

Abstract
In Saccharomyces cerevisiae, vesicles that carry proteins from the ER to the Golgi compartment are encapsulated by COPII coat proteins. We identified mutations in ten genes, designated LST (lethal with sec-thirteen), that were lethal in combination with the COPII mutation sec13-1. LST1 showed synthetic-lethal interactions with the complete set of COPII genes, indicating that LST1 encodes a new COPII function. LST1 codes for a protein similar in sequence to the COPII subunit Sec24p. Like Sec24p, Lst1p is a peripheral ER membrane protein that binds to the COPII subunit Sec23p. Chromosomal deletion of LST1 is not lethal, but inhibits transport of the plasma membrane proton-ATPase (Pma1p) to the cell surface, causing poor growth on media of low pH. Localization by both immunofluorescence microscopy and cell fractionation shows that the export of Pma1p from the ER is impaired in lst1Δ mutants. Transport of other proteins from the ER was not affected by lst1Δ, nor was Pma1p transport found to be particularly sensitive to other COPII defects. Together, these findings suggest that a specialized form of the COPII coat subunit, with Lst1p in place of Sec24p, is used for the efficient packaging of Pma1p into vesicles derived from the ER.
Keywords: endoplasmic reticulum, transport vesicle, COPII, proton-ATPase, Saccharomyces cerevisiae
The plasma membrane proton-ATPase (Pma1p)1 is an essential integral membrane protein that couples ATP hydrolysis to the translocation of protons across the plasma membrane (Serrano et al., 1986). The proton gradient generated by Pma1p then drives the uptake of nutrients, such as amino acids, from the extracellular medium (Vallejo and Serrano, 1989). A second physiological function of Pma1p is to maintain the cytosol at a neutral pH. In medium of low pH, the growth rate is limited by the amount of cellular Pma1p (McCusker et al., 1987; Portillo and Serrano, 1989). Pma1p transport to the cell surface depends upon the secretory pathway defined by the sec genes (Brada and Schekman, 1988; Chang and Slayman, 1991). Pma1p is one of the most abundant cargo molecules of the secretory pathway, constituting 25–50% of the total plasma membrane protein (Serrano, 1991). Because of its abundance and physiological importance, one might expect that yeast cells would have specialized mechanisms to ensure efficient transport of Pma1p through the secretory pathway. Such a function has been suggested for two proteins, Ast1p and Ast2p, in the transport of Pma1p from the Golgi compartment to the plasma membrane (Chang and Fink, 1995). For early steps in the secretory pathway, proteins that are specifically required for the transport of Pma1p have not yet been identified.
Proteins destined for the plasma membrane are transported from the ER to the Golgi compartment by vesicles coated with a set of proteins known as COPII (Barlowe et al., 1994). These COPII coats are thought to cause the deformation of the membrane into a vesicle and to recruit cargo molecules into vesicle buds (reviewed by Schekman and Orci, 1996). The stepwise recruitment and assembly of the COPII coat onto the membrane is thought to occur as follows. Action of the ER resident membrane protein Sec12p, a guanine nucleotide exchange factor for Sar1p, causes Sar1p to bind to the ER membrane (Barlowe and Schekman, 1993). Membrane-associated Sar1p, in turn, recruits the soluble Sec23p/Sec24p and Sec13p/Sec31p complexes (Matsuoka et al., 1998). Sec16p resides on the ER membrane and binds to both the Sec23p/Sec24p and Sec13p/Sec31p complexes, likely organizing their assembly onto the membrane (Espenshade et al., 1995; Gimeno et al., 1996; Shaywitz et al., 1997). To examine the role of different COPII coat subunits in recruitment of cargo molecules to vesicles, partially assembled COPII complexes have been tested for their ability to associate with cargo proteins. Association of a membrane-bound complex of Sar1p and Sec23p/Sec24p with integral membrane proteins indicates that cargo proteins may laterally partition into the vesicle membrane by virtue of their affinity for the Sec23p/Sec24p protein complex (Aridor et al., 1998; Kuehn et al., 1998).
An early indication that the COPII coat subunits would physically interact came from specific genetic interactions between mutations in COPII genes. When temperature-sensitive mutations in COPII genes are combined, the resulting double mutants are almost always more growth- restrictive than the component single mutations, and are usually inviable at 24°C. These synthetic-lethal interactions are restricted to genes involved in COPII vesicle formation and do not occur when mutations in genes required for vesicle formation are combined with genes required for vesicle fusion (Kaiser and Schekman, 1990). The specificity of this type of genetic interaction suggested that synthetic lethality with known COPII mutations would be a useful criterion to identify new mutations involved in the assembly of the COPII coat.
We screened for mutations that were lethal with the COPII mutation sec13-1 and identified ten LST genes (lethal with sec-thirteen). As we describe elsewhere, most of the LST genes are related to an unanticipated role for SEC13 in the regulated delivery of specific amino acid permeases to the cell surface (Roberg et al., 1997a,b). Accordingly, these LST genes display synthetic-lethal interactions with SEC13, but not with the other COPII genes. On the other hand, mutations in LST1 were lethal with the full set of mutations defective in COPII vesicle budding, but not with mutations defective in vesicle fusion, indicating that LST1 does participate in vesicle budding at the ER. Here we show that LST1 encodes a homologue of the COPII subunit, Sec24p, and that Lst1p is a peripheral membrane protein localized to the ER that can form complexes with Sec23p. The LST1 gene is not essential, but by examination of the phenotypes of lst1Δ mutants we show that LST1 is required for the efficient export of Pma1p from the ER to the Golgi compartment. These results suggest a specialized form of the vesicle coat that is responsible for recruitment of Pma1p into COPII-coated vesicles.
Materials and Methods
Media, Strains, and Plasmids
The Saccharomyces cerevisiae strains used in this study are listed in Table I. Rich medium (YPD) and supplemented minimal medium (SMM) were prepared according to Kaiser et al. (1994). To evaluate growth at low pH, YPD was adjusted to pH 3.8 with HCl (this medium remained at pH 3.8 throughout the growth of a yeast culture). For some experiments, SMM was buffered to pH 6.5 using 50 mM MOPS and 50 mM MES. Genetic manipulations were performed according to standard protocols (Kaiser et al., 1994). DNA manipulations were carried out as described in Sambrook et al. (1989). pAF70 carries the SEC24 gene in the centromere vector pCT3 (URA3; Gimeno et al., 1996). pKR34 and pKR41 carry the 3.8-kb KpnI/SalI fragment containing the SEC24 gene from pAF70 in the 2μ vectors pRS426 (URA3) and pRS425 (LEU2), respectively. pKR17 carries the LST1 gene on a 3.5-kb fragment in the centromere vector pRS316 (URA3). A subclone of the LST1 gene from pKR17 into the 2μ vector pRS426 gave yeast transformants at a very low efficiency because of the toxicity of LST1 sequences when present at high copy. To study the toxic effects of LST1, pKR35 was constructed which contains the entire LST1 coding sequence expressed from pGAL1 on pCD43 (URA3). pKR35 will prevent growth under conditions of full induction on galactose medium, establishing that overexpression of Lst1p is toxic to yeast cells. Under conditions of partial induction of pGAL1–LST1 in cells grown on raffinose, pKR35 will complement lst1Δ::LEU2 for growth on acidic medium. This shows that the LST1 open reading frame carried on pKR35 still posses LST1 function.
Table I.
S. cerevisiae Strains
| Strain | Genotype | Source or reference | ||
|---|---|---|---|---|
| CUY563 | MAT a ade2-101 ade3-24 leu2-3,112 ura3-52 | T. Huffaker (Cornell University) | ||
| CUY564 | MATα ade2-101 ade3-2 leu2-3,112 ura3-52 | T. Huffaker (Cornell University) | ||
| EGY40 | MATα ura3-52 leu2 his3 trp1 | Golemis and Brent, 1992 | ||
| CKY45 | MATα sec13-1 his4-619 ura3-52 | Kaiser Lab Collection | ||
| CKY50 | MATα sec16-2 his4-619 ura3-52 | Kaiser Lab Collection | ||
| CKY54 | MATα sec17-1 his4-619 ura3-52 | Kaiser Lab Collection | ||
| CKY58 | MATα sec18-1 his4-619 ura3-52 | Kaiser Lab Collection | ||
| CKY78 | MATα sec23-1 his4-619 ura3-52 | Kaiser Lab Collection | ||
| CKY348 | MAT a/α leu2-3/leu2-3 ura3-52/ura3-52 | Kaiser Lab Collection | ||
| CKY423 | MATα sec13-1 ade2-101 ade3-24 leu2-3,112 ura3-52 [pKR4] | |||
| CKY424 | MAT a sec13-1 ade2-101 ade3-24 leu2-3,112 ura3-52 [pKR4] | |||
| CKY426 | MAT a lst1-1sec13-1 ade2-101 ade3-24 leu2-3,112 ura3-52 [pKR4] | |||
| CKY435 | MAT a lst1-1 sec13-1::[SEC13, URA3] ade2-101 ade3-24 leu2-3,112 ura3-52 | |||
| CKY436 | MAT a lst2-1 sec13-1::[SEC13, URA3] ade2-101 ade3-24 leu2-3,112 ura3-52 | |||
| CKY437 | MAT a lst3-1 sec13-1::[SEC13, URA3] ade2-101 ade3-24 leu2-3,112 ura3-52 | |||
| CKY438 | MAT a lst4-1 sec13-1::[SEC13, URA3] ade2-101 ade3-24 leu2-3,112 ura3-52 | |||
| CKY439 | MAT a lst5-1 sec13-1::[SEC13, URA3] ade2-101 ade3-24 leu2-3,112 ura3-52 | |||
| CKY440 | MAT a lst6-1 sec13-1::[SEC13, URA3] ade2-101 ade3-24 leu2-3,112 ura3-52 | |||
| CKY441 | MAT a lst7-1 sec13-1::[SEC13, URA3] ade2-101 ade3-24 leu2-3,112 ura3-52 | |||
| CKY442 | MAT a lst8-1 sec13-1::[SEC13, URA3] ade2-101 ade3-24 leu2-3,112 ura3-52 | |||
| CKY443 | MAT a prototroph | Kaiser Lab Collection | ||
| CKY473 | MAT a leu2-3,112 ura3-52 Gal+ | Kaiser Lab Collection | ||
| CKY534 | MATα lst1Δ::LEU2 leu2-3,112 ura3-52 [pKR17HA] | |||
| CKY535 | MAT a lst1Δ::LEU2 leu2-3,112 ura3-52 [pKR17HA] | |||
| CKY536 | MAT a lst1Δ::LEU2 ura3-52 leu2-3,112 | |||
| CKY540 | MAT a leu2-3,112 ura3-52 [pNV31] | |||
| CKY541 | MAT a sec12-4 ura3-52 [pNV31] | |||
| CKY542 | MAT a lst1Δ::LEU2 leu2-3,112 ura3-52 [pNV31] | |||
| CKY552 | MATα lst1Δ::LEU2 leu2-3,112 ura3-52 |
All strains are from this study unless otherwise indicated.
Epitope-tagged LST1 was constructed as follows. First, the NotI site in the polylinker of pKR17 was deleted with a 350-bp SmaI/NaeI fragment (pKR17Δ), and then a 12-bp linker carrying a NotI site (1127; New England BioLabs) was inserted at the Eco47III site (at codon 13 of LST1) of pKR17Δ to make pKR17N. pKR17HA carries the 100-bp NotI fragment from pGTEPI (Tyers et al., 1993), which encodes three tandem copies of the hemagglutinin (HA1) epitope, inserted into the NotI site of pKR17N. Restriction analysis using sites flanking the point of insertion revealed that two 100-bp inserts (six HA epitopes) were present in pKR17HA. pKR17HA was transformed into CKY536 to make CKY535 (MAT a lst1Δ:: LEU2 leu2-3, 112 ura3-52 [pKR17HA]).
Synthetic-lethal Screen
The following plasmids and strains were constructed for use in the sec13-1 synthetic-lethal screen. The plasmid pKR1 carries SEC13 on a 1.8-kb SalI/ BamHI fragment excised from pCK1313 (Pryer et al., 1993), inserted into pRS316 (Sikorski and Heiter, 1989). pKR4 carries the same 1.8-kb SalI/ BamHI fragment and a 3.8-kb NheI/BamHI fragment containing ADE3 from pDK255, both inserted into the vector pRS315 (Sikorski and Heiter, 1989). CUY563 and CKY45 were crossed to produce a MAT a ade2 ade3 leu2 ura3 sec13-1 segregant, which was transformed with pKR4 to give CKY423. The mating type of CKY423 was switched by ectopic expression of the HO gene (Herskowitz and Jensen, 1991) to give CKY424.
Cultures of CKY423 and CKY424 were mutagenized by irradiation with a germicidal UV lamp at a dose resulting in 10% cell survival. Mutagenized cells were plated on YPD at a density of 150 colonies per plate. After 5 d of growth at 24°C, colonies with a solid red color and no white sectors were selected for further analysis. The dependence of the nonsectoring phenotype on the sec13-1 mutation was tested by transforming candidate mutants with either pKR1 or pRS316. Strains that sectored after transformation with pKR1, but not after transformation with pRS316, were scored as sec13-1–dependent.
Complementation tests were performed by mating mutants isolated from CKY423 with those isolated from CKY424. Zygotes isolated by micromanipulation were scored for their ability to form sectored colonies on YPD plates. The genes defined by these complementation groups were designated LST. All lst mutant strains were backcrossed to a parental strain twice.
The lst sec13-1 double mutants were converted to lst single mutants by integration of a wild-type copy of SEC13 at the sec13-1 locus, using the integrating plasmid p1312 (SEC13 URA3; Pryer et al., 1993). The integrants were grown on YPD and cells from white sectors (indicating loss of pKR4) were isolated. The integration of a wild-type copy of SEC13 was confirmed by the ability of the cells from white sectors to grow at 36°C, a temperature that is restrictive for the sec13-1 mutation. Owing to the poor growth of lst9 strains, we were not able to construct a lst9 single mutant by this method.
To test for synthetic-lethal interactions between lst mutations and mutations in sec genes, lst mutants CKY435 (lst1-1), CKY436 (lst2-1), CKY437 (lst3-1), CKY438 (lst4-1), CKY439 (lst5-1), CKY440 (lst6-1), CKY441 (lst7-1), and CKY442 (lst8-1) were crossed to the sec mutants CKY45 (sec13-1), CKY50 (sec16-2), CKY78 (sec23-1), and CKY450 (sec31-2). Inviability of a given lst sec double mutant was inferred from crosses where lethality segregated as a two-gene trait (most tetrads giving a segregation pattern of 1:3 for lethality), an outcome that was easily detectable since crosses to wild-type typically gave >95% spore viability. The segregation pattern of the sec mutation in the surviving sister spores was used as an additional test to establish that the inviable spores always carried the sec mutation, and therefore were not the result of random spore death.
Construction of lst1Δ Mutants
Replacement of the chromosomal LST1 gene with an allele disrupted with the LEU2 gene was constructed as follows. pKR18 carries the 5′ half of LST1 on a 2.0-kb Xho1/HindIII fragment inserted into pRS316. A 2.0-kb HindIII/BamHI fragment containing the LEU2 gene from plasmid pJJ252 (Jones and Prakash, 1990) and a 250-bp BclI/SacI fragment from the 3′ noncoding region of LST1 were inserted into pKR18 to construct pKR28. The NH2-terminal coding region of LST1 (except for codons 1–13) was removed by deleting a 1.7-kb Eco47III/MscI fragment from pKR28 to generate pKR28Δ. The lst1Δ::LEU2 construct, liberated from pKR28Δ by digestion with XhoI, was transformed into the wild-type diploid strain CKY348 (MAT a /α leu2-3,112/leu2-3,112 ura3-52/ura3-52). On sporulation and dissection, this diploid gave four viable spore clones, and haploid segregants carrying lst1Δ::LEU2 were confirmed by Southern blotting. One such segregant was further backcrossed to our S288C genetic background to give strains CKY536 and CKY542.
Proton Efflux from Intact Yeast Cells
Pma1p activity was assayed by proton efflux from intact cells into the external medium. Cells were grown to exponential phase in YPD at 37°C, washed, and then stored in deionized water at 4°C overnight. Cell number was measured by light scattering, and a total of 25 A600 units (∼5 × 108 cells) was suspended in 5 ml of 100 mM KCl, 10 mM glycine, pH 4.0. The pH of the cell suspension was measured using a combination electrode at 25°C with constant stirring. Once the pH had stabilized (∼10 min), glucose was added to a final concentration of 40 mM and the ensuing drop in pH was recorded at 30-s intervals over 15 min. In comparison of wild-type (CKY443) and lst1Δ (CKY536) strains, both suspensions had identical cell concentration as measured by light scattering, and showed the same response to calibration pulses with HCl.
Immunofluorescence Microscopy
The intracellular location of Pma1p in wild-type (CKY443) and lst1Δ (CKY536) cells was examined by indirect immunofluorescence microscopy using techniques described previously (Pringle et al., 1991; Espenshade et al., 1995). Strains were grown exponentially in SMM medium, pH 7.2, at 30°C. Cells were fixed in 3.7% formaldehyde and then converted to spheroplasts by digestion with lyticase. Both primary and secondary antibody incubations were for 1 h at 25°C. Affinity-purified anti-Pma1p antibody was prepared as follows. A crude preparation of yeast membranes was resolved by preparative SDS-PAGE, and after transfer of proteins to a nitrocellulose membrane by electrophoresis, the strip of membrane that contained Pma1p was excised. Rabbit antiserum to Pma1p was applied to the nitrocellulose strip, and after the strip was washed with 20 mM Tris, pH 7.5, 150 mM NaCl, 0.5% Tween 20, the bound antibody was eluted with 100 mM glycine, pH 2.8, 500 mM NaCl, 0.5% Tween 20. Affinity-purified Pma1p was used at a 1:100 dilution and FITC-conjugated anti-rabbit IgG was used at 1:200 dilution. Mounting medium was supplemented with 4′,6-diamidino-2-phenylindole (DAPI). Micrographs were taken with a Nikon Eclipse TE300 microscope with a Hamamatsu Orca C4742-95 CCD camera.
For the localization of Lst1p-HA, CKY535 was grown on SMM to exponential phase and prepared as described above. For visualization of Lst1p-HA, the 12CA5 antibody (Berkeley Antibody Co., Inc.) was used at a 1:5,000 dilution and FITC-conjugated goat anti–mouse IgG was used at a 1:50 dilution. Rabbit anti–Kar2p polyclonal serum (a gift of M. Rose, Princeton University, Princeton, NJ) was used at a 1:1,000 dilution and rhodamine-conjugated goat anti–rabbit IgG was used at a 1:200 dilution. Samples were viewed and imaged using a Nikon Optiphot 2 microscope and a Photometric ImagePoint CCD camera. Images were recorded using IP-Lab software (Molecular Dynamics, Inc.).
Cell Fractionation
Cell organelles were fractionated on equilibrium density gradients as previously described (Roberg et al., 1997a). Cultures were grown exponentially at 24°C and then shifted to 37°C for 3 h. 1.6 × 109 cells were collected by centrifugation and suspended in 0.5 ml STE10 (10% [wt/wt] sucrose, 10 mM Tris-HCl, pH 7.6, 10 mM EDTA) with a protease inhibitor cocktail (1 mM PMSF, 0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin, 2 μg/ml aprotinin) and lysed by vortexing with glass beads. An additional 1 ml of STE10 was added, and the lysate was cleared of unbroken cells and large cell debris by centrifugation at 300 g for 2 min. The cleared extract (300 μl) was layered on top of a 5-ml, 20–60% linear sucrose gradient in TE (10 mM Tris-HCl, pH 7.6, 10 mM EDTA) prepared for an SW50.1 rotor (Beckman Instruments, Inc.). Samples were centrifuged 100,000 g for 18 h at 4°C and fractions of 300 μl were collected from the top of the gradient. Protein was precipitated from each fraction by the addition of 100 μl of 0.15% deoxycholate and 100 μl of 72% trichloroacetic acid. Protein pellets were collected by centrifugation at 13,000 g, washed with cold acetone, and then solubilized in ESB (60 mM Tris-HCl, pH 6.8, 100 mM DTT, 2% SDS, 10% glycerol, 0.02% bromophenol blue). Pma1p, Gas1p, and Sec61p were resolved by SDS-PAGE and were detected by immunoblotting. The relative amount of each protein in cell fractions was determined by densitometry using an Ultroscan 2202 (LKB Instruments, Inc.). The Golgi GDPase activity was assayed in gradient fractions before protein precipitation using standard methods (Abeijon et al., 1989).
The subcellular distribution of Lst1p-HA was examined using techniques described previously (Espenshade et al., 1995). CKY535 carrying pKR17HA, which expresses Lst1p-HA, was grown to exponential phase in SMM without uracil. 2 × 109 cells were harvested, converted to spheroplasts, and then gently lysed by glass beads in 500 μl of cell lysis buffer (20 mM MES, pH 6.5, 100 mM NaCl, 5 mM MgCl2) including the protease inhibitor cocktail. The cell extract was sequentially centrifuged at 500 g for 20 min, 10,000 g for 20 min, and 150,000 g for 60 min, to give one soluble and three particulate fractions.
Release of Lst1p-HA from the particulate fraction was examined by treating cell extracts with 500 mM NaCl, 100 mM sodium carbonate, pH 11.5, 2.5 M urea, or 1% Triton X-100. After 1 h of incubation at 4°C, samples were centrifuged at 50,000 g for 30 min to separate soluble and particulate fractions. Fractions from both experiments were solubilized in sample buffer and analyzed by immunoblotting.
Immunoblotting
Samples of 10–30 μl in ESB were resolved by SDS-PAGE and immunoblotting was conducted according to standard protocols (Harlow and Lane, 1988). For transfer of Lst1p to nitrocellulose membranes, 0.1% SDS was included in the transfer buffer. The following antibodies were used: mouse monoclonal 12CA5 anti-HA at 1:1,000 dilution; rabbit anti-Pma1p (a gift of A. Chang, Albert Einstein College of Medicine, Bronx, NY) at 1:500 dilution; rabbit anti-Gas1p (a gift of H. Riezman, University of Basel, Basel, Switzerland) at 1:10,000 dilution; rabbit anti-Sec61p (a gift of R. Schekman, University of California, Berkeley, CA) at 1:3,000 dilution; rabbit anti-Gdh2p (a gift of B. Magasanik, Massachusetts Institute of Technology, Cambridge, MA) at 1:1,000 dilution; HRP-coupled sheep anti–mouse Ig and HRP-coupled sheep anti–rabbit Ig (Nycomed Amersham Corp.) at 1:10,000 dilution. Blots were developed using chemiluminescence detection system (Nycomed Amersham Corp.).
Pulse–Chase Kinetics of Invertase Maturation
The strains used for radiolabeling all carried the plasmid pNV31, which carries the SUC2 gene under the constitutive TPI1 promoter (a gift of M. Lewis, Medical Research Council Laboratories of Molecular Biology, Cambridge, UK). Wild-type (CKY540) and lst1Δ (CKY542) strains were grown in SMM without methionine (buffered with 50 mM MES and 50 mM MOPS to pH 6.5) at 24°C to exponential phase, and then shifted to 37°C for 3 h before labeling. A sec12-4 strain (CKY541) was similarly grown to exponential phase at 24°C, but was shifted to 37°C 5 min before the addition of label. Radiolabeling and immunoprecipitation of invertase was performed as previously described (Gimeno et al., 1995; Elrod-Erickson and Kaiser, 1996).
Two-Hybrid Interactions
The yeast two-hybrid assay was used to test potential protein–protein interactions as previously described (Gyuris et al., 1993; Bartel and Fields, 1995). Interactions were tested between either Lst1p or Sec24p fused to the LexA DNA-binding domain and Sec23p fused to an acidic transcriptional activation domain. The following plasmids were used: pPE81 carries SEC23 fused to the acidic activation domain of pJG4-5 (Espenshade et al., 1995); pRH286 carries SEC24 (codons 34–926) fused to the lexA DNA-binding domain in pEG202 (Gimeno et al., 1996); pKR37 carries LST1 fused to the lexA DNA-binding domain in pGilda (a derivative of pEG202 with pGAL1; provided by D. Shaywitz).
Combinations of control and fusion protein plasmids, along with the reporter plasmid pSH18-34, were transformed into the strain EGY40 (Golemis and Brent, 1992). Strains were grown exponentially in SMM with 2% raffinose as the carbon source. Galactose was added to a concentration of 2%, and incubation was continued for 10 h to induce fusion proteins expressed from pGAL1. Assays for β-galactosidase activity were performed on cells lysed by disruption with glass beads (Rose and Botstein, 1983). Activity was normalized to total protein determined by the Bradford assay (Bio-Rad Laboratories).
Binding of Lst1p to Sec23p
A gene fusion expressing Lst1p fused to glutathione S-transferase (GST) was constructed by inserting the 3.0-kb BamHI/XhoI fragment of pKR17HA into pRD56 (a gift of R. Deshaies, California Institute of Technology, Pasadena, CA) to construct pRH254, which gives GST–Lst1p-HA (amino acids 14–927 of Lst1p) fusion expressed from pGAL1. pPE123 is the SEC23 gene expressed from pGAL1 in pRS315 (Gimeno et al., 1996). Binding interactions were tested from extracts of CKY473 transformed with pRH254 (GST–Lst1p-HA) and either pCD43 (vector) or pPE123 (Sec23p).
Cells were grown to exponential phase in SMM with 2% raffinose, galactose was added to 2%, and incubation was continued for 2 h at 30°C to induce pGAL1 expression. 5 × 108 cells were converted to spheroplasts as previously described (Espenshade et al., 1995) and then gently lysed using glass beads in IP buffer (20 mM Hepes-KOH, pH 6.8, 80 mM KOAc, 5 mM magnesium acetate, 0.02% Triton X-100) containing the protease inhibitor cocktail. The extract was diluted to 1 ml with IP buffer, and membranes were collected by centrifugation at 500 g for 20 min. This pellet was extracted with 1 ml of IP buffer and 600 mM NaCl for 10 min at 0°C to release membrane-bound protein complexes. After clarification by centrifugation at 90,000 g for 10 min, the extract was diluted threefold with IP buffer, and a 1-ml aliquot was removed and incubated at room temperature for 1 h with glutathione Sepharose 4B beads (Pharmacia Biotech, Inc.). The beads were washed twice with 200 mM NaCl, 20 mM Hepes-KOH, pH 6.8, 80 mM KOAc, 5 mM magnesium acetate, 0.02% Triton X-100, and once in IP buffer without Triton X-100. Proteins were released from glutathione Sepharose 4B beads by solubilization in ESB. Samples of total lysate were prepared by adding 2× ESB to an equal amount of the diluted extract from the salt washed membranes. Samples were analyzed by immunoblots probed with anti-Sec23p antibody.
For analysis of the membrane association of GST–Lst1p-HA and Sec23p, cells expressing GST–Lst1p-HA, Sec23p, or both GST–Lst1p-HA and Sec23p from pGAL1, were grown in 2% raffinose and then induced by the addition of 2% galactose as described above. 2 h after induction, 2 × 107 cells were collected by centrifugation and resuspended in 20 μl of cell lysis buffer (20 mM MES, pH 6.5, 100 mM NaCl, 5 mM MgCl2) with protease inhibitor cocktail. Cells were lysed by vigorous agitation with glass beads and an additional 500 μl of lysis buffer was added. The lysate was cleared of unlysed cells and large cell debris by centrifugation at 300 g for 3 min. 50 μl of the supernatant was reserved for a total extract sample and the remainder was centrifuged to pellet ER membranes at 10,000 g for 30 min at 4°C in a microcentrifuge. An equal number of cell equivalents of total extract, membrane-pellet, and supernatant fractions was solubilized in ESB and analyzed by immunoblotting. The cytosolic protein Gdh2p was found only in the soluble fractions, demonstrating cell lysis was complete (data not shown).
Results
Mutations Synthetically Lethal with sec13-1
To find new genes required for the budding of COPII vesicles, we screened for mutations that displayed synthetic lethality with the COPII mutation sec13-1 using a plasmid sectoring assay (Roberg et al., 1997b). Strain CKY423 has the chromosomal mutations ade2 ade3 sec13-1 and harbors the plasmid pKR4, which carries wild-type copies of SEC13 and ADE3. This strain accumulates a red pigment because of the ade2 mutation, but the spontaneous loss of pKR4 during the growth of a colony gives white sectors of ade2 ade3 segregants. In this strain, a mutation that is lethal with sec13-1 will produce a nonsectoring colony. Mutagenesis of CKY423 and the isogenic strain of opposite mating type, CKY424, yielded 139 nonsectoring mutants (Fig. 1). These strains were then tested for restored ability to sector after transformation with pKR1, which carries wild-type SEC13, but lacks the ADE3 gene. By this test, 57 of the mutants had synthetic-lethal mutations that could be rescued by wild-type SEC13. In backcrosses, 52 mutants gave a segregation pattern indicating that the trait was due to a single nuclear mutation (Fig. 1).
Figure 1.

Colony-sectoring screen for mutations that are lethal with sec13-1. CKY423 (ade2 ade3 leu2 ura3 sec13-1 [pKR4: SEC13, ADE3]) can lose the plasmid pKR4 when grown at 24°C on YPD, to give ade2 ade3 segregants that form white sectors within a red colony. Mutagenized cells that have acquired an lst mutation cannot grow without the pKR4 plasmid and form nonsectoring, solid red colonies. Of 132 nonsectoring colonies, the sectoring in 57 was restored by transformation with a second SEC13-bearing plasmid (pKR1).
Matings between mutants identified 11 complementation groups using colony sectoring of the diploid as the criterion for allelic complementation. These complementation groups were designated LST (Table II). One of the complementation groups was shown to comprise recessive lethal mutations in the SEC13 gene itself (Roberg et al., 1997b). Tests for rescue of the nonsectoring phenotype by plasmids carrying known sec genes showed that LST10 was SEC16 (Roberg et al., 1997b).
Table II.
Mutations Lethal with sec13-1
| Gene | Number of alleles | |
|---|---|---|
| LST1 | 11 | |
| LST2 | 6 | |
| LST3 | 4 | |
| LST4 | 5 | |
| LST5 | 5 | |
| LST6 | 1 | |
| LST7 | 1 | |
| LST8 | 1 | |
| LST9 | 1 | |
| LST10 (SEC16) | 2 |
Synthetic Interactions of lst Mutations
To perform further genetic tests on the lst mutations, the lst sec13-1 double mutants were converted to lst single mutants by integration of a wild-type copy of SEC13 at the sec13-1 locus (Materials and Methods). Representative lst single mutants were then crossed to sec16, sec23, and sec31 mutants. For mutations in LST2, LST3, LST4, LST5, LST7, and LST8, only crosses to sec13-1 gave a segregation pattern indicative of a synthetic-lethal interaction (Table III). We have subsequently shown that these LST genes relate to a function of SEC13 in the sorting of amino acid permeases in the late secretory pathway, and analysis of these genes is described elsewhere (Roberg et al., 1997a,b). Mutations in LST1 were inviable when combined with sec16, sec23, and sec31 mutations, and mutations in LST6 were inviable with sec16 and sec31 (Table III). Importantly, mutations in LST1 and LST6 did not show synthetic lethality in parallel crosses to mutations in SEC17 or SEC18, genes required for fusion of COPII vesicles. Given that synthetic-lethal interactions usually occur between mutations in genes involved in the same step of the secretory pathway, the tests for genetic interactions indicated that LST1, and probably also LST6, participate in vesicle budding from the ER.
Table III.
Growth of lst sec Double Mutants at 24°C
| sec13-1 | sec16-2 | sec23-1 | sec31-1 | sec17-1 | sec18-1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| lst1-1 | − | − | − | − | + | + | ||||||
| lst2-1 | − | + | + | + | ND | ND | ||||||
| lst3-1 | − | + | + | + | ND | ND | ||||||
| lst4-1 | +/− | + | + | + | ND | ND | ||||||
| lst5-1 | − | + | + | + | ND | ND | ||||||
| lst6-1 | − | +/− − | + | − | + | + | ||||||
| lst7-1 | − | + | + | + | ND | ND | ||||||
| lst8-1 | − | + | + | + | ND | ND |
Growth is represented in decreasing order by: + > +/− > +/−− > −. ND, not determined.
Lst1p Is Homologous to Sec24p
The LST1 gene was isolated by its ability to restore sectoring to CKY426 (MAT a sec13-1 ade2 ade3 leu2 ura3 [pKR4]), a strain that forms solid red, nonsectoring colonies because of the presence of the lst1-1 mutation. CKY426 was transformed with yeast genomic libraries. 34 colonies that regained the ability to form white sectors were identified among 97,000 Ura+ transformants. We expected this screen to yield plasmids carrying either SEC13 or LST1. About half of the complementing plasmids were shown to carry SEC13 by restriction site mapping and by the ability to complement the temperature sensitivity of sec13-1. The restriction maps of the remaining rescuing plasmids show that they represent two unrelated chromosomal regions. The clones p21-31 and p77-2 were selected as representatives of each region. The genomic sequence from p77-2 (a clone in the pλYES vector; Elledge et al., 1991) was inserted as an XhoI fragment into the integrating vector pRS306 to produce pKR20. For chromosomal integration, pKR20 was linearized by digestion with HpaI and transformed into CUY564 (MATα ade2 ade3 leu2 ura3). The resulting strain was crossed to the lst1-1 mutant CKY426 (MAT a lst1-1 sec13-1 ade2 ade3 leu2 ura3 [pKR4]). After sporulation and dissection, the integrated pKR20 was found to be completely linked to the LST1 locus: sectoring segregated 2:2 and all sectored colonies were Ura+, whereas all nonsectored colonies were Ura−. Thus, p77-2 carries the LST1 gene. In parallel, the genomic sequence from p21-31 (a clone in the pCT3 vector; Thompson et al., 1993) was inserted as an EcoRI/HindIII fragment into pRS306 to produce pKR7. pKR7 was integrated at its chromosomal locus after linearization with MscI and was then checked for linkage to lst1-1. Tetrad analysis showed that pKR7 was not linked to LST1 and we concluded that pKR7 carries an unlinked suppressor gene.
The 3.5-kb insert of p77-2 was inserted into the XhoI site of the centromeric vector pRS316 to construct pKR17. The base sequence of this insert was determined and found to contain a single open reading frame encoding a protein of 929 amino acids. This sequence corresponds to the open reading frame YHR098c located on chromosome VIII (Saccharomyces Genome Database, Cherry et al., 1997). The predicted amino acid sequence of LST1 shows significant similarity to SEC24 (YIL109C). The two proteins share 23% sequence identity that extends over most of their length (Fig. 2), suggesting that Lst1p may have a function similar to that of Sec24p as a subunit of the COPII vesicle coat.
Figure 2.

Comparison of LST1 and SEC24 sequences. Identities are indicated by solid lines and similarities are indicated by dotted lines. Overall amino acid identity is 23%.
Phenotypes of lst1Δ
One copy of the LST1 gene in the wild-type diploid strain CKY348 was disrupted to generate a lst1Δ::LEU2/LST1 heterozygote. Sporulation and dissection of this diploid gave >95% spore viability on YPD medium and the LEU2 marker segregated 2:2, showing that LST1 is not essential for growth. A lst1Δ::LEU2 mutant spore clone was crossed to sec mutants to test for synthetic lethality. In these crosses, both the temperature sensitivity of the sec mutation and the lst1Δ allele marked by LEU2 could be followed independently. In crosses of lst1Δ to sec12, sec13, sec16, sec23, sec24, or sec31 mutants, inviability segregated as a two-gene trait (segregation patterns for dead:viable spore clones were 2:2, 1:3, and 0:4). Tests of the genotype of the surviving sister spore clones showed that the inviable spores in these crosses were always lst1Δ sec double mutants. Crosses between lst1Δ and sec17 or sec18 produced viable double mutants. These findings confirmed and extended our earlier tests for synthetic lethality with lst1-1, and demonstrated that lst1Δ was synthetically lethal with all the known genes required for COPII vesicle formation, but not with genes required for vesicle fusion.
We evaluated the growth of lst1Δ::LEU2 mutants under a variety of conditions. On YPD, the lst1Δ::LEU2 strain grew, as well as an isogenic wild-type strain at temperatures ranging from 14 to 37°C. However, on SMM the lst1Δ::LEU2 strain grew poorly at temperatures above 30°C. Since YPD (pH 6.5) and SMM (pH 3.8) differed markedly in pH, we suspected that lst1Δ mutants may be particularly sensitive to an acidic environment, and we tested the effect of pH on the growth of lst1Δ mutants. Although lst1Δ mutants grew as well as wild-type on YPD at all temperatures, when YPD was brought to pH 3.8, lst1Δ mutants grew much more slowly than wild-type at 37°C (Fig. 3 A). Conversely, on SMM buffered to pH 6.5, lst1Δ and wild-type grew even at 37°C (data not shown). These results demonstrated that at high temperature, growth of the lst1Δ mutant was sensitive to acidic conditions.
Figure 3.
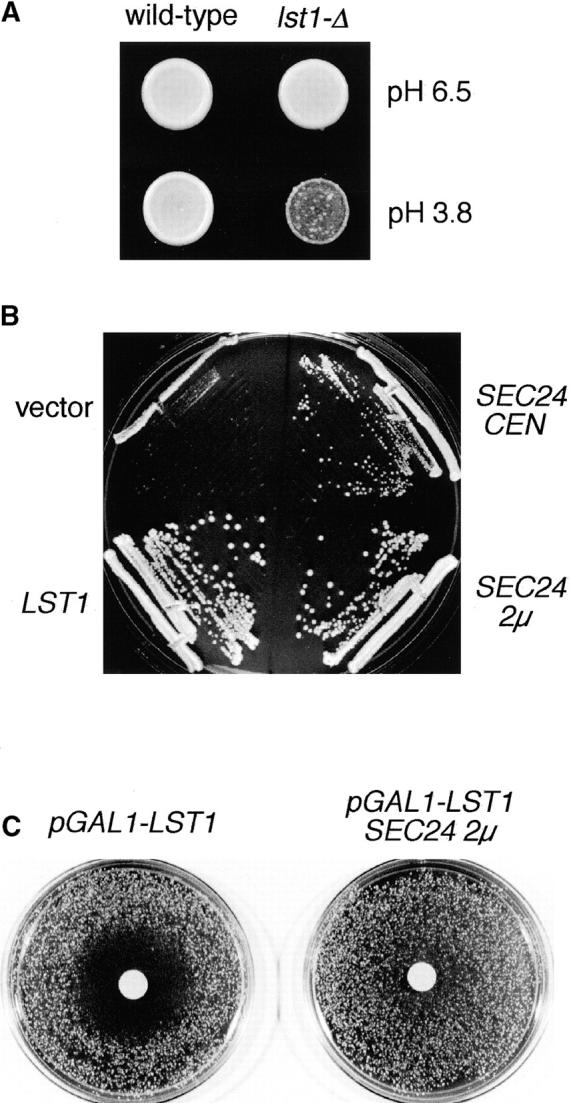
Functional relationships between LST1 and SEC24. (A) Sensitivity of lst1Δ mutants to acidic medium. Equal numbers of wild-type (CKY443) or lst1Δ::LEU2 (CKY534) cells were spotted onto YPD medium, pH 6.5, or acidic YPD medium (brought to pH 3.8 by the addition of HCl). Plates were photographed after incubation at 37°C for 2 d. (B) A lst1Δ::LEU2 strain (CKY552) was transformed with: vector only, pRS316; LST1 on a centromeric plasmid, pKR17; SEC24 on a centromeric plasmid, pAF70; or SEC24 on a 2μ plasmid, pKR34; and streaked onto YPD medium, pH 3.8. Colonies were photographed after growth at 37°C for 2 d. (C) A wild-type strain (CKY473) was transformed either with a plasmid carrying pGAL1–LST1 (pKR35) and vector control (pRS425), or with pKR35 and SEC24 on a 2μ plasmid (pKR41). Transformants were plated at a density of 800 cells/cm2 on SMM plates containing 2% raffinose and then 3 mg galactose solution was placed on a sterile 1-cm filter on top of the lawn. The plates were photographed after growth at 30°C for 2 d.
Having identified conditions where LST1 was needed for growth, we investigated whether overexpression of SEC24 could supply the function lost in lst1Δ. Some restoration of function was indicated by the ability of an lst1Δ mutant to grow on acidic medium when provided with extra copies of SEC24 on either centromeric or 2μ plasmids (Fig. 3 B). These findings imply some functional overlap between LST1 and SEC24. In parallel tests for suppression, we found that the genes SEC12, SEC13, SEC31, or SEC23, when expressed from 2μ plasmids, could not restore the ability of an lst1Δ mutant to grow on acidic medium. We found that the lst1Δ mutation caused a selective defect in the trafficking of Pma1p from the ER, and we also examined the ability of overexpressed SEC24 to suppress this phenotype caused by the lst1Δ mutation. By immunofluorescence microscopy, the proper localization of Pma1p to the cell surface was restored in an lst1Δ strain that also carried SEC24 on a 2μ plasmid (see Fig. 5).
Figure 5.

Pma1p accumulates the ER in lst1Δ cells and this accumulation is suppressed by overexpression of SEC24. Cells grown in SMM at 30°C were fixed with formaldehyde and then stained for immunofluorescence microscopy with affinity-purified anti-Pma1p antibody and FITC-conjugated secondary antibody. The same fields of cells, stained with DAPI to label the nuclear DNA, are also shown. Top panels, montage of lst1Δ cells (CKY536 carrying the empty vector pRS316); middle panels, genotypically wild-type cells (CKY536 carrying the LST1 plasmid pKR17); bottom panels, lst1Δ cells suppressed by SEC24 (CKY536 carrying the 2μ SEC24 plasmid pKR34). Bar, 5 μm.
In an attempt to test the effect of overexpression of LST1, we found that LST1 on a 2μ plasmid severely impaired growth of wild-type yeast cells. To examine the response of cells to different doses of Lst1p, we designed a way to express different levels of Lst1p according to the amount of galactose in the growth medium. A wild-type strain (CKY473) carrying a plasmid that expressed LST1 from pGAL1 (pKR35) was spread on an SMM plate with 2% raffinose, a carbon source that allows yeast growth without repression of the GAL1 promoter. When these cells are exposed to a gradient of galactose concentrations, from 3 mg of galactose in a filter disk on top of the lawn, growth was inhibited in a halo 1.5 cm beyond the edge of the filter (Fig. 3 C). A strain that did not contain pKR35 grew uniformly up to the edge of the filter, showing that the galactose itself was not inhibitory. Given the similarity of Lst1p to Sec24p, we asked whether the overexpression of SEC24 could compensate for overexpression of LST1. Cells carrying both the pGAL1–LST1 plasmid (pKR35) and the SEC24 gene on a 2μ plasmid (pKR41) were tested in an identical halo assay, and were found to be resistant to the effect of galactose (Fig. 3 C). Suppression by SEC24 appeared to be specific, since parallel tests of 2μ plasmids carrying SEC12, SEC13, SEC31, or SEC23 failed to show suppression. It is worth noting that SEC23 expressed from a 2μ plasmid significantly slows the growth of our yeast strains. Any suppression afforded by overexpression of SEC23 might be counteracted by this inherent toxicity of SEC23. A simple conclusion that can be drawn from these overexpression studies is that too great of a stoichiometric excess of Lst1p over Sec24p is lethal. This observation can be explained if Lst1p and Sec24p compete with one another in the assembly of vesicle coat complexes and that excess Lst1p causes sequestration of vesicle components into complexes that fail to satisfy some essential function of COPII.
lst1Δ Diminishes the Activity of the Plasma Membrane Proton-ATPase
The sensitivity of lst1Δ mutants to low pH suggested the involvement of Pma1p, which has been shown to be the limiting cell component for growth on acidic medium (McCusker et al., 1987; Portillo and Serrano, 1989). The dependence of Pma1p activity on LST1 was supported by the observation that lst1Δ mutants exhibited an unusual morphology characteristic of pma1 mutants. When lst1Δ mutants were grown in low pH (SMM or YPD brought to pH 3.8) at 37°C, ∼10% of the cells formed multibudded rosettes; in some cases, as many as 15 daughters radiated from a single large mother cell (Fig. 4 A). The unseparated daughter cells contained nuclei that could be stained with DAPI and the daughter cells could be separated from their mothers by micromanipulation, indicating they had completed cytokinesis. Cells depleted of Pma1p produce similar multibudded cells with attached daughters that had completed cytokinesis. In this case, multibudded rosettes are thought to form because a mother cell formed with sufficient Pma1p in the plasma membrane will continue to bud, whereas daughter cells formed after Pma1p transport is compromised will have insufficient Pma1p to form buds themselves (Cid et al., 1987). The morphology of lst1Δ cells grown at relatively high pH (YPD or SMM buffered to pH 6.5) at 37°C appeared normal, with few cells having more than one attached daughter.
Figure 4.


Pma1p defects caused by lst1Δ. (A) lst1Δ cells (CKY534) were photographed using differential interference contrast microscopy after growth at 37°C on YPD, pH 3.8. A montage of multibudded cells is shown. Cells of this type comprise ∼10% of a lst1Δ culture, but are never seen in wild-type grown under the same conditions. Bar, 10 μm. (B) Reduced capacity for proton pumping by lst1Δ cells. Wild-type (CKY443) and lst1Δ (CKY536) were grown to exponential phase in YPD medium, pH 6.8, at 37°C. Cells were incubated in water overnight and then suspended in 10 mM glycine buffer at pH 4.0. Proton efflux from the cells after addition of glucose was recorded as a decrease in the pH of the external medium. Based on the average rate of change in pH over the first 5 min after glucose addition, lst1Δ cells exhibited 65% the rate of proton efflux as wild-type.
As a more direct test of the effect of lst1Δ on the activity of Pma1p, we measured the capacity of mutant cells to pump protons into the external medium. Wild-type and lst1Δ strains were cultured in YPD at 37°C, conditions under which both strains grow equally well. After starvation by prolonged incubation in water, the cells were placed in a weakly buffered medium and proton efflux on addition of glucose was measured as a drop in extracellular pH. For both wild-type and lst1Δ strains, addition of glucose produced a sharp decline in pH (after a 30-s lag), which began to level off after ∼5 min (Fig. 4 B). Although the responses of wild-type and lst1Δ cells were qualitatively similar, proton efflux from lst1Δ cells was compromised: in the first 5 min after addition of glucose the rate of change in pH produced by the lst1Δ mutant was 65% of that of wild-type. These findings indicate that the lst1Δ mutant grown at 37°C has about half of the Pma1p activity as wild-type cells.
LST1 Is Required for Efficient Transport of Pma1p Out of the ER
To determine whether the reduced Pma1p activity in lst1Δ mutants was due to a defect in the transport of Pma1p to the cell surface, we compared the localization of Pma1p in wild-type and lst1Δ mutant cells by immunofluorescence microscopy. Cells were grown at 30°C in YPD medium to avoid possible secondary effects due to the pH sensitivity of lst1Δ mutants. In lst1Δ cells, Pma1p was located primarily at the nuclear periphery and at the cellular rim, indicating that a large proportion of Pma1p remains in the ER (Fig. 5). This pattern of localization differed markedly from the surface localization of Pma1p in wild-type cells incubated at 30°C (Fig. 5) or in lst1Δ cells incubated at 24°C (data not shown).
We also examined the subcellular distribution of Pma1p in lst1Δ cells by cell fractionation. Lysates from cells grown at 37°C for 3 h were fractionated on sucrose density gradients under conditions where the ER and plasma membrane are well separated on the basis of their buoyant density. Pma1p from wild-type cells was located in dense fractions of the gradient in a peak that was coincident with that of Gas1p, a GPI-linked plasma membrane protein (Nuoffer et al., 1991). In contrast, <35% of the total Pma1p from lst1Δ cells coincided with the plasma membrane marked by Gas1p protein and the majority of Pma1p was located in fractions containing the ER (Fig. 6). Interestingly, the ER from lst1Δ mutants (marked by Sec61p) reproducibly resolved into two peaks of different density, suggesting that accumulation of Pma1p segregates ER membranes into subdomains of relatively high and low density. Given that most of the Pma1p was located in the ER peak of higher density, it is possible that the density of the ER had been increased because of the accumulation of Pma1p. A similar increase in density of a portion of the ER is caused when folding mutants of PMA1 are retained within the ER (Harris et al., 1994).
Figure 6.

Cell fraction to localize Pma1p in lst1Δ cells. Wild-type (CKY443) and lst1Δ (CKY536) cells were grown in YPD at 24°C and then were shifted to 37°C for 3 h. Cell lysates were fractionated on density gradients of 20–60% sucrose. Relative levels of Pma1p, Gas1p (plasma membrane marker), and Sec61p (ER marker) in each fraction were quantitated by immunoblotting and densitometry. GDPase (Golgi compartment marker) was determined by enzymatic assay.
The fact that transport of Pma1p, but not of Gas1p, was affected by deletion of LST1 suggested that LST1 may be specifically required for the export of Pma1p from the ER. The absence of a general protein secretion defect in lst1Δ mutants was implied by the normal growth of lst1Δ mutants at 37°C in medium of pH 6.5 (the doubling time of both lst1Δ and wild-type was 1.75 h in YPD), indicating a normal rate of expansion of the plasma membrane. As a specific test for the rate of ER to Golgi transport, pulse– chase experiments were performed to follow the rate of maturation of invertase from its core glycosylated ER form to the Golgi and secreted forms. No delay in invertase transport was observed in lst1Δ mutants that had been grown at 37°C for 3 h, conditions that caused the accumulation of Pma1p (Fig. 7). Similarly, no defect in the maturation of carboxypeptidase Y from the ER form to the Golgi and vacuolar forms of the enzyme could be detected (data not shown).
Figure 7.

Transport of invertase is not affected by lst1Δ. Wild-type (CKY540), lst1Δ (CKY542), and sec12-4 (CKY541) strains expressing invertase from the constitutive pTPI1–SUC2 fusion, were grown to exponential phase at 24°C in SMM medium, pH 6.5, without methionine. Wild-type and lst1Δ strains were shifted to 37°C, grown for 3 h, and the sec12-4 (CKY541) strain was shifted to 37°C 5 min before labeling. Cells were pulse-labeled with [35S]methionine and cysteine for 5 min and then chased by the addition of an excess of unlabeled methionine and cysteine. Invertase was immunoprecipitated from labeled extracts and resolved by SDS-PAGE. Positions of the core glycosylated ER form and mature Golgi and secreted forms of invertase are indicated.
We also considered the possibility that transport of Pma1p may be particularly sensitive to any subtle defect in vesicle formation. We addressed this possibility by examining the localization of Pma1p in sec24-1 and sec31-2 mutant cells at the semipermissive temperature of 28°C. Although the growth rate of both mutants was compromised at this temperature (doubling time on YPD: 2.9 h for sec24 and 2.4 h for sec31, as compared with 1.7 h for wild-type), no accumulation of Pma1p was detected in the perinuclear region of either mutant by immunofluorescence microscopy (data not shown). Thus, partial defects in COPII functions did not lead to the extensive accumulation of Pma1p in the ER that was observed for lst1Δ mutants. Taken together, comparisons between the lst1Δ mutation and COPII gene mutations indicate that the lst1Δ mutation is unusual in its ability to inhibit Pma1p exit from the ER without interfering with the transport of other cargo proteins.
Localization of Lst1p
To examine the intracellular distribution of Lst1p, an epitope-tagged derivative was constructed by inserting six copies of the 10–amino acid HA near the NH2 terminus of Lst1p. The HA-tagged LST1 was functional, as demonstrated by its ability to complement lst1-1 in a sectoring assay, and to restore the ability of a lst1Δ mutant to grow on acidic medium at 37°C (not shown). In cells expressing Lst1p-HA that were fixed for immunofluorescence microscopy, staining was found primarily at the nuclear periphery (Fig. 8). No signal was seen in cells expressing untagged Lst1p, verifying that the origin of the staining pattern was due to Lst1p-HA. Although Lst1p-HA staining largely coincided with the ER marker Kar2p, there were subtle differences in their patterns of localization: Kar2p appeared uniformly, distributed around the nuclear periphery, whereas Lst1p-HA staining had a more punctate appearance indicating that Lst1p might be concentrated in particular regions of the ER. In addition, weak punctate staining was observed throughout the cell body, some of which may correspond to ER membranes near the cell periphery.
Figure 8.

Immunolocalization of Lst1p-HA. CKY535 (MATa lst1Δ::LEU2 leu2-3,112 ura3-52 [pKR17HA]) expressing Lst1p-HA from a centromeric plasmid was fixed and labeled with mouse anti-HA, FITC-conjugated anti–mouse antibodies, rabbit anti-Kar2p, and rhodamine-conjugated anti–rabbit antibodies. Nuclear DNA was visualized by DAPI staining. Cell bodies were visualized by differential interference contrast microscopy (DIC). Bar, 1 μm.
The intracellular distribution of Lst1p was also examined by subcellular fractionation. Cells expressing Lst1p-HA were converted to spheroplasts and then gently lysed. This cell lysate was subjected to differential centrifugation and most of Lst1p-HA was found to pellet at either 500 g or 10,000 g (Fig. 9 A). All of the soluble marker protein Gdh2p (Miller and Magasanik, 1990) was found in the 150,000 g supernatant fraction, indicating complete cell lysis (data not shown). The association of the Lst1p protein with the sedimenting fraction was analyzed by chemical treatment of cell lysates before centrifugation at 50,000 g. Incubation of cell extracts in 1% Triton X-100, 2.5 M urea, 100 mM sodium carbonate, pH 11.5, or 500 mM NaCl resulted in the release of a portion of the Lst1p-HA into the soluble fraction (Fig. 9 B). The partial dissociation of Lst1p-HA from the sedimenting fraction by these agents suggested that Lst1p is a peripheral membrane protein that adheres tightly to the membrane.
Figure 9.

The intracellular distribution of Lst1p. (A) Cells expressing Lst1p-HA from a centromeric plasmid (CKY535) were gently lysed and subjected to sequential centrifugation steps, giving 500 g, 10,000 g, and 150,000 g pellet fractions (P) and a 150,000 g supernatant fraction (S). Each sample contains extract from the same number of cells. (B) Cell lysates were treated for 1 h at 4°C with either 2.5 M urea, 500 mM NaCl, 100 mM sodium carbonate (pH 11.5), or 1% Triton X-100. Pellet (P) and supernatant (S) fractions were then separated by centrifugation at 50,000 g. Lst1p-HA was detected by SDS-PAGE and immunoblotting with anti-HA antibody.
Lst1p Binds Sec23p
Sec24p was first identified as a protein that formed a 400-kD complex with Sec23p (Hicke et al., 1992). Because of the similarity of Lst1p to Sec24p, we investigated whether Lst1p could also bind to Sec23p. To assay potential interactions by the yeast two-hybrid assay, LST1 was fused to the lexA DNA-binding domain (pKR37) and SEC23 was fused to an acidic activation domain (pPE81). Interaction between the two fusion proteins was tested by assaying for activation of a lacZ reporter gene. Induction of β-galactosidase was observed when the LST1 and SEC23 fusions were coexpressed, but not when expressed alone (Table IV). The level of induction caused by interaction of LST1 and SEC23 was similar to that seen for interaction of SEC24 and SEC23 (Gimeno et al., 1996).
Table IV.
Two-Hybrid Interaction between LST1 and SEC23
| β-galactosidase activity | ||||||
|---|---|---|---|---|---|---|
| LST1 | SEC24 | No fusion | ||||
| SEC23 | 395 ± 8 | 629 ± 1 | 24.4 ± 0.1 | |||
| No fusion | 30.4 ± 4.0 | 25.3 ± 1.4 | 44.8 ± 5.3 | |||
Fusions to the LexA DNA-binding domain and to a transcriptional activation domain were induced by growth in galactose for 10 h. Activities shown are the mean from five independent transformants. Units of β-galactosidase activity are nmol/mg × min.
To confirm the interaction between Lst1p and Sec23p, association of these proteins was examined in yeast cell extracts. The coding sequence of LST-1-HA (codons 14–927) was fused to GST and expressed in yeast from the pGAL1 promoter. SEC23 was also expressed from pGAL1. Since both proteins are largely associated with intracellular membranes (Fig. 10 B), membranes prepared from cells overexpressing both Sec23p and GST–Lst1p-HA were first extracted with 600 mM NaCl to release protein complexes from the membrane, the salt extracts were clarified by centrifugation at 90,000 g, and diluted to give a final concentration of 200 mM NaCl. GST–Lst1p-HA was isolated from the extracts by affinity to glutathione Sepharose beads. Sec23p was found in association with GST–Lst1p-Ha, but not in control extracts prepared from cells expressing Sec23p and GST alone (Fig. 10 A). Together, these experiments show that Lst1p, like Sec24p, can form a complex with Sec23p.
Figure 10.

Lst1p/–Sec23p complex is membrane associated. (A) Affinity isolation of Lst1p/–Sec23p complexes. GST–Lst1p-HA or GST alone was coexpressed with Sec23p and isolated by affinity to glutathione Sepharose beads. Proteins bound to glutathione beads were loaded in lanes 2 and 4. One-sixth of the total lysate was loaded in lanes 1 and 3. (B) GST–LST1-HA, SEC23, or both were expressed from the GAL1 promoter. Cell lysates were cleared of cell debris by centrifugation at 300 g for 2 min. Pellet (P) and supernatant (S) fractions from cleared cell lysates were separated by centrifugation at 10,000 g for 30 min. An aliquot of the total cleared lysate (T) was removed before centrifugation. An equal number of cell equivalents were loaded for each sample. The GST–Lst1p-HA fusion was detected using anti-HA antibodies. For both A and B, the Sec23p protein was detected using anti-Sec23p antibodies.
Sec23p and Sec24p have been shown to assemble onto the ER membrane as a complex (Matsuoka et al., 1998). While working out conditions to optimize recovery of Sec23p bound to GST–Lst1p-HA, we discovered that assembly of an Lst1p/Sec23p complex appears to enhance the association of both proteins with the ER membrane. When both GST–Lst1p-HA and Sec23p were overexpressed in the same cell, >60% of the Sec23p, and 70% of the GST–Lst1p-HA were found in a fraction that pelleted at 10,000 g (Fig. 10 B). This pellet contains most of the ER, as marked by the ER membrane protein Sec61p (data not shown). When material that pelleted at 10,000 g was suspended in 60% sucrose and applied to the bottom of a sucrose density gradient, >90% of the GST– Lst1p-HA and Sec23p cofractionated with the ER resident membrane protein, Sec61p, at a density corresponding to 45% sucrose, showing that GST–Lst1p-HA and Sec23p were associated with membranes (data not shown). In contrast to the case when Sec23p and GST–Lst1p-HA were expressed together, <10% of the Sec23p pelleted at 10,000 g in lysates from a strain overexpressing Sec23p alone. Similarly, <20% of the GST–Lst1p-HA pelleted at 10,000 g in lysates from a strain expressing GST–Lst1p-HA alone (Fig. 10 B). Thus, when either Sec23p or GST–Lst1p-HA was overexpressed alone, most of the overexpressed protein was soluble, but when the proteins were expressed together, most of the proteins were associated with the ER membranes. These data support the observation that Lst1p can form a complex with Sec23p, and that the Lst1p/ Sec23p complex has affinity for ER membranes.
Discussion
By screening for mutants that exhibited synthetic-lethal genetic interactions with the COPII mutation sec13-1, we identified the LST1 gene. Subsequent genetic tests showed that lst1Δ is lethal when combined with mutations in genes required for COPII vesicle budding from the ER (SEC12, SEC13, SEC16, SEC23, SEC24, and SEC31), but lst1Δ is not lethal when combined with mutations in genes that are required for vesicle fusion with the Golgi compartment (SEC17 and SEC18). This pattern of genetic interactions indicated that LST1 participates in the process of vesicle budding from the ER, an expectation that was born out by the examination of the LST1 gene and its product. The following observations indicate a role for Lst1p as part of a COPII-like vesicle coat: (I) LST1 encodes a 90-kD protein that is homologous to the COPII-coat subunit Sec24p. The two proteins share 23% amino acid identity over their entire lengths. (II) Lst1p is a peripheral ER membrane protein as shown by immunofluorescence microscopy and cell fractionation. (III) Lst1p, like Sec24p, can bind to Sec23p as shown by tests for two-hybrid interaction and affinity purification of a complex of GST–Lst1p and Sec23p. (IV) Assembly of the Sec23p–Lst1p complex appears to enhance the membrane association of both Lst1p and Sec23p: when both proteins are overexpressed together, most associate with membranes, whereas either protein overexpressed alone is mostly cytosolic. (V) Although strains with chromosomal deletion of LST1 are viable and appear normal for secretion of marker proteins, these mutants show a pronounced accumulation of Pma1p in the ER, indicating a selective defect in ER to Golgi traffic. Based on these findings, we propose that Lst1p takes the place of Sec24p in a specialized COPII coat complex that is used for the recruitment of Pma1p into vesicles.
Strains carrying lst1Δ have the phenotypic hallmarks of a deficiency in Pma1p activity, including sensitivity to growth in an acidic environment, the formation of multibudded cells, and a decreased rate of proton efflux from intact cells. All three traits are expressed only at temperatures of 30°C and above, indicating that LST1 is only required for Pma1p activity at high temperature. Localization of Pma1p in lst1Δ cells by immunofluorescence and sucrose density cell fractionation demonstrate that the transport of Pma1p from the ER is compromised in lst1Δ at 37°C.
Export of Pma1p from the ER cannot be completely dependent on Lst1p, since Pma1p transport appears normal in lst1Δ mutants at 24°C. Even at 37°C, the block in Pma1p transport may not be complete since ∼35% of the total Pma1p fractionates with the plasma membrane, although some of the Pma1p detected in the plasma membrane in this experiment was probably synthesized before the shift to restrictive temperature. Therefore, it seems likely that Lst1p and Sec24p share the burden of transporting Pma1p from the ER. At 24°C, it appears that Sec24p (or some other protein) can compensate for the absence of Lst1p, but at temperatures of 30°C or higher, compensation is no longer possible unless extra copies of Sec24p are provided by expression from a multicopy plasmid.
The transport defect caused by deletion of LST1 appears to be specific for Pma1p. Under conditions where a defect in Pma1p transport was observed in lst1Δ mutants, transport of Gas1p, carboxypeptidase Y, and invertase was unaffected. Using growth as a more general assay for trafficking defects, we found that lst1Δ mutants grew at an identical rate to wild-type at 37°C when we compensated for the defect in Pma1p transport by using media at pH 6.5. This indicates that rate of expansion of the plasma membrane, including the transport of all essential plasma membrane proteins, is not significantly affected by the absence of LST1.
We also considered the possibility that there may be differences among cargo molecules in their response to general defects in the protein transport machinery. Of particular concern was the possibility that Pma1p transport might be particularly sensitive to slowed ER to Golgi transport, such that a defect in transport too subtle to have an effect on our standard marker proteins might have a significant effect on the rate of transport of Pma1p. If this were the case, partial defects in other COPII components should also interfere with Pma1p transport. Therefore, we examined sec24 and sec31 mutants, but could find no evidence for a defect in Pma1p transport, even at semipermissive temperatures where the rate of growth was inhibited. Although Pma1p was the only essential protein for which we could detect a transport defect in lst1 mutants, a defect in the transport of any nonessential protein could have been overlooked by our analysis.
Factors required for the transport of specific membrane proteins have been documented in a number of other cases. The SHR3 gene encodes an ER resident protein that is required for the transport of amino acid permeases out of the ER, but is not required for the transport of a variety of other proteins (Ljungdahl et al., 1992; Kuehn et al., 1996). A set of ER proteins, Vma12p, Vma21p, and Vma22p, are required for transport from the ER of the integral membrane subunit of the vacuolar ATPase (Hill and Stevens, 1994, 1995; Jackson and Stevens, 1997). Similarly, mutational studies have shown that the small ER membrane protein Erv14p is specifically required for transport of the plasma membrane protein Axl2p out of the ER (Powers and Barlowe, 1998). Finally, Ast1p has been suggested to be a factor specifically needed for the transport of Pma1p from the Golgi compartment to the plasma membrane (Chang and Fink, 1995). In all of these cases, the question remains whether Shr3p, the Vma proteins, Erv14p, or Ast1p act directly in vesicular transport of their respective cargo molecules, or whether they are primarily involved in protein folding and influence protein sorting indirectly through quality control mechanisms. Because Lst1p appears to be a component of a vesicle coat, Lst1p seems more likely to have a direct role in the sorting of Pma1p rather than in its folding.
Expression of a variety of dominant PMA1 mutations can cause accumulation of both mutant and wild-type Pma1p in proliferated ER (Harris et al., 1994; Portillo, 1997). Similarly, the transport of wild-type Pma1p from the ER is blocked when PMA2 (an isoform of PMA1) or plant plasma membrane proton-ATPases are overexpressed in yeast (Villalba et al., 1992; Supply et al., 1994; de Kerchove d'Exaerde et al., 1995). One proposal was that a special factor may be required for the transport of Pma1p from the ER in a manner analogous to the requirement for Shr3p in the transport of amino acid permeases (Supply et al., 1994). The specific role of Lst1p in the transport of Pma1p suggests that it may be the factor depleted by the expression of dominant forms of Pma1p. In the future, it may be possible to test this idea by evaluating the ability of Lst1p overexpression to reverse the effects of dominant PMA1 mutations.
The mechanism by which Lst1p acts in the transport of Pma1p may be inferred from recent studies examining the recruitment of cargo molecules into COPII vesicles. Using ER-derived microsomes and purified COPII components, Kuehn et al. (1998) have shown that the Sec23p/Sec24p complex, along with Sar1p, associate with amino acid permeases and other integral membrane protein that are destined for the plasma membrane. In parallel experiments using mammalian microsomes, mammalian Sec23p/Sec24p and Sar1p were found to bind to microsomal membranes and form a complex that contains the cargo protein VSV-G (Aridor et al., 1998). The conclusion from both experimental systems is that the Sec23p/Sec24p complex contains specific binding sites for the capture of membrane cargo proteins within the plane of the ER membrane. Based on the data presented here, Lst1p appears to be an isoform of Sec24p that is adapted for selection of Pma1p. This provides the first evidence that Sec24 family members carry information specifying the type of cargo molecules that are accepted by ER-derived vesicles.
We have looked for association of Lst1p with ER-derived vesicles, but under the conditions of an in vitro budding reaction, a large quantity of Lst1p-HA is released from the membrane in soluble form. Soluble Lst1p-HA gives a high background in vesicle fractions preventing us from reliably determining whether there is a specific association of Lst1p with vesicles. In future experiments, it may be possible to isolate vesicles coated with Lst1p by performing an in vitro budding reaction using purified cytosolic components, including a purified complex of Lst1p and Sec23p. It may also be possible to determine whether vesicles that are formed using a Sec23p/Lst1p complex more efficiently incorporate Pma1p than vesicles formed using the Sec23p/Sec24p complex. Finally, it will be of interest to determine if there is direct binding of Lst1p to Pma1p.
The identification of a Sec24p homologue that also acts in transport from the ER raises the possibility that the coats of ER-derived vesicles may be heterogeneous. It is possible that Sec23p/Lst1p complexes act to form a class of vesicle that is distinct from those formed by Sec23p/ Sec24p complexes. Alternatively, it is possible that the two complexes assemble together forming vesicles with coats of mixed composition. The identification of additional homologues of Sec23p and Sec24p suggest the existence of coats with even greater combinatorial complexity. We have identified a third Sec24p family member, which we call Iss1p, as a protein that binds to Sec16p. Iss1p (YNL049c) also binds Sec23p and appears to be associated with the ER membrane (Gimeno, 1996). In addition, the Saccharomyces genome contains an uncharacterized open reading frame (YHR035w) that is 21% identical to Sec23p (Saccharomyces Genome Database, Cherry et al., 1997). If each of the Sec23p and Sec24p homologues carry different determinants for cargo selection, and if mixed coats can form, the possible combinations of Sec23p and Sec24p homologues should allow the formation of a wide variety of COPII-like vesicles with different capacities to carry different cargo molecules.
Acknowledgments
We thank the members of the Kaiser lab for their technical assistance, advice, and encouragement, and especially we thank A. Frand for providing pAF70. We thank A. Chang, M. Rose, B. Magasanik, R. Schekman, and H. Riezman for their generous gifts of antibodies, and M. Lewis for providing the pNV31 plasmid. We also thank R. Schekman for providing us with the sequence of Sec24p before publication.
This work was supported by a grant from the National Institute of General Medical Sciences and the Searle Scholars Program to C.A. Kaiser, a National Institutes of Health predoctoral traineeship to K.J. Roberg, a National Science Foundation predoctoral fellowship to P. Espenshade, and a Merck predoctoral fellowship to R.E. Gimeno. C.A. Kaiser is a Lucille P. Markey Scholar, and this work was funded in part by the Lucille P. Markey Charitable Trust.
Abbreviations used in this paper
- DAPI
4′,6-diamidino-2-phenylindole
- GST
glutathione S-transferase
- HA
hemagglutinin epitope
- LST
lethal with sec-thirteen
- Pma1p
plasma membrane proton-ATPase
- SMM
supplemented minimal medium
- YPD
rich medium
References
- Abeijon C, Orlean P, Robbins PW, Hirschberg CB. Topography of glycosylation in yeast: characterization of GDP mannose transport and lumenal guanosine diphosphatase activities in Golgi-like vesicles. Proc Natl Acad Sci USA. 1989;86:6935–6939. doi: 10.1073/pnas.86.18.6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M, Weissman J, Bannykh S, Nuoffer C, Balch WE. Cargo selection by the COPII budding machinery during export from the ER. J Cell Biol. 1998;141:61–70. doi: 10.1083/jcb.141.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C, Schekman R. SEC12encodes a guanine-nucleotide- exchange factor essential for transport vesicle budding from the ER. Nature. 1993;365:347–349. doi: 10.1038/365347a0. [DOI] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Bartel PL, Fields S. Analyzing protein–protein interactions using two-hybrid system. Methods Enzymol. 1995;254:241–263. doi: 10.1016/0076-6879(95)54018-0. [DOI] [PubMed] [Google Scholar]
- Brada D, Schekman R. Coincident localization of secretory and plasma membrane proteins in organelles of the yeast secretory pathway. J Bacteriol. 1988;170:2775–2783. doi: 10.1128/jb.170.6.2775-2783.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Slayman CW. Maturation of the yeast plasma membrane [H+]ATPase involves phosphorylation during intracellular transport. J Cell Biol. 1991;115:289–295. doi: 10.1083/jcb.115.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Fink GR. Targeting of the yeast plasma membrane [H+]ATPase: a novel gene AST1prevents mislocalization of mutant ATPase to the vacuole. J Cell Biol. 1995;128:39–49. doi: 10.1083/jcb.128.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry, J.M., C. Adler, C. Ball, S. Dwight, S. Chervitz, Y. Jia, G. Juvik, S. Weng, and D. Botstein. 1997. Saccharomyces Genome Database. http:// genome-www.stanford.edu/Saccharomyces/ [DOI] [PMC free article] [PubMed]
- Cid A, Perona R, Serrano R. Replacement of the promoter of the yeast plasma membrane ATPase gene by a galactose-dependent promoter and its physiological consequences. Curr Genet. 1987;12:105–110. doi: 10.1007/BF00434664. [DOI] [PubMed] [Google Scholar]
- de Kerchove d'Exaerde, A., P. Supply, J.P. Dufour, P. Bogaerts, D. Thines, A. Goffeau, and M. Boutry. Functional complementation of a null mutation of the yeast Saccharomyces cerevisiaeplasma membrane H(+)-ATPase by a plant H(+)-ATPase gene. J Biol Chem. 1995;270:23828–23837. doi: 10.1074/jbc.270.40.23828. [DOI] [PubMed] [Google Scholar]
- Elledge SJ, Mulligan JT, Ramer SW, Spottswood M, Davis RW. Lambda YES: a multifunctional cDNA expression vector for the isolation of genes by complementation of yeast and Escherichia colimutations. Proc Natl Acad Sci USA. 1991;88:1731–1735. doi: 10.1073/pnas.88.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod-Erickson MJ, Kaiser CA. Genes that control the fidelity of endoplasmic reticulum to Golgi transport identified as suppressors of vesicle budding mutations. Mol Biol Cell. 1996;7:1043–1058. doi: 10.1091/mbc.7.7.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espenshade P, Gimeno RE, Holzmacher E, Teung P, Kaiser CA. Yeast SEC16gene encodes a multidomain vesicle coat protein that interacts with Sec23p. J Cell Biol. 1995;131:311–324. doi: 10.1083/jcb.131.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno, R.E. 1996. Proteins that interact with Sec16p during COPII vesicle formation in Saccharomyces cerevisiae. Ph.D. thesis. Massachusetts Institute of Technology, Cambridge, Massachusetts. 215 pp.
- Gimeno RE, Espenshade P, Kaiser CA. SED4encodes a yeast endoplasmic reticulum protein that binds Sec16p and participates in vesicle formation. J Cell Biol. 1995;131:325–338. doi: 10.1083/jcb.131.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno RE, Espenshade P, Kaiser CA. COPII coat subunit interactions: Sec24p and Sec23p bind to adjacent regions of Sec16p. Mol Cell Biol. 1996;7:1815–1823. doi: 10.1091/mbc.7.11.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golemis EA, Brent R. Fused protein domains inhibit DNA binding by LexA. Mol Cell Biol. 1992;12:3006–3014. doi: 10.1128/mcb.12.7.3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyuris J, Golemis E, Chertkov H, Brent R. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Harris SL, Na S, Zhu X, Seto YD, Perlin DS, Teem JH, Haber JE. Dominant lethal mutations in the plasma membrane H+-ATPase gene of Saccharomyces cerevisiae. . J Biol Chem. 1994;266:24439–24445. doi: 10.1073/pnas.91.22.10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz I, Jensen RE. Putting the HO gene to work: practical uses for mating-type switching. Methods Enzymol. 1991;155:132–146. doi: 10.1016/0076-6879(91)94011-z. [DOI] [PubMed] [Google Scholar]
- Hicke L, Yoshihisa T, Schekman R. Sec23p and a novel 105-kDa protein function as a multimeric complex to promote vesicle budding and protein transport from the endoplasmic reticulum. Mol Biol Cell. 1992;3:667–676. doi: 10.1091/mbc.3.6.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill KJ, Stevens TH. Vma21p is a yeast membrane protein retained in the endoplasmic reticulum by a di-lysine motif and is required for the assembly of the vacuolar H+-ATPase complex. Mol Biol Cell. 1994;5:1039–1050. doi: 10.1091/mbc.5.9.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill KJ, Stevens TH. Vma22p is a novel endoplasmic reticulum- associated protein required for assembly of the yeast vacuolar H+-ATPase complex. J Biol Chem. 1995;270:22329–22336. doi: 10.1074/jbc.270.38.22329. [DOI] [PubMed] [Google Scholar]
- Jackson DD, Stevens TH. VMA12 encodes a yeast endoplasmic reticulum protein required for vacuolar H+-ATPase assembly. J Biol Chem. 1997;272:25928–25934. doi: 10.1074/jbc.272.41.25928. [DOI] [PubMed] [Google Scholar]
- Jones JS, Prakash L. Yeast Saccharomyces cerevisiaeselectable markers in pUC18 polylinkers. Yeast. 1990;6:363–366. doi: 10.1002/yea.320060502. [DOI] [PubMed] [Google Scholar]
- Kaiser CA, Schekman R. Distinct sets of SECgenes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]
- Kaiser, C.A., S. Michaelis, and A. Mitchell. 1994. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Kuehn MJ, Schekman R, Ljungdahl P. Amino acid permeases require COPII components and the ER resident membrane-protein Shr3p for packaging into transport vesicles in vitro. J Cell Biol. 1996;135:585–595. doi: 10.1083/jcb.135.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn MJ, Herrmann JM, Schekman R. COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Science. 1998;391:187–190. doi: 10.1038/34438. [DOI] [PubMed] [Google Scholar]
- Lawrence CW. Classical mutagenesis techniques. Methods Enzymol. 1991;155:132–146. doi: 10.1016/0076-6879(91)94021-4. [DOI] [PubMed] [Google Scholar]
- Ljungdahl PO, Gimeno CJ, Styles CA, Fink GR. SHR3: a novel component of the secretory pathway specifically required for localization of amino acid permeases in yeast. Cell. 1992;71:463–478. doi: 10.1016/0092-8674(92)90515-e. [DOI] [PubMed] [Google Scholar]
- Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically-defined liposomes. Cell. 1998;93:263–275. doi: 10.1016/s0092-8674(00)81577-9. [DOI] [PubMed] [Google Scholar]
- McCusker JH, Perlin DS, Haber JE. Pleiotropic plasma membrane ATPase mutations of Saccharomyces cerevisiae. . Mol Cell Biol. 1987;7:4082–4088. doi: 10.1128/mcb.7.11.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S, Magasanik B. Role of NAD-linked glutamate dehydrogenase in nitrogen metabolism in Saccharomyces cerevisiae. . J Bacteriol. 1990;172:4927–4935. doi: 10.1128/jb.172.9.4927-4935.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuoffer C, Jeno P, Conzelmann A, Riezman H. Determinants for glycophospholipid anchoring of the Saccharomyces cerevisiaeGAS1 protein to the plasma membrane. Mol Cell Biol. 1991;11:27–37. doi: 10.1128/mcb.11.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portillo F. Characterization of dominant lethal mutations in the yeast plasma-membrane H+-ATPase gene. FEBS Lett. 1997;402:136–140. doi: 10.1016/s0014-5793(96)01515-3. [DOI] [PubMed] [Google Scholar]
- Portillo F, Serrano R. Growth control strength and active site of yeast plasma membrane ATPase studied by site-directed mutagenesis. Eur J Biochem. 1989;186:501–507. doi: 10.1111/j.1432-1033.1989.tb15235.x. [DOI] [PubMed] [Google Scholar]
- Powers J, Barlowe C. Transport of axl2p depends on erv14p, an ER-vesicle protein related to the Drosophilacornichon gene product. J Cell Biol. 1998;142:1209–1222. doi: 10.1083/jcb.142.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle JR, Adams AEM, Drubin DG, Haarer BK. Immuno-fluorescence methods for yeast. Methods Enzymol. 1991;194:565–602. doi: 10.1016/0076-6879(91)94043-c. [DOI] [PubMed] [Google Scholar]
- Pryer NK, Salama NR, Schekman R, Kaiser CA. Cytosolic Sec13p complex is required for vesicle formation from the endoplasmic reticulum in vitro. J Cell Biol. 1993;120:865–875. doi: 10.1083/jcb.120.4.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberg KJ, Rowley N, Kaiser CA. Physiological regulation of membrane protein sorting in the Golgi of Saccharomyces cerevisiae. . J Cell Biol. 1997a;137:1469–1482. doi: 10.1083/jcb.137.7.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberg KJ, Bickel S, Rowley N, Kaiser CA. Control of amino-acid permease sorting in the late secretory pathway of Saccharomyces cerevisiae by SEC13, LST4, LST7, and LST8. . Genetics. 1997b;147:1569–1584. doi: 10.1093/genetics/147.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M, Botstein D. Construction and use of gene fusions lacZ(β-galactosidase) which are expressed in yeast. Methods Enzymol. 1983;101:167–180. doi: 10.1016/0076-6879(83)01012-5. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Serrano, R. 1991. Transport across yeast vacuolar and plasma membranes. In The Molecular and Cellular Biology of the Yeast Saccharomyces: Genome Dynamics, Protein Synthesis, and Energetics. J.R. Broach, E.W. Jones, and J.R. Pringle, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 523–585.
- Serrano R, Kieland-Brandt MC, Fink GR. Yeast plasma membrane ATPase is essential for growth and has homology with (Na+ + K+), K+- and Ca2+-ATPase. Nature. 1986;319:689–693. doi: 10.1038/319689a0. [DOI] [PubMed] [Google Scholar]
- Shaywitz DA, Espenshade PJ, Gimeno RE, Kaiser CA. COPII subunit interactions in the assembly of the vesicle coat. J Biol Chem. 1997;272:25413–25416. doi: 10.1074/jbc.272.41.25413. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuffle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply P, Wach A, Thines-Sempoux D, Goffeau A. Proliferation of intracellular structures upon overexpression of the PMA2 ATPase in Saccharomyces cerevisiae. . J Biol Chem. 1994;268:19753–19759. [PubMed] [Google Scholar]
- Thompson CM, Koleske AJ, Chao DM, Young R. A multisubunit complex associated with the RNA polymerase II CTD and TATA-binding protein in yeast. Cell. 1993;73:1361–1375. doi: 10.1016/0092-8674(93)90362-t. [DOI] [PubMed] [Google Scholar]
- Tyers M, Tokiwa G, Futcher B. Comparison of the Saccharomyces cerevisiaeG1 cyclins: Cln3 may be an upstream activator of Cln1, Cln2, and other cyclins. EMBO (Eur Mol Biol Organ) J. 1993;12:1955–1968. doi: 10.1002/j.1460-2075.1993.tb05845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo CG, Serrano R. Physiology of mutants with reduced expression of plasma membrane H+-ATPase. Yeast. 1989;5:307–319. doi: 10.1002/yea.320050411. [DOI] [PubMed] [Google Scholar]
- Villalba JM, Palmgren MG, Berberian GE, Ferguson C, Serrano R. Functional expression of plant plasma membrane H(+)-ATPase in yeast endoplasmic reticulum. J Biol Chem. 1992;267:12341–12349. [PubMed] [Google Scholar]