

Abstract
Many cells express more than one integrin receptor for extracellular matrix, and in vivo these receptors may be simultaneously engaged. Ligation of one integrin may influence the behavior of others on the cell, a phenomenon we have called integrin crosstalk. Ligation of the integrin αvβ3 inhibits both phagocytosis and migration mediated by α5β1 on the same cell, and the β3 cytoplasmic tail is necessary and sufficient for this regulation of α5β1. Ligation of α5β1 activates the calcium- and calmodulin-dependent protein kinase II (CamKII). This activation is required for α5β1-mediated phagocytosis and migration. Simultaneous ligation of αvβ3 or expression of a chimeric molecule with a free β3 cytoplasmic tail prevents α5β1-mediated activation of CamKII. Expression of a constitutively active CamKII restores α5β1 functions blocked by αvβ3-initiated integrin crosstalk. Thus, αvβ3 inhibition of α5β1 activation of CamKII is required for its role in integrin crosstalk. Structure-function analysis of the β3 cytoplasmic tail demonstrates a requirement for Ser752 in β3-mediated suppression of CamKII activation, while crosstalk is independent of Tyr747 and Tyr759, implicating Ser752, but not β3 tyrosine phosphorylation in initiation of the αvβ3 signal for integrin crosstalk.
Keywords: integrin, vitronectin, kinase, cross- talk, signaling
Dynamic interaction of cells with the complex protein mixtures found in the extracellular matrix occurs during many biologic and pathologic processes including development, wound healing, hemostasis, metastasis, inflammation, and thrombosis (13, 18). Most cells express multiple integrin receptors capable of interaction with the numerous ligands found in complex tissues. The simultaneous ligation of multiple integrins mandates coordination of the resulting signals. The coordination of integrin signaling into a hierarchy with a net effect on cell behavior has been called integrin crosstalk (2, 3, 17).
Numerous examples of integrin crosstalk have been reported. The common theme of these reports lies in the regulation of the function of one integrin (the target) as a result of coligation of a second integrin (the transducer) on the same cell. Examples of integrin crosstalk have been demonstrated in numerous primary cell types and cell lines including macrophages (2), T cells (17, 23), smooth muscle cells (1), neutrophils (10), monocytes (15), umbilical vein endothelial cells (20), malignant astrocytomas (16), CHO cells (7, 9), K562 cells (2, 3), and embryonic kidney 293 cells (20). Crosstalk may be initiated by transducing integrins belonging to the β1 (11, 15, 16, 22), β2 (17, 23), or β3 (1–3, 7, 9, 10, 20) family with targets in any of these families as well. Integrin functions affected by crosstalk include phagocytosis (2, 3, 10), soluble ligand binding (7, 15), adhesion (9, 17, 23), migration (1, 17, 20), gene expression (11), and receptor-mediated endocytosis (16).
It is important to note that all reported cases of integrin crosstalk are unidirectional, that is, ligation of the target integrin does not affect the transducer integrin. In many cases, the transducing integrin is much less highly expressed than the target integrin (2, 3). This suggests a hypothesis that the receptor pairs involved in crosstalk are not simply competing for interaction with a signaling molecule, but rather that ligation of the transducing integrin initiates a unidirectional signaling cascade which affects the function of the target integrin. The molecular mechanisms of integrin crosstalk remain undetermined. With a single exception (20), crosstalk signals from the transducing integrin require the cytoplasmic tail of the β-subunit, and where it has been examined, the β-subunit cytoplasmic tail has been sufficient for initiation of signaling (3).
We have previously described integrin crosstalk between αvβ3 and α5β1 in macrophages and in a K562 cell transfection model of macrophage integrins (2, 3). We have shown that ligation of αvβ3 inhibits α5β1-mediated phagocytosis, which requires the high affinity state of the integrin, without affecting α5β1-mediated adhesion, which is independent of the high affinity state of the integrin. The cytoplasmic tail of β3 is necessary and sufficient for this crosstalk. αvβ3-mediated inhibition of α5β1 phagocytosis occurs at a step subsequent to α5β1 binding of ligand and is reversed by H7, a pharmacologic inhibitor of serine/ threonine kinases.
In this report, we define a molecular mechanism required for αvβ3-to-α5β1 crosstalk. Ligation of α5β1 enhances the activity of the calcium/calmodulin-dependent protein kinase II (CamKII)1. This increase in CamKII activity is required for α5β1-dependent migration as well as α5β1-dependent phagocytosis. Simultaneous ligation of αvβ3 inhibits α5β1 activation of CamKII activity, thus blocking α5β1 migration and phagocytosis. Mutational analysis of the β3 cytoplasmic tail demonstrates that Ser752 is required for both αvβ3-initiated inhibitory crosstalk to α5β1 and αvβ3 suppression of CamKII activity, while tyrosine phosphorylation of the β3 cytoplasmic tail has no effect on this activity. These results describe a potential molecular pathway for integrin crosstalk that involves integrin regulation of CamKII activity.
Materials and Methods
Cells and β3 cDNA Mutation
Human peripheral blood monocyte-derived macrophages were prepared as previously described (2). The human erythroleukemic cell line K562 transfected with cDNA encoding αvβ3 (Kαvβ3), αvβ3 in which the tyrosine residue at position 747 or 759 was mutated to phenylalanine (Kαvβ3Y747F and Kαvβ3Y759F, respectively) and Tacβ3 (chimera of the extracellular and transmembrane domain of the IL2 receptor α-chain [Tac subunit] and the cytoplasmic tail of β3, KTacβ3) were derived and maintained as described (2, 3). In addition K562 cells were similarly transfected with cDNA encoding Tacβ3 in which the serine residue at position 752 was replaced with proline (KTacβ3S-P), cysteine (KTacβ3S-C), alanine (KTacβ3S-A), or glutamic acid (KTacβ3S-E). Expression of all Tacβ3S-X chimeras was equivalent to Tacβ3 (3) as determined by flow cytometry as described (2; see Table I). For construction of Tacβ3S-X, the HindIII and XhoI fragment of pTacβ3 encoding the CT of β3 (3) was ligated into HindIII-XhoI–digested pBluescript (Stratagene), creating pBSKSPB3TAIL. This contruct was subjected to PCR using a 5′ T7 oligonucleotide (Stratagene) with the 3′ oligonucleotide (5′-CCCCCCTCGAGTTAAGTGCCCCGGTACGTGATATTGGTGAAGGT-XXX-CGTGGC-3′) where XXX is AGG for S752P, ACA for S752C, GCT for S752A, and TTC for S752E. The resulting products were digested with HindIII-XhoI and ligated into pTacβ3 digested with HindIII-XhoI, creating pTacβ3S-P, pTacβ3S-C, pTacβ3S-A, and pTacβ3S-E, respectively.
Table I.
Integrin Expression in Transfected K562 Cells
| P5D2 (β1) | AP3 (β3) | 4E3 (Tac) | mAb16 (α5) | IC12 (αv) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| K562 | 9.83 | 0.37 | 0.42 | 13.2 | 0.22 | |||||
| Kαvβ3 | 10.1 | 29.2 | 0.44 | 13.0 | 31.4 | |||||
| Kαvβ3Y747F | 9.99 | 29.7 | 0.44 | 12.8 | 30.9 | |||||
| Kαvβ3Y759F | 9.87 | 30.0 | 0.37 | 13.1 | 29.7 | |||||
| Kαvβ3S752A | 10.7 | 29.8 | 0.44 | 12.6 | 29.9 | |||||
| KTacβ3 | 11.1 | 0.45 | 8.77 | 13.0 | 0.33 | |||||
| KTacβ3S-P | 10.5 | 0.57 | 9.32 | 13.3 | 0.34 | |||||
| KTacβ3S-A | 9.89 | 0.49 | 8.91 | 12.5 | 0.26 | |||||
| KTacβ3S-E | 11.0 | 0.57 | 9.74 | 12.8 | 0.29 | |||||
| KTacβ3S-C | 9.87 | 0.46 | 11.2 | 13.2 | 0.41 | |||||
| KTacβ5 | 10.3 | 0.67 | 9.88 | 13.1 | 0.31 |
K562 cells also were transfected with full-length αvβ3 in which the serine at position 752 of β3 was mutated to alanine as described for the mutation of β3 tyrosine residues (4). In brief, nested PCR was performed on pBLY100 using the overlapping oligonucleotides 5′-GAGGCCACGCCTACCTTCACCAATATCACG-3′ and 5′-CTCCGGTGCGGATGGAAGTGGTTATAGTGC-3′ encoding the S-A mutation with oligonucleotides in the mutation cassette (4). After the nested PCR reaction, the wild-type β3 CT was replaced with the S-A mutant CT by NdeI-NheI restriction. Transfection, selection, and fluorescent cell sorting for expression levels of αvβ3S752A equivalent to wild-type αvβ3 was as described previously, resulting in Kαvβ3S752A (Table I). Modified cDNAs were verified by dideoxy nucleotide sequencing.
Phagocytosis, Adhesion, and Migration
Phagocytosis assays were performed as described (2) by flow cytometry using either FITC-FN– or FITC-mAb16 (anti-α5)–coated 3.0-μm beads. Data are presented as a Phagocytic Index, the number of beads internalized per 100 cells.
Chemotaxis assays were performed in modified Boyden chambers (Neuroprobe) using 14.0-μM polycarbonate filters as described (19). Vitronectin (VN), fibronectin (FN), and BSA were added to basal chambers at 5 μg/ml and mAb at 10 μg/ml were added to apical chambers coincident with cells. Cells in Iscove's Modified Eagle's Medium (IMDM) adjusted to 1 mM Ca2+ and 1 mM Mg2+ with 0.5% human serum albumin and 2.0 mM Mn2+Cl were incubated for 4 h at 37°C in a humidified 5% CO2 atmosphere for migration. Migration was quantitated by counting the number of cells per high power field on the underside of the filter after Giemsa staining.
Adhesion assays were performed as described in FN-coated (10 μg/ml) microtiter wells (2). Data are presented as the percent of added cells adherent after 1 h at 37°C.
CamKII Activity Assay
Transfected K562 cells or monocyte-derived macrophages were stimulated as described in the text, washed once by centrifugation in ice-cold IMDM and suspended in ice-cold homgenization buffer containing Hepes (50 mM), EDTA (4 mM), EGTA (2 mM), sucrose (0.25 M), dithiothreitol (1 mM), phenylmethylsulfonyl fluoride (0.2 mM), Na3VO4 (2.0 mM), NaF (5.0 mM), phenyl-arsine-oxide (10.0 mM), and leupeptin (10 μg/ml), pH 7.5. Suspended cells were sonicated on ice and assayed for CamKII activity against the synthetic substrate autocamtide II (KKALRRQETVDAL) (21). An aliquot of cell extracts was used for protein determination by BCA. Parallel aliquots were assayed for CamKII activity in a 25-μl reaction mixture containing Hepes (50 mM), magnesium acetate (10 mM), Na3VO4 (2.0 mM), NaF (5.0 mM), phenyl-arsine-oxide (10.0 mM), CaCl2 (1 mM), calmodulin (0.1 μM; Sigma Chemical Co.), autocamtide II (20 μM), and γ-[32P]ATP (0.1 mM, 3,000 cpm/pmol). The reaction was initiated by ATP addition and terminated by addition of trichloroacetic acid to a final concentration of 10%. The reaction mixture was centrifuged through phosphocellulose separation units (Pierce) and washed as described (26). CamKII activation results from phosphorylation events that result in kinase activity which is no longer dependent upon exogenous calcium or calmodulin. CamKII activity in cellular extracts was measured by quantitating the incorporation of radioactive phosphate into a synthetic CamKII substrate (autocamtide-2) in the presence (calcium/calmodulin-independent + calcium/calmodulin-dependent activity) or absence (calcium/calmodulin-independent activity) of calcium and calmodulin. The activation of CamKII (autonomous activity) is expressed as a direct percentage of the total cellular CamKII activity (1) in which:
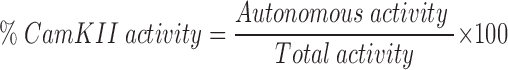 |
where autonomous activity equals the CamKII activity without calcium or calmodulin and total activity equals CamKII activity with calcium or calmodulin.
CamKII expression levels in transfected cell lines was assessed by immunoprecipitation as previously described, followed by Western blot analysis (1).
Infection of K562 Cells with Adenovirus Encoding Constitutively Active CamKII
Kαvβ3, KTacβ3, and KTacβ5 were infected with a replication defective adenovirus in which the E1 region was replaced with the CMV early promoter and the cDNA for a constitutively active CamKII (AdCMV.CKIID3) or β-galactosidase (AdCMV.gal) and viral stocks propagated and titered as described (1). Transfected K562 cells at 5 × 106/ml in IMDM were infected with recombinant adenovirus at a multiplicity of infection of 100 for 1 h followed by the addition of normal growth medium to dilute cells to a concentration of 0.5 × 106/ml. After 4–6 h, cells were harvested for analysis of CamKII activity or functional assay as described in Results. Viability of all infected cell types exceeded 85% at the initiation, and 70% at the conclusion of experimental time courses.
Proteins and Antibodies
FN was purified by gelatin affinity and VN by heparin affinity as previously described (2, 3). Monoclonal antibodies 7G2 (anti–human β3), W6/ 32 (anti–human HLA), IC12 (anti–human αv), AP3 (anti–human β3), P1F6 (anti–human β5), 4E3 (anti-IL2Rα, gp55, TAC), and mAb16 (anti– human α5) have been previously described and were used in excess at 5.0 μg/ml unless otherwise indicated (2, 3).
Reagents
The kinase inhibitors H7 (50 nM), KN62 (2.5 μM), KN04 (5.0 μM), KT5926 (20 nM), and ML-9 (2 μM) were included in some assays where indicated and all were from LC Laboratories (Woburn, MA). All other reagents were from Sigma Chemical Co. unless otherwise indicated.
Data Presentation
Data are presented as the mean ± SEM from at least three replicates for all studies. Significance was determined by analysis of variance followed by Duncan's comparison testing. A minimum confidence interval of 95% was employed for all studies.
Results
αvβ3 Crosstalk Regulates α5β1-mediated Migration
We have previously described a phenomenon, termed integrin crosstalk, in which ligation of αvβ3 prevents α5β1-mediated phagocytosis in macrophages and in K562 cells expressing transfected αvβ3. To determine if integrin crosstalk regulated α5β1 functions other than phagocytosis, we evaluated the effects of αvβ3 ligation on the migration of K562 cells on the α5β1 ligand FN. K562 cells did not migrate specifically to FN in IMDM containing 1 mM Ca2+ and 1 mM Mg2+. However, addition of 2 mM Mn2+ or the α5β1 conformation-stabilizing mAbs 8A2 or A1A5 at 5.0 μg/ml greatly enhanced the FN-specific migration of these cells, consistent with a requirement for high affinity α5β1 in migration (data not shown). As shown in Fig. 1 A, the migration of untransfected K562 to FN in the presence of 2 mM Mn2+ was enhanced sixfold over migration to the nonspecific protein casein; this migration was completely inhibited by mAb to α5β1 (data not shown). We also examined αvβ3-mediated migration in K562 expressing this transfected integrin in addition to the endogenous α5β1. Kαvβ3 migrated in response to VN (Fig. 1 A); migration response to VN was inhibited by mAb to αv or β3 (data not shown). However, migration of Kαvβ3 to FN was severely impaired compared with untransfected or mock transfected K562 (Fig. 1 A). Migration of Kαvβ3 to FN was restored by the addition of the ser/thr kinase inhibitor H7 (50 nM), while addition of H7 had no effect on Kαvβ3 migration to VN (data not shown). Restored migration of Kαvβ3 to FN in the presence of H7 was completely inhibited by mAb to β1 (data not shown). These results completely parallel the previously described αvβ3-mediated crosstalk which inhibits α5β1-mediated phagocytosis (3) and support the hypothesis that the coligation of αvβ3 by FN regulates α5β1-mediated K562 cell migration to FN because this function, like phagocytosis, requires a high affinity form of α5β1.
Figure 1.


αvβ3 crosstalk regulates α5β1-mediated migration. Untransfected K562 cells, Kαvβ3, KTacβ3, and KTacβ5 were permitted to migrate to vitronectin (VN), fibronectin (Fn) or casein through 14.0-μM pores in a modified Boyden chamber apparatus as described in Materials and Methods (A). For structure-function analysis of integrin crosstalk, untransfected K562 cells and K562 cells expressing transfected wild-type αvβ3 (Kαvβ3) or αvβ3 bearing single amino acid mutations Tyr747-Phe (Kαvβ3Y747F), Tyr759-Phe (Kαvβ3Y759F) or Ser752-Ala (Kαvβ3S752A) were assayed for migration to Fn, VN, or casein as described in Materials and Methods (B). Migration was quantitated manually by counting cells adherent to the basal side of the filter and reported as the number of cells per well. Shown are mean ± SEM of four determinations with no fewer than eight replicates.
To demonstrate definitively that αvβ3 regulation of α5β1-mediated migration was another example of integrin crosstalk, we examined migration to FN in KTacβ3 and KTacβ5, K562 cells expressing chimeric molecules comprised of the extracellular domain of the IL2 receptor and the cytoplasmic tail domain of the β3 or β5 integrin, respectively. Expression of Tacβ3, but not Tacβ5, leads to constitutive inhibition of α5β1-mediated phagocytosis in K562 cells (3; see Fig. 7 B). Expression of Tacβ3, but not Tacβ5 (Fig. 1 A) or Tac lacking a cytoplasmic tail (KTacNT, Fig. 2 A), completely inhibited α5β1-mediated migration to FN. The constitutive inhibition of migration to FN in KTacβ3 was reversed by the addition of 50 nM H7 (Fig. 2 A). These studies demonstrate that α5β1-mediated migration and α5β1-mediated phagocytosis are similarly regulated by αvβ3 or the isolated β3 CT and that this regulation is dependent upon a ser/thr kinase regulated by H7. These data suggest that both α5β1-mediated migration and α5β1-mediated phagocytosis are regulated by αvβ3-initiated crosstalk.
Figure 2.
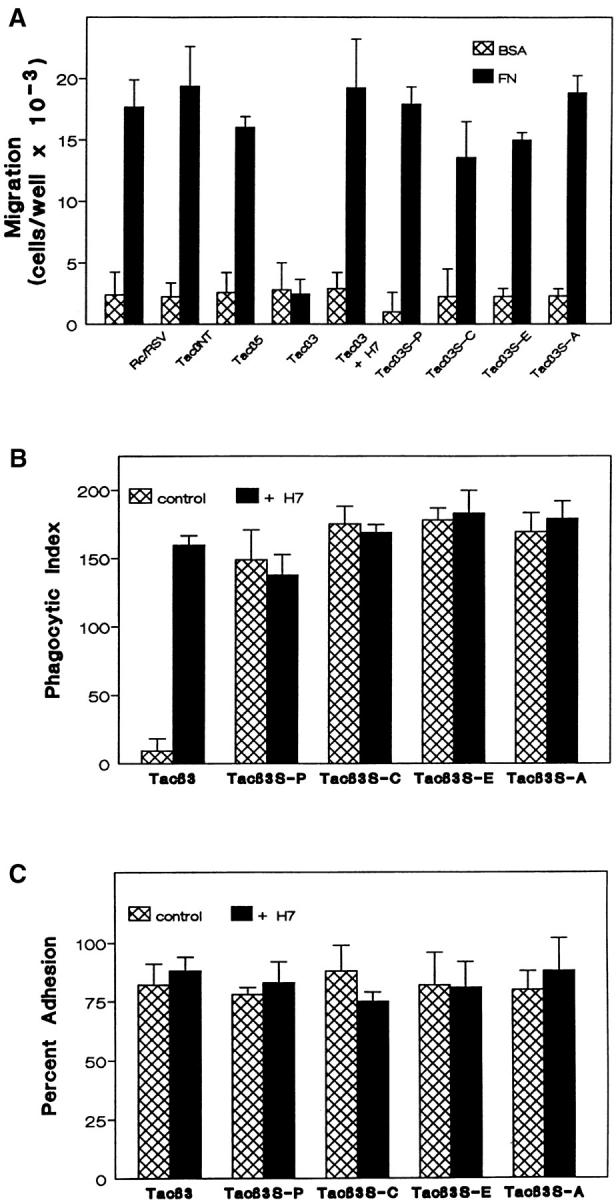
Role of β3 Ser752 in αvβ3 crosstalk. K562 cells transfected with vector alone (Rc/RSV) or a chimeric molecule composed of the extracellular and transmembrane domains of the IL2 receptor (Tac) and the cytoplasmic tail of integrin subunits; no tail (TacNT), β3 (Tacβ3), β5 (Tacβ5), or β3 in which Ser752 had been mutated to Pro (Tacβ3S-P), Cys (Tacβ3S-C), Ala (Tacβ3S-A), or Glu (Tacβ3S-E) were evaluated for their ability to migrate to FN (A), phagocytose FN opsonized beads (B), and adhere to FN-coated tissue culture plastic (C) as described in Materials and Methods. Parallel samples were treated with 50 nM H7 as indicated. Shown are mean ± SEM of no fewer than three determinations performed in triplicate.
β3 Ser752 Is Required for β3 Crosstalk
We have previously demonstrated that expression of the isolated β3 cytoplasmic tail is sufficient for initiation of αvβ3 crosstalk (Fig. 1 A and reference 12). To further delineate the required sequence elements of this unique regulatory pathway, we introduced point mutations in the β3 cytoplasmic tail and analyzed their effects upon αvβ3-initiated crosstalk to α5β1-mediated migration.
In a spontaneously occurring Glanzmann's Thrombasthenia mutation, the serine residue at position 752 of the β3 CT is mutated to proline (6). This mutation results in loss of platelet β3 function and a severe bleeding disorder. In vitro study has shown that Ser752 of the β3 CT is required for the conformational change associated with elevated affinity of β3 for ligand (8). To test whether Ser752 also is required for integrin crosstalk, we expressed an αvβ3 receptor in K562 cells in which Ser752 of β3 was mutated to Ala (Fig. 1 B). While the ligation of wild-type αvβ3 blocked α5β1-mediated migration on FN (Fig. 1 B), the S752A mutant migrated as well as the untransfected cells. In addition, the S752A mutant migrated as well as wild-type αvβ3 on VN (Fig. 1 B), consistent with reports that this mutation does not affect ligand binding by β3 integrins (8). This demonstrates that failure of the S752A mutant to initiate crosstalk did not result from an inability to recognize ligand.
Recently, a tyrosine in the β3 cytoplasmic tail, Tyr747, has been implicated in activation-dependent αvβ3 adhesion to VN (4). In contrast to the S752A mutation, Y747F had no effect on αvβ3-initiated integrin crosstalk (Fig. 1 B). Consistent with the previous report of a requirement for this tyrosine in firm adhesion, the Y747F mutation did abolish migration of Kαvβ3Y747F to VN (Fig. 1 B). Mutation of Tyr759 to Phe (Y759F) did not affect either crosstalk or the migration function of αvβ3. These data demonstrate that the crosstalk signaling and adhesive functions of αvβ3 have distinct and independent sequence requirements in the β3 cytoplasmic tail.
To evaluate further the requirement for β3 S752 in integrin crosstalk, additional mutations at that position were made in the consititutively inhibitory Tacβ3 construct. Mutation of Ser752 to Glu, Pro, or Cys as well as Ala abolished the inhibitory activity of Tacβ3 on α5β1-dependent migration (Fig. 2 A) and α5β1-dependent phagocytosis (Fig. 2 B). Like the wild-type β3 cytoplasmic tail, none of the mutants affected K562 binding to FN-coated surfaces, a function that does not require the high affinity state of α5β1 (Fig. 2 C). The addition of H7 reversed the Tacβ3 inhibition of α5β1-mediated migration and phagocytosis (Fig. 2, A and B).
α5β1 and αvβ3 Differentially Regulate CamKII
αvβ3 ligation inhibits the α5β1 high affinity functions of phagocytosis and migration, without effect upon α5β1-mediated adhesion. Alterations in α5β1 affinity can be regulated by calcineurin, a calcium/calmodulin-dependent phosphatase and CamKII (calcium/calmodulin-dependent protein kinase II; reference 1). Recently, inhibition of CamKII activity by αvβ3 ligation in smooth muscle cells was reported (1). Therefore, we evaluated αvβ3 regulation of CamKII as a potential mediator of αvβ3-initiated crosstalk.
CamKII activity was measured in human monocyte-derived macrophages in the presence and absence of an α5β1-specific phagocytosis target (mAb-16–coated latex beads) (3). Ligation of macrophage α5β1 with mAb-16 beads enhanced CamKII activity twofold, while ligation with a control target (W6/32 beads) had no effect (Fig. 3 A). Both basal and stimulated CamKII activities were decreased by the CamKII inhibitor KN62, but not the structurally related, but non-inhibitory KN04. Ligation of αvβ3 with soluble mAb 7G2 prevented the rise in CamKII activity induced by mAb-16 beads (Fig. 3 A). No additional decrease in CamKII activity was detected when KN62 and 7G2 were combined. Thus, β3 ligation prevented the α5β1-induced rise in CamKII activity.
Figure 3.

Regulation of CamKII activity by α5β1 and αvβ3. Human monocyte-derived macrophages (A) or K562 cells expressing transfected αvβ3 (Kαvβ3, B) or integrin β5 (KTacβ5) or β3 (KTacβ3) cytoplasmic tail chimeras (C) were assayed for activity of the calcium- and calmodulin-dependent protein kinase II against a synthetic substrate as described in Materials and Methods. Cells were either left unstimulated (no beads) or presented with control phagocytosis targets (W6/32 beads) or with an α5β1-specific phagocytosis target (mAb-16 beads) in the presence and absence of KN62 (2.5 μM), KN04 (5.0 μM) or 5.0 μg/ml mAb 7G2 either individually or in combinations as indicated. D shows the inhibition of CamKII activity in Kαvβ3 after α5β1 ligation with mAb-16 beads by soluble mAb 7G2 (5.0 μg/ml), 7G2 Fab′ (3.8 μg/ml), and 2.0 mM GRGDSP peptide and the lack of inhibition by mAb P1F6 or 2 mM GRGESP. Regulated CamKII activity is presented as the percent of total activity as described in Materials and Methods. Shown are mean ± SEM for no fewer than three determinations in each group of all panels.
To explore further the hypothesis that CamKII mediates αvβ3 regulation of α5β1, we evaluated the regulation of CamKII in Kαvβ3. Binding of mAb-16 beads to Kαvβ3, and to vector-transfected K562 (data not shown), resulted in an increase in CamKII activity (Fig. 3 B) that was not seen when Kαvβ3 were incubated with W6/32 beads that bound to the cells equivalently. As in macrophages, the α5β1-mediated rise in CamKII activity was prevented by ligation of αvβ3 with soluble mAb 7G2 (Fig. 3 B) and by 7G2 Fab fragments or Arg-Gly-Asp peptide (Fig. 3 D). As seen in macrophages, inhibition of the α5β1-induced increase in CamKII activity by αvβ3 ligation was blocked by KN62, but not KN04.
Previously we have demonstrated that the cytoplasmic tail of β3 is both necessary and sufficient for αvβ3 inhibitory crosstalk to α5β1 (reference 3 and Fig. 2, A and B). In the presence of mAb-16 beads, expression of Tacβ3, but not Tacβ5, prevented the α5β1-mediated rise in CamKII activity (Fig. 3 C). These results indicate that α5β1 and αvβ3 differentially regulate CamKII activity in macrophages and K562 cells.
To determine if the failure of the Kαvβ3S752A to initiate crosstalk was related to an inability to regulate CamKII, we evaluated CamKII activity after mAb-16 bead binding in K562 cells transfected with wild-type αvβ3 and the S752A and Y747F mutants. While ligation of αvβ3 with the β3-specific mAb 7G2 suppressed mAb-16 bead-induced activation of CamKII, mutation of Ser752 of β3 prevented the suppression of CamKII activity seen upon αvβ3 ligation (Fig. 4 A). However, mutation of Tyr747 or Tyr759 (data not shown) did not affect αvβ3 regulation of CamKII. Thus, Ser752 is required for both αvβ3 inhibitory crosstalk to α5β1 (Fig. 2) and αvβ3 regulation of CamKII (Fig. 4).
Figure 4.


Role of β3 Ser752 in αvβ3 regulation of CamKII. Untransfected K562 cells or K562 cells expressing transfected wild-type αvβ3 (Kαvβ3) or αvβ3 in which Tyr747 was mutated to Phe (Kαvβ3Y747F) or in which Ser752 was mutated to Ala (Kαvβ3S752A) were assayed for CamKII activity (A) as described in Materials and Methods, after α5β1 ligation with mAb-16 beads in the presence or absence of the αvβ3 ligand 7G2 (5.0 μg/ml). Shown are mean ± SEM of three determinations with no fewer than three replicates. Also as described in Materials and Methods, total cellular CamKII (see arrow) from transfected K562 cell populations used in this study was recovered by immunoprecipitation and revealed by Western blotting (B). For untransfected K562 cells, cell lysates were cleared by immunoprecipitation with anti-CamKII twice (cleared 2×) or once (cleared 1×), before analysis of the final immunoprecipitation to ensure that recovery of protein was complete. Shown is a representative study.
To determine the effect of αvβ3 and mutant β3 on expression of CamKII, immunoprecipitates of CamKII were analyzed by Western blot with CamKII-specific Ab. As shown in Fig. 4 B, cellular expression of CamKII (see arrow) was unchanged by the expression of αvβ3 and mutants in transfected K562 cells.
Role of CamKII in αvβ3 Crosstalk to α5β1
Suppression of the α5β1-dependent increase in CamKII activity by αvβ3 ligation or by Tacβ3 expression suggested that CamKII regulation could have a role in αvβ3 crosstalk to α5β1. To determine the role of CamKII in αvβ3 crosstalk to α5β1, Kαvβ3 cells were incubated with the α5β1 phagocytosis target, mAb-16 beads, or control target, P1F6 (anti-αvβ5) beads. Phagocytosis was measured in the presence and absence of 7G2 to ligate αvβ3, the CamKII inhibitor KN62, or control KN04. As reported previously, phagocytosis via α5β1 was inhibited upon αvβ3 ligation with mAb 7G2 (2, 3). α5β1 phagocytosis also was inhibited by KN62 (Fig. 5 A), but not KN04. Combining 7G2 and KN62 resulted in no further decrease in α5β1 phagocytosis. Under all conditions, there was no significant internalization of P1F6 beads.
Figure 5.
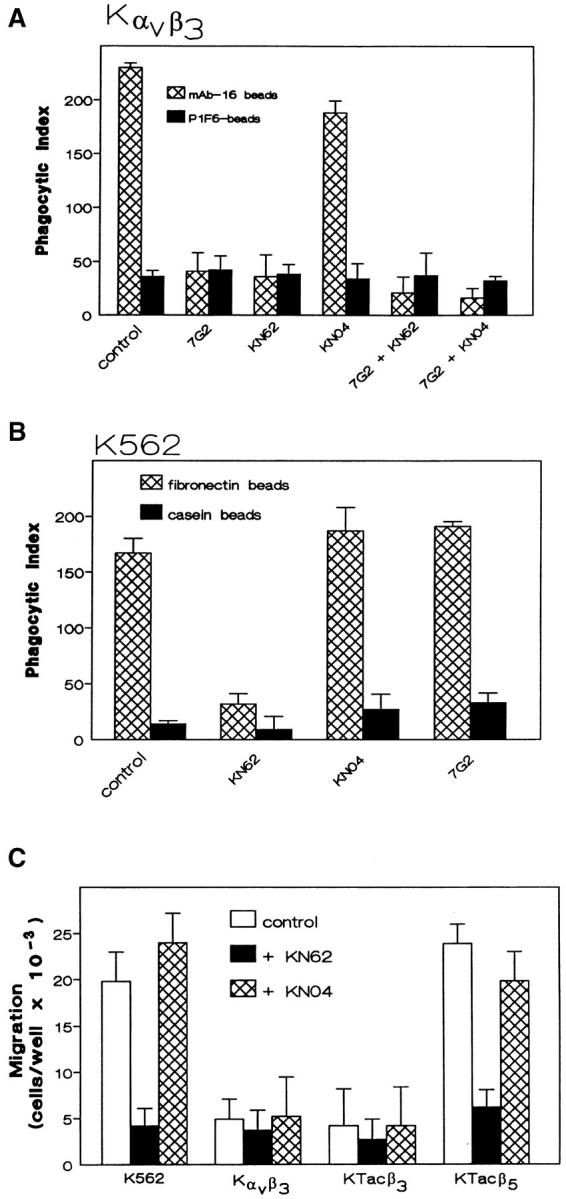
Effects of CamKII inhibitors on α5β1 function and αvβ3 crosstalk. Phagocytosis of the α5β1-specific target mAb-16 beads, or control target P1F6-beads was evaluated in K562 expressing αvβ3 in the presence and absence of αvβ3 ligation by mAb 7G2 (5.0 μg/ml) and/or the CamKII inhibitor KN62 (2.5 μM) or control KN04 (5.0 μM) (A) as described in Materials and Methods. Shown are mean ± SEM of three determinations with four replicates each. α5β1-mediated phagocytosis of FN beads was evaluated in untransfected K562 cells in the presence and absence of KN62 (2.5 μM), KN04 (5.0 μM), or the β3-specific mAb 7G2 (5.0 μg/ml) (B) as described in Materials and Methods. Shown are the mean ± SEM of two determinations with three replicates each. In C, the migration of untransfected K562 cells, Kαvβ3, KTacβ3, and KTacβ5 to FN was assessed in the presence and absence of KN62 (2.5 μM) or KN04 (5.0 μM) as described in Materials and Methods. Shown are mean ± SEM of four determinations with at least three replicates each.
To determine the dependence of α5β1 phagocytosis on CamKII activation, we evaluated phagocytosis in untransfected K562 cells which express α5β1, but not αvβ3. The absence of αvβ3 in these cells permitted the use of FN-coated beads as a phagocytosis target for α5β1 rather than the more selective mAb-16 beads used when αvβ3 is present. K562 phagocytosis of FN-coated beads via α5β1 was inhibited by the CamKII inhibitor KN62 (Fig. 5 B), but not the control KN04. Thus, enhanced CamKII activity, initiated by α5β1 binding of mAb-16 beads, appears to be required for α5β1 phagocytosis.
These data support the hypothesis that ligation of α5β1 stimulates CamKII activity and that αvβ3-mediated suppression of this activity is at least in part responsible for its inhibition of α5β1-mediated phagocytosis. To demonstrate that a similar mechanism was responsible for the inhibitory β3 crosstalk to α5β1 during migration, we evaluated the effects of the CamKII inhibitor KN62 on K562 cell migration in response to FN. KN62, but not the inactive analogue KN04, inhibited the FN-induced migration of mock transfected K562 cells and KTacβ5 (Fig. 5 C). The presence of KN62 did not further attenuate the minimal migration of Kαvβ3 or KTacβ3 cells.
Constitutively Active CamKII Overcomes αvβ3-Inhibitory Integrin Crosstalk
To test the hypothesis that αvβ3 crosstalk to α5β1 was a result of CamKII downregulation by β3, K562 cells were infected with an adenovirus-directing expression of a constitutively active form of CamKII (1). Expression of this construct in untransfected K562 cells resulted in an eightfold increase in CamKII activity over a control viral construct encoding β-galactosidase (Fig. 6 A).
Figure 6.


Constitutively active CamKII reverses αvβ3 inhibitory crosstalk. Untransfected K562 cells (A), or KTacβ3 and KTacβ5 (B) were infected with an adenovirus encoding either β-galactosidase (AdCMV.gal) or a constitutively active form of CamKII (AdCMV.CKIID3). CamKII activity against a synthetic substrate was assayed in the presence and absence of calmodulin and the CamKII inhibitor KN62 (2.5 μM) as described in Materials and Methods and reported as the CamKII activity per mg of total cellular protein (A). Migration of KTacβ3 and KTacβ5 expressing either β-galactosidase or constitutively active CamKII to FN was assessed as described in Materials and Methods (B). Shown are mean ± SEM of three determinations performed in triplicate.
Next, KTacβ3 and KTacβ5 infected with virus encoding either β-galactosidase or constitutively active CamKII were assayed for their ability to migrate in response to FN. Expression of the active kinase specifically overcame the constitutive inhibition of α5β1-mediated migration in KTacβ3, without any effect on migration in KTacβ5 (Fig. 6 B). Thus, expression of active CamKII overcame αvβ3-mediated suppression of α5β1 high affinity functions. Unfortunately, safety concerns precluded testing the effect of the constitutively active CamKII in the phagocytosis assay.
Discussion
Integrin crosstalk is an important mechanism for coordinating signals from multiple simultaneously ligated integrins on a single cell for a functional response to extracellular matrix. Although sometimes called “transdominant inhibition,” crosstalk may induce, as well as suppress functions of the target integrin, so we believe the more general term, preferable (9). Although the number of examples of integrin crosstalk has rapidly expanded in the past few years, little is known concerning the molecular mechanisms by which one integrin affects the function of another. K562 cells have proved a valuable model for examination of integrin crosstalk because these cells express a single integrin, α5β1, permitting a wide variety of genetic experiments exploring the basis of integrin crosstalk. In this system, we have previously shown that ligation of transfected αvβ3 inhibits the high affinity phagocytic function of α5β1 without effect upon low affinity α5β1-mediated adhesion and that the β3 cytoplasmic tail is both necessary and sufficient for this effect. We now have used this model to explore the biochemical mechanisms involved in crosstalk. Based on a previous report, we examined a potential role for CamKII in αvβ3-mediated suppression of the high affinity functions of α5β1, and performed structure-function analysis of the β3 cytoplasmic tail to further delineate the required structures for this unique signaling event.
In this study, we show that either αvβ3 ligation or expression of the isolated β3 cytoplasmic tail exerts an inhibitory effect upon α5β1-mediated migration as well as phagocytosis. Since both α5β1 migration and phagocytosis are events that require the high affinity state of the integrin, and since the αvβ3-mediated inhibition of α5β1 is reversed by KN62 in both cases, these data suggest that a common signaling mechanism is responsible for these crosstalk events.
Based on the data in this report, we propose the hypothesis that CamKII, a ser/thr kinase with multiple intracellular substrates, is an important regulator of α5β1 function and a target of integrin crosstalk. First, ligation of α5β1 by specific antibody- or ligand-coated beads enhances the activity of CamKII in both macrophages and K562 cells. Second, activation of CamKII by ligation of α5β1 is required for both phagocytosis and migration. In contrast CamKII inhibitors do not affect adhesion which can be effected by low affinity α5β1. Thus, the requirement for CamKII activation appears to be specific for the high affinity functions of α5β1.
Coligation of αvβ3, or exposure of the isolated β3 cytoplasmic tail, prevents α5β1-induced rise in CamKII activity. Since the β3 integrin and the CamKII inhibitor have the same effect on α5β1 function, the data suggest that suppression of the ability of α5β1 to activate CamKII may be an important mechanism of integrin crosstalk. A role for CamKII suppression in integrin crosstalk is supported by the reversal of crosstalk inhibition of migration with constitutively active CamKII. Thus, our data support the hypothesis that α5β1-mediated CamKII activation is required for the high affinity functions of migration and phagocytosis and that αvβ3-activated crosstalk suppresses these functions through inhibition of CamKII activation. Thus, α5β1 and αvβ3 have opposing effects on CamKII activity.
Neither the upstream events regulating CamKII nor its downstream effector are yet known. Tyrosine kinase inhibitors have no effect either on the high-affinity functions of α5β1 or on suppression by αvβ3, suggesting the possibility that the entire pathway is independent of the well-known effects of integrin ligation on several tyrosine kinases (2, 3). Indeed, the independence of integrin crosstalk from the phosphorylation of Tyr747 further suggests that the signaling involved in the regulation of CamKII may be completely independent of these pathways. A recent report by Wu et al. (25) demonstrates that ligation of α5β1 and αvβ3 have opposite effects on plasma membrane calcium channel activity. Since calcium is an important regulator of CamKII, this voltage gated calcium channel may be important in the differential regulation of CamKII by these two integrins. Based on our preliminary pharmacologic data, one likely effector for CamKII in α5β1 high affinity function is myosin light chain kinase (MLCK). MLCK inhibitors KT5926 and ML9 both reverse αvβ3 inhibition of α5β1-mediated phagocytosis and migration without affecting inhibition of CamKII activation by αvβ3 ligation (Blystone, S.D., and E.J. Brown, unpublished data). MLCK phosphorylation by CamKII is known to inhibit MLCK activity, leading presumably to decreased myosin-induced cell traction (21). This integrin-mediated modulation of myosin function is consistent with the known role for myosin in phagocytosis and migration.
Analysis of structural requirements in the β3 cytoplasmic tail in αvβ3-mediated crosstalk reveals that Ser752 of the β3 cytoplasmic tail is required for inhibition of CamKII and for initiation of integrin crosstalk, while crosstalk is independent of either of the β3 cytoplasmic tail tyrosines. The requirement for β3 Ser752 in crosstalk is unexpected. The importance of Ser752 was suggested by a mutation to proline in a patient with Glanzmann's Thrombasthenia which abolished high affinity binding of fibrinogen by platelet αIIbβ3 (6, 8). However, detailed analysis has shown that mutation of Ser752 to Ala does not affect ligand binding by αIIbβ3 (8). The failure of the Ser752 to Ala β3 mutation to affect ligand binding is supported by our studies in K562 which demonstrate normal adhesion, normal migration (Fig. 2, B and C), and normal generation of the ligand-induced binding site (LIBS) recognized by the antibody LIBS-1 in response to RGD peptide in this mutant (data not shown). In contrast, integrin crosstalk is entirely abolished by the S752A mutation, as it is by mutation to Pro (the original Glanzmann's mutation), to Glu (to mimic a potential phosphorylation), and to Cys (as a conservative mutation). Thus, it appears that Ser is absolutely required at this position. While this suggests the possibility of Ser phosphorylation in integrin crosstalk, we have been unable to demonstrate such phosphorylation so far. In contrast, Tyr747, which is absolutely required for stimulated adhesion and for αvβ3-mediated migration (Fig. 1) in K562 cells, is not involved in integrin crosstalk. Thus, these two amino acids, closely spaced in the relatively short cytoplasmic domain of one chain of an integrin, mediate two entirely distinct signaling cascades.
In a recent report, Bouvard et al. (5) showed that increased CamKII levels resulted in a decrease in the affinity of α5β1 for FN. In this in vitro system, CamKII and the phosphatase calcineurin regulate α5β1 affinity. Because the β3 suppression of α5β1 phagocytosis occurs subsequent to α5β1 binding of ligand (3), it is possible that repeated cycling of α5β1 affinity is required for phagocytosis and migration. Binding of ligand-coated beads to high affinity α5β1 would activate CamKII, which would then decrease integrin affinity. This hypothesis predicts that integrin crosstalk from αvβ3 which blocks CamKII activation would prevent α5β1 movement to the low affinity state. This is entirely consistent with reports of receptor activation rather than inactivation by integrin crosstalk (14, 24) which measured ligand binding rather than functions that require affinity modulation.
Finally, these data demonstrate that, while increased CamKII activity is required for α5β1-mediated phagocytosis and migration, αvβ3 can perform these same functions independent of any increase in CamKII. This is a startling example of the diversity of signaling and function among the integrins. It suggests that there may be fundamental differences within this family of closely related receptors in how they mediate even their most basic functions. While many studies have emphasized common features of integrin α- and β-chains in association with cytoskeleton, calreticulin, and signaling molecules, the differences between αvβ3 and α5β1 in requirements for phagocytosis and migration suggest that there will be profound differences among integrins even as they perform similar functions.
Acknowledgments
The authors wish to thank all contributors of cDNAs and antibodies used in these studies and the members of the Brown laboratory for suggestions and advice on the performance of these studies.
Abbreviations used in this paper
- CamKII
calcium/calmodulin-dependent protein kinase II
- FN
fibronectin
- IMDM
Iscove's Modified Eagle's Medium
- LIBS
ligand-induced binding site
- MLCK
myosin light chain kinase
- VN
vitronectin
Footnotes
S.D. Blystone is an investigator of the Arthritis Foundation. This work was supported by grants AI24674 and GM38330 from the National Institutes of Health to E.J. Brown. During these studies, S.D. Blystone was a recipient of NRSA AI08990-02 and a grant from the Lucille P. Markey Foundation.
References
- 1.Bilato C, Curto K, Monticone RE, Pauly RR, White AJ, Crow MT. The inhibition of vascular smooth muscle cell migration by peptide and antibody agonists of the αv/β3integrin complex is reversed by activated calcium/calmodulin-dependent protein kinase II. J Clin Invest. 1997;100:693–704. doi: 10.1172/JCI119582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blystone SD, Graham IL, Lindberg FP, Brown EJ. Integrin αV/β3 differentially regulates adhesive and phagocytic functions of the fibronectin receptor αv/β1 . J Cell Biol. 1994;127:1129–1137. doi: 10.1083/jcb.127.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blystone SD, Lindberg FP, LaFlamme SE, Brown EJ. Integrin β3 cytoplasmic tail is necessary and sufficient for regulation of alpha5/beta1 phagocytosis by αV/β3and integrin associated protein. J Cell Biol. 1995;130:745–754. doi: 10.1083/jcb.130.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blystone SD, Williams MP, Slater SE, Brown EJ. Requirement of integrin β3 tyrosine 747 for β3 tyrosine phosphorylation and regulation of αV/β3avidity. J Biol Chem. 1997;272:28757–28761. doi: 10.1074/jbc.272.45.28757. [DOI] [PubMed] [Google Scholar]
- 5.Bouvard D, Molla A, Block MR. Calcium/calmodulin-dependent protein kinase II controls α5/β1integrin-mediated inside-out signaling. J Cell Sci. 1998;111:657–665. doi: 10.1242/jcs.111.5.657. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y-P, Djaffar I, Pidard D, Steiner B, Cieutat A-M, Caen JP, Rosa J-P. Ser-752→ Pro mutation in the cytoplasmic domain of integrin β3 subunit and defective activation of platelet integrin αIIbβ3 (glycoprotein IIb-IIIa) in a variant of . Glanzmann thrombasthenia Proc Natl Acad Sci USA. 1992;89:10169–10173. doi: 10.1073/pnas.89.21.10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y-P, O'Toole TE, Shipley T, Forsyth J, LaFlamme SE, Yamada KM, Shattil SJ, Ginsberg MH. “Inside-out” signal transduction inhibited by isolated integrin cytoplasmic domains. J Biol Chem. 1994;269:18307–18310. [PubMed] [Google Scholar]
- 8.Chen Y-P, O'Toole TE, Ylänne J, Rosa J-P, Ginsberg MH. A point mutation in the integrin β3 cytoplasmic domain (S752→ P) impairs bidirectional signaling through αIIbβ3(platelet glycoprotein IIb-IIIa) Blood. 1994;84:1857–1865. [PubMed] [Google Scholar]
- 9.Diaz-Gonzalez F, Forsyth J, Steiner B, Ginsberg MH. Trans-dominant inhibition of integrin function. Mol Biol Cell. 1996;7:1939–1951. doi: 10.1091/mbc.7.12.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gresham HD, Goodwin JL, Anderson DC, Brown EJ. A novel member of the integrin receptor family mediates Arg-Gly-Asp-stimulated neutrophil phagocytosis. J Cell Biol. 1989;108:1935–1943. doi: 10.1083/jcb.108.5.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huhtala P, Humphries MJ, McCarthy JB, Tremble PM, Werb Z, Damsky CH. Cooperative signalling by alpha5/beta1 and alpha4/ beta1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J Cell Biol. 1995;129:867–879. doi: 10.1083/jcb.129.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunt TK, Knighton DR, Thakral KK, Goodson WH, Andrews WS. Studies on inflammation and wound healing: Angiogenesis and collagen synthesis stimulated in vivo by resident and activated wound macrophages. Surgery. 1984;96:48–54. [PubMed] [Google Scholar]
- 13.Hynes RO. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 14.Ishibashi Y, Claus S, Relman DA. Bordetella pertussisfilamentous hemagglutinin interacts with a leukocyte signal transduction complex and stimulates bacterial adherence to monocyte CR3 (CD11b/ CD18) J Exp Med. 1994;180:1225–1233. doi: 10.1084/jem.180.4.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pacifici R, Roman J, Kimble R, Civitelli R, Brownfield CM, Bizzarri C. Ligand binding to monocyte α5/β1 integrin activates the α2/β1 receptor via the α5subunit cytoplasmic domain and protein kinase C. J Immunol. 1994;153:2222–2233. [PubMed] [Google Scholar]
- 16.Pijuan-Thompson V, Gladson CL. Ligation of integrin α5/β1 is required for internalization of vitronectin by integrin αV/β1 . J Biol Chem. 1997;272:2736–2743. doi: 10.1074/jbc.272.5.2736. [DOI] [PubMed] [Google Scholar]
- 17.Porter JC, Hogg N. Integrin cross talk: activation of lymphocyte function-associated antigen-1 on human T cells alters α4/β1- and α5/β1-mediated function. J Cell Biol. 1997;138:1437–1447. doi: 10.1083/jcb.138.6.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruoslahti E. Integrins. J Clin Invest. 1991;87:1–5. doi: 10.1172/JCI114957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senior RM, Gresham HD, Griffin GL, Brown EJ, Chung AE. Entactin stimulates neutrophil adhesion and chemotaxis through interactions between its Arg-Gly-Asp (RGD) domain and the leukocyte response integrin (LRI) J Clin Invest. 1992;90:2251–2257. doi: 10.1172/JCI116111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon KO, Nutt EM, Abraham DG, Rodan GA, Duong LT. The α2/β1 integrin regulates α5/β1-mediated cell migration toward fibronectin. J Biol Chem. 1997;272:29380–29389. doi: 10.1074/jbc.272.46.29380. [DOI] [PubMed] [Google Scholar]
- 21.Tansey MG, Word RA, Hidaka H, Singer HA, Schworer CM, Kamm KE, Stull JT. Phosphorylation of myosin light chain kinase by the multifunctional calmodulin-dependent kinase II in smooth muscle cells. J Biol Chem. 1992;267:12511–12516. [PubMed] [Google Scholar]
- 22.Turner CE, Miller JT. Primary sequence of paxillin contains putative SH2 and SH3 domain binding motifs and multiple LIM domains: identification of a vinculin and pp125FAKbinding region. J Cell Sci. 1994;107:1583–1591. doi: 10.1242/jcs.107.6.1583. [DOI] [PubMed] [Google Scholar]
- 23.van Kooyk Y, Van de Wiel-van Kemenade E, Weder P, Huijbens RJF, Figdor CG. Lymphocyte function-associated antigen 1 dominates very late antigen 4 in binding of activated T cells to endothelium. J Exp Med. 1993;177:185–190. doi: 10.1084/jem.177.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Strijp JAG, Russell DG, Tuomanen E, Brown EJ, Wright SD. Ligand specificity of purified complement receptor type 3 (CD11b/CD18, Mac-1, αM β2): indirect effects of an Arg-Gly-Asp sequence. J Immunol. 1993;151:3324–3336. [PubMed] [Google Scholar]
- 25.Wu X, Mogford JE, Platts SH, Davis GE, Meininger GA, Davis MJ. Modulation of calcium currents in arteriolar smooth muscle by αV/β3 and α5/β1integrin ligands. J Cell Biol. 1998;143:241–252. doi: 10.1083/jcb.143.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou MJ, Lublin DM, Link DC, Brown EJ. Distinct tyrosine kinase activation and Triton X-100 insolubility upon FcRII or FcRIIIB ligation in human polymorphonuclear leukocytes. J Biol Chem. 1995;270:13553–13560. doi: 10.1074/jbc.270.22.13553. [DOI] [PubMed] [Google Scholar]