

Abstract
Laminin 5 regulates anchorage and motility of epithelial cells through integrins α6β4 and α3β1, respectively. We used targeted disruption of the LAMA3 gene, which encodes the α3 subunit of laminin 5 and other isoforms, to examine developmental functions that are regulated by adhesion to the basement membrane (BM). In homozygous null animals, profound epithelial abnormalities were detected that resulted in neonatal lethality, consistent with removal of all α3-laminin isoforms from epithelial BMs. Alterations in three different cellular functions were identified. First, using a novel tissue adhesion assay, we found that the mutant BM could not induce stable adhesion by integrin α6β4, consistent with the presence of junctional blisters and abnormal hemidesmosomes. In the absence of laminin 5 function, we were able to detect a new ligand for integrin α3β1 in the epidermal BM, suggesting that basal keratinocytes can utilize integrin α3β1 to interact with an alternative ligand. Second, we identified a survival defect in mutant epithelial cells that could be rescued by exogenous laminin 5, collagen, or an antibody against integrin α6β4, suggesting that signaling through β1 or β4 integrins is sufficient for survival. Third, we detected abnormalities in ameloblast differentiation in developing mutant incisors indicating that events downstream of adhesion are affected in mutant animals. These results indicate that laminin 5 has an important role in regulating tissue organization, gene expression, and survival of epithelium.
Keywords: laminin 5, integrins, cell adhesion, epithelial cells, junctional epidermolysis bullosa
Laminins are multifunctional extracellular matrix (ECM)1 proteins that regulate adhesion, motility, gene expression, and apoptosis. Genetically distinct α, β, and γ subunits of laminin form specialized heterotrimers that maintain the function of neuromuscular junctions (Noakes et al., 1995a), striated muscle (Xu et al., 1994; Helbling-Leclerc et al., 1995), renal glomeruli (Noakes et al., 1995b), and skin (Verrando et al., 1987; Carter et al., 1991; Rousselle et al., 1991). We have focused on skin to examine regulatory functions in the epidermis mediated by adhesion to the basement membrane (BM), which led to the identification of laminin 5 (α3β3γ2) as the major adhesive ligand present in the BM of stratified squamous epithelium (Carter et al., 1991). Laminin 5 differentially regulates anchorage and motility of epithelial cells through integrins α6β4 and α3β1, respectively (Carter et al., 1990; Xia et al., 1996; Goldfinger et al., 1998). Proper regulation of these functions is essential for normal wound repair that requires keratinocyte activation followed by cell motility for reepithelialization of the epidermis. Upregulation of laminin α3 chain mRNA and protein indicates that newly synthesized laminin 5 may be required for cell migration and repair of the BM in vivo (Ryan et al., 1994; Lampe, 1998; Nguyen and Carter, manuscript in preparation).
To investigate the role of laminin 5 in migratory and homeostatic epithelium, we performed targeted ablation of the LAMA3 gene, which encodes the α3 subunit of laminin 5. Laminin 5 is expressed in a variety of epithelial tissues (Carter et al., 1991; Aberdam et al., 1994) indicating that LAMA3 null animals can be used to study multiple developmental systems. Biochemical studies have identified the α3 chain as a subunit in laminin 6 (Marinkovich et al., 1992; Gil et al., 1994) and laminin 7 (Champliaud et al., 1996), suggesting that other laminin trimers derived from the LAMA3 gene can be analyzed for loss of function. Furthermore, because basal keratinocytes use laminin 5 as a preferred ligand (Carter et al., 1990), we anticipated that removal of laminin 5 might allow for the identification of other BM ligands that could not be detected in the presence of laminin 5.
In this report, we show that LAMA3 null animals develop a lethal blistering condition similar to human junctional epidermolysis bullosa (JEB; Christiano and Uitto, 1996). Our results demonstrate that ablation of the LAMA3 gene perturbs the formation of hemidesmosomes (HDs) in homeostatic epithelium and disrupts the functional interaction between laminin 5 and integrin α6β4. We found that in the absence of laminin 5 basal cells can utilize integrin α3β1 to interact with an alternative BM ligand. Despite evidence for an alternative α3β1 ligand and the presence of multiple laminin isoforms in the BM of mutant skin, homozygous null animals fail to survive. In vitro studies indicate that laminin 5 deficient epithelial cells have a survival disadvantage when compared with wild-type cells and we identified conditions that will allow for the rescue of mutant cells. Finally, our data indicate that events downstream of adhesion are effected in mutant animals. In particular, we show that ameloblast differentiation is impaired in the developing incisors of mutant animals. These studies provide a basis to examine the influence of endogenous and exogenous laminin 5 on cell survival and gene expression in epithelium. In addition, because all laminin trimers containing the α3 chain have been removed in mutant animals, this animal model has implications for widespread defects in BM stemming from the removal of laminins 5–7.
Materials and Methods
Materials
Restriction enzymes, TaqI polymerase, and RadPrime DNA labeling system was purchased from GIBCO BRL. The Biotechnology Core Lab (Fred Hutchinson Cancer Research Center, Seattle, WA) prepared oligonucleotide primers used for PCR. The multiple tissue Northern blot and hybridization buffer were purchased from CLONTECH Laboratories, Inc. FBS was from Summit. Other cell culture reagents included BSA, trypsin, and hydrocortisone (Sigma Chemical Co.); cholera toxin (Calbiochem-Novabiochem Corp.); aminoguanidine (Aldrich Chemical Co.); EGF (Collaborative Biomedical Products); and keratinocyte growth medium (KGM; Clonetics).
Cloning of the Murine LAMA3 Gene
A 600-bp cDNA clone (Ep-1) corresponding to the helical region of the human α3 laminin chain (Ryan et al., 1994) was used to screen a murine fetal kidney library. Based on sequence homology, the resulting cDNAs were confirmed to be the murine equivalent of the α3-laminin chain. The clones were later found to be compatible with published sequence for the murine laminin α3 chain (Galliano et al., 1995). The α3 laminin cDNAs were then used to screen a 129Sv genomic library (kindly provided by Dr. Phil Soriano, Fred Hutchinson Cancer Center, Seattle, WA) and multiple clones corresponding to the LAMA3 gene were identified.
Targeted Disruption of the LAMA3 Gene
A 1.2-kb NsiI/SacI genomic fragment that contained the murine equivalent of exon A3 from the LAMA3 gene (Pulkkinen et al., 1998a) was replaced with a neo cassette driven by the PGK promoter. The construct was flanked 5′ and 3′ by a 0.8-kb BglII/NsiI fragment and a 5-kb SacI/SacI fragment, respectively. Both flanking sequences were derived from genomic fragments corresponding to the LAMA3 gene. A PGK-driven diphtheria toxin expression cassette was placed 5′ to the 0.8-Kb BglII/NsiI fragment. The construct was linearized with XhoI and electroporated into embryonic stem (ES) cells. Colonies were selected for G418 resistance and screened for homologous recombination using PCR as described (Soriano et al., 1991). The PCR strategy included an oligonucleotide from the neo gene (5′ TCGCAGCGCATCGCCTTCTA 3′) and an oligonucleotide specific for the LAMA3 gene (5′ AACCCTGGCTAGTCTGGAAC 3′) upstream of the 0.8-kb NsiI/BglII fragment used in the targeting construct. PCR was performed in a DNA Thermal Cycler (Perkin-Elmer Corp.) for 40 cycles as follows: 93°C for 30 s; 55°C for 30 s; 65°C for 3 min. Positive clones were further characterized by Southern blot analysis using genomic DNA digested with NsiI and hybridized with an XbaI fragment that is 5′ to the PGKneo insert. Tissue culture and blastocyst injections were performed as previously described (Soriano et al., 1991).
Histology and Immunohistochemistry
For histology, samples were fixed using 10% formalin, rinsed in PBS, dehydrated through graded alcohol, and embedded in paraffin. Sections were stained with hematoxylin and eosin. For immunohistochemistry, frozen tissues were embedded directly in Tissue-Tek O.C.T. Compound (Sakura Finetek USA, Inc.). Cryostat sections (8–10 microns) were extracted with 1% Triton in PBS and fixed with 2% formaldehyde in PBS (20 min). Tissue sections were either stained using a Vectastain ABC kit (Vector Labs) or processed for immunofluorescence. For the ABC kit, sections were blocked with 10% goat serum, incubated with primary antibody for 2 h, washed, incubated with a biotinylated secondary antibody, washed, incubated with peroxidase-conjugated avidin, washed, and developed using diaminobenzidine plus nickel chloride. For immunofluorescence, tissue sections were blocked, incubated with primary antibody for 2 h, washed, incubated with FITC or rhodamine-conjugated secondary antibodies, and washed. Sections were mounted in a solution containing 25 mg/ml of 1,4-diazobicyclo-(2,2,2)octane in glycerol (Johnson et al., 1982) and visualized for immunofluorescence using a Zeiss Microscope.
Electron Microscopy
Tissue was fixed with half strength Karnovsky's fixative plus 0.1% tannic acid, rinsed in 0.1 M cacodylate buffer, and post-fixed in 2% osmium tetroxide (Sakai and Keene, 1994). Samples were dehydrated in graded ethanols and propyleneoxide and then embedded in Polybed 812 resin. Thin sections (80–90 nm) were cut and stained with uranyl acetate. A JEOL 100-SX electron microscope was used for examining and photographing the samples.
Culturing of Mouse Epidermal Keratinocytes
Neonatal mouse pups were killed by decapitation, rinsed in 70% ethanol and PBS, and skinned. The skin was digested in 0.25% trypsin overnight at 4°C for 14 h. The epidermis was separated from the dermis and placed in N-medium according to the protocol of Hager et al. (1999). N-medium is MEM (0.06 mM Ca2+) plus 7.3% chelexed FBS supplemented with culture supernatant from freshly isolated fibroblasts. N-medium also contains hydrocortisone, cholera toxin, aminoguanidine, and EGF. Cells are released from the epidermis into N-medium by shaking. The cells were directly seeded onto dishes that were untreated or coated with 10 μg/ml of type IV collagen (Collaborative Biochemical Products; Becton Dickinson Labware). Immortalized mouse epidermal keratinocytes (MEKs) were cultured in KGM containing 0.06 mM calcium chloride (Clonetics Corp.).
Tissue Adhesion Assays
Adhesion assays on tissue sections were as follows: cryostat sections of split skin from wild-type and mutant animals were attached to the lid of a petri dish. Tissue sections were washed with PBS and blocked with 0.5% BSA. Trypsinized human foreskin keratinocytes (HFKs; Carter et al., 1991) that had been resuspended in KGM (Clonetics Corp.) were labeled with calcein-AM (Molecular Probes, Inc.) for 15 min at room temperature (Lampe, 1998). Cells were washed with PBS and incubated with tissue sections for 1 h in the presence or absence of inhibitory antibodies. After incubation, cells were gently washed once using PBS, fixed with 2% formaldehyde in 0.1 M sucrose cacodylate buffer for 20 min, washed three times in PBS, and rinsed with distilled water. Sections were mounted with a solution containing 25 mg/ml of 1,4-diazobicyclo-(2,2,2)octane in glycerol (Johnson et al., 1982) and visualized for fluorescence using a Zeiss microscope.
Antibodies
Rat mAbs against mouse β1 and γ1 laminin were purchased from Chemicon International, Inc. A rat mAb against β4 integrin was purchased from PharMingen. Dr. Takashi Hashimoto (Kurume University, Kurume, Fukuoka, Japan) kindly supplied human mAb 5E against bullous pemphigoid antigen 230 (BP230; Hashimoto et al., 1993). Dr. Eva Engvall (Burnham Institute, La Jolla, CA) provided polyclonal antibodies for α1 and α2 laminin chains, which were made against the E3 fragment of EHS laminin and recombinant domain VI from the α2 chain, respectively. A polyclonal antibody prepared against recombinant α5 laminin was prepared by Dr. Jeffery Miner (Washington University, St. Louis, MO) as previously described (Miner et al., 1997). A polyclonal antiserum that was prepared against laminin 5 isolated from rat 804G cells was supplied by Dr. Jonathon Jones (Northwestern University, Chicago, IL; Langhofer et al., 1993). A mouse mAb, D3-4, that cross-reacts with mouse laminin 5 was prepared in this lab by Susana Gil as previously described (Gil et al., 1994). Secondary antibodies were purchased from Vector Labs, Southern Biotechnology Associates Inc., and Jackson ImmunoResearch Laboratories, Inc.
Results
Analysis and Strategy for Targeted Disruption of the LAMA3 Gene
Northern blot analysis showed that two major transcripts for the laminin α3 chain are expressed in multiple mouse tissues (Fig. 1). The schematic illustration in Fig. 1 indicates that multiple laminin trimers can be derived from these gene products. Therefore, to introduce a mutation into the LAMA3 gene, we developed a strategy that would ablate all possible α3-laminin trimers. We removed the murine equivalent of exon A3 of the LAMA3 gene (Pulkkinen et al., 1998a) and flanking sequence from the 5′ NsiI site (Ns*) to the 3′ SacI site (S*) replacing it with a PGK-driven neomycin (neo) cassette (Fig. 2 A). The coding sequence of exon A3 is common to both the α3a and α3b transcripts (Ryan et al., 1994). Consequently, removal of exon A3 will cause a frame shift mutation followed by a premature stop codon that will disrupt the α3a and α3b transcripts produced by the LAMA3 gene (Ryan et al., 1994).
Figure 1.

Transcripts produced by the LAMA3 gene. A Northern blot containing polyA+ RNA from adult mouse tissues was hybridized with a probe overlapping with the α-helical domain of the α3a and α3b transcripts. Brain and lung express 10- and 6-kb transcripts, while liver and kidney express primarily the 6-kb transcript. Expression of α3-laminin mRNA was not detectable in adult mouse spleen, skeletal muscle, or testes. A faint signal for the 10-kb transcript was present in heart. The 10-kb transcript corresponds to a full-length α3b polypeptide chain (Ryan et al., 1996; Doliana et al., 1997; Miner et al., 1997) while the 6-kb transcript is expressed as a truncated α3a chain, lacking the NH2-terminal protein domains (Ryan et al., 1994). Extended arrows point to possible laminin trimers synthesized from full-length and truncated α3 chains. Removal of exon A3 (Fig. 2) will ablate the α3-laminin chain at the start of the helical region (short arrow) preventing the assembly of laminin 5 and other laminin isoforms. Trimers for laminin 5 and 6 have been identified (Carter et al., 1991; Marinkovich et al., 1992; Gil et al., 1994). The composition of trimers derived from the full-length α3b chain has not been characterized, so the proposed structure is indicated as laminin X.
Figure 2.

(A) Strategy for homologous recombination. Exon A3 of the murine LAMA3 gene and flanking sequence from the 5′ NsiI site (Ns*) to the 3′ SacI site (S*) were replaced by a neomycin (neo) cassette. The removal of exon A3 will cause a frameshift resulting in a premature stop codon that will disrupt the α3a and α3b transcripts of the LAMA3 gene. (B) Southern blot of ES cells. Genomic DNA from wild-type (control) and recombinant (ES 5-2, 15-4, and 19-1) ES cells was isolated and digested with NsiI. The wild-type ES cells (control lane) contain only a 3.5-kb NsiI fragment corresponding to the normal LAMA3 gene. In contrast, the recombinant ES cells contain an additional 4.4-kb NsiI fragment, which corresponds to the mutated LAMA3 gene (ES 5-2, 15-4, and 19-1 lanes). (C) PCR analysis of wild-type and mutant pups. Primers designed to detect the wild-type and mutant alleles were used to genotype the wild-type and affected littermates. The affected animals were homozygous for the mutant allele (α3−/α3−, lanes 2 and 3) while the unaffected littermates were either homozygous (α3+/α3+, lane 1) or heterozygous (α3+/ α3−, lane 4) for the wild-type allele. Genomic DNA from heterozygous ES cells was used as a control (ES 5-2, lane 5).
The targeting construct (Fig. 2 A) was introduced into 129Sv ES cells by electroporation and homologous recombination occurred at a frequency of 12%. Southern blot analysis on genomic DNA digested with NsiI confirmed the presence of the mutant allele (Fig. 2 B). Five clones that contained the mutant allele were used for blastocyst injection resulting in the generation of multiple chimeric animals. Chimeric animals generated from ES clones 5-2, 15-4, and 19-1 (Fig. 2 B) were crossed with C57BL/6J females. ES-agouti coat color identified pups with germline transmission of the mutant gene. Mice heterozygous for the LAMA3 mutation were crossed to obtain homozygous null offspring. Homozygous null animals derived from ES clones 5-2, 15-4, and 19-1 displayed similar phenotypes, so we continued our studies with the animals generated from ES clone 5-2. PCR analysis was used to determine the genotype of the offspring by using primers designed to detect the wild-type and mutant alleles (Fig. 2 C).
Phenotype of LAMA3 Null Mice
Homozygous α3−/− null animals appeared indistinguishable from α3+/− and α3+/+ wild-type littermates at birth (Fig. 3 A). After birth, homozygous α3−/− null animals, referred to as mutant animals, develop progressive blistering of the forepaws, limbs, and oral mucosa (Fig. 3, B and E). 25% of the newborn pups were homozygous null, suggesting that failure to express α3 did not cause embryonic lethality in the C57BL/6J genetic background. However, the animal in Fig. 3 C showed more extensive blistering than most of the mutant littermates, suggesting that the skin phenotype may be more severe in some of the affected animals. In most cases, the limbs and paws of the mutant animals were visibly red and bleeding (Fig. 3 F), even when blistering lesions were not detected. It was noted that the content of the mutant stomach was substantially smaller (Fig. 3 D) and the mutant animals weighed 40–50% less than wild-type littermates. The presence of milk in the intestine of mutant animals indicates that there was no gastric obstruction, such as pyloric atresia, which can occur in JEB patients with mutations in integrin α6β4 (Vidal et al., 1995; Brown et al., 1996). The mutant animals died 2–3 d after birth from a failure to thrive, possibly caused by dehydration and malnutrition.
Figure 3.

Phenotype of the null animals. (A) Newborn mutant (α3−/−) and wild-type (α3+/+) pups before blister formation. Arrow indicates reduced milk content in the stomach of the mutant pup. (B) Progression of the phenotype in an affected pup 48 h after birth. Blistering of the forelimbs and oral mucosa is shown. (C) A 48 h mutant pup with severe blistering showing loss of epidermis over a significant percentage of the head and body. (D) Milk in the gastrointestinal tract of the wild-type (α3+/+) and mutant (α3−/−) pup indicates that there is not an obstruction between the stomach and intestine (pyloric atresia) in mutant pups. The reduced stomach size and milk content was consistently observed in mutant pups. (E) Front paw of a mutant pup in the early stages of blistering, ∼6–8 h after birth. The digits of the forelimbs are not detectable because the epidermis has formed a cuff over the paw. (F) Bleeding, indicated by the arrow, was frequently detected in the forelimbs of the mutant animals, even when blistering was not detectable.
Paraffin-embedded sections of skin from wild-type and mutant animals were analyzed using hematoxylin and eosin staining (Fig. 4, A–C). The lesions present in mutant skin were confirmed to be junctional blisters caused by a separation at the dermal–epidermal junction (Fig. 4 C). The epidermis of mutant skin showed distinct organizational changes in lesional areas when compared with nonlesional areas. In lesional areas, the epidermis contained clusters of cells in the superbasal layer that maintained an undifferentiated morphology (Fig. 4 C) similar to the pearl cells described in β4 null animals (Dowling et al., 1996). It was noted that the basal cells present in lesional regions were sparse and contained flattened nuclei (Fig. 4 C). These alterations contrasted with the organization of the epidermis in nonlesional regions. In nonlesional regions of mutant skin (Fig. 4 B), the morphology of the basal cells and the organization of the epidermis were similar to that of the wild-type skin (Fig. 4 A).
Figure 4.
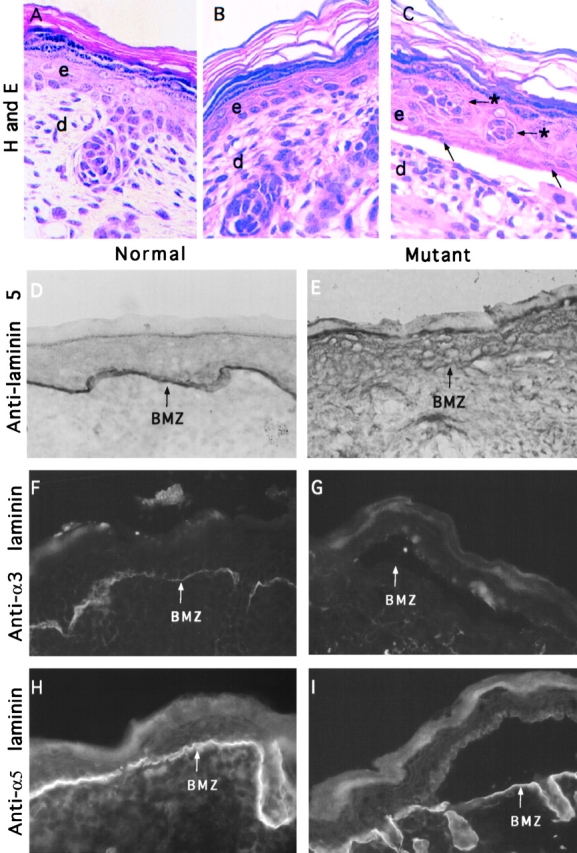
Junctional blisters and laminin expression in mutant skin. Normal (A) and mutant (B and C) skin was fixed in 4% formalin, paraffin embedded, and sections were stained with hematoxylin and eosin. (C) A junctional blister present in the mutant skin that is characterized by a separation of the epidermis (e) from the dermis (d) at the BMZ. Basal cells in the lesion appear sparse and flattened (arrows). Arrows with asterisks mark cell clusters that appear undifferentiated in the superbasal cell layer (C). Cyrostat sections of OCT-embedded skin sections from normal (D, F, and H) and mutant skin (E, G, and I) were extracted with 1% Triton X-100, fixed with 2% formaldehyde, and immunostained with an antilaminin 5 polyclonal antibody (D and E), anti-α3 laminin mAb (F and G), and anti-α5 laminin polyclonal antibody (H and I). D and E were immunostained using peroxidase-conjugated secondary antibodies, whereas F–I were visualized using either FITC- or rhodamine-conjugated secondary antibodies. The BMZ is marked with arrows. The absence of staining for laminin 5 (E) or α3-laminin (G) shows that all laminin trimers containing the α3 chain have been removed from the BMZ of mutant skin. As expected, normal skin is positive for laminin 5 (D) and the α3 subunit of laminin 5 and 6 (F). Expression of α5-laminin trimers are present in normal (H) and mutant skin (I).
Laminin Expression in Wild-type and Mutant Skin
Immunostaining confirmed the absence of laminin 5 from the epidermal BM of mutant skin (Fig. 4 E), consistent with the blistering phenotype. Using mAb D3-4, we sought to examine the expression of all α3-laminin heterotrimers in skin. mAb D3-4 immunoprecipitates multiple α3-containing heterotrimers from human keratinocytes (Gil et al., 1994), including laminin 5 (α3β3γ2) and laminin 6 (α3β1γ1), suggesting that it interacts with α3 chain. Immunostaining showed that mAb D3-4 was positive in wild-type skin, but absent from the epidermal BM of mutant skin (Fig. 4, F and G). The absence of staining in the mutant BM with mAb D3-4 (Fig. 4 G) shows that we have removed all laminin trimers containing the α3 chain from the BM of mutant skin. We also found that α3-laminin was absent from multiple tissues in late stage mutant embryos including the tissues that were shown by Northern blot analysis to express the α3a and α3b transcripts (data not shown). In contrast, immunostaining showed that the expression of the β1 and γ1 laminin chains were maintained in the wild-type and mutant tissues (Table I). The β1 and γ1 subunits of laminin form trimers with different α chains to generate tissue-specific laminin isoforms (Engvall et al., 1990; Miner et al., 1997). Therefore, we used antibodies against the α1-, α2-, and α5-laminin chains to identify which laminin isoforms are present in the epidermal BM of wild-type and mutant skin. Strong staining for α5 laminin is maintained in wild-type (Fig. 4 H) and mutant (Fig. 4 I) skin, indicating that laminin 10 (α5β1γ1) or 11 (α5β2γ1) may be present in the epidermal BM. The results, summarized in Table I, indicate that multiple laminin isoforms remain in the BM of mutant skin. However, none of the other laminins are sufficient to stabilize the adhesion of epithelium to the BM.
Table I.
Laminin Expression in Mouse Skin
| Laminin chain | Normal | Mutant | ||
|---|---|---|---|---|
| β1 | + | + | ||
| γ1 | + | + | ||
| α1 | + | + | ||
| α2 | + (weak) | + (weak) | ||
| α3 | + | − | ||
| α5 | + | + |
Structure and Composition of HDs
The skin phenotype of mutant animals is consistent with an autosomal recessive disorder in humans known as junctional epidermolysis bullosa-gravis (JEB-G; Christiano and Uitto, 1996). The loss of anchorage function in mutant epidermis led us to examine the organization and composition of HDs in mutant skin. HDs link the laminin 5-rich BM to the keratin cytoskeletal through integrin α6β4 (Carter et al., 1990; Stepp et al., 1990). BP230 stabilizes the adhesion plaque on the cytoplasmic side (Tanaka et al., 1991). Immunohistochemical staining showed that integrin α6β4 and BP230 are expressed and are polarized in the basal cells of wild-type (Fig. 5, A and B) and mutant (Fig. 5, C and D) skin. Integrin α6β4 and BP230 both displayed an unexpected discontinuous organization in blistered and nonblistered regions of mutant skin when compared with the wild-type (Fig. 5, E and F). This discontinuity suggests that the absence of laminin 5 effects organization of HD components in basal cells, even in nonlesional areas. Furthermore, these results identify a novel phenotypic alteration in HDs that may be diagnostic in cell adhesion defects stemming from abnormalities in the basement membrane zone (BMZ).
Figure 5.
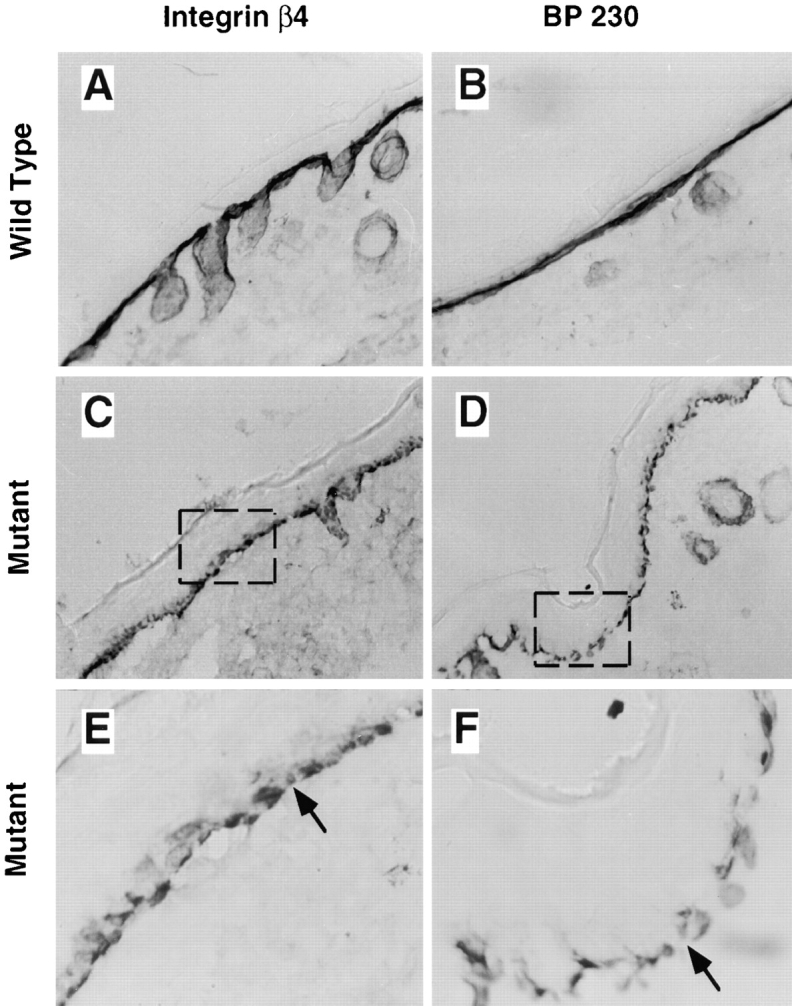
Expression of HD components in normal and mutant skin. Cyrostat sections of OCT-embedded skin sections from normal (A and B) and mutant skin (C–F) were extracted with 1% Triton X-100, fixed with 2% formaldehyde, and immunostained with anti-integrin β4 (A, C, and E) or anti-BP230 (B, D, and F). E and F are higher magnification views of the boxed region in C and D to show the discontinuous expression of integrin β4 (E) and BP230 (F) in mutant skin.
We used transmission electron microscopy to examine the ultrastructure of HDs in tissue. HDs appear normal in wild-type animals, containing a basal and subbasal electron dense plaque (Fig. 6, A and B). Anchoring filaments extend from the basal lamina toward the HD. In the cytoplasm, keratin filaments accumulate near the subbasal plaque (Fig. 6 B). This fine structural organization was lacking in mutant animals. The epidermis of mutant skin contained electron dense plaques at the plasma membrane, but lacked discernible subbasal plaques (Fig. 6, C and D). Intermittently, rudimentary HDs formed at the plasma membrane in the mutant animals (Fig. 6 D). Consistent with our immunostaining results, there was discontinuity in the formation of electron dense plaques resulting in a complete absence of HDs in some regions of the basal cells in mutant skin (Fig. 6 D). Taken together, these findings indicate that the formation and stability of continuous HDs at the basal surface requires laminin 5, whereas basal polarization of integrin α6β4 and BP230 proceed in the absence of laminin 5.
Figure 6.
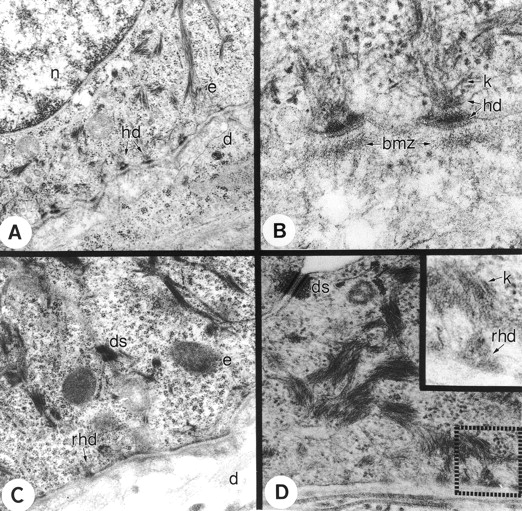
Electron micrograph of wild-type and mutant skin. A and B show the ultrastructure of wild-type mouse skin. (A) HDs (hd) are clearly defined in the wild-type mouse skin. (B) Higher magnification shows the outer and inner electron dense plaque of the HDs (hd) with keratin filaments (k) running toward the HDs. Beneath the HDs is the BMZ, which contains laminin 5. The mutant skin (C and D) contains rudimentary HDs (rhd) that are not well-defined. Higher magnification (inset in D) shows that the HDs in the mutant skin are more diffuse and less well-defined.
Detection of a Ligand for Integrin α3β1
To evaluate the adhesive properties of the BM in vivo, short term adhesion assays were done on cryostat sections of split skin from wild-type and mutant animals (Gil, S.G., T.A. Brown, and W.G. Carter, manuscript in preparation). Previous studies have indicated that the function of β4 integrin can be more effectively evaluated when the function of β1 integrins are suppressed (Niessen et al., 1994; Xia et al., 1996). Therefore, we used cytochalasin D to suppress the function of β1 integrins by inhibiting the organization of the actin cytoskeleton, which allowed the function of β1 and β4 integrins to be distinguished. Tissue adhesion assays were done using human foreskin keratinocytes (HFKs) instead of mouse keratinocytes because some of the inhibitory antibodies do not cross-react with mouse cells. Fig. 7 A shows that HFKs adhere well to the BM of wild-type tissue regardless of whether they are untreated (Fig. 7 A, a) or pretreated with cytochalasin D (Fig. 7 A, b). The cytochalasin D-resistant adhesion could be blocked using an inhibitory antibody against α6 integrin (Fig. 7 A, c) indicating that this interaction is dependent on integrin α6β4. In contrast, pretreatment of HFKs with cytochalasin D resulted in complete inhibition of adhesion to the BM of mutant tissue (Fig. 7 A, h) demonstrating the loss of a functional interaction between integrin α6β4 and laminin 5. However, in untreated HFKs, we were able to detect adhesion to the mutant BM (Fig. 7 A, g), suggesting that a ligand for β1 integrin is present in the BM of mutant tissue. This led us to investigate which β1-integrin receptor was required for adhesion to the mutant BM. The results showed that mAb P1B5, an inhibitory antibody against integrin α3, significantly reduced adhesion to the BM of mutant tissue (Fig. 7 B, h) when compared with the control (Fig. 7 A, g). A comparison of several representative areas indicated that the inhibition observed in the presence of P1B5 was ∼80%. A combination of anti-α3 and anti-α6 inhibitory antibodies allowed for complete inhibition of adhesion to the BM of mutant skin (Fig. 7 B, i). The nature of any contribution from integrin α6, if significant, awaits further investigation. It is sufficient that in the presence of mAb P1B5 adhesion of HFKs to the mutant BM is reduced by >80% demonstrating that a ligand for integrin α3β1 is detectable in laminin 5 deficient tissue. In contrast, mAb P1B5 (Fig. 7 B, b) did not effect adhesion of HFKs to the wild-type BM due to the interaction of laminin 5 with integrin α6β4. This was confirmed by using a combination of GoH3 and P1B5, which completely blocked adhesion of HFKs to the wild-type tissue (Fig. 7 B, c). Therefore, in mutant tissue, we were able to detect a ligand for integrin α3β1 that could not be detected in wild-type tissue because of the dominant interaction of laminin 5 with integrin α3β1 and integrin α6β4. These results provide evidence that other endogenous BM proteins can serve as a ligand for α3β1, but not for the anchorage function mediated by integrin α6β4 in the epidermis.
Figure 7.

Tissue adhesion assay. Cryostat sections containing split skin from wild-type (a–f) and mutant (g–l) animals were incubated with normal HFKs that previously had been labeled with fluorescent dye. (A) Normal HFKs were either untreated (control) or pretreated with cytochalasin D (cyto D) and incubated with wild-type (a–f) or mutant (g–i) skin sections in the absence or presence of GoH3 as indicated. Fluorescent images for normal (a–c) and mutant (g–l) split skin are shown above the corresponding phase-contrast photo (d–f, j–l). Phase-contrast photos contain the dermis and BMZ. The epidermis has been separated from the BMZ and is not visible in most photos. Labeled HFKs are adherent to the BM of wild-type split skin in the untreated control (a) and cytochalasin D treated samples (b), but not in samples treated with a combination of cytochalasin D + GoH3 (c). Incubation of mutant split skin with HFKs shows that only untreated HFKs can bind (g). Pretreatment with cytochalasin D was sufficient to inhibit adhesion to the mutant BM, regardless of whether the assay was done in the absence (h) or presence (i) of GoH3. (B) Labeled HFKs were incubated with wild-type split skin (a–f) or mutant split skin (g–l) in the absence of inhibitory antibodies (a, d, g, and j), in the presence of P1B5 (b, e, h, and k), or in the presence of P1B5 + GoH3 (c, f, i, and l). Adhesion of HFKs to the BM of wild-type split skin is detectable in the absence (a and d) or presence (b and e) of P1B5, but not in the presence of P1B5 + GoH3 (c and f). Adhesion of HFKs to mutant BM can be inhibited with P1B5 alone (h and k) or with P1B5 + GoH3 (i and j). In the control, HFKs adhere well to the mutant BM in the absence of inhibitory antibodies (g and j). The phase-contrast photos for normal (d and e) and mutant (j) tissue shows that cell adhesion is to the BM.
Reduced Survival of Laminin 5 Deficient Keratinocytes
MEKs were isolated from wild-type and mutant skin. The yield of cells and seeding density was comparable for normal and mutant MEKs. However, in contrast to normal MEKs, mutant MEKs failed to survive when plated on an untreated culture dish (Fig. 8, compare A and B). Survival of mutant MEKs was restored when cells were plated on an exogenous ligand such as collagen (Fig. 8, compare B and D). These results show that ablation of laminin 5 is sufficient to prevent the survival of mutant MEKs in the absence of an exogenous ligand. This concept was further reinforced with the generation of a laminin 5 deficient cell line. Using the E6/E7 transforming genes from papilloma virus (Kaur et al., 1989) we immortalized wild-type and mutant epithelial cells. The mutant epithelial cells remained dependent on exogenous ligand for survival and this dependence was confirmed in a growth curve (Fig. 9). Using flow cytometry, there was no accumulation of a pre-G1 peak (Dou et al., 1995), suggesting that extensive apoptosis was not occurring in the mutant cells (data not shown). Studies were initiated to identify receptor/ligand interactions that could rescue the survival defect in mutant MEKs. Laminin 5-enriched ECM (Fig. 8, compare F and H) or immobilized anti-integrin β4 antibody (Fig. 8, compare F and J) were able to rescue mutant MEKs, suggesting that interactions of laminin 5 with integrin α6β4 contribute to the survival of epithelial cells. Because keratinocytes use integrin α3β1 and α6β4 to interact with laminin 5, both receptors may be contributing to the enhanced survival observed on laminin 5 (Fig. 8, compare F and H). We could not evaluate the contribution from integrin α3β1 separately because antibodies that react with mouse are not available. However, rescue of mutant MEKs on collagen (Fig. 9) or immobilized anti-α2 antibody (data not shown) indicate that ligation of β1 integrins is also sufficient to rescue survival of mutant MEKs. In contrast to mutant cells, the MEKs derived from normal animals retained a high survival rate and characteristic epithelial morphology regardless of whether they were plated on untreated culture dishes, exogenous ligand, or immobilized antibody (Fig. 8 E, G, I, and Fig. 9). Therefore, within the limits of this assay, we found that ligation of β1 or β4 integrins are sufficient to rescue the survival defect resulting from ablation of laminin 5. These results confirm the proposal that laminin 5 secreted into the ECM by cultured keratinocytes is the primary adhesive ligand produced by these cells, even though an additional ligand for integrin α3β1 resides in the BM.
Figure 8.

Survival of primary and immortalized mouse epithelial cells. Primary MEKs isolated from wild-type (A and C) and mutant (B and D) skin were plated on untreated tissue culture (TC) dishes (A and B) or dishes coated with 10 μg/ml of collagen type IV (Col; C and D). Mutant MEKs were unable to survive on untreated TC dishes (B), but could be rescued by plating on collagen (D). Immortalized MEKs derived from wild-type (E, G, and I) or mutant epithelial cells (F, H, and J) were plated on untreated TC dishes (E and F), laminin 5-enriched ECM (G and H), or immobilized anti-β4 antibody (I and J). The mutant MEKs did not grow on untreated TC dishes (F), but could be rescued on ECM (H) or immobilized anti-β4 antibodies (J). MEKs derived from wild-type animals survived independent of exogenous ligand (compare E, G, and I).
Figure 9.
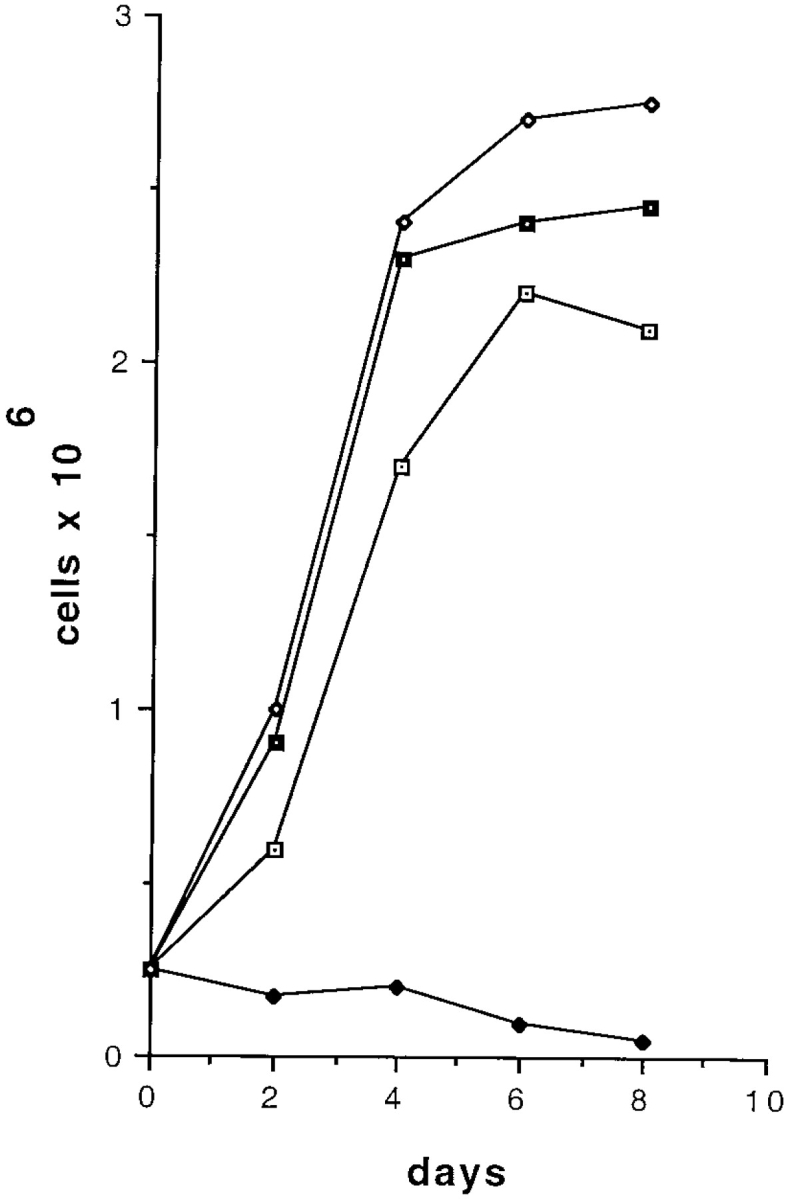
Growth curve of epithelial cells. Laminin 5-expressing and laminin 5 deficient epithelial cell lines were grown in KGM (60 μM Ca2+) on either collagen (10 μg/ml) or untreated tissue culture dishes. Laminin 5-expressing cells grew on collagen (filled squares) and untreated tissue culture dishes (open diamonds). Laminin 5 deficient cells failed to grow on untreated tissue culture dishes (filled diamonds), but could be rescued by plating on collagen (open squares).
Ameloblast Differentiation Is Dependent on Laminin 5
Laminin 5 is expressed in a variety of epithelial BMs. In skin, we observed severe defects in basal keratinocytes resulting in abnormal HDs and loss of anchorage function in the BM of the mutant epidermis. We predicted that if laminin 5 were involved in late stage differentiation we would be able to detect abnormalities in other target organs that develop late in gestation. Examination of cross-sections from neonatal mice revealed gross abnormalities in the developing incisors of mutant animals. Therefore, developing incisors of wild-type and mutant animals were selected for further investigation. Cross-sections from medial to lateral regions of the head were taken to evaluate histogenesis of the incisors at different stages of maturation (Fig. 10 A). Progressive differentiation is identifiable from the base of the tooth (region I) to the tip (region III and VI). The stages of differentiation are outlined for ameloblasts (Fig. 10, B and C), which are the specialized epithelial population that deposit enamel on one side of the developing rodent incisor (Fig. 10 C, a). Mitotic preameloblasts of the inner dental epithelium located at the base of the tooth in region I (Fig. 10, B and C) develop into post-mitotic, secretory ameloblasts in region II (Fig. 10, B and C) and produce the enamel layer of the tooth. A sharp boundary is visible at the junction between the ameloblasts and the stratum intermedium (Fig. 10 F, boundary between a and si). This boundary corresponds to cytoplasmic filaments, not a BM. The BM is located between the developing tooth and the ameloblast (Fig. 10 F, bm). As a result, the nuclei of the ameloblast become polarized away from the BM (Fig. 10 G). Consistently, immunostaining with mAb D3-4 showed intense deposition of the laminin α3 chain between the secretory ameloblasts and the enamel boundary in the wild-type incisor (Fig. 10 D). Positive staining with mAb D3-4 was absent from a comparable region of the mutant tooth (Fig. 10 E) confirming the removal of laminin α3 chain trimers from the developing incisor.
Figure 10.
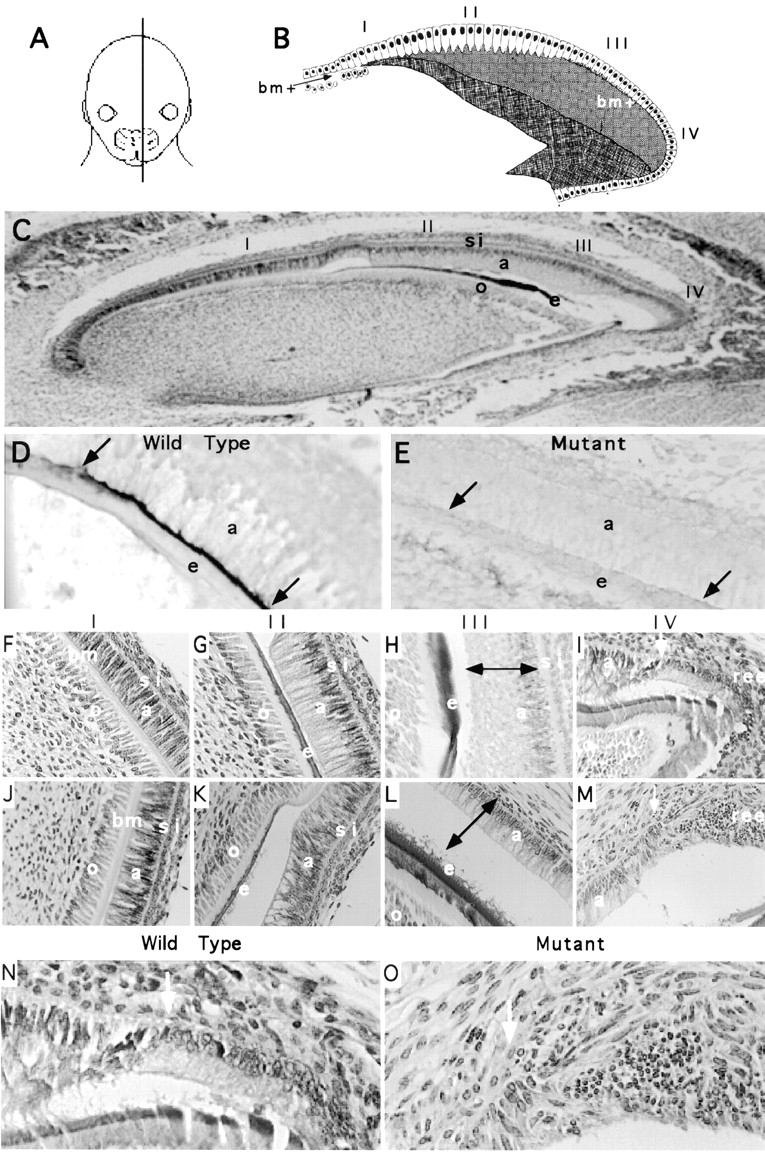
Analysis of developing incisors. Neonatal pups were killed by decapitation, heads were cut at the midline, fixed in 10% formalin, and processed for paraffin embedding. (A) Cross-sections were taken from medial to lateral regions of the head. Cartoon (B) and hematoxylin- and eosin-stained section (C) show the progression of differentiation occurring in the inner dental epithelium from the base of the tooth (region I) to the tip (region III and VI). Mitotic preameloblasts of the inner dental epithelium present in the base of region I develop into post-mitotic, secretory ameloblasts in region II and produce the enamel layer of the tooth. A single layer of cells, referred to as the stratum intermedium (si), is adjacent to the ameloblasts (a). The discrete line visible between the stratum intermedium and the ameloblasts is not BM. The BM is located between the tooth and the differentiating ameloblasts (see F and G). During enamel secretion (region II), the BM is absent, but it reforms during ameloblast maturation (region III and IV). Immunostaining with mAb D3-4 shows that the laminin α3 chain is expressed during ameloblast differentiation in the wild-type (D), but not the mutant (E) tooth. Cross-sections from comparable regions of wild-type (F–I and N) and mutant (J–M and O) teeth are shown for region I (F and J), region II (G and K), region III (H and L), and region IV (I and M). The double arrows in H and L are the same size to emphasize the reduced size of the mutant ameloblasts. N and O are enlargements of I and M, respectively. The white arrow marks the location where the boundary between the stratum intermedium and the ameloblasts is no longer distinguishable. At this precise location, the organization of the mutant epithelium becomes extremely disorganized (compare N and O). Abbreviations are as follows: a, ameloblasts; o, odonoblasts; bm, basement membrane; si, stratum intermedium; e, enamel; ree, reduced enamel epithelium. The cartoon in B was modified from Eisenmann (1989).
Cross-sections from comparable regions of the wild-type teeth (Fig. 10, F–I and N) and mutant teeth (Fig. 10, J–M and O) were evaluated. In wild-type incisors, secretory ameloblasts of region II were discernible as elongated epithelial cells with Tomes' processes extending toward the deposited enamel (Fig. 10 G). Differentiation continued normally with the appearance of ruffled edge mature ameloblasts in region III (Fig. 10 H) followed by the formation of the reduced enamel epithelium in region IV (Fig. 10, I and N). A discrete morphological change occurs in region IV: the stratum intermedium no longer marks a discrete boundary (Fig. 10 N, arrow) because the ameloblasts, stratum intermedium, stellate reticulum, and outer dental epithelium become incorporated into the stratified epithelium referred to as the reduced enamel epithelium.
In mutant animals, incisor development appeared normal until the onset of enamel secretion. Presecretory ameloblasts in region I of wild-type (Fig. 10 F) and mutant animals (Fig. 10 J) are indistinguishable. At the onset of enamel secretion in region II, the ameloblasts of mutant incisors (Fig. 10 K) were shorter with visible undulations at the edges when compared with wild-type incisors (Fig. 10 G). As differentiation proceeded, the ameloblasts of the mutant incisor continued to be reduced in size relative to the wild-type teeth (Fig. 10, H and L; compare height of ameloblasts relative to double arrows in H and L) and enamel deposition did not appear normal. The enamel edge of mutant incisors appeared frayed in comparison to the enamel deposited in wild-type incisors (Fig. 10, compare H and L). The abnormal appearance and size of the mutant ameloblasts made it difficult to distinguish the transition from secretory ameloblasts of region II to mature ameloblasts of region III. In region IV, where the ameloblasts and the adjacent stratum intermedium form the reduced enamel epithelium, tissue organization was completely disrupted (Fig. 10, compare N to O). This disorganization corresponded precisely with the point where the stratum intermedium no longer formed a discrete boundary (Fig. 10 O, arrow). At this junction between region III and IV the reduced enamel epithelium should form a stratified epithelium. This does not occur in the mutant tissue (Fig. 10 O). These results define a role for the α3 subunit of laminin 5 in murine tooth development and provide a biological basis for the hypoplastic enamel that has been described in human JEB-G (Wright et al., 1993).
Discussion
Laminin 5 Regulates the Organization of HDs
Our results show that loss of α6β4-laminin 5 anchorage functions have profound effects on HD organization. Discontinuities in localization of integrin α6β4 and BP230 were so prominent in mutant tissue that they could easily be detected at the light microscope level by immunohistochemical staining with anti-β4, -α6, or -BP230 antibodies. Analysis with other cellular markers, such as keratin-1 or keratin-14, did not display alterations that would allow us to distinguish between wild-type and mutant skin (data not shown). In contrast, the discontinuous staining of α6β4 and BP230 was reproducible in 100% of the homozygous null pups allowing us to readily identify wild-type and mutant skin. Because the discontinuity of β4-integrin and BP230 staining occurred in both lesional and nonlesional regions, our data suggests that HD alterations are a primary consequence of laminin 5 deficiency rather than a secondary effect caused by the lesion. Consistently, a subpopulation of patients lacking either laminin 5 or bullous pemphigoid antigen 180 (BP180) display a similar disorganization of HD proteins (Brown, T.A., and W.G. Carter, unpublished observation), suggesting that this phenotypic alteration may have diagnostic value in patients with structural abnormalities in the BMZ. It was noteworthy that the discontinuity of α6β4 and BP230 appeared to occur at cell–cell boundaries, suggesting that stability of cell–cell junctions may be reduced in these regions. The notion that cell-substrate adhesion can regulate interactions at cell– cell junctions has been established in epithelial cells. Tiam1/Rac signaling in epithelial cells can promote either cell–cell adhesion or cell migration depending on the type of matrix used for cell adhesion (Sander et al., 1998). Similarly, laminin 5 interactions with integrin α3β1 selectively promote intercellular communication of basal keratinocytes through gap junctions (Lampe, 1998).
Ablation of LAMA3 Results in JEB and Survival Defects
Ablation of the LAMA3 gene causes a phenotype similar to a lethal variant of human epidermolysis bullosa, JEB-G. Clinical features of JEB-G include mechanical fragility of the skin, growth retardation, oral erosions, gastrointestinal and genitourinary tract involvement, dental abnormalities, hypoplastic HDs, and high morbidity (Fine et al., 1991). JEB-G is an autosomal recessive disorder that has been associated with mutations in the LAMA3 (Kivirikko et al., 1995), LAMB3 (Pulkkinen et al., 1994b), and LAMC2 (Pulkkinen et al., 1994a) genes of laminin 5. Mutations in the LAMA3 gene have been documented in only a small fraction of the JEB-G cases, which led us to wonder if null mutations in the LAMA3 gene would result in embryonic lethality, particularly since we removed all trimers containing the α3 chain from the BM, including laminins 5–7. On the contrary, we found that mice homozygous for the null mutation were born at the expected frequency of 25%, suggesting that embryonic lethality did not occur in the C57/BL6 genetic background. The reduced number of patients with mutations in the LAMA3 gene relative to the other genes that encode laminin 5 may be due in part to a reported hotspot in the LAMB3 gene (Kivirikko et al., 1996).
In vitro studies on primary and immortalized keratinocytes (Figs. 8 and 9) have indicated that laminin 5 contributes to keratinocyte survival, which may have relevance to pathology of human JEB-G. Jonkman et al. (1997) described an individual who is mosaic for mutations in the COL17A1 gene encoding BP180, a component of HDs. Surprisingly, the subpopulation of keratinocytes from this mosaic individual that express BP180 display a survival advantage in culture and in the skin of the individual (Jonkman et al., 1997). Thus, both laminin 5 and BP180 provide a survival advantage for keratinocytes. Curiously, keratinocytes from individuals with JEB-pyloric atresia with inherited defects in β4 appear to survive in culture as well as or better than wild-type keratinocytes (Gil and Carter, unpublished observation). Additional experiments will be necessary to determine if the survival advantage observed in wild-type cells is due to a direct effect of laminin 5 on cell cycle regulation through integrins. It has been shown that adhesion of primary keratinocytes to laminin 5 promotes entry into the cell cycle through signaling pathways that are generated by ligation of integrin α6β4 (Mainiero et al., 1997; Murgia et al., 1998). Laminin 5 may also promote cell proliferation through a second signaling pathway involving integrin α3β1 (Gonzales et al., 1999). Consistently, our results indicate that exogenous ligands are not adequate for longterm survival of mutant MEKs (Ryan, M.C., unpublished observation), suggesting that the survival contributions from laminin 5 may not simply be due to adhesion. Whether or not exogenous laminin 5 is sufficient to rescue defective cellular functions caused by the absence of endogenous laminin 5 remains to be determined, particularly since endogenous and exogenous laminin 5 may have different biological functions. Exogenous laminin 5 is a scatter factor for carcinoma cells that do not make endogenous laminin 5, but not for carcinomas that deposit endogenous laminin 5 (Kikkawa et al., 1994). Similarly, we have observed that keratinocytes from JEB-G patients with defective laminin 5 expression and MEKs from LAMA3 null animals will both scatter in response to exogenous laminin 5 while normal cells do not (Gil, S.G., M.C. Ryan, and W.G. Carter, unpublished observation). Future studies will determine if exogenous laminin 5 or transfection with α3-laminin cDNAs can rescue cellular defects stemming from the removal of laminin 5.
Relationship to Other Knock-Out Animals
The integrity of the epidermis of LAMA3 null animals remained intact during development and birth indicating that adhesion independent of laminin 5 may provide sufficient developmental instruction and stability for survival before birth. Junctional blisters developed in the affected pups several hours after birth and were usually restricted to the forepaws, limbs, and oral mucosa. A similar blistering phenotype was found in mice carrying a disruption in the β3 subunit of laminin 5 that was caused by insertion of an intracisternal-A particle into the LAMB3 gene (Kuster et al., 1997). The relatively restricted blistering phenotype of the LAMA3 null animals contrasts with the phenotype of pups lacking integrin α6β4, the anchorage receptor for laminin 5. Pups lacking either integrin α6 or β4 displayed extensive blisters over the entire body surface and died within hours of birth (Dowling et al., 1996; Georges-Labouesse et al., 1996; van der Neut et al., 1996). The extensive skin fragility may have been caused by weakening in the cytoplasm and at the BMZ that resulted in the formation of both simplex and junctional blisters in these animals (Dowling et al., 1996; Georges-Labouesse et al., 1996; van der Neut et al., 1996). The epithelium in β4 null animals was also susceptible to apoptotic cell death. Using a tunnel staining assay (data not shown), we did not detect apoptosis in LAMA3 null animals. The accelerated and heightened severity of the blistering in the β4 null animals may also have contributed to the onset of apoptosis in these animals. We noted organization changes in the epidermis of LAMA3 null animals that were similar to the β4 null animals. In particular, we identified cell clusters that appeared undifferentiated in the superbasal cell layer similar to the pearl cells described in the β4 null animals (Dowling et al., 1996). Immunostaining of skin with anti-β4 antibodies identified positive staining in the superbasal cell layer (data not shown), suggesting that cellular differentiation in lesional regions of the epidermis may be abnormal in LAMA3 null animals.
Laminin 5 can regulate both anchorage and motility of epithelial cells through integrin α6β4 and α3β1, respectively (Carter et al., 1990; Xia et al., 1996; Goldfinger et al., 1998). Consistently, analysis of skin from integrin α3 null animals has revealed alterations in the epidermis stemming from the absence of integrin α3β1 function (DiPersio et al., 1997). In particular, the integrin α3 null animals showed a disorganized BM and blister formation at the dermal–epidermal junction (DiPersio et al., 1997). Similar to LAMA3 null animals, bleeding was identifiable in the paws of integrin α3 null pups (Hodivala-Dilke et al., 1998). Whether these two observations are connected remains to be determined. The bleeding that occurred in the paws of the LAMA3 null pups was detectable even before a blister had formed, suggesting that it was a primary defect. The bleeding may be an indication of an abnormality stemming from the role of α3-laminin in an alternative trimer such as laminin 6 or 7.
The BM of Mutant Tissue Supports β1-Integrin Function, but Not α6β4-Anchorage
Using a novel adhesion assay to directly assay BM function in vivo we found that the mutant BM could no longer induce adhesion by integrin α6β4. These results confirm that functional interactions between integrin α6β4 and laminin 5 have been eliminated in homozygous null animals. This data has implications for the organism as a whole because it indicates that integrin α6β4 may be unligated or no longer functional in multiple tissues, which may contribute to the neonatal lethality in homozygous null animals. In contrast to loss of α6β4 function, cell adhesion via integrin α3β1 was retained in the mutant BM. Because we have demonstrated that laminin 5 is absent from the mutant BM, the most logical conclusion is that we are detecting an alternative ligand for integrin α3β1 that is present in the BM of mutant skin. Our immunostaining experiments have identified several laminin isoforms which may be candidate ligands for integrin α3β1 in the epidermal BM (Table I). In a recent study that compared the ligand binding activities of different laminin isoforms, laminin 10/11 was identified as a potent substrate for adhesion of lung carcinoma cells via integrin α3β1 (Kikkawa et al., 1998). The α3β1-mediated adhesion to laminin 10/11 was comparable to laminin 5 and found to be greater than adhesion to laminin 1 or laminin 2/4 (Kikkawa et al., 1998). Likewise, we have shown that adhesion of HFKs via integrin α3β1 is better on laminin 5 than on laminin 1, suggesting that laminin 1 does not significantly contribute to adhesion of keratinocytes in vivo (Carter et al., 1990). Accordingly, laminin 10/11 or a new laminin isoform may contribute to α3β1-mediated adhesion and will be investigated as a possible second ligand for integrin α3β1 in epidermis.
Laminin 5 Regulates Late Stage Differentiation of Epithelium
We have established a role for laminin 5 in late stage differentiation of ameloblasts in developing incisors of mutant animals. The phenotypic alterations found in mutant incisors are consistent with the dental abnormalities and enamel hypoplasia described for JEB-G patients (Brain and Wigglesworth, 1968; Gardner and Hudson, 1975). Enamel hypoplasia has also been reported for an epidermolysis bullosa patient with a confirmed mutation in the ITGB4 gene (Pulkkinen et al., 1998b), implicating a role for integrin α6β4 in amelogenesis. In our studies, ameloblast abnormalities were first detected at the onset of enamel secretion and continued throughout ameloblast differentiation, culminating in the disorganization of the reduced enamel epithelium. The phenotypic alterations coincided nicely with the deposition of α3-laminin trimers in the wild-type incisor where we identified positive staining for the laminin α3 chain along the edge of the differentiating ameloblasts. No staining was detected in a comparable region of the mutant incisor, confirming the absence of laminin 5 in the mutant tooth. Positive staining for integrin β4 (data not shown) suggests that the ameloblast differentiation may be dependent on laminin 5 interactions with integrin α6β4. The deposition of laminin α3 chain in the wild-type tooth is consistent with recent studies that have shown that the subunits of laminin 5 are expressed in differentiating ameloblasts, even during enamel secretion when laminin 1 expression has disappeared (Salmivirta et al., 1997; K. Yoshiba et al., 1998; N. Yoshiba et al., 1998). It is interesting that abnormalities were detected in the mutant tooth at the onset of enamel secretion because ultrastructural analysis of developing teeth have shown that the basal lamina disappears during the secretory stage of amelogenesis and then reforms during ameloblast maturation (Smith, 1998). Our results suggest that laminin 5 has a unique role in regulation of ameloblast differentiation and that the requirement for laminin 5 may begin at the onset of enamel secretion. Furthermore, the disorganization that occurred in the reduced enamel epithelium emphasizes a role for laminin 5 in the maintenance of stratified epithelium.
Acknowledgments
We wish to thank Drs. Phil Soriano and Kevin Foley for helpful advice on preparing the targeting construct, Dr. Leigh Anderson for assistance on the analysis of developing incisors, Katrina Buglai and Josephine Hidalgo for expert technical assistance, Linda O'Neal for preparation and staining of sections used for histology, Dr. Phil Fleckman and Barbara Hager for advice on growing MEKs, Liz Caldwell and Judy Groombridge for assistance with electron microscopy, and Image Analysis for help with figure preparation. We thank Dr. Paul Lampe for critical reading of the manuscript.
We are grateful to Drs. Jonathan Jones, Jeffrey Miner, Takashi Hashimoto, and Eva Engvall for providing antibodies.
Abbreviations used in this paper
- BM
basement membrane
- BMZ
basement membrane zone
- BP180
bullous pemphigoid antigen 180
- BP230
bullous pemphigoid antigen 230
- ECM
extracellular matrix
- ES
embryonic stem
- HD
hemidesmosome
- HFK
human foreskin keratinocyte
- JEB-G
junctional epidermolysis bullosa gravis
- KGM
keratinocyte growth medium
- MEK
mouse epidermal keratinocyte
Footnotes
The authors would like to acknowledge financial support from the National Institutes of Health Grants CA49259 and AR-21557 to W.G. Carter and the Dermatology Foundation (M.C. Ryan).
References
- Aberdam D, Galliano MF, Vailly J, Pulkkinen L, Bonifas J, Christiano AM, Tryggvason K, Uitto J, Epstein EH, Jr, Ortonne JP, et al. Herlitz's junctional epidermolysis bullosa is linked to mutations in the gene (LAMC2) for the gamma 2 subunit of nicein/kalinin (LAMININ-5) Nat Genet. 1994;6:299–304. doi: 10.1038/ng0394-299. [DOI] [PubMed] [Google Scholar]
- Brain EB, Wigglesworth JS. Developing teeth in epidermolysis bullosa hereditaria letalis. A histological study. Br Dent J. 1968;124:255–260. [PubMed] [Google Scholar]
- Brown TA, Gil SG, Sybert VP, Lestringant GG, Tadini G, Caputo R, Carter WG. Defective integrin alpha 6 beta 4 expression in the skin of patients with junctional epidermolysis bullosa and pyloric atresia [published erratum appears in J. Invest. Dermatol.1997. 108:237] J Invest Dermatol. 1996;107:384–391. doi: 10.1111/1523-1747.ep12363370. [DOI] [PubMed] [Google Scholar]
- Carter WG, Kaur P, Gil SG, Gahr PJ, Wayner EA. Distinct functions for integrins α3β1 in focal adhesions and α6β4/bullous pemphigoid antigen in a new stable anchoring contact (SAC) of keratinocytes: relationship to hemidesmosomes. J Cell Biol. 1990;111:3141–3154. doi: 10.1083/jcb.111.6.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter WG, Ryan MC, Gahr PJ. Epiligrin, a new cell adhesion ligand for integrin α3β1 in epithelial basement membranes. Cell. 1991;65:599–610. doi: 10.1016/0092-8674(91)90092-d. [DOI] [PubMed] [Google Scholar]
- Champliaud MF, Lunstrum GP, Rousselle P, Nishiyama T, Keene DR, Burgeson RE. Human amnion contains a novel laminin variant, laminin 7, which like laminin 6, covalently associates with laminin 5 to promote stable epithelial–stromal attachment. J Cell Biol. 1996;132:1189–1198. doi: 10.1083/jcb.132.6.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiano AM, Uitto J. Molecular complexity of the cutaneous basement membrane zone. Revelations from the paradigms of epidermolysis bullosa. Exp Dermatol. 1996;5:1–11. doi: 10.1111/j.1600-0625.1996.tb00086.x. [DOI] [PubMed] [Google Scholar]
- DiPersio CM, Hodivala-Dilke KM, Jaenisch R, Kreidberg JA, Hynes RO. α3β1 integrin is required for normal development of the epidermal basement membrane. J Cell Biol. 1997;137:729–742. doi: 10.1083/jcb.137.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doliana R, Bellina I, Bucciotti F, Mongiat M, Perris R, Colombatti A. The human alpha3b is a ‘full-sized' laminin chain variant with a more widespread tissue expression than the truncated alpha3a. FEBS Lett. 1997;417:65–70. doi: 10.1016/s0014-5793(97)01251-9. [DOI] [PubMed] [Google Scholar]
- Dou QP, An B, Will PL. Induction of a retinoblastoma phosphatase activity by anticancer drugs accompanies p53-independent G1 arrest and apoptosis. Proc Natl Acad Sci USA. 1995;92:9019–9023. doi: 10.1073/pnas.92.20.9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling J, Yu QC, Fuchs E. Beta4 integrin is required for hemidesmosome formation, cell adhesion, and cell survival. J Cell Biol. 1996;134:559–572. doi: 10.1083/jcb.134.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmann, D.R. 1989. Amelogenesis. In Oral Histology: Development, Structure, and Function. A.R. Ten Cate, editor. Mosby Publishing, St. Louis, MO. 197–212.
- Engvall E, Earwicker D, Haaparanta T, Ruoslahti E, Sanes JR. Distribution and isolation of four laminin variants; tissue restricted distribution of heterotrimers assembled from five different subunits. Cell Regul. 1990;1:731–740. doi: 10.1091/mbc.1.10.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine JD, Bauer EA, Briggaman RA, Carter DM, Eady RA, Esterly NB, Holbrook KA, Hurwitz S, Johnson L, Lin A, et al. Revised clinical and laboratory criteria for subtypes of inherited epidermolysis bullosa. A consensus report by the Subcommittee on Diagnosis and Classification of the National Epidermolysis Bullosa Registry. J Am Acad Dermatol. 1991;24:119–135. doi: 10.1016/0190-9622(91)70021-s. [DOI] [PubMed] [Google Scholar]
- Galliano MF, Aberdam D, Aguzzi A, Ortonne JP, Meneguzzi G. Cloning and complete primary structure of the mouse laminin alpha 3 chain. Distinct expression pattern of the laminin alpha 3A and alpha 3B chain isoforms. J Biol Chem. 1995;270:21820–21826. doi: 10.1074/jbc.270.37.21820. [DOI] [PubMed] [Google Scholar]
- Gardner DG, Hudson CD. The disturbances in odontogenesis in epidermolysis bullosa hereditaria letalis. Oral Surg Oral Med Oral Pathol. 1975;40:483–493. doi: 10.1016/0030-4220(75)90246-7. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Messaddeq N, Yehia G, Cadalbert L, Dierich A, Le Meur M. Absence of integrin alpha 6 leads to epidermolysis bullosa and neonatal death in mice. Nat Genet. 1996;13:370–373. doi: 10.1038/ng0796-370. [DOI] [PubMed] [Google Scholar]
- Gil SG, Brown TA, Ryan MC, Carter WG. Junctional epidermolysis bullosis: defects in expression of epiligrin/nicein/kalinin and integrin beta 4 that inhibit hemidesmosome formation. J Invest Dermatol. 1994;103:31S–38S. doi: 10.1111/1523-1747.ep12398953. [DOI] [PubMed] [Google Scholar]
- Goldfinger LE, Stack MS, Jones JC. Processing of laminin-5 and its functional consequences: role of plasmin and tissue-type plasminogen activator. J Cell Biol. 1998;141:255–265. doi: 10.1083/jcb.141.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales M, Haan K, Baker SE, Fitchmun M, Todorov I, Weitzman S, Jones JCR. A cell signal pathway involving laminin-5, alpha 3 beta 1 integrin, and mitogen-activated protein kinase can regulate epithelial cell proliferation. Mol Biol Cell. 1999;10:259–270. doi: 10.1091/mbc.10.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager, B., J.R. Bickenbach, and P. Fleckman. 1999. Long-term culture of murine epidermal keratinocytes. J. Invest. Dermatol. In press. [DOI] [PubMed]
- Hashimoto T, Amagai M, Ebihara T, Gamou S, Shimizu N, Tsubata T, Hasegawa A, Miki K, Nishikawa T. Further analysis of epitopes for human monoclonal anti-basement membrane zone antibodies produced by stable human hybridoma cell lines constructed with Epstein-Barr virus transformants. J Invest Dermatol. 1993;100:310–315. doi: 10.1111/1523-1747.ep12469916. [DOI] [PubMed] [Google Scholar]
- Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tome FM, Schwartz K, Fardeau M, Tryggvason K, et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. 1995;11:216–218. doi: 10.1038/ng1095-216. [DOI] [PubMed] [Google Scholar]
- Hodivala-Dilke KM, DiPersio CM, Kreidberg JA, Hynes RO. Novel roles for α3β1 integrin as a regulator of cytoskeletal assembly and as a trans-dominant inhibitor of integrin receptor function in mouse keratinocytes. J Cell Biol. 1998;142:1357–1369. doi: 10.1083/jcb.142.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GD, Davidson RS, McNamee KC, Russell G, Goodwin D, Holborow EJ. Fading of immunofluorescence during microscopy: a study of the phenomenon and its remedy. J Immunol Meth. 1982;55:231–242. doi: 10.1016/0022-1759(82)90035-7. [DOI] [PubMed] [Google Scholar]
- Jonkman MF, Scheffer H, Stulp R, Pas HH, Nijenhuis M, Heeres K, Owaribe K, Pulkkinen L, Uitto J. Revertant mosaicism in epidermolysis bullosa caused by mitotic gene conversion. Cell. 1997;88:543–551. doi: 10.1016/s0092-8674(00)81894-2. [DOI] [PubMed] [Google Scholar]
- Kaur P, McDougall JK, Cone R. Immortalisation of primary human epithelial cells by cloned cervical carcinoma DNA containing human papillomavirus 16 E6/7 ORFs. J Gen Virol. 1989;70:1261–1266. doi: 10.1099/0022-1317-70-5-1261. [DOI] [PubMed] [Google Scholar]
- Kikkawa Y, Umeda M, Miyazaki K. Marked stimulation of cell adhesion and motility by ladsin, a laminin-like scatter factor. J Biochem. 1994;116:862–869. doi: 10.1093/oxfordjournals.jbchem.a124608. [DOI] [PubMed] [Google Scholar]
- Kikkawa Y, Sanzen N, Sekiguchi K. Isolation and characterization of laminin-10/11 secreted by human lung carcinoma cells: laminin-10/11 mediates cell adhesion through integrin alpha-3-beta-1. J Biol Chem. 1998;273:15854–15859. doi: 10.1074/jbc.273.25.15854. [DOI] [PubMed] [Google Scholar]
- Kivirikko S, McGrath JA, Baudoin C, Aberdam D, Ciatti S, Dunnill MG, McMillan JR, Eady RA, Ortonne JP, Meneguzzi G, et al. A homozygous nonsense mutation in the alpha 3 chain gene of laminin 5 (LAMA3) in lethal (Herlitz) junctional epidermolysis bullosa. Hum Mol Genet. 1995;4:959–962. doi: 10.1093/hmg/4.5.959. [DOI] [PubMed] [Google Scholar]
- Kivirikko S, McGrath JA, Pulkkinen L, Uitto J, Christiano AM. Mutational hotspots in the LAMB3 gene in the lethal (Herlitz) type of junctional epidermolysis bullosa. Hum Mol Genet. 1996;5:231–237. doi: 10.1093/hmg/5.2.231. [DOI] [PubMed] [Google Scholar]
- Kuster JE, Guarnieri MH, Ault JG, Flaherty L, Swiatek PJ. IAP insertion in the murine LamB3 gene results in junctional epidermolysis bullosa. Mamm Genome. 1997;8:673–681. doi: 10.1007/s003359900535. [DOI] [PubMed] [Google Scholar]
- Lampe PD, Nguyen BP, Gil S, Usui M, Olerud J, Takada Y, Carter WG. Cellular interaction of integrin α3β1 with laminin 5 promotes gap junctional communication. J Cell Biol. 1998;143:1735–1747. doi: 10.1083/jcb.143.6.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhofer M, Hopkinson SB, Jones JC. The matrix secreted by 804G cells contains laminin-related components that participate in hemidesmosome assembly in vitro. J Cell Sci. 1993;105:753–764. doi: 10.1242/jcs.105.3.753. [DOI] [PubMed] [Google Scholar]
- Mainiero F, Murgia C, Wary KK, Curatola AM, Pepe A, Blumemberg M, Westwick JK, Der CJ, Giancotti FG. The coupling of alpha6beta4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. EMBO (Eur Mol Biol Organ) J. 1997;16:2365–2375. doi: 10.1093/emboj/16.9.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinkovich MP, Lunstrum GP, Keene DR, Burgeson RE. The dermal–epidermal junction of human skin contains a novel laminin variant. J Cell Biol. 1992;119:695–703. doi: 10.1083/jcb.119.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jenkins NA, Copeland NG, Sanes JR. The laminin α chains: expression, developmental transitions, and chromosomal locations of α1-5, identification of heterotrimeric laminins 8-11, and cloning of a novel α3 isoform. J Cell Biol. 1997;137:685–701. doi: 10.1083/jcb.137.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgia C, Blaikie P, Kim N, Dans M, Petrie HT, Giancotti FG. Cell cycle and adhesion defects in mice carrying a targeted deletion of the integrin beta4 cytoplasmic domain. EMBO (Eur Mol Biol Organ) J. 1998;17:3940–3951. doi: 10.1093/emboj/17.14.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen CM, Hogervorst F, Jaspars LH, de Melker AA, Delwel GO, Hulsman EH, Kuikman I, Sonnenberg A. The alpha 6 beta 4 integrin is a receptor for both laminin and kalinin. Exp Cell Res. 1994;211:360–367. doi: 10.1006/excr.1994.1099. [DOI] [PubMed] [Google Scholar]
- Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/ laminin beta 2. Nature. 1995a;374:258–262. doi: 10.1038/374258a0. [DOI] [PubMed] [Google Scholar]
- Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP. The renal glomerulus of mice lacking s-laminin/laminin beta 2: nephrosis despite molecular compensation by laminin beta 1. Nat Genet. 1995b;10:400–406. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- Pulkkinen L, Christiano AM, Airenne T, Haakana H, Tryggvason K, Uitto J. Mutations in the gamma 2 chain gene (LAMC2) of kalinin/ laminin 5 in the junctional forms of epidermolysis bullosa. Nat Genet. 1994a;6:293–297. doi: 10.1038/ng0394-293. [DOI] [PubMed] [Google Scholar]
- Pulkkinen L, Christiano AM, Gerecke D, Wagman DW, Burgeson RE, Pittelkow MR, Uitto J. A homozygous nonsense mutation in the beta 3 chain gene of laminin 5 (LAMB3) in Herlitz junctional epidermolysis bullosa. Genomics. 1994b;24:357–360. doi: 10.1006/geno.1994.1627. [DOI] [PubMed] [Google Scholar]
- Pulkkinen L, Cserhalmi-Friedman PB, Tang M, Ryan MC, Uitto J, Christiano AM. Molecular analysis of the human laminin alpha3a chain gene (LAMA3a): a strategy for mutation identification and DNA-based prenatal diagnosis in Herlitz junctional epidermolysis bullosa. Lab Invest. 1998a;78:1067–1076. [PubMed] [Google Scholar]
- Pulkkinen L, Kim DU, Uitto J. Epidermolysis bullosa with pyloric atresia: novel mutations in the beta4 integrin gene (ITGB4) Am J Pathol. 1998b;152:157–166. [PMC free article] [PubMed] [Google Scholar]
- Rousselle P, Lunstrum GP, Keene DR, Burgeson RE. Kalinin: an epithelium-specific basement membrane adhesion molecule that is a component of anchoring filaments. J Cell Biol. 1991;114:567–576. doi: 10.1083/jcb.114.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MC, Tizard R, VonDevanter DR, Carter WG. Cloning of the LamA3 gene encoding the α3 chain of the adhesive ligand epiligrin: expression in wound repair. J Biol Chem. 1994;269:22779–22787. [PubMed] [Google Scholar]
- Ryan MC, Christiano AM, Engvall E, Wewer UM, Miner JH, Sanes JR, Burgeson RE. The functions of laminins: lessons from in vivo studies. Matrix Biol. 1996;15:369–381. doi: 10.1016/s0945-053x(96)90157-2. [DOI] [PubMed] [Google Scholar]
- Sakai LY, Keene DR. Fibrillin: monomers and microfibrils. Meth Enzymol. 1994;245:29–52. doi: 10.1016/0076-6879(94)45004-8. [DOI] [PubMed] [Google Scholar]
- Salmivirta K, Sorokin LM, Ekblom P. Differential expression of laminin alpha chains during murine tooth development. Dev Dyn. 1997;210:206–215. doi: 10.1002/(SICI)1097-0177(199711)210:3<206::AID-AJA2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Sander EE, van Delft S, ten Klooster JP, Reid T, van der Kammen RA, Michiels F, Collard JG. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell–cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J Cell Biol. 1998;143:1385–1398. doi: 10.1083/jcb.143.5.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE. Cellular and chemical events during enamel maturation. Crit Rev Oral Biol Med. 1998;9:128–161. doi: 10.1177/10454411980090020101. [DOI] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- Stepp MA, Spurr-Michaud S, Tisdale A, Elwell J, Gipson IK. Alpha 6 beta 4 integrin heterodimer is a component of hemidesmosomes. Proc Natl Acad Sci USA. 1990;87:8970–8974. doi: 10.1073/pnas.87.22.8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Parry DA, Klaus-Kovtun V, Steinert PM, Stanley JR. Comparison of molecularly cloned bullous pemphigoid antigen to desmoplakin I confirms that they define a new family of cell adhesion junction plaque proteins. J Biol Chem. 1991;266:12555–12559. [PubMed] [Google Scholar]
- van der Neut R, Krimpenfort P, Calafat J, Niessen CM, Sonnenberg A. Epithelial detachment due to absence of hemidesmosomes in integrin beta 4 null mice. Nat Genet. 1996;13:366–369. doi: 10.1038/ng0796-366. [DOI] [PubMed] [Google Scholar]
- Verrando P, Hsi BL, Yeh CJ, Pisani A, Serieys N, Ortonne JP. Monoclonal antibody GB3, a new probe for the study of human basement membranes and hemidesmosomes. Exp Cell Res. 1987;170:116–128. doi: 10.1016/0014-4827(87)90121-2. [DOI] [PubMed] [Google Scholar]
- Vidal F, Aberdam D, Miquel C, Christiano AM, Pulkkinen L, Uitto J, Ortonne JP, Meneguzzi G. Integrin beta4 mutations associated with junctional epidermolysis bullosa with pyloric atresia. Nat Genet. 1995;10:229–234. doi: 10.1038/ng0695-229. [DOI] [PubMed] [Google Scholar]
- Wright JT, Johnson LB, Fine JD. Development defects of enamel in humans with hereditary epidermolysis bullosa. Arch Oral Biol. 1993;38:945–955. doi: 10.1016/0003-9969(93)90107-w. [DOI] [PubMed] [Google Scholar]
- Xia Y, Gil SG, Carter WG. Anchorage mediated by integrin α6β4 to laminin 5 (epiligrin) regulates tyrosine phosphorylation of a membrane-associated 80-kD protein. J Cell Biol. 1996;132:727–740. doi: 10.1083/jcb.132.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Christmas P, Wu XR, Wewer UM, Engvall E. Defective muscle basement membrane and lack of M-laminin in the dystrophic dy/dy mouse. Proc Natl Acad Sci USA. 1994;91:5572–5576. doi: 10.1073/pnas.91.12.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiba K, Yoshiba N, Aberdam D, Meneguzzi G, Perrin-Schmitt F, Stoetzel C, Ruch JV, Lesot H. Expression and localization of laminin-5 subunits during mouse tooth development. Dev Dyn. 1998;211:164–176. doi: 10.1002/(SICI)1097-0177(199802)211:2<164::AID-AJA5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Yoshiba N, Yoshiba K, Aberdam D, Meneguzzi G, Perrin-Schmitt F, Stoetzel C, Ruch JV, Lesot H. Expression and localization of laminin-5 subunits in the mouse incisor. Cell and Tissue Research. 1998;292:143–149. doi: 10.1007/s004410051044. [DOI] [PubMed] [Google Scholar]