

Abstract
Cells of the yeast Saccharomyces cerevisiae choose bud sites in a manner that is dependent upon cell type: a and α cells select axial sites; a/α cells utilize bipolar sites. Mutants specifically defective in axial budding were isolated from an α strain using pseudohyphal growth as an assay. We found that a and α mutants defective in the previously identified PMT4 gene exhibit unipolar, rather than axial budding: mother cells choose axial bud sites, but daughter cells do not. PMT4 encodes a protein mannosyl transferase (pmt) required for O-linked glycosylation of some secretory and cell surface proteins (Immervoll, T., M. Gentzsch, and W. Tanner. 1995. Yeast. 11:1345–1351). We demonstrate that Axl2/Bud10p, which is required for the axial budding pattern, is an O-linked glycoprotein and is incompletely glycosylated, unstable, and mislocalized in cells lacking PMT4. Overexpression of AXL2 can partially restore proper bud-site selection to pmt4 mutants. These data indicate that Axl2/Bud10p is glycosylated by Pmt4p and that O-linked glycosylation increases Axl2/ Bud10p activity in daughter cells, apparently by enhancing its stability and promoting its localization to the plasma membrane.
Keywords: O-glycosylation, PMT4, AXL2/BUD10, budding, polarity
Yeast cells grow by budding, a specialized form of cell division. Bud emergence occurs in the G1 phase of the cell cycle at predictable locations on the cell surface. Several proteins required for bud emergence, including actin and the rho-type GTPase, Cdc42p (Adams and Pringle, 1984; Ziman et al., 1993), assemble at this site. Subsequently, the actin cytoskeleton is polarized towards this site, which leads to polarized secretion, cell surface growth, and formation of a bud (Tkacz and Lampen, 1973; Field and Schekman, 1980; Adams and Pringle, 1984; Novick and Botstein, 1985). Cytokinesis later occurs at the position of bud emergence, resulting in a mother and a daughter cell. Thus, when cells choose bud sites, they fix the orientation of the actin cytoskeleton, the polarity of secretion, and the position for cytokinesis.
The position of bud assembly is specified by a set of bud-site selection genes and by cell type (Hicks et al., 1977; Bender and Pringle, 1989; Chant and Herskowitz, 1991; Chant et al., 1991; Valtz and Herskowitz, 1996; Zahner et al., 1996). a and α cells choose sites in an axial pattern, in which cells bud adjacent to the previous division site. a/α cells choose bipolar sites, in which daughter cells (cells that have not budded previously) typically bud distal to the previous division site; mother cells (cells that have budded previously) choose sites proximal or distal to the previous division site (Freifelder, 1960; Chant and Pringle, 1995). The ability of cells to exhibit predictable budding patterns suggests that intracellular landmarks direct the position of bud emergence (Chant and Herskowitz, 1991; Snyder et al., 1991). The behavior of two classes of mutants is consistent with this landmark hypothesis. Mutations in BUD3, BUD4, AXL1, or AXL2/BUD10 disrupt the axial budding pattern, but have no effect on the bipolar budding pattern of a/α cells (Chant and Herskowitz, 1991; Chant et al., 1991; Fujita et al., 1994; Halme et al., 1996; Roemer et al., 1996). In contrast, mutations in BNI1, SPA2, PEA2, BUD6, BUD7, BUD8, or BUD9 disrupt the bipolar budding pattern, but have no effect on the axial budding pattern of a and α cells (Valtz and Herskowitz, 1996; Zahner et al., 1996; Amberg et al., 1997). The first set of genes is specifically required for the axial budding pattern, whereas the second set is specifically required for the bipolar budding pattern.
For cells budding in the axial pattern, the bud site used in one cell cycle specifies an adjacent position for bud emergence in the next cell cycle (Chant and Pringle, 1995). Bud3p, Bud4p, and Axl2/Bud10p are located in a cortical position at the mother-bud neck from mitosis of one cell cycle to G1 of the next (Chant et al., 1995; Halme et al., 1996; Roemer et al., 1996; Sanders and Herskowitz, 1996). Thus, during G1, Bud3p, Bud4p, and Axl2/Bud10p behave as spatial remnants of the position of bud emergence in the previous cell cycle and promote adjacent bud emergence in the new cell cycle. These cortical factors are proposed to organize the cytoskeleton by recruiting downstream proteins required for bud emergence, such as Cdc24p and Bem1p, to that site (Sloat and Pringle, 1978; Bender and Pringle, 1991). Indeed, Rsr1p/Bud1p, required for both axial and bipolar budding, binds to bud emergence proteins Cdc24p and Bem1p (Zheng et al., 1995; Park et al., 1997), and Bud6p interacts with actin in the yeast two-hybrid assay (Amberg et al., 1995, 1997).
Axl2/Bud10p is a type I integral membrane protein that is thought to provide spatial information for the axial pattern (Halme et al., 1996; Roemer et al., 1996). It traverses the secretory pathway to reach its final destination at the plasma membrane and contains N-linked oligosaccharides. Axl2/Bud10 has a receptor-like topology: a single transmembrane domain and substantial intra- and extracellular domains. The large NH2-terminal domain is extracellular, rich in serine and threonine residues, and is postulated to interact with the cell wall. During G1, Axl2/Bud10p is present first at the presumptive bud site and later at the periphery of the bud. As the bud grows, Axl2/Bud10p is found in an apparent ring structure on the bud side of the mother-bud neck. It later forms an additional apparent ring on the mother side of the mother-bud neck during mitosis. Initial localization of Axl2/Bud10p to the neck requires septin proteins (Roemer et al., 1996). Subsequent organization of Axl2/Bud10p into rings at the neck requires Bud3p (Halme et al., 1996).
In the present study, we describe a requirement for O-glycosylation in bud-site selection. Yeast has seven protein mannosyl transferases (pmt)1, encoded by PMT genes, that catalyze the initial step of O-linked glycosylation (Tanner and Lehle, 1987; Gentzsch and Tanner, 1996). These enzymes transfer a mannosyl residue from a dolichol-phosphate donor onto serine or threonine residues of substrate proteins in the endoplasmic reticulum (Haselbeck and Tanner, 1983; Strahl-Bolsinger and Tanner, 1991; Immervoll et al., 1995; Lussier et al., 1995; Gentzsch and Tanner, 1996). Further modification of these O-linked oligosaccharides occurs in the Golgi apparatus (Haselbeck and Tanner, 1983; Herscovics and Orlean, 1993). Pmts are required for cell wall integrity, cell morphology, and coordination of the nuclear and morphological cell cycles (Gentzsch and Tanner, 1996). We show here that mutations in the mannosyl transferase gene PMT4 affect the axial budding pattern of a and α cells. The requirement of Pmt4p for bud-site selection appears to result from its O-linked glycosylation of Axl2/Bud10p.
Materials and Methods
Materials
HA11 mAb, used to detect the hemagglutinin epitope (HA) in an Axl2/ Bud10p-HA fusion protein, was from Berkeley Antibody Company. Polylysine, protease inhibitors (phenylmethylsulfonyl fluoride, benzamidine, antipain, leupeptin, and pepstatin), calcofluor white, and protein standards were from Sigma Chemical Co. HRP-coupled sheep anti–mouse antibodies were from Nycomed Amersham, Inc. Peptide N-glycosidase F (PNGase) and tunicamycin were from Boehringer Mannheim Corp.
Microbiological and Molecular Biological Methods
Strains are described in Table I, and plasmids in Table II. DNA and yeast genetic manipulations were performed as described (Ausubel et al., 1987; Sambrook et al., 1989; Rose et al., 1990). Synthetic low ammonium histidine dextrose (SLAHD) solid medium for assaying pseudohyphal growth was prepared as described by Gimeno et al. (1992). Microcolony budding assays were performed as previously described (Chant and Herskowitz, 1991; Sanders and Herskowitz, 1996).
Table I.
Strains Used in this Study
| Strain | Genotype | Source | ||
|---|---|---|---|---|
| IH2531 | α ura3-52 | Gimeno et al., 1992 | ||
| SY96* | α ura3-52 | Fink Laboratory | ||
| SY77* | α hmra::URA3 | This study | ||
| SY167* | α bad15 ura3 | This study | ||
| SY172* | α bad43 ura3 | This study | ||
| SY171* | α bud4-3 | This study | ||
| SY202 | a/α bad15/bad15 ura3/+ his4/+ trp1/+ (isogenic with SY204) | This study | ||
| SY204 | a/α ura3/+ his4/+ trp1/+ (isogenic with SY202) | This study | ||
| MY16* | α leu2 ura3-52 | M. Maxon, IH Lab | ||
| SY448* | α leu2 pmt4::LEU2 | This study | ||
| MY13* | a leu2 | M. Maxon, IH Lab | ||
| SY447* | a leu2 pmt4::LEU2 | This study | ||
| SY463 | a/α leu2/leu2 | This study | ||
| SY464 | a/α pmt4::LEU2/pmt4::LEU2 leu2/leu2 | This study | ||
| SY414 | α leu2-3 ura3-52 his3-Δ200 lys2-801 trp1-Δ901 suc2-Δ9 | Tanner Lab Collection | ||
| SY655‡ | α pmt1::URA3 | Tanner Lab Collection | ||
| SY656‡ | α pmt2::LEU2 | Tanner Lab Collection | ||
| SY657‡ | α pmt3::HIS3 | Tanner Lab Collection | ||
| SY415‡ | α pmt4::TRP1 | Tanner Lab Collection | ||
| SY658‡ | α pmt5::URA3 | Tanner Lab Collection | ||
| SY659‡ | α pmt6::URA3 | Tanner Lab Collection | ||
| IH1783 | a trpl leu2 ura3 his4 can1 | IH Lab Collection | ||
| SY450§ | a pmt4::LEU2 | This study | ||
| SY748§ | mat::URA3 pmt4::LEU2 | This study | ||
| IH1785§ | matal-50 | IH Lab Collection | ||
| SY221 | matal-50 bad15 his4 leu2 ura3 | This study | ||
| MGY60‡ | α axl2::AXL2-HA | This study | ||
| MGY61‡ | α pmt1::HIS3 axl2::AXL2-HA | This study | ||
| MGY62‡ | α pmt2::LEU2 axl2::AXL2-HA | This study | ||
| MGY63‡ | α pmt3::HIS3 axl2::AXL2-HA | This study | ||
| MGY64‡ | α pmt4::TRP1 axl2::AXL2-HA | This study | ||
| MGY65‡ | α pmt5::URA3 axl2::AXL2-HA | This study | ||
| MGY66‡ | α pmt6::LYS2 axl2::AXL2-HA | This study | ||
| YWO333 | α ade2-1o his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 | Dieter Wolf | ||
| YWO339‖ | a pep4Δ::HIS3 prb1::LEU2 | Dieter Wolf | ||
| YWO340‖ | a pep4Δ::HIS3 prb1::hisG prc1::hisG | Dieter Wolf | ||
| MGY69‖ | α axl2::AXL2-HA | This study | ||
| MGY70‖ | a pep4Δ::HIS3 prb1::LEU2 axl2::AXL2-HA | This study | ||
| MGY71‖ | a pep4Δ::HIS3 prb1::hisG prc1::hisG axl2::AXL2-HA | This study | ||
| MGY72‖ | α pmt4Δ::TRP1 axl2::AXL2-HA | This study | ||
| MGY73‖ | a pmt4::TRP1 pep4Δ::HIS3 prb1::LEU2, axl2::AXL2-HA | This study | ||
| MGY74‖ | a pmt4::TRP1 pep4::HIS3 prb1::hisG prc1::hisG axl2::AXL2-HA | This study | ||
| MGY67‡ | α axl2::AXL2-HA ubc7::LEU2 | This study | ||
| MGY68‡ | α pmt4::TRP1 axl2::AXL2-HA ubc7::LEU2 | This study | ||
| MGY75 ¶ | axl2::AXL2-HA | This study | ||
| MGY76 ¶ | α ura3-52 leu2-3,112 suc2-Δ9 sec7axl2::AXL2-HA pmt4::LEU2 | This study | ||
| MGY77† | axl2::AXL2-HA | This study | ||
| MGY78† | α ura3-52 leu2-3,112 suc2-Δ9 sec18axl2::AXL2-HA pmt4::LEU2 | This study |
Isogenic to IH2531, except where noted.
Isogenic to SY414, except where noted.
Isogenic to IH1783, except where noted.
Isogenic to YWO333, except where noted.
Isogenic to SEY5018, except where noted.
Isogenic to SEY5188, except where noted.
Table II.
Plasmids Used in this Study
| Plasmid | Description | Source | ||
|---|---|---|---|---|
| pDR58 | hmra::URA3 knock-out construct | D. Rivier, University of Illinois | ||
| pSS51 | pmt4::LEU2 knock-out construct | This study | ||
| pSS52 | pmt4::TRP1 knock-out construct | This study | ||
| pAH14 | AXLBUD10-GFP in pRS314 | Halme et al., 1996 | ||
| pRS314 | CEN ARS vector containing TRP1 | Sikorski and Hieter, 1989 | ||
| pAXL2-HA | Encodes AXL2-HA; inYIp5 | Roemer et al., 1996 | ||
| YEp24-AXL2 | 2μ AXL2 | Roemer et al., 1996 | ||
| B1,B5,B7,B9 | Original bad15 complementing clones in YCp50 containing ORFs YJR140C-YJR147W | This study; Rose et al., 1987 | ||
| pSS44 | B7 lacking the ∼4.5 kB SphI fragment (one SphI site was from the plasmid vector), | This study | ||
| contains ORFs YJR140C-YJR143C (PMT4) | ||||
| pSS45 | ClaI-HpaI fragment of B7 cloned into the ClaI-NruI sites of Ycp50, | This study | ||
| contains all of YJR142W and a portion of YJR143C (PMT4) | ||||
| pSS46 | EcoRI-BglII fragment of pSS44 in the EcoRI-BamHI sites of YCp50, | This study | ||
| contains all of YJR140C, YJR141W, and a portion of YJR142W | ||||
| pSS47 | pSS44 lacking the HindIII fragment, contains all of ORFs YJR142W and YFR143C (PMT4) | This study | ||
| pSS48 | HindIII-SalI fragment of pSS47 cloned into the HindIII-SalI sites of pBluescript (Stratagene) | This study | ||
| pSS49 | pSS48 lacking the PMT4 ORF (see Materials and Methods) | This study |
Isolation of Axial-defective Mutants
Construction of SY77.
The HMRa gene of IH2531 (α ura3-52) was disrupted by introduction of pDR58 digested with EcoRV (Loo et al., 1995), creating SY77. The disruption was confirmed by Southern blot analysis.
Mutagenesis.
A culture of SY77 was grown to saturation in rich medium, resuspended in water, and UV-irradiated in separate aliquots to 0.3–24% survival. Mutagenized cells were plated on solid SLAHD medium to induce pseudohyphal growth (Gimeno et al., 1992) and incubated at 30°C for 2 wk in the dark. Of ∼70,000 surviving colonies, 407 exhibited the pseudohyphal colony morphology. Microcolony budding assays were performed on the 107 colonies that retested for pseudohyphal growth. 23 of these mutants exhibited >30% bipolar budding and were studied further.
Genetic Tests.
Mutants were tested for single-gene segregation by analyzing the budding pattern of the meiotic progeny of crosses to IH2393 (a Bud+). Mutants whose progeny exhibited 2:2 axial/bipolar budding were studied further. Because a/α strains do not exhibit the axial budding pattern, dominance or recessiveness was assessed by analyzing mata1/MATα diploid progeny (phenotypically α) formed by crosses between the mutants and a mata1 strain (IH1785). Recessive mutations yielded an axial budding pattern in the mata1/MATα diploids; dominant or semidominant mutations yielded a bipolar or partially bipolar phenotype. Mutants defective in known genes required for axial budding were identified by complementation tests using plasmids carrying BUD3, BUD4, or AXL1, by mating to bud3, bud4, axl1, or axl2 strains, and, for new BUD4 and AXL1 alleles, by linkage tests. Complementation tests among the mutants were performed by analyzing diploids (phenotypically α) formed by crossing mata1 and α mutant strains. If the resultant diploid exhibited axial budding, the two mutations were assigned to different complementation groups. If the resultant diploid exhibited bipolar budding, the two mutations were assigned to the same complementation group.
Cloning BAD15/PMT4
The bud-site selection axial determinant BAD15/PMT4 gene was cloned by complementation of the pseudohyphal growth phenotype. SY167 (α bad15-1 ura3) was transformed with a low copy yeast library marked with URA3 (Rose et al., 1987). Approximately 10,200 transformants were patched onto SLAHD plates in two concentric rings of 25 (because the pseudohyphal phenotype in a and α cells was sensitive to position on the plate, as well as the number of colonies per plate). Plates were incubated for 2 wk at 30°C and scored for pseudohyphae by microscopic examination at 30×. Nine transformant colonies failed to exhibit pseudohyphal growth and were assayed for budding pattern. Plasmid DNA isolated from the three colonies that exhibited axial budding contained overlapping inserts by restriction analysis.
Complete Deletion of PMT4
The pmt4Δ constructs replaced the entire PMT4 coding sequence with the LEU2 or TRP1 gene. The HindIII–SalI fragment of pSS47, which contains PMT4 and YJR142W, was cloned into the vector pBluescript (Stratagene), creating pSS48. All sequences of pSS48, except the PMT4 open reading frame (ORF), were amplified using PCR with divergent primers (OSS8 5′-CCGaggcctCTTTTTCTGTTCACTAACCACAAACGAACTG and OSS9 5′-CCGaggcctCTGTACTTCTGTGGACTGTCAGAAAATATCTTGG; a StuI restriction site is denoted in lower case) that hybridized just upstream of the PMT4 start codon and downstream of the stop codon. The resulting linear fragment was cleaved at the introduced StuI sites and circularized, yielding a plasmid (pSS49) that contained the PMT4 flanking regions separated by the StuI site. The LEU2 gene was then introduced into the StuI site to form pSS51, with the direction of LEU2 transcription opposite that of PMT4. The resulting disruption fragment was excised from pSS51 by digestion with HindIII and SphI and used to replace the chromosomal PMT4 gene by one-step gene replacement (Rothstein, 1983). The presence of the pmt4 deletion was confirmed by colony PCR with the following oligonucleotides: OSS10, derived from LEU2: 5′-TTAGACCGCTCGGCCAAACAACC; and OSS11, derived from the PMT4 3′ sequences not present in the pSS49 plasmid, 5′-GGCCATACCATGAAGTATACC; amplified a ∼0.8-kB fragment in pmt4:: LEU2 cells. Control oligonucleotides: OSS7, derived from the PMT4 3′ flanking sequences, 5′-CCGAGGCCTCTGTACTTCTGTGGACTGTCAGAAAATATCTTGG; and OSS11 amplified a ∼0.5-kB fragment in Pmt+ cells. To create a pmt4::TRP1 allele, the HincII–StuI fragment of YPP14, containing TRP1, was introduced into the StuI site of pSS49. The resultant plasmid, pSS52, was digested with ClaI and SphI and used to disrupt the chromosomal copy of PMT4 by one-step gene replacement (Rothstein, 1983). The presence of the pmt4 deletion was confirmed by analyzing bud-site selection pattern.
Membrane Extracts and Western Blot Analysis
Yeast cells grown overnight and harvested in log phase were washed with 50 mM Tris-HCl, pH 7.5, 50 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, and disrupted with glass beads in the same buffer containing 20 μg/ml antipain, 1 μg/ml leupeptin, and 1 μg/ml pepstatin. The cell wall fraction was removed by centrifugation at 1,500 g. The resulting supernatant was centrifuged at 48,000 g to generate a membrane pellet. 5 to 10 μg of membrane protein was subjected to SDS-PAGE using 8 or 10% SDS-polyacrylamide gels. Proteins were transferred to nitrocellulose membranes, blocked in TBS containing 0.1% Tween 20 and 4% nonfat dry milk, and decorated with a 1:1,000 dilution of HA antibodies in TBS containing 0.1% Tween 20 and 1% nonfat dry milk. Axl2/Bud10p-HA was visualized using the Nycomed Amersham ECL protein detection kit.
Peptide N-Glycosidase F Treatment
Membrane pellets were resuspended in 1% SDS, 1% β-mercaptoethanol, 35 mM EDTA, and heated to 95°C for 3 min. 10 vol of incubation buffer (50 mM potassium phosphate buffer, pH 6.8, 35 mM EDTA, 1% β-mercaptoethanol, 0.55% octyl glucoside) was added, followed by a second incubation at 95°C for 3 min. Protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 20 μg/ml antipain, 1 μg/ml leupeptin, and 1 μg/ml pepstatin) were added, and the reaction mixture was divided into two parts. PNGase was added to one aliquot, and both reaction mixtures were incubated at 37°C for 12 to 16 h.
Hydrogen Fluoride Treatment
O- and N-linked sugars were removed by anhydrous hydrogen fluoride (HF) cleavage (Mort and Lamport, 1977). Membrane fractions (50 μg protein) were lyophilized and dried in a desiccator containing solid P2O5. 250 μl HF was added and samples were treated for 80 min at 0°C. HF was subsequently removed by evaporation in a nitrogen stream.
Tunicamycin Treatment
Yeast cells were grown overnight to early log phase, harvested, resuspended in fresh medium, and grown for 30 min at 30°C. Tunicamycin (10 μg/ml) was added, cells were incubated at 30°C for 60 min, and then subjected to membrane extraction as described in Membrane Extracts and Western Blot Analysis.
Microscopy
To observe the autofluorescence of a fusion protein containing Axl2/ Bud10p and green fluorescent protein (GFP), cells in log phase were mounted on 0.5% agarose pads under coverslips sealed with pink or red nail polish. To observe bud scars, cells were stained with calcofluor as described by Pringle (1991). Cells were visualized using a Zeiss Axioplan microscope with the FITC filter set and photographed with a three-chip cooled CCD camera.
Results
Use of the Pseudohyphal Phenotype to Isolate Bud-site Selection Mutants
To identify genes required specifically for the axial budding pattern, we searched for mutants of α cells that exhibited a bipolar, rather than axial, budding pattern. This screening method took advantage of the relationship between pseudohyphal growth and budding pattern. Pseudohyphal growth is exhibited by cells with a bipolar budding pattern, such as a/α cells (Gimeno et al., 1992), as well as a bud4 and α bud4 mutants in the Σ1278b strain background (Sanders and Herskowitz, 1996). In contrast, cells that bud in the axial pattern, such as α strain SY77 (α ura3-52 hmra::URA3), do not exhibit pseudohyphal growth. We screened ∼70,000 colonies derived from UV-irradiated SY77 cells for the pseudohyphal growth phenotype. Budding patterns of the 107 mutants that displayed the pseudohyphal phenotype were assayed using a microcolony assay (see Materials and Methods). Ten BAD mutants exhibited bipolar budding patterns upon retesting and were shown to be defective in single genes by segregation analysis (see Materials and Methods). Of these, four were defective in BUD4, one was defective in BUD3, and one in AXL1 (Chant and Herskowitz, 1991; Fujita et al., 1994). Complementation tests revealed that the four remaining mutants, bad15, bad43, bad54, and bad85, defined three additional complementation groups. We show here that the complementation group identified by bad15 and bad43 corresponds to the previously identified gene PMT4.
bad15 Mutants Have a Daughter-specific Defect in Bud-site Selection
bad15 mutant cells exhibited a novel budding pattern that is neither axial nor bipolar (Table III, line 3). In the axial pattern, cells bud adjacent to the previous division site (Chant and Pringle, 1995; Table III, lines 1 and 7). In the bipolar pattern, daughter cells typically bud distal to the previous division site, whereas mother cells bud either near the division site or distal to the previous division site (Chant and Pringle, 1995; Table III, lines 2 and 6). bad15 mutants (and to a lesser extent, bad43 mutants) exhibited a distinctive, unipolar budding pattern in which daughter cells budded distal to the division site, as in the bipolar pattern, and mother cells exhibited a typical axial pattern, budding adjacent to the previous division site (Table III, lines 3 and 5). The similar phenotypes of bad15 and bad43 mutants suggested that they might be defective in the same gene. Indeed, these mutants failed to complement (Table III, line 9). To determine whether the bad15 mutation affected bipolar budding, a/α bad15/bad15 diploids were constructed and analyzed. These strains exhibited normal bipolar budding (Table III, compare lines 2 and 4). These data indicated that the bad15 mutation affected only the axial budding pattern and primarily the behavior of daughter cells. This conclusion was supported by observing the budding patterns of bad15 cells over several generations. In one experiment in which eleven separate pedigrees were followed, 38/39 bad15 mother cells budded towards the previous division site, whereas 27/28 bad15 daughter cells budded away from the previous division site. Nine daughter cells in this experiment divided to become mother cells that divided again. In all cases, these cells budded towards the previous division site in the next cell cycle when they became mothers. These data demonstrated that the bad15 budding defect was exhibited only by daughter cells and not by mother cells.
Table III.
Mutants Defective in bad15 Display a Daughter-specific Defect in Bud-site Selection
| % Class I | % Class II | % Class III | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Relevant genotype |

|
% Class IV Other | n | |||||||
| 1. α BAD15 | 98 | 1 | <1 | <1 | 200 | |||||
| 2. a/α BAD15/BAD15 | 7 | 33 | 53 | 7 | 600 | |||||
| 3. α bad15 | 21 | 71 | 5 | 4 | 300 | |||||
| 4. a/α bad15/bad15 | 9 | 20 | 63 | 8 | 300 | |||||
| 5. α bad43 | 22 | 51 | 21 | 6 | 100 | |||||
| 6. α bud4 | 10 | 27 | 40 | 24 | 300 | |||||
| Relevant genotype | % Class I | % Classes II, III, IV | ||||||||
| 7. a 1 −/α bad15/+ | 93 | 7 | 100 | |||||||
| 8. a 1 −/α bad15/bad15 | 15 | 85 | 100 | |||||||
| 9. a 1 −/α bad43/bad15 | 12 | 88 | 100 | |||||||
Budding pattern was determined by analyzing the morphology of microcolonies (see Materials and Methods). Class I, axial pattern, mother and daughter cells budded adjacent to the previous division site; Class II, mother cells budded near the previous division site, daughter cells budded distal to the previous division site; Class III, both mother and daughter cells budded distal to the previous division site; Class IV, all other microcolony morphologies. Class II and III together represent the bipolar budding pattern. The mother cell is drawn on the left. Strains used were: SY77, line 1; SY204, line 2; SY167, line 3; SY202, line 4; SY172, line 5; SY171, line 6; SY221 × IH2531, line 7; SY221 × SY167, line 8; and SY221 × SY172, line 9.
To determine whether bad15 mother cells exhibited normal axial budding, bud scars were analyzed by calcofluor staining. Since bad15 is an allele of PMT4 (see below), a complete deletion of PMT4 was used for this study. Cells with a single bud or bud scar and a visible birth scar were scored. Consistent with the microcolony data, which suggest that daughter cells budded incorrectly, only 29% of pmt4 cells placed the first bud or bud scar adjacent to the birth scar. Rather, 66% of pmt4 cells placed the first bud or bud scar opposite the birth scar (n = 100), as is typical of the first bud in the bipolar pattern (Chant and Pringle, 1995). In contrast, 94% of wild-type (WT) cells positioned a bud or bud scar adjacent to the birth scar (n = 100), as is typical of the axial budding pattern (Chant and Pringle, 1995). However, after the initial incorrect bud-site selection event, pmt4 mother cells budded in an axial pattern (Fig. 1, top, compare PMT4 to pmt4::LEU2). 99% of the bud scars in both WT and pmt4 cells with two bud scars or a bud and a bud scar were adjacent (n = 100 in both cases), as is typical of the axial budding pattern (Chant and Pringle, 1995). In addition, 96% of the bud scars in WT and 92% of the bud scars in pmt4 cells with three or more scars were adjacent (n = 300). The calcofluor staining pattern of WT and pmt4 cells with three or more bud scars was indistinguishable, suggesting that pmt4 cells bud axially after an initial incorrect bud-site selection choice.
Figure 1.

bad15/pmt4 mother cells bud in an axial pattern. WT cells (IH1783, PMT4) or cells lacking the BAD15/PMT4 gene (SY450, pmt4::LEU2) were stained with calcofluor. Cells with two bud scars (top) or ≥3 bud scars (bottom) were counted and photographed.
bad15 Mutants Are Defective in the PMT4 Gene
The BAD15 gene was cloned by screening for reversal of the pseudohyphal behavior of bad15 strain SY167. Only subclones containing the PMT4 gene were able to complement the bud-site selection defect of bad15 mutants (Fig. 2; Table IV, lines 1–4). Pmt4p is one of seven yeast enzymes that initiate addition of O-linked oligosaccharides to proteins (Gentzsch and Tanner, 1996). BAD15 was confirmed to be allelic to PMT4 from the following experiments. First, pmt4 mutants failed to complement the bud-site selection defect of bad15 mutants, although both pmt4 and bad15 mutations were recessive (Table IV, lines 5, 6 and 7). Second, the same spectrum of proteins underglycosylated in pmt4 mutants was underglycosylated in bad15 and bad43 mutants (data not shown). Third, the bud-site selection phenotype of strains with mutations in PMT4 was identical to that of bad15 and bad43 mutants: all exhibited an axial budding defect that was daughter-specific, resulting in unipolar, rather than axial or bipolar budding (Table V, lines 8–13). Fourth, BAD15 and PMT4 exhibited tight genetic linkage: only two axial-budding recombinants were found in 13 tetrads (52 segregants) in crosses between α bad15 (SY167) and a pmt4::LEU2 (SY450).
Figure 2.

The budding pattern defect of the bad15 mutant is complemented by plasmids containing PMT4. SY167 cells (α bad15) were transformed with the indicated plasmids. A, B7; B, pSS44; C, pSS45; D, pSS47; E, pSS46. Black lines indicate DNA present in the clones. Arrows indicate the direction of transcription of individual ORFs. 1, YJR140C; 2, YJR141W; 3, YJR142W; 4, YJR144W; 5, YJR145C; 6, YJR146W; 7, YJR147W. Budding pattern of the transformants was scored using a microcolony assay (Materials and Methods).
Table IV.
bad15 Mutants Are Defective in PMT4 Activity
| Relevant genotype | Plasmid | % Class I | % Classes II, III, IV | n | ||||
|---|---|---|---|---|---|---|---|---|
| 1. BAD15 | YCp50 | 80 | 20 | 200 | ||||
| 2. BAD15 | PMT4 | 79 | 21 | 100 | ||||
| 3. bad15 | YCp50 | 7 | 93 | 171 | ||||
| 4. bad15 | PMT4 | 76 | 24 | 301 | ||||
| 5. mat::URA3/α pmt4::LEU2/bad15 | none | 25 | 75 | 300 | ||||
| 6. a 1 −/α bad15/BAD15 | none | 92 | 8 | 300 | ||||
| 7. mat::URA3/α pmt4::LEU2/PMT4 | none | 97 | 3 | 300 |
Budding pattern was determined by analyzing the morphology of microcolonies as described in the legend to Table III (see also Materials and Methods). Strains used were: IH2531, lines 1 and 2; SY167, lines 3 and 4; SY748 × SY167, line 5; IH1785 × SY167, line 6; and SY748 × SY96, line 7.
Table V.
Mutants Defective in PMT4, but Not other PMT Genes, Have Altered Budding Patterns
| Relevant genotype | % Class I | % Class II | % Classes III & IV | n | ||||
|---|---|---|---|---|---|---|---|---|
| 1. Pmt+ | 93 | 7 | <1 | 200 | ||||
| 2. pmt1 | 90 | 5 | 5 | 200 | ||||
| 3. pmt2 | 97 | 2 | 1 | 200 | ||||
| 4. pmt3 | 91 | 6 | 3 | 200 | ||||
| 5. pmt4 | 59 | 38 | 3 | 200 | ||||
| 6. pmt5 | 92 | 6 | 2 | 200 | ||||
| 7. pmt6 | 90 | 8 | 2 | 116 | ||||
| 8. a Pmt+ | 96 | 2 | 2 | 200 | ||||
| 9. a pmt4::LEU2 | 5 | 86 | 9 | 200 | ||||
| 10. α Pmt+ | 96 | 3 | 1 | 155 | ||||
| 11. α pmt4::LEU2 | 13 | 77 | 10 | 179 | ||||
| 12. a/α Pmt+ | 16 | 27 | 57 | 200 | ||||
| 13. a/α pmt4::LEU2/pmt4::LEU2 | 1 | 43 | 56 | 600 |
Budding pattern was determined by analyzing the morphology of microcolonies as described in the legend to Table III (see also Materials and Methods). Strains used were: SY414, line 1; SY655, line 2; SY656, line 3; SY657, line 4; SY415, line 5; SY658, line 6; SY659, line 7; MY13, line 8; SY447, line 9; MY16, line 10; SY448, line 11; SY463, line 12; and SY464, line 13.
The seven yeast pmt enzymes have different substrate specificities: for example, O-glycosylation of Bar1p and Cts1p is severely affected by mutations in PMT1 and PMT2, but not by mutations in PMT4. In contrast, O-glycosylation of Kex2p and Gas1p/Gpg1p/gp115 is severely affected by mutations in PMT4, but not by mutations in PMT1 and PMT2 (Gentzsch and Tanner, 1996, 1997). To determine if PMT4 is unique among PMT genes in being required for normal budding pattern, mutants defective in each of the PMT genes were analyzed. Of the six pmt mutants tested, only pmt4 mutants exhibited altered budding patterns (Table V, lines 1–7). The difference in penetrance of the pmt4 phenotype in the experiments shown in Tables III and V was due to differences in the strain backgrounds (data not shown).
Pmt4p Promotes Addition of O-linked Oligosaccharides to Axl2/Bud10p
Since Pmt4p is a protein mannosyl transferase, we hypothesized that it was involved in O-glycosylation of a protein required for axial budding. Because PMT activity is confined to the ER, only proteins that pass through the ER can be glycosylated by these enzymes. Of all the proteins known to be involved in axial bud-site selection, only Axl2/Bud10p transits the secretory pathway (Roemer et al., 1996) and thus, is a candidate for O-glycosylation. We have used an epitope-tagged allele, AXL2/BUD10-HA, which complements axl2/bud10 mutations (Roemer et al., 1996), to test whether Axl2/Bud10p is a substrate for Pmt4p.
Because a lack of O-linked sugars might cause a change in the apparent molecular weight of Axl2/Bud10p, its mobility in pmt mutants was examined by SDS-PAGE (Fig. 3). Full-length Axl2/Bud10p (∼205 kD) was found in all pmt mutants, with the exception of pmt4 (Fig. 3 A), in which smaller fragments (∼43–59 kD) accumulated. Since the predicted molecular weight of the Axl2/Bud10p ORF is ∼91 kD, these data indicated that Axl2/Bud10p had undergone proteolytic cleavage. These 43–59-kD fragments, found exclusively in the membrane fraction (M. Gentzsch and W. Tanner, unpublished data), apparently lacked N-linked oligosaccharides, as their mobility did not change after treatment with PNGase (Fig. 3 B).
Figure 3.

Axl2/Bud10p in pmt4 mutants is degraded, membrane-associated, and lacks N-linked sugars. (A) Membrane extracts were prepared from WT (MGY60) and pmt mutant cells (MGY61-66) producing AXL2-HA (Material and Methods). Extracts were separated by SDS-PAGE and visualized by immunoblotting using anti-HA antibodies. (B) Extracts from WT (MGY60) and a pmt4 mutant (MGY64) were treated with PNGase. Samples were separated by SDS-PAGE and visualized by immunoblotting.
Fortuitously, studies of Axl2/Bud10p in the presence of tunicamycin revealed a stabilized form of the protein in pmt4 cells (Fig. 4 A). We do not understand the nature of this tunicamycin-induced stabilization. In the presence of tunicamycin, Axl2/Bud10p from WT cells migrated at ∼125 kD. The absence of fully glycosylated (∼205 kD) Axl2/Bud10p suggests that its half-life is short since all is cleared within the 60 min tunicamycin treatment. Axl2/ Bud10p from pmt4 mutants migrated at ∼114 kD under the same conditions, indicating that Axl2/Bud10p in pmt4 mutants lacked a posttranslational modification. The modification was characterized by subjecting the Axl2/Bud10p species from tunicamycin-treated WT and pmt4 cells to treatment with HF to release remaining carbohydrates (O-linked sugars; Fig. 4 B). HF treatment of WT extracts resulted in a further reduction in molecular weight of Axl2/Bud10p to ∼100 kD, indicating that it is an O-glycosylated protein (Fig. 4 B, compare WT + HF to WT-HF). After treatment with HF, Axl2/Bud10p from pmt4 cells comigrated with the Axl2/Bud10p from WT cells at ∼100 kD (Fig. 4 B, compare + HF samples). The shift in SDS-PAGE mobility of the 114-kD form of Axl2/Bud10p from pmt4 mutants indicated that it possessed some O-linked oligosaccharide (Fig. 4 B, compare pmt4 samples). These observations indicate that Axl2/Bud10p from pmt4 mutants and from WT cells both contained O-linked oligosaccharides, but that Axl2/Bud10p received fewer O-linked oligosaccharides in pmt4 cells than in WT cells. Axl2/Bud10p received ∼25 kD of O-linked oligosaccharides: ∼13 kD are Pmt4p-independent; the remainder are Pmt4p-dependent.
Figure 4.

Axl2/Bud10p is stabilized in pmt4 cells treated with tunicamycin and is incompletely O-glycosylated. (A) WT (MGY60) and pmt mutant cells (MGY61-66) were treated with tunicamycin (Materials and Methods). Membrane extracts were prepared, separated by SDS-PAGE, and visualized by immunoblotting. (B) Membranes of tunicamycin-treated WT (MGY60) and pmt4 cells (MGY64) were subjected to further deglycosylation by treatment with HF. Samples were separated by SDS-PAGE and visualized by immunoblotting.
In contrast to its behavior in pmt4 strains, Axl2/Bud10p from tunicamycin-treated pmt1, pmt2, pmt3, pmt5, and pmt6 strains was almost indistinguishable in size from WT Axl2/Bud10p (Fig. 4 A). Longer SDS-PAGE runs, which separated proteins of high molecular weight, showed that Axl2/Bud 10p was slightly smaller in pmt1 and pmt2 strains (data not shown). Addition of the Pmt4p-independent O-linked oligosaccharides present on Axl2/Bud10p in pmt4 mutants may be added by Pmt1p and Pmt2p.
Degradation of Axl2/Bud10p Probably Occurs in the Golgi Apparatus
To determine the subcellular compartment in which proteolysis of Axl2/Bud10p occurred in pmt4 cells, Axl2/ Bud10p was analyzed in strains with temperature-sensitive mutations in sec18 and sec7, which disrupt secretory traffic from the ER to the Golgi apparatus and through the Golgi apparatus, respectively (Novick et al., 1981). Axl2/Bud10p was degraded in pmt4 sec18 cells grown at permissive temperature (Fig. 5, left, 25°C). Incubation of cells at 37°C resulted in accumulation of full-length, nondegraded Axl2/ Bud10p (Fig. 5, left, 37°C). In contrast, Axl2/Bud10p was degraded at both 25°C and 37°C in the sec7 pmt4 strain (Fig. 5, right). These data suggested that degradation of Axl2/Bud10p occurred after the secretory block imposed by sec18 and before the block imposed by sec7. Thus, degradation of Axl2/Bud10p most likely occurred in the Golgi apparatus.
Figure 5.

Degradation of Axl2/Bud10p in pmt4 mutants occurs in the Golgi apparatus. Mutant strains, pmt4 sec18 (MGY78) and pmt4 sec7 (MGY76), were grown at the permissive temperature (25°C) or at 37°C to induce the secretory block. Membrane extracts were prepared, separated by SDS-PAGE, and visualized by immunoblotting.
If Axl2/Bud10p is degraded in the Golgi apparatus, its cleavage should be independent of vacuolar proteases. That prediction was borne out. Mutations in the genes encoding vacuolar proteases Pep4p, Prb1p, and Prc1p did not prevent degradation of full-length Axl2/Bud10p in pmt4 mutants (Fig. 6 A). Considerably more of the 43–59-kD Axl2/Bud10p species was found in pmt4 pep4 prb1 and pmt4 pep4 prb1 prc1 mutants than in pmt4 mutants alone, suggesting that vacuolar proteases further degrade the 43– 59-kD fragments. Degradation of Axl2/Bud10p in pmt4 mutants was unaffected by mutation of UBC7, which encodes a ubiquitin-conjugating enzyme that participates in degradation of ER proteins (Biederer et al., 1997), again suggesting that the cleavage of Axl2/Bud10p does not occur in the ER (Fig. 6 B).
Figure 6.

Initial cleavage of Axl2/Bud10p in pmt4 mutants does not depend on vacuolar proteases or the ER-linked proteolytic system. (A) Membrane extracts were prepared, separated by SDS-PAGE, and visualized by immunoblotting from PMT4 or pmt4 cells possessing (MGY69 or MGY72) or lacking (MGY70, 71, 73, 74) vacuolar proteases Pep4, Prb1p, and Prc1p. (B) Membrane extracts were prepared, separated by SDS-PAGE, and visualized by immunoblotting from PMT4 or pmt4 cells possessing (MGY69 or MGY72) or lacking (MGY67 or 68) ubiquitin conjugating enzyme Ubc7p.
Axl2/Bud10p Is Improperly Localized in pmt4 Mutants
Localization of Axl2/Bud10p is dependent upon cell cycle stage (Halme et al., 1996; Roemer et al., 1996). In early G1 cells, Axl2/Bud10p is found in a patch at the presumptive bud site; it is later found at the periphery of the bud. By the time of the G2/M transition, Axl2/Bud10p is observed on the bud side of the mother-bud neck and is subsequently found on both sides of the mother-bud neck. Axl2/ Bud10p persists at the neck region of mother and daughter cells past cytokinesis. We made similar observations in our Pmt+ strain background using cells that produce a fusion of Axl2/Bud10p to the GFP (Halme et al., 1996; Fig. 7, a–f). 97% of fluorescent WT cells in an asynchronous population exhibited strong staining that fell into one of three classes. Almost 50% of cells (202/407) exhibited staining at bud periphery (Fig. 7, a and b). 30% of cells (121/407) exhibited staining predominantly at the bud side of the mother-bud neck (Fig. 7, c and d). 17% exhibited strong staining that was symmetrically distributed with respect to the mother-bud neck (Fig. 7, e and f). Though we detected no difference in the distribution of cells with respect to the cell cycle in pmt4 mutants versus WT cells, only 10% of pmt4 cells (37/369) exhibited staining that fell into the three classes defined by WT cells. Most mutant cells exhibited a strikingly different localization pattern (Fig. 7, g–l): 58% (216/369) exhibited a punctate intracellular pattern (Fig. 7, g, h, i, k, and l) that resembled the Golgi apparatus (Franzusoff et al., 1991; Redding et al., 1991). Such staining was seen in only 3% of Pmt+ cells. 13% of pmt4 cells with these punctate intracellular patches also exhibited relatively normal staining at the bud periphery (28/216; Fig. 7, g [right cell] and h). Many pmt4 cells (206/369) accumulated Axl2/Bud10p at the neck. However, unlike WT cells, the neck staining was enriched on, or was specific to, the mother side of the mother-bud neck (78/369 cells; Fig. 7, i and j) or faint, but equally distributed on either side of the neck (121/369 cells; Fig. 7, k and l). None of the neck staining in WT cells was observed in either of these two categories. Localization of the Bud4 protein was unaffected by mutation of pmt4 (data not shown).
Figure 7.
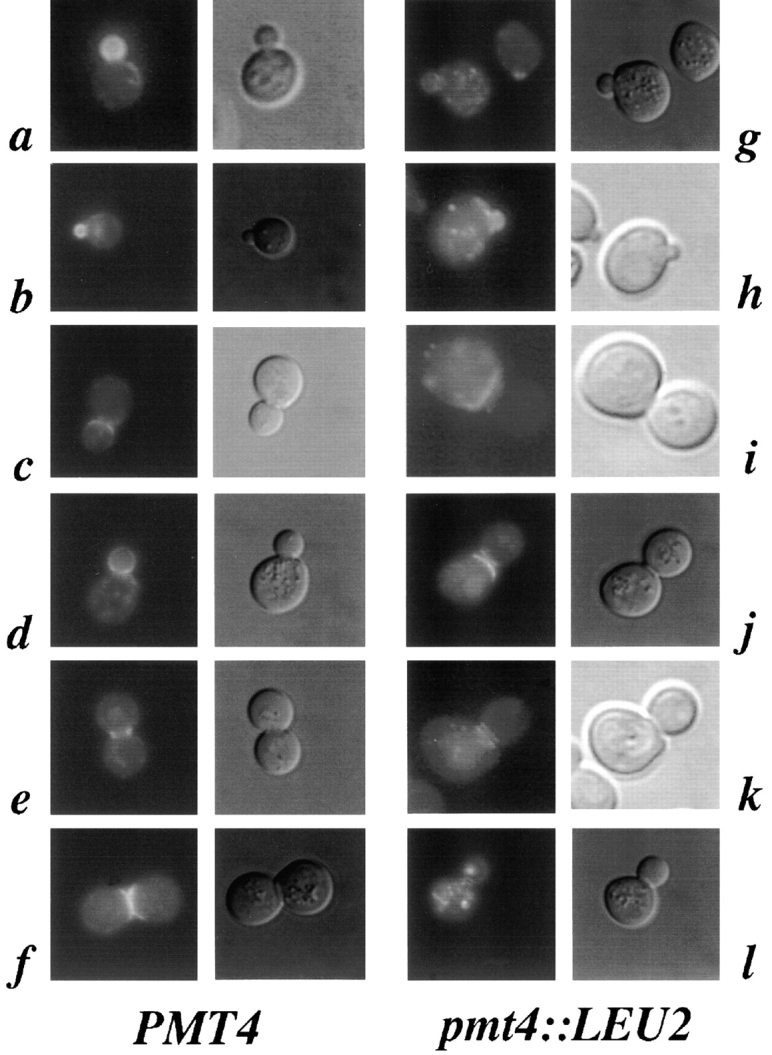
Axl2/Bud10p is improperly localized in pmt4 mutants. IH1783 cells (PMT4, a–f) or SY450 cells (pmt4::LEU2, g–l) carried pAH14, a low-copy plasmid encoding Axl2/Bud10p-GFP. Cells were grown to early log phase, sonicated, and visualized by fluorescence microscopy.
Overexpression of AXL2/BUD10 Partially Restores Axial Bud-site Selection to pmt4 Mutants
If Axl2/Bud10p activity is decreased due to the lack of O-linked oligosaccharides, overexpression of AXL2/BUD10 might partially restore the axial budding pattern to pmt4 mutants. To test this possibility, pmt4 and WT strains were transformed with YEp24-AXL2, which carries AXL2/ BUD10 on a high copy plasmid. This plasmid slightly decreased the axial budding of WT cells, from 93 to 76% axial. In contrast, YEp24-AXL2 partially rescued the unipolar budding pattern of pmt4 mutants to axial, increasing axial budding from 5 to 40%.
Discussion
Axl2/Bud10p is a transmembrane glycoprotein required for yeast cells to exhibit the axial budding pattern (Halme et al., 1996; Roemer et al., 1996). Like Bud3p and Bud4p (Chant et al., 1995; Sanders and Herskowitz, 1996), it is proposed to function as an intracellular landmark: Axl2/ Bud10p localizes to both sides of the mother-bud neck for part of the cell cycle and is inherited by both mother and daughter cells at the position of bud emergence in the previous cell cycle. Through the isolation and characterization of mutants with altered bud-site selection, we have uncovered a requirement for O-linked glycosylation by the mannosyl transferase Pmt4p, which presumably acts directly on Axl2/Bud10p. Though O-linked glycosylation has been assumed to be important for protein function, identifying specific roles for this modification on particular proteins has been elusive. We suggest that O-linked glycosylation of Axl2/Bud10p promotes its movement through the secretory pathway, prevents its degradation, and allows Axl2/Bud10p to function on the cell surface to assemble components required for bud emergence.
O-linked Glycosylation by Pmt4p Enhances the Stability of Axl2/Bud10p
Axl2/Bud10p acquires O-linked oligosaccharides, some of which are dependent upon Pmt4p. Only the Pmt4p-dependent sugars appear to be important for Axl2/Bud10p function. Because Axl2/Bud10p in pmt4 mutants contains some O-linked oligosaccharide, it is clear that the simple presence of O-linked oligosaccharide is not sufficient to facilitate Axl2/Bud10p protein function. It appears that, as with protein phosphorylation, the amount of O-linked oligosaccharide or the specific sites of modification are important for function. Studies of Axl2/Bud10p provide an opportunity to identify the determinants for recognition by Pmt4p and other PMT proteins. Because proteins, such as Kex2p and Gas1p/gp115/Ggp1p are glycosylated primarily by Pmt4p (Gentzsch and Tanner, 1997), comparison of Kex2p, Gas1p/gp115/Ggp1p, and Axl2/Bud10p may help to identify consensus acceptor sequences for Pmt4p-dependent O-linked glycosylation. All three proteins have extracellular domains rich in serine and threonine residues that are close to the membrane (Mizuno et al., 1988; Vai et al., 1991; Halme et al., 1996; Roemer et al., 1996). These domains are glycosylated in the case of Kex2p and Gas1p/ gp115/Ggp1p (Fuller et al., 1989; Wilcox and Fuller, 1991; Gatti et al., 1994; Gentzsch and Tanner, 1997), and perhaps Axl2/Bud10p as well. Serine- and threonine-rich domains close to the membrane may be recognized specifically by Pmt4p.
Pmt4p is required for the stability of Axl2/Bud10p. Axl2/Bud10p is a large, type I integral protein of the plasma membrane. In pmt4 mutants, Axl2/Bud10p is cleaved to produce stable membrane-associated fragments of ∼43–59 kD, which appear to be the cytoplasmic-transmembrane segment of Axl2/Bud10p. This structure is inferred from three lines of evidence. First, the fragments are recognized by antibodies specific for an epitope tag at the COOH terminus of Axl2/Bud10p. Second, as these fragments are membrane-associated, they presumably also contain the transmembrane domain. Third, these fragments lack both N- and O-linked oligosaccharides, which should be present solely in the extracellular domain. Roemer et al. (1996) determined that Axl2/Bud1p is a type I membrane protein by demonstrating that the COOH terminus of Axl2/Bud10p is refractory to degradation by proteases added extracellularly. The apparent molecular weight of the protease-resistant fragments (a collection of fragments 50–55 kD) is similar to that of the fragments we observe in pmt4 mutants (43–59 kD), adding further credence to the notion that the fragments contain the COOH terminus of Axl2/Bud10p joined to the transmembrane domain. We do not know the fate of the remaining ∼40% of Axl2/Bud10p, which includes the NH2-terminal segment.
Two lines of evidence suggest that the initial degradation of Axl2/Bud10p in pmt4 mutants occurs in the Golgi apparatus. First, proteolytic cleavage of Axl2/Bud10p occurs in sec7 pmt4, but not in sec18 pmt4 mutants, suggesting that cleavage requires transit to the Golgi apparatus and occurs even when further transport is inhibited. Consistent with this notion, cleavage of Axl2/Bud10p to the ∼55-kD fragments does not require vacuolar proteases. The accumulation of these ∼55-kD fragments in pep4 prb1 and pep4 prb1 prc1 cells suggested that once generated, these fragments are degraded in the vacuole.
Previous studies have revealed an effect of O-glycosylation on protein stability. Axl2/Bud10p is stabilized in pmt4 mutants grown in the presence of the N-linked glycosylation inhibitor, tunicamycin. We do not know the reason for this stabilization. One possibility is that in the absence of N-linked sugars, Axl2/Bud10p cannot reach the Golgi apparatus, where its initial degradation occurs. However, Axl2/Bud10p localization in pmt4 mutants appears the same in the presence versus absence of tunicamycin (data not shown). We favor a second possibility, that the conformation of Axl2/Bud10p lacking its N-linked sugars makes it a poorer substrate for proteolytic degradation. Several proteins, including the low density lipoprotein (LDL) receptor, decay accelerating factor, major envelope protein of Epstein-Barr virus, Kex2p, and neurotrophin receptor are less stable when they lack O-linked oligosaccharides (Kozarsky et al., 1988; Fuller et al., 1989; Reddy et al., 1989; Monlauzeur et al., 1998). Not all O-linked oligosaccharides are important for protein function, however: Gas1p/Ggp1p/gp115 and human chorionic gonadotropin lacking O-linked oligosaccharides (Matzuk et al., 1987; Gatti et al., 1994) are indistinguishable in stability from their glycosylated counterparts.
Instability of Axl2/Bud10p and its failure to accumulate efficiently at the cell surface without modification by Pmt4p are probably coupled, though it is unclear which is the primary consequence of the O-glycosylation defect. Perhaps unmodified Axl2/Bud10p is recognized as a misfolded protein and targeted for destruction by a quality control system of the Golgi apparatus. Resulting proteolytic fragments might be inefficiently targeted to the plasma membrane. Another possibility is that absence of Pmt4p-dependent modifications delays movement of Axl2/ Bud10p through the secretory pathway, increasing the probability of cleavage by a resident Golgi protease. Some Axl2/Bud10p does assemble at the bud periphery and neck in pmt4 cells, though the amount is reduced with respect to WT cells. As for the LDL receptor (Kozarsky et al., 1988), the lack of O-linked oligosaccharide may also affect Axl2/Bud10p stability once it reaches the plasma membrane. Axl2/Bud10p seems to be inherently unstable.
PMT4 Is Required for Proper Budding Only in Daughter Cells
Because cells defective in PMT4 exhibit a bud-site selection defect, we initially predicted the existence of a Pmt4p substrate protein important for accurate budding. Our studies indicate that Axl2/Bud10p is the Pmt4p substrate relevant to bud-site selection: Axl2/Bud10p is O-glycosylated, unstable and mislocalized in pmt4 mutants, and its overproduction can partially rescue the pmt4 phenotype. The requirement for Pmt4p only in daughter cells might be explained in a variety of ways. In principle, Axl2/ Bud10p might be required only in daughter cells, or its glycosylation might be necessary only in daughter cells. Because Axl2/Bud10p is required for bud-site selection in both mother and daughter cells (Halme et al., 1996; Roemer et al., 1996), the first explanation appears to be incorrect. Therefore, we entertain a second possibility, that O-linked glycosylation of Axl2/Bud10p by Pmt4p is required only in daughter cells. Perhaps O-glycosylation is not needed to stabilize Axl2/Bud10p in mother cells. This explanation is unlikely, since a single, partially degraded population of Axl2/Bud10p is detected by Western blot (Fig. 3 A), and intracellular patches of Axl2/Bud10p accumulate in both mother and daughter cells (Fig. 7). Another possibility is that mother cells can accommodate the reduction of Axl2/Bud10p activity in pmt4 mutants, whereas daughter cells cannot. We favor this third possibility.
The differences between mother and daughter cells in the requirement for PMT4 may arise from differences in the path by which Axl2/Bud10p reaches the mother-bud neck (Fig. 8). Axl2/Bud10p is thought to become localized to the bud side of the mother-bud neck by reorganization of protein in cells with a small bud (Fig. 8 A, 2 and 3). Localization of Axl2/Bud10p at the mother side of the mother-bud neck is hypothesized to occur subsequently by new synthesis within the mother cell (Roemer et al., 1996; Fig. 8 A, 3). After cytokinesis, both mother and daughter cells inherit a patch of Axl2/Bud10p, which promotes utilization of axial bud sites. In pmt4 mutants, we propose that Axl2/Bud10p is at a reduced level, so that the activity in the daughter side of the mother-bud neck is inadequate (Fig. 8 B). In contrast, newly synthesized Axl2/Bud10p, which becomes localized to the mother side of the mother-bud neck (Fig. 8 B, 3), is proposed to be adequate for promoting utilization of axial bud sites after cytokinesis (Fig. 8 B, 4). In other words, Axl2-Bud10p used for axial budding in daughter cells must persist from S phase until the next G1 phase, whereas that in mother cells need persist only from late mitosis until the next G1 phase. That daughter cells exhibit a longer G1 phase than do mother cells might also make it harder for Axl2/Bud10p to persist until it is needed again in daughter cells. Support for the proposal that Axl2-Bud10p is differentially affected in mother and daughter cell compartments comes from the observation that Axl2-Bud10p can be observed preferentially on the mother side of the mother-bud neck in a substantial fraction of pmt4 mutant cells (21%), a distribution that is rare in WT cells (0.25%). For the 33% of pmt4 cells that exhibited faint Axl2/Bud10p staining on both sides of the mother-bud neck, we argue that this level of Axl2/ Bud10p is sufficient for bud-site selection in mother cells, but is inadequate in daughter cells.
Figure 8.

The daughter cell-specific budding pattern phenotype of pmt4 mutants may be explained by reduced activity of Axl2/Bud10p in both mother and daughter cells. (A) WT cells. 1, Axl2/Bud10p (black crescent) is localized at the periphery of a WT bud. 2, Axl2/ Bud10p is reorganized to the bud side of the mother-bud neck. 3, New synthesis of Axl2/ Bud10p leads to its accumulation on the mother side of the mother-bud neck. 4, After cytokinesis, both mother and daughter cells inherit a patch of Axl2/Bud10p. The mother cell initiates bud formation adjacent to the residual patch of Axl2/ Bud10p. 5, The daughter cell initiates bud formation adjacent to the residual patch of Axl2/ Bud10p. The residual patches of Axl2/Bud10p disappear (hatched rectangles). (B) pmt4 cells: 1, Axl2/Bud10p (black crescent) is localized at the periphery of a pmt4 mutant bud. Axl2/Bud10p is also found in punctate intracellular patches (black circles). 2, Axl2/Bud10p is reorganized to the bud side of the mother-bud neck. The reduced accumulation of Axl2/Bud10p to the bud neck is represented by the small size of the black rectangle (compare to A 2). 3, New synthesis of Axl2/Bud10p leads to its accumulation on the mother side of the mother-bud neck. The unstable Axl2/Bud10p disappears from the bud side of the mother-bud neck. 4, After cytokinesis, only mother cells inherit a patch of Axl2/Bud10p. The mother cell initiates bud formation adjacent to the residual patch of Axl2/Bud10p. 5, The daughter cell initiates bud formation using bipolar information, since it lacks a residual patch of Axl2/Bud10p. The residual patch of Axl2/Bud10p disappears from the mother cell (hatched rectangles).
Several functional differences between mother and daughter cells have been described previously. Mother cells can switch mating type, whereas daughter cells cannot (Strathern and Herskowitz, 1979); mother cells specifically inherit plasmids containing autonomously replicating sequences (ARS) and lacking centromere sequences, whereas daughter cells do not (Murray and Szostak, 1983); and mother cells budding in the bipolar pattern may bud distal or proximal to the birth site, whereas new daughter cells bud primarily at distal sites (Chant and Pringle, 1995). O-glycosylated cell-surface molecules certainly contribute to selection of bud sites in a/α cells: both Bud8p and Bud9p appear to be O-glycosylated (Zahner et al., 1996; N. Pagé, H. Bussey, J.R. Pringle, personal communication). Here, we have observed that the integrity of an O-glycosylated landmark protein can differentially affect the behavior of mother and daughter cells and generate an asymmetric cell division. Asymmetrically located landmarks that are anchored to the cell wall by O-glycosylated proteins might contribute to the asymmetric switching of mating type and plasmid inheritance as well.
Acknowledgments
We thank David Rivier, Mary Maxon, Carlos Gimeno, Gerry Fink, Merrilyn Mitchelitch, John Chant, Terry Roemer, Mike Snyder, Gunther Daum, Dieter Wolf, and Thomas Sommer for strains and plasmids. We are grateful to Ludwig Lehle for assistance with the HF experiments, Chi Tu for analysis of bad mutants, Tom Wang for demonstrating the allelism between BAD15 and BAD43, Ramon Tabtiang for sequencing the ends of the B7 clone, and Aji Kron for assistance in figure preparation. We appreciate discussions with members of the Herskowitz laboratory and Anita Sil, Frank Solomon, Steve Bell, and Ramon Tabtiang for comments on the manuscript.
This work was supported by a postdoctoral fellowship from the Helen Hay Whitney Foundation and a Burroughs Wellcome Career Award in the Biomedical Sciences to S.L. Sanders, a grant from the Deutsche Forschungsgemeinschaft (SFB521) to W. Tanner, and a National Institutes of Health research grant (GM48052) to I. Herskowitz.
Abbreviations used in this paper: BAD
bud-site selection axial determinant
- GFP
green fluorescent protein
- HA
hemagglutinin epitope
- HF
hydrogen fluoride
- ORF
open reading frame
- pmt
protein mannosyl transferase
- PNGase
peptide N-glycosidase F
- SLAHD
synthetic low ammonium histidine dextrose
- WT
wild-type
Footnotes
S.L. Sanders' present address is Department of Biology, Massachusetts Institute of Technology and Howard Hughes Medical Institute, 31 Ames Street, Cambridge, MA 02139.
M. Gentzsch's present address is Mayo Clinic Scottsdale, S.C. Johnson Medical Research Center, 13400 East Shea Boulevard, Scottsdale, AZ 85259.
References
- Adams AEM, Pringle JR. Localization of actin and tubulin in wild-type and morphogenetic-mutant Saccharomyces cerevisiae. . J Cell Biol. 1984;98:934–945. doi: 10.1083/jcb.98.3.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg DC, Basart E, Botstein D. Defining protein interactions with yeast actin in vivo. Nature Struct Biol. 1995;2:28–35. doi: 10.1038/nsb0195-28. [DOI] [PubMed] [Google Scholar]
- Amberg DC, Zahner JE, Mulholland JW, Pringle JR, Botstein D. Aip3p/Bud6p, a yeast actin-interacting protein that is involved in morphogenesis and the selection of bipolar budding sites. Mol Biol Cell. 1997;8:729–753. doi: 10.1091/mbc.8.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1987. Current Protocols in Molecular Biology. Wiley Interscience.
- Bender A, Pringle JR. Multicopy suppression of the cdc24 budding defect in yeast by CDC42 and three newly identified genes included the ras-related RSR1. . Proc Natl Acad Sci USA. 1989;86:9976–9980. doi: 10.1073/pnas.86.24.9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Pringle JR. Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. . Mol Cell Biol. 1991;11:1295–1305. doi: 10.1128/mcb.11.3.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Volkwein C, Sommer T. Role of Cue1p in ubiquitination and degradation at the ER surface. Science. 1997;278:1806–1809. doi: 10.1126/science.278.5344.1806. [DOI] [PubMed] [Google Scholar]
- Chant J, Herskowitz I. Genetic control of bud-site selection in yeast by a set of gene products that comprise a morphogenetic pathway. Cell. 1991;65:1203–1212. doi: 10.1016/0092-8674(91)90015-q. [DOI] [PubMed] [Google Scholar]
- Chant J, Pringle JR. Patterns of bud-site selection in the yeast Saccharomyces cerevisiae. . J Cell Biol. 1995;129:751–765. doi: 10.1083/jcb.129.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chant J, Corrado K, Pringle JR, Herskowitz I. Yeast BUD5, encoding a putative GDP–GTP exchange factor, is necessary for bud site selection and interacts with bud formation gene BEM1. . Cell. 1991;65:1213–1224. doi: 10.1016/0092-8674(91)90016-r. [DOI] [PubMed] [Google Scholar]
- Chant J, Mischke M, Mitchell E, Herskowitz I, Pringle JR. Role of Bud3p in producing the axial budding pattern of yeast. J Cell Biol. 1995;129:767–778. doi: 10.1083/jcb.129.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field C, Schekman R. Localized secretion of acid phosphatase reflects the pattern of cell surface growth in Saccharomyces cerevisiae. . J Cell Biol. 1980;86:123–128. doi: 10.1083/jcb.86.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzusoff A, Redding K, Crosby J, Fuller RS, Schekman R. Localization of components involved in protein transport and processing through the yeast Golgi apparatus. J Cell Biol. 1991;112:27–37. doi: 10.1083/jcb.112.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freifelder D. Bud position in Saccharomyces cerevisiae. . J Bacteriol. 1960;80:567–568. doi: 10.1128/jb.80.4.567-568.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita A, Oka C, Arikawa Y, Katagai T, Tonouchi A, Kuhara S, Misumi Y. A yeast gene necessary for bud-site selection encodes a protein similar to insulin-degrading enzymes. Nature. 1994;372:567–570. doi: 10.1038/372567a0. [DOI] [PubMed] [Google Scholar]
- Fuller RS, Brake A, Thorner J. Yeast prohormone processing enzyme (KEX2 gene product) is a Ca2+-dependent serine protease. Proc Natl Acad Sci USA. 1989;86:1434–1438. doi: 10.1073/pnas.86.5.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti E, Popolo L, Vai M, Rota N, Alberghina L. O-linked oligosaccharides in yeast glycosyl phosphatidylinositol-anchored protein gp115 are clustered in a serine-rich region not essential for its function. J Biol Chem. 1994;269:19695–19700. [PubMed] [Google Scholar]
- Gentzsch M, Tanner W. The PMT gene family: protein O-glycosylation in Saccharomyces cerevisiaeis vital. EMBO (Eur Mol Biol Organ) J. 1996;15:5752–5759. [PMC free article] [PubMed] [Google Scholar]
- Gentzsch M, Tanner W. Protein-O-glycosylation in yeast: protein-specific mannosyltransferases. Glycobiology. 1997;7:481–486. doi: 10.1093/glycob/7.4.481. [DOI] [PubMed] [Google Scholar]
- Gimeno CJ, Ljungdahl PO, Styles CA, Fink GR. Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: regulation by starvation and RAS. . Cell. 1992;68:1077–1090. doi: 10.1016/0092-8674(92)90079-r. [DOI] [PubMed] [Google Scholar]
- Halme A, Michelitch M, Mitchell EL, Chant J. Bud10p directs axial cell polarization in budding yeast and resembles a transmembrane receptor. Curr Biol. 1996;6:570–579. doi: 10.1016/s0960-9822(02)00543-2. [DOI] [PubMed] [Google Scholar]
- Haselbeck A, Tanner W. O-glycosylation in Saccharomyces cerevisiaeis initiated at the endoplasmic reticulum. FEBS Lett. 1983;158:335–338. doi: 10.1016/0014-5793(83)80608-5. [DOI] [PubMed] [Google Scholar]
- Herscovics A, Orlean P. Glycoprotein biosynthesis in yeast. FASEB J. 1993;7:540–550. doi: 10.1096/fasebj.7.6.8472892. [DOI] [PubMed] [Google Scholar]
- Hicks JB, Strathern JN, Herskowitz I. Interconversion of yeast mating types. III. Action of the homothallism (HO)gene in cells homozygous for the mating type locus. Genetics. 1977;85:395–405. doi: 10.1093/genetics/85.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immervoll T, Gentzsch M, Tanner W. PMT3 and PMT4, two new members of the protein-O-mannosyltransferase gene family of Saccharomyces cerevisiae. . Yeast. 1995;11:1345–1351. doi: 10.1002/yea.320111403. [DOI] [PubMed] [Google Scholar]
- Kozarsky K, Kingsley D, Krieger M. Use of a mutant cell line to study the kinetics and function of O-linked glycosylation of low density lipoprotein receptors. Proc Natl Acad Sci USA. 1988;85:4335–4339. doi: 10.1073/pnas.85.12.4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo S, Laurenson P, Foss M, Dillin A, Rine J. Roles of ABF1, NPL3, and YCL54 in silencing in Saccharomyces cerevisiae. . Genetics. 1995;141:889–902. doi: 10.1093/genetics/141.3.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier M, Gentzsch M, Sdicu AM, Bussey H, Tanner W. Protein O-glycosylation in yeast. The PMT2 gene specifies a second protein O-mannosyltransferase that functions in addition to the PMT1-encoded activity. J Biol Chem. 1995;270:2770–2775. doi: 10.1074/jbc.270.6.2770. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Krieger M, Corless CL, Boime I. Effects of preventing O-glycosylation on the secretion of human chorionic gonadotropoin in Chinese hamster ovary cells. Proc Natl Acad Sci USA. 1987;84:6354–6358. doi: 10.1073/pnas.84.18.6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K, Nakamura T, Ohshima T, Tanaka S, Matsuo H. Yeast KEX2gene encodes an endopeptidase homologous to subtilisin-like serine proteases. Biochem Biophys Res Commun. 1988;156:246–254. doi: 10.1016/s0006-291x(88)80832-5. [DOI] [PubMed] [Google Scholar]
- Monlauzeur L, Breuza L, Bivic AL. Putative O-glycosylation sites and a membrane anchor are necessary for apical delivery of the human neurotrophin receptor in Caco-2 cells. J Biol Chem. 1998;273:30263–30270. doi: 10.1074/jbc.273.46.30263. [DOI] [PubMed] [Google Scholar]
- Mort AJ, Lamport DT. Anhydrous hydrogen fluoride deglycosylates glycoproteins. Analyt Biochem. 1977;82:289–309. doi: 10.1016/0003-2697(77)90165-8. [DOI] [PubMed] [Google Scholar]
- Murray AW, Szostak JW. Pedigree analysis of plasmid segregation in yeast. Cell. 1983;34:961–970. doi: 10.1016/0092-8674(83)90553-6. [DOI] [PubMed] [Google Scholar]
- Novick P, Botstein D. Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell. 1985;40:405–416. doi: 10.1016/0092-8674(85)90154-0. [DOI] [PubMed] [Google Scholar]
- Novick P, Ferro S, Schekman R. Order of events in the yeast secretory pathway. Cell. 1981;25:461–469. doi: 10.1016/0092-8674(81)90064-7. [DOI] [PubMed] [Google Scholar]
- Park H-O, Bi E, Pringle JR, Herskowitz I. Two active states of the Ras-related Bud1/Rsr1 protein bind to different effectors to determine yeast cell polarity. Proc Natl Acad Sci USA. 1997;94:4463–4468. doi: 10.1073/pnas.94.9.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle JR. Staining of bud scars and other cell wall chitin with calcofluor. Methods Enzymol. 1991;194:732–735. doi: 10.1016/0076-6879(91)94055-h. [DOI] [PubMed] [Google Scholar]
- Redding K, Holcomb C, Fuller RS. Immunolocalization of Kex2 protease identifies a putative late Golgi compartment in the yeast Saccharomyces cerevisiae. . J Cell Biol. 1991;113:527–538. doi: 10.1083/jcb.113.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P, Caras I, Krieger M. Effects of O-linked glycosylation on the cell surface expression and stability of decay-accelerating factor, a glycophospholipid-anchored membrane protein. J Biol Chem. 1989;264:17329–17336. [PubMed] [Google Scholar]
- Roemer T, Madden K, Chang JT, Snyder M. Selection of axial growth sites in yeast requires Axl2p, a novel plasma membrane glycoprotein. Genes Dev. 1996;10:777–793. doi: 10.1101/gad.10.7.777. [DOI] [PubMed] [Google Scholar]
- Rose MD, Novick P, Thomas JH, Botstein D, Fink GR. A Saccharomyces cerevisiaegenomic plasmid bank based on a centromere-containing shuttle vector. Gene. 1987;60:237–243. doi: 10.1016/0378-1119(87)90232-0. [DOI] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Hieter. 1990. Methods in Yeast Genetics: A Laboratory Course. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Rothstein RJ. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sanders SL, Herskowitz I. The Bud4 protein of yeast, required for axial budding, is localized to the mother/BUD neck in a cell cycle-dependent manner. J Cell Biol. 1996;134:413–427. doi: 10.1083/jcb.134.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloat BF, Pringle JR. A mutant of yeast defective in cellular morphogenesis. Science. 1978;200:1171–1173. doi: 10.1126/science.349694. [DOI] [PubMed] [Google Scholar]
- Snyder M, Gehrung S, Page BD. Studies concerning the temporal and genetic control of cell polarity in Saccharomyces cerevisiae. . J Cell Biol. 1991;114:515–532. doi: 10.1083/jcb.114.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl-Bolsinger S, Tanner W. Protein O-glycosylation in Saccharomyces cerevisiae.Purification and characterization of the dolichyl-phosphate-D-mannose-protein O-D-mannosyltransferase. Eur J Biochem. 1991;196:185–190. doi: 10.1111/j.1432-1033.1991.tb15802.x. [DOI] [PubMed] [Google Scholar]
- Strathern JN, Herskowitz I. Asymmetry and directionality in production of new cell types during clonal growth: the switching pattern of homothallic yeast. Cell. 1979;17:371–381. doi: 10.1016/0092-8674(79)90163-6. [DOI] [PubMed] [Google Scholar]
- Tanner W, Lehle L. Protein glycosylation in yeast. Biochim Biophys Acta. 1987;906:81–99. doi: 10.1016/0304-4157(87)90006-2. [DOI] [PubMed] [Google Scholar]
- Tkacz J, Lampen J. Surface distribution of invertase on growing Saccharomyces cerevisiae. . J Bacteriol. 1973;113:1073–1075. doi: 10.1128/jb.113.2.1073-1075.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vai M, Gatti E, Lacana E, Popolo L, Alberghina L. Isolation and deduced amino acid sequence of the gene encoding gp115, a yeast glycophospholipid-anchored protein containing a serine-rich region. J Biol Chem. 1991;266:12242–12248. [PubMed] [Google Scholar]
- Valtz N, Herskowitz I. Pea2 protein of yeast is localized to sites of polarized growth and is required for efficient mating and bipolar budding. J Cell Biol. 1996;135:725–739. doi: 10.1083/jcb.135.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CA, Fuller RS. Posttranslational processing of the prohormone-cleaving Kex2 protease in the Saccharomyces cerevisiaesecretory pathway. J Cell Biol. 1991;115:297–307. doi: 10.1083/jcb.115.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahner JE, Harkins HA, Pringle JR. Genetic analysis of the bipolar pattern of bud site selection in the yeast Saccharomyces cerevisiae. . Mol Cell Biol. 1996;16:1857–1870. doi: 10.1128/mcb.16.4.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Bender A, Cerione RA. Interactions among proteins involved in bud-site selection and bud-site assembly in Saccharomyces cerevisiae. . J Biol Chem. 1995;270:626–630. doi: 10.1074/jbc.270.2.626. [DOI] [PubMed] [Google Scholar]
- Ziman M, Preuss D, Mulholland J, O'Brien JM, Botstein D, Johnson DI. Subcellular localization of Cdc42p, a Saccharomyces cerevisiaeGTP-binding protein involved in the control of cell polarity. Mol Biol Cell. 1993;4:1307–1316. doi: 10.1091/mbc.4.12.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]