, we performed a similar experiment but assayed for p64 by bandshift complementation. On a gel filtration column, we observed a clean shift in the elution of p64 from fraction 12 to fraction 10 (Fig. 4 b). Thus, the p58-shifted p64 elutes in the same fraction as the p64-shifted p58, confirming the reliability of the methodology. The calculated native mass of the putative p58/p64/ p23Skp1 complex was ∼200 kD (data not shown), consistent with the binding of one p58-p23Skp1 heterodimer (77 kD) to one p64 homodimer (132 kD). From these data we conclude that p58, p23Skp1, and p64 can form a stable heterotetramer in solution in the absence of p110 and centromeric DNA. Moreover, when other CBF3 proteins are present in excess, essentially all of the p58 is found in a p58/p23Skp1/p64 hetero-oligomeric complex.
Figure 4.

Associations among CBF3 proteins. Insect cell extracts containing CBF3 proteins were applied to Superose 12 gel filtration columns and assayed for p58 activity (a and c) or for p64 activity (b, d, and e) using bandshift complementation assays. In these samples, the total amount of insect cell lysate was kept constant to ensure that changes in apparent molecular mass arose from interactions among CBF3 proteins and not as a consequence of binding to contaminating insect proteins. (a) Elution profile of p58 activity from a column to which extract containing p23Skp1, p58, and p64 (solid line) or, for comparison, extract containing only p23Skp1 and p58 (dotted line), was applied. (b) Elution profile of p64 activity from a column to which a mixture of extracts from cells coexpressing p23Skp1 and p58 and cells expressing p64 (solid line) or, for comparison, extract containing only p64 (dotted line), was applied. (c) Elution profile of p58 activity from a column to which extract containing p23Skp1, p58, and p110 (solid line) or, for comparison, extract containing p23Skp1 and p58 only (dotted line), was applied. (d) Elution profile of p64 activity from a column to which a mixture of extracts from cells expressing p64 and cells expressing p110 (solid line) or, for comparison, extract containing p64 alone (dotted line) was applied. (e) p64 activity in an eluate from a Superose 12 column to which a mixture of extracts from cells expressing p64, cells expressing p110, and cells coexpressing p23Skp1 and p58 was applied (solid line). For comparison, a p64 activity profile is shown (dotted lines) for a column to which only the p64-containing extract was applied. The shift of p64 activity to fraction 5 represents the formation of a multiprotein complex likely containing all four CBF3 subunits (see text for details). (f) Profile of the sedimentation of CBF3-CDEIII complexes in a 15–35% glycerol gradient. CBF3-DNA complexes were generated by incubating extracts from insect cells coexpressing p23Skp1, p58, p64, and p110 with radiolabeled 56 base CDEIII probe. Fractions from the gradient were analyzed by loading them directly onto bandshift gels.
Applying the same logic, we looked for the association of p58/p23Skp1 with p110 by coexpressing the three proteins and assaying for p58 activity in gel filtration eluates. We observed that the coexpression of p58 with p110 shifted a fraction of the p58 activity to a larger sized complex (Fig. 4 c). Multiple earlier-eluting peaks were present but a significant amount of the p58 was not altered in mobility. We conclude that p58 and p23Skp1 can bind to p110 in solution. The existence of several peaks on the gel filtration column may reflect the presence of multiple p58-p23Skp1-p110 complexes or it may arise from the formation of a single relatively unstable complex that partially dissociates during chromatography.
Finally, we mixed extracts from cells expressing p64 and p110, and, by determining the position of active p64 on a gel filtration column, examined whether p64 and p110 would bind to each other in solution. We could detect no difference in the elution behavior of p64 in the presence or absence of p110 (Fig. 4 d). In addition, the elution of p23Skp1 from columns was not altered by coexpression with either p64 or p110 (data not shown). While these are negative results, they suggest that p23Skp1, p64, and p110 are unable to form subcomplexes in the absence of p58.
p58, p64, and p110 Form a Complex in the Absence of CEN DNA
Having determined that several different p58-containing CBF3 subcomplexes can form, we asked whether all four CBF3 proteins would associate in the absence of centromeric DNA (as discussed below, we cannot always be certain that p23Skp1 is present in p58-containing complexes, but our data suggest that the majority of p58 is p23Skp1-associated). It has been shown previously that p58, p64, and p110 coelute from a gel filtration column (Lechner and Carbon, 1991), but these earlier experiments did not demonstrate that the proteins were associated in a single complex. To test for the formation of a p58/p64/p110 complex, we looked for a shift in p64 mobility on columns that could arise only from the formation of a three-way p58-p64-p110 complex (or a four-way complex also containing p23Skp1). When extracts containing p23Skp1, p58, p110, and limiting amounts of p64 were analyzed on a Superose 12 column, one population of p64 peaked at fraction 12, the position of p64 alone, and a second population peaked at fraction 5 (Fig. 4 e). Because p58/p23Skp1/p64 complexes peak in fraction 10 (Fig. 4 b), the peak of p64 activity in fraction 5 must represent a complex containing p58, p64, p110, and probably also p23Skp1 (although we cannot rigorously prove, even with GST/p23Skp1, that p23Skp1 is present in the complex). From these data we conclude that a CBF3 complex containing three, and probably four, subunits can form in the absence of CEN DNA.
CBF3 Forms a 15 S Complex with DNA
What is the relationship between the CBF3 subcomplexes detected in solution and DNA-bound CBF3? To investigate this, extracts from insect cells expressing all four CBF3 proteins were incubated with a 56-bp probe spanning CDEIII and the migration of the resulting DNA-protein complexes on sizing columns and glycerol gradients was determined by analyzing fractions directly on nondenaturing bandshift gels. We were unable to recover activity from a gel filtration column, perhaps because the complex dissociated during chromatography. However, following fractionation on velocity gradients, we observed a single peak of CBF3-CDEIII with a sedimentation coefficient of 15.3 S (Fig. 4 f). These data show that CBF3 forms a single compact structure on a 56-bp fragment of CDEIII DNA. As we will discuss in detail in the discussion, these data can be combined with CBF3-DNA protein cross-linking that we have reported previously (Espelin et al., 1997) to generate a preliminary but compelling model for the architecture of CBF3.
Mapping p58 Domains Responsible for p64 and p110 Binding
To determine the significance of the protein-protein interactions we had detected among CBF3 proteins, we wanted to disrupt them one-by-one first in vitro and then in yeast. We focused on mapping binding domains in p58 because the hydrodynamic data suggest that p58 forms a core subunit responsible for mediating interactions among the other three CBF3 proteins. Mapping binding domains involves a simpler assay than hydrodynamic measurements. We fused GST to the amino termini of p23Skp1, p64, and p110 and expressed the resulting fusion proteins in either bacteria or yeast. Purified fusion proteins were incubated with p58 translated in reticulocyte lysate, and the amount of p58 associated with the GST fusion was quantitated following GST capture on glutathione-Sepharose. Using this assay, we observed p58 binding to GSTp23Skp1, GSTp64, and to GSTp110 but not to GST alone or to a series of p110 truncations (Fig. 5 a; data not shown). In a typical reaction, 15–20% of the input p58 bound to GSTp23Skp1 or to GSTp64 following a 4-h incubation at 4°C; binding to GSTp110 was considerably less efficient but still well above background levels. The selectivity in the binding of p58 to the CBF3-GST fusions relative to GST alone and the relatively low concentrations of the soluble and immobilized proteins (∼0.25 pM soluble p58 and 0.2 μM immobilized GST fusion in a reaction with a total protein concentration of 10 mg/ml) confirm that the binding of p58 to p23Skp1, p64, and p110 is specific and of high affinity.
Figure 5.

Mapping p58 sequences required for CBF3 interactions. (a) In vitro binding assay. In vitro translated 35S-p58 was incubated with either GSTp23Skp1, GSTp64, GST-p110, or GST alone immobilized on glutathione-Sepharose beads. The beads were spun down, washed and the bead-bound 35S-p58 was detected by SDS-PAGE and autoradiography. Fourfold more p58 was incubated with the beads (lanes 2–5) than was run as the input (lane 1). The experiment in lane 5 was run separately but is scaled to match the exposure of lanes 1–4. (b) Fragments of p58 were translated in vitro in reticulocyte lysates and tested for their ability to bind to GSTp23Skp1 or GSTp64 expressed in bacteria or to GSTp110 expressed in yeast. Due to variations in the efficiency with which different p58 fragments were translated, the amount of bound p58 was normalized to the amount of input p58 and the results are depicted as follows: +++, >25% bound; ++, 11–25% bound; +, 5–10% bound; and −, <5% bound.
To map regions of the p58 protein responsible for binding to other CBF3 proteins, a series of NH2- and COOH-terminally deleted p58 proteins was translated in vitro and incubated with bead-bound GSTp23Skp1, GSTp64, or GSTp110 (summarized in Fig. 5 b). There was considerable variability in the efficiency with which different p58 fragments were translated, so the extent of binding to p58 was expressed in a manner that corrects for translation efficiency. Both the NH2-terminal p58 truncation (139–479) and the COOH-terminal truncation (1–336) bound as efficiently to GSTp64 as to full-length p58 (Fig. 5 b). This finding suggested that a region sufficient for p64 binding might lie in the sequence common to the two deletions. This was confirmed by using an internal fragment spanning residues 139–336. The 197–amino acid polypeptide containing p58 residues 139–336 is the smallest linear fragment of p58 to which we can detect efficient p64 binding: nine smaller derivatives of p58 were inactive in p64 binding. GSTp110 bound efficiently to p58(1–336) but not to p58(1–271) or p58(139–336) (Fig. 5 b). We conclude from these data that p64 binds to p58 in a region between amino acids 139 and 336. The p110 binding site appears to span a region that lies in the primary structure of p58 just COOH-terminal to the p64 binding site, but partially overlaps it.
The F-box in p58 Is Necessary but Not Sufficient for p23Skp1 Binding
p58 contains an apparent match to the F-box consensus, a sequence thought to constitute a binding site for p23Skp1 (Bai et al., 1996). The failure of an F-box–deleted p58 protein, p58(61–479), to bind to p23Skp1 but not to GSTp64 or GSTp110 shows that the F-box is required for p58-p23Skp1 association (Fig. 5 b). To probe sequence requirements in the F-box, 7 single and double point mutations were introduced into residues that are highly conserved among F-box proteins, and 10 mutations were introduced into nonconserved but charged residues. Mutations that changed conserved F-box amino acids (L12A, L12D, P13A, L12A/ P13A, V20A, Y21R, and L24A) abolished the binding of p58 to p23Skp1 (Fig. 6 a, lanes 6, 10, 14, 18, 26, and 30; see also summary in Fig. 6 b). These mutations did not appear to disrupt the overall structure of p58, because binding to p64 was unaffected (Fig. 6 a, lanes 8, 12, 16, 20, 24, 28, and 32). None of the mutations made at nonconserved but charged residues had any effect on p58 binding to either p23Skp1 or to p64 (Fig. 6 a, lanes 33–64). We conclude that the F-box in p58 is required for p23Skp1 binding and that this binding primarily involves amino acids conserved among F-box family members. The majority of these conserved F-box residues are nonpolar, suggesting that interaction of p23Skp1 with p58 is mediated largely by hydrophobic contacts.
Figure 6.
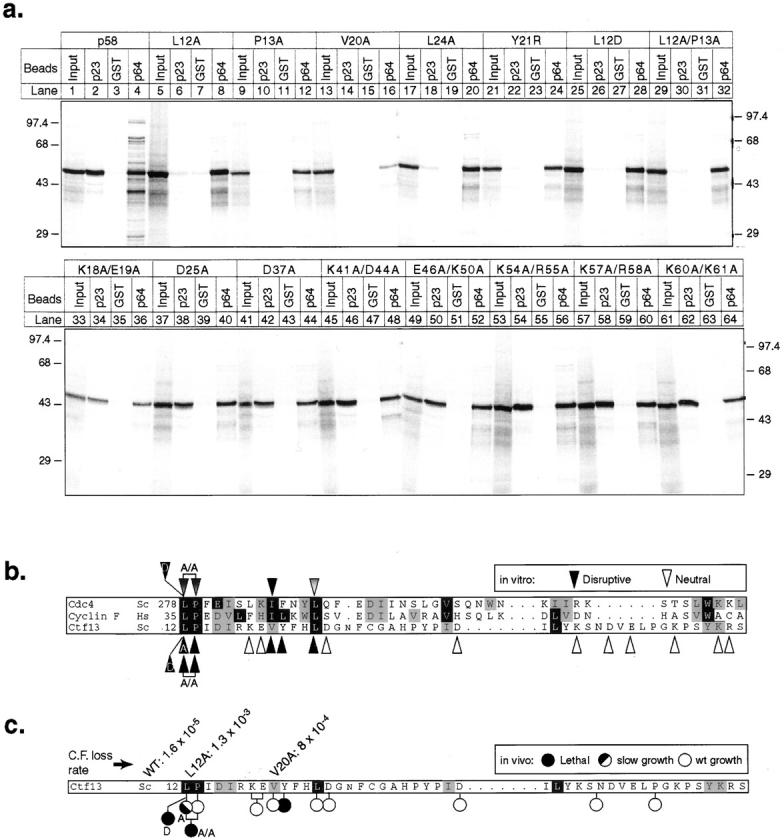
p58 contains a functional F-box that is required for binding to p23Skp1. (a) p58 variants containing point mutations in the F-box were translated in rabbit reticulocyte lysates and assayed for their ability to bind to GSTp23Skp1, GSTp64, or GST alone using the in vitro binding assay described in Fig. 1 a. (b) Alignment of p58 with the F-box proteins Cdc4p and Cyclin F. The consensus residues in the F-box are indicated by the dark (highly conserved) or gray (somewhat conserved) boxes. Note that not all of the “consensus” residues are found in the p58 F-box (p58-Y21 for example is in a conserved position, but does not match the consensus leucine). Black arrows indicate point mutations that abolished p58/p23Skp1 interactions in vitro and white arrows indicate point mutations that had no effect (as shown in a). Brackets and the A/A notation indicate the introduction of a double point mutation (L12A/P13A) into p58. p58 mutations are indicated below the alignment and Cdc4p mutations above. (c) Sequence of p58 showing the positions of F-box mutants that were tested for their ability to function in vivo. Mutant alleles inviable in vivo are marked with black circles (L12D, L12A-P13A, Y21R), alleles that exhibited wild-type growth in vivo are marked with white circles (P13A, K18A/E19A, V20A, L24A, D25A, D36A, K40A/D43A, E45A/K49A, K53A/R54A, K56A/R57A, and K59A/K60A), and a single allele that exhibited slow growth is marked with a black/white circle (L12A). From the class of mutants that were viable but showed no p58-p23Skp1 interaction in vitro, the L12A and V20A were selected for further analysis. The rate of loss of a nonessential tester chromosome (per generation) is shown for these mutant proteins and for a control wild-type strain.
To compare our in vitro observations to data obtained in cells, we examined the ability of p58 proteins carrying F-box mutations to function in vivo in place of wild-type p58. Mutant p58 genes were integrated at the leu2 locus in yeast strains carrying a deletion of the gene encoding p58 (using plasmid shuffling, see Materials and Methods). Of the mutations that impaired p58-p23Skp1 binding in vitro, we found that three mutations in consensus F-box residues (L12D, L12A-P13A, and Y21A) either prevented or severely impaired cell growth, and that two others caused a 10–100-fold increase in the rate of chromosome loss (L12A, V20A; Fig. 6 c). In contrast, mutations in nonconsensus F-box residues had no effect on cell growth (Fig. 6 c, white circles). We conclude from these observations that the F-box in p58 is required for function in vivo, presumably because p23Skp1-p58 binding is required for p58 activation in cells just as it is in vitro.
Is the p58 F-box sufficient for p23Skp1 binding? We observed that an NH2-terminal fragment of p58 encompassing the F-box alone (residues 1–58) did not bind to p23Skp1 either in reticulocyte lysates (Fig. 5 b) or in yeast extracts (Fig. 7 b). Binding of p23Skp1 was observed to p58 proteins lacking the extreme COOH terminus (1–412) but not to more extensively truncated p58 derivatives. We conclude that the F-box in p58 is not sufficient for binding to p23Skp1 and that at least one additional region of p58, probably located between residues 336 and 412, is also required.
Figure 7.

Probing p58 interactions in yeast. (a) p58 activity in eluates from a Superose 12 column to which extracts from p23Skp1/p58 coexpressing insect cells (open circles), p23Skp1/ p58/p64 coexpressing insect cells (open squares), or wild-type yeast cells (solid line) were applied. The peak of p58 activity in the yeast extract overlaps the peak of the recombinant p23Skp1-p58-p64 co-complex. (b) Full-length or truncated p58 was fused to GST and expressed from the GAL1 promoter in yeast. GSTp58 and associated proteins were purified using glutathione-Sepharose, and the Sepharose-bound proteins analyzed by immunoblotting using anti-p23Skp1, anti-p64 or anti-GST as indicated (lanes 1–4). A similar analysis of proteins bound to GSTp110 was performed as a negative control (lane 5). The ability of each p58 fragment or mutant to complement a p58 deletion in vivo is indicated in the bottom panel. Unfused p58(L12A/P13A) protein (note 1) was unable to complement function in vivo; the GSTp58(L12A/P13A) fusion protein was not tested.
CBF3 Subcomplexes in Yeast
Having mapped regions of p58 involved in binding to p23Skp1, p64, and p110 in vitro, we wanted to determine the consequences of disrupting various interactions for p58 activation and destruction in cells. First, it was necessary to determine that the contacts we had mapped using recombinant proteins were also important in yeast. We therefore examined the state of association of p58 in wild-type yeast by fractionation on a gel filtration column. We observed that the bulk of activated p58 was present in a peak centered on fraction 10, the same position as recombinant p58-p23Skp1-p64 (Fig. 7 a). Some activated p58 in yeast extracts eluted earlier from the column, indicating that a portion of the protein is probably bound to p110 or to other unidentified proteins. Thus, p58 in wild-type yeast is predominantly complexed with p64 and very little is present as a simple p23Skp1-p58 heterodimer. Consistent with this, Western blotting shows that GSTp58 is associated with p23Skp1 and with p64 when isolated from yeast extracts (Fig. 7 b, lane 1).
Next, we examined various mutated p58 proteins in yeast. As expected from our in vitro data, a double point mutation in the p58 F-box (L12A/P13A; Fig. 7 b, lane 2) abolishes binding to p23Skp1 but not to p64. p58-p64 association was eliminated, however, in the GSTp58(Δ139–336) protein by the deletion of the presumed p64 binding site between residues 139 and 336 (compare Fig 7 b, lanes 1 and 3). These data show that the p58 sequences determined to be important for protein-protein interactions among recombinant CBF3 subunits are also required in vivo. In particular, we find (a) that the p58 F-box is required for the binding of p23Skp1 to p58 in vivo but that it is not sufficient (Fig. 7 b, lane 4) and (b) that an internal region of p58 between residues 139 and 336 is required for p64 binding.
Stabilization of p58 by p64 Binding
To estimate the half-life of p58 when it is a part of various binary complexes containing other CBF3 subunits, we raised the levels of the complexes in cells by overexpressing p58 and either p23Skp1, p64, or p110 under control of the GAL1 promoter in otherwise wild-type cells. In all cases, Western blotting confirmed that CBF3 proteins were being expressed at elevated levels (data not shown). The task of comparing the half-lives of full-length and truncated p58 proteins was made easier by fusing p58 to GST. GST is normally very stable in yeast (half-life of >5 h; Fig. 8 a) but the half-life of GSTp58, ∼15 min, was indistinguishable from that of wild-type p58 (Kaplan et al., 1997), showing that the instability of p58 can be transferred to a fused GST moiety. The stability of p58 was measured by placing the protein under the control of the GAL1 promoter, repressing transcription by the addition of glucose, preparing cell extracts at various times following transfer to glucose media, and then determining GSTp58 levels by Western blotting. The relative stability of p23Skp1, p64, and p110 (half-lives of >5 h; Kaplan et al., 1997) ensured that they were present at similar levels at the start and end of the p58 transcriptional pulse-chase. We observed that overexpression of p64, but not p23Skp1 or p110, increased the half-life of GSTp58 dramatically (compare Fig. 8, b and c). To rule out the possibility that p64, which is a DNA-binding protein, was altering p58 transcription, we measured p58 message levels by Northern blotting in wild-type cells and in cells overexpressing p64 and determined that they were similar (data not shown). We conclude from these findings that the overexpression of p64 has a direct effect on the half-life of p58 protein.
Figure 8.

GSTp58 is stabilized in vivo by the overexpression of p64 but not p110 or p23Skp1. (a) Promoter shut-off experiments were used to compare the stability of GSTp58 (squares) to GST alone (circles). Yeast cells containing plasmid borne GAL1-GSTp58 and GAL1-GST were grown in galactose media, a sample was chilled and reserved for time 0, and the remaining cells were washed into glucose media, grown at 30°C, and aliquots removed to ice at 30 and 90 min following addition of glucose. Extracts were prepared and GST fused proteins purified on glutathione-Sepharose from 2 mg of total yeast protein by binding to glutathione-Sepharose. Bead-bound GST fusions were loaded in duplicate lanes on SDS-containing gels and GST proteins were detected by immunoblotting with an anti-GST antibody and 125I-labeled protein A. The rate of degradation of HA-tagged p58 (HAp58) is also plotted for comparison (diamonds; see Kaplan et al., 1997). (b) Stability of GSTp58 in a wild-type strain (circles) or a strain containing an integrated copy of GAL1-p64 (squares) as determined by promoter shut-off. (c) Stability of GSTp58 in a strain containing an integrated copy of GAL1-p23 (squares) or GAL1-p110 (circles). (d) Stability of the GAL1-GSTp58(Δ139–336) proteins, which does not bind to p64, in a wild-type strain (circles) or a strain containing an integrated copy of GAL1-p64 (squares). (e) Rate of degradation of GST fusions to an internal deletion of p58 lacking a putative PEST sequence (Δ211–230; circles), to p58 carrying a double point mutation in the F-box (L12A/P13A, squares), (f) to the p58 F-box (1–58; circles) or to an NH2-terminal truncation of p58 lacking the F-box (139–479; diamonds). Each data point represents the average of several independent determinations.
Next, we asked whether overexpressed p64 was in fact forming a complex with GSTp58 and whether complex formation was required for p58 stabilization. We isolated GSTp58 on glutathione beads and measured the level of bound p64 by Western blotting (see below). This showed that the amount of GSTp58/p64 complex was three- to fourfold higher in cells overexpressing p64 than in wild-type cells. To demonstrate that the formation of the GSTp58/p64 complex was required for p58 stabilization, we analyzed the GSTp58(Δ139–336) deletion, which does not bind to p64 (Fig. 7 b). At steady state, GSTp58(Δ139– 336) was two- to threefold less abundant than wild-type p58 and was slightly less stable. Importantly, the half-life of GSTp58(Δ139–336) was unaltered by p64 overexpression (Fig. 8 d) demonstrating that p64 exerts its effect on p58 by directly binding to the p58 protein.
Multiple Pools of Active p58
Why are the kinetics of p58 degradation biphasic? In cells overexpressing p64, p58 levels fell as rapidly as in wild-type cells (t 1/2 15 min) between 0 and 30 min but then remained essentially constant for >2 h (a similar effect was observed with unfused p58 in the presence of overexpressed p64, data not shown). The simplest explanation is that there are two pools of p58 in the cell, free p58 and p64-associated p58, and that the half-life of the p64-associated p58 is substantially longer than that of free p58. If this hypothesis is correct, the levels of the p58/p64 complex should remain nearly constant following a transcriptional shut-off, even as the levels of total p58 protein fall rapidly (with a half-life of 15 min). If the hypothesis is wrong, both free p58 and p64-bound p58 should be degraded with similar kinetics. As a test, we expressed GSTp58 under the control of the GAL1 promoter and then isolated GSTp58 and tightly bound p64 on glutathione-Sepharose at various times after transcriptional shut-off. The levels of bead-bound GSTp58 and p64 were determined by Western blotting. We observed that the amount of GSTp58 fell sixfold in 90 min, consistent with the previously determined 15 min half-life, but the amount of p58/p64 complex fell by <20% (Fig. 9, a and b). Furthermore, in cells overexpressing p64, a similar phenomenon was observed, except that the absolute levels of p58/p64 were four- to fivefold higher and p58 was much longer lived (Fig. 9 c). We interpret this to mean that there is indeed a special pool of p64-bound p58 that is more stable than the free p58 (Fig. 9 d).
Figure 9.
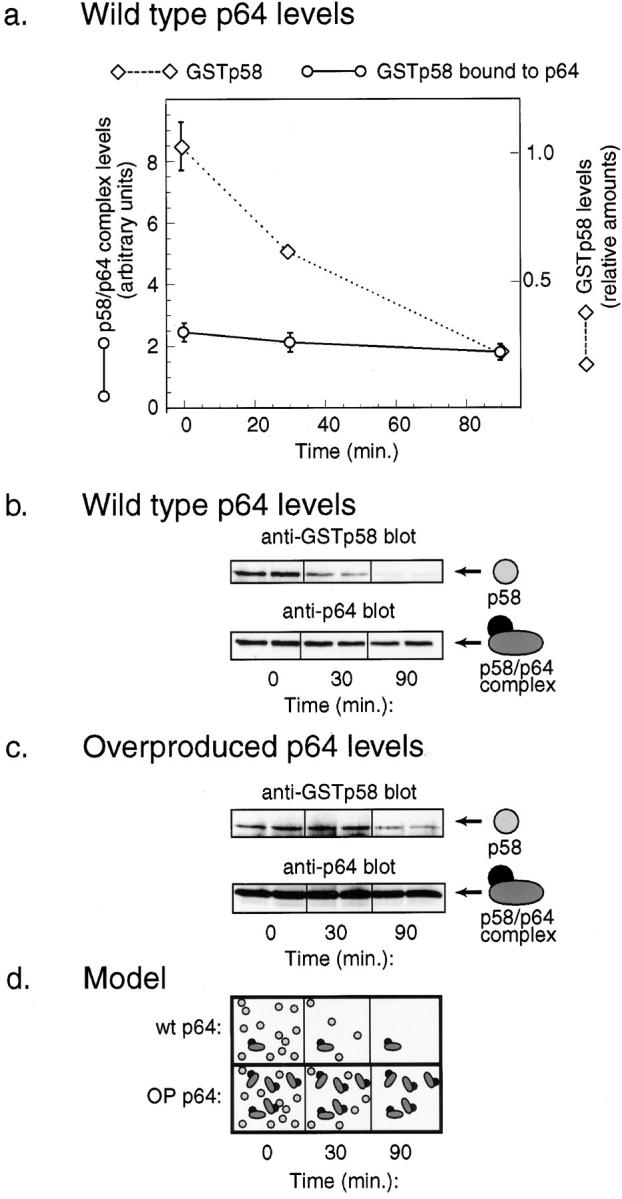
Selective stabilization of p58 that is p64-associated. (a) Rate of degradation of total GSTp58 (diamonds) compared to degradation of GSTp58 complexed with p64 (circles). The amount of total GSTp58 was determined by blotting as described in Fig. 8. The amount of GSTp58 in complex with p64 was determined by isolating GSTp58 on glutathione-Sepharose and then blotting for p64. (b) Western blot of the data shown in part a. (c) Degradation of total GSTp58 and p64-associated GSTp58 in cells overexpressing p64. (d) Model interpreting the data in a and c showing two pools of p58: an unstable pool of free p58 (circles) and a stable pool of p64-bound p58 (oval and circle).
Thus, we have shown that the overexpression of p64 stabilizes p58. This effect is specific to p64, and is not observed with overexpressed p23 or p110 (Fig. 8, b and c). Moreover, the stabilization of p58 requires the formation of a p58/p64 complex (Fig. 8 d). From these data we conclude that the stabilization of the p58 polypeptide is linked to its assembly into a p58/p64 CBF3 subcomplex that, while not competent to bind DNA on its own (Kaplan et al., 1997), is probably an intermediate in kinetochore formation.
Instability is conferred on proteins such as mitotic cyclins by a discrete destruction box that is necessary and sufficient for promoting ubiquitin-mediated degradation. However, in many other proteins, instability determinants are found throughout a protein. To explore instability determinants in p58, we determined the half-lives of truncated p58 variants following fusion to GST. Because the p23Skp1-containing SCF complex is required for the destruction of a number of cell cycle regulators in yeast, including G1 cyclins (Willems et al., 1996) and the p34Cdc28 kinase inhibitor Sic1p (Bai et al., 1996; Feldman et al., 1997; Skowyra et al., 1997; Verma et al., 1997), we wondered whether p23Skp1 binding was required for p58 degradation. We found that a p58 variant lacking the F-box, p58(139–479) was unstable (Fig. 8 e), as was p58 with a double point mutant in the F-box p58 (L12A/P13A; Fig. 8 f). In addition, an apparent PEST sequence, identified by the PEST-FIND algorithm of Rechsteiner and Rogers (1996) as lying between residues 211 and 231, was not necessary for p58 instability (Fig. 8 f). Intriguingly, however, the p58 F-box (residues 1–58) was sufficient to confer instability on GST (Fig. 8 e). In all, we examined seven different fusions between GST and p58 and observed that all had a half-life of 15–20 min (Fig. 8 and data not shown). Considered together, these data show that sequences conferring rapid degradation on p58 are present at multiple locations in the p58 polypeptide and that neither p23Skp1 binding nor PEST sequences are required.
Discussion
CBF3 is an essential component of the yeast kinetochore that binds to the critical CDEIII region of centromeric DNA. An important goal of this work was to probe the structure of the CBF3 proteins and to investigate how they associate to form a functional CBF3 complex. To this end, we have used hydrodynamic methods to determine the stoichiometries and approximate physical dimensions of individual CBF3 proteins and of complexes containing two or more different proteins. The primary advantages of using calibrated gradients and columns to examine complex formation are that one can follow active subsets of proteins using functional assays and that the stoichiometries of various complexes can be determined directly. In addition, however, we have confirmed the existence of the critical protein-protein interactions using pull-down assays from reticulocyte lysates and from yeast extracts.
We have found that all of the CBF3 proteins are multimeric. p110 exists as an elongated homodimer, p64 forms a homodimer, and p23Skp1 and p58 form a stable heterodimer. Protein hydrodynamics yield a fairly crude picture of a protein's structure, but our calculations for the stoichiometries of individual subunits are close to integral, as one would hope, and estimates for the stoichiometries of various binary and ternary complexes yield numbers that are the sums of the masses of the individual subunits. We therefore believe that the information is sufficiently precise to build a low resolution model for the CBF3 complex. To represent this information pictorially, we have modeled the CBF3 proteins as ellipsoids (with circular cross-sections; see Fig. 10 a).
Figure 10.

Models for individual CBF3 subunits and for the CBF3-CDEIII DNA complex. (a) Representation of CBF3 subunits as ellipsoids (with circular cross-sections) whose longest axis is equal to twice the Stokes radius and whose shorter axis is calculated using the experimentally determined mass and a typical value for protein density. p64 is shown as having two domains. Interactions that have been detected among CBF3 subunits are shown with heavy arrows. X denotes those interactions that have not been be detected in solution. (b) Enlarged cross-section of the CBF3-DNA complex showing the interaction of p58 and p64 with conserved bases in CDEIII. The boxes mark bases that are absolutely conserved among all 16 yeast centromeres and arrows indicate the positions of the putative p64 half-sites. Cross-linking data are summarized from Espelin et al. (1997). Below the enlargement is the proposed arrangement of CBF3 subunits on CDEIII DNA. A 184-bp piece of DNA with an extended CBF3 complex is shown. For a sense of scale, the dotted circles show the approximate diameter of a nucleosome and a microtubule in cross-section. The dotted box indicates the region of the model used to generate the cross-sectional view shown in section c. A model for organization of the core CBF3-CDEIII complex on 56 bp of centromeric DNA. The complex contains one p58 subunit, one p23Skp1, two p64s, and two p110s. The calculated mass of this complex (450 kD) and the experimentally determined sedimentation coefficient of 15 S yield a Stokes radius of 70 Å. CDEIII has been bent at an arbitrary position to conform to this 70 Å length. (d) Model for the regulation of CBF3 formation by p58. Newly synthesized p58 is activated by phosphorylation in a p23Skp1-dependent manner. Both active and inactive p58 are subject to ubiquitination and subsequent degradation. p58 bound by p64 assumes a conformational state in which it is resistant to degradation. However, p64-bound p58 remains in equilibrium with the free p58. Association of the p58/p64 complex with p110 and centromeric DNA leads to the formation of a stable complex that dissociates only very slowly.
p58 Is a Central Component of CBF3
In solution, p58 forms a complex with p23Skp1, p64, and p110, but these proteins do not appear to associate stably with each other in the absence of p58. This suggests that p58 forms the core of CBF3 and mediates stable interactions among the other CBF3 proteins (Fig. 10 a). The primary complex formed by native p58 in yeast extracts and recombinant p58 appears to be a stable heterotrimeric assembly containing one copy of p23Skp1, one copy of p58, and two copies of p64. Reflecting its role as a bridging subunit, p58 can be divided into distinct functional regions. The amino terminus of p58 contains a fairly good match to the F-box consensus (Bai et al., 1996) that is necessary but not sufficient for binding to p23Skp1; a COOH-terminal region of p58 is also required. The binding of p64 and p110 to p58 appears to be mediated by sequences that lie approximately in the center of the p58 polypeptide (residues 139–336). The observation that the p23Skp1 binding sites do not overlap p64 and p110 binding sites is consistent with our finding that p58 binds independently to p23Skp1, p64, and p110, and, at the resolution of our current assay, the presence of p23Skp1 neither reduces nor enhances p64 and p110 binding (data not shown). However, preliminary data obtained from the analysis of recombinant CBF3 suggest that the efficient formation of a four-protein CBF3 complex requires p23Skp1-mediated activation of p58, perhaps because unactivated p58 assumes a structure in which p64 and p110 exclude each other from a shared binding site (Kaplan, K., and P.K. Sorger, unpublished observations).
Binding Sites for p23Skp1 in p58
At least 10 proteins from S. cerevisiae contain potential F-boxes (Bai et al., 1996). These proteins function in physiologically diverse pathways including methionine biosynthesis (Patton et al., 1998), glucose repression (Li and Johnston, 1997; Kishi et al., 1998), protein ubiquitination (Bai et al., 1996; Feldman et al., 1997; Skowyra et al., 1997; Verma et al., 1997), and chromosome segregation (Connelly and Hieter, 1996). In three yeast proteins, Cdc4p (Bai et al., 1996), Grr1p (Li and Johnston, 1997; Kishi et al., 1998), and p58 (this work), and in one human protein, Skp2 (Lisztwan et al., 1998; Lyapina et al., 1998), point mutations or deletions in the F-box have been shown to impair p23Skp1 binding. In p58, we have examined 15 point mutations in a 45-residue sequence that spans the F-box and shown that 6 mutations abolish p23Skp1 binding in vitro. Three of these mutations also severely impair cell growth and several others increase the rate of chromosome loss. All of the F-box residues shown by mutagenesis to be important for p23Skp1 binding are nonpolar, suggesting that the interaction of p23Skp1 with the F-box is mediated primarily by hydrophobic contacts. It is not clear whether the COOH-terminal region of p58 that is also required for p23Skp1 binding has a counterpart in other F-box proteins. p58 is not detectably similar in sequence to Cdc4p and Grr1 outside the F-box.
Does p64 Function as a Traditional Zinc Cluster Protein?
The amino terminus of p64 contains a Zn2Cys6 zinc cluster (Lechner, 1994) that in Gal4p contains six cysteines coordinating two zinc ions and fits into the major groove of DNA (Marmorstein et al., 1992; Marmorstein and Harrison, 1994; Swaminathan et al., 1997). Well-characterized members of the fungal zinc cluster family bind to DNA as dimers, and recognize sequences that contain either direct or inverted repeats of the DNA sequence CCG (Marmorstein et al., 1992; Marmorstein and Harrison, 1994; Zhang and Guarente, 1994; Swaminathan et al., 1997). In Gal4p, dimerization is mediated in part by coiled coils found in an arm that lies COOH-terminal to the zinc cluster. We have found that p64 is also dimeric, and that its zinc cluster is linked to the remainder of the p64 protein by a proteolytically accessible sequence that may represent a linker arm. Despite these similarities with Gal4p, p64 is unique among studied zinc cluster proteins in that it does not bind to DNA except as part of a multiprotein complex. Does this reflect an inherent property of p64 or is it a feature (such as low affinity) of the CDEIII binding site? DNA-protein cross-linking (Espelin et al., 1997) shows that p64 contacts a CCG sequence at the center of CDEIII that is essential for centromere function (Hegemann and Fleig, 1993) and a TGT (or ACA on the bottom strand) 12 bp to the left that is essential for CBF3 binding (Fig. 2 e and Fig. 10 b). Among Zn2Cys6 zinc cluster proteins, the requirement for two intact CCGs is not absolute: in vitro selection yielded Hap1p binding sites that contain a single intact CCG and a second degenerate CCG in which only the central C is conserved (Zhang and Guarente, 1994). When CDEIII is mutated to generate sequences with two “perfect” CCG sites in various orientations, not only is p64 unable to bind to the mutant CDEIII on its own, but CBF3 binding is largely abolished (Fig. 2 e). We therefore postulate that p64 binds to CDEIII only in the context of a CBF3 complex not because CDEIII contains a low-affinity binding site, but because free p64 is inactive.
Modeling the CBF3-DNA Complex
To build a preliminary structural model for the CBF3-DNA complex we have combined the protein data presented in this paper with earlier results from DNA-protein cross-linking (Espelin et al., 1997). Based on the stoichiometries of the CBF3 proteins in solution, the simplest composition for CBF3 bound to a 56-bp CDEIII fragment is one copy of p23, one p58, two p64s, and two p110s. The stoichiometries and apparent shapes of the CBF3 proteins are consistent with the pattern of DNA-protein cross-linking. For example, p64 cross-links to two putative half-sites about 40 Å apart, p58 cross-links at bases midway between the p64 cross-link sites, and p110 is apparently in contact with an extended region of CDEIII, consistent with its elongated shape (Fig. 10 c). Thus, the close association observed in solution between p58 and p64 also exists on DNA. The predicted mass of a CBF3 complex with the composition shown in Fig. 10 c is 450 kD (including DNA). Although problems with the analysis of CBF3-DNA complexes on gel filtration columns have prevented us from confirming this mass by hydrodynamic methods, we can use the measured 15 S sedimentation coefficient and the 450-kD predicted mass to arrive at a proposed Stokes radius for DNA-bound CBF3 of 70 Å. This radius implies that the CDEIII DNA is bent and we have therefore bent CDEIII at an arbitrary position in our model.
The CBF3-DNA core complex we have modeled is the smallest complex that can associate stably with DNA. We have detected additional binding sites for p110 in the centromeric DNA that lies outside the 56-bp fragment used here (Espelin et al., 1997) and believe that the CBF3-DNA structure that assembles in cells is considerably more complicated than the structure shown in Fig. 10. In addition, yeast kinetochores contain other DNA-binding proteins including CBF1 and specialized histones (for review see Clarke, 1998) and these proteins probably interact with CBF3. Nevertheless, much of the sequence specificity in CBF3-centromere interaction lies in the core complex. Thus, the structural model we have developed is a critical first step in determining the architecture of the larger yeast kinetochore. In the future, by probing longer fragments of centromeric DNA, including additional kinetochore proteins and using methods such as atomic force microscopy that are suitable for the analysis of large DNA-protein complexes, we intend to extend our structural model beyond the core CBF3 complex.
Assembly-dependent Stabilization of p58
p58 is subject to both positive and negative regulation: it is activated by phosphorylation and degraded by the proteosome (Kaplan et al., 1997). We have proposed previously that coupled activation and destruction of p58 may control the amount of active CBF3 and thereby increase the fidelity of kinetochore assembly. We imagine an editing process in which p58, correctly incorporated into functional CBF3 and assembled on centromeric DNA, is protected from degradation, whereas soluble, unassembled p58 is degraded. Implicit in this proposal are two key postulates: that p58 plays a critical role in CBF3 assembly and that assembly regulates p58 stability. Evidence for the first postulate is the finding that p58 constitutes the structural core of the CBF3 complex. Because p23Skp1, p64, and p110 form stable complexes in solution only with p58 and not with each other, the regulation of p58 abundance and activity directly controls that state of association among all of the CBF3 subunits.
Evidence for the second postulate is the observation that raising the intracellular concentration of the p58/p64 complex (but not other p58-containing complexes) greatly stabilizes p58. A similar type of regulation has been observed with the mating type specific transcription factor a1/α2 (Johnson et al., 1998). When not associated with a1 (in a cells for example), α2 is ubiquitinated and rapidly degraded. Similarly, a1 is rapidly degraded when not associated with α2. However, when assembled into an a1/α2 complex, both a1 and α2 are protected from proteolysis. One of the instability determinants in α2 lies in the α helix that mediates interaction with a1; thus, the stabilization of α2 by a1 involves a direct masking of an instability determinant. A similar mechanism may be operating in p58 but we do not yet have enough data to localize the key regulatory sequences.
We propose the following pathway for CBF3 assembly (Fig. 10 d). Newly synthesized p58 is activated by phosphorylation in a p23Skp1-dependent manner. Both active and inactive p58 are subject to ubiquitination by SCFcdc4, and thus to degradation by the 26 S proteosome (Kaplan et al., 1997). Those p58 molecules that bind to p64 are protected from degradation, perhaps because they are no longer substrates for SCFcdc4. The p58/p64 complex is not particularly stable and can dissociate into free p58 and p64, again exposing p58 to the degradation pathway. However, p58/p64 complexes that bind to p110 and to centromeric DNA assemble into a CBF3 complex that dissociates only very slowly from DNA (the rate of dissociation of DNA-bound CBF3 is at least 3 h in vitro; Espelin et al., 1997). Thus, the half-life of p58 increases as CBF3 assembles, but it is only when a high-affinity CBF3 complex forms on centromeric DNA that p58 is fully protected from degradation.
To our knowledge, there are few structures in a cell for which the copy number of active complexes must be as closely controlled as the number of active kinetochores. We believe that the high selectivity of kinetochore assembly is ensured not only by the intrinsic sequence specificity of CBF3-CDEIII interactions, but also by the regulation of CBF3 abundance and activity. This regulation of abundance and activity appears to comprise an assembly-dependent editing mechanism that balances two competing demands. Following the replication of chromosomes in S-phase, sufficient active CBF3 must be present to occupy the centromeres on all chromosomes. However, CBF3 must be maintained at low concentrations to reduce the possibility of ectopic kinetochore formation and the generation of highly unstable dicentric chromosomes. Linking p58 stability to CBF3 assembly and to DNA binding would couple the number of active CBF3 complexes to the number of centromeres.
Acknowledgments
This work was supported by a Fellowship from Merck and Co. to I.D. Russell, by the Lucille P. Markey Foundation and the Searle Scholars Progam, and by Travel Grant CRG.960163 from the North Atlantic Treaty Organization, and grant GM51464 from the National Institutes of Health.
Note Added in Proof:
Pietrasanta et al. have recently used AFM to examine CBF3-DNA complexes in vitro. Their estimated molecular mass for the complex agrees well with values reported here. They also show directly that CBF3 bends DNA as our results would suggest (Pietrasanta, L.I., D. Thrower, W. Hsieh, S. Rao, O. Stemmann, J. Lechner, J. Carbon, and H. Hansma. 1999. Probing the Saccharomyces cerevisiae centromeric DNA (CEN DNA)-binding factor 3 (CBF3) kinetochore complex by using atomic force microscopy. Proc. Natl. Acad. Sci. USA. 96:3757–3762.
Footnotes
We are particularly indebted to Tony Hyman, Bob Sauer, Steve Bell, Caroline Shamu, Ken Kaplan, and Liz Alcamo for help with experiments and with this manuscript; and to C. Correll and R. Deshaies for generous gifts of reagents.
The first two authors contributed equally to this work.
References
- Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Clarke L. Centromeres: proteins, protein complexes, and repeated domains at centromeres of simple eukaryotes. Curr Opin Genet Dev. 1998;8:212–218. doi: 10.1016/s0959-437x(98)80143-3. [DOI] [PubMed] [Google Scholar]
- Connelly C, Hieter P. Budding yeast SKP1encodes an evolutionarily conserved kinetochore protein required for cell cycle progression. Cell. 1996;86:275–285. doi: 10.1016/S0092-8674(00)80099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doheny KF, Sorger PK, Hyman AA, Tugendreich S, Spencer F, Hieter P. Identification of essential components of the S. cerevisiaekinetochore. Cell. 1993;73:761–774. doi: 10.1016/0092-8674(93)90255-O. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espelin CW, Kaplan KB, Sorger PK. Probing the architecture of a simple kinetochore using DNA–protein crosslinking. J Cell Biol. 1997;139:1383–1396. doi: 10.1083/jcb.139.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman RM, Correll CC, Kaplan KB, Deshaies RJ. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- Fitzgerald-Hayes M, Clarke L, Carbon J. Nucleotide sequence comparisons and functional analysis of yeast centromere DNAs. Cell. 1982;29:235–244. doi: 10.1016/0092-8674(82)90108-8. [DOI] [PubMed] [Google Scholar]
- Goh PY, Kilmartin JV. NDC10: a gene involved in chromosome segregation in Saccharomyces cerevisiae. . J Cell Biol. 1993;121:503–512. doi: 10.1083/jcb.121.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie, C., and G.R. Fink. 1991. Guide to Yeast Genetics and Molecular Biology. Academic Press, Inc., San Diego, CA. 933 pp.
- Hegemann JH, Fleig UN. The centromere of budding yeast. Bioessays. 1993;15:451–460. doi: 10.1002/bies.950150704. [DOI] [PubMed] [Google Scholar]
- Hieter P, Mann C, Snyder M, Davis RW. Mitotic stability of yeast chromosomes: a colony color assay that measures nondisjunction and chromosome loss. Cell. 1985a;40:381–392. doi: 10.1016/0092-8674(85)90152-7. [DOI] [PubMed] [Google Scholar]
- Hieter P, Pridmore D, Hegemann J, Thomas M, Davis R, Phillippsen P. Functional selection and analysis of yeast centromeric DNA. Cell. 1985b;42:913–921. doi: 10.1016/0092-8674(85)90287-9. [DOI] [PubMed] [Google Scholar]
- Jiang W, Lechner J, Carbon J. Isolation and characterization of a gene (CBF2) specifying a protein component of the budding yeast centromere. J Cell Biol. 1993;121:513–519. doi: 10.1083/jcb.121.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PR, Swanson R, Rakhilina L, Hochstrasser M. Degradation signal masking by heterodimerization of MATalpha2 and MATa1 blocks their mutual destruction by the ubiquitin-proteasome pathway. Cell. 1998;94:217–227. doi: 10.1016/s0092-8674(00)81421-x. [DOI] [PubMed] [Google Scholar]
- Kaplan, K.B., and P.K. Sorger. 1997. Purification of sequence-specific DNA-binding proteins. In Protein Function: A Practical Approach. T.E. Creighton, editor. Oxford University Press, Oxford. 245–278.
- Kaplan KB, Hyman AA, Sorger PK. Regulating the yeast kinetochore by ubiquitin-dependent degradation and Skp1p-mediated phosphorylation. Cell. 1997;91:491–500. doi: 10.1016/s0092-8674(00)80435-3. [DOI] [PubMed] [Google Scholar]
- Kingsbury J, Koshland D. Centromere function on minichromosomes isolated from budding yeast. Mol Biol Cell. 1993;4:859–870. doi: 10.1091/mbc.4.8.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi T, Seno T, Yamao F. Grr1 functions in the ubiquitin pathway in Saccharomyces cerevisiaethrough association with Skp1. Mol Gen Genet. 1998;257:143–148. doi: 10.1007/s004380050633. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Bebenek K, McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- Lechner J. A zinc finger protein, essential for chromosome segregation, constitutes a putative DNA binding subunit of the Saccharomyces cerevisiaekinetochore complex Cbf3. EMBO (Eur Mol Biol Organ) J. 1994;13:5203–5211. doi: 10.1002/j.1460-2075.1994.tb06851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner J, Carbon J. A 240kd multisubunit protein complex, CBF3, is a major component of the budding yeast centromere. Cell. 1991;64:717–726. doi: 10.1016/0092-8674(91)90501-o. [DOI] [PubMed] [Google Scholar]
- Li FN, Johnston M. Grr1 of Saccharomyces cerevisiaeis connected to the ubiquitin proteolysis machinery through Skp1: coupling glucose sensing to gene expression and the cell cycle. EMBO (Eur Mol Biol Organ) J. 1997;16:5629–5638. doi: 10.1093/emboj/16.18.5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisztwan J, Marti A, Sutterluty H, Gstaiger M, Wirbelauer C, Krek W. Association of human CUL-1 and ubiquitin-conjugating enzyme CDC34 with the F-box protein p45(SKP2): evidence for evolutionary conservation in the subunit composition of the CDC34–SCF pathway. EMBO (Eur Mol Biol Organ) J. 1998;17:368–383. doi: 10.1093/emboj/17.2.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyapina SA, Correll CC, Kipreos ET, Deshaies RJ. Human CUL1 forms an evolutionarily conserved ubiquitin ligase complex (SCF) with SKP1 and an F-box protein. Proc Natl Acad Sci USA. 1998;95:7451–7456. doi: 10.1073/pnas.95.13.7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein R, Harrison SC. Crystal structure of a PPR1-DNA complex: DNA recognition by proteins containing a Zn2Cys6 binuclear cluster. Genes Dev. 1994;8:2504–2512. doi: 10.1101/gad.8.20.2504. [DOI] [PubMed] [Google Scholar]
- Marmorstein R, Carey M, Ptashne M, Harrison SC. DNA recognition by GAL4: structure of a protein-DNA complex. Nature. 1992;356:408–414. doi: 10.1038/356408a0. [DOI] [PubMed] [Google Scholar]
- Patton EE, Willems AR, Sa D, Kuras L, Thomas D, Craig KL, Tyers M. Cdc53 is a scaffold protein for multiple Cdc34/Skp1/F-box proteincomplexes that regulate cell division and methionine biosynthesis in yeast. Genes Dev. 1998;12:692–705. doi: 10.1101/gad.12.5.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;7:267–271. [PubMed] [Google Scholar]
- Schjerling P, Holmberg S. Comparative amino acid sequence analysis of the C6 zinc cluster family of transcriptional regulators. Nucleic Acids Res. 1996;24:4599–4607. doi: 10.1093/nar/24.23.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel LM, Monty KJ. Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim Biophys Acta. 1966;112:346–362. doi: 10.1016/0926-6585(66)90333-5. [DOI] [PubMed] [Google Scholar]
- Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Sober, H.A. 1970. Handbook of Biochemistry. Chemical Rubber Co. Cleveland, OH. 1,479 pp.
- Sorger PK, Severin FF, Hyman AA. Factors required for the binding of reassembled yeast kinetochores to microtubules in vitro. J Cell Biol. 1994;127:995–1008. doi: 10.1083/jcb.127.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmann O, Lechner J. The Saccharomyces cerevisiaekinetochore contains a cyclin-CDK complexing homologue, as identified by in vitro reconstitution. EMBO (Eur Mol Biol Organ) J. 1996;15:3611–3620. [PMC free article] [PubMed] [Google Scholar]
- Strunnikov AV, Kingsbury J, Koshland D. CEP3 encodes a centromere protein of Saccharomyces cerevisiae. . J Cell Biol. 1995;128:749–760. doi: 10.1083/jcb.128.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan K, Flynn P, Reece RJ, Marmorstein R. Crystal structure of a PUT3-DNA complex reveals a novel mechanism for DNA recognition by a protein containing a Zn2Cys6 binuclear cluster. Nat Struct Biol. 1997;4:751–759. doi: 10.1038/nsb0997-751. [DOI] [PubMed] [Google Scholar]
- Verma R, Feldman RM, Deshaies RJ. SIC1 is ubiquitinated in vitro by a pathway that requires CDC4, CDC34, and cyclin/CDK activities. Mol Biol Cell. 1997;8:1427–1437. doi: 10.1091/mbc.8.8.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems AR, Lanker S, Patton EE, Craig KL, Nason TF, Mathias N, Kobayashi R, Wittenberg C, Tyers M. Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway. Cell. 1996;86:453–463. doi: 10.1016/s0092-8674(00)80118-x. [DOI] [PubMed] [Google Scholar]
- Zhang L, Guarente L. The yeast activator HAP1—a GAL4 family member—binds DNA in a directly repeated orientation. Genes Dev. 1994;8:2110–2119. doi: 10.1101/gad.8.17.2110. [DOI] [PubMed] [Google Scholar]