

Abstract
α-Dystroglycan (α-DG) is a laminin-binding protein and member of a glycoprotein complex associated with dystrophin that has been implicated in the etiology of several muscular dystrophies. To study the function of DG, C2 myoblasts were transfected stably with an antisense DG expression construct. Myotubes from two resulting clones (11F and 11E) had at least a 40–50% and 80–90% reduction, respectively, in α-DG but normal or near normal levels of α-sarcoglycan, integrin β1 subunit, acetylcholine receptors (AChRs), and muscle-specific kinase (MuSK) when compared with parental C2 cells or three clones (11A, 9B, and 10C) which went through the same transfection and selection procedures but expressed normal levels of α-DG. Antisense DG-expressing myoblasts proliferate at the same rate as parental C2 cells and differentiate into myotubes, however, a gradual loss of cells was observed in these cultures. This loss correlates with increased apoptosis as indicated by greater numbers of nuclei with condensed chromatin and more nuclei labeled by the TUNEL method. Moreover, there was no sign of increased membrane permeability to Trypan blue as would be expected with necrosis. Unlike parental C2 myotubes, 11F and 11E myotubes had very little laminin (LN) on their surfaces; LN instead tended to accumulate on the substratum between myotubes. Exogenous LN bound to C2 myotubes and was redistributed into plaques along with α-DG on their surfaces but far fewer LN/α-DG plaques were seen after LN addition to 11F or 11E myotubes. These results suggest that α-DG is a functional LN receptor in situ which is required for deposition of LN on the cell and, further, implicate α-DG in the maintenance of myotube viability.
Keywords: dystrophin associated-proteins, laminin, apoptosis
α-dystroglycan (α-DG)1 is a component of the dystrophin associated glycoprotein complex (DGC) which, in muscle, is thought to serve as a transmembrane link between the submembraneous cytoskeleton and the basal lamina (Campbell and Kahl, 1989; Yoshida and Ozawa, 1990; Ervasti and Campbell, 1991). Other components of the complex include at least seven transmembrane glycoproteins, β-DG, sarcospan (Crosbie et al., 1997), α-, β-, γ-, δ-, and ε-sarcoglycan (SG; McNally et al., 1998) as well as the syntrophins, a family of three intracellular PDZ domain containing proteins (for review see Carbonetto and Lindenbaum, 1995). α- and β-DG are derived from cleavage of a common polypeptide precursor (Ibraghimov-Beskrovnaya et al., 1992) and remain associated with one another on the cell surface (Bowe et al., 1994). β-DG associates with either dystrophin or its autosomal homologue utrophin via an SH3 domain-binding region in its COOH terminus (Jung et al., 1995). This region has also been shown to interact with the adapter protein Grb 2 (Yang et al., 1995) raising the possibility that modulation of DG-cytoskeletal interactions may be mediated by a signaling pathway involving small GTP-binding proteins.
In contrast to the transmembrane protein β-DG, α-DG is a heavily glycosylated, mucin-like protein (Smalheiser and Kim, 1995) anchored on the extracellular surface of the myotube. In vitro studies have demonstrated that α-DG can bind the extracellular matrix component laminin (LN) with high affinity (Smalheiser and Schwartz, 1987; Douville et al., 1988; Ibraghimov-Beskrovnaya et al., 1992; Gee et al., 1993). LN is a heterotrimer of α, β, and γ chains, each of which is a member of multigene families. α-DG binds to the last two globular domains in the COOH-terminal extension of the LN α1 and α2 chains (Gee et al., 1993). α-DG has been shown to colocalize with LN in skeletal and cardiac muscle (Klietsch et al., 1993) and a number of other cells including peripheral nerve (Yamada et al., 1994), astrocytes, Purkinje neurons (Tian et al., 1996), and kidney epithelium (Durbeej et al., 1995). During muscle development, α-DG upregulation coincides temporally with the onset of innervation and ECM deposition (Leschziner, A., and S. Carbonetto, unpublished observations). These findings are consistent with the hypothesis that α-DG is a receptor in situ linking LN in the ECM to the subsarcolemmal cytoskeleton and thus may be important in the organization of these extracellular and subplasmalemmal networks. The existence of this putative transmembrane linkage is further supported by genetic evidence. For example, naturally occurring mutations in mice and humans that alter the expression of dystrophin and secondarily the DGC give rise to Duchenne and Becker muscular dystrophies, which are characterized by severe progressive damage to the sarcolemma (reviewed in Campbell, 1995; Worton, 1995). The phenotypes of these lesions bear considerable resemblance to those resulting from mutations affecting the gene encoding the α2 chain of LN 2 (α2β1γ1; merosin), which is enriched in the ECM of skeletal muscle (Xu et al., 1994; Sunada et al., 1995), or mutations affecting expression of the SG complex (Roberds et al., 1994; Bönnemann et al., 1995; Jung et al., 1996a,b; Nigro et al., 1996; Carrie et al., 1997). This convergence of biochemical and genetic evidence underscores the importance of interactions between the ECM and the DGC in the maintenance of sarcolemmal integrity.
To date, no naturally occurring mutations in α-DG have been identified which would substantiate and elucidate its role as an ECM receptor. Recently, targeted mutations abolishing DG expression in mice have been shown to result in early embryonic lethality due to disruption of the Reichert's membrane (Williamson et al., 1997; Côté, P., M. Lindenbaum, and S. Carbonetto. 1997. Mol. Biol. Cell. 8:222a). This indicates that DG expression is absolutely required for embryonic survival and strongly implicates DG in the maintenance of Reichert's basement membrane but sheds little light on its function(s) in more differentiated tissues such as skeletal muscle.
To test the assertion that α-DG is a LN receptor required for the elaboration of the ECM of muscle, we have perturbed its expression with stable transfection of C2 myoblasts with an antisense DG expression construct. In the process, we have generated two clonal cell lines 11F and 11E that express 40–50% and 10–20%, respectively, of the levels of α-DG protein in parental C2 cells after differentiation. These cells maintain the ability to fuse and form multinucleate myotubes and express normal or near normal levels of other DGC components including α-SG, but have greatly reduced levels of LN expression on their surfaces relative to C2 cells. This reduction correlates well with the level of α-DG in these clones. We also show that the residual, unbound α-DG on the surface of 11F myotubes is redistributed upon addition of exogenous LN. However, little or no binding by exogenous LN is seen in 11E myotubes, which express the lowest levels of α-DG. After transfer to fusion medium there is an increase in cell death and increased numbers of apoptotic nuclei in 11F and 11E cultures, which again correlates with levels of α-DG expressed in these clones. In 11E cells the integrity of the plasma membrane is not obviously compromised, as revealed by exclusion of the vital dye Trypan blue, and is consistent with apoptotic, not necrotic, cell death. We conclude that α-DG serves as a LN receptor in muscle and that interactions between the ECM and the DGC are required for maintenance of muscle viability.
Materials and Methods
Materials
LN was purified from Engelbreth-Holm-Swarm (EHS) tumor by the method of Timpl et al. (1982). SDS-PAGE of purified preparations shows bands at ∼215 kD (β and γ chains) and ∼400 kD (α chain). The latter was not recognized by an antiserum to LN α2 chain (a gift from Dr. Peter Yurchenco) but did react with an anti-LN antiserum produced by immunizing rabbits with purified EHS tumor LN. This antiserum recognizes all three subunits of LN 1 (α1, β1, and γ1), but does not cross react with agrin or LN α2 chain. Anti-LN IgG was purified by chromatography on Affigel Blue (BioRad Laboratories) according to the manufacturer's instructions. An antiserum to LN α2 chain was raised against the recombinant G domain of this chain and does not cross-react with LN α1 chain. mAb 5D3 to LN (Life Technologies) has been previously characterized (Abrahamson et al., 1989). Antiserum to the integrin β1 subunit was generated by immunizing rabbits with purified rat integrin β1 subunit (Tawil et al., 1990). mAb IIH6 recognizes a unique carbohydrate epitope on α-DG while the antiserum to fusion protein B recognizes a portion of the core protein of both α- and β-DG. mAb NCL–β-DG is directed against the last 15 amino acids of the COOH terminus of β-DG (Novocastra Laboratories Ltd.). The antiserum to α-SG, raised against a peptide corresponding to the last 19 amino acids of the COOH-terminal domain of α-SG, specifically recognizes a single protein of ∼50 kD in purified DGC from rabbit muscle and in crude protein extracts from skeletal muscle and C2 cells. mAb HUC1-1 to muscle actin was purchased from ICN Pharmaceuticals.
Plasmid Construction
A 1.8-kb fragment of the mouse DG cDNA extending from approximately −100 bp (5′ to the translation start site) to the HindIII site situated at +1,725 bp was removed by digestion of a mouse DG cDNA subclone in bluescript SK(−) with NotI-HindIII. This Not1-HindIII fragment was then subcloned in the antisense orientation into the pRcCMV expression vector (Invitrogen) using standard subcloning techniques (Sambrook et al., 1989). Before transfection, the plasmid was linearized by digestion with BglII.
Cell Culture and Transfection
C2 cells were plated on 10-cm tissue culture plastic dishes (Falcon) maintained at 37°C, 8% CO2 atmosphere, in growth medium consisting of DMEM (low glucose; Life Technologies) supplemented with 20% FBS (heat inactivated; Life Technologies), 0.5% chick embryo extract (ultrafiltered; Life Technologies), and penicillin/streptomycin (Life Technologies).
Stable transfections were carried out by calcium phosphate coprecipitation after methods of Yoshihara and Hall (1993). In brief, when C2 myoblasts reached 70% confluence, they were harvested by treatment with trypsin/EDTA, replated at a 1:20 dilution on fresh 10-cm dishes and allowed to attach overnight. On day 2, the medium was changed 3 h before transfection and DNA/Ca2PO4 coprecipitate, prepared according to published procedures (Graham and Van der Eb, 1973; Wigler et al., 1979), and was added directly to the culture medium (5 mg of linearized plasmid per dish). Cells were returned to the incubator for 16 h, and on the following day, the medium and precipitate were removed. The cells were washed briefly with Dulbecco's PBS plus 0.5 mM EDTA to remove excess precipitate. Fresh medium was added and 24–36 h after the beginning of transfection it was replaced with selection medium consisting of growth medium supplemented with G418 (750 μg/ml active concentration; Life Technologies). Selection was carried out for up to 10 d, or until all cells of an equivalent, untransfected culture were killed. At that point, drug-resistant C2 cell colonies could easily be seen on transfected plates. Colonies were then picked and expanded for further characterization. Stable clones were maintained in growth medium supplemented with 70 μg/ml active G418. Low (6–20) passage 11F, 11E, 9B, 10C, 11A, and control C2 cells were cultured on tissue culture plastic dishes (Falcon) coated with 0.15% gelatin and maintained in growth medium until confluent. Cultures were switched to fusion medium (DMEM high glucose, 1% horse serum) and allowed to differentiate for an additional four days. Some cultures were treated with 12 nM LN on the third day of fusion.
SDS-PAGE and Western Blotting
Control and transfected C2 clones differentiated into myotubes for 3–5 d were washed three times with ice-cold Ca/Mg-free PBS, then extracted into 0.2-ml/10-cm dish of 1× SDS sample buffer (50 mM Tris-HCl, pH 6.8, 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol) preheated to 85°C. The remaining extract was scraped and transferred to a 1.5-ml microcentrifuge tube, and heated at 95°C for 5 min. It was subsequently passed five times through a 30-gauge syringe needle and centrifuged at 16,000 g for 10 min to remove insoluble debris. A portion of the lysate was precipitated with trichloroacetic acid/deoxycholate for protein determination (Peterson, 1977). For some experiments, cultured myotubes were subjected to subcellular fractionation generating soluble fractions and KCl-washed light and heavy microsomes (Ohlendieck et al., 1991). To assess expression of the integrin β1 subunit by Western blotting, cells were scraped in ice-cold PBS and membrane proteins were detergent extracted in 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% (vol/vol) Triton X-100, 20 mM N-ethylmaleimide, Complete protease inhibitor cocktail (Boehringer Mannheim) for 10 min on ice. Protein concentration was determined (Peterson, 1977) and extracts were diluted in 5× SDS sample buffer without dithiothreitol. Cellular proteins released into the culture medium were assayed by collecting medium from two 10-cm dishes of differentiated cells. The medium was pooled, concentrated ∼100-fold and partially purified using Centricon Plus-20 centrifugal filter devices (Millipore). All samples were assayed for protein content (Peterson, 1977) before electrophoresis on 7.5 or 10% SDS–polyacrylamide mini gels (0.75-mm thickness; BioRad Laboratories) at 20-mA constant current for 1 h. Fractionated proteins were electroblotted onto nitrocellulose membranes (BA-S 75; Schleicher and Schuell) under standard conditions (100-V constant voltage for 1 h). Blots were stained with Ponceau-S red to assess transfer, then incubated in 10 ml of Blotto (10 mM Tris, pH 7.5, 150 mM NaCl, 1% Tween 20, 5% dried skim milk powder) at room temperature for 1 h and subsequently in an appropriate dilution of primary antibody in 4–5 ml Blotto for 1 h at room temperature with constant agitation. Primary antibodies used in Western blotting included: mAb IIH6 culture supernatant (1:10); antiserum to fusion protein B (1:50); antiserum to α-SG (1:1,000); mAb to β-DG (1:350); antiserum to LN (1:100); antiserum to the integrin β1 subunit (1:5,000); mAb to sarcomeric actin (1:1,000). After incubation with the primary antibody, blots were washed 4× 15 min in TBS-Tween (10 mm Tris, pH 7.5, 150 mM NaCl, 1% Tween 20), then incubated with the appropriate HRP-labeled secondary antibody diluted in Blotto for 1 h at room temperature with constant agitation. Blots were washed in TBS-Tween (4× 15 min) and labeled bands were visualized by enhanced chemiluminescence (Mandel/ NEN Life Science Products) after exposure to x-ray film (Hyperfilm-ECL; Amersham Life Sciences). Blots were often stripped in 0.2 M glycine, 0.1% (vol/vol) Tween 20, pH 2.5, for 30 min, rinsed in PBS and reprobed with mAb HUC1-1 to muscle actin to take into account differences in the levels of DGC and integrin expression due to differences in the proportion of differentiated myotubes among cultures (see text).
Microscopy and TUNEL Assay
Cultures of confluent myoblasts (day 0), or of cells allowed to differentiate for 2 or 4 d, were washed twice with PBS then fixed and stained with Coomassie blue (25% propanol, 10% acetic acid, 0.1% Coomassie brilliant blue) for 10 min at room temperature. Cultures were washed twice in PBS, air dried and visualized in brightfield on a Zeiss Axioskop.
For immunocytochemistry, myotube cultures were fixed with 2% paraformaldehyde in 0.1 M phosphate buffer, pH 7.2, for 20 min, rinsed three times with PBS, blocked for 1 h in PBS + 1% horse serum, then incubated overnight at 4°C with the primary antibody. For immunostaining of live cells, the primary antibody was added directly to the culture medium and cells were incubated for 30 min at 37°C in 8% CO2, then fixed as described above. Cells were then washed and incubated with the appropriate biotinylated secondary antibody for 1 h, followed by fluorescein-conjugated streptavidin and rhodamine-conjugated α-bungarotoxin (α-BTX; Molecular Probes) for 20 min at room temperature. Staining was visualized with epifluorescence illumination on a Zeiss Axioskop. Primary antibodies used include: mAb IIH6 ascites, 1:100 dilution; antiserum to LN 1, 1:50 dilution; mAb 5D3, 1:100 dilution; antiserum to LN α2 chain, 1:150 dilution; antiserum to the integrin β1 subunit, 1:100 dilution.
To assess the level of apoptosis in myotube cultures, cells were differentiated for 3 d, then fixed sequentially in 2% formaldehyde and 4% neutral-buffered formalin for 10 min each at room temperature and washed twice in PBS, pH 7.4. Cultures were permeabilized by incubation in either 0.1% Triton X-100 for 15 min at room temperature or in ethanol/acetic alcohol (1:4) for 5 min at –20°C, then processed for TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling; Apoptag Plus In Situ Apoptosis Detection Kit; Oncor). Negative controls were run for each experiment by omitting the anti-digoxigenin antibody or the terminal deoxynucleotidyl transferase enzyme. Positive control slides were provided with the kit. Cultures were counterstained with 0.1 μg/ml DAPI to visualize the total number of nuclei present and to confirm by morphological criteria that TUNEL stained nuclei were not mitotic or necrotic. The number of apoptotic nuclei and the total number of nuclei were counted with a 63× objective on a Zeiss Axioskop. 20 fields were quantified per coverslip and three coverslips were analyzed per cell type and per experiment. Collapsed myotubes and masses of dead cells were not included in the quantification since the number of labeled nuclei could not be accurately determined.
Cell Loss, Proliferation, and Membrane Integrity Assays
C2, 11F, 11E, 9B, and 10C cells were cultured in 96-well plates (Falcon) coated with 0.15% gelatin and assayed at day 0 (confluent myoblasts), and days 2 to 4 in fusion medium. To determine cell number, plates were thoroughly washed with PBS, frozen at −80°C and labeled with the Blue DNA assay kit (Molecular Probes). Hoechst fluorescence was measured with a Cytofluor 2300 fluorometer using a 360 nm excitation/460 nm emission filter set. Empty wells coated with gelatin were used as controls for background fluorescence. Proliferating cells were labeled using the colorimetric cell proliferation ELISA kit (Boehringer Mannheim). In brief, cells were incubated for 2 h in medium containing 10 μM bromodeoxyuridine (BrdU), fixed for 30 min at room temperature, incubated for 2 h with the anti-BrdU antibody, washed, and incubated for 5 min in the substrate solution. The reaction was stopped with 1 M sulfuric acid and the plates were immediately read with an ELISA plate reader at 450 nm using a reference wavelength of 690 nm. Wells where the BrdU was omitted were used as control for nonspecific labeling of the antibody. Plates for the cell loss and proliferation assays were prepared on the same day and from the same aliquot of cells. Four replicate wells were prepared per cell type and per plate. Proliferation is expressed as a ratio of the average number of cells per well as determined by Hoechst staining on an age-matched plate.
To assay membrane integrity cells differentiated for 4 d on tissue culture dishes coated with gelatin were washed in serum-free DMEM and incubated for 1 min in 0.2% Trypan blue in DMEM. Cells were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer for 10 min at room temperature, washed in PBS and then dehydrated in 70% and 95% ethanol for 1 min each. Cultures were counterstained with eosin (0.1% eosin in 95% ethanol, 2.8 μl/ml concentrated acetic acid) for 1 min then washed three times 1 min each in 100% ethanol. Cultures were visualized on a Zeiss Axioskop. Blue nuclei were counted with a 40× objective for three sets of 10-cm dishes (10–15 fields/dish) for C2 and 11E cells. Large aggregates of dead cells and cell debris were not included in the quantification.
Statistical Analyses
Statistical significance for cell loss, proliferation, and membrane integrity assays was determined for three to four independent experiments using a simple ANOVA test followed by the Fisher's t-test. The n represents the individual experiments. For statistical analysis of the percentage of apoptotic nuclei, each experiment was evaluated separately since the permeabilization procedure used was not always the same. A simple ANOVA test was also used followed by the Fisher's t-test. The n represents the number of dishes used (three for each experiment). The correlation between cell number and α-DG expression for C2, 11F, and 11E cells was determined using a simple regression analysis followed by Fisher's z-test (null hypothesis: correlation coefficient = 0). The relative cell number was obtained from quantification of Hoechst fluorescence and the average levels of α-DG expression for each cell type were assessed by densitometry from seven individual Western blots. In each Western blot, extracts from all three cell types had been assayed side by side.
Results
Characterization of Stably Transfected C2 Clones Expressing Antisense α-DG cDNA
To assess the role of α-DG in the deposition and organization of muscle ECM, the C2 muscle cell line was transfected with a DG cDNA fragment (Fig. 1 A) in the antisense orientation under the control of the cytomegalovirus promoter (Yoshihara and Hall, 1993). G418-resistant clones were selected and screened by examining the levels of α-DG expression relative to untransfected controls or cells transfected with the vector alone. Five clones, 11E, 11F, 9B, 10C, and 11A were chosen for further characterization. Control C2 cells, and antisense clones were cultured to confluence then induced to fuse into myotubes as described in Materials and Methods. After 4–5 d in culture, the myotubes were extracted directly in SDS sample buffer and equal protein loads were fractionated by SDS PAGE and transferred onto nitrocellulose membranes. Fig. 1 B shows a representative blot probed with mAb IIH6, which recognizes a carbohydrate epitope on α-DG (Ervasti and Campbell, 1993). Control C2 cells show a typically broad band of muscle α-DG extending from 130–160 kD. By contrast, only low levels of α-DG expression were detected in 11F cells and even lower levels were detected in 11E cells (this blot is greatly overexposed to allow for visualization of α-DG expression in the 11E line). Nearly normal levels of α-DG expression were detected in the 9B, 10C, and 11A clones (Fig. 1 C) possibly as a result of low expression of the antisense construct, and these clones were used as additional controls for any nonspecific effects resulting from the transfection or selection procedures. Densitometric analysis of films from Western blots exposed over a range of times to insure that the emulsion was not overexposed, revealed a 50–60% decrease in α-DG in 11F cultures and an 80–90% decrease in 11E cultures. The blot in Fig. 1 B was also probed with an antiserum raised against α-SG, one of the four transmembrane components of the SG complex which is associated with, but distinct from, the DG complex (Yoshida et al., 1994). No reduction in the expression of this 50-kD protein was seen in either α-DG antisense clone compared with C2 cells (Fig. 1 B). A similar result was obtained for β-SG (not shown). Since mutations affecting the expression of any one member of the SG complex can lead to the loss of all other members from the sarcolemma (Roberds et al., 1994; Bönnemann et al., 1995; Noguchi et al., 1995; Jung et al., 1996a,b), we deduced that the SG complex is unaffected in the antisense clones. This result is also in agreement with the reported relative independence of the DG and SG complexes (Roberds et al., 1994; Yoshida et al., 1994; Bönnemann et al., 1995; Jung et al., 1996a,b). In addition, Western blots of extracts probed for LN (Fig. 4 B), collagen IV and perlecan (Montanaro, F., and S. Carbonetto, unpublished data), integrin β1 (Fig. 5 B) AChR α, δ, and γ subunits, or for the MuSK receptor tyrosine kinase (Jacobson et al., 1998) showed no reduction in expression of these proteins in 11F and 11E cells, nor in 9B, 10C, and 11A cells, confirming the specificity of the antisense construct.
Figure 1.

Molecular characterization of antisense DG-expressing C2 myotubes. (A) Map of antisense DG expression vector (Materials and Methods). (B) Equal amounts of proteins extracted from C2, 11F, and 11E myotube cultures were probed with mAb IIH6, directed against a carbohydrate epitope specific to α-DG. Densitometric analysis of similar blots revealed consistent 50–60% and 80–90% decreases in α-DG expression in 11F and 11E extracts, respectively, compared with C2 extracts. The same blot was also probed with an antiserum specific for α-SG. Densitometry of this protein revealed no change in its expression in the antisense-expressing clones. (C) Western blot with mAb IIH6 indicating that the expression of α-DG is not significantly affected in the 9B, 10C, and 11A antisense clones. (D) Western blot with an antiserum to the DG core protein, confirming the decreased expression of α-DG in 11F and 11E myotube cultures. (E) C2, 11F, and 11E myotubes were fractionated into soluble and KCL-washed, light microsome fractions (Materials and Methods) and equal protein loads of each fraction were probed for the presence of α-DG with mAb IIH6. The presence of α-DG in the microsomal rather than soluble fractions is consistent with proper targeting of the residual α-DG to membranes in 11F and 11E cells. (F) Western blots probed for β-DG (43 kD) showing its decreased expression in 11F and 11E clones, but not 9B, 10C, or 11A clones.
Figure 4.
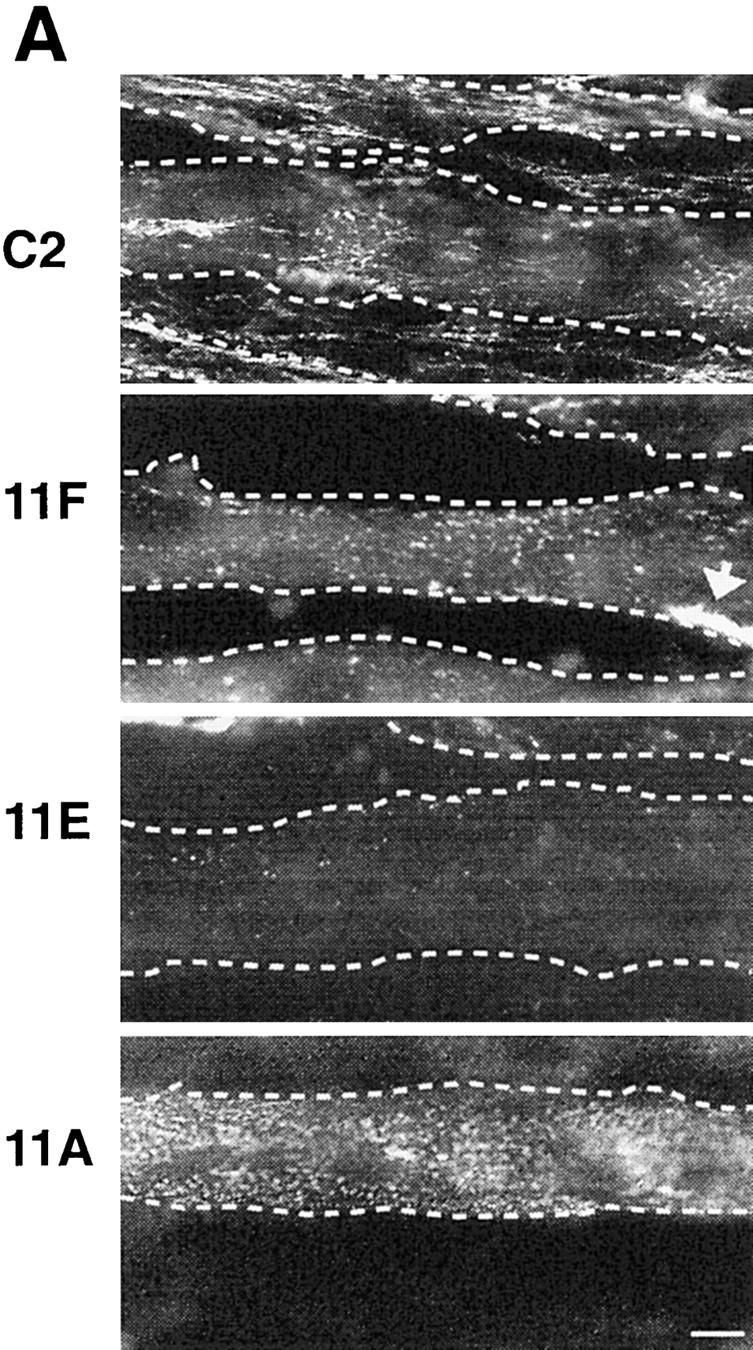

LN expression in C2, 11F, and 11E cultures. (A) Differentiated cultures of C2, 11A, 11F, and 11E myotubes were double labeled with an antiserum recognizing all three chains of mouse LN-1 (α1β1γ1) and rhodamine-conjugated α-bungarotoxin (not shown). LN immunoreactivity on C2 and 11A myotubes is a composite pattern of dots, fine linear deposits and large patches. 9B and 10C myotubes showed an identical LN distribution on their surface (not shown). LN immunoreactivity between C2 myotubes is associated with myoblasts. In contrast, large portions of 11F myotubes are devoid of LN and alternate with regions showing a punctate pattern of immunoreactivity. The few LN patches present always correspond to AChR clusters (arrow). Essentially no LN immunoreactivity was detected on 11E myotubes. Few myoblasts are present in cultures of 11F and 11E cells and large LN deposits are often present in-between myotubes. These are not shown here since their intense fluorescence obscured the labeling on the surface of myotubes at this high magnification. The dotted lines show the myotube outline. Bar, 10 μm. (B) Cultures from C2, 11F, 11E, 9B, 10C, and 11A myotubes were directly extracted in reducing sample buffer (C) and the culture medium (M) was concentrated ∼100-fold. Equal amounts of protein were probed with an antiserum that recognizes the α1, β1, and γ1 chains of LN. Decreased expression of α- and β-DG in 11F and 11E cells did not result in decreased expression of LN by the cells (C) or increased secretion in the culture medium (M).
Figure 5.
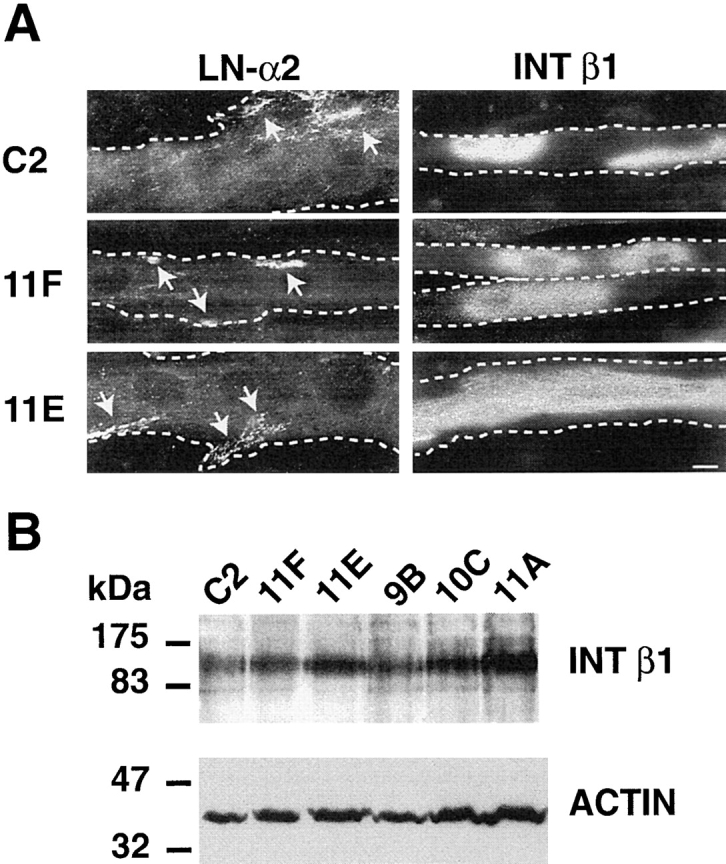
Expression of LN α2 chain and integrin β1 subunit in C2, 11F, and 11E cells. (A) Cultures of C2, 11F, and 11E myotubes were double labeled with rhodamine-conjugated α-bungarotoxin and with antisera to either the LN α2 chain found in merosin, or the integrin β1 subunit. LN α2 chain is present at very low level on the surface of C2 myotubes and is concentrated at AChR clusters (arrows) on C2, 11F, and 11E myotubes. The integrin β1 subunit is expressed in large oval patches on the surface of C2 and 11F myotubes, and is often found in long strips running the length of 11E myotubes. The dotted lines show the myotube outline. Bar, 10 μm. (B) Cultures from C2, 11F, 11E, 9B, 10C, and 11A cultures were detergent extracted. Proteins were separated by SDS-PAGE and blots were simultaneously probed with an antiserum to the integrin β1 subunit and a mAb to muscle actin, to compensate for differences in the degree of differentiation among cultures. No decrease in expression of the integrin β1 subunit could be detected in the antisense clones compared with C2 cultures; rather the expression of this integrin subunit appears slightly increased in 11E cultures.
Since mAb IIH6 is known to recognize a carbohydrate epitope on α-DG involved in binding to LN (Ervasti and Campbell, 1993), it was important to demonstrate that the defect in the antisense-expressing clones was not simply due to aberrant glycosylation of α-DG. Therefore, similar blots were probed with an antiserum to an α-DG fusion protein (anti-fusion protein B) which recognizes the DG core protein (Ibraghimov-Beskrovnaya et al., 1992). For C2 cells as well as 11F and 11E antisense clones, the relative intensity of labeling by this antiserum is very similar, albeit generally weaker (Fig. 1 D), to that observed with mAb IIH6 (Fig. 1 B) suggesting that the decreased expression of α-DG in the 11E and 11F clones is due to decreased levels of α-DG polypeptide and not merely altered glycosylation. To confirm further that the expression of membrane associated α-DG was similarly altered, C2, 11F, and 11E myotubes were fractionated into soluble and KCl-washed light microsome fractions, then analyzed by SDS-PAGE and Western blotting with mAb IIH6. The decrease in membrane associated α-DG seen in the antisense clones relative to control C2 cells (Fig. 1 E) closely parallels that seen for the levels of total α-DG (Fig. 1 B). Little, if any, expression of α-DG was detected in the soluble fraction (Fig. 1 E) or heavy microsome fraction (data not shown) of either control C2 cells or antisense clones, suggesting that the latter are not defective in the localization of α-DG to the cell membrane. Consistent with this β-DG, like its cotranscript α-DG, is also reduced by 70 and 88% in 11F and 11E cells, respectively, but not significantly in 9B, 10C, and 11A cells, when compared with C2 cells (Fig. 1 F). This confirms that DG synthesis is reduced in antisense-expressing cell lines and that the reduced levels of α-DG do not result simply from shedding into the medium.
Morphological Characterization of Antisense-expressing Myoblasts and Myotubes
α-DG is a high affinity LN binding protein in vitro (Smalheiser and Schwartz, 1987; Douville et al., 1988; Ibraghimov-Beskrovnaya et al., 1992; Gee et al., 1993) and colocalizes with LN in muscle cells (Klietsch et al., 1993; Cohen et al., 1997). Since LN isoforms have been reported to play an important role in myoblast fusion (Foster et al., 1987; Öcalan et al., 1988; von der Mark and Öcalan, 1989; Schuler and Sorokin, 1995; Vachon et al., 1996), we compared the ability of C2, 11F, 11E, 9B, 10C, and 11A myoblasts to differentiate into myotubes. In low density cultures 11F and 11E myoblasts appeared somewhat flatter and more spread than control C2 myoblasts. However, the proliferation rate and survival of 11F and 11E myoblasts in these cultures were indistinguishable from C2 myoblasts. In cultures approaching confluence little morphological difference could be seen between C2, 11F, and 11E myoblasts (Fig. 2). One day after transfer to fusion medium, myoblasts assumed a more elongate shape and on the second day of differentiation numerous thin myotubes could be seen in C2 as well as in 11F and 11E cultures (Fig. 2). After 4 d of differentiation, many large myotubes were found in all cultures (Fig. 2) indicating that 11F and 11E myoblasts can differentiate into myotubes with the same time course as C2 cells although 11F and 11E cultures had lower densities of myotubes (discussed below). 9B, 10C, and 11A cells also differentiated normally within the same time frame as C2 cells but did not show any appreciable decrease in cell density with differentiation. 11A myoblasts seemed to replicate more slowly compared with the other clones and usually required 3 d to reach confluence rather than 2 d like C2 cells and all the other clones, although, once confluence was reached, differentiation into myotubes proceeded at the same rate as C2 cells. It should be noted that after 4 d in fusion medium, cultures of 11F, 9B, 10C, and 11A cells had a very low proportion of myoblasts compared with C2 cells. This may be a reflection of heterogeneity in the parental C2 line relative to the transfected myoblast clones. Thus, substantial depletion of α-DG in 11F and 11E cells does not obviously affect these aspects of muscle cell differentiation in agreement with studies implicating integrins as the major receptors mediating the effects of ECM on myoblast alignment and fusion (Chung and Kang, 1990; MacCalman et al., 1992; Collo et al., 1993; George-Weinstein et al., 1993; Blaschuk and Holland, 1994; Sastry et al., 1996; Blaschuk et al., 1997). However, even in the 11E cell line α-DG expression is not completely abolished and it remains possible that the residual α-DG is sufficient to play a role in the differentiation of myoblasts into myotubes.
Figure 2.

Morphology of C2, 11F, and 11E cells. Cultures of C2, 11F, and 11E cells were stained with Coomassie blue at day 0, or 2 or 4 d in fusion medium. Cell morphology as well as differentiation of the antisense-expressing clones resembles that of C2 cells, apart from a decreased density of myotubes most obvious in 11E cultures at 4 d. Bar, 250 μm.
Expression of α-DG, LN, and Integrin β1 Subunits in Cultured C2 and Antisense-expressing Myotubes
In addition to α-DG, several LN receptors of the integrin superfamily such as the α7β1, α9β1, and α3β1 integrins are expressed in skeletal muscle (Collo et al., 1993; Palmer et al., 1993; de Melker et al., 1997) raising questions as to the relative contributions of integrins and α-DG in LN deposition on myotubes. As a first step in exploring this issue, C2 cells were immunostained with mAb IIH6 to α-DG or with antibodies recognizing several LN isoforms and the distribution of these proteins was compared in DG-deficient clones. In C2, 11F, and 11E cells, α-DG immunoreactivity is punctate and uniformly distributed over the myotube surface (Fig. 3). However, the intensity of staining is greatly decreased on the surfaces of 11F and even more so on 11E myotubes relative to C2 myotubes (Fig. 3; the top photomicrograph for C2 cells and the two corresponding ones for 11F and 11E cells were taken at the same exposure to illustrate this point). A similar pattern was obtained with live cultures labeled with this antibody (data not shown) confirming that the residual α-DG is properly targeted to the cell surface in the antisense cell lines. In all cell types, dense patches of α-DG immunoreactivity were found to be associated with spontaneous AChR clusters (Montanaro et al., 1998; unpublished observations). Thus, the antisense cDNA construct affects the amount of α-DG on the surface of myotubes but not its distribution.
Figure 3.
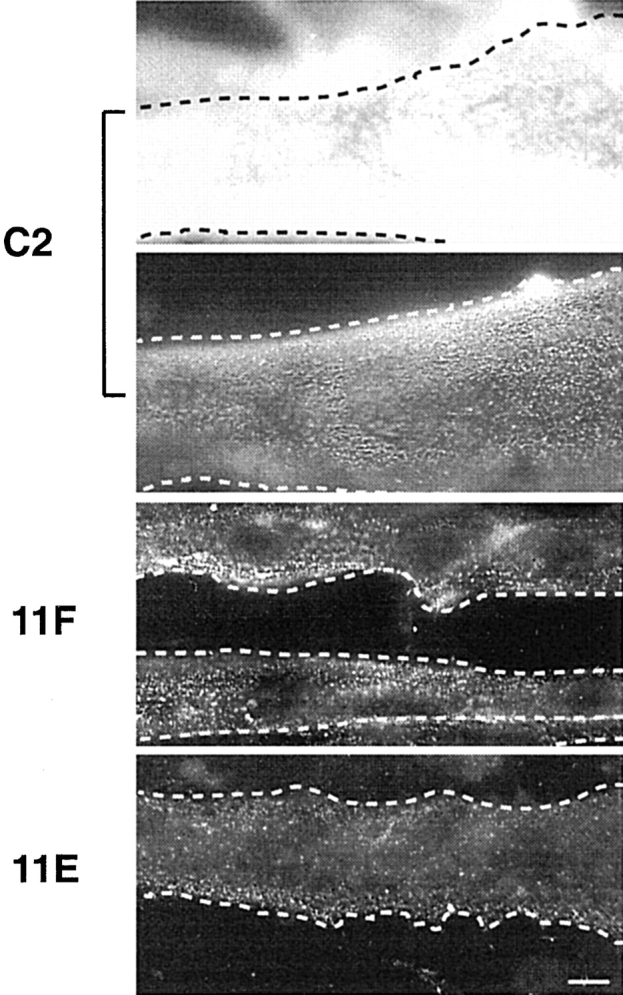
α-DG expression on the surface of C2, 11F, and 11E myotubes. Cultures of C2, 11F, and 11E myotubes were immunostained with mAb IIH6 to α-DG. For the same exposure time, α-DG staining in C2 myotubes (C2, top) was extremely intense compared with 11F and especially 11E cells. Since the specific signal was amplified using the biotin-streptavidin system (see Materials and Methods), differences in fluorescence intensity are qualitative, not quantitative. A shorter exposure time for C2 cells (C2, bottom) revealed a fine punctate immunoreactive pattern on the myotube surface. In 11F and 11E cells the density but not the pattern of expression of α-DG was affected. The dotted lines show the myotube outline. Bar, 10 μm.
In adult skeletal muscle, LN α2 and γ1 chains are expressed both synaptically and extrasynaptically, LN β1 chain is excluded from synaptic regions, while LN α4, α5, and β2 chains are only found at the NMJ (Sanes et al., 1990; Patton et al., 1997). To map the distribution of most LN heterotrimers expressed in C2 cells, we used an anti-LN antiserum to LN α1, β1, and γ1 chains as well as the rat mAb 5D3 (Abrahamson et al., 1989) which recognizes mouse LN α1, α2, β1, and γ1 chains. Both antibodies gave similar results, showing a patchy distribution of LN on the surface of C2 myotubes with some larger aggregates (Fig. 4 A). Since α-DG is diffusely distributed on the surface of C2 myotubes and there appears to be a pool of α-DG unbound to LN, it is difficult to determine from their distributions alone whether LN and α-DG may be interacting with one another. Nevertheless, decreased expression of α-DG in 11F and 11E cells leads to a dramatic reduction in LN immunoreactivity on the myotube surface (Fig. 4 A) suggesting that LN is bound to α-DG. Occasionally, dense accumulations of LN immunoreactivity were seen on the surfaces of 11F and, much less frequently, on 11E myotubes and these often coincided with densities of AChRs (Montanaro et al., 1998). 11A, 9B, and 10C myotubes expressed LN on their surface in a pattern and amount similar to C2 myotubes (Fig. 4 A). Western blots of total extracts of C2, 11A, 9B, 10C, 11F, and 11E myotubes probed with the anti-LN antiserum showed similar levels of LN expression (Fig. 4 B) indicating that there is no defect in the biosynthesis of LN per se in the DG-deficient clones. Furthermore, very little LN is released into the culture medium (Fig. 4 B), and no difference could be seen in the pattern of LN staining between cultures immunolabeled live or after fixation indicating that 11F and 11E myotubes do not accumulate LN intracellularly. Rather, most of the LN in 11F and especially in 11E cells is deposited on the surface of the dish in large, irregularly shaped deposits found between live cells (not shown). This paucity of surface LN and the extensive extracellular deposition of LN observed in cultures of antisense-expressing myotubes suggest a deficit in the ability of LN to bind to myotubes deficient in α-DG.
Because of the high expression of LN α2 chain in skeletal muscle and its involvement in some types of muscular dystrophy in both humans and mice, we looked specifically at the distribution of this LN chain in C2 cells and DG antisense clones. In C2, 11F, and 11E cells LN α2 chain immunoreactivity was detected only on the surface of myotubes and never on the culture dish. In contrast with its abundance in mature skeletal muscle, LN α2 is rather sparse in C2 cells and could not be detected by Western blot on crude protein extracts (data not shown) but has been detected after enrichment by immunoprecipitation (Vachon et al., 1996). LN α2 immunoreactivity on the cell surface was associated with AChR clusters (Fig. 5 A, arrows) in C2, 11F, and 11E myotubes. Outside of these clusters, LN α2 immunoreactivity was diffuse and faint.
Integrin localization in C2 cells was determined with an antiserum to the β1 subunit which is common to all integrin heterodimers expressed in muscle that recognize LN (Collo et al., 1993; Palmer et al., 1993; de Melker et al., 1997). In all cultures, myotubes were intensely labeled, whereas myoblasts showed a much fainter punctate staining pattern. For C2 as well as 11F myotubes the integrin β1 subunit either had a diffuse punctate pattern or was found in large, intensely immunoreactive, oval patches (Fig. 5 A). In 11E cells, a strip of intense immunoreactivity occasionally ran the length of the myotube. In cultures double labeled with α-BTX, immunoreactivity for the integrin β1 subunit only rarely overlapped with AChR clusters (Montanaro et al., 1998), indicating that this receptor is not likely to be responsible for the presence of LN at these sites. To further investigate any differences in the levels of expression of this integrin subunit, we used Western blotting to simultaneously probe the same blot for expression of the integrin β1 subunit and sarcomeric actin (Fig. 5 B). Expression of sarcomeric actin has been shown to increase with muscle differentiation (Sawtell and Lessard, 1989) and is therefore an indicator of the relative proportion of myotubes versus myoblasts in our cultures. Under these conditions, Western blotting confirmed that all clones express similar levels of the integrin β1 subunit, indicating that β1 integrins are not upregulated to compensate for decreased DG expression in 11F and 11E cells. Conversely, the integrin β1 subunit is not downregulated in 11F and 11E cells indicating that the decrease in LN immunoreactivity on the surface of 11F and 11E myotubes is not due to a lack of integrin expression and that integrins are unable to compensate for decreased α-DG expression. In summary, the level of surface LN correlates well with α-DG levels in C2, 11F, and 11E myotubes, suggesting that α-DG is responsible for the assembly of an LN-rich ECM on the surface of myotubes. However, the amount of α-DG staining appeared more diffuse and extensive than that of LN, in C2 and, to a lesser extent, 11F myotubes, possibly because a significant proportion of α-DG was either free or else bound to another ligand, such as agrin (Bowe et al., 1994; Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994).
Effect of Exogenous LN Addition on α-DG–deficient Cell Lines
As a further test of the ability of α-DG to function as a LN receptor, we assessed whether excess surface α-DG was capable of binding exogenously added LN and whether such binding could redistribute α-DG on the myotube surface as shown previously for Xenopus myocytes (Cohen et al., 1997). C2 and antisense-expressing myotubes were incubated overnight in the presence of 12 nM EHS purified LN 1 (α1β1γ1), then fixed and immunostained for α-DG or LN. A frequent feature of the LN treated C2 myotubes was the presence of large plaques of α-DG immunoreactivity (Fig. 6; arrows) as well as similar plaques of LN immunoreactivity (Fig. 6, arrows) suggesting that exogenous LN can bind to and reorganize the unbound pool of α-DG on the myotube surface (Cohen et al., 1997). Plaques of α-DG and LN immunoreactivity were also seen on the surface of LN-treated 11F myotubes but these were of smaller size and much less frequent (Fig. 6, arrows). No such plaques were found on LN-treated 11E myotubes, nor did exogenous LN bind well to 11E myotubes, consistent with the relative abundance of α-DG in these clones. This correlation of LN binding with α-DG expression and the redistribution of α-DG by exogenous LN suggest strongly that α-DG is a functional LN receptor. As noted previously (Cohen et al., 1997), the aggregation of α-DG by LN may be an early step in ECM assembly, which, from our data, appears to be dependent on the level of α-DG expression.
Figure 6.
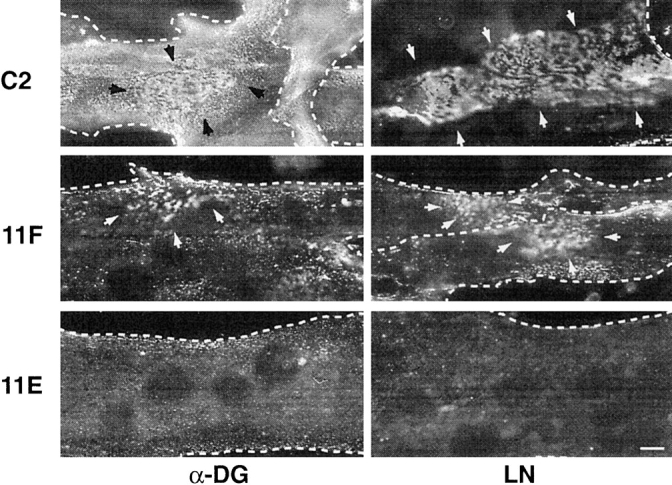
Addition of exogenous LN results in aggregates of α-DG in C2 and 11F but not 11E myotubes. After treatment with exogenous LN (12 nM), the distribution of surface α-DG and LN on C2, 11F, and 11E was visualized with mAb IIH6 and the anti-LN antiserum. LN was found to elicit the formation of plaques (arrows) of surface α-DG and LN in C2 myotubes. A few such plaques were seen on 11F myotubes (arrows) but their area and number are greatly reduced relative to C2 myotubes. No α-DG or LN plaques were seen on 11E myotubes, which did not seem able to bind the exogenous LN. Photomicrographs for α-DG and LN are from separate fields. Dotted lines show the myotube outline. Bar, 10 μm.
α-DG Is Involved in Cell Survival
Although antisense clones were able to differentiate normally (Fig. 2), a consistently higher degree of cell loss was observed after differentiation in 11F and 11E cultures. DAPI staining of cultures after 4 d in fusion medium confirmed a decreased number of nuclei in 11F and 11E cultures compared with C2 cultures (Fig. 7, DAPI). The degree and time of onset of cell loss in the antisense clones was quantified spectrofluorometrically after staining DNA with Hoechst dye. For the first 24 hours in fusion medium, C2, 11F, and 11E cultures have comparable numbers of cells. Subsequently, significant drops in cell number compared with C2 cultures are first detected at day 2 in 11E cultures and at day 3 in 11F cultures (Fig. 8 A; P < 0.0001) and by day 4, cultures of 11F and 11E cells emit 45 and 65% less Hoechst fluorescence, respectively, when compared with C2 cultures (Fig. 7, DAPI, and Fig. 8 A). Statistical analysis revealed a linear correlation between cell number in C2, 11F, and 11E cultures at day 4 and the amount of α-DG expressed (R 2 = 0.80; P < 0.0001). In contrast, 9B and 10C clones behaved essentially like C2 cells. 11A cells also showed an increase in cell number similar to C2 cells but were not included here because the slightly slower proliferation rate of 11A myoblasts led to an asynchronous differentiation of these cultures compared with the other clones rendering any comparison difficult. Thus the level of α-DG expression correlates with the amount as well as the time of onset of cell loss during differentiation of myoblasts into myotubes.
Figure 7.
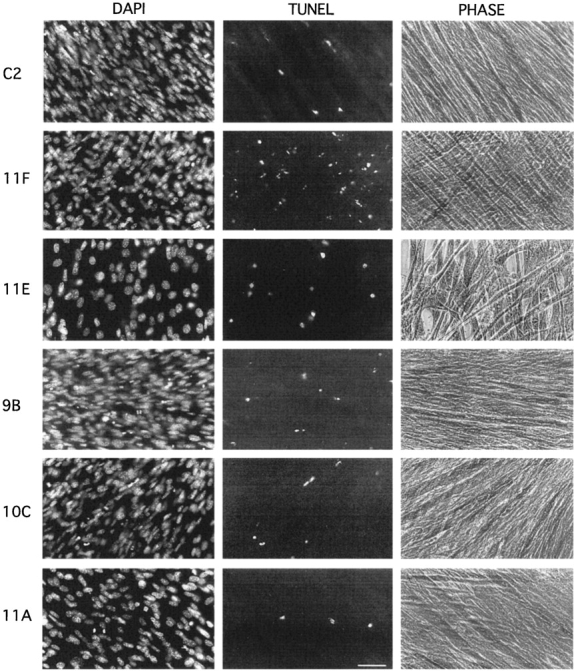
Decreased levels of α-DG expression correlate with increased cell death. DAPI staining and TUNEL labeling of day 4 cultures of C2, 11F, 11E, 9B, 10C, and 11A cells reveal differences in cell density and in the level of apoptotic cell death between the clones. 11E cultures show a marked decrease in the number of adherent cells compared with all other clones. Bar, 100 μm.
Figure 8.
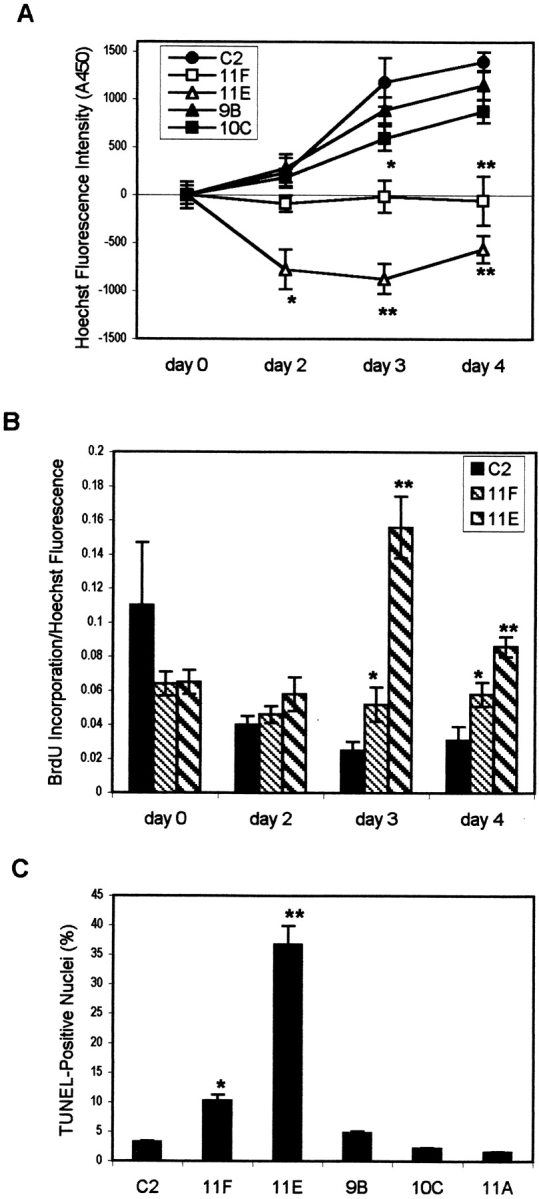
Quantification of cell loss and apoptotic cell death. (A) Spectrofluorometric quantification (Materials and Methods) of cell number after transfer to fusion medium beginning with day 0 at which time the culture is a monolayer of confluent myoblasts. A significant decrease in the number of cells in α-DG–deficient cells is detectable from day 2 (11E) in fusion medium and is maintained until day 4. In contrast, 9B and 10C clones that express normal levels of α-DG do not show a marked decrease in cell number. Values represent mean ± SEM from two experiments. Values for day 0 were used as a reference for each cell line. (B) Proportion of proliferating cells as determined by BrdU incorporation (Materials and Methods). Decreased expression of α-DG in 11F and 11E cells is not associated with a decreased ability of these cells to proliferate. Values represent mean ± SEM from three experiments. (C) Percentage of TUNEL-labeled nuclei relative to the total number of nuclei as determined by DAPI staining. Data are from one representative experiment. Values represent means ± SEM derived from three separate dishes; 20 fields were quantified per dish using a 63× objective. * and ** represent statistically significant differences (P < 0.01 and P < 0.001, respectively).
Since the number of cells increases with differentiation in control cultures of C2, 9B, and 10C cells, it was important to establish whether the decreased cell number in DG-deficient clones was due to actual cell loss or a deficiency in proliferation. BrdU incorporation, as measured by a colorimetric assay, was used to determine proliferative activity. When relative proliferation rates were expressed as a ratio of BrdU incorporation over total cell number as determined by Hoechst fluorescence, 11F and 11E cells did not have lower proliferation rates than C2, 9B, or 10C cells (Fig. 8 B). In fact the loss of cells at day 2 in 11E cultures (Fig. 8 A) seems to cause a drop in cell density sufficient to stimulate proliferation of the remaining myoblasts as indicated by the increase in BrdU incorporation in 11E cultures at day 3 (Fig. 8 B). This increased proliferation translates into a small increase in cell number at day 4 (Fig. 8 A). The reduction in cell number observed in both α-DG–deficient clones is therefore unlikely to be due to decreased proliferation rates but reflects a genuine loss of cells in these cultures due to increased apoptosis or necrosis.
Visual inspection of DAPI-labeled cultures revealed the presence of a greater number of nuclei with condensed chromatin in cultures of antisense clones compared with control cells. Since apoptotic cell death leads to chromatin condensation, we used the TUNEL method to assay for apoptosis (Fig. 7, TUNEL). To verify the specificity of this method in our assay, we counterstained the cultures with the nuclear dye DAPI allowing us to also identify apoptotic cells by morphological criteria. We found that cells with marked condensation of chromatin and cytoplasm (apoptotic cells) as well as cytoplasmic fragments with condensed chromatin (apoptotic bodies) were intensely labeled by the TUNEL method. Generally, labeled cells were in the latest stages of apoptotic cell death and had often assumed a round morphology. This method labeled both individual, poorly attached cells and nuclei within aggregates. It was therefore difficult to determine whether TUNEL-positive nuclei belonged to former myoblasts or myotubes. More rarely, nuclei of adherent cells were labeled, albeit less intensely. These nuclei were often found within myoblasts but were occasionally also seen in myotubes. Since in most cases TUNEL-positive nuclei could not be definitely assigned to myoblasts or myotubes, we quantified the number of apoptotic nuclei without attributing them to a particular cell type (Fig. 8 C). A small proportion of nuclei was TUNEL-positive in differentiating C2, 11A, 10C, and 9B cultures, while a larger number of nuclei, often within rounded, poorly attached cells, were labeled in day 2 and older cultures of 11F and 11E cells (Fig. 7, TUNEL). At day 3, 11F, and 11E cultures have 3 and 11 times more TUNEL-positive nuclei compared with cultures of C2 cells (Fig. 8 C). These data indicate that reduced α-DG expression results in a persistent loss of myoblasts after transfer to fusion medium and a correspondingly significant increase in apoptotic cell death in both myoblasts and myotubes.
Decreased Levels of α-DG Expression Are Not Associated with Loss of Membrane Integrity
Studies of dy/dy and mdx mice suggest that loss of membrane integrity is linked to a loss of interaction between dystrophin and the actin cytoskeleton, rather than a decreased cell surface expression of the DGC (Straub et al., 1997). However, recent studies have provided evidence that mutations affecting the expression of SGs can result in a loss of membrane integrity (Duclos et al., 1998; Hack et al., 1998). Since we perturbed the expression of DG without affecting the SGs, we set out to determine the role, if any, of DG in the maintenance of membrane integrity. We used the vital dye Trypan blue to visualize myoblasts or portions of myotubes where membrane integrity was compromised. In our hands, Trypan blue stained nuclei much more intensely than the cell cytoplasm and allowed a direct visualization of the extent of membrane damage on each multinucleated myotube. In cultures of C2, 11F, and 11E cells maintained 3 d in fusion medium cell death has peaked (Fig. 8 A) and Trypan blue labeled the nuclei of some myoblasts (Fig. 9 B). At this time myotubes often had one or two blue nuclei, indicating a localized loss of membrane integrity and only rarely were all nuclei in a myotube labeled. Notably, there were no significant differences between C2 and 11E cultures in either the number of myotubes with one or more nuclei stained with Trypan blue (Fig. 9 A) or in the proportion of myoblasts versus myotubes with blue nuclei (Fig. 9 B). Therefore, a substantial reduction of α-DG expression at the surface of myotubes does not lead to a loss of membrane integrity.
Figure 9.


Decreased levels of α-DG expression do not correlate with loss of membrane integrity. Live C2 and 11E cultures were stained with the vital dye Trypan blue that is normally excluded from cells with an intact plasma membrane. (A) Quantification of the percentage of myotubes with one or more nuclei stained with Trypan blue. No statistically significant difference was found between C2 and 11E myotubes (ANOVA, Fisher's t-test). 11F cells were not assayed. Data represent the mean ± SEM from three separate experiments. (B) Quantification of the percentage of Trypan blue stained nuclei belonging to myoblasts versus myotubes. No significant differences were found for either myoblasts or myotubes between C2 (open bars) and 11E (shaded bars) cultures (ANOVA, Fisher's t-test). Data represent the mean ± SEM from three separate experiments.
Discussion
In muscle, the DGC is thought to link two proteinaceous matrices, the ECM and the subplasmalemmal cytoskeleton, providing structural support for the interposed plasma membrane. According to this widely held model α- and β-DG associate to form the core of the DGC with α-DG bound to LN (Smalheiser and Schwartz, 1987; Douville et al., 1988; Ibraghimov-Beskrovnaya et al., 1992; Gee et al., 1993; Smalheiser and Kim, 1995; Yoshida et al., 1994) and β-DG to dystrophin (Suzuki et al., 1994; Jung et al., 1995). To investigate functional aspects of this model, we have perturbed the expression of α- and β-DG by generating muscle cell lines stably transfected with an antisense DG cDNA expression construct. After several transfections and screening of many stable lines we identified two clones of C2 cells in which expression of DG was reduced significantly i.e., 40–50% and 80–90%, respectively. The two DG-deficient cell lines retain the ability to fuse and form myotubes, and express near normal levels of α- and β-SG, two other DGC proteins. Similarly, expression of other membrane proteins such as the AChR, and the MuSK receptor tyrosine kinase (Jacobson et al., 1998) and β1 integrin (Fig. 4) are indistinguishable from parental C2 cells. Three other clones, 9B, 10C, and 11A, which were subjected to the same transfection and antibiotic selection and have wild-type levels of α- and β-DG, fuse normally and have no obvious cell loss indicating that the altered phenotype of 11E and 11F cells is not a trivial outcome of transfection and antibiotic selection. These observations argue that 11E and 11F cells differ only in their abnormally low expression of DG. An alternative possibility, especially in light of the relatively small number of clones we were able to isolate, is that an unidentified mutation in the antisense-expressing cell lines affects the glycosylation of α-DG so that it is poorly detected by mAb IIH6 (which recognizes, at least in part glycosylated epitopes; Ervasti and Campbell, 1993). In fact, we do see a slightly faster migration during SDS-PAGE of α-DG from 11F and 11E myotubes (Fig. 1 B). This may well be due to altered glycosylation since most of the apparent mass of α-DG on SDS-PAGE is due to carbohydrate moieties (Ibraghimov-Beskrovnaya et al., 1992; Smalheiser and Kim, 1995; Chiba et al., 1997). However, an antibody directed against the core protein (anti-fusion protein B antiserum) also reveals a decrease of at least 50–80% in α-DG levels, which is equivalent to that seen with mAb IIH6 (Fig. 1 C). Thus, α-DG appears to be expressed at low levels in 11F and 11E cells. That the residual α-DG is recognized about equally by a polyclonal antiserum to DG and by mAb IIH6 which is to a binding site on α-DG (Ervasti and Campbell, 1993; Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994; Durbeej et al., 1995) suggests that the small shift in electrophoretic mobility of α-DG in the antisense clones does not affect the interaction of α-DG with LN. The concomitant decrease of β-DG by 70–90% in 11F and 11E cells compared with control C2 cells, further suggests that any possible defects in glycosylation do not lead to excessive shedding of α-DG into the medium of 11F and 11E cells.
α-DG Is a Functional LN Receptor In Situ That Is Involved in ECM Assembly on Muscle Cells
To date there have been no reports of any human or animal myopathy linked to mutations in the DG gene. Mice null for the DG gene die very early in development (Williamson et al., 1997; Côté, P., M. Lindenbaum, and S. Carbonetto. 1997. Mol. Biol. Cell. 8:222a) suggesting that mutations which compromise its expression in tissues other than skeletal muscle may lead to embryonic lethality in humans. Previous studies indicate that α-DG functions as a receptor for two ECM proteins agrin (Gee et al., 1994; Campanelli et al., 1994; Bowe et al., 1994; Sugiyama et al., 1994; Cohen et al., 1995) and LN. For LN, this includes observations on: (a) binding of LN to α-DG isolated from muscle and nervous system (Smalheiser and Schwartz, 1987; Ibraghimov-Beskrovnaya, 1992; Gee et al., 1993; Yamada et al., 1994, 1996); (b) colocalization of LN with α-DG in muscle and other tissues (Klietsch et al., 1993; Yamada et al., 1994; Durbeej et al., 1995; Cohen et al., 1997); (c) inhibition of LN-dependent differentiation in kidney by a mAb IIH6 to α-DG (Durbeej et al., 1995); (d) coprecipitation of LN with dystrophin in cultured muscle cells (Dickson et al., 1992); (e) disruption of Reichert's basement membrane in mice rendered null for the DG gene (Williamson et al., 1997; Côté, P., M. Lindenbaum, and S. Carbonetto. 1997. Mol. Biol. Cell. 8:222a). More recently, Henry and Campbell (1998) have implicated DG in basement membrane assembly in cultured embryoid bodies, though it is unclear whether this is mediated uniquely by α-DG binding to LN or also to other ECM molecules such as agrin (Bowe et al., 1994; Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994; Cohen et al., 1995) and perlecan (Peng et al., 1998). In support of the hypothesis that α-DG is a LN receptor in muscle, we find that lower α-DG levels in 11E and 11F cells result in a corresponding reduction in the level of exogenous LN bound to the surfaces of these lines when compared with parental C2 cells. Moreover, there is a clear decrease in the deposition of endogenous LN on the surface of myotubes. This is not obviously a result of reduced synthesis and secretion of LN since DG-deficient cells have normal levels of LN (Fig. 4 C) and the ECM which forms on the culture substratum between the myotubes appears equivalently LN-rich in DG-deficient and parental C2 cells. The loss of surface LN in 11F and 11E myotubes does not appear to be a secondary consequence of a general disruption of the ECM since the distribution of collagen IV is not significantly affected (Montanaro, F., and S. Carbonetto, unpublished observations). Instead, DG deficiency leads to a rather selective loss of LN from the myotube ECM implicating α-DG as a functional LN receptor necessary for proper ECM assembly in skeletal muscle.
Vachon et al. (1997) have suggested recently that the “de facto receptor” for LN 2 (α2β1γ1) in skeletal muscle is the α7β1 integrin. They report a decrease of the α7β1 integrin in human and mouse muscular dystrophies where mutations in the LN α2 chain lead to loss of LN 2 or expression of a truncated form. They further note that α-DG expression is unaffected in these instances concluding that it is not necessary for LN assembly in vivo. However in the dy/dy and dy2J mutant mice there is a compensatory upregulation of LN 8 (α4β1γ1) which replaces LN 2 (Patton et al., 1997) and could be responsible for the maintained expression of the DGC at the cell surface. Alternatively, expression of the DGC might be more dependent on its association with dystrophin than with its extracellular ligands. In fact, muscle cells in culture have a pool of surface α-DG that is not bound to LN (Fig. 5; Cohen et al., 1997). Similarly, in the dystrophin mutant mdx mouse, the observation that LN is present in the muscle ECM in spite of the dramatic decrease in the DGC at the cell surface, does not eliminate α-DG as a LN receptor in skeletal muscle. For example, although the α7β1 integrin is expressed at wild-type levels in mdx mice (Vachon et al., 1997), a large fraction of LN in skeletal muscle in these mice and DMD patients is unusually soluble indicative of a weak binding to the muscle cell surface (Dickson et al., 1992). Indeed, an early sign of pathology in DMD is the separation of the basement membrane from the muscle cell surface (Carpenter and Karpati, 1979). Furthermore, no obvious colocalization of LN and the integrin β1 subunit is observed in cultures of C2 cells or of primary myotubes from mouse or human (Fig. 4; Dickson et al., 1992) and in C2 myotubes the distribution of the LN α2 chain (Fig. 4) does not match that reported for the α7 integrin subunit (Vachon et al., 1997). More importantly, in mice null for the integrin α7 gene the distribution of LN in skeletal muscle appears normal and the progressive muscle degeneration seems to be predominantly due to defects at the myotendinous junction (Mayer et al., 1997). Thus, as Mayer et al. (1997), we also propose integrins and α-DG may function as independent receptor complexes which together provide a link between LN and the muscle membrane that is necessary for muscle homeostasis.
α-DG Is Involved in Muscle Cell Viability in Culture
The DGC has been postulated to act as a superstructure for the muscle cell surface, and its loss has been correlated with disruption of the plasma membrane, necrosis, and in some cases apoptosis (Rosalki, 1989; D'Amore et al., 1994; Matsuda et al., 1995; Tidball et al., 1995; Tews and Goebel, 1997).
In vivo, apoptosis could be a secondary consequence of the inflammation caused by the constant degeneration and regeneration of muscle fibers that occurs in the absence of dystrophin. Indeed, apoptotic cell death in skeletal muscle of the mdx mouse appears to be mainly caused by activated inflammatory cells that infiltrate the muscle mass and subsequent release of the cytotoxic protein perforin (Spencer et al., 1997). However, our studies and those of Vachon et al. (1996) suggest that other apoptotic pathways might also be activated. Vachon et al. (1996) have studied the effect of LN 2 (α2β1γ1; merosin) on myotube stability in human and mouse muscle cell lines in culture. Several spontaneous variants deficient in LN 2 expression were cloned and found to have increased myotube degeneration after fusion. Addition of exogenous LN 2 increased myotube numbers in all clones and transfection with a human LN α2 chain cDNA decreased myotube degeneration and the abnormally high level of apoptosis in these cell lines. In our studies, decreased expression of α-DG disrupts LN expression on the surface of myotubes, and we expected to see a similar pattern of myotube degeneration. While there are some apoptotic nuclei within adherent myotubes, most TUNEL-positive nuclei were found within amorphous, loosely adherent masses, which could have been detached or “collapsed” myotubes, or clumps of dead myoblasts. Possibly, deficiency of α-DG could cause myotubes to detach more readily than a reduction in the expression of a single LN isoform. Indeed, α-DG–deficient cells would have impaired binding to most if not all LN heterotrimers expressed by muscle cells, as well as agrin (Bowe et al., 1994; Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994; Cohen et al., 1995) and possibly perlecan (Peng et al., 1998). However, Henry and Campbell (1998) reported that absence of DG in embryonic stem cells affects basal lamina assembly but not adhesion to a LN-coated substratum. Similarly, preliminary results show no detectable difference in the ability of 11F and 11E myoblasts to adhere to LN-coated dishes compared with control myoblasts. Vachon et al. (1996) also reported that myoblasts from LN-deficient clonal variants have a reduced ability to fuse. In our studies, 11E and 11F cells fuse and differentiate normally indicating that α-DG may not mediate the effects of LN on myoblast fusion and that these are likely integrin mediated (Chung and Kang, 1990; MacCalman et al., 1992; Collo et al., 1993; George-Weinstein et al., 1993; Blaschuk and Holland, 1994; Sastry et al., 1996; Blaschuk et al., 1997). However myoblast survival was compromised in both clones after serum withdrawal from the onset of differentiation (Fig. 8 A). Serum withdrawal is typically associated with cell death, and we observed a decrease in the number of cells in all cultures within 24 h of switching to fusion medium. While C2 cells and all control clones had completely recovered after 48 h, 11F and 11E cells continued to be lost. Although death of DG-deficient myoblasts could be due to loss of adhesion, we located some adherent myoblasts that were TUNEL-positive indicating that apoptotic cell death can precede cell detachment. It is interesting to note that during differentiation of C2 myoblasts, the level of α-DG expression increases dramatically and its glycosylation changes (Leschziner, A., and S. Carbonetto, unpublished observations) suggesting that it might play a particular function at this developmental stage. Our results suggest that DG is important for myoblast survival during differentiation in culture and we speculate that loss of DG in vivo might affect muscle regeneration by satellite cells.
Straub et al. (1997) have studied the permeability of muscle fibers to the vital dye Evans blue in dystrophic mice. In mdx mice skeletal myofibers have increased permeability to this hydrophilic dye most likely through disruptions in their plasma membranes which allow serum proteins to enter these presumably necrotic cells (Straub et al., 1997). In contrast, dy/dy mice with mutations in the LN α2 chain develop a more severe muscular dystrophy with a minor loss in membrane integrity as reflected by a relative impermeability to Evans blue and by normal levels of creatine kinase in the serum. In both dy/dy mice and patients with merosin-deficient congenital muscular dystrophy skeletal myofibers are apoptotic (Miyagoe et al., 1997; Tews and Goebel, 1997). Furthermore, recent studies have shown a strong correlation between loss of membrane integrity and lack of SGs at the muscle cell surface (Duclos et al., 1998; Hack et al., 1998). In our studies, decreased expression of DG does not appear to affect the expression of α- and β-SG. We are currently investigating whether these SGs are correctly expressed at the sarcolemma of DG-deficient myotubes as would be expected from observations that membrane integrity is not compromised in myotubes from both 11F and 11E cells (Fig. 9). Our results support the notion that the DG and SG complexes are expressed independently of one another in muscle cells and that they perform distinct functions vis à vis membrane integrity.
Mechanism of α-DG–mediated Apoptosis
The data presented here raise questions about how α-DG, a peripheral membrane protein, may transduce an extracellular signal to suppress apoptosis in either myoblasts or myotubes. α-DG is bound tightly to its transmembrane partner β-DG (Bowe et al., 1994; Yoshida et al., 1994) which, in turn, can interact with the SH2/SH3 domain containing adapter protein Grb 2 (Yang et al., 1995). An additional, second messenger, nitric oxide synthase, associates with syntrophins (Brenman et al., 1995, 1996), equipping the DGC further for intracellular signaling. Thus, one can envision a scenario wherein these signaling intermediates are activated by LN binding to α-DG transmitting a signal to the cell interior via β-DG. To our knowledge, however, there is no precedent for an extracellular peripheral membrane protein like α-DG transmitting signals in this manner.
Although no evidence is presently available for the involvement of the DG complex in the initiation of signaling, DG has been shown to be involved in the aggregation of acetylcholine receptors and to act downstream of the muscle specific tyrosine kinase receptor MuSK (Jacobson et al., 1998). Apoptosis or its prevention is often associated with activation of receptors to growth factors or ECM molecules. DG could be part of a cell surface signaling complex and/or act downstream of other tyrosine kinase receptors. LN/α-DG complexes on the myotube surface are involved in the assembly of a heterogeneous basal lamina that would include constituents of the ECM (e.g., glial growth factor, agrin, collagen) which directly activate receptor tyrosine kinases (Jo et al., 1994; Glass et al., 1996; Shrivastava et al., 1997; Vogel et al., 1997) or act as coreceptors for them (e.g., perlecan; Aviezer et al., 1994). Their inclusion in a two dimensional array of ECM in close proximity to the plasma membrane may facilitate activation of these or similar receptors inhibiting cell death. Also, the assembly of an ECM of proteins occurs coincident with assembly of an intracellular network which may “trap” and concentrate receptor tyrosine kinases activating them in a ligand-independent manner.
Integrins may offer another route for intracellular signaling after LN binding to α-DG. Integrin expression changes dramatically during muscle differentiation (Gullberg et al., 1995), affecting the passage from a proliferative to a quiescent state in myoblasts (Sastry et al., 1996), and promoting fusion (Rosen et al., 1992; Sastry et al., 1996). In mature myotubes, Vachon et al. (1996) speculate that the LN receptors responsible for regulating apoptosis in their studies are members of the integrin superfamily viz. α7β1. Integrins are well-known to be involved in apoptosis in nonmuscle cells where loss of integrin-mediated adhesion results in the downregulation of Bcl 2 and upregulation of Bax expression (Zhang et al., 1995; Stromblad et al., 1996), as well as activation of the ICE protease cascade (Guan, 1997), all of which are intermediates in apoptotic cell death. Interestingly, α-DG accumulates at focal adhesions and colocalizes with the integrin β1 subunit when fibroblasts are grown on a LN substrate (Belkin and Smalheiser, 1996). Furthermore, Yoshida et al. (1998) recently reported evidence for cross-talk between integrins and the DGC. In their studies the α5β1 fibronectin receptor in L6 myoblasts can associate with dystrophin and the DGC. Fibronectin, or amino acid mimetics of fibronectin, stimulate phosphorylation of α- and γ-SG. Perhaps, similar cross-talk occurs between α7β1 heterodimer, and the DGC. α-DG binds to the last 2 globular domains in the LN α1 and α2 chains, a region distal to that of any known integrin-binding site so that LN bound to an integrin should be able to bind α-DG or vice versa. Binding of LN to α-DG stimulates aggregation of α/β-DG on muscle cells (Fig. 5; Cohen et al., 1997) which may increase the chance of interacting with an integrin. Thus, α-DG, in addition to its function as a structural element of the cell surface may, by stimulating ECM assembly, enhance the interaction of ligands embedded in the ECM with integrins and other transmembrane receptors that suppress apoptosis.
In conclusion, our data provide strong evidence that α-DG functions in muscle cells as a LN receptor which mediates ECM assembly. Furthermore, they indicate that the DGC regulates apoptosis in culture, which may have important implications for novel functions of the DGC as a signaling complex.
Acknowledgments
We thank Drs. Kevin Campbell (HHMI, University of Iowa) and Peter Yurchenco (UMDNJ) for their generous gifts of antibodies to α-DG and LN-2, respectively. We also thank Chris Jacobson for generously contributing panel C of Fig. 1, and Cathy Lan for her technical assistance with protein extraction and Western blotting.
Abbreviations used in this paper
- α-DG
α dystroglycan
- AChR
acetylcholine receptor
- BrdU
bromodeoxyuridine
- BTX
bungarotoxin
- DAPI
4′,6-diamidino-2-phenylindole
- DGC
dystrophin associated glycoprotein complex
- EHS
Engelbreth-Holm-Swarm
- LN
laminin
- SG
sarcoglycan
- TUNEL
terminal deoxylnucleotidyl transferase-mediated dUTP nick end labeling
Footnotes
This research was supported by grants to S. Carbonetto from the Muscular Dystrophy Association (US), and the Medical Research Council (Canada). F. Montanaro was the recipient of a studentship from the Canadian National Centers of Excellence.
The first two authors contributed equally to this work.
References
- Abrahamson DR, Irwin MH, St. John PL, Perry EW, Accavitti MA, Heck LW, Couchman JR. Selective immunoreactivities of kidney basement membranes to monoclonal antibodies against laminin: localization of the end of the long arm and the short arms to discrete microdomains. J Cell Biol. 1989;109:3477–3491. doi: 10.1083/jcb.109.6.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviezer D, Hecht D, Safran M, Eisinger M, David G, Yayon A. Perlecan, a basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell. 1994;79:1005–1013. doi: 10.1016/0092-8674(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Belkin AM, Smalheiser NR. Localization of cranin (dystroglycan) at sites of cell-matrix and cell-cell contact: recruitment to focal adhesion is dependent upon extracellular ligands. Cell Adhes Commun. 1996;4:281–296. doi: 10.3109/15419069609010772. [DOI] [PubMed] [Google Scholar]
- Blaschuk KL, Holland PC. The regulation of α5β1 integrin expression in human muscle cells. Dev Biol. 1994;164:475–483. doi: 10.1006/dbio.1994.1217. [DOI] [PubMed] [Google Scholar]
- Blaschuk KL, Guerin C, Holland PC. Myoblast αvβ3 integrin levels are controlled by transcriptional regulation of expression of the β3 subunit and down-regulation of β3 subunit expression is required for skeletal muscle cell differentiation. Dev Biol. 1997;184:266–277. doi: 10.1006/dbio.1997.8527. [DOI] [PubMed] [Google Scholar]
- Bönnemann CG, Modi R, Noguchi S, Mizuno Y, Yoshida M, Gussoni E, McNally EM, Duggan DJ, Angelini C, Hoffman EP, et al. β-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet. 1995;11:266–273. doi: 10.1038/ng1195-266. [DOI] [PubMed] [Google Scholar]
- Bowe MA, Deyst KA, Leszyk JD, Fallon JR. Identification and purification of an agrin receptor from Torpedo post-synaptic membranes: a heterodimeric complex related to the dystroglycans. Neuron. 1994;12:1173–1180. doi: 10.1016/0896-6273(94)90324-7. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Xia H, Aldope K, Brendt DS. Nitric oxide synthase is complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- Campanelli JT, Roberds SL, Campbell KP, Scheller RH. A role for dystrophin associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell. 1994;77:663–674. doi: 10.1016/0092-8674(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Campbell KP. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338:259–262. doi: 10.1038/338259a0. [DOI] [PubMed] [Google Scholar]
- Carbonetto S, Lindenbaum MH. The basement membrane of the neuromuscular junction: a synaptic mediatrix. Curr Opin Neurobiol. 1995;5:596–605. doi: 10.1016/0959-4388(95)80064-6. [DOI] [PubMed] [Google Scholar]
- Carpenter S, Karpati G. Duchenne muscular dystrophy: plasma membrane loss initiates muscle cell necrosis unless it is repaired. Brain. 1979;102:147–161. doi: 10.1093/brain/102.1.147. [DOI] [PubMed] [Google Scholar]
- Carrie A, Piccolo F, Leturcq F, de Toma C, Azibi K, Beldjord C, Vallat JM, Merlini L, Voit T, Sewry C, et al. Mutational diversity and hot spots in the α-sarcoglycan gene in autosomal recessive muscular dystrophy (LGMD2D) J Med Genet. 1997;34:470–475. doi: 10.1136/jmg.34.6.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba A, Matsumura K, Yamada H, Inazu T, Shimizu T, Kusunoki S, Kanazawa I, Kobata A, Endo T. Structures of sialylated O-linked oligosaccharides of bovine peripheral nerve α-dystroglycan. J Biol Chem. 1997;272:2156–2162. doi: 10.1074/jbc.272.4.2156. [DOI] [PubMed] [Google Scholar]
- Chung CY, Kang M-S. Correlation between fibronectin and its receptor in chick myoblast differentiation. J Cell Physiol. 1990;142:392–400. doi: 10.1002/jcp.1041420224. [DOI] [PubMed] [Google Scholar]
- Cohen MW, Jacobson C, Godfrey EW, Campbell KP, Carbonetto S. Distribution of α-dystroglycan during embryonic nerve-muscle synaptogenesis. J Cell Biol. 1995;129:1093–1101. doi: 10.1083/jcb.129.4.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MW, Jacobson C, Yurchenco PD, Morris GE, Carbonetto S. Laminin-induced clustering of dystroglycan on embryonic muscle cells: comparison with agrin-induced clustering. J Cell Biol. 1997;136:1047–1058. doi: 10.1083/jcb.136.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collo G, Starr L, Quaranta V. A new isoform of the laminin receptor α7β1 is developmentally regulated in skeletal muscle. J Biol Chem. 1993;268:19019–19024. [PubMed] [Google Scholar]
- Crosbie RH, Heighway J, Venzke DP, Lee JC, Campbell KP. Sarcospan, the 25-kDa transmembrane component of the dystrophin-glycoprotein complex. J Biol Chem. 1997;272:31221–31224. doi: 10.1074/jbc.272.50.31221. [DOI] [PubMed] [Google Scholar]
- D'Amore PA, Brown RH, Jr, Ku PT, Hoffman EP, Watanabe H, Arahata K, Ishihara T, Folkman J. Elevated basic fibroblast growth factor in the serum of patients with Duchenne muscular dystrophy. Ann Neurol. 1994;35:362–365. doi: 10.1002/ana.410350320. [DOI] [PubMed] [Google Scholar]
- de Melker AA, Sterk LM, Delwel GO, Fles DL, Daams H, Weening JJ, Sonnenberg A. The A and B variants of the α3 integrin subunit: tissue distribution and functional characterization. Lab Invest. 1997;76:547–563. [PubMed] [Google Scholar]
- Dickson G, Azad A, Morris GE, Simon H, Noursadeghi M, Walsh FS. Co-localization and molecular association of dystrophin with laminin at the surface of mouse and human myotubes. J Cell Sci. 1992;103:1223–1233. doi: 10.1242/jcs.103.4.1223. [DOI] [PubMed] [Google Scholar]
- Douville PJ, Harvey WJ, Carbonetto S. Isolation and partial characterization of high affinity laminin receptors in neural cells. J Biol Chem. 1988;263:14964–14969. [PubMed] [Google Scholar]
- Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, Durbeej M, Lebakken CS, Ettinger AJ, van der Meulen J, et al. Progressive muscular dystrophy in α-sarcoglycan-deficient mice. J Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M, Larsson E, Ibraghimov-Beskrovnaya O, Roberds SL, Campbell KP, Ekblom P. Non-muscle α-dystroglycan is involved in epithelial development. J Cell Biol. 1995;130:79–91. doi: 10.1083/jcb.130.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster RF, Thompson JM, Kaufman SJ. A laminin substrate promotes myogenesis in rat skeletal muscle cultures: analysis of replication and development using anti-desmin and anti-BrdUrd monoclonal antibodies. Dev Biol. 1987;122:11–20. doi: 10.1016/0012-1606(87)90327-7. [DOI] [PubMed] [Google Scholar]
- Gee SH, Blacher RW, Douville PJ, Provost PR, Yurchenco PD, Carbonetto S. Laminin-binding protein 120 from brain is closely related to the dystrophin-associated glycoprotein, dystroglycan, and binds with high affinity to the major heparin binding domain of laminin. J Biol Chem. 1993;268:14972–14980. [PubMed] [Google Scholar]
- Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-α, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- George-Weinstein M, Foster RF, Gerhart JV, Kaufman SJ. In vitro and in vivo expression of α7 integrin and desmin define the primary and secondary myogenic lineages. Dev Biol. 1993;156:209–229. doi: 10.1006/dbio.1993.1071. [DOI] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattson K, Burden SJ, et al. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- Graham FL, Van der Eb AJ. A new technique for the assay of infectivity of human adenovirus DNA. Virol. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- Guan J-L. Role of focal adhesion kinase in integrin signaling. Int J Biochem. 1997;29:1085–1095. doi: 10.1016/s1357-2725(97)00051-4. [DOI] [PubMed] [Google Scholar]
- Gullberg D, Sjoberg G, Velling T, Sejersen T. Analysis of fibronectin and vitronectin receptors on human fetal skeletal muscle cells upon differentiation. Exp Cell Res. 1995;220:112–123. doi: 10.1006/excr.1995.1297. [DOI] [PubMed] [Google Scholar]
- Hack AA, Ly CL, Jiang F, Clendenin CJ, Sigrist KS, Wollmann RL, McNally EM. γ-Sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J Cell Biol. 1998;142:1279–1287. doi: 10.1083/jcb.142.5.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry MD, Campbell KP. A role for dystroglycan in basement membrane assembly. Cell. 1998;95:859–870. doi: 10.1016/s0092-8674(00)81708-0. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Jacobson C, Montanaro F, Lindenbaum M, Carbonetto S, Ferns M. Alpha-dystroglycan functions in acetylcholine receptor aggregation but is not a coreceptor for agrin-MuSK signaling. J Neurosci. 1998;18:6340–6348. doi: 10.1523/JNEUROSCI.18-16-06340.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo SA, Zhu X, Marchionni MA, Burden SJ. Neuregulins are concentrated at nerve-muscle synapses and activate ACh-receptor gene expression. Nature. 1994;373:158–161. doi: 10.1038/373158a0. [DOI] [PubMed] [Google Scholar]
- Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP. Identification and characterization of the dystrophin anchoring site on β-dystroglycan. J Biol Chem. 1995;270:27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- Jung D, Duclos F, Apostol B, Straub V, Lee JC, Allamand V, Venzke DP, Sunada Y, Moomaw CR, Leveille CJ, et al. Characterization of δ-sarcoglycan, a novel component of the oligomeric sarcoglycan complex involved in limb-girdle muscular dystrophy. J Biol Chem. 1996a;271:32321–32329. doi: 10.1074/jbc.271.50.32321. [DOI] [PubMed] [Google Scholar]
- Jung D, Leturcq F, Sunada Y, Duclos F, Tomé FMS, Moomaw C, Merlini L, Azibi K, Chaouch M, Slaughter C, et al. Absence of γ-sarcoglycan (35 DAG) in autosomal recessive muscular dystrophy linked to chromosome 13q12. FEBS Lett. 1996b;381:15–20. doi: 10.1016/0014-5793(96)00056-7. [DOI] [PubMed] [Google Scholar]
- Klietsch R, Ervasti JM, Arnold W, Campbell KP, Jorgensen AO. Dystrophin-glycoprotein complex and laminin colocalize to the sarcolemma and transverse tubules of cardiac muscle. Circ Res. 1993;72:349–360. doi: 10.1161/01.res.72.2.349. [DOI] [PubMed] [Google Scholar]
- MacCalman CD, Bardeesy N, Holland PC, Blaschuk OW. Noncoordinate developmental regulation of N-cadherin, N-CAM, integrin, and fibronectin mRNA levels during myoblast terminal differentiation. Dev Dyn. 1992;195:127–132. doi: 10.1002/aja.1001950207. [DOI] [PubMed] [Google Scholar]
- Matsuda R, Nishikawa A, Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J Biochem. 1995;118:959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- Mayer U, Saher G, Fässler R, Bornemann A, Echtermeyer F, von der Mark H, Miosge N, Pöschl E, von der Mark K. Absence of integrin α7 causes a new form of muscular dystrophy. Nat Genet. 1997;17:318–323. doi: 10.1038/ng1197-318. [DOI] [PubMed] [Google Scholar]
- McNally EM, Ly CT, Kunkel LM. Human ε-sarcoglycan is highly related to α-sarcoglycan (adhalin), the limb girdle muscular dystrophy 2D gene. FEBS Lett. 1998;422:27–32. doi: 10.1016/s0014-5793(97)01593-7. [DOI] [PubMed] [Google Scholar]
- Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, Nabeshima Y, Takeda S. Laminin α2 chain-null mutant mice by targeted disruption of the Lama2gene: a new model of merosin (laminin-2)- deficient congenital muscular dystrophy. FEBS Lett. 1997;415:33–39. doi: 10.1016/s0014-5793(97)01007-7. [DOI] [PubMed] [Google Scholar]
- Montanaro F, Gee SH, Jacobson C, Lindenbaum MH, Carbonetto S. Laminin and α-dystroglycan mediate acetylcholine receptor aggregation via a MuSK-independent pathway. J Neurosci. 1998;18:1250–1260. doi: 10.1523/JNEUROSCI.18-04-01250.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro V, de Sa E, Moreira, Piluso G, Vainzof M, Belsito A, Politano L, Puca AA, Passos-Bueno MR, Zatz M. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the δ-sarcoglycan gene. Nat Genet. 1996;14:195–198. doi: 10.1038/ng1096-195. [DOI] [PubMed] [Google Scholar]
- Noguchi S, McNelly EM, Othmane KB, Hagiwara Y, Mizuno Y, Yoshida M, Yamamoto H, Bönnemann CG, Gussoni E, Denton PH, et al. Mutations in the dystrophin-associated protein γ-sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270:819–822. doi: 10.1126/science.270.5237.819. [DOI] [PubMed] [Google Scholar]
- Öcalan M, Goodman SL, Kühl U, Hauschka SD, von der Mark K. Laminin alters cell shape and stimulates motility and proliferation of murine skeletal myoblasts. Dev Biol. 1988;125:158–167. doi: 10.1016/0012-1606(88)90068-1. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K, Ervasti JM, Snook JB, Campbell KP. Dystrophin- glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma. J Cell Biol. 1991;112:135–148. doi: 10.1083/jcb.112.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer EL, Ruegg C, Ferrano R, Pytela R, Sheppard D. Sequence and tissue distribution of the integrin α9 subunit, a novel partner of β1 that is widely distributed in epithelia and muscle. J Cell Biol. 1993;123:1289–1297. doi: 10.1083/jcb.123.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of the developing, adult, and mutant mice. J Cell Biol. 1997;139:1507–1521. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng HB, Ali AA, Daggett DF, Rauvala H, Hassell JR, Smalheiser NR. The relationship between perlecan and dystroglycan and its implication in the formation of the neuromuscular junction. Cell Adhes Commun. 1998;5:475–489. doi: 10.3109/15419069809005605. [DOI] [PubMed] [Google Scholar]
- Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- Roberds SL, Leturcq F, Allamand V, Piccolo F, Jeanpierre M, Anderson RD, Lim LE, Lee JC, Tomé FMS, Romero NB, et al. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell. 1994;78:625–633. doi: 10.1016/0092-8674(94)90527-4. [DOI] [PubMed] [Google Scholar]
- Rosalki SB. Serum enzymes in disease of skeletal muscle. Clin Lab Med. 1989;9:767–781. [PubMed] [Google Scholar]
- Rosen GD, Sanes JR, LaChance R, Cunningham JM, Roman J, Dean DC. Roles for the integrin VLA-4 and its counter receptor VCAM-1 in myogenesis. Cell. 1992;69:1107–1119. doi: 10.1016/0092-8674(92)90633-n. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning. A Laboratory Manual. 2nd Edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. Section 1.53.
- Sanes JR, Engvall E, Butkowski R, Hunter DD. Molecular heterogeneity of basal laminae: isoforms of laminin and collagen IV at the neuromuscular junction and elsewhere. J Cell Biol. 1990;111:1685–1699. doi: 10.1083/jcb.111.4.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry SK, Lakonishok M, Thomas DA, Muschler J, Horwitz AF. Integrin subunit ratios, cytoplasmic domains, and growth factor synergy regulate muscle proliferation and differentiation. J Cell Biol. 1996;133:169–184. doi: 10.1083/jcb.133.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Lessard JL. Cellular distribution of smooth muscle actins during mammalian embryogenesis: expression of the α-vascular but not the γ-enteric isoform in differentiating striated myocytes. J Cell Biol. 1989;109:2929–2937. doi: 10.1083/jcb.109.6.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler F, Sorokin LM. Expression of laminin isoforms in mouse myogenic cells in vitro and in vivo. J Cell Sci. 1995;108:3795–3805. doi: 10.1242/jcs.108.12.3795. [DOI] [PubMed] [Google Scholar]
- Shrivastava A, Radziejewski C, Campbell E, Kovac L, McGlynn M, Ryan TE, Davies S, Goldfarb MP, Glass DJ, Lemke G, Yancopoulos GD. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell. 1997;1:25–34. doi: 10.1016/s1097-2765(00)80004-0. [DOI] [PubMed] [Google Scholar]
- Smalheiser NR, Schwartz NB. Cranin, a laminin-binding protein of cell membranes. Proc Natl Acad Sci USA. 1987;84:6457–6461. doi: 10.1073/pnas.84.18.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalheiser NR, Kim E. Purification of cranin, a laminin-binding membrane protein: identity with dystroglycan and reassessment of its carbohydrate moieties. J Biol Chem. 1995;270:15425–15433. doi: 10.1074/jbc.270.25.15425. [DOI] [PubMed] [Google Scholar]
- Spencer MJ, Walsh CM, Dorshkind KA, Rodriguez EM, Tidball JT. Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity. J Clin Invest. 1997;99:2745–2751. doi: 10.1172/JCI119464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromblad S, Becker JC, Yebra M, Brooks PC, Cheresh DA. Suppression of p53 activity and p21WAF1/CIP1 expression by vascular cell integrin αvβ3 during angiogenesis. J Clin Invest. 1996;98:426–433. doi: 10.1172/JCI118808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama J, Bowen DC, Hall ZW. Dystroglycan binds nerve and muscle agrin. Neuron. 1994;13:103–115. doi: 10.1016/0896-6273(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Sunada Y, Bernier SM, Utani A, Yamada Y, Campbell KP. Identification of a novel mutant transcript of laminin α2 chain responsible for muscular dystrophy and dysmyelination in dy2Jmice. Human Mol Genet. 1995;4:1055–1061. doi: 10.1093/hmg/4.6.1055. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yoshida M, Hayashi K, Mizuno Y, Hagiwara Y, Ozawa E. Molecular organization at the glycoprotein-complex-binding site of dystrophin. Eur J Biochem. 1994;220:283–292. doi: 10.1111/j.1432-1033.1994.tb18624.x. [DOI] [PubMed] [Google Scholar]
- Tawil N, Houde M, Blacher R, Esch F, Reichardt LF, Turner DC, Carbonetto S. α1β1 integrin heterodimer functions as dual laminin/collagen receptor in neural cells. Biochemistry. 1990;29:6450–6544. doi: 10.1021/bi00479a028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tews DS, Goebel HH. DNA-fragmentation and expression of apoptosis-related proteins in muscular dystrophies. Neuropath Appl Neurobiol. 1997;23:331–338. [PubMed] [Google Scholar]
- Tian M, Jacobson C, Gee SH, Campbell KP, Carbonetto S, Jucker M. Dystroglycan in the cerebellum is a laminin α2-chain binding protein at the glial-vascular interface and is expressed in Purkinje cells. Eur J Neurosci. 1996;8:2739–2747. doi: 10.1111/j.1460-9568.1996.tb01568.x. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Albrecht DE, Lokensgard BE, Spencer MJ. Apoptosis precedes necrosis in dystrophin-deficient muscle. J Cell Sci. 1995;108:2197–2204. doi: 10.1242/jcs.108.6.2197. [DOI] [PubMed] [Google Scholar]
- Timpl R, Rohde H, Risteli L, Ott U, Robey PG, Martin GR. Laminin. Methods Enzymol. 1982;82:831–838. doi: 10.1016/0076-6879(82)82104-6. [DOI] [PubMed] [Google Scholar]
- Vachon PH, Loechel F, Xu H, Wewer UM, Engvall E. Merosin and laminin in myogenesis; requirement for merosin in myotube stability and survival. J Cell Biol. 1996;134:1483–1479. doi: 10.1083/jcb.134.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon PH, Xu H, Liu L, Loechel F, Hayashi Y, Arahata K, Reed JC, Wewer U, Engvall E. Integrins (α7β1) in muscle function and survival: disrupted expression in merosin-deficient congenital muscular dystrophy. J Clin Invest. 1997;100:1870–1881. doi: 10.1172/JCI119716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1:13–23. doi: 10.1016/s1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- von der Mark H, Öcalan M. The differentiation and redifferentiation of myoblasts is triggered by fibronectin and laminin. Differentiation. 1989;40:150–157. doi: 10.1111/j.1432-0436.1989.tb00823.x. [DOI] [PubMed] [Google Scholar]
- Wigler M, Pellicer A, Silverstein S, Axel R, Urlaub G, Chasin L. DNA mediated transfer of the adenine-phosphoribosyltransferase locus into mammalian cells. Proc Natl Acad Sci USA. 1979;76:1373–1376. doi: 10.1073/pnas.76.3.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y, Ibraghimov-Beskrovnaya O, Campbell KP. Dystroglycan is essential for early embryonic development: disruption of Reichert's membrane in DAG1-null mice. Human Mol Genet. 1997;6:831–841. doi: 10.1093/hmg/6.6.831. [DOI] [PubMed] [Google Scholar]
- Worton R. Muscular dystrophies: Diseases of the dystrophin-glycoprotein complex. Science. 1995;270:755–756. doi: 10.1126/science.270.5237.755. [DOI] [PubMed] [Google Scholar]
- Xu H, Christmas P, Wu X-R, Wewer UM, Engvall E. Defective muscle basement membrane and lack of M-laminin in the dystrophic dy/dy mouse. Proc Natl Acad Sci USA. 1994;91:5572–5576. doi: 10.1073/pnas.91.12.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Shimizu T, Tanaka T, Campbell KP, Matsumura K. Dystroglycan is a binding protein of laminin and merosin in peripheral nerve. FEBS Lett. 1994;352:49–53. doi: 10.1016/0014-5793(94)00917-1. [DOI] [PubMed] [Google Scholar]
- Yamada H, Chiba A, Endo T, Kobata A, Anderson LVB, Hori H, Fukuta-Ohi H, Kanazawa I, Campbell KP, Shimizu T, Matsumura K. Characterization of dystroglycan-laminin interaction in peripheral nerve. J Neurochem. 1996;66:1518–1524. doi: 10.1046/j.1471-4159.1996.66041518.x. [DOI] [PubMed] [Google Scholar]
- Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP. SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem. 1995;270:1–4. doi: 10.1074/jbc.270.20.11711. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem (Tokyo) 1990;108:748–752. doi: 10.1093/oxfordjournals.jbchem.a123276. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Suzuki A, Yamamoto H, Noguchi S, Mizuno Y, Ozawa E. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by n-octyl-D-glucoside. Eur J Biochem. 1994;222:1055–1061. doi: 10.1111/j.1432-1033.1994.tb18958.x. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Pan Y, Hanada H, Iwata Y, Shigekawa M. Bidirectional signaling between sarcoglycans and the integrin adhesion system in cultured L6 myocytes. J Biol Chem. 1998;273:1583–1590. doi: 10.1074/jbc.273.3.1583. [DOI] [PubMed] [Google Scholar]
- Yoshihara CM, Hall ZW. Increased expression of the 43-kD protein disrupts acetylcholine receptor clustering in myotubes. J Cell Biol. 1993;122:169–179. doi: 10.1083/jcb.122.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Vuori K, Reed JC, Ruoslahti E. The α5β1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc Natl Acad Sci USA. 1995;92:6161–6165. doi: 10.1073/pnas.92.13.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]