

Abstract
Using fluorescence in situ hybridization we show striking differences in nuclear position, chromosome morphology, and interactions with nuclear substructure for human chromosomes 18 and 19. Human chromosome 19 is shown to adopt a more internal position in the nucleus than chromosome 18 and to be more extensively associated with the nuclear matrix. The more peripheral localization of chromosome 18 is established early in the cell cycle and is maintained thereafter. We show that the preferential localization of chromosomes 18 and 19 in the nucleus is reflected in the orientation of translocation chromosomes in the nucleus. Lastly, we show that the inhibition of transcription can have gross, but reversible, effects on chromosome architecture. Our data demonstrate that the distribution of genomic sequences between chromosomes has implications for nuclear structure and we discuss our findings in relation to a model of the human nucleus that is functionally compartmentalized.
Keywords: chromosome territories, genome organization, nuclear compartmentalization, transcription, translocations
The arrangement of human chromosomes on the mitotic spindle could reflect an ordered arrangement of chromosomes at interphase (Nagele et al., 1995). Whole chromosomes occupy discrete territories in the nucleus (Cremer et al., 1993 and references therein). Nevertheless, with the exception of the inactive X (Xi)1 that is positioned against the nuclear membrane, and the rDNA-containing chromosomes that are intrinsic to the nucleolus, there has been little evidence that other whole mammalian chromosomes regularly adopt defined addresses within the nucleus (Manuelidis, 1990).
Here, we investigate whether the two small human chromosomes 18 and 19 (HSA18 and HSA19) adopt different dispositions within the nucleus by fluorescence in situ hybridization (FISH). There are several reasons why these two chromosomes make for interesting comparisons: HSA18 and 19 are similarly sized DNA molecules, containing 85 Mb and 67 Mb of DNA, and so comprising 2.6 and 2% of the physical length of the genome, respectively (Morton, 1991), yet these two chromosomes have contrasting functional and structural characteristics. HSA19 contains a high density of CpG islands (Craig and Bickmore, 1994) and repeats of the Alu family (Korenberg and Rykowski, 1988), replicates most of its DNA in the early part of S phase (Dutrillaux et al., 1976), and has abundant hyperacetylated histone H4 in its chromatin (Jeppesen and Turner, 1993). HSA19 also has the highest observed/ expected ratio of gene-based marker assignments of any human autosome (Cross et al., 1997; Deloukas et al., 1998) with 3.7% of such markers assigned to it. In contrast, HSA18 has far fewer gene assignments than expected for its size (Cross et al., 1997; Deloukas et al., 1998), with only 1.7% of gene-based markers mapped to it. It also has a low CpG island density, a high concentration of the L1 family of repeats, and a high proportion of late replicating DNA (Dutrillaux et al., 1976; Korenberg and Rykowski, 1988; Craig and Bickmore, 1994). Little hyperacetylated H4 is detected on this chromosome (Jeppesen and Turner, 1993).
We compare the nuclear position and chromosome morphology of human chromosomes 18 and 19 through the cell cycle and in different cell types and visualize the association of DNA from each chromosome with substructure of the nucleus. The effects of inhibiting transcription and histone deacetylation on nuclear organization are assessed and we have used a reciprocal t(18;19) to study the behavior of portions of chromosomes 18 and 19 that have been moved to new genomic locations. We show that human chromosomes 18 and 19, and portions thereof, occupy distinct nuclear compartments and that their associations with nuclear substructure are dramatically different. We also demonstrate reversible changes in chromosome morphology, accompanying the inhibition of transcription, that differ between the two chromosomes. Our demonstration that, within the nucleus, specific human chromosomes can have quite different dispositions from each other illustrates the importance of transposing linear maps of the genome into the nuclear space.
Materials and Methods
Cell Culture, Cell Cycle Fractionation, and Preparation of Nuclear Halos
Human primary lymphocytes were stimulated with phytohemagglutinin; both lymphocytes and lymphoblasts (46XY) were grown in RPMI + 10% FCS. Human primary fibroblasts (less than passage 10) and HT1080 cells were grown in DMEM + 10% FCS. To identify cells in S phase, BrdU was added to cultures at 0.1 mM. To irreversibly inhibit transcription, actinomycin D (AMD) was added 2 h before harvest to 5 μg/ml. To reversibly inhibit RNA pol II cells were cultured with 50 μg/ml 5,6-dichloro-β-d-ribofuranosylbenzimidazole (DRB) for 5 h (Haaf and Ward, 1996). Inhibition was relieved by culturing the cells for a further 1 h in the absence of drug. To inhibit histone deacetylase activity, cells were cultured in the presence of 10 ng/ml Trichostatin A (TSA) for 2 h (Bickmore and Carothers, 1995).
Cells were fixed in 3:1 methanol/acetic acid (MAA) using standard procedures. Human lymphoblasts, swollen in 0.075 M KCl, were also cytocentrifuged onto slides and fixed with 4% paraformaldehyde (pFa) made up in 120 mM KCl, 20 mM NaCl, 10 mM Tris HCl, pH 8.0, 0.5 mM EDTA, 0.1% Triton X-100. To preserve three dimensional (3D) nuclear structure, HT1080 cells and primary fibroblasts were grown on slides and fixed with 4% pFa for 10–20 min.
An exponential culture of lymphoblastoid cells was labeled with 0.1 mM BrdU for 45 min before harvest. 1.25 × 108 cells in 10 ml of PBS, 1% FCS, 0.3 mM EDTA, 0.1% glucose were loaded at 11 ml/min into a 5-ml elutriation chamber of a JE5.0 centrifugal elutriator (Beckman) at 2,500 rpm. Fractions were taken at increasing flow rates up to a maximum of 60 ml/ min; the last fraction was collected at 60 ml/min, 2,000 rpm. Fractions were assayed by FACS® after staining with propidium iodide (PI), by nuclear size, and by their pattern of BrdU incorporation.
Interphase nuclei from lymphoblasts, prepared in polyamine buffer, were incubated in extraction buffer (Bickmore and Oghene, 1996) containing increasing concentrations of NaCl (0.5, 1.0, 1.2, and 1.8 M). Slides were then fixed twice in MAA before FISH.
Preparation of Chromosome Painting Probes
Chromosome paints for HSA18 and 19 were prepared in a variety of different ways and their specificity was confirmed by FISH to mitotic chromosomes. A chromosome 19 paint was prepared by human-specific inter-Alu PCR from a Chinese hamster–human monochromosome 19 hybrid cell line GM10449A and labeled by nick translation. To test that this probe detected the entirety of HSA19, total DNA from the hybrid cell line was also nick translated and a commercially available HSA19 paint, produced by both inter-Alu and inter-LI PCR (Oncor) was also tested. Chromosome paints prepared by the amplification of total DNA from microdissected HSA19p and q arms were also used (Guan et al., 1996).
A total chromosome 18 paint was prepared from FACS®-sorted chromosomes digested with MseI, and ligated to catch-linkers (5′-TACCGTTAAGCGTCAATCATGG-3′ [CH18-1] and 5′-CCATGATTGACGCTTAACGG-3′ [CH18-2]), provided by S. Cross. CH18-2 was used as a primer for PCR in the presence of biotin- or digoxigenin-labeled dUTP for 35 cycles. Total DNA from the Chinese hamster–human somatic cell hybrid GM10110, a commercially available chromosome 18 paint (Oncor) and microdissected 18p and q arm paints (Guan et al., 1996) were also tested.
The area taken up by FISH signal with paints from different sources was assessed in 50 nuclei. No significant differences were found between paints for the same whole chromosome prepared in different ways but detected with the same fluorochrome. However, when signals from the same paints labeled with both biotin and digoxigenin and cohybridized were compared, Texas red (TR) signals occupied a larger proportion of the nuclear area than did FITC signals. Therefore, it was important that comparisons made were with the same fluorochrome.
FISH and Immunofluorescence
FISH on 3:1 fixed nuclei was as described previously (Bickmore and Carothers, 1995). Where flattened cells had been fixed in pFa for 15 min, slides were treated with 0.07 M NaOH-EtOH (2:5) for 5 min before denaturation. 3D-preserved cells were fixed in 4% pFa for 10 min, washed in PBS, and then permeabilized with 0.5% saponin, 0.5% Triton X-100 in PBS for 10 min at room temperature. The slides were incubated in 20% glycerol-PBS for 30 min and then subjected to <5 repeated freeze–thaw cycles in liquid N2. Before hybridization these slides were treated with 0.1 M HCl for 5 min at room temperature and with 100 μg/ml RNase A at 37°C for 20 min. Slides were then denatured at 75°C in 70% formamide, 2× SSC, pH 7.0, for 3 min followed by 1 min in 50% formamide, 2× SSC, pH 7.0 (Kurz et al., 1996). 150 ng of chromosome paint and 10–30 μg CotI DNA, or 200 ng labeled P1-BAC DNAs and 3 μg Cot 1 DNA, were used per slide. Biotinylated probes were detected using fluorochrome-conjugated avidin (FITC or TR) (Vector), followed by biotinylated antiavidin (Vector) and a final layer of fluorochrome-conjugated avidin. Digoxigenin-labeled probes were detected with sequential layers of FITC-conjugated antidigoxigenin (BCL) and FITC-conjugated anti–sheep (Vector).
To combine immunofluorescence with FISH in cells fixed with 4% pFa, after detection of the hybridization signals, slides were incubated for 1 h at room temperature, or at 4°C overnight, with a 1:10 dilution of rabbit anti-pKi67 MIB-1 antibody (Dianova) and a monoclonal antibody LN43.2 (gift of B. Lane) directed against B-type lamin. These were detected with TR anti–rabbit and AMCA anti–mouse secondary antibodies (Vector). Slides were counterstained with either 1 μg/ml 4,6-diamidino-2-phenylindole (DAPI) or 0.2 μg/ml PI and examined either using a Zeiss Axioplan fluorescence microscope, equipped with a triple band-pass filter (Chroma 81000), or with a BioRad-MRC 600 confocal laser scanning microscope fitted with an argon laser and a dual filter set (FITC and PI). For random selection of nuclei for analysis, images were taken of consecutive nuclei that presented in a spiral scan pattern from the center of the slide and which did not touch adjacent nuclei. Gray scale images from the Axioplan were collected with a CCD camera (Photometrics), pseudocolored, and merged using Digital Scientific SmartCapture extension to IPLab Spectrum.
Image Analysis
Using IPLab Spectrum software, scripts were written to analyze the data from flattened specimens. In the first, most applicable to the analysis of circular (spherical) nuclei, the DAPI image was segmented and the area and centroid coordinates calculated. The mean FITC and TR pixel intensities within the area of the DAPI segmented nucleus were calculated and subtracted from the FITC and TR images to remove background. A region of interest was then defined manually around the signal. The signal within this region was then segmented and the area and signal intensity weighted centroid coordinates calculated. The area of the signal was normalized for nuclear size by dividing by the nuclear area (see Table II). The DAPI image was converted to binary form. Using the coordinates of the signal weighted centroid as the center, an appropriately sized segmentation disc was adjusted by dilation and erosion until a single pixel with zero intensity was determined. This was taken as the nearest edge of the nucleus to the signal. The signal segment was converted to binary form, a chord was drawn from the centroid of the signal to the nearest edge of the nucleus, and the coordinates established for the first pixel with zero intensity. This was taken to be the edge of the hybridization signal closest to nuclear periphery. These coordinates were used to determine the relative distances between either the edge of the hybridization signal, or the weighted center of the signal, and the nearest edge of the nucleus and also between the center of the signal and the center of the nucleus (see Table I).
Table II.
Relative Area of HSA18 and 19 Chromosome Territories
| Cell type | Cell cycle stage or treatment | Proportion of nuclear area occupied by chromosome | Area HSA19/ HSA18 | |||||
|---|---|---|---|---|---|---|---|---|
| 18 | 19 | |||||||
| Lympho- | C-Metaphase | N/A | N/A | 0.92 | ||||
| cytes | Interphase | 5.7±0.2 | 9.2±0.3 | 1.60(P < 0.00) | ||||
| Lympho- | C-Metaphase | N/A | N/A | 0.90 | ||||
| blasts | Interphase | 5.3±0.1 | 6.8±0.3 | 1.28(P < 0.00) | ||||
| p + q arms | 1.4 + 3.0±0.1 | 2.5 + 2.9±0.2 | 1.23(P < 0.00) | |||||
| Interphase (4% pFA) | 5.0±0.2 | 6.2±0.2 | 1.24(P < 0.00) | |||||
| G1 | 6.1±0.1 | 7.3±0.2 | 1.20(P < 0.00) | |||||
| Early S | 5.5±0.2 | 9.3±0.3 | 1.70(P < 0.00) | |||||
| Late S | 5.4±0.1 | 7.7±0.2 | 1.43(P < 0.00) | |||||
| G2 | 4.8±0.1 | 6.6±0.2 | 1.40(P < 0.00) | |||||
| AMD | 5.4±0.2 | 5.6±0.2 | 1.03(P < 0.40) | |||||
| DRB | 5.8±0.2 | 5.2±0.2 | 0.90(P < 0.04) | |||||
| DRB release | 5.3±0.1 | 5.8±0.2 | 1.09(P < 0.11) | |||||
| TSA | 4.9±0.1 | 7.4±0.3 | 1.51(P < 0.00) | |||||
Territories from 25 metaphase spreads or 50 2D nuclei were analyzed in each case. The mean proportion of nuclear area (area signal ÷ nuclear area in pixels) and the standard error of the mean are shown where applicable. The ratio of areas for C-metaphase chromosomes 19/18 is similar to the ratio of lengths for these chromosomes previously recorded (Van Dyke et al., 1986). All cells were treated with hypotonic and fixed with MAA unless otherwise indicated. The significance of the difference in area between signals from chromosomes 18 and 19 was assessed using a two-sample, two-tailed distribution Student's t test. N/A, not applicable.
Table I.
Relative Nuclear Positions of HSA18 and 19
| Cell type | Cell cycle stage/preparation | Normalized edge signal to edge nucleus (÷ by NA) | Normalized center signal to edge nucleus (÷ by NA) | Normalized center signal to center nucleus (÷ by NA) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 18 | 19 | 18 | 19 | 18 | 19 | |||||||||
| Lymphoblastoid | Interphase (4% pFa) | 0.09 | 0.12 | 0.20 | 0.25 | 0.35 | 0.30 | |||||||
| Interphase (MAA) | 0.08 | 0.18 | 0.18 | 0.32 | 0.38 | 0.25 | ||||||||
| p arm | 0.15 | 0.27±0.02 | 0.19 | 0.32±0.02 | 0.36 | 0.23±0.02 | ||||||||
| q arm | 0.13 | 0.29±0.02 | 0.18 | 0.34±0.02 | 0.38 | 0.22±0.02 | ||||||||
| G1 | 0.08 | 0.18 | 0.19 | 0.32 | 0.38 | 0.25 | ||||||||
| Early S | 0.10 | 0.16 | 0.21 | 0.29 | 0.35 | 0.24 | ||||||||
| Late S | 0.13 | 0.18 | 0.24 | 0.32 | 0.31 | 0.23 | ||||||||
| G2 | 0.11 | 0.16 | 0.22 | 0.30 | 0.34 | 0.26 | ||||||||
| AMD | 0.09 | 0.17 | 0.20 | 0.30 | 0.35 | 0.25 | ||||||||
| DRB | 0.15 | 0.24 | 0.21 | 0.32 | 0.34 | 0.24 | ||||||||
| DRB release | 0.14 | 0.23 | 0.20 | 0.31 | 0.35 | 0.24 | ||||||||
| TSA | 0.09 | 0.19 | 0.19 | 0.32 | 0.37 | 0.24 | ||||||||
| Normal lymphocytes | 0.07 | 0.16 | 0.17 | 0.32 | 0.39 | 0.23 | ||||||||
| t(18;19) lymphocytes | Normal chromosomes | 0.06 | 0.18 | 0.19±0.02 | 0.35 | 0.38±0.02 | 0.21 | |||||||
| Derived chromosomes | 0.09±0.02 | 0.15 | 0.18±0.02 | 0.28 | 0.38±0.03 | 0.29 | ||||||||
Measurements made (in pixels) from 50 2D nuclei for the distance between the edge of the chromosome territory to the edge of the nucleus, and between the center of gravity of the chromosome territory to the center of the nucleus, in lymphoblastoid cells fixed either with 4% pFa or MAA, and from both asynchronous cultures as well as from different cell cycle fractions separated by centrifugal elutriation. Cell cycle stage was assessed both by FACS® analysis and by staining patterns with BrdU (see Fig. 3 b). Measurements were also made with 2D nuclei from lymphoblasts treated with inhibitors of transcription (AMD or DRB) and histone deacetylation (TSA). Nuclei within standard cytogenetic preparations of normal lymphocytes and those from a t(18;19) were also measured. All distances were standardized by dividing by the square root of nuclear area (NA) as an estimate of the nuclear radius. All standard errors of the mean were ≤0.01 unless otherwise indicated. The significance of the difference in position between HSA18 and 19 was assessed using a two-sample, two-tailed distribution Student's t test. P < 0.000 in all cases. The significance of the difference in position between the derived and normal chromosomes in the t(18;19) was similarly assessed, but P < 0.059 for normal and derived 18s and 0.11 for the chromosomes 19.
The erosion analysis script segmented the DAPI signal and recorded the area and centroid coordinates. The area was divided into concentric shells (1–5) of equal area from the periphery of the nucleus to the center. Background was removed from the FISH signal by subtraction of the mean signal pixel intensity within the segmented nucleus, as described above. The proportion of FISH signal and DAPI fluorescence was then calculated for each shell (see Fig. 1).
Figure 1.

Human chromosomes 18 and 19 interphase territories. 2D preparations of nuclei, swollen in hypotonic and fixed either with MAA (a–d) or with 4% pFa (e), were hybridized with HSA18 and 19 paints. In the central panels of a–e HSA18 and 19 paints were biotinylated and detected with avidin-FITC (green). In the left-hand panels of a and b, paints were labeled either with biotin and detected with TR (red) or with digoxigenin and detected with FITC (green). All nuclei were counterstained with DAPI (blue). (a) Primary lymphocytes; (b and e) lymphoblastoid cell line; (c) primary fibroblasts; (d) HT1080 fibrosarcoma cells. Bars, 2 μm. On the right, histograms show the mean proportion of DAPI stain (blue bars), and HSA18 (filled bars) and HSA19 (open bars) hybridization signals in each of the five concentric shells of equal area eroded from the periphery (1) to the center (5) of 50 segmented nuclei. Error bars show SEM.
For confocal analysis nuclei were scanned and imaged using BioRad Comos software. Images from four high resolution scans were averaged using a Kalman electronic filter and 0.5 or 1 μm optical sections were taken. The positioning of chromosomes in relation to the nuclear periphery was assessed manually from the PI staining using display software in IP LabSpectrum. The area of hybridization signal in optical sections was measured as a proportion of the nuclear area as defined by the counterstain.
Results
Chromosomes 18 and 19 Occupy Contrasting Positions within the Nucleus
Chromosome paints for HSA18 and 19 were used in FISH to two-dimensional (2D) preparations of nuclei, swollen in hypotonic, and fixed in MAA, from asynchronous cultures of two primary and two transformed human cell types (Fig. 1, a–d), and also to nuclei swollen in hypotonic and then fixed with 4% pFa (Fig. 1 e). In all of these cases HSA18 and 19 were seen to adopt different positions within the nucleus, and when nuclei were cohybridized with differently labeled paints for HSA18 and 19 (Fig. 1, a and b) signals for both chromosomes 18 appeared to be in a more peripheral location than those of HSA19.
Using flattened specimens enabled the capture and analysis of images from large numbers of nuclei (∼50 in each case) hybridized either with biotinylated HSA18 or HSA19 (Fig. 1) and subsequent statistical evaluation of the relative nuclear positions of these two chromosomes. In the circular (spherical) nuclei of lymphocytes and lymphoblasts the distances between both the edge, and the signal intensity weighted center of the chromosome territory, to the edge of the nucleus were measured, as was the distance between the center of the nucleus and the signal intensity weighted center of the chromosome territory (Table I). Signals for both chromosomes 18 were significantly closer to the nuclear periphery in lymphocytes and lymphoblasts than those of HSA19 and, conversely, chromosomes 19 were located significantly closer to the center of the nuclear area than HSA18s. These data were independent of whether total PCR amplified chromosome 19p and q arm DNA was used as a probe or whether the DNA was labeled by inter-Alu PCR (Table I).
The nuclei of fibroblasts and fibrosarcoma cells are ellipsoid, so the analyses in Table I were not appropriate. 2D nuclei from four cell types were examined by erosion. The nuclear area was divided into concentric shells (1–5) of equal area from the periphery of the nucleus to the center. The proportion of FISH signal and DAPI fluorescence was calculated for each shell. The absolute values of these are influenced by the geometric properties of the nucleus in each cell type and the definition of the nuclear periphery afforded by the DAPI counterstain, but a comparison of the proportion of the DAPI stain or hybridization signal from HSA18 and 19 paints in each concentric nuclear shell (Fig. 1) indicates that chromosome 18 is more peripheral than HSA19 within the diploid nuclei of all the cell types and in nuclei fixed with either MAA or pFa. The proportion of hybridization signal from HSA18 in the outermost shell (shell 1) was always greater than, or similar to, the proportion of DAPI stain located there, whereas the proportion of HSA19 located in this shell was always significantly less than the proportion of the DAPI stain. Conversely, in the most central shell (shell 5) the proportion of HSA19 signal found there was always greater than that of the DAPI stain. The same relative positions of HSA18 and 19 were also seen in fibrosarcoma cells carrying trisomies for either HSA18 or 19 (the population of these cells has a variable karyotype) (Fig. 1 d).
Extrapolation of data from 2D preparations assumes that the relative organization of the intact nucleus is not significantly perturbed. In Drosophila, a comparison of data collected from hypotonically treated and squashed nuclei, with those collected from 3D-preserved nuclei, has confirmed the validity of this assumption (Csink and Henikoff, 1998). However, we still considered it important to confirm our observations in 3D human nuclei that had not been subject to hypotonic swelling. Also, signals that appear to be in the center of flattened nuclei might be located close to the periphery on either the top or bottom surface of the nucleus. Therefore, hybridization signals for HSA18 and 19 were examined in optical sections through the nuclei of primary fibroblasts and HT1080 cells that had been fixed with pFa, using confocal laser scanning microscopy (Fig. 2). In 94% of cells (n = 78 cells, 156 chromosomes), a substantial amount of the signals from both chromosomes 18 was coincident with the nuclear periphery, as defined from the DNA counterstain (e.g., Fig. 2 a). By contrast, in 16% of cells hybridized with an inter-Alu HSA19 paint (n = 56 cells, 112 chromosomes) and sectioned at 0.5-μm intervals, and in 20% of cells hybridized with total HSA19 DNA (n = 35 cells, 70 chromosomes) (1-μm sections) we could not detect any signal from either chromosome 19 adjacent to the nuclear periphery at this level of resolution (e.g., Fig. 2 d). Hence our observations in 3D-preserved cells, that had been fixed with pFa and not subjected to hypotonic swelling, are consistent with the conclusions of our analysis of 2D specimens. It was also interesting to note that in the z sections of 3D-preserved fibroblasts, 65.5% of chromosomes 18 were close to the lateral edge of confocal midsections (e.g., both chromosomes in Fig. 2 a). Proximity of chromosomes 18 to the upper or lower edges of the nucleus could be seen (note the green hybridization signals within the outer optical sections of Fig. 2 b). Where chromosomes 19 did appear close to the nuclear periphery this was often at the top or bottom surface of the nucleus.
Figure 2.

Subnuclear localization of HSA18 and 19 in optical sections through 3D-preserved nuclei. Confocal z series (1 μm) of hybridization to 4% pFa–fixed 3D human dermal fibroblasts with paints for either HSA18 (a and b) or HSA19 (c and d), prepared from randomly amplified total DNA from each chromosome and detected with FITC (green-yellow) and counterstained with PI (red). Cells were either untreated (a and c) or treated with DRB (b and d). Bars, 10 μm.
The demands of preserving nuclear structure yet allowing hybridization to denatured target DNA are conflicting. However, it has been shown that in cells fixed with pFa, permeabilized, and then processed for FISH using the method described here, that centromeres remain in the same spatial position before and after the FISH procedure (Cremer et al., 1993; Kurz et al., 1996). To confirm that some components of nuclear compartments and the nuclear periphery were not disrupted by this procedure, FISH with HSA18 and 19 paints was combined with immunofluorescence for a B-type lamin, a component of the nuclear lamina (Gerace et al., 1978), and for pKi-67, a protein that locates from satellite DNA to the nucleolus from mid-late G1 (Fig. 3 a) (Bridger et al., 1998). Between 50 and 100 nuclei were examined and the majority of chromosomes 18, but not 19, were located close to the nuclear lamina, as detected with anti–lamin B.
Figure 3.
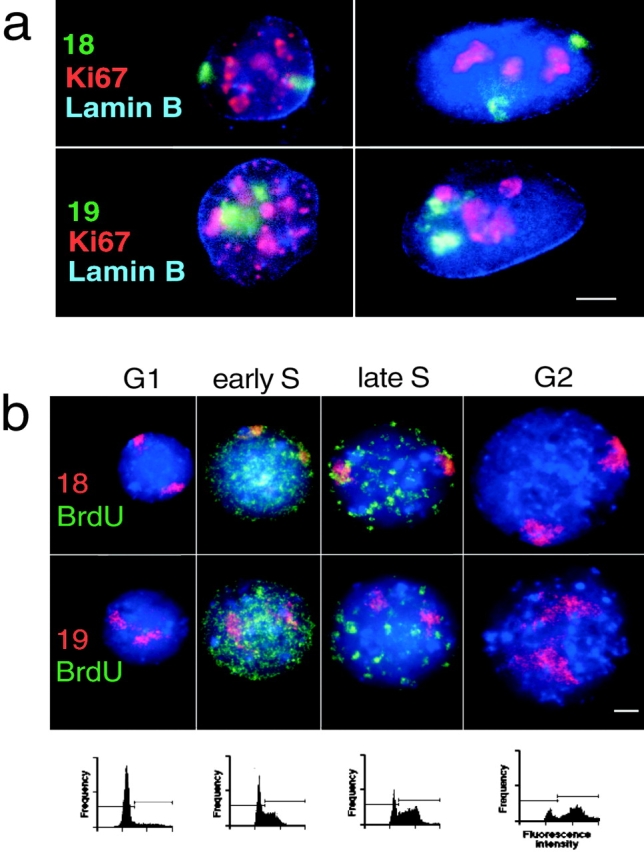
HSA18 and 19 territories through the cell cycle. (a) Combined FISH and immunofluorescence in 4% pFa–fixed 3D fibroblasts with either chromosome 18 or 19 paints (green) and with antibodies against pKi-67 (red) and B-type lamin (blue). The cells on the left are in early stages of G1 and those on the right in mid G1, S, or G2 based on their pattern of pKi-67 staining (Bridger et al., 1998). Bar, 5 μm. (b) FISH for HSA18 or 19 (red) in flattened preparations of MAA-fixed lymphoblast nuclei, pulsed with BrdU (green), and separated by centrifugal elutriation. Blue is DAPI. Bar, 2 μm. FACS® analyses of PI-stained nuclei from elutriated fractions chosen to represent G1, early S, late S, and G2 are shown beneath each panel. Horizontal bars on the FACS® profiles indicate the gating for cells in G1 (left) and for those in S and G2 (right).
Chromosome 18 and 19 Localization Is Established Early in the Cell Cycle
The pKi-67 antigen coats chromosomes in mitosis and for most of interphase (from mid G1 through to G2) it accumulates in the nucleolus. However, in very early G1 pKi-67 is distributed in nuclear foci that correspond with blocks of heterochromatin (Bridger et al., 1998). In combined immunofluorescence-FISH of 3D preparations of human fibroblasts HSA18 was located close to the lamina both in cells with a nucleolar pattern of pKi-67 distribution and also in those cells with a pKi-67 staining pattern indicative of early G1. At these same stages human chromosome 19 appeared to be in a more internal region of the nucleus (Fig. 3 a). Hence, the difference in subnuclear compartmentalization of HSA18 and 19 is established very early in the cell cycle, probably as cells exit telophase.
HSA18 and 19 territories were also examined in the 2D nuclei from exponentially growing human lymphoblasts, pulsed with BrdU, and fractionated by centrifugal elutriation (Fig. 3 b). 50 nuclei from each cell cycle stage fraction were examined. The relative positions of HSA18 and 19 were maintained throughout the cell cycle. Chromosome 18 was more peripheral than HSA19 even in cells that, on the basis of their fractionation and pattern of BrdU staining, appeared to be in late S phase (Fig. 3 b and Table I). This suggests that the relative nuclear positions of chromosomes 18 and 19 are maintained before, during, and after chromosome replication.
Human Chromosome 19 Occupies a Less Compact Chromosome Territory than Chromosome 18
Paralleling their DNA content, chromosomes 18 at C-metaphase have a mean area 10% greater than chromosomes 19 (Table II). However, HSA19 hybridization signal appeared to occupy a larger proportion of nuclear area in 2D preparations than did HSA18 (Fig. 1). The normalized area of hybridization signals for both homologues of HSA18 and 19 were measured in lymphocytes and lymphoblasts (Table II and Fig. 4 a). HSA18 occupied a significantly smaller fraction of nuclear area than HSA19, in flattened preparations of both MAA- and pFa-fixed nuclei, independent of the method of labeling of HSA19. The differences in the proportion of nuclear area occupied by whole chromosome paints for HSA18 and 19 were also seen when the signals from chromosome paints specific for the p or q arms of the chromosomes were compared (Table II and Fig. 5, a–d) (Guan et al., 1996). For example, although the q arm of the submetacentric chromosome 18 is estimated to contain 65 Mb of DNA, it occupied the same proportion of the nuclear area as the estimated 37 Mb of DNA in the q arm of metacentric chromosome 19 (Table II).
Figure 4.

Dependence of territory sizes on transcription and histone deacetylation. (a) Frequency histograms of the proportion of nuclear area occupied by hybridization signals (detected with FITC) of HSA18 (filled bars) and 19 (open bars) in 50 swollen and flattened MAA-fixed lymphoblastoid nuclei. (b) Histograms of the proportion of summed nuclear area occupied by hybridization signals (detected with FITC) of HSA18 (filled bars) and 19 (open bars) through the confocal sections of 5–10 fibroblast nuclei fixed with 4% pFa. Vertical solid and dashed lines show mean proportionate areas for HSA18 and 19, respectively. Cells were either untreated, or treated with AMD, DRB, or TSA before harvest. DRB-treated cells were also grown for 1 h in the absence of DRB (DRB release).
Figure 5.
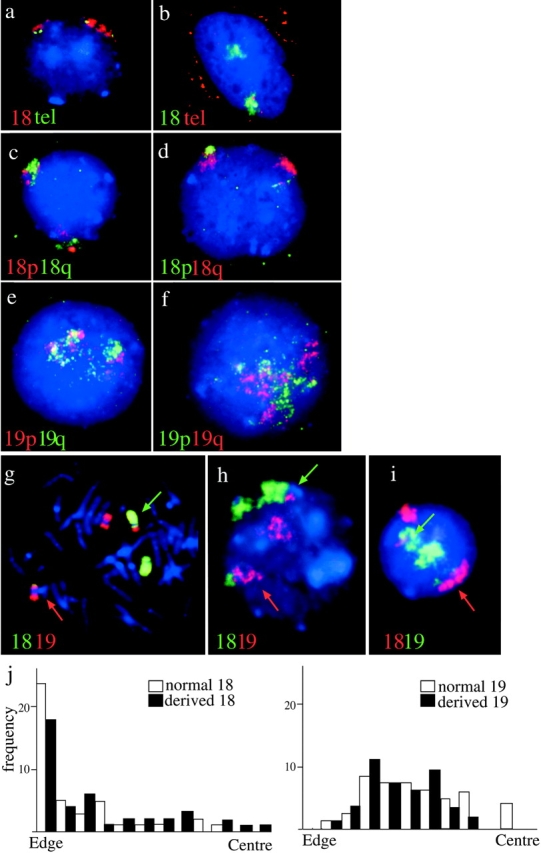
The orientation of chromosomes 18 and 19 in normal nuclei and those from a t(18;19). (a and b) Lymphoblast nuclei cohybridized with paints specific for 18p and q arms (Guan et al., 1996). 18p is in red in a and in green in b. 18q is in the reciprocal color in each case, as indicated. DAPI counterstain is blue. (c and d) Lymphoblast nuclei cohybridized with paints specific for 19p and q arms (Guan et al., 1996). 19p is in red in c and in green in d. 19q is in the reciprocal color in each case, as indicated. DAPI counterstain is blue. (e) Flattened primary lymphocytes hybridized simultaneously with HSA18 paint (red) and telomeric clones 52M11 and 75F20 (green) specific for 18pter and qter, respectively. DAPI counterstain is blue. (f) 3D-preserved nuclei cohybridized with HSA18 paint (green) and telomeric clones 52M11 and 75F20 (red). Red signal in the cytoplasm is from endogenous biotin. Telomere signals are apparent with only one of the territories, those associated with the other territory are in a different focal plane. (g) FISH to a metaphase spread from an individual with t(18; 19)(p11;p13) with chromosome 18 material shown in green and 19 in red. The appropriately colored arrows indicate the derived chromosomes. (h and i) Interphase nuclei from t(18;19) cells. HSA18-derived material is detected in green in h and in red in i. HSA19 is detected in the reciprocal color in each panel as indicated. As in g, appropriately colored arrows indicate the derived chromosomes. A line was drawn from the center to the edge of the nucleus passing through each derived chromosome. A second line, perpendicular to the first, was put through the middle of the signal and it was ascertained which side of this line the translocated portion was found. (j) Histograms of the position of the edge of the signal in relation to the edge or the center of the nucleus, in 50 t(18;19) nuclei, for both the normal (open bars) and derived (filled bars) chromosomes 18 and 19. There is no significant difference in the positions of derived and normal chromosomes (P < 0.059 for HSA18 and P < 0.110 for HSA19).
Variance in the proportion of nuclear area occupied by the two chromosomes 18 within a single nucleus was similar to that of chromosomes 18 in different nuclei (0.007 vs. 0.010, P < 0.079). However, for HSA19 territories there was significantly less variance between homologues within the same nucleus (0.016) than between chromosomes in different nuclei (0.040, P < 0.001) indicating that chromosome 19 compaction may vary between cells that, for example, are at different stages of the cell cycle to a larger extent than chromosomes 18. In elutriated fractions of lymphoblasts, areas of HSA18 signals were always smaller than those of chromosome 19 and the proportion of nuclear area that they occupied decreased from G1 to G2. In contrast, HSA19 occupied the largest proportion of nuclear area in early S phase (Table II).
Signals from chromosome 19 also appeared to be more dispersed and irregular than those of HSA18 in both flattened nuclei (Fig. 1) and in optical sections of 3D nuclei (Fig. 2). We do not have the facilities necessary to directly compute the volumes of HSA18 and 19 in 3D reconstructions. However, we analyzed the sum of the areas occupied by segmented hybridization signal from each chromosome as a proportion of the sum of nuclear areas in 1-μm sections through 10 nuclei (20 chromosomes 18 and 19 each). By this analysis, chromosomes 18 and 19 occupied 3.4 ± 0.3% and 5.0 ± 0.3%, respectively, of the summed nuclear areas of these 3D-preserved cells (Fig. 4 b). The proportion of the summed nuclear areas occupied by signals from chromosomes 19 is significantly larger (P < 0.001) than that occupied by chromosome 18, consistent with the analyses performed on flattened specimens (Table II).
Transcription and Histone Acetylation Affect Territory Compaction but Not Nuclear Position
To analyze the effects of transcription on the territories of HSA18 and 19, lymphoblasts were treated with AMD, an irreversible inhibitor of RNA pols I and II. To assess the drug's effectiveness a sample of cells, before and after treatment, was examined by immunofluorescence with an antibody against snRNPs (data not shown) (Carmo-Fonseca et al., 1992; Bregman et al., 1995). In AMD-treated cells the chromosome 19 territory, but not that of HSA18, occupied a significantly smaller proportion of the nuclear area than in untreated cells (Table II and Fig. 4 a). The adenosine analogue DRB is a reversible and specific inhibitor of RNA polymerase II. The effect of DRB, and its removal, on lymphoblast and fibroblast cells was assessed by the redistribution of nuclear and nucleolar antigens by immunofluorescence (data not shown) (Scheer et al., 1984; Bridger et al., 1998). In the presence of DRB HSA19 territories occupied a smaller proportion of the nuclear area in flattened specimens than those of HSA18 and this compaction of HSA19 was partially reversed after removal of DRB for 1 h (Table II and Fig. 4 a). This was confirmed by analyzing the proportion of nuclear area occupied by signal from each territory summed across confocal 1-μm optical sections of 3D-preserved cells fixed in pFa. Whereas in untreated cells the summed chromosome 19 territory areas were 1.5 times larger than those of HSA18, in DRB-treated cells chromosome 19 appeared to be only 0.33 times the size of chromosome 18 (n = 10 cells each) (Fig. 4 b). No significant differences between the summed areas of chromosomes 18 within treated and untreated cells were detected (P < 0.3), however, chromosomes 19 in DRB-treated cells appeared to be significantly smaller than those in untreated controls (P < 0.000).
By contrast, treatment of cultures with the histone deacetylase inhibitor TSA, to concentrations known to influence replication timing within lymphoblast cells (Bickmore and Carothers, 1995), enhanced the differences in HSA18 and 19 areas (Table II and Fig. 4 a). There was no change in the relative chromosome positions within the nucleus when transcription was inhibited with AMD or DRB, or in the presence of TSA (Table I).
The Orientation of Normal and Translocation Chromosomes
Subnuclear localization of specific human chromosomes may result from differences in DNA sequences distributed along the chromosome arms, or at the centromeres or telomeres of each chromosome. To determine whether both arms of chromosome 18 lie close to the nuclear periphery, and whether both 19p and q are equidistant from the center of the nucleus, paints specific for either 18p or q and 19p and q were cohybridized to flattened lymphoblast nuclei (Guan et al., 1996). Both 18p and q appeared in association with the edge of the nucleus (Fig. 5, a and b) and both 19p and q were found in a more central location (Fig. 5, c and d). Examination of the distances between the edge of the nucleus and the edge or intensity weighted signal center for the p and q arms, and between the center of the nucleus and the intensity weighted signal centers (Table I) confirmed that the relative subnuclear localization observed for HSA18 and 19 reflects properties of both the p and q arms equally. To establish the orientation of chromosome 18 with respect to the nuclear periphery, HSA18 paint was also cohybridized with P1 52M11 and BAC 75F20, specific for 18pter and 18qter, respectively (gift of J. Fantes). Most (63–65%) territories in flattened nuclei fixed in MAA had the p or q telomere at one or the other end of the territory (n = 98). In most other cases the telomere appeared to be on the surface of the territory facing the nuclear interior; localization to the surface of the territory adjacent to the nuclear envelope was rare (7–11%) (Fig. 5 e). This was also the case in 3D-preserved nuclei (Fig. 5 f) and suggests that HSA18 might contact the nuclear periphery through the bulk of its chromosome arms, not specifically through either telomeric end.
Are the positions of HSA18 and 19 within the nucleus therefore confined to the intact chromosomes or can they be conferred by subchromosomal regions? Chromosomes and nuclei in standard cytogenetic preparations from an asymptomatic individual with a balanced reciprocal translocation t(18;19)(p11;p13) were analyzed. We estimate that 20 Mb of material from 18p (24% of HSA18) and 15 Mb of 19p (23% of HSA19) have been exchanged in this translocation (Fig. 5 g). The nuclear disposition of both the normal and derived chromosomes was analyzed (Fig. 5, h and i). Although the derived chromosome 18 tended to be less peripheral than the normal 18, this difference was not statistically significant (Table I, P < 0.059), neither was the apparently more central location of the normal 19 over that of the derived 19 significant (P < 0.11) (Table I and Fig. 5 j). However, there was a striking difference in the relative orientations of the two derived chromosomes. The translocated portion of 19p was more central than the remainder of the derived 18 in almost 80% of nuclei. Conversely, nearly 90% of translocated 18ps were peripheral to the derived 19 material. Hence the characteristic subnuclear localization of HSA18 and 19 can be conferred by 15–20-Mb subchromosomal regions and is not dependent on the centromeric heterochromatin from each chromosome. As no viable cells are available from this t(18;19) individual we could not analyze the localization of the derived chromosomes in 3D-preserved cells.
Chromosomes 18 and 19 Have Different Associations with Nuclear Substructure
The nuclear scaffold and matrix are nuclear substructures that include the lamina, residual nucleoli, and a proteinaceous network pervading the nuclear volume and that are left after extraction of soluble nuclear proteins with the detergent-like salt lithium diiodosalicylate (LIS) or with high salt (Mirkovitch et al., 1984; Luderus et al., 1992).
Nuclei were extracted with increasing concentrations of NaCl before fixation in MAA and hybridization with HSA18 and 19 paints (Fig. 6). At low salt concentrations (0.5 M) no substantial release of DNA from the nucleus was seen. At 1 M NaCl, residual nuclei surrounded by halos of released DNA were revealed by DAPI staining and FISH exposed striking differences in behavior of HSA18 and 19. Most chromosome 18 DNA was released up to 3 μm away from the nuclear remnants, whereas the detectable chromosome 19 DNA remained in the center of the nucleus. As salt concentration increased up to 1.8 M, release of HSA18 became more exaggerated, extending for 7 μm away from the confines of nuclei, within which chromosome 19 was still tightly restrained (Fig. 6). These differences in chromosome behavior were unchanged in nuclei from cells that had been treated with AMD to inhibit transcription.
Figure 6.
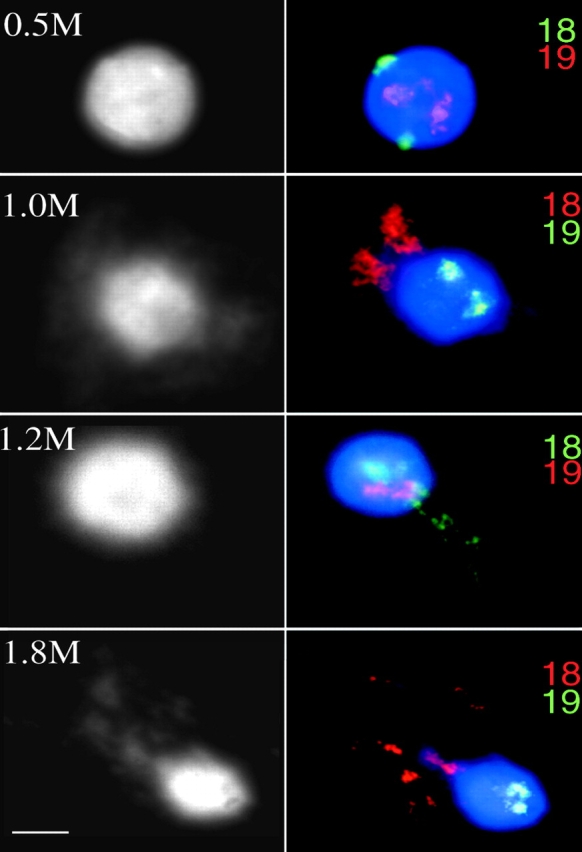
Associations of HSA18 and 19 with nuclear substructure. Lymphoblastoid nuclei extracted with 0.5, 1.0, 1.2, and 1.8 M NaCl, fixed with MAA, and hybridized with alternately labeled chromosome 18 and 19 paints, the color of which is indicated at the right of each panel. DNA was stained with DAPI (shown in blue on the right and in black and white on the left). Chromosome 19 material is retained within the residual nucleus. HSA18 is released into the DNA halo at high salt concentrations. Bar, 2 μm.
Discussion
In this paper we have examined and compared the subnuclear localization of human chromosomes 18 and 19. In different primary and transformed human cells, and in both flattened specimens and in cells fixed to preserve 3D structure, we observed distinct dispositions toward either the interior or periphery of the nucleus for both the p and q arms of chromosomes 19 and 18, respectively (Figs. 1, 2, and 5 and Table I).
Specific parts of chromosomes may adopt different orientations during the cell cycle (Ferguson and Ward, 1992; Vourc'h et al., 1993; Brown et al., 1997; Csink and Henikoff, 1998; Li et al., 1998), but it was not known whether entire mammalian chromosomes move or change their state of condensation. Measurements of chromatin movements in vivo suggest that significant passive diffusion of chromatin could occur during the course of interphase but predict that whole chromosomes are largely constrained within a limited subregion of the nucleus (Marshall et al., 1997). Therefore, specific subnuclear localization of DNA segments might be established most effectively as cells exit mitosis and before interphase nuclear architecture is fully formed. The distinctive arrangements of HSA18 and 19, that we have described here, are established early in the cell cycle and are maintained throughout interphase (Fig. 3). However, the difference in spatial localization between HSA18 and 19 is at its smallest during late S phase (Table I). This may reflect some movement of HSA18 to a more internal location accompanying the replication of its DNA (Li et al., 1998).
In addition to the differences between HSA18 and 19 in subnuclear localization, we have also demonstrated differences in the proportion of nuclear area that these two human chromosomes occupy in 2D preparations. The larger proportion of nuclear area occupied by HSA19, as compared with that of HSA18, is in contrast to the larger physical size (in bp) of the latter chromosome (Morton, 1991) and its greater size in metaphase preparations (Table II). Since much of HSA18 has the characteristics typical of G-band chromosome regions, whereas HSA19 is more R-band–like in its properties (Dutrillaux et al., 1976; Korenberg and Rykowski, 1988; Jeppesen and Turner, 1993; Craig and Bickmore, 1994), our data are consistent with the regional differences in chromatin compaction at the 0.1–1.5-Mb level that have been recorded between G- and R-band regions of the human genome in nuclei prepared in similar ways to those described here (Yokota et al., 1997). The proportion of nuclear area occupied by HSA19 reached a peak in early S phase. This might reflect HSA19 chromatin decondensation preceding its replication (Csink and Henikoff, 1998) or merely the doubling of DNA content for this gene-rich chromosome before most of the rest of the genome.
The apparently larger area of HSA19 within flattened nuclei is also seen in 3D-preserved nuclei (Fig. 4 b). While sophisticated computational algorithms are necessary to accurately compute chromosome volumes within the nuclear space (Eils et al., 1996; Visser et al., 1998), our simplified approach of adding together the chromosome signal area and nuclear area in series of optical sections taken through nuclei suggests that chromosome 19 may occupy a larger volume within the human nucleus than chromosomes 18 and hence be less condensed. Hybridization signals seen with paints for HSA19 also appeared to us to have a more irregular and scattered character than those from HSA18 (e.g., Fig. 1). The chromosome territory of the active X (Xa) is similarly more rutted in appearance than that of Xi (Eils et al., 1996); however, the volumes calculated for the territories occupied by Xa and Xi in optical sections are, in fact, the same despite the more compact appearance of Xi in flattened preparations (Eils et al., 1996). It remains to be determined whether the actual nuclear volumes occupied by HSA18 and 19 are different from each other.
We have also shown here that transcription affects the topology of chromosome territories since the larger apparent size of HSA19 is only seen in the presence of transcription by RNA polymerase II (Fig. 4), i.e., in untreated cells or in cells in which the inhibition of transcription by DRB has been relieved. In the absence of transcription (AMD or DRB treatments) HSA19 occupies a compact territory similar in size, or smaller, than that of HSA18 (Table II and Fig. 4). We do not see the gross disruption of chromosome territories in the presence of DRB that was reported previously (Haaf and Ward, 1996), except in a small minority of both treated and untreated cells that we believe are dying cells. We have no explanation for this discrepancy, but different cell types may have different tolerances and responses to the same concentrations of drugs.
The enhanced differences in areas of HSA19 and 18 we recorded when histone deacetylation was inhibited with TSA (Table II and Fig. 4 a) suggest that levels of steady-state histone acetylation influence the gross architecture of chromosome territories. However, chromosome position within the nucleus is independent of transcription and histone acetylation activities (Table I).
More genomic sequences partitioning with the operationally defined nuclear matrix (MARs) or nuclear scaffold (SARs) derive from HSA18 than from 19 (Craig et al., 1997). This, together with the proximity of HSA18 to the lamina (Fig. 3 a), a component of the nuclear matrix, lead us to expect that there might be a tight association of this chromosome with the matrix. However, in Drosophila, sequences isolated as SARs do not correspond with loci at the nuclear periphery (Marshall et al., 1996) and so the relationship between MARs-SARs and sequences that visibly remain inside of the residual nucleus after extraction, rather than in the surrounding halo of DNA loops, is not clear. Indeed, we saw very little retention of chromosome 18 DNA within nuclear matrices in contrast to the retention of chromosome 19 within the bounds of residual nuclei (Fig. 6). The degree of extension of chromosome 18 sequences varied between nuclei, but within individual nuclei the two homologues behaved similarly (Fig. 6). It has been reported that ∼16 kb of inactive DNA can decondense to cover ∼5 μm in extracted nuclei (Gerdes et al., 1994); therefore, the 85 Mb of chromosomes 18 extruded from nuclei with high salt retain a substantial degree of higher order structure that is independent of interactions with the nucleus.
RNA is an important component of the nuclear matrix, and active genes associate with residual nuclei and not with the nuclear halo (Gerdes et al., 1994). However, retention of HSA19 within the residual nucleus and release of HSA18 was seen in the absence of transcription (AMD treatment). The more central location of chromosome 19 in the human nucleus may be mediated by substantive and transcription-independent association with, as yet unidentified, nuclear proteins that resist extraction from the nucleus with high salt. Jackson and Pombo (1998) have demonstrated that early replicating DNA is retained within the residual nucleus of salt-extracted human cells and, indeed, the bulk of HSA19 replicates earlier in S phase than does HSA18 (Dutrillaux et al., 1976).
Subnuclear localization of HSA18 and 19 is not determined by the centromeres of the chromosomes since distinctive localization is retained by regions (<20 Mb) of the chromosome arms of HSA18 and 19 that are translocated to the reciprocal-derived chromosome (Table I and Fig. 5, g–j). We also find no evidence that the telomeres of the chromosomes are attached at the nuclear periphery of human nuclei (Fig. 5, e and f) as has been observed in simpler eukaryotes (Hiraoka et al., 1990; Funabiki et al., 1993; Gotta et al., 1996). We conclude that differences in the overall composition of bulk chromosome 18 and 19 DNA sequences may play a direct role in the nuclear destiny of these two chromosomes and the genes placed upon them.
It has been argued that lack of phenotypic abnormalities in individuals with balanced translocations is evidence that spatial arrangement of different chromosomes in the nucleus is not functionally important. This was based on the assumption that such translocations disrupt the normal nuclear location of chromosome domains (Haaf and Schmid, 1991; Qumsiyeh, 1995). However, we have shown that this is not necessarily the case to any significant degree (Table I and Fig. 5, g–j). The diffusion constraints on chromatin movement in vivo (Marshall et al., 1997) mean that the physical proximity of different chromosomes in interphase, and their interactions with nuclear substructure, may be important in determining the likelihood of any two chromosomes meeting and exchanging material in a translocation (Qumsiyeh, 1995). The most recurrent chromosome rearrangements in humans are Robertsonian translocations between the acrocentric rDNA–carrying chromosomes that are known to be physically close to one another in both interphase and metaphase (Kaplan et al., 1993). We surveyed two large databases cataloguing balanced translocations in humans (http://www.hgmp.mrc. ac.uk/local-data/Cad_Start.html and http://mendel.imag.fr/BACH/transloc/carto/) and found that t(18;19) is indeed very rare in the human population when compared with other translocations among small chromosomes.
The segregation of different chromosomes of the karyotype with different functional characteristics, described here, is reminiscent of the extreme genome separation seen in plant hybrids (Leitch et al., 1991). What are the biological consequences of this type of compartmentalization? The chromosomal and nuclear position of a gene can influence its activity (Brown et al., 1997; Andrulis et al., 1998) and the position of a gene within the nucleus can be dictated by the sequences it is joined to on the chromosome (Csink and Henikoff, 1996; Dernburg et al., 1996). The edge of the nucleus is also a place where genes are repressed in many eukaryotes (Bridger and Bickmore, 1998). Condensed heterochromatin and later replicating DNA tend to concentrate toward the nuclear periphery in many vertebrates (Rae and Franke, 1972; Fox et al., 1991; Kill et al., 1991; Ferreira et al., 1997), whereas early replicating DNA and poly(A) RNA partition toward the nuclear interior (Carter et al., 1993). At the level of individual loci some, but not all, active mammalian genes have been found predominantly within the nuclear interior, whereas inactive genes have been located at the nuclear and nucleolar peripheries (Xing et al., 1995). The number of gene-based markers that has been assigned to HSA18 is small in comparison to those located on HSA19 (Craig and Bickmore, 1994; Deloukas et al., 1998). Because of their location within the nuclear space, the relatively small number of genes located on human chromosome 18 might be habituated to very different types of transcriptional regulation to those on HSA19. Flexibility of expression of exogenous genes placed into these two different chromosome environments may also differ. This might impose constraints on the chromosomal position of genes through evolution.
Acknowledgments
We thank Sally Cross (ICMB, Edinburgh) and Nigel Carter (Sanger Centre, Cambridge) for flow-sorted chromosomes and catch-linked DNAs. Hongen Zhang, Michael Bittner, and Jeffrey Trent (Bethesda) provided us with 18 and 19 p and q arm–specific paints. Pat Jacobs (Wessex Regional Genetics Laboratory, Salisbury) kindly provided invaluable fixed material from a t(18;19) and Judy Fantes (Chicago) gave us P1 and BAC clones for 18pter and qter. Birgit Lane (Dundee) provided us with the anti–B-type lamin antibody LN43.2. We are indebted to Andrew Carothers for input into data analysis and the statistical interpretation of our data and to the MRC HGU Photography Department for preparation of figures.
Abbreviations used in this paper
- 2D
two dimensional
- 3D
three dimensional
- AMD
actinomycin D
- DAPI
4,6-diamidino-2-phenylindole
- DRB
5,6-dichloro-β-d-ribofuranosylbenzimidazole
- FISH
fluorescence in situ hybridization
- HSA18
human chromosome 18
- HSA19
human chromosome 19
- MAA
3:1 methanol/acetic acid
- pFa
paraformaldehyde
- PI
propidium iodide
- TSA
Trichostatin A
- Xa
active X chromosome
- Xi
inactive X chromosome
Footnotes
W.A. Bickmore is a Centennial Fellow in Human Genetics of the James S. McDonnell Foundation.
J.A. Croft and J.M. Bridger contributed equally to this work.
References
- Andrulis ED, Neiman AM, Zappulla DC, Sternglanz R. Perinuclear localization of chromatin facilitates transcriptional silencing. Nature. 1998;394:592–595. doi: 10.1038/29100. [DOI] [PubMed] [Google Scholar]
- Bickmore WA, Carothers AD. Factors affecting the timing and imprinting of replication on a mammalian chromosome. J Cell Sci. 1995;108:2801–2809. doi: 10.1242/jcs.108.8.2801. [DOI] [PubMed] [Google Scholar]
- Bickmore WA, Oghene K. Visualizing the spatial relationships between defined DNA sequences and the axial region of extracted metaphase chromosomes. Cell. 1996;84:95–104. doi: 10.1016/s0092-8674(00)80996-4. [DOI] [PubMed] [Google Scholar]
- Bregman DB, Du L, van der Zee S, Warren SL. Transcription-dependent redistribution of the large subunit of RNA polymerase II to discrete nuclear domains. J Cell Biol. 1995;129:287–298. doi: 10.1083/jcb.129.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridger JM, Bickmore WA. Putting the genome on the map. Trends Genet. 1998;14:403–409. doi: 10.1016/s0168-9525(98)01572-8. [DOI] [PubMed] [Google Scholar]
- Bridger JM, Kill IR, Lichter P. Association of pKi-67 with satellite DNA of the human genome in early G1cells. Chrom Res. 1998;6:13–24. doi: 10.1023/a:1009210206855. [DOI] [PubMed] [Google Scholar]
- Brown KE, Guest SS, Smale ST, Hahm K, Merkenschlager M, Fisher AG. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell. 1997;91:845–854. doi: 10.1016/s0092-8674(00)80472-9. [DOI] [PubMed] [Google Scholar]
- Carmo-Fonseca M, Pepperkok R, Carvalho MT, Lamond AI. Transcription-dependent colocalization of the U1, U2, U4/U6 and U5 snRNPs in coiled bodies. J Cell Biol. 1992;117:1–14. doi: 10.1083/jcb.117.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter KC, Bowman D, Carrington W, Fogarty K, McNeil JA, Fay FS, Lawrence JB. A three-dimensional view of precursor messenger RNA metabolism within the mammalian nucleus. Science. 1993;259:1330–1335. doi: 10.1126/science.8446902. [DOI] [PubMed] [Google Scholar]
- Craig JM, Bickmore WA. The distribution of CpG islands in mammalian chromosomes. Nat Genet. 1994;7:376–382. doi: 10.1038/ng0794-376. [DOI] [PubMed] [Google Scholar]
- Craig JM, Boyle S, Perry P, Bickmore WA. Scaffold attachments within the human genome. J Cell Sci. 1997;110:2673–2682. doi: 10.1242/jcs.110.21.2673. [DOI] [PubMed] [Google Scholar]
- Cremer T, Kurz A, Zirbel R, Dietzel S, Rinke B, Schrock E, Speicher MR, Mathieu U, Jauch A, Emmerich P, et al. Role of chromosome territories in the functional compartmentalization of the cell nucleus. Cold Spring Harbor Symp Quant Biol. 1993;58:777–792. doi: 10.1101/sqb.1993.058.01.085. [DOI] [PubMed] [Google Scholar]
- Cross SH, Lee M, Clark VH, Craig JM, Bird AP, Bickmore WA. The chromosomal distribution of CpG islands in the mouse: evidence for genome scrambling in the rodent lineage. Genomics. 1997;40:454–461. doi: 10.1006/geno.1996.4598. [DOI] [PubMed] [Google Scholar]
- Csink AK, Henikoff S. Genetic modification of heterochromatic association and nuclear organization in Drosophila. . Nature. 1996;381:529–531. doi: 10.1038/381529a0. [DOI] [PubMed] [Google Scholar]
- Csink AK, Henikoff S. Large-scale chromosomal movements during interphase progression in Drosophila. . J Cell Biol. 1998;143:13–22. doi: 10.1083/jcb.143.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deloukas P, Schuler GD, Gyapay G, Beasley EM, Soderlund C, Rodriguez-Tome P, Hui L, Matise TC, McKusick KB, Beckmann JS, et al. A physical map of 30,000 human genes. Science. 1998;282:744–746. doi: 10.1126/science.282.5389.744. [DOI] [PubMed] [Google Scholar]
- Dernburg AF, Broman KW, Fung JC, Marshall WF, Philips J, Agard DA, Sedat JW. Perturbation of nuclear architecture by long-distance chromosome interactions. Cell. 1996;85:745–759. doi: 10.1016/s0092-8674(00)81240-4. [DOI] [PubMed] [Google Scholar]
- Dutrillaux B, Couturier J, Richer C-L, Viegas-Pequignot E. Sequence of DNA replication in 277 R- and Q-bands of human chromosomes using a BrdU treatment. Chromosoma. 1976;58:51–61. doi: 10.1007/BF00293440. [DOI] [PubMed] [Google Scholar]
- Eils R, Dietzel S, Bertin E, Schrock E, Speicher MR, Reid T, Robert-Nicoud M, Cremer C, Cremer T. Three-dimensional reconstruction of painted human interphase chromosomes: active and inactive X chromosome territories have similar volumes but differ in shape and surface structure. J Cell Biol. 1996;135:1427–1440. doi: 10.1083/jcb.135.6.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson M, Ward D. Cell cycle dependent chromosomal movement in pre-mitotic human T-lymphocyte nuclei. Chromosoma. 1992;101:557–565. doi: 10.1007/BF00660315. [DOI] [PubMed] [Google Scholar]
- Ferreira J, Paolella G, Ramos C, Lamond AI. Spatial organization of large-scale chromatin domains in the nucleus: a magnified view of single chromosome territories. J Cell Biol. 1997;139:1597–1610. doi: 10.1083/jcb.139.7.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MH, Arndt-Jovin DJ, Jovin TM, Baumann PH, Robert-Nicoud M. Spatial and temporal distribution of DNA replication sites localized by immunofluorescence and confocal microscopy in mouse fibroblasts. J Cell Sci. 1991;99:247–253. doi: 10.1242/jcs.99.2.247. [DOI] [PubMed] [Google Scholar]
- Funabiki H, Hagan I, Usawa S, Yanagida M. Cell cycle dependent specific positioning and clustering of centromeres and telomeres in fission yeast. J Cell Biol. 1993;121:961–976. doi: 10.1083/jcb.121.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerace L, Blum A, Blobel G. Immunocytochemical localization of the major polypeptides of the nuclear pore complex-lamina fraction. Interphase and mitotic distribution. J Cell Biol. 1978;79:546–566. doi: 10.1083/jcb.79.2.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes MG, Carter K, Moens PT, Lawrence JB. Dynamic changes in the higher level chromatin organization of specific sequences revealed by in situ hybridization to nuclear halos. J Cell Biol. 1994;126:289–304. doi: 10.1083/jcb.126.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotta M, Laroche T, Formenton A, Maillet L, Shertan H, Gasser SM. Cytological evidence for the clustering of telomeres and their colocalization with Rap1, Sir3 and Sir4 proteins in wild-type S. cerevisiae. . J Cell Biol. 1996;134:1349–1363. doi: 10.1083/jcb.134.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XY, Zhang H, Bittner M, Jiang Y, Meltzer P, Trent J. Chromosome arm painting probes. Nat Genet. 1996;12:10–11. doi: 10.1038/ng0196-10. [DOI] [PubMed] [Google Scholar]
- Haaf T, Schmid M. Chromosome topology in mammalian interphase nuclei. Exp Cell Res. 1991;192:325–332. doi: 10.1016/0014-4827(91)90048-y. [DOI] [PubMed] [Google Scholar]
- Haaf T, Ward DC. Inhibition of RNA polymerase II transcription causes chromatin decondensation, loss of nucleolar structure and dispersion of chromosome territories. Exp Cell Res. 1996;224:163–173. doi: 10.1006/excr.1996.0124. [DOI] [PubMed] [Google Scholar]
- Hiraoka Y, Agard DA, Sedat JW. Temporal and spatial coordination of chromosome movement, spindle formation, and nuclear envelope breakdown during prometaphase in Drosophila melanogaster. . J Cell Biol. 1990;11:2815–2828. doi: 10.1083/jcb.111.6.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen P, Turner BM. The inactive X chromosome in female mammals is distinguished by a lack of histone H4 acetylation, a cytogenetic marker for gene expression. Cell. 1993;74:281–289. doi: 10.1016/0092-8674(93)90419-q. [DOI] [PubMed] [Google Scholar]
- Kaplan FS, Murray J, Sylvester JE, Gonzales IL, O'Connor JP, Doering JL, Muenke M, Emanuel BS, Zasloff MA. The topographic organization of repetitive DNA in the human nucleolus. Genomics. 1993;15:123–132. doi: 10.1006/geno.1993.1020. [DOI] [PubMed] [Google Scholar]
- Kill IR, Bridger JM, Campbell KHS, Maldonado-Codina G, Hutchison CJ. The timing of the formation and usage of replicase clusters in S-phase nuclei of human diploid fibroblasts. J Cell Sci. 1991;100:869–876. doi: 10.1242/jcs.100.4.869. [DOI] [PubMed] [Google Scholar]
- Korenberg JR, Rykowski MC. Human genome organization: Alu, Lines, and the molecular structure of metaphase chromosome bands. Cell. 1988;53:391–400. doi: 10.1016/0092-8674(88)90159-6. [DOI] [PubMed] [Google Scholar]
- Kurz A, Lampel S, Nickolenko JE, Bradl J, Benner A, Zirbel RM, Cremer T, Lichter P. Active and inactive genes localize preferentially in the periphery of chromosome territories. J Cell Biol. 1996;135:1195–1205. doi: 10.1083/jcb.135.5.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch AR, Schwarzacher T, Mosgoller W, Bennet MD, Heslop-Harrison JS. Parental genomes are separated throughout the cell cycle in a plant hybrid. Chromosoma. 1991;101:206–213. [Google Scholar]
- Li G, Sudlow G, Belmont AS. Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining region: precise choreography of condensation/decondensation and nuclear positioning. J Cell Biol. 1998;143:13–22. doi: 10.1083/jcb.140.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luderus MEE, de Graaf A, Mattia E, den Blaauwen JL, Grande MA, de Jong L, van Driel R. Binding of matrix attachment regions to lamin B1. Cell. 1992;70:949–959. doi: 10.1016/0092-8674(92)90245-8. [DOI] [PubMed] [Google Scholar]
- Manuelidis L. A view of interphase chromosomes. Science. 1990;250:1533–1540. doi: 10.1126/science.2274784. [DOI] [PubMed] [Google Scholar]
- Marshall WF, Dernburg AF, Harmon B, Agard DA, Sedat JW. Specific interactions of chromatin with the nuclear envelope: positional determination within the nucleus in Drosophila melanogaster. . Mol Biol Cell. 1996;7:825–842. doi: 10.1091/mbc.7.5.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WF, Straight A, Marko JF, Swedlow J, Dernburg A, Belmont A, Murray AW, Agard DA, Sedat JW. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr Biol. 1997;7:930–939. doi: 10.1016/s0960-9822(06)00412-x. [DOI] [PubMed] [Google Scholar]
- Mirkovitch J, Mirault M-E, Laemmli UK. Organization of the higher-order chromatin loop: specific DNA attachment sites on nuclear scaffold. Cell. 1984;39:223–232. doi: 10.1016/0092-8674(84)90208-3. [DOI] [PubMed] [Google Scholar]
- Morton NE. Parameters of the human genome. Proc Natl Acad Sci USA. 1991;88:7474–7476. doi: 10.1073/pnas.88.17.7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagele R, Freeman T, McMorrow L, Lee H. Precise spatial positioning of chromosomes during prometaphase: evidence for chromosomal order. Science. 1995;270:1831–1835. doi: 10.1126/science.270.5243.1831. [DOI] [PubMed] [Google Scholar]
- Qumsiyeh MB. Impact of rearrangements on function and position of chromosomes in the interphase nucleus and on human genetic disorders. Chrom Res. 1995;3:455–465. doi: 10.1007/BF00713959. [DOI] [PubMed] [Google Scholar]
- Rae PM, Franke WW. The interphase distribution of satellite DNA-containing heterochromatin in mouse nuclei. Chromosoma. 1972;39:443–456. doi: 10.1007/BF00326177. [DOI] [PubMed] [Google Scholar]
- Scheer U, Hugle B, Hazan R, Rose KM. Drug-induced dispersal of transcribed rRNA genes and transcriptional products: immunolocalization and silver staining of different nucleolar components in rat cells treated with 5,6-dichloro-beta-d-ribofuranosylbenzimidazole. J Cell Biol. 1984;99:672–679. doi: 10.1083/jcb.99.2.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke DL, Worsham MJ, Fisher LJ, Weiss L. The centromere index and relative length of human high-resolution G-banded chromosomes. Hum Genet. 1986;73:130–132. doi: 10.1007/BF00291602. [DOI] [PubMed] [Google Scholar]
- Visser AE, Eils R, Jauch A, Little G, Bakker PJ, Cremer T, Aten JA. Spatial distributions of early and late replicating chromatin in interphase chromosome territories. Exp Cell Res. 1998;243:398–407. doi: 10.1006/excr.1998.4144. [DOI] [PubMed] [Google Scholar]
- Vourc'h C, Taruscio D, Boyle AL, Ward DC. Cell cycle-dependent distribution of telomeres, centromeres, and chromosome-specific subsatellite domains in the interphase nucleus of mouse lymphocytes. Exp Cell Res. 1993;205:142–151. doi: 10.1006/excr.1993.1068. [DOI] [PubMed] [Google Scholar]
- Xing Y, Johnson CV, Moen PT, McNeil JA, Lawrence J. Nonrandom gene organization: structural arrangements of specific pre-mRNA transcription and splicing with SC-35 domains. J Cell Biol. 1995;131:1635–1647. doi: 10.1083/jcb.131.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota H, Singer MJ, van den Engh GJ, Trask BJ. Regional differences in the compaction of chromatin in human G0/G1interphase nuclei. Chrom Res. 1997;5:157–166. doi: 10.1023/a:1018438729203. [DOI] [PubMed] [Google Scholar]