

Abstract
Ezrin/radixin/moesin (ERM) proteins have been thought to play a central role in the organization of cortical actin-based cytoskeletons including microvillar formation through cross-linking actin filaments and integral membrane proteins such as CD43, CD44, and ICAM-2. To examine the functions of these ERM-binding membrane proteins (ERMBMPs) in cortical morphogenesis, we overexpressed ERMBMPs (the extracellular domain of E-cadherin fused with the transmembrane/cytoplasmic domain of CD43, CD44, or ICAM-2) in various cultured cells. In cultured fibroblasts such as L and CV-1 cells, their overexpression significantly induced microvillar elongation, recruiting ERM proteins and actin filaments. When the ERM-binding domains were truncated from these molecules, their ability to induce microvillar elongation became undetectable. In contrast, in cultured epithelial cells such as MTD-1A and A431 cells, the overexpression of ERMBMPs did not elongate microvilli. However, in the presence of EGF, overexpression of ERMBMPs induced remarkable microvillar elongation in A431 cells. These results indicated that ERMBMPs function as organizing centers for cortical morphogenesis by organizing microvilli in collaboration with activated ERM proteins. Furthermore, immunodetection with a phosphorylated ERM-specific antibody and site-directed mutagenesis suggested that ERM proteins phosphorylated at their COOH-terminal threonine residue represent activated ERM proteins.
Keywords: ezrin, radixin, moesin, ERM, microvilli
Plasma membranes can be classified into two domains: cell adhesion and free surface domains. In the former domain, their outer surface interacts with extracellular matrix or plasma membranes of adjacent cells, while in the latter it is covered with fluid components. The actin cytoskeleton is essential for the morphogenesis of both domains. In the cell adhesion domain, especially in integrin-based focal adhesions and cadherin-based cell-to-cell adherens junctions, how actin filaments are involved in the morphogenesis of these membrane domains has been examined in detail (Geiger, 1983; Gumbiner, 1996). The formation of the cell adhesion domain is initiated by binding of specific ligands to cell adhesion molecules, integrins, or cadherins. These activated adhesion molecules are thought to recruit specific cytoplasmic proteins such as talin, vinculin, paxillin, α-actinin, tensin, ZO-1, and various signaling molecules, which may nucleate and organize actin-based cytoskeletons underlying the cell adhesion domain (Yonemura et al., 1995; Burridge et al., 1997; Yamada and Geiger, 1997).
The free surface domain is characterized by various types of cellular protrusions such as microvilli, filopodia, and lamellipodia which contain abundant actin filaments (Bretscher, 1991; Condeelis, 1993; Hitt and Luna, 1994; Mooseker and Cheney, 1995; Mitchison and Cramer, 1996). These cellular protrusions are known to be formed and destroyed dynamically in response to various signals (Chinkers et al., 1979; Stossel, 1993; Greenberg, 1995; Forte and Yao, 1996; Hallett, 1997; Tapon and Hall, 1997). Compared to focal adhesions and cell-to-cell adherens junctions, however, our knowledge regarding the molecular sequences responsible for their morphogenesis is still limited. Experimentally, the formation of cellular protrusions was induced by overexpressing several cytoskeletal proteins. Overexpression of villin, an actin-bundling protein identified in intestinal microvilli, resulted in marked elongation of microvilli in CV-1 cells (Friederich et al., 1989). Also, when T-plastin, another actin-bundling protein, was overexpressed, microvilli were elongated in LLC-PK1 cells (Arpin et al., 1994b). Pleckstrin, which interacts with lipids, promoted the formation of membrane projections in Cos-1 cells (Ma et al., 1997). However, there have been no reports indicating the existence of integral membrane proteins which function as organizing centers for the morphogenesis of cellular protrusions as integrins do in the formation of focal adhesions.
Ezrin/radixin/moesin (ERM)1 proteins are localized mainly just beneath the plasma membranes of cellular protrusions such as microvilli, and are thought to function as cross-linkers between plasma membranes and actin filaments (Arpin et al., 1994a; Bretscher et al., 1997; Tsukita et al., 1997a,b). ERM proteins bind to actin filaments mainly at their COOH-terminal domain (Algrain et al., 1993; Turunen et al., 1994; Henry et al., 1995; Martin et al., 1995; Pestonjamasp et al., 1995), and at their NH2-terminal half they associate with the cytoplasmic domains of several integral membrane proteins such as CD43, CD44, and ICAM-1, -2, and -3 (Algrain et al., 1993; Yonemura et al., 1993; Tsukita et al., 1994; Henry et al., 1995; Helander et al., 1996; Hirao et al., 1996; Serrador et al., 1997; Heiska et al., 1998). Recently, a positively charged amino acid cluster in the juxta-membrane portion was identified as an ERM protein-binding region in CD43, CD44, and ICAM-2 (Legg and Isacke, 1998; Yonemura et al., 1998). As an alternative to the ERM protein-plasma membrane interaction, ezrin was reported to indirectly bind to Na+/H+ exchanger 3 (NHE3) via a cytoplasmic phosphoprotein called EBP50 or NHE-RF (Reczek et al., 1997; Yun et al., 1997; Murthy et al., 1998).
Soluble ERM proteins in the cytoplasm are dormant in terms of their cross-linking activity through mutual intramolecular and/or intermolecular association of their NH2- and COOH-terminal halves. Some cellular signals are thought to activate these dormant ERM proteins by exposing their halves to allow them to interact with integral membrane proteins and actin filaments (Berryman et al., 1995; Bretscher et al., 1995; Gary and Bretscher, 1995; Hirao et al., 1996; Matsui et al., 1998). These activated ERM proteins have been shown to be directly involved in the morphogenesis of the free surface domain of plasma membranes, especially in the organization of microvilli. For example, overexpression of the COOH-terminal half of ezrin or radixin induced abnormally long cellular protrusions (Henry et al., 1995; Martin et al., 1995), and overexpression of the NH2-terminal half of ezrin prevented formation of microvilli (Crepaldi et al., 1997). Furthermore, the suppression of ERM protein expression with antisense oligonucleotides in cultured cells resulted in complete disappearance of microvilli (Takeuchi et al., 1994). At the initial stage of anoxic injury and the apoptotic process, most microvilli disappear from the cell surface, and in both cases ERM proteins are translocated from microvilli to the cytoplasm with their concomitant dephosphorylation (Chen et al., 1995; Kondo et al., 1997).
Considering that in focal adhesions activated integrins recruit talin, vinculin, paxillin, etc., to organize the underlying actin-based cytoskeleton, the question has naturally arisen whether ERM-binding membrane proteins (ERMBMPs) such as CD44, CD43, and ICAM-2 function as organizers for the recruitment of ERM proteins and actin filaments to organize microvilli. In the present study, we overexpressed ERMBMPs (the extracellular domain of E-cadherin fused with the transmembrane/cytoplasmic domain of CD43, CD44, or ICAM-2) in several cell lines, and found that these proteins induce formation and elongation of microvilli in collaboration with activated ERM proteins. This study has led to a better understanding of the molecular sequences of the morphogenesis of the free surface domain of plasma membranes.
Materials and Methods
Cells and Antibodies
CV-1 monkey kidney cells (Jensen et al., 1964) and A431 human epidermoid cells (Giard et al., 1973) were obtained from Health Science Research Resources Bank. Mouse fibroblastic L cells (Earle, 1943) and MTD-1A epithelial cells (Enami et al., 1984) were provided by M. Takeichi (Kyoto University, Kyoto, Japan). These cells were cultured in DME supplemented with 10% FCS.
Mouse anti-ERM mAb (CR22) with higher affinity to moesin than to ezrin/radixin (Sato et al., 1991), rat anti-ezrin mAb (M11), rat anti-radixin mAb (R2-1) and rat anti-moesin mAb (M22) (Takeuchi et al., 1994), rat anti–COOH-terminal phosphorylated ERM (CPERM) mAb (297S) specific for phosphorylated ERM proteins at T567, T564, or T558 for ezrin, radixin, or moesin, respectively (Matsui et al., 1998), and rabbit anti-ERM polyclonal antibodies (pAbs) (TK88 and TK89) (Kondo et al., 1997) were used. TK88 was raised in a rabbit against synthesized peptides corresponding to the mouse ERM proteins' common sequence (293–302) and detected all ERM proteins. Rabbit anti–E-cadherin pAb (Nagafuchi et al., 1987) and rat anti–E-cadherin mAb (ECCD-2; Shirayoshi et al., 1986) were provided by M. Takeichi. IM7.8.1 is a rat anti–mouse CD44 mAb (Trowbridge et al., 1982). Mouse anti–phosphotyrosine mAb (clone 4G10) and mouse anti-vesicular stomatitis virus glycoprotein G (VSVG) mAb (clone P5D4) were purchased from Upstate Biotechnology Inc. and Sigma Chemical Co., respectively.
Mammalian Expression Vectors and Transfection
Some vectors used in this study were described previously (Yonemura et al., 1993, 1998). E-43, E-44, or E-ICAM-2 represents a chimeric molecule consisting of the extracellular domain of mouse E-cadherin and transmembrane/cytoplasmic domain of rat CD43, mouse CD44, or mouse ICAM-2, respectively. The extracellular domain of E-cadherin was used as an epitope tag to detect the cell surface expression of each chimeric molecule in L fibroblasts expressing no detectable E-cadherin. Several mutant proteins, E-43/1-31, E-44/1-19, E-44/20-70, and E-43/KRR:NGG, were described in detail previously (Yonemura et al., 1998). To obtain cDNA encoding VSVG-tagged ezrin (pSK/mEz-VSVG), an oligonucleotide encoding 11 COOH-terminal amino acids of VSVG was ligated into the 3′ terminal end of mouse ezrin cDNA (Funayama et al., 1991) according to Algrain et al. (1993). A cDNA fragment of pSK/mEz-VSVG was inserted into the HindIII-HpaI fragment of the pβact-CAT9 (Fregien and Davidson, 1986) to construct its expression vector (pA/mEz-VSVG). Plasmids encoding ezrin mutants on the COOH-terminal threonine (T567) were generated by PCR with appropriate primers from pSK/mEz-VSVG. Alanine or aspartic acid was substituted for T567 (pA/mEz-T/A-VSVG or pA/mEz-T/D-VSVG, respectively).
Cells were transfected with DNA using Lipofectin or Lipofectamine (GIBCO BRL). Cultured cells on coverslips were washed twice with Opti-MEM (GIBCO BRL), and incubated for 3–5 h with 1 ml Opti-MEM containing 1 μg of plasmid DNAs and 10 μl of the reagents, followed by addition of 3 ml of normal medium containing FCS. Cells were then cultured for 3–5 d. Since transfection with Lipofectin or Lipofectamine occasionally caused damage to the cell surface structures of transfected cells, cells were also transfected by microinjection using a set of manipulators (MN-188 and MO-189; Narishige) connected to an Eppendorf microinjector 5242 (Eppendorf, Inc.). Expression vectors in injection buffer (100 mM KCl, 10 mM Hepes buffer, pH 7.5) were injected into the nuclei of cells cultured on coverslips. Cells were examined 12–24 h after injection.
EGF Treatment of A431 Cells
Subconfluent A431 cells on coverslips in DME with 10% FCS were transferred to DMEM without FCS and cultured for 6–12 h. Recombinant human EGF (GIBCO BRL) was added at a final concentration of 100 ng/ml. Cells were incubated for 30 s, and quickly fixed and processed for immunofluorescence microscopy or SDS-PAGE followed by immunoblotting. In some experiments, cells were microinjected with expression vectors, allowed to express proteins for 6–18 h, serum-starved, treated with EGF, and then processed for immunofluorescence microscopy.
Immunofluorescence Microscopy
L and CV-1 Cells.
Cells were fixed with 1–4% fresh formaldehyde in 0.1 M Hepes buffer (pH 7.5) for 15 min. After three washes with PBS containing 30 mM glycine (G-PBS), cells were soaked in blocking solution (G-PBS containing 2% normal goat serum) for 5 min and incubated with anti– E-cadherin mAb (ECCD-2) diluted with the blocking solution for 10 min. Cells were then washed three times with G-PBS, treated with 0.2% Triton X-100 in G-PBS for 10 min, and washed with G-PBS. Cells were then soaked in blocking solution for 10 min, incubated with CR22 for 30 min, washed three times with G-PBS, and incubated with secondary antibodies for 30 min. DTAF-conjugated goat anti–rat IgG antibody (Chemicon) and Cy3-conjugated goat anti–mouse IgG antibody (Amersham International) were used as secondary antibodies. For triple labeling, FITC-phalloidin (Molecular Probes, Inc.), Cy3-conjugated goat anti–rat IgG antibody (Amersham International), and Cy5-conjugated goat anti–mouse IgG antibody (Amersham International) were used. Cells were washed three times, then mounted in 90% glycerol-PBS containing 0.1% para-phenylenediamine and 1% n-propylgalate. Specimens were observed using a Zeiss Axiophot photomicroscope (Carl Zeiss) with appropriate combinations of filters and mirrors. Photographs were taken on T-MAX 400 film (Eastman Kodak Co.), or recorded with a cooled CCD camera (SenSys 0400, 768 × 512 pixels; Photometrics) controlled by a Power Macintosh 7600/132 and the software package IPLab Spectrum V3.1 (Signal Analytics Corp.).
MTD-1A Cells.
Cells were fixed and doubly stained with anti–E-cadherin mAb (ECCD-2) and anti-ERM mAb (CR22) as described above. Alternatively, cells were microinjected with expression vectors, washed with cold culture medium the next day, and incubated with cold hybridoma culture medium containing anti-CD44 mAb (IM7.8.1) and anti– E-cadherin pAb for 15 min at 4°C. After washing twice with cold culture medium, cells were fixed with 1% formaldehyde in 0.1 M Hepes buffer (pH 7.5) for 15 min at room temperature. Cells were washed, permeabilized, and soaked in blocking solution. Cy2-conjugated goat anti–rabbit IgG antibody (Amersham International) and Cy3-conjugated goat anti– rat IgG antibody were used as secondary antibodies.
A431 Cells.
For double labeling of cells with 297S and TK88 to detect CPERMs and total ERM proteins, respectively, cells were fixed with 10% trichloroacetic acid for 15 min on ice (Hayashi et al., 1999). In this case, TK89 which also detected all ERM proteins was not used, since the epitope for 297S is overlapped with that for TK89. To label cells with ECCD-2, TK89, P5D4, and/or FITC-phalloidin, they were fixed with 4% formaldehyde in 0.1 M Hepes buffer (pH 7.5) for 15 min. Cells were washed, permeabilized, and soaked in blocking solution. Cy2-conjugated goat anti–rabbit IgG antibody, Cy2-conjugated donkey anti–mouse IgG antibody (Jackson ImmunoResearch Laboratories, Inc.), Cy3-conjugated goat anti–mouse IgG (Amersham), Cy3-conjugated goat anti–rat IgG antibody, and/or Cy5-conjugated goat anti-mouse IgG antibody were used as secondary antibodies. In A431 cells, cell surface expression of E-cadherin-chimeric molecules was hard to detect because these cells express considerable amounts of endogenous E-cadherin. However, E-cadherin-chimeric molecules could be detected by staining cells with ECCD-2 after permeabilization with Triton X-100 because these molecules, but not endogenous E-cadherin, were accumulated in the cytoplasm in large amounts.
Immunoscanning Electron Microscopy
Polystyrene latex beads with a sulfate group on their surface (0.1 μm FluoSpheres; Molecular Probes) were coated with avidin according to the method of Molday et al. (1974). 1 d after microinjection with the expression vector encoding E-43, CV-1 cells on coverslips were fixed with 4% formaldehyde in 0.1 M Hepes buffer (pH 7.5) for 20 min, washed with G-PBS three times, soaked in blocking solution for 10 min, and then incubated in blocking solution containing anti–E-cadherin mAb, ECCD-2 for 20 min. Cells were washed with G-PBS, incubated with biotinylated goat anti–rat IgG antibody (Amersham International) in blocking solution, and then washed with G-PBS. Samples were incubated with the avidin-coated beads suspended in blocking solution for 1 h. Cells were washed with G-PBS and fixed with 2.5% glutaraldehyde in 0.1 M Hepes buffer (pH 7.5) for 2 h followed by postfixation with the same buffer containing 1% OsO4 at 4°C for 1 h. Samples were then washed with distilled water, dehydrated through a graded series of ethanol, transferred into isoamyl acetate, and dried in a critical point drier (Hitachi Co.) after substitution with liquid CO2. Dried samples were coated with gold using a gold sputter coater (Eiko Engineering), and were examined under a scanning electron microscope (model S-800; Hitachi Co.).
Immunoblotting
Subconfluent A431 cells cultured on a 35-mm dish were serum-starved for 6 h as described above. With or without EGF treatment (100 ng/ml) for 30 s, cells were washed with ice-cold PBS twice, and then recovered in 200 μl of SDS-PAGE sample buffer by scraping. Proteins in 20 μl of the sample were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Blots were first incubated with anti-phosphotyrosine mAb (4G10), which were localized using the enhanced chemiluminescence Western blotting detection system (Amersham). Bound antibodies were then removed from the membranes by incubating with 7 M guanidine-HCl, 50 μM EDTA, 20 mM 2-mercaptoethanol for 10 min at room temperature. These membranes were incubated with anti-CPERM mAb (297S) to detect threonine-phosphorylated ERM proteins, then bound mAb was again removed. Finally, these membranes were incubated with anti-ERM protein pAb (TK89) to total ERM proteins.
Results
Overexpression of ERMBMPs in Fibroblastic Cells
In our previous study, we transfected L cells with cDNAs encoding several chimeric molecules containing the extracellular domain of E-cadherin and the transmembrane/cytoplasmic domain of CD43, CD44, or ICAM-2 (E-43, E-44, or E-ICAM-2, respectively), and showed that these chimeric molecules were concentrated in microvilli together with ERM proteins (Yonemura et al., 1998). Then, we attempted to examine the abilities of these chimeric molecules to elongate microvilli using stable transfectants. However, the microvillar elongation was not clearly observed in these cells, probably because the stable transfectants expressed relatively low levels of chimeric molecules. Next, we examined the effects of transient overexpression of these chimeric molecules on L cell surface morphology.
The expression and localization of each chimeric molecule was detected with anti–E-cadherin mAb, ECCD-2, which recognized the extracellular domain of E-cadherin, and microvilli were specifically visualized by immunofluorescence staining with anti-ERM protein mAb, CR22. As shown in Fig. 1, a and b, when the expression level of E-43 was sufficiently high, expressed E-43 was not only colocalized with ERM proteins in microvilli but also caused elongation of microvilli, as compared to nontransfected L cells (Fig. 1 g). The number of microvilli did not appear to increase. Overexpression of E-44 or E-ICAM-2 gave the same results (Fig. 1, c–f). Overexpression of E-43/1-31 (a deletion mutant of E-43 lacking the COOH-terminal region but which carries the juxta-membrane ERM-binding region of CD43 cytoplasmic domain; see Yonemura et al., 1998) also induced microvillar elongation (data not shown). Immunofluorescence microscopy with ezrin-, radixin-, or moesin-specific mAbs revealed that in all cases ezrin, radixin, and moesin were recruited to the elongated microvilli without any bias (data not shown).
Figure 1.

Microvillar elongation in L fibroblasts overexpressing ERMBMPs. L transfectants transiently expressing E-cadherin chimeric molecules with the transmembrane/cytoplasmic domain of CD43 (E-43, a and b), CD44 (E-44, c and d), or ICAM-2 (E-ICAM-2, e and f) were doubly stained with anti– E-cadherin mAb (a, c, and e), and anti-ERM mAb (b, d, and f). In these cells, microvilli were significantly elongated, where both ERMBMPs and ERM proteins were recruited (inset, higher magnification). Cells were transfected by microinjection. As a control, parental L cells were stained with anti-ERM mAb (g). Bar, a–g, 10 μm; insets, 2.5 μm.
Two distinct control experiments were performed. First, wild-type E-cadherin was introduced into L cells. As previously reported (Yonemura et al., 1998), E-cadherin was distributed diffusely over the cell surface (Fig. 2 a), and the length and number of ERM-positive microvilli were not changed (Fig. 2 b). Second, two chimeric molecules lacking the ERM-binding ability were introduced into L cells: E-44/20-70 in which the juxta-membrane ERM-binding region was deleted from the E-44 cytoplasmic domain and E-43/KRR:NGG in which the juxta-membrane ERM-binding region of E-43 was disrupted by site-directed mutation (see Yonemura et al., 1998). These chimeric molecules did not cause elongation of microvilli in transient transfectants (Fig. 2, c–f).
Figure 2.

L fibroblasts overexpressing membrane proteins lacking the ERM-binding ability. L transfectants transiently expressing wild-type E-cadherin (E-cad, a and b), E-44/20-70, a mutant E-44 lacking the juxta-membrane ERM-binding region (E-44/20-70, c and d), or E-43/KRR:NGG, a mutant E-43 bearing site-directed disrupted ERM-binding region (E-43/ KRR:NGG, e and f), were doubly stained with anti–E-cadherin mAb (a, c, and e) and anti-ERM mAb (b, d, and f). No elongated microvilli were observed. Cells were transfected by microinjection (a, b, e, and f) or by lipofection (c and d). Bar, a–f, 10 μm; insets, 2.5 μm.
Furthermore, the effects of overexpression of E-43 on the cortical actin cytoskeleton organization were examined (Fig. 3). In the dorsal cortex of nontransfected L cells, stress fibers and relatively short microvillar core actin bundles were observed (arrow in Fig. 3 b). In the dorsal cortex of E-43–expressing cells, stress fibers were disrupted with concomitant formation of relatively long microvillar actin bundles (asterisk in Fig. 3 b), whereas ventral stress fibers were not affected (data not shown). In E-cadherin–overexpressing cells, dorsal stress fibers were not affected (data not shown).
Figure 3.
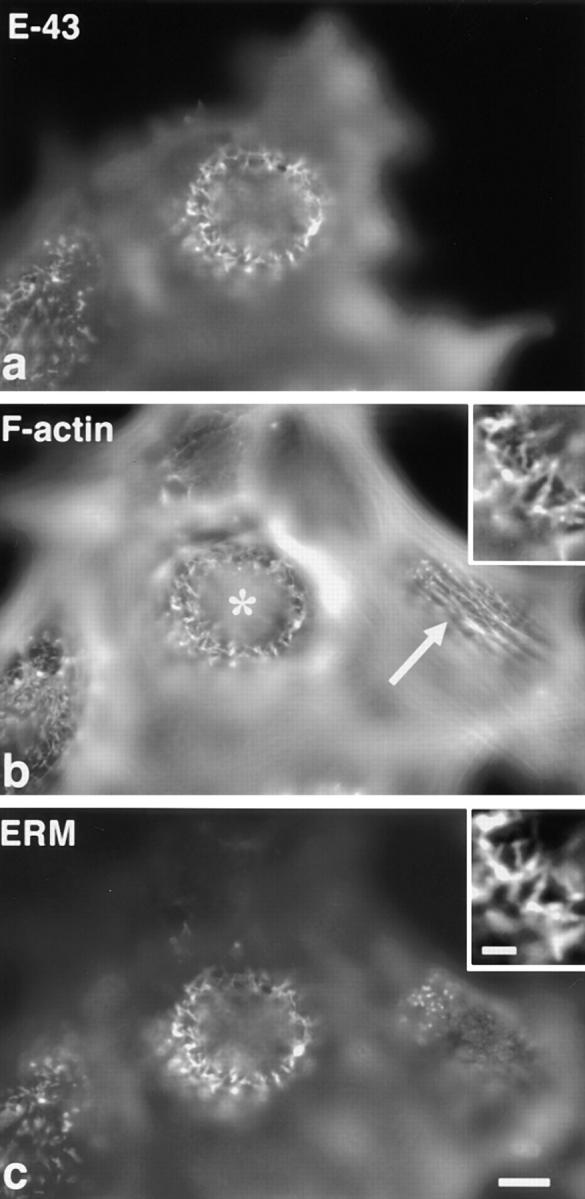
Changes in cortical actin filament organization in L fibroblasts transiently overexpressing E-43. Cells were triply stained with anti–E-cadherin mAb (E-43, a), FITC-phalloidin (F-actin, b), and anti-ERM mAb (ERM, c). Nontransfected cells (compare a with b) were characterized by dorsal stress fibers (arrow in b) and short microvilli (c), whereas in E-43–overexpressing cells long microvillar core actin bundles were induced with concomitant disruption of dorsal stress fibers (asterisk in b). The precise colocalization of F-actin and ERM proteins in elongated microvilli in E-43–expressing cells is shown at higher magnification in the insets. Cells were transfected by microinjection. Bar, a–c, 10 μm; insets, 2 μm.
Next, another fibroblastic cell line, CV-1, was used. CV-1 cells bear short microvilli and were used to examine the effects of villin on microvillar elongation (Friederich et al., 1989). When E-43 was transiently introduced into CV-1 cells, microvillar elongation was more clearly demonstrated than in L cells (Fig. 4). The length of microvilli was then quantitatively compared between nontransfected and E-43–expressing CV-1 cells using anti–ERM mAb-stained images (Fig. 5). In nontransfected cells, microvilli were mostly shorter than 1 μm (0.53 ± 0.33 μm; n = 320), whereas in E-43–expressing cells most microvilli were 3–4 μm in length (3.5 ± 1.4 μm; n = 300). These results are comparable with those in villin-induced microvilli (2.4 μm) (Friederich et al., 1989). Finally, the surface morphology of these cells was examined by immunoscanning electron microscopy (Fig. 6). To detect cells transiently expressing E-43, the extracellular domain of E-cadherin was detected with anti–E-cadherin mAb labeled with biotinylated secondary antibody followed by avidin-conjugated beads. The surface of nontransfected cells lacking bead labeling was characterized by numerous short microvilli (Fig. 6 a). In contrast, many elongated microvilli were observed on the surface of E-43–expressing cells together with scattered beads (Fig. 6 b).
Figure 4.
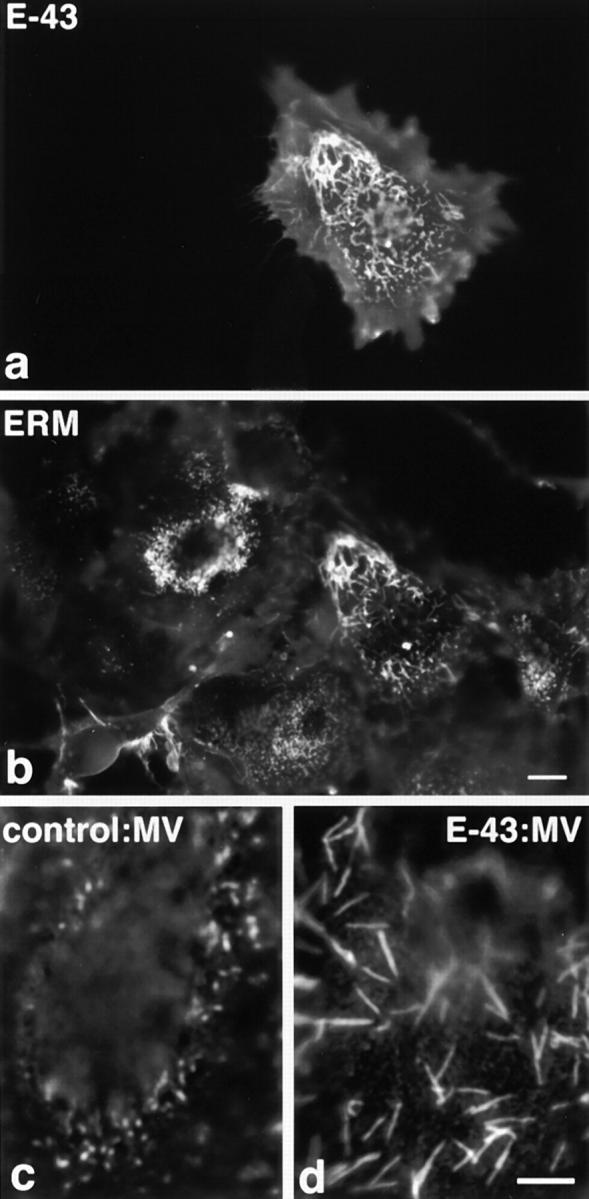
Microvillar elongation in CV-1 fibroblasts transiently overexpressing E-43. Cells were doubly stained with anti–E-cadherin mAb (E-43, a) and anti-ERM mAb (ERM, b). In E-43– expressing CV-1 cells, microvilli were significantly elongated by recruiting E-43 as well as ERM proteins as compared to surrounding nontransfected cells (a and b). E-43 overexpression did not appear to increase the density of microvilli. Microvillar elongation was more clearly demonstrated at higher magnification when nontransfected cells (c) and E-43–overexpressing cells (d) were stained with anti-ERM mAb. Cells were transfected by microinjection. Bars, a and b, 10 μm; c and d, 5 μm.
Figure 5.

Length distribution of microvilli in E-43–expressing (open box) and nontransfected (closed box) CV-1 cells. The length of each microvillus was measured from anti-ERM mAb-stained images (see Fig. 4 b). In nontransfected cells, microvilli were relatively short (0.53 ± 0.33 μm; n = 320), whereas in E-43– overexpressing cells they were significantly elongated (3.5 ± 1.4 μm; n = 300). Cells were transfected by lipofection.
Figure 6.

Scanning electron microscopic surface images of nontransfected (a) and E-43–overexpressing (b) CV-1 cells. E-43– expressing cells were identified by surface labeling by 0.1-μm beads (arrows) which recognized the extracellular domain of E-cadherin (see Materials and Methods in detail). Note the significant difference in microvillar length between a and b. Cells were transfected by microinjection. Bar, 1 μm.
Overexpression of ERMBMPs in Epithelial Cells
When E-43 was introduced into cultured MTD-1A epithelial cells, E-43 was concentrated at ERM-positive microvilli at their apical membrane surface, but did not induce apparent elongation of microvilli (Fig. 7, a and b). Instead, in these E-43–overexpressing cells, the endogenous microvillar integral membrane protein, CD44, was significantly excluded from microvilli (Fig. 7, c and d). Overexpression of E-44 also gave the same results, whereas that of E-cadherin did not affect the microvillar localization of endogenous CD44 (Fig. 7, e–h). This effect appeared to be dependent on the level of E-43 or E-44 expression. In cultured fibroblastic L cells, E-43, E-44, or E-ICAM-2 did not cause exclusion of endogenous CD44 from microvilli (data not shown).
Figure 7.
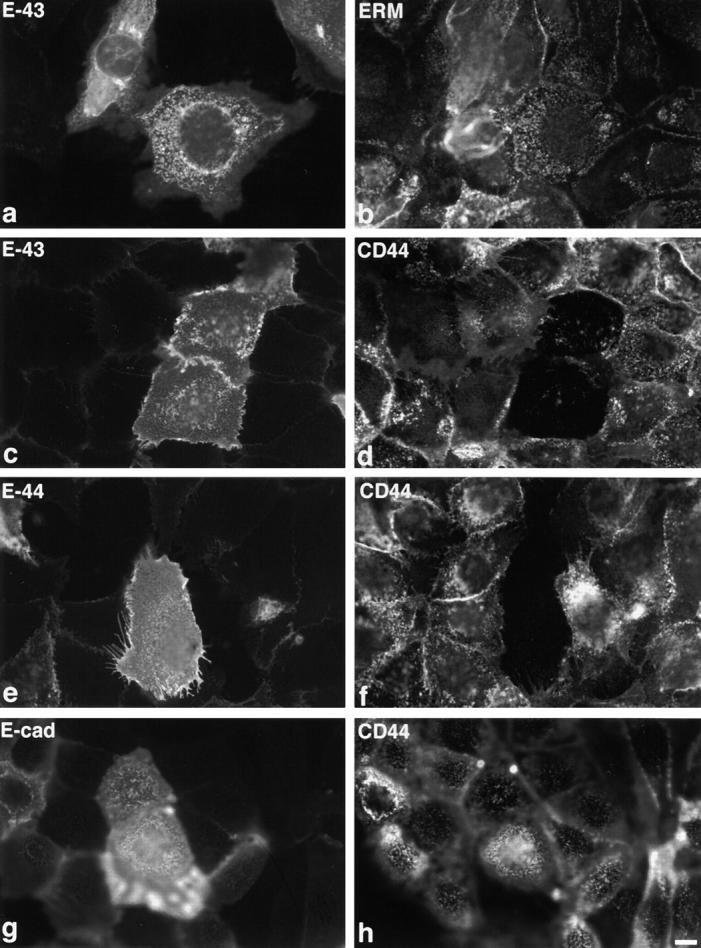
Transient overexpression of E-43 or E-44 in MTD-1A epithelial cells. Cells were doubly stained with anti–E-cadherin mAb (a) and anti-ERM mAb (b) or with anti–E-cadherin pAb (c, e, and g) and anti-CD44 mAb (d, f, and h). Overexpression of E-43 (a–d) or E-44 (e and f) did not induce microvillar elongation (a and b), but caused exclusion of CD44 from preexisting microvilli (c–f). Overexpression of E-cadherin (g and h) did not affect the distribution of CD44. Cells were transfected by microinjection. Bar, 10 μm.
Another epithelial cell line, A431, was examined. Similar to MTD-1A cells, overexpression of E-43 (Fig. 8, a and b), E-44, or E-ICAM-2 (data not shown) did not induce elongation of microvilli. In A431 cells, EGF treatment was reported to induce morphological changes in the cell surface such as microvillar elongation and ruffling with concomitant activation of ezrin (Chinkers et al., 1979; Bretscher, 1989; Franck et al., 1993; Berryman et al., 1995). Interestingly, as shown in Fig. 8, c and d, when E-43–overexpressing cells were treated with EGF, within 30 s significantly longer microvilli appeared on the cell surface as compared to surrounding nontransfected cells. Ezrin was clearly recruited into and concentrated in these overelongated microvilli. This type of EGF-dependent overelongation of microvilli was not observed in A431 cells expressing E-43/KRR:NGG that has no binding ability to ERM proteins (Fig. 8, e and f).
Figure 8.

Transient overexpression of E-43 or E-43/KRR:NGG in A431 cells. Cells were doubly stained with anti–E-cadherin pAb (a and c)/anti-ezrin mAb, M11 (b and d), or anti–E-cadherin mAb, ECCD-2 (e)/anti-ERM pAb, TK89 (f) in the absence (a and b) or presence (c–f) of EGF. In this experiment, cells were stained with antibodies after Triton X-100 treatment, resulting in intense cytoplasmic staining with anti–E-cadherin (a, c, and e). M11 did not bind to ezrin in the cytoplasm strongly (b and d). When cells were exposed to EGF for 30 s, even in nontransfected cells, short ezrin-positive microvilli appeared on the cell surface, but E-43– expressing cells elongated significantly longer ezrin-positive microvilli (d). In sharp contrast, cells expressing E-43/KRR: NGG, which lacks the binding ability to ERM proteins, elongated short ERM-positive microvilli at the similar level to the surrounding nontransfected cells (f) at 30 s after EGF treatment. Cells were transfected by microinjection. Bar, 10 μm.
ERM Proteins Phosphorylated at the COOH-terminal Threonine as Activated Forms
The difference in the effects of overexpression of ERMBMPs between fibroblastic and epithelial cells suggested that in fibroblasts ERM proteins are more efficiently activated than in epithelial cells (see Fig. 11). If this is the case, it is likely that in A431 cells EGF effectively activates ERM proteins. ERM proteins phosphorylated at the COOH-terminal threonine residue (T567, T564, and T558 in ezrin, radixin, and moesin, respectively) have been proposed to function as active forms in cross-linking ERMBMPs to actin filaments (Nakamura et al., 1995; Matsui et al., 1998; Oshiro et al., 1998; Pietromonaco et al., 1998; Shaw et al., 1998; Simons et al., 1998; Hayashi et al., 1999). We examined the effects of EGF on the threonine phosphorylation of ERM proteins in A431 cells (Fig. 9).
Figure 11.
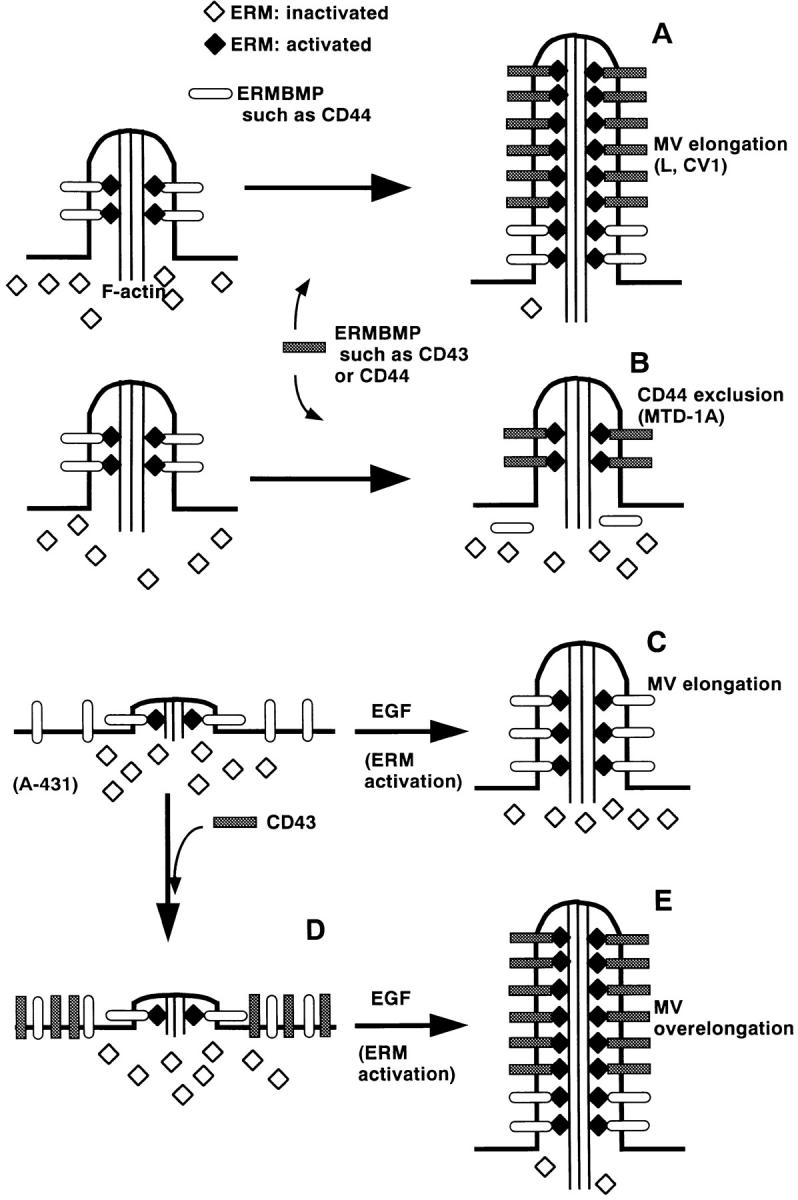
Possible involvement of ERMBMPs and activated ERM proteins in organizing microvilli. (A) Fibroblastic cells such as L cells and CV-1 cells bear short microvilli on their surface. In this model, ERM proteins were effectively activated in response to increases in amounts of ERMBMP in these cells. Overexpressed ERMBMPs such as E-43, E-44, and E-ICAM-2 then recruit these activated ERM proteins and actin filaments to elongate microvilli. (B) Epithelial cells such as MTD-1A cells bear short microvilli on their surface. The activation process of ERM proteins is postulated to be suppressed in these cells. Due to the limited amount of activated ERM proteins, overexpressed ERMBMPs do not elongate microvilli but compete out endogenous CD44 from preexisting microvilli. (C) A431 cells carry only very short microvilli in the absence of EGF. The activation process of ERM proteins is also postulated to be suppressed in these epithelial cells. EGF treatment released this suppression mechanism, resulting in an increase in the amount of activated ERM proteins, which are recruited to the cytoplasmic domain of endogenous ERMBMPs to elongate microvilli. (D) Even when E-43 is overexpressed in A431 cells in the absence of EGF, microvilli are not elongated due to the suppression of the activation process of ERM proteins. (E) When E-43 is overexpressed in A431 cells in the presence of EGF, increased amounts of activated ERM proteins are recruited to the overexpressed E-43 to cause overelongation of microvilli.
Figure 9.

Phosphorylation of the COOH-terminal threonine of ERM proteins in EGF-treated A431 cells. (A) Serum-starved A431 cells were incubated with EGF for 30 s, then immediately they were applied onto SDS-PAGE followed by immunoblotting with anti-phosphotyrosine mAb (4G10) (upper panel, P-Y), a mAb (297S) specific for CPERM (middle panel, CP-ERM), or anti-ERM pAb (TK89) (lower panel, ERM). The same nitrocellulose membrane was repeatedly probed with each antibody, confirming that ezrin and moesin were predominant in A431 cells, that ezrin but not moesin was tyrosine-phosphorylated, and that both ezrin and moesin were phosphorylated at the COOH-terminal threonine residue. Similar immunoblotting was performed also in MTD-1A cells and L cells. EGF-R, EGF receptor; E, ezrin; R, radixin; M, moesin. Bars indicate molecular masses of 200, 116, 97, 66, 45, 31, and 21 kD from the top, respectively. (B) Immunofluorescence images of serum-starved A431 cells doubly stained with mAb 297S specific for CPERM (a and c) and TK88 specific for all ERM proteins (b and d) before (a and b) and 30 s after (c and d) EGF treatment. CPERM appeared in large amounts at microvilli and cell–cell borders only in EGF-treated cells (c). Some amount of total ERM proteins appeared to be translocated from the cytoplasm to microvilli after EGF treatment (c and d). Bar, 10 μm.
As reported previously (Hunter and Cooper, 1981; Bretscher, 1989; Franck et al., 1993), immunoblotting of EGF-treated A431 cells with anti-phosphotyrosine mAb revealed that ezrin as well as EGF receptor were heavily tyrosine-phosphorylated (Fig. 9 A, upper panel). In A431 cells with or without EGF treatment, anti-ERM pAb (TK89) identified almost equal amounts of ezrin and moesin, but only a trace amount of radixin (Fig. 9 A, lower panel). Previously, we generated mAb 297S which specifically recognized CPERMs (Matsui et al., 1998). Immunoblotting with this mAb clearly revealed that the amount of CPERMs was increased in A431 cells after EGF treatment (Fig. 9 A, middle panel). Importantly, not only ezrin but also moesin were phosphorylated at their COOH-terminal threonine residues. MTD-1A cells and L cells contained CPERMs in comparable amounts to EGF-treated A431 cells (Fig. 9 A).
We then compared the behavior of CPERMs with that of total ERM proteins by immunofluorescence microscopy during the activation of ERM proteins induced by EGF treatment (Fig. 9 B). Before EGF treatment, CPERMs were hardly detected with some exceptional signals from the cell surface (Fig. 9 B, panel a). Total ERM proteins were distributed diffusely in the cytoplasm and in a punctate manner on the dorsal surface (Fig. 9 B, panel b). When cells were treated with EGF for 30 s, the staining intensity of CPERMs was increased markedly on cell surface structures including elongating microvilli but not in the cytoplasm (Fig. 9 B, panel c). In contrast, the total ERM proteins were still detected in large amounts in the cytoplasm in addition to the cell surface (Fig. 9 B, panel d). These observations favored the suggestion that CPERMs represent activated ERM proteins.
To further evaluate this suggestion experimentally, we performed site-directed mutagenesis on ezrin which was predominant among ERM proteins in A431 cells. The COOH-terminally VSVG-tagged ezrin (Ez-VSVG) was shown previously to behave similarly to endogenous ezrin (Algrain et al., 1993; Crepaldi et al., 1997). Also, in serum-starved A431 cells Ez-VSVG was codistributed with endogenous ERM proteins (Fig. 10, a and b). When the COOH-terminal threonine residue of ezrin (T567) was mutated to alanine (Ez-T/A-VSVG; nonphosphorylatable ezrin), the expressed mutant ezrin was again codistributed with endogenous ezrin in the cytoplasm as well as on the cell surface of serum-starved A431 cells (Fig. 10, c and d). In sharp contrast, when T567 was mutated to aspartic acid (Ez-T/D-VSVG), which was expected to mimic the T567-phosphorylated form of ezrin, the expressed mutant was preferably enriched at the cell surface to elongate microvilli in serum-starved A431 cells (Fig. 10 e). These elongated microvilli contained actin filaments along their length (Fig. 10, g and h), and the expressed Ez-T/D-VSVG often formed large aggregates on the cell surface or in the cytoplasm where actin filaments were recruited (arrows in Fig. 10, g and h). In contrast, when Ez-T/D-VSVG was introduced into L cells, no elongation of microvilli was detected (data not shown).
Figure 10.

Localization of site-directed mutants of ezrin in serum-starved A431 cells. VSVG-tagged ezrin (Ez-VSVG) (a and b), Ez-VSVG mutant which T567 was mutated to alanine (Ez-T/A-VSVG) (c and d), or Ez-VSVG mutant which T567 was mutated to aspartic acid (Ez-T/D-VSVG) (e–h) was transfected to A431 cells by microinjection. Cells were doubly stained with anti-VSVG mAb (P5D4) (a, c, and e)/anti-ERM pAb (TK89) (b, d, and f), or anti-VSVG mAb (P5D4) (g)/ FITC-phalloidin (h). Ez-VSVG and Ez-T/A-VSVG did not affect cell morphology and were codistributed with endogenous ERM proteins diffusely in the cytoplasm as well as on the cell surface (a–d). In contrast, Ez-T/ D-VSVG induced microvillar elongation and was localized exclusively at the cell surface structures such as microvilli (e and f). Actin filaments were recruited to Ez-T/D-VSVG– positive elongated microvilli throughout their length (inset, g and h). Occasionally, on the cell surface or intracellular vesicles overexpressed Ez-T/D-VSVG formed large aggregates that recruited actin filaments (arrows in g and h). Bar, 10 μm; insets, 2.5 μm.
Discussion
Although microvilli are ubiquitous membrane surface structures, their physiological functions and the molecular mechanism behind their organization have not been fully elucidated. It has been reported that overexpression of some cytoskeletal or lipid-binding proteins results in the elongation of microvilli (Friederich et al., 1989; Arpin et al., 1994b; Ma et al., 1997), but no information regarding the involvement of integral membrane proteins in the microvillar organization is available. In this study, we first showed that overexpression of several ERMBMPs such as E-43, E-44, and E-ICAM-2 induced the elongation of microvilli in fibroblasts such as L and CV-1 cells. This activity of these integral membrane proteins was dependent on their direct binding to ERM proteins, since the removal of the ERM-binding domains from their cytoplasmic domains resulted in loss of activity. Furthermore, as shown in Fig. 3, this ERMBMP-dependent elongation of microvilli was associated with disruption of the dorsal stress fibers, suggesting that the expressed ERMBMPs recruited not only ERM proteins but also actin filaments. Of course, the possibility cannot be excluded that unidentified cytoplasmic proteins other than ERM proteins are also involved in such integral membrane protein-induced microvillar organization in some types of cells. In human RPM-MC melanoma cells, CD44 mutants lacking the ezrin-binding domain were reported to still localize at microvilli together with ezrin (Legg and Isacke, 1998), and in some types of tissues CD44 and ERM proteins are not necessarily colocalized (Berryman et al., 1995; von Andrian et al., 1995; Nakamura and Ozawa, 1996).
Interestingly, in epithelial cells such as MTD-1A and A431 cells, these ERMBMPs did not induce the elongation of microvilli. Instead, they caused exclusion of endogenous CD44 from preexisting microvilli in MTD-1A cells. This differential behavior of ERMBMPs in epithelial and fibroblastic cells could be explained if the pool size of activated ERM proteins is larger in fibroblastic cells than in epithelial cells. However, immunoblotting did not identify any significant difference in the amount of CPERMs (which may correspond to activated ERM proteins as discussed below) between fibroblastic L cells and epithelial MTD-1A cells (Fig. 9 A). Thus, as shown schematically in Fig. 11, A and B, we postulated that in epithelial cells used in this study the ability to convert ERM proteins from inactive form to active form was suppressed as compared to fibroblastic cells. In response to increases in the amount of ERMBMPs, ERM proteins in the cytoplasm of fibroblasts would be efficiently activated with their concomitant recruitment to organize microvilli. In contrast, since in epithelial cells the activation of ERM proteins in response to increases in the amount of ERMBMPs was suppressed, the introduced ERMBMPs would compete out endogenous CD44 from the membrane-bound activated ERM proteins in preexisting microvilli. This interpretation was supported by the observations in EGF-treated A431 cells (Fig. 11, C–E). Bretscher and colleagues reported that EGF treatment increased the levels of tyrosine phosphorylation and oligomerization of ezrin in A431 cells, resulting in the elongation of microvilli and membrane ruffles by recruiting ezrin (Bretscher, 1989; Berryman et al., 1995). These findings suggest that in these cells, EGF directly or indirectly released the suppression of the ezrin (and probably ERM protein) activation process (Fig. 11 C). The present results indicated that in these EGF-treated A431 cells, in which ERM proteins could be efficiently activated in response to increases in the amount of ERMBMPs, the overexpression of E-43 caused marked elongation of microvilli (Fig. 11, D and E). Furthermore, introduction of Ez-T/D-VSVG, which mimicked activated ERM proteins, into serum-starved A431 cells but not L cells induced microvillar elongation. These findings suggest that in L cells the elongation of microvilli is regulated by the amount of ERMBMPs, whereas in serum-starved A431 cells it is regulated by the activation of ERM proteins as shown in Fig. 11. Based on these observations, we concluded that ERMBMPs such as CD43, CD44, and ICAM-2 can serve as organizing centers for microvilli in collaboration with activated ERM proteins. Of course, the role of ERMBMPs remains undetermined in the normal organization of microvilli in a variety of cells because our conclusion resulted from overexpression experiments. In support of our model, lymphocytes from patients with Wiskott-Aldrich syndrome, where CD43 is defective, were relatively devoid of microvilli (Kenney et al., 1986).
The question has naturally arisen as to the nature of the activated state of ERM proteins and how they are activated. Ezrin has been reported to be tyrosine-, serine-, and also threonine-phosphorylated in vivo. Ezrin is heavily tyrosine-phosphorylated in response to EGF in A431 and to hepatocyte growth factor in LLC-PK1 cells (Gould et al., 1986; Bretscher, 1989; Berryman et al., 1995; Crepaldi et al., 1997). However, tyrosine phosphorylation of ezrin did not alter the association of ezrin with the cytoskeleton (Gould et al., 1986; Egerton et al., 1992). Furthermore, even when two tyrosine residues in ezrin that are known to be major phosphorylation sites by EGF receptor (Krieg and Hunter, 1992) were mutated to phenylalanine, transfected LLC-PK1 cells became unresponsive to hepatocyte growth factor in terms of cell motility and tubulogenesis, but the localization of ezrin at microvilli was not affected (Crepaldi et al., 1997). These findings suggest that tyrosine phosphorylation of ezrin (and probably ERM proteins) is not required for ezrin activation as a membrane-cytoskeleton linker.
Several lines of evidence suggest that serine/threonine phosphorylation of ERM proteins is associated with their activation (Urushidani et al., 1989; Hanzel et al., 1991; Chen et al., 1995; Kondo et al., 1997; Oshiro et al., 1998; Shaw et al., 1998). Moesin was reported to be threonine-phosphorylated at its COOH terminus during thrombin activation in platelets (Nakamura et al., 1995). Phosphorylation at the COOH-terminal threonine residue was shown to affect the direct interaction between the NH2- and COOH-terminal halves of radixin in vitro (Matsui et al., 1998), suggesting that this phosphorylation keeps ERM proteins in an opened (activated) form. The results of our previous investigations and of the present study indicated that CPERMs represent the activated ERM proteins shown in Fig. 11. Immunofluorescence microscopy with a mAb specific for CPERMs showed that they were highly concentrated at microvilli of cultured fibroblasts (Hayashi et al., 1999) and also at elongated microvilli of L and CV-1 cells overexpressing ERMBMPs (data not shown). When A431 cells were treated with EGF, levels of CPERMs were shown to be increased by immunoblotting, which were concentrated in elongating microvilli on the cell surface (Fig. 9), although it remains unclear how the activation of EGF receptor tyrosine kinase resulted in the phosphorylation of COOH-terminal threonine of ERM proteins. As expected, CPERMs were also highly concentrated at overelongated microvilli in EGF-treated A431 cells overexpressing ERMBMPs (data not shown). Furthermore, transfection experiments with site-directed mutants of ezrin revealed that Ez-T/D-VSVG (which mimicked CPERMs) was preferably recruited to the plasma membranes of serum-starved A431 cells to elongate microvilli. Similar results with mutated moesin were recently reported in COS7 cells (Oshiro et al., 1998). In contrast, Ez-T/A-VSVG, a nonphosphorylatable form at the COOH- terminal threonine, was codistributed with endogenous ERM proteins at the cytoplasm as well as plasma membranes (Fig. 10). Since ERM proteins form dimers or oligomers (Berryman et al., 1995; Bretscher et al., 1995), the localization of Ez-T/A-VSVG, i.e., inactivated form of ERM protein, on plasma membranes can be explained if these mutant molecules are incorporated into oligomers of endogenous ERM proteins.
Current knowledge regarding how ERM proteins are activated, i.e., how the COOH-terminal threonine residues of ERM proteins are phosphorylated in vivo, is still limited. Rho-associated kinase and/or protein kinase C-θ are good candidates for the kinase responsible for production of CPERMs in vivo (Matsui et al., 1998; Oshiro et al., 1998; Pietromonaco et al., 1998). Although protein kinase C-θ but not Rho-associated kinase was shown in vitro to phosphorylate full-length ezrin and moesin to directly open the closed form of ERM proteins (Simons et al., 1998), further analyses would be required to determine conclusively which serine/threonine kinase by itself functions as an activator for ERM proteins in vivo. Phosphoinositides such as phosphatidylinositol 4,5-bisphosphate were also thought to be key factors in opening of the closed ERM proteins (Niggli et al., 1995; Hirao et al., 1996). Further detailed analyses of how ERM proteins are activated in response to increases in amounts of ERMBMPs in fibroblasts and how this activation process is suppressed in epithelial cells will lead to a better understanding of the molecular mechanism of cell surface morphogenesis including the formation and destruction of microvilli in general.
Acknowledgments
We thank all the members of our laboratory (Department of Cell Biology, Faculty of Medicine, Kyoto University) for helpful discussions. We would like to express our appreciation to Mr. H. Maebashi (Laboratory of Electron Microscopy, National Institute for Physiological Sciences) for his assistance with scanning electron microscopy. The anti–E-cadherin pAb, mAb, and the plasmids pBATEM2 and pβ actCAT9 were gifts from Dr. M. Takeichi. We are also grateful to Dr. M. Miyasaka (Osaka University, Osaka, Japan) for his generous gift of anti–mouse CD44 mAb, IM7.8.1.
This study was supported in part by a Grant-in-Aid for Cancer Research and Grant-in-Aid for Scientific Research (A) from the Ministry of Education, Science and Culture of Japan.
Abbreviations used in this paper
- CPERM
ERM protein phosphorylated at the COOH-terminal threonine residues
- ERM
ezrin/radixin/ moesin
- ERMBMP
ERM-binding membrane protein
- pAb
polyclonal antibody
- VSVG
vesicular stomatitis virus glycoprotein G
References
- Algrain M, Turunen O, Vaheri A, Louvard D, Arpin M. Ezrin contains cytoskeleton and membrane binding domains accounting for its proposed role as a membrane-cytoskeletal linker. J Cell Biol. 1993;120:129–140. doi: 10.1083/jcb.120.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpin M, Algrain M, Louvard D. Membrane-actin microfilament connections: an increasing diversity of players related to band 4.1. Curr Opin Cell Biol. 1994a;6:136–141. doi: 10.1016/0955-0674(94)90127-9. [DOI] [PubMed] [Google Scholar]
- Arpin M, Friederich E, Algrain M, Vernel F, Louvard D. Functional differences between L- and T-plastin isoforms. J Cell Biol. 1994b;127:1995–2008. doi: 10.1083/jcb.127.6.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryman M, Gary R, Bretscher A. Ezrin oligomers are major cytoskeletal components of placental microvilli: a proposal for their involvement in cortical morphogenesis. J Cell Biol. 1995;131:1231–1242. doi: 10.1083/jcb.131.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A. Rapid phosphorylation and reorganization of ezrin and spectrin accompany morphological changes induced in A-431 cells by epidermal growth factor. J Cell Biol. 1989;108:921–930. doi: 10.1083/jcb.108.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A. Microfilament structure and function in the cortical cytoskeleton. Annu Rev Cell Biol. 1991;7:337–374. doi: 10.1146/annurev.cb.07.110191.002005. [DOI] [PubMed] [Google Scholar]
- Bretscher A, Gary R, Berryman M. Soluble ezrin purified from placenta exists as stable monomers and elongated dimers with masked C-terminal ezrin-radixin-moesin association domains. Biochemistry. 1995;34:16830–16837. doi: 10.1021/bi00051a034. [DOI] [PubMed] [Google Scholar]
- Bretscher A, Reczek D, Berryman M. Ezrin: a protein requiring conformational activation to link microfilaments to the plasma membrane in the assembly of cell surface structures. J Cell Sci. 1997;110:3011–3018. doi: 10.1242/jcs.110.24.3011. [DOI] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M, Zhong C. Focal adhesion assembly. Trends Cell Biol. 1997;7:342–346. doi: 10.1016/S0962-8924(97)01127-6. [DOI] [PubMed] [Google Scholar]
- Chen J, Cohn JA, Mandel LJ. Dephosphorylation of ezrin as an early event in renal microvillar breakdown and anoxic injury. Proc Natl Acad Sci USA. 1995;92:7495–7499. doi: 10.1073/pnas.92.16.7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinkers M, McKanna JA, Cohen S. Rapid induction of morphological changes in human carcinoma cells A-431 by epidermal growth factor. J Cell Biol. 1979;83:260–265. doi: 10.1083/jcb.83.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J. Life at the leading edge: the formation of cell protrusions. Annu Rev Cell Biol. 1993;9:411–444. doi: 10.1146/annurev.cb.09.110193.002211. [DOI] [PubMed] [Google Scholar]
- Crepaldi T, Gautreau A, Comoglio PM, Louvard D, Arpin M. Ezrin is an effector of hepatocyte growth factor-mediated migration and morphogenesis in epithelial cells. J Cell Biol. 1997;138:423–434. doi: 10.1083/jcb.138.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earle WR. Production of malignancy in vitro. IV. The mouse fibroblast cultures and changes seen in the living cells. J Natl Cancer Inst. 1943;4:165–212. [Google Scholar]
- Egerton M, Burgess WH, Chen D, Druker BJ, Bretscher A, Samelson LE. Identification of ezrin as an 81-kDa tyrosine-phosphorylated protein in T cells. J Immunol. 1992;149:1847–1852. [PubMed] [Google Scholar]
- Enami J, Enami S, Koga M. Isolation of an insulin-responsive preadipose cell line and a mammary tumor-producing, dome-forming epithelial cell line from a mouse mammary tumor. Dev Growth Differ. 1984;26:223–234. doi: 10.1111/j.1440-169X.1984.00223.x. [DOI] [PubMed] [Google Scholar]
- Forte JG, Yao X. The membrane-recruitment-and-recycling hypothesis of gastric HCl secretion. Trends Cell Biol. 1996;6:45–48. doi: 10.1016/0962-8924(96)81009-9. [DOI] [PubMed] [Google Scholar]
- Franck Z, Gary R, Bretscher A. Moesin, like ezrin, colocalizes with actin in the cortical cytoskeleton in cultured cells, but its expression is more variable. J Cell Sci. 1993;105:219–231. doi: 10.1242/jcs.105.1.219. [DOI] [PubMed] [Google Scholar]
- Fregien N, Davidson N. Activating elements in the promotor region of the chicken β-actin gene. Gene. 1986;48:1–11. doi: 10.1016/0378-1119(86)90346-x. [DOI] [PubMed] [Google Scholar]
- Friederich E, Huet C, Arpin M, Louvard D. Villin induces microvilli growth and actin redistribution in transfected fibroblasts. Cell. 1989;59:461–475. doi: 10.1016/0092-8674(89)90030-5. [DOI] [PubMed] [Google Scholar]
- Funayama N, Nagafuchi A, Sato N, Tsukita Sa, Tsukita Sh. Radixin is a novel member of the band 4.1 superfamily. J Cell Biol. 1991;115:1039–1048. doi: 10.1083/jcb.115.4.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary R, Bretscher A. Ezrin self-association involves binding of an N-terminal domain to a normally masked C-terminal domain that includes the F-actin binding site. Mol Biol Cell. 1995;6:1061–1075. doi: 10.1091/mbc.6.8.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B. Membrane-cytoskeleton interaction. Biochim Biophys Acta. 1983;737:305–341. doi: 10.1016/0304-4157(83)90005-9. [DOI] [PubMed] [Google Scholar]
- Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, Parks WP. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–1423. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- Gould KL, Cooper JA, Bretscher A, Hunter T. The protein-tyrosine kinase substrate, p81, is homologous to a chicken microvillar core protein. J Cell Biol. 1986;102:660–669. doi: 10.1083/jcb.102.2.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg S. Signal transduction of phagocytosis. Trends Cell Biol. 1995;5:93–98. doi: 10.1016/s0962-8924(00)88957-6. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Hallett MB. Controlling the molecular motor of neutrophil chemotaxis. Bioessays. 1997;19:615–621. doi: 10.1002/bies.950190712. [DOI] [PubMed] [Google Scholar]
- Hanzel D, Reggio H, Bretscher A, Forte JG, Mangeat P. The secretion-stimulated 80K phosphoprotein of parietal cells is ezrin, and has properties of membrane cytoskeletal linker in the induced apical microvilli. EMBO (Eur Mol Biol Organ) J. 1991;10:2363–2373. doi: 10.1002/j.1460-2075.1991.tb07775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Yonemura S, Matsui T, Tsukita Sa, Tsukita Sh. Immunofluorescence detection of ezrin/radixin/moesin (ERM) proteins with their carboxyl-terminal threonine phosphorylated in cultured cells and tissues: application of a novel fixation protocol using trichloroacetic acid (TCA) as a fixative. J Cell Sci. 1999;112:1149–1158. doi: 10.1242/jcs.112.8.1149. [DOI] [PubMed] [Google Scholar]
- Heiska L, Alfthan K, Gröholm M, Vilja P, Vaheri A, Carpen O. Association of ezrin with intercellular adhesion molecule-1 and -2 (ICAM-1 and ICAM-2) J Biol Chem. 1998;273:21893–21900. doi: 10.1074/jbc.273.34.21893. [DOI] [PubMed] [Google Scholar]
- Helander TS, Carpén O, Turunen O, Kovanen PE, Vaheri A, Timonen T. ICAM-2 redistributed by ezrin as a target for killer cells. Nature. 1996;382:265–268. doi: 10.1038/382265a0. [DOI] [PubMed] [Google Scholar]
- Henry MD, Gonzalez C, Agosti, Solomon F. Molecular dissection of radixin: distinct and interdependent functions of the amino- and carboxyl-terminal domains. J Cell Biol. 1995;129:1007–1022. doi: 10.1083/jcb.129.4.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao M, Sato N, Kondo T, Yonemura S, Monden M, Sasaki T, Takai Y, Tsukita Sh, Tsukita Sa. Regulation mechanism of ERM protein/ plasma membrane association: possible involvement of phosphatidylinositol turnover and rho-dependent signaling pathway. J Cell Biol. 1996;135:37–52. doi: 10.1083/jcb.135.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitt AL, Luna E. Membrane interactions with the actin cytoskeleton. Curr Opin Cell Biol. 1994;6:120–130. doi: 10.1016/0955-0674(94)90125-2. [DOI] [PubMed] [Google Scholar]
- Hunter T, Cooper JA. Epidermal growth factor induces rapid tyrosine phosphorylation of proteins in A431 human tumor cells. Cell. 1981;24:741–752. doi: 10.1016/0092-8674(81)90100-8. [DOI] [PubMed] [Google Scholar]
- Jensen FC, Girardi AJ, Girden RV, Koprowski H. Infection of human and simian tissue cultures with rous sarcoma virus. Proc Natl Acad Sci USA. 1964;52:53–59. doi: 10.1073/pnas.52.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney D, Cairns L, Remold-O'Donnell E, Peterson J, Rosen FS, Parkman R. Morphological abnormalities in the lymphocytes of patients with the Wiskott-Aldrich syndrome. Blood. 1986;68:1329–1332. [PubMed] [Google Scholar]
- Kondo T, Takeuchi K, Doi Y, Yonemura S, Nagata S, Tsukita Sh, Tsukita Sa. ERM (ezrin/radixin/moesin)-based molecular mechanism of microvillar breakdown at an early stage of apoptosis. J Cell Biol. 1997;139:749–758. doi: 10.1083/jcb.139.3.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg J, Hunter T. Identification of the two major epidermal growth factor-induced tyrosine phosphorylation sites in the microvillar core protein ezrin. J Biol Chem. 1992;267:19258–19265. [PubMed] [Google Scholar]
- Legg JW, Isacke CM. Identification and function analysis of the ezrin-binding site in the hyaluronan receptor, CD44. Curr Biol. 1998;8:705–708. doi: 10.1016/s0960-9822(98)70277-5. [DOI] [PubMed] [Google Scholar]
- Ma AD, Brass LF, Abrams CS. Pleckstrin associates with plasma membranes and induces the formation of membrane projections: requirements for phosphorylation and the NH2-terminal PH domain. J Cell Biol. 1997;136:1071–1079. doi: 10.1083/jcb.136.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Andréoli C, Sahuquet A, Montcourrier P, Algrain M, Mangeat P. Ezrin NH2-terminal domain inhibits the cell extension activity of the COOH-terminal domain. J Cell Biol. 1995;128:1081–1093. doi: 10.1083/jcb.128.6.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita Sa, Tsukita Sh. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol. 1998;140:647–657. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Molday RS, Dreyer WJ, Rembaum A, Yen SPS. New immunolatex spheres: visual markers of antigens on lymphocytes for scanning electron microscope. J Cell Biol. 1974;64:75–88. doi: 10.1083/jcb.64.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooseker MS, Cheney RE. Unconventional myosins. Annu Rev Cell Biol. 1995;11:633–675. doi: 10.1146/annurev.cb.11.110195.003221. [DOI] [PubMed] [Google Scholar]
- Murthy A, Gonzalez-Agosti C, Cordero E, Pinney D, Candia C, Solomon F, Gusella J, Ramesh V. NHE-RF, a regulatory cofactor for Na(+)-H+ exchange, is a common interactor for merlin and ERM (MERM) proteins. J Biol Chem. 1998;273:1273–1276. doi: 10.1074/jbc.273.3.1273. [DOI] [PubMed] [Google Scholar]
- Nagafuchi A, Shirayoshi Y, Okazaki K, Yasuda K, Takeichi M. Transformation of cell adhesion properties by exogenously introduced E-cadherin cDNA. Nature. 1987;329:341–343. doi: 10.1038/329341a0. [DOI] [PubMed] [Google Scholar]
- Nakamura F, Amieva MR, Furthmayr H. Phosphorylation of threonine 558 in the carboxyl-terminal actin-binding domain of moesin by thrombin activation of human platelets. J Biol Chem. 1995;270:31377–31385. doi: 10.1074/jbc.270.52.31377. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Ozawa H. Immunolocalization of CD44 and ERM family in osteoblasts and osteoclasts in mouse tibiae. J Bone Miner Res. 1996;11:1715–1722. doi: 10.1002/jbmr.5650111115. [DOI] [PubMed] [Google Scholar]
- Niggli V, Andréoli C, Roy C, Mangeat P. Identification of a phosphatidylinositol-4,5-bisphosphate-binding domain in the N-terminal region of ezrin. FEBS Lett. 1995;376:172–176. doi: 10.1016/0014-5793(95)01270-1. [DOI] [PubMed] [Google Scholar]
- Oshiro N, Fukata Y, Kaibuchi K. Phosphorylation of moesin by rho-associated kinase (Rho-kinase) plays a crucial role in the formation of microvilli-like structures. J Biol Chem. 1998;273:34663–34666. doi: 10.1074/jbc.273.52.34663. [DOI] [PubMed] [Google Scholar]
- Pestonjamasp K, Amieva MR, Strassel CP, Bauseef WM, Furthmayr H, Luna EJ. Moesin, ezrin, and p205 are actin-binding proteins associated with neutrophil plasma membranes. Mol Biol Cell. 1995;6:247–259. doi: 10.1091/mbc.6.3.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietromonaco SF, Simons PC, Altman A, Elias L. Protein kinase C-θ phosphorylation of moesin in the actin-binding sequence. J Biol Chem. 1998;273:7594–7603. doi: 10.1074/jbc.273.13.7594. [DOI] [PubMed] [Google Scholar]
- Reczek D, Berryman M, Bretscher A. Identification of EBP-50: a PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J Cell Biol. 1997;139:169–179. doi: 10.1083/jcb.139.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Yonemura S, Obinata T, Tsukita Sa, Tsukita Sh. Radixin, a barbed end-capping actin-modulating protein, is concentrated at the cleavage furrow during cytokinesis. J Cell Biol. 1991;113:321–330. doi: 10.1083/jcb.113.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrador JM, Alonso-Lebrero JL, del Pozo MA, Furthmayr H, Schwartz-Albiez R, Calvo J, Lozano F, Sánchez-Madrid F. Moesin interacts with the cytoplasmic region of intercellular adhesion molecule-3 and is redistributed to the uropod of T lymphocytes during cell polarization. J Cell Biol. 1997;138:1409–1423. doi: 10.1083/jcb.138.6.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Henry M, Solomon F, Jacks T. Rho-A-dependent phosphorylation and relocalization of ERM proteins into apical membrane/actin protrusions in fibroblasts. Mol Biol Cell. 1998;9:403–419. doi: 10.1091/mbc.9.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayoshi Y, Nose A, Iwasaki K, Takeichi M. N-linked oligosaccharides are not involved in the function of a cell-cell binding glycoprotein E-cadherin. Cell Struct Funct. 1986;11:245–252. doi: 10.1247/csf.11.245. [DOI] [PubMed] [Google Scholar]
- Simons PC, Pietromonaco SF, Reczek D, Bretscher A, Elias L. C-terminal threonine phosphorylation activates ERM proteins to link the cell's cortical lipid bilayer to the cytoskeleton. Biochem Biophys Res Commun. 1998;253:561–565. doi: 10.1006/bbrc.1998.9823. [DOI] [PubMed] [Google Scholar]
- Stossel TP. On the crawling of animal cells. Science. 1993;260:1086–1094. doi: 10.1126/science.8493552. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Sato N, Kasahara H, Funayama N, Nagafuchi A, Yonemura S, Tsukita Sa, Tsukita Sh. Perturbation of cell adhesion and microvilli formation by antisense oligonucleotides to ERM family members. J Cell Biol. 1994;125:1371–1384. doi: 10.1083/jcb.125.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N, Hall A. Rho, Rac and Cdc43 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- Trowbridge IS, Lesley J, Schulte R, Hyman R, Trotter J. Biochemical characterization and cellular distribution of a polymorphic, murine cell-surface glycoprotein expressed on lymphoid tissues. Immunogenetics. 1982;15:299–312. doi: 10.1007/BF00364338. [DOI] [PubMed] [Google Scholar]
- Tsukita Sa, Oishi K, Sato N, Sagara J, Kawai A, Tsukita Sh. ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin-based cytoskeletons. J Cell Biol. 1994;126:391–401. doi: 10.1083/jcb.126.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita Sa, Yonemura S, Tsukita Sh. ERM proteins: head-to-tail regulation of actin-plasma membrane interaction. Trends Biochem Sci. 1997a;22:53–58. doi: 10.1016/s0968-0004(96)10071-2. [DOI] [PubMed] [Google Scholar]
- Tsukita Sa, Yonemura S, Tsukita Sh. ERM (ezrin/radixin/moesin) family: from cytoskeleton to signal transduction. Curr Opin Cell Biol. 1997b;9:70–75. doi: 10.1016/s0955-0674(97)80154-8. [DOI] [PubMed] [Google Scholar]
- Turunen O, Wahlstrom T, Vaheri A. Ezrin has a COOH-terminal actin-binding site that is conserved in the ezrin protein family. J Cell Biol. 1994;126:1445–1453. doi: 10.1083/jcb.126.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urushidani T, Hanzel DK, Forte JG. Characterization of an 80-kDa phosphoprotein involved in parietal cell stimulation. Am J Physiol. 1989;256:G1070–G1081. doi: 10.1152/ajpgi.1989.256.6.G1070. [DOI] [PubMed] [Google Scholar]
- von Andrian UH, Hasslen SR, Nelson RD, Erlandsen SL, Butcher EC. A central role for microvillus receptor presentation in leukocyte adhesion under flow. Cell. 1995;82:989–999. doi: 10.1016/0092-8674(95)90278-3. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Geiger B. Molecular interactions in cell adhesion complexes. Curr Opin Cell Biol. 1997;9:76–85. doi: 10.1016/s0955-0674(97)80155-x. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Nagafuchi A, Sato N, Tsukita Sh. Concentration of an integral membrane protein, CD43(leukosialin, sialophorin), in the cleavage furrow through the interaction of its cytoplasmic domain with actin-based cytoskeletons. J Cell Biol. 1993;120:437–449. doi: 10.1083/jcb.120.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S, Itho M, Nagafuchi A, Tsukita Sh. Cell-to-cell adherens junction formation and actin filament organization: similarities and differences between non-polarized fibroblasts and polarized epithelial cells. J Cell Sci. 1995;108:127–142. doi: 10.1242/jcs.108.1.127. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Hirao M, Doi Y, Takahashi N, Kondo T, Tsukita Sa, Tsukita Sh. Ezrin/radixin/moesin (ERM) proteins bind to a positively charged amino acid cluster in the juxta-membrane cytoplasmic domain of CD44, CD43, and ICAM-2. J Cell Biol. 1998;140:885–895. doi: 10.1083/jcb.140.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CHC, Oh S, Zizak M, Steplock D, Tsao S, Tse C-M, Weinman EJ, Donowitz M. c-AMP-mediated inhibition of the epithelial brush border Na+/H+exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci USA. 1997;94:3010–3015. doi: 10.1073/pnas.94.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]